
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImaging of nanoparticle uptake and kinetics of intracellular trafficking in individual cells†
Natalia
Vtyurina
a,
Christoffer
Åberg
 *b and
Anna
Salvati
*a
*b and
Anna
Salvati
*a
aDepartment of Nanomedicine & Drug Targeting, Groningen Research Institute of Pharmacy, University of Groningen, Antonius Deusinglaan 1, 9713AV Groningen, the Netherlands. E-mail: a.salvati@rug.nl
bDepartment of Pharmaceutical Analysis, Groningen Research Institute of Pharmacy, University of Groningen, Antonius Deusinglaan 1, 9713AV Groningen, the Netherlands. E-mail: christoffer.aberg@rug.nl
First published on 25th May 2021
Abstract
Live cell imaging is a powerful tool to understand how nano-sized objects, such as the drug carriers used for nanomedicine applications, are taken up and trafficked by cells. Here we visualized human HeLa cells as they took up and trafficked nanoparticles of different sizes and quantified nanoparticle colocalization with different fluorescently-labelled intracellular compartments over time. This allowed us to obtain kinetic profiles of nanoparticle transport towards the lysosomes in individual cells. With a simple theoretical model, we determined the typical departure time of nanoparticles from the cell membrane and typical lysosome arrival time. We compared these kinetics parameters for nanoparticles of different sizes and determined how they vary in individual cells. We also performed a similar analysis for early endocytic compartments through which nanoparticles transit and discuss challenges in quantifying the colocalization in this case. The results show a high variability in intracellular trafficking kinetics between individual cells. Additionally, they help us to understand how nanoparticle properties affect their cellular uptake and intracellular distribution kinetics.
Introduction
Nanomedicines must successfully overcome multiple extracellular barriers throughout their journey in the body before reaching their target. Once at their target, intracellular barriers can limit their efficacy: these include the cell membrane, and – following uptake – the early sorting and trafficking compartments, among others.1–4 Even after targeted uptake and efficient internalization, other aspects related to their intracellular trafficking can also affect nanomedicine efficacy. These include their intracellular distribution and the kinetics of the processes involved, such as, for instance, the time between uptake and arrival to a certain intracellular location.2,5Unlike small molecules that access the cytosol via active transporters or passive diffusion across the cell membrane, nanoparticles enter cells by energy-dependent endocytic mechanisms.6–8 Following endocytosis, they are enclosed into endosomal compartments and are typically trafficked to the lysosomes.1,7,9–11 However, the details of the uptake mechanisms and intracellular processes are often not clarified, including how nanoparticle properties affect them.1,12 This knowledge can help the design of targeted nanomedicines.2,5,13
Within this context, we aimed at gaining a better understanding of nanoparticle behaviour at the subcellular level by visualizing living cells as they internalized and trafficked nanoparticles. Several methods can be used to study transport inside cells, each presenting some advantages and limitations. Many methods rely on perturbing transport in cells in different ways, for instance using chemical inhibitors or by silencing or knocking down the expression of key proteins involved in uptake and transport.1,7,14,15 However, the perturbation may induce a different response of the cells compared to what is observed under physiological conditions.1,15 In contrast, live cell imaging allows following uptake and intracellular transport of nanoparticles in cells expressing fluorescent markers, but otherwise unperturbed.9,16 Furthermore, imaging live cells takes advantage of observing the processes of interest using both spatial and temporal information.17 For instance, Vercaturen et al. used dynamic colocalization of nanoparticles with different intracellular compartments to characterize the mechanism of uptake of polyplexes and their intracellular trafficking,16 while in the work of Sandin et al. different Rab GTPase proteins were labeled in HeLa cells to monitor trafficking of carboxylated polystyrene nanoparticles along early and late endosomal compartments.9
In line with these studies, here we expressed key fluorescently labeled markers involved in uptake and intracellular trafficking in live cells to visualize nanoparticles as they enter cells and are trafficked to their final destination in the lysosomes. By acquiring rapid dual-colour 3D confocal fluorescence microscopy images of the same cells over time, we determined the kinetics of nanoparticle uptake and trafficking through early to late stages of endocytosis in individual cells.
Additionally, by applying a simple theoretical model we determined the typical nanoparticle departure time from the membrane and the typical time required to arrive in the lysosomes and compared the results for nanoparticles of different sizes. By quantifying colocalization in the same (live) cells over time, we have been able to determine relevant intracellular rate constants in individual cells and study their variability. Thus, we used live cell imaging not only to determine whether nanoparticles are internalized and trafficked to certain compartments, but also to obtain the kinetics of the processes involved and analyze how these vary in individual cells and as a function of nanoparticle properties, such as size.
Results and discussion
Experimental procedure and panel of fluorescent markers
Fluorescence microscopy was used to follow nanoparticles in live cells over time and determine colocalization kinetics in a number of individual cells (up to 20). HeLa cells were selected as a common model for transport studies, widely used in both the endocytosis and nanomedicine fields.7,9,10,18 For imaging, so-called HeLa Kyoto cells are often preferred, as they are more resistant and show less displacement over time, making time-lapse acquisition easier.9,19,20 It has been shown that HeLa cells grown in different laboratories (such as the HeLa Kyoto) can change phenotype over time.21,22 Interestingly, this can affect also their intracellular trafficking behaviour, as observed for EGFR uptake.23 Thus, for comparison both HeLa and HeLa Kyoto cells were imaged after exposure to nanoparticles.In order to set up the method, carboxylated polystyrene nanoparticles were selected as a model, because of their high fluorescence, resistance to photobleaching and stability in biological media.9,24–26 As a first step, in order to probe how nanoparticle properties affect intracellular distribution kinetics, nanoparticles of different sizes were used. Size is one of the key parameters that can be modulated in nanomedicine design, together with many other nanoparticle properties, such as material, charge, hydrophobicity, stiffness, as well as surface functionalization and addition of different types of surface ligands. In order to form a human biomolecular corona appropriate for testing on human cells, the nanoparticles were dispersed in medium containing 4 mg ml−1 human serum, roughly corresponding to the amount of proteins in standard cell culture medium supplemented with 10% bovine serum.27,28 This is an excess of proteins compared to the available nanoparticle surface area (ESI Table S1†). Dynamic light scattering confirmed that for all nanoparticles homogeneous dispersions were obtained in both PBS and the cell culture medium with serum (Fig. S1 and Table S2†).
In order to visualize different intracellular compartments, cells were labelled or transfected with constructs to express fluorescently labelled proteins of interest. Next, cells were exposed to nanoparticles of different sizes (40, 100 or 200 nm) in the form of a short “pulse” (of 5, 10, and 15 min, respectively) after which the cells were “chased” by acquiring 3D images at various times and these were analysed to determine colocalization (Fig. 1A illustrates this workflow). More in detail, cells were labelled with LysoTracker to stain the acidic compartments of the cells (mainly lysosomes, together with late endosomes and multivesicular bodies) and determine intracellular trafficking kinetics to the lysosomes. Indeed, after active uptake via different endocytic mechanisms, most nanoparticles are trafficked via the endosomes towards the lysosomes, as illustrated in Fig. 1B.1,7,9,11 Additionally, cells were transfected with a series of endocytic and membrane trafficking proteins carrying fluorescent tags. These included Rab5 and EEA1 to visualize the early endosomes; and clathrin light chain, caveolin-1 and pMyrPalm to stain different endocytic compartments (also depicted in Fig. 1B, together with examples of images of cells with the different labels). This selection was made to monitor eventual association of the nanoparticles with clathrin, caveolae or macropinosomes (pMyrPalm) and gain some information on the potential uptake mechanism.29,30
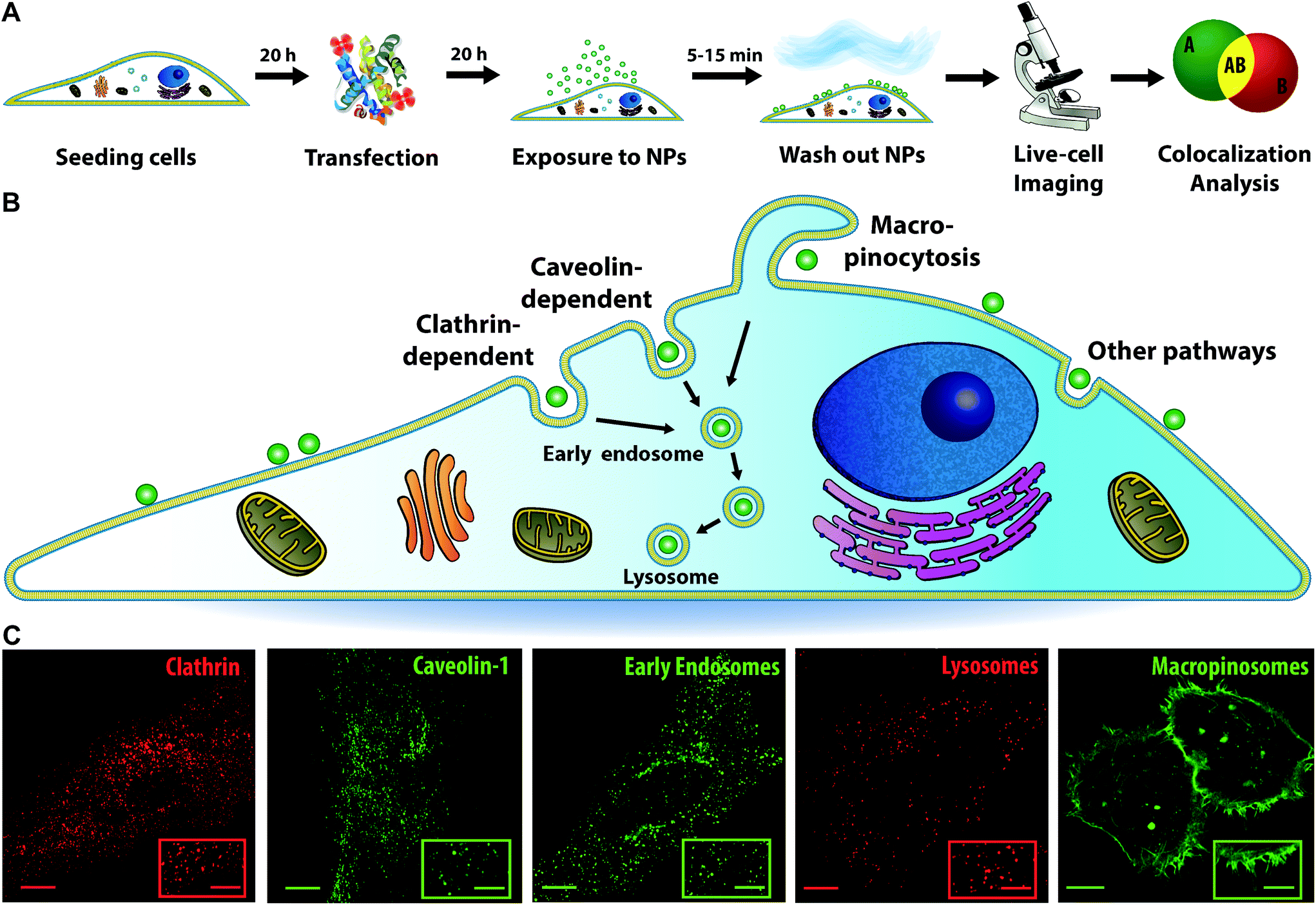 | ||
| Fig. 1 Experimental procedure and panel of key fluorescent markers involved in major endocytic pathways and membrane trafficking. (A) Cells were transfected with the plasmid of interest carrying a fluorescent tag or stained with LysoTracker. Next, cells were exposed to a “pulse” of nanoparticles, and “chased” over time by acquiring 3D images of individual cells, followed by colocalization analysis. (B) Schematic representation of a cell exposed to nanoparticles (green spheres), showing different uptake pathways (clathrin- and caveolin-dependent, macropinocytosis and other clathrin- and caveolin-independent pathways), and nanoparticle trafficking through an early endosome towards a lysosome. (C) Images of cells labelled with the selected panel of fluorescent markers (the insets show enlarged views). From left to right: HeLa cells expressing clathrin (mRFP-Clc), caveolin-1 (Cav1-mEGFP), Rab5 to stain early endosomes (Rab5-GFP), a cell labelled with LysoTracker to stain cell acidic compartments (mainly lysosomes) and HeLa Kyoto cells expressing pMyrPalm to visualize macropinocytosis. Scale bar: 10 μm (main) and 5 μm (inset). | ||
Quantitative analysis of nanoparticle uptake and intracellular trafficking to the lysosomes
As a first step, we followed nanoparticle transport to LysoTracker-positive compartments (for simplicity hereafter referred to as lysosomes). Rapid 3D time-lapse images of several individual cells were recorded every 10–20 min for up to 6 h (Fig. 2A). The LysoTracker channel was used to detect cell borders (Fig. 2B) and generate a 3D mask for each individual cell (Fig. 2C). This mask was applied to both the lysosome and nanoparticle channels (Fig. 2D) to exclude adjacent cells and eventual clusters of nanoparticles adsorbed to the glass close to the cell membrane. Then, nanoparticles and lysosomes were identified as spherical objects (Fig. 2E–G), followed by colocalization analysis to determine the fraction of nanoparticle objects colocalized with lysosomes (Fig. 2H). Determining colocalization in this way from 3D stacks acquired every 10–20 minutes allowed us to follow the same cells, live, over time for up to 6 h, thus monitoring the whole process of nanoparticle accumulation towards the lysosomes. This is in contrast to trajectory-based colocalization,16 which requires much faster and continuous illumination, usually associated with photo-toxicity.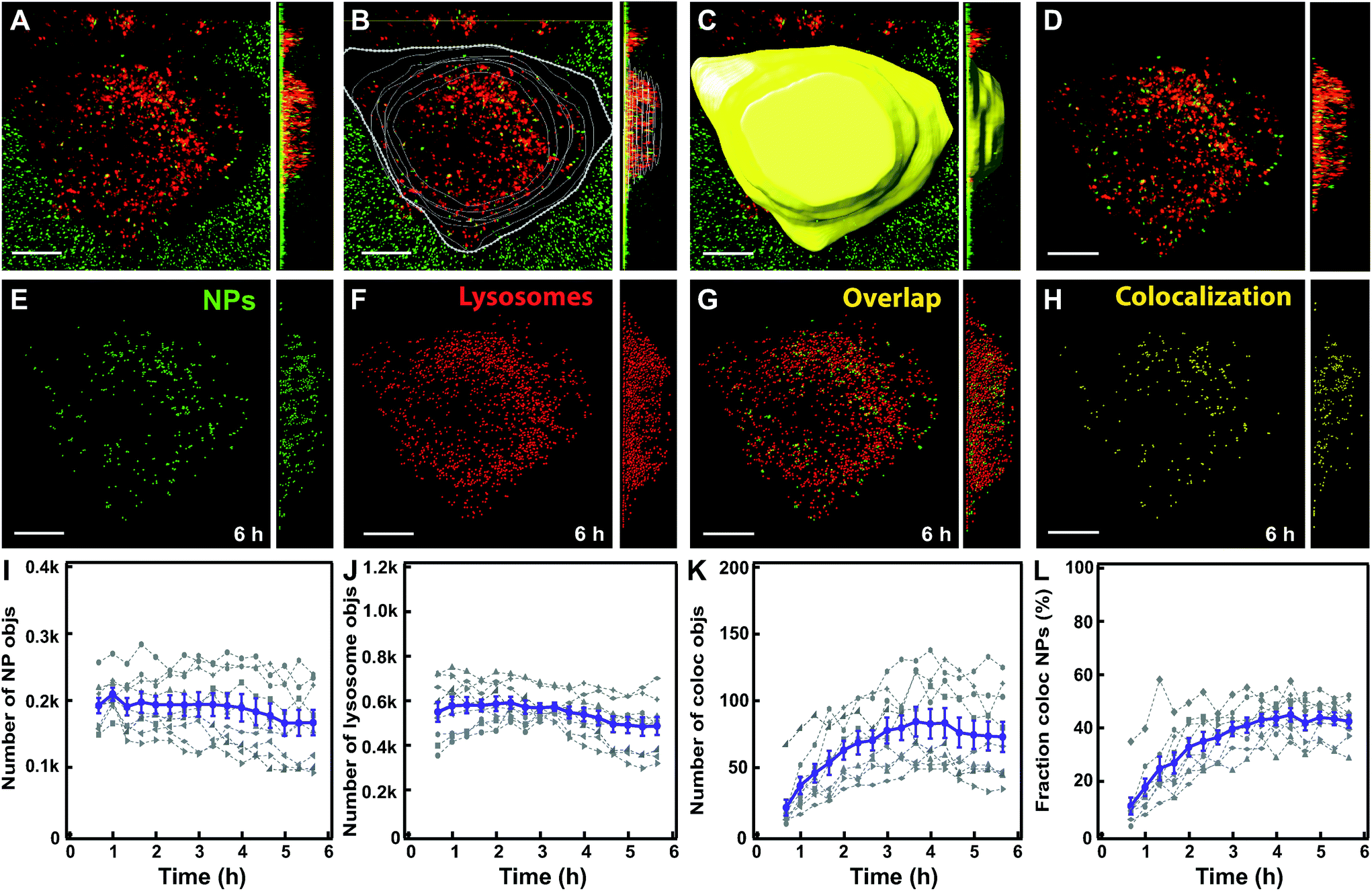 | ||
| Fig. 2 Workflow of image analysis to determine nanoparticle colocalization with lysosomes. (A) HeLa Kyoto cell with lysosomes stained by LysoTracker (red) and exposed for 10 min to 300 μg ml−1 of 100 nm carboxylated polystyrene nanoparticles (green). An example of an xy image and, on the right, the yz projection across the full cell volume of the raw 3D stack of images are shown. (B) Cell contours were manually defined based on the LysoTracker channel. (C) A 3D cell mask was generated. (D) The cell mask was applied to the nanoparticle and LysoTracker fluorescence channels to define the region of interest for the analysis. (E) Detected nanoparticle objects (green). (F) Detected lysosome objects (red). (G) Overlap of nanoparticle objects (green) and lysosome objects (red). (H) Detected nanoparticle objects that colocalize with lysosome objects (yellow). (I) Number of detected nanoparticle objects over time. (J) Number of detected lysosome objects. (K) Number of nanoparticle objects colocalized with lysosome objects. (L) Fraction of nanoparticle objects colocalized with lysosome objects. The results for individual cells are shown in grey and the mean and standard error over 8 cells are shown in purple. Scale bar 10 μm. Additional results for all individual cells are shown in Fig. S5.† | ||
Note also that what was identified as a nanoparticle “object” may contain several nanoparticles too close to be resolved optically. Movie S1† shows an example of a 3D reconstruction of a cell and the analysis performed and Fig. 2I–L show the data generated in 8 individual cells and their average.
The results showed that the number of nanoparticle objects and lysosomes remained relatively stable over the 6 h imaging period (Fig. 2I and J), confirming that with optimized imaging settings photobleaching was minimal. Instead, the number and fraction of nanoparticle objects colocalizing with lysosomes increased over time and plateaued within 6 h (Fig. 2K and L). This confirmed nanoparticle trafficking to the lysosomes, as previously observed for these and most other nanoparticles.1,7,9,10
Kinetics of nanoparticles arriving to lysosomes and cell-to-cell variability
We repeated this procedure to evaluate the trafficking of 40, 100 and 200 nm nanoparticles to the lysosomes in HeLa and HeLa Kyoto cells (Fig. 3 and 4 and Fig. S2–S9;† note in particular that the ESI figures† show the results for all individual cells). We started with 100 nm nanoparticles in HeLa Kyoto cells (Fig. 3A) and attempted to fit a simple model to the results from individual cells. Denoting the number of nanoparticles at the membrane and in the lysosomes at time t by Nm (t) and Nl (t), respectively, we modelled the kinetics of nanoparticles arriving to lysosomes with the following simple differential equation system:31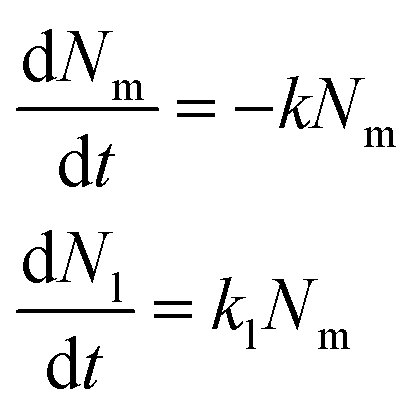 | (1) |
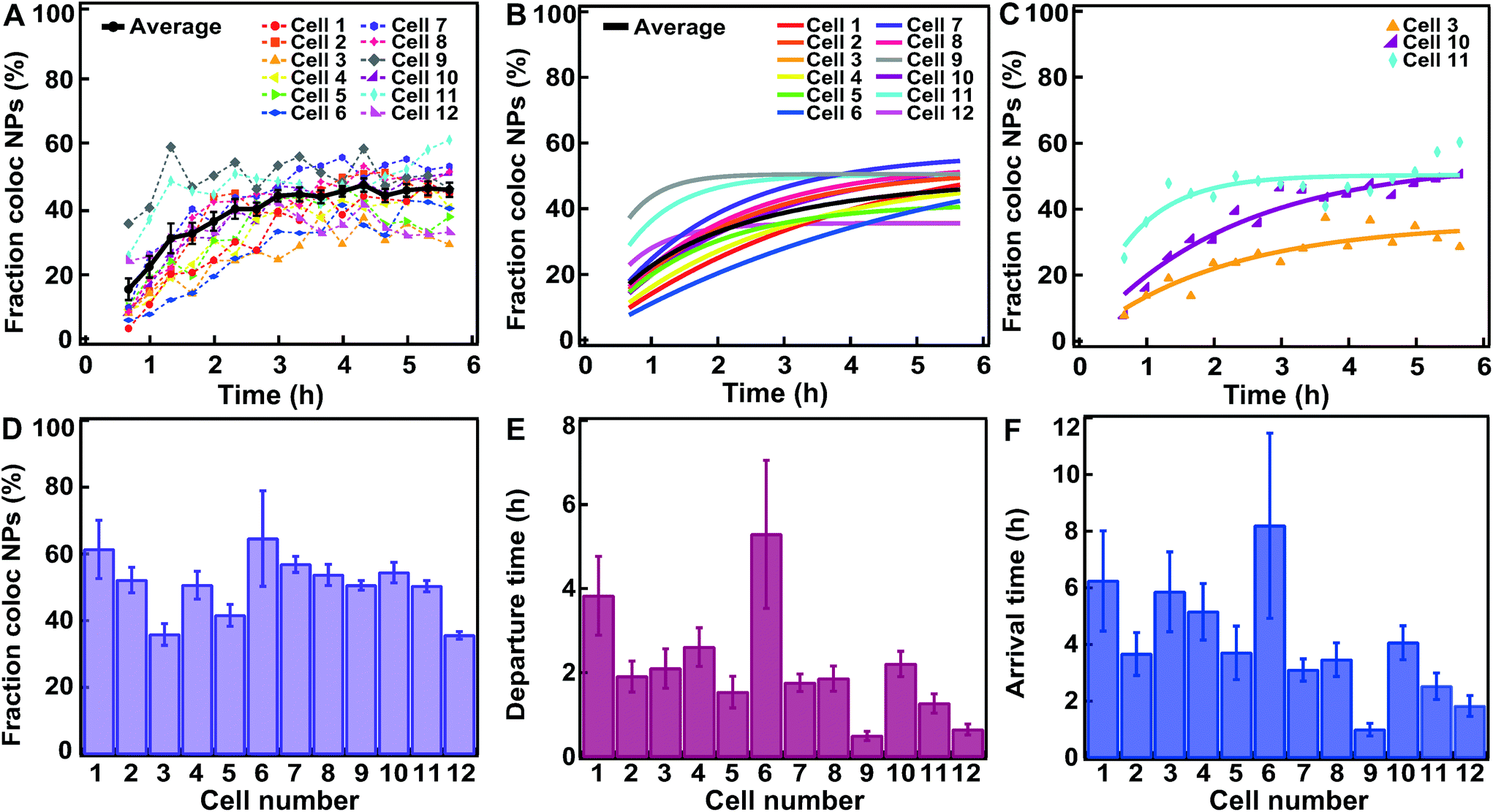 | ||
| Fig. 3 Fitting a kinetic model to data for HeLa Kyoto cells exposed for 10 min to 300 μg ml−1 of 100 nm carboxylated polystyrene nanoparticles. (A) Fraction of nanoparticle objects colocalized with lysosome objects in individual cells (colors) and averaged over cells (black). (B) Fit of eqn (2) to the fraction of nanoparticle objects colocalized with lysosome objects in individual cells (colored) and averaged over all cells (black). Averages show the mean and standard error over 12 individual cells. (C) Three example fits of eqn (2) (solid line) to fraction of nanoparticle objects colocalized with lysosome objects (symbols) in individual cells. (D) Fraction of nanoparticle objects that ultimately end up colocalized with lysosome objects (fl) from the fits to individual cells shown in B. (E) Departure time of nanoparticle objects from the membrane (τ) from the fits shown in B. (F) Nanoparticle object arrival time to lysosomes (τl) calculated from the results shown in D and E. Error bars in panel D and E are uncertainties from the fit, while error bars in panel F are uncertainties propagated from those of fl and τ using Gauss’ formula. Additional results for all individual cells are shown in Fig. S5.† | ||
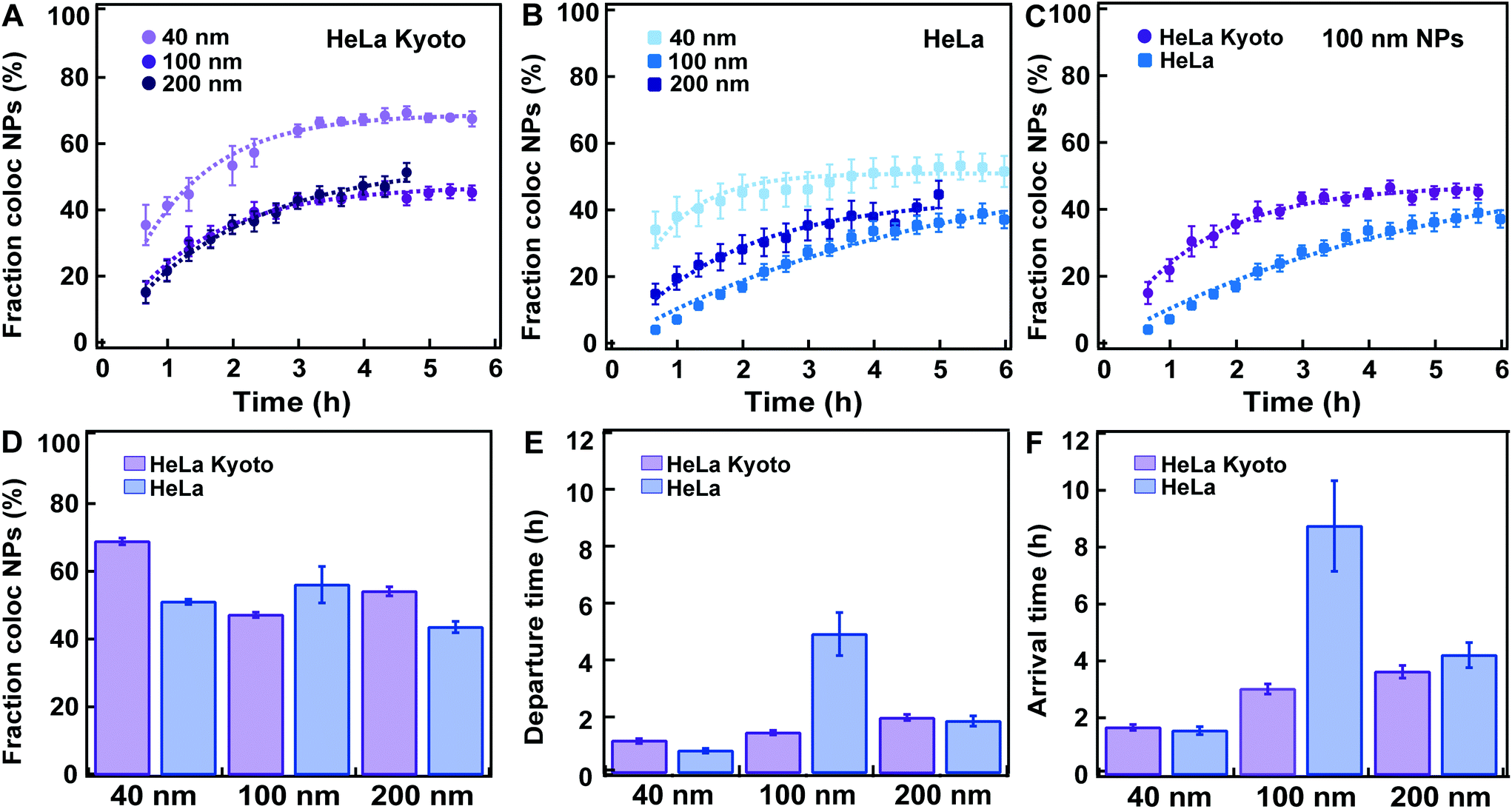 | ||
| Fig. 4 Fitting a kinetic model to data for HeLa Kyoto and HeLa cells exposed to 40, 100 and 200 nm carboxylated polystyrene nanoparticles. HeLa and HeLa Kyoto cells were exposed to 40, 100 and 200 nm carboxylated polystyrene nanoparticles and stained with LysoTracker, the cells were “chased” and nanoparticle colocalization with lysosomes was calculated (see Experimental section for details). Averages show the mean and standard error over various numbers of individual cells. (A) Average fraction of nanoparticle objects colocalized with lysosome objects: 40 (light purple), 100 (purple), 200 nm (dark purple) in HeLa Kyoto cells and corresponding model fits (dashed lines) (mean ± SE (n = 8–16)). (B) Average fraction of nanoparticle objects colocalized with lysosome objects: 40 (light blue), 100 (blue), 200 nm (dark blue) in HeLa cells (mean ± SE (n = 6–8)) and corresponding model fits (dashed lines). (C) Average fraction of nanoparticle objects colocalized with lysosome objects in HeLa Kyoto (purple circles) and HeLa (blue squares) cells exposed to 100 nm nanoparticles. (D) Fraction of nanoparticle objects that ultimately end up colocalized with lysosome objects fl extracted from the fits to averaged HeLa Kyoto (purple) and HeLa (blue) kinetic profiles shown in A and B. (E) Departure time of nanoparticle objects from the membrane (τ) extracted from the fits to averaged HeLa Kyoto (purple) and HeLa (blue) cell results shown in A and B. (F) Nanoparticle object arrival time to lysosomes (τl) calculated from the results shown in D and E. Error bars in panel D and E are uncertainties from the fit, while error bars in panel F are uncertainties propagated from those of fl and τ using Gauss’ formula. The results for all individual cells are shown in Fig. S4–S9.† | ||
Here k is the rate constant for the departure of nanoparticles from the membrane via all potential processes, while kl is the rate constant for a nanoparticle at the membrane arriving to a lysosome. We expected these two rate constants to be different, as not all nanoparticles at the membrane arrived to the lysosomes (Fig. 3A).
Given that cells were exposed to a “pulse” of nanoparticles, we made the idealized assumption that at t = 0 all nanoparticles were at the membrane and none in the lysosomes. Under this assumption, the solution to eqn (1) can be written as
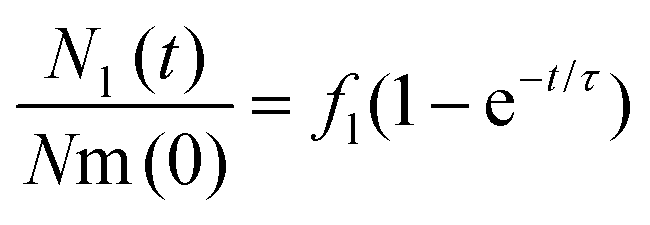 | (2) |
The left-hand side of this equation is the fraction of nanoparticles in the lysosomes as a function of time (Fig. 3A). It is characterized in terms of two parameters, namely fl, which is the fraction of nanoparticles that ultimately end up in the lysosomes (fl = kl/k) and τ, which is the typical time it takes nanoparticles to leave the membrane (τ = k−1). Thus, in this simple model the time-dependence is given at membrane level (in terms of τ or k) rather than along the endolysosomal pathway (in terms of kl). Fitting this simple model (eqn (2)) to data demonstrated that this model provides a good description of the data (Fig. 3A–C; and Fig. S5†) and it does so not only for the kinetics averaged over all cells, but also for individual cells.
From the fits, we also found the fraction of nanoparticles that ultimately end up in the lysosomes (fl; Fig. 3D) and the typical membrane departure time (τ; Fig. 3E). Compared to the latter, the more interesting parameter to describe the accumulation of the particles in lysosomes is the typical lysosome arrival time (τl = kl−1), which we can also calculate (τl = τ/fl). The typical lysosome arrival time τl (Fig. 3F) generally demonstrated the same trend as the membrane departure time (Fig. 3E). We also observed that the fraction of nanoparticles accumulating in the lysosomes, while certainly variable, was at least relatively uniform over the investigated cells (Fig. 3D). In contrast, the typical membrane departure (τ; Fig. 3E) and lysosomal arrival times (τl; Fig. 3F) were highly variable.
We note that we characterized the kinetics with a single transport step from membrane to lysosome. In a more extended model one could include further steps, for example, transport from membrane to early endosomes, from early endosomes to late endosomes and from late endosomes to lysosomes.31 In such an extended model, our current description would be an approximation. However, this simpler description was sufficient to describe the acquired data.
Kinetics of nanoparticle arrival to lysosomes for nanoparticles of different sizes
Similar data sets as shown in Fig. 3A were acquired for both HeLa Kyoto and HeLa cells labelled with LysoTracker and exposed to 40, 100 and 200 nm carboxylated polystyrene nanoparticles (Fig. 4 and Fig. S2–S9,† with the results for all individual cells). Given the different size and uptake efficiencies, cells were exposed to different amounts of nanoparticles for short “pulses” of time in order to allow a comparable number of nanoparticle objects to be detected inside cells, large enough for quantification of colocalization over time (see the Experimental section for details). For all nanoparticle sizes, in good agreement with other similar studies, the results showed an overall steady increase in the accumulation of nanoparticles in the lysosomes.9,16 Nanoparticles colocalizing with lysosomes were observed as early as 40 min after exposure, while a portion of nanoparticle objects was seemingly stuck on the cell edges for 3 h or longer. For all sizes the fraction of colocalized nanoparticle objects reached a plateau only several hours after the “pulse”. In all cases the fraction of nanoparticle objects colocalized with the lysosomes never reached 100%, but plateaued at lower values, between 40% up to a maximum of 70% in the case of the 40 nm nanoparticles in HeLa Kyoto cells (Fig. 4A). The same observation was made even when using cells expressing fluorescently labelled LAMP1 to identify the lysosomes, instead of LysoTracker (Movie S2†) or when different doses were applied (Fig. S2†). The colocalized fraction did not increase further even when waiting for up to 24 h “chase” (Fig. S3 and Movie S3†). Thus, a substantial amount of nanoparticles did not arrive to the lysosomes at all (Fig. 4A–C). Similar results were observed by Vercauteren et al.16 and Sandin et al.9 It will be important to determine precisely whereto these nanoparticles are transported, why some of them end up in a different subcellular compartment, and overall how cells regulate these processes.We then fitted the model to data averaged over all cells for each dataset. In agreement with the observed kinetic profiles (Fig. 4A–C), the fraction of nanoparticles that ultimately ended up in the lysosomes extracted from the applied model (fl) varied as a function of nanoparticle size in both cell lines (Fig. 4D). We stress that the fraction of nanoparticles that ultimately end up in the lysosomes is what one expects to have at the plateau, when the kinetic process is over. The majority of the kinetic curves in Fig. 4A–C have indeed plateaued, but not all. Specifically, we observe that the 100 nm particles in HeLa cells had not yet fully accumulated in the lysosomes when imaging was stopped (Fig. 4C). Nevertheless, the model allows us to extract the value we expect to observe at the plateau.
A variation was also observed for the membrane departure time (τ; Fig. 4E) and arrival time to the lysosomes (τl; Fig. 4F) for the different sizes. It would be interesting to study in more detail how the intracellular trafficking kinetics (membrane departure time and arrival time to the lysosomes) and the fraction of nanoparticles arriving into the lysosomes vary when cells are exposed to different amounts of nanoparticles. This could allow determining – for instance – whether uptake or intracellular trafficking compartments saturate and how the kinetics may be affected. The proposed methods and modelling can be used also to answer this kind of questions.
When comparing the results for the different nanoparticle sizes, the most prominent result appear to be that in HeLa cells, the 100 nm nanoparticles both leave the membrane and arrive in lysosomes far slower than the other sizes (Fig. 4E and F; blue). It is known that different uptake pathways sort the internalized cargoes into different early compartments, presumably with concomitant differences in intracellular processing kinetics, and then, in most cases, they converge to the lysosomes.11 Since it is likely that the nanoparticles of different sizes are internalized by cells via different mechanisms, this could be the underlying reason for the different kinetics. These results further illustrate how important it is for successful nanomedicine design to understand the details of nanoparticle uptake and intracellular trafficking. By changing nanomedicine design one expects not only uptake efficiency to be affected, but also the kinetics of intracellular trafficking and arrival to the lysosomes of the internalized material. However, the slower kinetics for the 100 nm nanoparticles could not be confirmed in HeLa Kyoto cells, where, if anything, there is an increase in both the departure and lysosomal arrival time with size (Fig. 4E and F; purple). It would be interesting to determine whether this comes from a different uptake mechanism in the two cell lines, as observed for instance for EGFR,23 or only a difference in the kinetics of these events for this particular nanoparticle size and type.
When comparing the results obtained in HeLa and HeLa Kyoto cells, no major differences were observed, with the noted exception of the 100 nm nanoparticles. However lysosomal colocalization seemed slightly higher in HeLa Kyoto, especially for the smaller 40 nm nanoparticles (Fig. 4D). We attempted to correlate the values of the kinetic parameters of individual cells with some basic characteristics of the cells. However, we could not find any obvious correlation between the fraction of colocalized nanoparticle objects, departure or arrival times and the amount of detected objects, cell volume and cell area (Fig. S4–9†).
In this context, we stress that we were able to fit the simple model to the kinetic data for individual cells (Fig. 3B and C, Fig. S4–S9†) rather than data averaged over cells. This allows extracting information on individual cell variability, another important aspect to consider. Such efforts would underpin a more detailed understanding of these processes that cannot be achieved at an average cell level. Indeed, average kinetic curves (Fig. 4A–C) implicitly hide a significant variability at the single-cell level, perhaps best appreciated by examining cell-to-cell variability in kinetic parameters (Fig. 3D–F and Fig. S4–S9† with the results on all individual cells). Given the significant effort in measuring the kinetics in individual cells in the way currently done (here or elsewhere),9,16,32–35 development of new methods with higher throughput is warranted. This could be achieved for instance by translating the approaches presented here to high content analysis, a high-throughput imaging method which is finding increasing applications in the nanomedicine field, including first examples of intracellular trafficking studies.36,37
We also note that even at the average cell level there are two conceivable averaging procedures: one may fit to the kinetic curves averaged over cells (Fig. 4A–C) or one may fit to kinetic curves of individual cells (Fig. 3B and C, Fig. S4–S9†) and subsequently average the resulting fitting parameters over cells. We performed both procedures, showing that overall the values and trends were comparable, but that the kinetic parameters extracted from averaged kinetic curves differ numerically from those extracted from individual cells (Fig. 4D–F and Fig. S10†). This is hardly surprising, but is nevertheless something to keep in mind when interpreting averaged kinetic curves.
Kinetic profiles of nanoparticles colocalizing with clathrin, caveolin-1 and Rab5
Similar imaging experiments and colocalization analysis were performed also on cells expressing markers of earlier compartments where nanoparticles may transit prior to arrival to the lysosomes. Thus, we used the same workflow (Fig. 2) to data sets acquired for HeLa Kyoto cells expressing fluorescently labeled clathrin, caveolin-1 and Rab5 exposed to 100 nm nanoparticles (Fig. 5).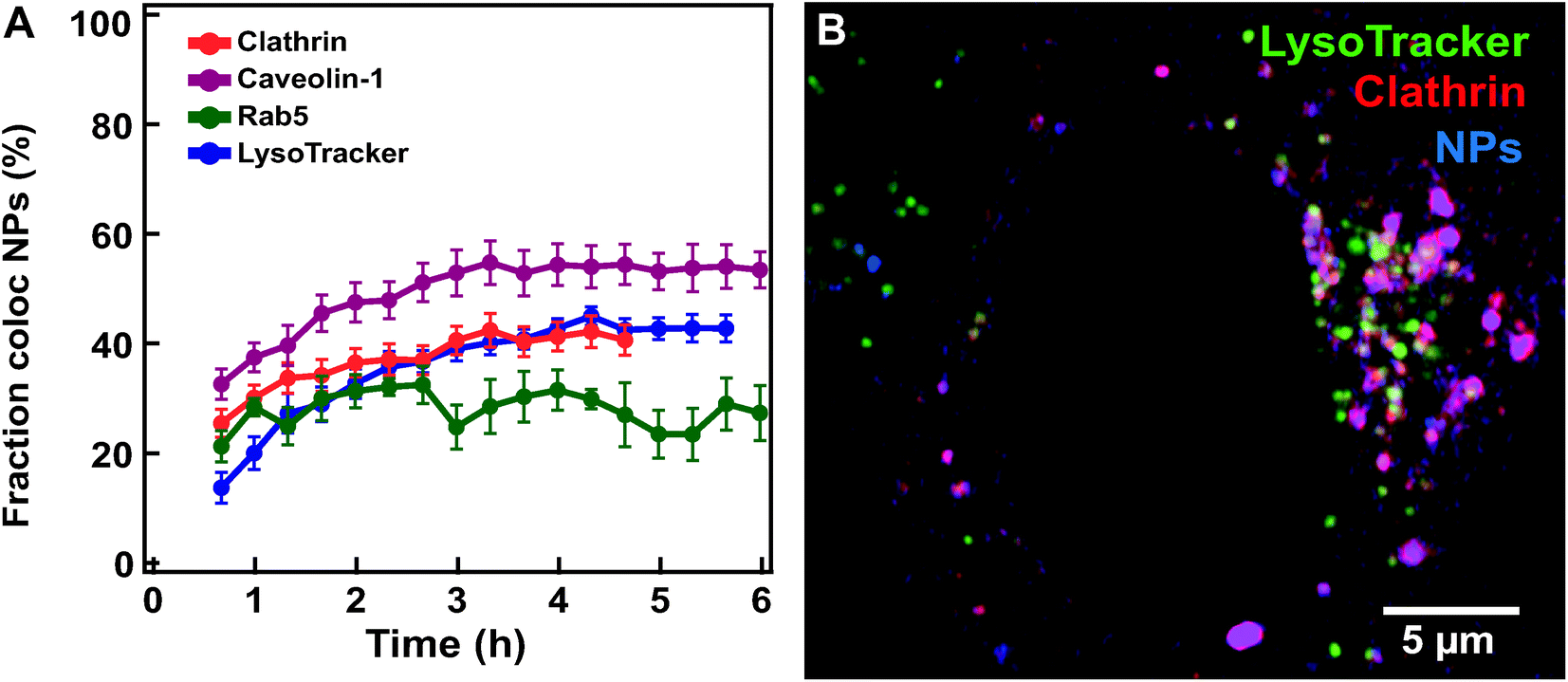 | ||
| Fig. 5 Nanoparticle colocalization with early compartments (labelled by clathrin, caveolin-1, Rab5 and LysoTracker. (A) Averaged fraction of nanoparticle objects colocalized with clathrin (red) (mean ± SE (n = 20)), caveolin-1 (purple) (n = 8), Rab5 (green) (n = 5) and lysosome objects (blue) (n = 12) in HeLa Kyoto cells exposed for 10 min to 300 μg ml−1 100 nm carboxylated polystyrene nanoparticles and then “chased”. (B) Confocal image of a HeLa Kyoto cell expressing fluorescently labelled clathrin (mRFP-Clc, red) with LysoTracker stained lysosomes (green) 6 h after exposure for 5 min to 75 μg ml−1 of 40 nm carboxylated polystyrene nanoparticles (blue). The results for all individual cells are shown in Fig. S11.† | ||
Nanoparticles were observed to populate all these compartments within the first few hours, later reaching an apparent plateau at time scales comparable to those observed for arrival to the lysosomes (Fig. 5A and; Fig. S11† for the results in all individual cells. Movie S4† shows an example of a cell expressing labelled clathrin). This is a known limitation when using similar fluorescent constructs.38 With these constructs, the total concentration of the protein of interest can become much larger than its endogenous level and overexpressed markers are transported into the lysosomes, as indeed confirmed by co-staining (Fig. 5B and Movie S4†).
To avoid such complications, it is preferable to ensure that the protein of interest is expressed from its genomic locus under its native promoter, for instance using CRISPR/Cas9 genome editing.39,40 To overcome this limitation, we restricted the colocalization analysis to the objects located at the cell edges by manually removing in 3D the interior of the cells where most lysosomes are found (Fig. 6A and B for HeLa Kyoto cells expressing labelled clathrin exposed to 100 nm nanoparticles; see extended Experimental section in ESI for details†). The fraction of nanoparticles colocalized with clathrin now remained stable over 6 h at around 20% (Fig. 6C and Fig. S12† for the results in all individual cells).
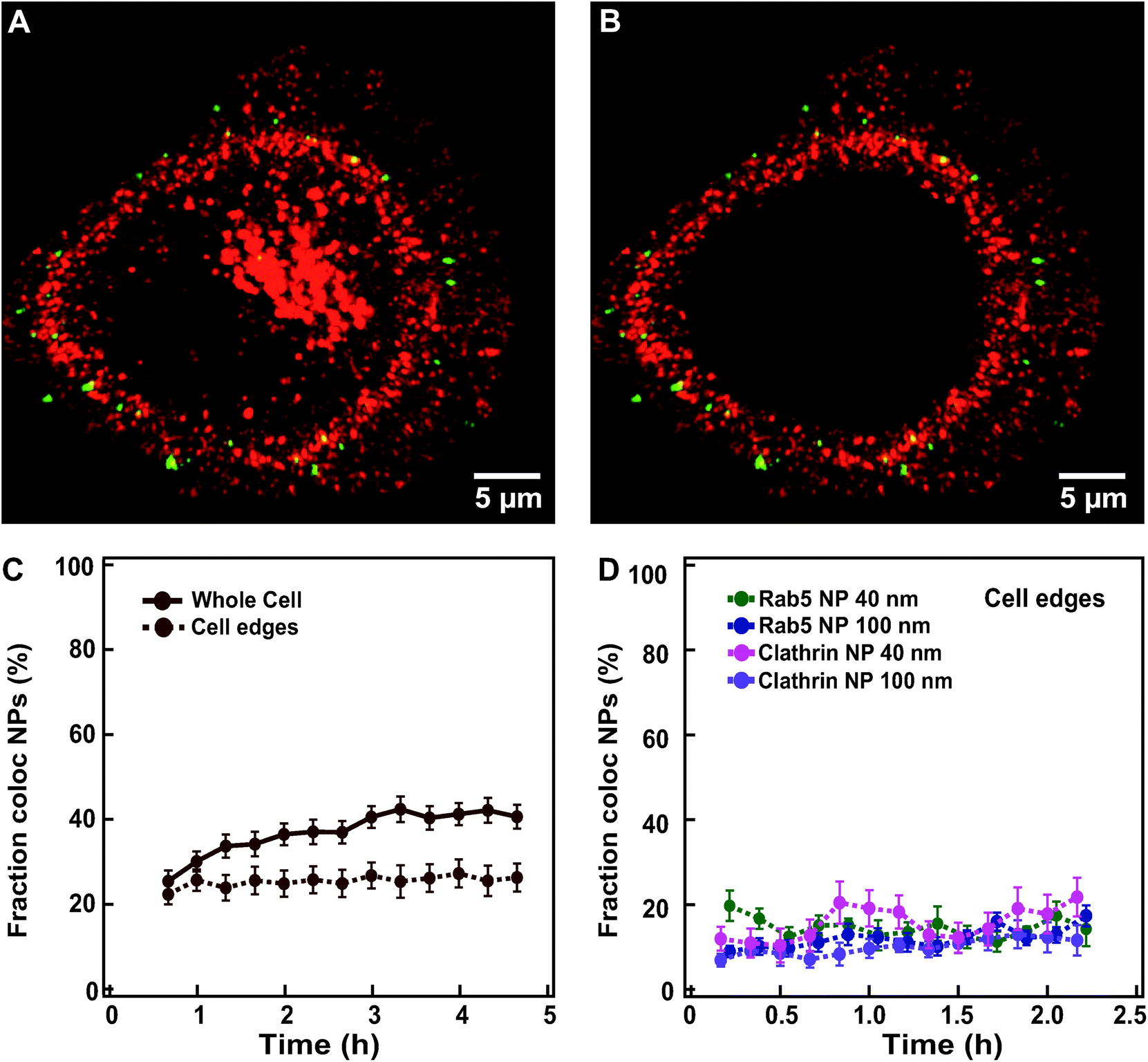 | ||
| Fig. 6 Overcoming overexpression limits when quantifying colocalization between nanoparticles and labelled endocytic proteins. (A) Example of an xy view of the 3D z-stack image of a HeLa Kyoto cell expressing fluorescently labelled clathrin (mRFP-Clc, red) and exposed for 10 min to 300 μg ml−1 of 100 nm nanoparticles (green). (B) The same cell as in A after removing the inner volume to restrict analysis to the cell borders. (C) Averaged fraction of nanoparticle objects colocalized with clathrin in “whole” HeLa Kyoto cells (solid line) exposed to 100 nm nanoparticles and “cell edges” region (dashed line) (mean ± SE (n = 20)). (D) Averaged fraction of colocalized nanoparticle objects at cell edges in HeLa Kyoto cells expressing fluorescently labelled Rab5 (green) or clathrin (pink) after exposure for 5 min to 75 μg ml−1 of 40 nm nanoparticles; and cells expressing fluorescently labelled Rab5 (blue) and clathrin (purple), after exposure for 10 min to 300 μg ml−1 of 100 nm nanoparticles (mean ± SE (n = 6–9)). Data in panel D are based on images acquired with a DeltaVision microscope (see Experimental section for details). The results for all individual cells are shown in Fig. S12.† | ||
Even when starting imaging earlier after addition of the nanoparticles and sampling every 10 min instead of 20 min, we did not observe a sharp transition of nanoparticles through these early compartments, but rather a stable colocalization at around 20% (Fig. 6D and Fig. S12† for the results in all individual cells). Similar results were also obtained for the 40 nm nanoparticles and cells expressing fluorescently labelled clathrin and Rab5 (Fig. 6D), suggesting that some nanoparticles are still present in these compartments even two hours after exposure. Additionally, movies acquired at faster rate in the first minutes of chase clearly showed some colocalization events with all of the markers tested, including clathrin, caveolin-1, pMyrPalm to follow macropinocytosis, and Rab5 and EEA1 for early endosomes (Movies S5–9† show an overview of typical events in labelled cells exposed to nanoparticles), suggesting that some nanoparticles do transit all of these compartments. A closer inspection of the images recorded also showed that events of colocalization of nanoparticles with these early endocytic markers could be observed both at early (∼15 min) “chase” time, but also at very late “chase” times (∼2.5 h). Some of these events may be related to technical issues, such as nanoparticles still present in the extracellular medium or stuck on the glass desorbing and becoming available for uptake at much later “chase” times. Additionally, some lysosomes are present also at cell edges and thus part of these colocalized objects may still be in the lysosomes. Nevertheless, similar to what was observed for nanoparticle departure time from the membrane or arrival time to the lysosomes, these late colocalization events may also suggest a very heterogeneous behaviour in the uptake events within a cell, with some nanoparticles stuck on the membrane for long time before their actual internalization, as indeed observed in the recorded movies.
The low colocalization observed with multiple endocytic compartments (Fig. 6C and D for clathrin and Movies S5–9† for all markers investigated) may also suggest that within the same cell nanoparticles may be internalized via different pathways. Other studies have reported similar observations.1,9,16,32,41,42 If multiple pathways are present, the source of this variability should be determined, as well as how cells decide which pathway to activate for each individual nanoparticle.
Conclusions
Imaging nanoparticle uptake and intracellular trafficking in live cells allowed us to determine important details of the intracellular kinetics of nanoparticles. Intracellular trafficking and its kinetics are some of the many factors affecting nanomedicine efficacy. As an example of this, we showed how by changing nanoparticle size, the fraction of nanoparticles arriving to the lysosomes and also the kinetics of arrival to lysosomes are affected. The same approaches can be used to investigate how the observed results translate to other cells or different nanoparticles, as well as how other nanoparticle properties aside from size affect intracellular trafficking kinetics and distribution. Additionally, by determining rate constants in individual cells we showed how these parameters can strongly vary in individual cells. Live cell imaging also showed that within the same cells some nanoparticles arrive to lysosomes within 15–30 min, while others require several hours. Similarly, for all nanoparticles, association with markers of different uptake mechanisms was observed and such events were observed early after exposure, as well as several hours later. Although further studies are needed to elucidate the mechanisms behind the observed variability, these are all important aspects that the nanomedicine field needs to tackle in order to gain a deeper understanding of how cells process these objects and to guide the design of nanomedicines with improved efficacy.Experimental section
Cell culture
All cell culture reagents were purchased from Gibco (Thermo Fisher Scientific) unless otherwise specified. The wild type human HeLa cells (Kyoto strain) were obtained from S. Narumiya (Kyoto University, Kyoto, Japan) and validated by a Multiplex human cell line authentication test (MCA). These cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). HeLa cells were purchased from ATCC (CCL-2) and cultured in Minimum Essential Medium (MEM) supplemented with 10% FBS. Cells were tested on a monthly basis to exclude mycoplasma contamination.Nanoparticles
Yellow-green and red carboxylated polystyrene nanoparticles (FluoSpheres) with mean diameter of 0.04 μm (red, green, dark red), 0.1 μm (red, green) and 0.2 μm (red, green) were purchased from Invitrogen (Thermo Fisher Scientific).Expression of fluorescent protein constructs and staining
The following constructs were used for transfecting cells: clathrin light chain mRFP-Clc43 (Addgene plasmids # 14435) and CAV1-mEGFP44 (Addgene plasmids # 27704) were purchased from Addgene. Rab5-GFP45 was kindly provided by J. Smit (Department of Medical Microbiology and Infection Prevention, University Medical Center Groningen). To visualize the acidic compartment of the cells (mainly lysosomes, together with late endosomes and multivesicular bodies), cells were stained with 100 nM LysoTracker Red or Green (Thermo Fisher Scientific) in DMEM supplemented with 10% FBS at 37 °C and incubated for 10 min prior to exposure to nanoparticles. To allow long-term imaging 10 nM LysoTracker was left in the solution during the experiments.Exposure of cells to nanoparticles
Typically, 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well were seeded in 4-well chambered 35 mm Petri dishes (Greiner Bio-One) in complete medium. 20 h after transfection with FuGene HD (Promega), cells were exposed to the nanoparticle dispersions prepared by dilution of the stock into DMEM supplemented with 4 mg ml−1 human serum (human serum from pooled donors, TCS BioSciences), roughly corresponding to the amount of proteins in standard cell culture medium supplemented with 10% serum. Cells were exposed for 5, 10 and 15 min to a “pulse” of 40, 100 and 200 nm nanoparticles (75 μg ml−1, 300 μg ml−1 and 1600 μg ml−1 respectively, unless otherwise specified). Then cells were washed 3 times with phenol-red free DMEM (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS pre-warmed at 37 °C. Next, dishes were placed at the microscope for imaging in a live cell chamber at 37 °C under 5% CO2 and individual cells were imaged for different times (“chase”) in fresh nanoparticle-free phenol-red free DMEM supplemented with 10% FBS containing 1:100 diluted antifade reagent (Invirogen, Thermo Fisher Scientific).
000 cells per well were seeded in 4-well chambered 35 mm Petri dishes (Greiner Bio-One) in complete medium. 20 h after transfection with FuGene HD (Promega), cells were exposed to the nanoparticle dispersions prepared by dilution of the stock into DMEM supplemented with 4 mg ml−1 human serum (human serum from pooled donors, TCS BioSciences), roughly corresponding to the amount of proteins in standard cell culture medium supplemented with 10% serum. Cells were exposed for 5, 10 and 15 min to a “pulse” of 40, 100 and 200 nm nanoparticles (75 μg ml−1, 300 μg ml−1 and 1600 μg ml−1 respectively, unless otherwise specified). Then cells were washed 3 times with phenol-red free DMEM (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS pre-warmed at 37 °C. Next, dishes were placed at the microscope for imaging in a live cell chamber at 37 °C under 5% CO2 and individual cells were imaged for different times (“chase”) in fresh nanoparticle-free phenol-red free DMEM supplemented with 10% FBS containing 1:100 diluted antifade reagent (Invirogen, Thermo Fisher Scientific).
Live-cell imaging and image analysis
Dual color visualization of cells was performed using a DeltaVision Elite fluorescence microscope (GE Healthcare), and a Laser Scanning Confocal Microscope LSM880 (Zeiss). Data was analyzed using Imaris 7.6.4 software (Bitplane). Full details are given in the ESI.†Author contributions
A. S. conceived and designed the project. N. V. performed all experiments and all image analysis. C. Å. developed the kinetic model and performed the statistical analysis. N. V. and C. Å. applied the kinetic model to data. N. V. wrote the original draft. A. S. and C. Å. reviewed and edited the manuscript. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the ERC grant no. 637614 to A.S. The Microscopy & Imaging Centre of the University Medical Centre Groningen is kindly acknowledged for providing access to instruments and technical support. The EMBO Short-Term Fellowship and the Van Leersum Travel Grant of the Royal Netherlands Academy of Arts and Sciences are acknowledged for financing a research visit of N. V. to the NorMIC Advanced Light Microscopy Facility at University of Oslo within the Euro-BioImaging research infrastructure. We thank the EuroBioImaging facility “NorMIC Oslo” led by Prof. Oddmund Bakke for the use of their equipment, and are grateful for the expert assistance of Frode Skjeldal and Linda Haugen.References
- T. G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- S. Muro, J. Controlled Release, 2012, 164, 125–137 CrossRef CAS PubMed.
- J. Suh, M. Dawson and J. Hanes, Adv. Drug Delivery Rev., 2005, 57, 63–78 CrossRef CAS PubMed.
- B. Yameen, W. I. Choi, C. Vilos, A. Swami, J. J. Shi and O. C. Farokhzad, J. Controlled Release, 2014, 190, 485–499 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- J. A. Varela, M. G. Bexiga, C. Åberg, J. C. Simpson and K. A. Dawson, J. Nanobiotechnol., 2012, 10, 39 CrossRef CAS PubMed.
- P. Sandin, L. W. Fitzpatrick, J. C. Simpson and K. A. Dawson, ACS Nano, 2012, 6, 1513–1521 CrossRef CAS PubMed.
- J. A. Kim, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2011, 7, 62–68 CrossRef PubMed.
- J. J. Rennick, A. P. R. Johnston and R. G. Parton, Nat. Nanotechnol., 2021, 16, 266–276 CrossRef CAS PubMed.
- V. Francia, D. Montizaan and A. Salvati, Beilstein J. Nanotechnol., 2020, 11, 338–353 CrossRef CAS PubMed.
- Editorial, Nat. Biotechnol., 2014, 32, 961–961 CrossRef PubMed.
- D. Vercauteren, R. E. Vandenbroucke, A. T. Jones, J. Rejman, J. Demeester, S. C. De Smedt, N. N. Sanders and K. Braeckmans, Mol. Ther., 2010, 18, 561–569 CrossRef CAS PubMed.
- M. Al Soraj, L. He, K. Peynshaert, J. Cousaert, D. Vercauteren, K. Braeckmans, S. C. De Smedt and A. T. Jones, J. Controlled Release, 2012, 161, 132–141 CrossRef CAS PubMed.
- D. Vercauteren, H. Deschout, K. Remaut, J. F. Engbersen, A. T. Jones, J. Demeester, S. C. De Smedt and K. Braeckmans, ACS Nano, 2011, 5, 7874–7884 CrossRef CAS PubMed.
- J. A. Varela, C. Åberg, J. C. Simpson and K. A. Dawson, Small, 2015, 11, 2026–2031 CrossRef CAS PubMed.
- A. Kucera, M. Borg Distefano, A. Berg Larsen, F. Skjeldal, U. Repnik, O. Bakke and C. Progida, Traffic, 2016, 17, 211–229 CrossRef CAS PubMed.
- D. H. Murray, M. Jahnel, J. Lauer, M. J. Avellaneda, N. Brouilly, A. Cezanne, H. Morales-Navarrete, E. D. Perini, C. Ferguson, A. N. Lupas, Y. Kalaidzidis, R. G. Parton, S. W. Grill and M. Zerial, Nature, 2016, 537, 107–111 CrossRef CAS PubMed.
- J. C. Simpson, B. Joggerst, V. Laketa, F. Verissimo, C. Cetin, H. Erfle, M. G. Bexiga, V. R. Singan, J. K. Heriche, B. Neumann, A. Mateos, J. Blake, S. Bechtel, V. Benes, S. Wiemann, J. Ellenberg and R. Pepperkok, Nat. Cell Biol., 2012, 14, 764–774 CrossRef CAS PubMed.
- Y. Liu, Y. Mi, T. Mueller, S. Kreibich, E. G. Williams, A. Van Drogen, C. Borel, M. Frank, P. L. Germain, I. Bludau, M. Mehnert, M. Seifert, M. Emmenlauer, I. Sorg, F. Bezrukov, F. S. Bena, H. Zhou, C. Dehio, G. Testa, J. Saez-Rodriguez, S. E. Antonarakis, W. D. Hardt and R. Aebersold, Nat. Biotechnol., 2019, 37, 314–322 CrossRef CAS PubMed.
- A. Frattini, M. Fabbri, R. Valli, E. De Paoli, G. Montalbano, L. Gribaldo, F. Pasquali and E. Maserati, Sci. Rep., 2015, 5, 15377 CrossRef CAS PubMed.
- S. Sigismund, V. Algisi, G. Nappo, A. Conte, R. Pascolutti, A. Cuomo, T. Bonaldi, E. Argenzio, L. G. Verhoef, E. Maspero, F. Bianchi, F. Capuani, A. Ciliberto, S. Polo and P. P. Di Fiore, EMBO J., 2013, 32, 2140–2157 CrossRef CAS PubMed.
- A. Salvati, C. Åberg, T. dos Santos, J. Varela, P. Pinto, I. Lynch and K. A. Dawson, Nanomedicine, 2011, 7, 818–826 CrossRef CAS PubMed.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Åberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
- T. dos Santos, I. Lynch, A. Salvati and K. A. Dawson, Small, 2011, 7, 3341–3349 CrossRef CAS PubMed.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- S. Schottler, K. Klein, K. Landfester and V. Mailander, Nanoscale, 2016, 8, 5526–5536 RSC.
- K. Sandvig, S. Pust, T. Skotland and B. van Deurs, Curr. Opin. Cell Biol., 2011, 23, 413–420 CrossRef CAS PubMed.
- G. J. Doherty and H. T. McMahon, Annu. Rev. Biochem., 2009, 78, 857–902 CrossRef CAS PubMed.
- C. Åberg, Nanoscale Adv., 2021, 3, 2196–2212 RSC.
- J. Gilleron, W. Querbes, A. Zeigerer, A. Borodovsky, G. Marsico, U. Schubert, K. Manygoats, S. Seifert, C. Andree, M. Stoter, H. Epstein-Barash, L. Zhang, V. Koteliansky, K. Fitzgerald, E. Fava, M. Bickle, Y. Kalaidzidis, A. Akinc, M. Maier and M. Zerial, Nat. Biotechnol., 2013, 31, 638–646 CrossRef CAS PubMed.
- L. Yang, L. Shang and G. U. Nienhaus, Nanoscale, 2013, 5, 1537–1543 RSC.
- J. Reinholz, C. Diesler, S. Schottler, M. Kokkinopoulou, S. Ritz, K. Landfester and V. Mailander, Acta Biomater., 2018, 71, 432–443 CrossRef CAS PubMed.
- M. Liu, Q. Li, L. Liang, J. Li, K. Wang, J. Li, M. Lv, N. Chen, H. Song, J. Lee, J. Shi, L. Wang, R. Lal and C. Fan, Nat. Commun., 2017, 8, 15646 CrossRef CAS PubMed.
- G. Sahay, W. Querbes, C. Alabi, A. Eltoukhy, S. Sarkar, C. Zurenko, E. Karagiannis, K. Love, D. Chen, R. Zoncu, Y. Buganim, A. Schroeder, R. Langer and D. G. Anderson, Nat. Biotechnol., 2013, 31, 653–658 CrossRef CAS PubMed.
- D. J. Brayden, S.-A. Cryan, K. A. Dawson, P. J. O'Brien and J. C. Simpson, Drug Discovery Today, 2015, 20, 942–957 CrossRef CAS PubMed.
- P. I. H. Bastiaens and R. Pepperkok, Trends Biochem. Sci., 2000, 25, 631–637 CrossRef CAS PubMed.
- J. D. Sander and J. K. Joung, Nat. Biotechnol., 2014, 32, 347–355 CrossRef CAS PubMed.
- M. Ratz, I. Testa, S. W. Hell and S. Jakobs, Sci. Rep., 2015, 5, 9592 CrossRef CAS PubMed.
- R. Firdessa, T. A. Oelschlaeger and H. Moll, Eur. J. Cell Biol., 2014, 93, 323–337 CrossRef CAS PubMed.
- V. Francia, K. Yang, S. Deville, C. Reker-Smit, I. Nelissen and A. Salvati, ACS Nano, 2019, 13, 11107–11121 CrossRef CAS PubMed.
- A. Tagawa, A. Mezzacasa, A. Hayer, A. Longatti, L. Pelkmans and A. Helenius, J. Cell Biol., 2005, 170, 769–779 CrossRef CAS PubMed.
- A. Hayer, M. Stoeber, C. Bissig and A. Helenius, Traffic, 2010, 11, 361–382 CrossRef CAS PubMed.
- H. M. van der Schaar, M. J. Rust, C. Chen, H. van der Ende-Metselaar, J. Wilschut, X. Zhuang and J. M. Smit, PLoS Pathog., 2008, 4, e1000244 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Extended experimental methods, additional results of nanoparticle characterization and colocalization analysis in individual cells for all cells, nanoparticles and marker tested, movies of live cells expressing the different markers and exposed to nanoparticles. See DOI: 10.1039/d1nr00901j |
| This journal is © The Royal Society of Chemistry 2021 |