
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom temperature synthesized solid solution AuFe nanoparticles and their transformation into Au/Fe Janus nanocrystals†
Maria V.
Efremova‡
 *ab,
Marina
Spasova
c,
Markus
Heidelmann
d,
Ivan S.
Grebennikov
b,
Zi-An
Li
c,
Anastasiia S.
Garanina
b,
Iana O.
Tcareva
b,
Alexander G.
Savchenko
b,
Michael
Farle
c,
Natalia L.
Klyachko
af,
Alexander G.
Majouga
be and
Ulf
Wiedwald
*bc
*ab,
Marina
Spasova
c,
Markus
Heidelmann
d,
Ivan S.
Grebennikov
b,
Zi-An
Li
c,
Anastasiia S.
Garanina
b,
Iana O.
Tcareva
b,
Alexander G.
Savchenko
b,
Michael
Farle
c,
Natalia L.
Klyachko
af,
Alexander G.
Majouga
be and
Ulf
Wiedwald
*bc
aDepartment of Chemistry, Lomonosov Moscow State University, Moscow, 119991, Russian Federation
bNational University of Science and Technology “MISIS”, Moscow, 119049, Russian Federation
cFaculty of Physics and Center for Nanointegration Duisburg-Essen, University of Duisburg-Essen, Duisburg, 47057 Germany. E-mail: ulf.wiedwald@uni-due.de
dICAN - Interdisciplinary Center for Analytics on the Nanoscale and Center for Nanointegration Duisburg-Essen, University of Duisburg-Essen, Duisburg, 47057 Germany
eD. Mendeleev University of Chemical Technology of Russia, Moscow, 125047, Russian Federation
fG.R. Derzhavin Tambov State University, Tambov, 392000, Russian Federation
First published on 6th May 2021
Abstract
Solid solution AuFe nanoparticles were synthesized for the first time under ambient conditions by an adapted method previously established for the Fe3O4–Au core–shell morphology. These AuFe particles preserved the fcc structure of Au incorporated with paramagnetic Fe atoms. The metastable AuFe can be segregated by transformation into Janus Au/Fe particles with bcc Fe and fcc Au upon annealing. The ferromagnetic Fe was epitaxially grown on low index fcc Au planes. This preparation route delivers new perspective materials for magnetoplasmonics and biomedical applications and suggests the reconsideration of existing protocols for magnetite–gold core–shell synthesis.
Introduction
Magnetite and gold nanoparticles (NPs) are the materials of choice for a vast variety of biomedical applications due to their size-dependent and controllable physicochemical properties and biocompatibility.1–5 Hybrid magnetite–gold NPs combine the magnetic and plasmonic properties, which make them one of the best candidates for tumor theranostics,6–8 multimodal magnetic resonance imaging/computer tomography (MRI/CT) imaging,9–11 MRI-guided radiosensitization,12 enhanced hyperthermia,13,14 and other applications.15–18 For these purposes, core–shell magnetite–gold NPs are especially attractive if the gold shell completely covers the magnetic core and protects the latter from oxidation, minimizes its possible toxicity, and enables the functionalization with sulfur-containing ligands (mainly thiols).19–21 To date, various synthetic approaches have been developed to generate such NPs.22–25 Among the most common methods is the iterative reduction of hydrogen tetrachloroaurate with hydroxylamine hydrochloride.26 Despite promising in vitro and in vivo applications of magnetite–gold NPs obtained by this method,27–29 the authors often do not provide detailed structural and magnetic characterization of these nanomaterials. In particular, unambiguous evidence of the core–shell structure is still pending whereas it was presented for other core-shell systems like magnetite–silica NPs.30,31 From the chemical standpoint, at least partial dissolution of Fe ions from Fe3O4 cores can be expected considering the harsh acidic environment during Au salt reduction. In the current study, we have investigated this approach in detail and provided a comprehensive step-by-step characterization of the final product after the removal of uncoated Fe3O4 seeds.Despite previous reports, we do not observe core-shell morphologies but find gold-rich solid solution AuFe NPs. In the phase diagram, the solubility of Fe in Au is extremely low with a limit of 1–2 mass% at 25 °C.32 Nonetheless, solid solutions of Fe and Au can be prepared by non-equilibrium techniques like gas phase condensation33,34 and pulsed laser ablation in liquids.35–37 In the present study, we have described the synthesis of AuFe solid solution nanocrystals, for the first time under ambient conditions, using the methods of colloidal chemistry. Interestingly, the solid solution is metastable at room temperature forming Fe-rich regions in the Au matrix during storage. By in situ annealing experiments in transmission electron microscopy (TEM) and vibrating sample magnetometry (VSM), we have proved the segregation of metallic Fe from the AuFe solid solution finally forming Au/Fe Janus NPs. Therefore, this study provides new insights into the established synthesis of core–shell NPs suggesting completely different underlying mechanisms and final structures. It also facilitates the reassessment of possible applications of such NPs leading to new materials for magnetoplasmonics.
Experimental
Materials
Iron chloride(II) tetrahydrate (FeCl2·4H2O, 98%), iron(III) chloride (FeCl3, anhydrous, 97%), hydrogen tetrachloroaurate(III) trihydrate (HAuCl4·3H2O, 99.9%), sodium citrate dihydrate (Na3C6H5O7·2H2O, 99%), hydroxylamine hydrochloride (NH2OH·HCl, 99%), ammonium hydroxide solution (NH3·H2O, 28%), nitric acid (HNO3, 69%), hydrochloric acid (HCl, 36%), perchloric acid (HClO4, 70%), iron standard for ICP (TraceCERT, 1000 mg L−1 Fe in nitric acid), and gold standard for ICP (TraceCERT, 1000 mg L−1 Au in hydrochloric acid) were purchased from Sigma-Aldrich. HS–PEG–COOH (2 kDa) was purchased from Nanocs Inc. SERVAPOR dialysis tubing, MWCO 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–14000 kDa, was purchased from SERVA. Water used in all experiments was deionized (18.2 MΩ cm−1, Millipore Milli-Q Academic System). All vessels were washed with hot solution of aqua regia and then rinsed with deionized water before synthesis.
000–14000 kDa, was purchased from SERVA. Water used in all experiments was deionized (18.2 MΩ cm−1, Millipore Milli-Q Academic System). All vessels were washed with hot solution of aqua regia and then rinsed with deionized water before synthesis.
Synthesis of Fe3O4 NPs
Fe3O4 NPs were prepared by co-precipitation of Fe(II) and Fe(III) salts according to the procedure described in ref. 38 with slight modifications. Briefly, 650 mg of FeCl3 and 398 mg of FeCl2·4H2O were dissolved in 5 mL of 2 M HCl, the obtained solution was added dropwise to 50 mL of 0.7 M NH3 solution in deionized H2O, and then the mixture was magnetically stirred for 30 minutes. After that, the NPs were magnetically decanted and washed with 50 mL of 2 M HClO4, and they were magnetically decanted again and dissolved in 50 mL of H2O for 30 minutes using an ultrasonic bath (BANDELIN SONOREX).Synthesis of AuFe NPs
AuFe NPs were prepared by room-temperature iterative reduction of chloroauric acid with hydroxylamine hydrochloride according to the general scheme described in ref. 26. 0.19 mL of previously obtained Fe3O4 NP solution (or 0.19 mL of H2O during the control experiment) was added to 7.31 mL of H2O in a flat-bottomed flask under magnetic stirring. Then 7.5 mL of 0.1 M sodium citrate solution was added, and the mixture was stirred for 10 minutes. After that the solution was diluted with 135 mL of H2O, and aliquots of 1% HAuCl4 solution and 0.2 M solution of NH2OH·HCl were incrementally added (with 10–15 minutes between additions) according to the scheme described in ref. 26.Purification and modification of AuFe NPs
20 mL of the as prepared freshly sonicated (30 min) AuFe NP dispersion were centrifuged using a MiniSpin Plus centrifuge at 7000 rpm (RCF 3300g) for 5 min. The supernatant was separated with a pipette; the residue was resuspended in 10 mL of H2O by sonication. Then, it was mixed with 10 mL of an aqueous HS–PEG–COOH solution (0.103 μmol of ligand) and stirred overnight. The unbound ligand was washed off by dialysis (3 times in 1 L H2O). For the investigation by inductively coupled plasma mass-spectrometry, AuFe NPs were additionally purified by acid treatment. 50 mL of freshly sonicated (30 min) AuFe NP dispersion were mixed with 10 mL of concentrated hydrochloric acid under vigorous stirring for 12 hours. Then the NPs were decanted, washed with 50 mL of H2O, decanted again and resuspended in 50 mL of H2O. For X-ray diffraction and magnetometry, AuFe NPs were desiccated using a rotary evaporator.Electron microscopy
TEM micrographs were obtained using a JEOL JEM-1400 (120 kV) microscope. Samples were prepared by casting and evaporating a droplet of aqueous solution of NPs onto a formvar-coated copper grid (300 mesh). The average diameter of NPs was calculated from TEM images by analyzing about 1000 NPs for each sample using ImageJ software (National Institutes of Health, USA). HRTEM micrographs were obtained using probe-side Cs-corrected JEOL JEM 2200FS and probe-side Cs-corrected JEM-ARM200F HR/Cold FEG microscopes, both operated at an acceleration voltage of 200 kV. Overview images were taken in conventional bright-field TEM mode while the high-angle annular dark-field STEM and bright-field HRTEM modes were used for the high-resolution micrographs. Samples were prepared by casting and evaporating a droplet of solution onto a carbon-coated copper grid (300 mesh). EDX elemental mapping was carried out in the scanning mode utilizing an Oxford X-max detector. For the annealing experiment, the NPs were deposited on a heating chip. The annealing experiment was carried out utilizing a Protochips Aduro in situ heating holder in the JEM 2200FS.X-ray diffraction
Patterns were measured from 2θ = 30° to 120° at a scan rate of 0.1° per step and 3 s per point using a X-ray powder diffractometer Rigaku Ultima IV with Co-Kα radiation and a graphite monochromator in the diffracted beam path. Quantitative XRD analysis (including crystal size evaluation by determination of the coherent scattering region) was performed using PHAN% and SPECTRUM programs developed by Physical Materials Science Department of NUST “MISiS” (modified Rietveld method) based on the minimization of the difference between the experimental spectrum and the model. For fitting the spectra, the lattice parameters, the amount of each phase and their crystallite sizes were optimized.Magnetometry
The dried powder of Fe3O4 or AuFe NPs (about 10 mg) was filled in synthetic capsules for vibrating sample magnetometry (VSM) and the hysteresis loops and temperature dependent magnetization were measured using a Quantum Design PPMS DynaCool system. For high temperature VSM, sample powder has been dispersed in Zircar cement and deposited on a heating stick.Visible absorption spectroscopy
AuFe NPs solution was examined with a Thermo Scientific Multiskan GO spectrometer at wavelengths from 400 nm to 800 nm.ICP-MS measurements
The iron and gold content in AuFe NPs were determined with an ELAN DRC II 9000 (PerkinElmer SCIEX) inductively coupled plasma mass spectrometer. A series of 5 calibration samples in the concentration range of 0.1–10 ppm were prepared by dilution of commercial ICP standards with diluted (to 2–5% HNO3) aqua regia. The samples were similarly digested with diluted aqua regia in the concentration range of 0.1–10 ppm.Cell cultures
PC-3 and LNCaP human prostate cancer cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPMI-1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco), 2 mM L-glutamine (Gibco), and RPMI vitamin solution (Sigma) at 37 °C in a humidified incubator supplied with 5% CO2. Both cell lines routinely tested negative for mycoplasma.MTS assay
PC-3 and LNCaP cells were seeded in a 96-well plate (10 × 103 cells per well) in a culture medium and cultivated at 37 °C under a humidified atmosphere containing 5% CO2. The cells were counted using an automatic cell counter EVE. After 24 h, the cells were washed with 100 μL of serum-free medium and incubated with 100 μL of AuFe NP serial dilutions. To evaluate the cytotoxicity of NPs, the standard MTS test39 was used. The solution of AuFe NPs in the concentration range 7.5–150.0 μg Au mL−1 was added to the cell culture medium. 48 h later, the cells were washed with 100 μL of serum-free medium and new 100 μL of culture medium with 20 μL of MTS reagent (CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay, Promega, USA) were added per well. After 4 h of incubation at 37 °C in darkness, the absorbance of the solution was measured at 490 nm using a Thermo Scientific Multiskan GO spectrometer. 1× PBS, 10% well volume, was used as a negative control, and 30% DMSO diluted in cell medium was used as a positive control.Results
Synthesis of NPs
Fe3O4 seeds were obtained by the co-precipitation of iron salts with ammonia solution as described in ref. 38. For the synthesis of AuFe NPs, we use a room-temperature iterative reduction of HAuCl4 with NH2OH·HCl in the presence of Fe3O4 seeds adapted from ref. 26. The process can be described as follows:| 4HAuCl4 + 6NH2OH·HCl → 4Au + 3N2O + 22HCl + 3H2O | (1) |
During the reaction, a considerable amount of hydrochloric acid (HCl) is produced. We believe that this can lead to at least partial dissolution of Fe ions from Fe3O4 seed NPs. While it is not a priori known whether Fe ions or Fe3O4 seeds serve as primary nucleation sites for Au reduction, the latter is clearly a heterogeneous nucleation process.26 To this end, we failed to obtain stable Au NPs when Fe3O4 seeds were initially excluded from the reaction mixture and replaced with an equal amount of deionized water. In that case, visible aggregates appeared at the bottom of the flask after the first addition of HAuCl4 and NH2OH·HCl.
After synthesis, the reaction mixture contains AuFe, Fe3O4, and Au NPs. For purification, we have developed a two-step approach based on centrifugation and subsequent HCl treatment of the final product. Centrifugation is a widely used method40 based on the selection of rotation speeds to precipitate larger-sized and heavier AuFe NPs while smaller and lighter Fe3O4 NPs (and, possibly, small Au NPs) remain in the supernatant. Yet, it only allows for mechanical separation of the reaction mixture and cannot ensure complete removal of the uncoated Fe3O4 NPs, especially when the latter are clustered or formed on the Au surface. Therefore, we supplemented a chemical “etching” step, namely, the treatment of NPs with 1.7 M HCl acid that dissolves Fe3O4 NPs, while the analogous examples involving 1 M HCl41 or 2 M HCl42 have been reported before. To keep the AuFe NPs stable in the colloidal solution, they are coated with PEG prior to the acid treatment (see Experimental section for details).
Morphology, structure, and composition of the NPs
Fig. 1 presents low-resolution TEM images. Fe3O4 seeds have roughly a spherical shape and an average diameter of 9 ± 2 nm (Fig. 1a and Fig. S1a†). After reduction of Au in the presence of iron oxide seeds we obtain a mixture of more electron-dense and less electron-dense NPs with or without Au, respectively (see Fig. S2†). Purification by centrifugation and acid treatment leaves predominantly electron-dense Au-rich and rather spherical NPs with an average diameter of 32 ± 5 nm in the colloidal solution (Fig. 1b and Fig. S1b†). The electron diffraction pattern (Fig. 1c) only shows the fcc Au structure in AuFe NPs without additional rings that can be assigned to any of the iron oxides (magnetite Fe3O4, maghemite γ-Fe2O3, etc.). This finding is confirmed by powder X-ray diffraction (XRD) patterns displayed in Fig. 1d. For initial seeds, all diffraction peaks correspond to Fe3O4 (100% volume fraction) while after the reaction with hydrogen tetrachloroaurate and subsequent purification, only fcc Au peaks (100% volume fraction) are detected.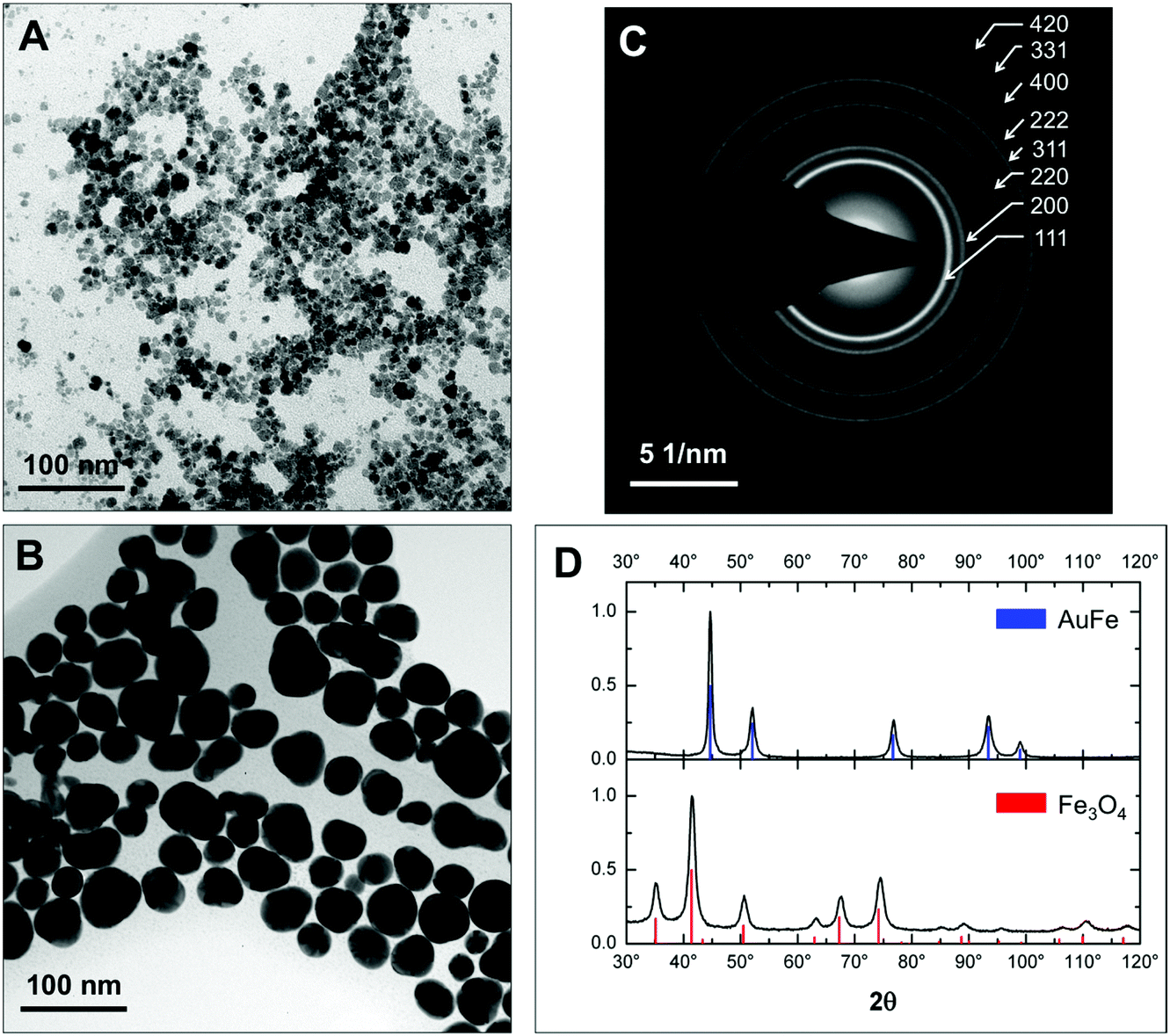 | ||
| Fig. 1 Morphology and crystalline structure of NPs. Bright-field TEM images of Fe3O4 NPs (A) and AuFe NPs after purification (B). (C) SAED pattern of (B). All diffraction rings can be ascribed to the fcc Au lattice as indicated in the image. (D) X-ray diffraction patterns of Fe3O4 seeds (bottom panel) and AuFe NPs after purification (top panel). The intensities are normalized to the strongest peak. The red and blue vertical lines represent the angular position and relative intensity of the reference powder diffractograms of the Fe3O4 and Au phases, respectively. | ||
By XRD, the lattice constants and crystallite sizes are extracted from the experimental data using Rietveld refinement with the reference data of Fe3O4 (ICDD PDF-2 no. 00-019-0629) and Au (ICDD PDF-2 no. 03-065-8601). Iron oxide seeds are monocrystalline with a crystallite size of 8 ± 1 nm matching well to the TEM diameter of 9 ± 2 nm. The lattice parameter of 0.8362 ± 0.0004 nm suggests a non-stoichiometric, intermediate state between Fe3O4 (a = 0.8397 nm) and γ-Fe2O3 (a = 0.8347 nm),43 which is further verified using room-temperature hysteresis data showing a saturation magnetization of 61.0 ± 0.1 A m2 kg−1 (Fig. S3a†) in agreement with the data for Fe3O4 NPs and their hybrids of similar size previously reported in the literature.44 For AuFe NPs, the lattice parameter of 0.4087 nm ± 0.0002 nm matches the volumetric Au value (a = 0.4087 nm),45 and a polycrystalline state is obtained (crystallite size of 15 ± 2 nm vs. 32 ± 5 nm given by TEM which is confirmed by high-resolution TEM shown below).
The composition of the NPs was determined by ICP-MS. After each purification step, the amount of Fe decreases. The AuFe NPs contain 6.60 ± 1.55 mass% Fe directly after synthesis, 0.70 ± 0.19 mass% Fe after purification via centrifugation and finally 0.26 ± 0.06 mass% Fe after etching with HCl.
Crystalline structure and elemental composition at the level of single NPs
Further insight into the structure of NPs is gained by high-angle annular dark field-scanning TEM (HAADF-STEM) and bright-field high resolution HRTEM imaging and energy-dispersive X-ray (EDX) elemental mapping as shown in Fig. 2. Most of the NPs are polycrystalline and contain several inclusions (up to 8 per NP) that appear as small spherical regions of different contrast with an average diameter of 3.4 ± 1.0 nm (Fig. 2a and b, size distribution in Fig. S4†). At higher magnification (Fig. 2c and d), one can also see an inhomogeneous contrast distribution within the NPs, which is especially evident in the darker region with ≈3 nm size. Nevertheless, it is clear from these images that the crystallographic planes and atomic columns of Au are not distorted even near or at inclusions. STEM-EDX elemental mapping at room temperature (Fig. 2e) confirms the presence of gold and a rather homogeneous distribution of Fe. Importantly, we revealed that there is no oxygen within the particles, excluding the possibility of a magnetite–gold core–shell structure.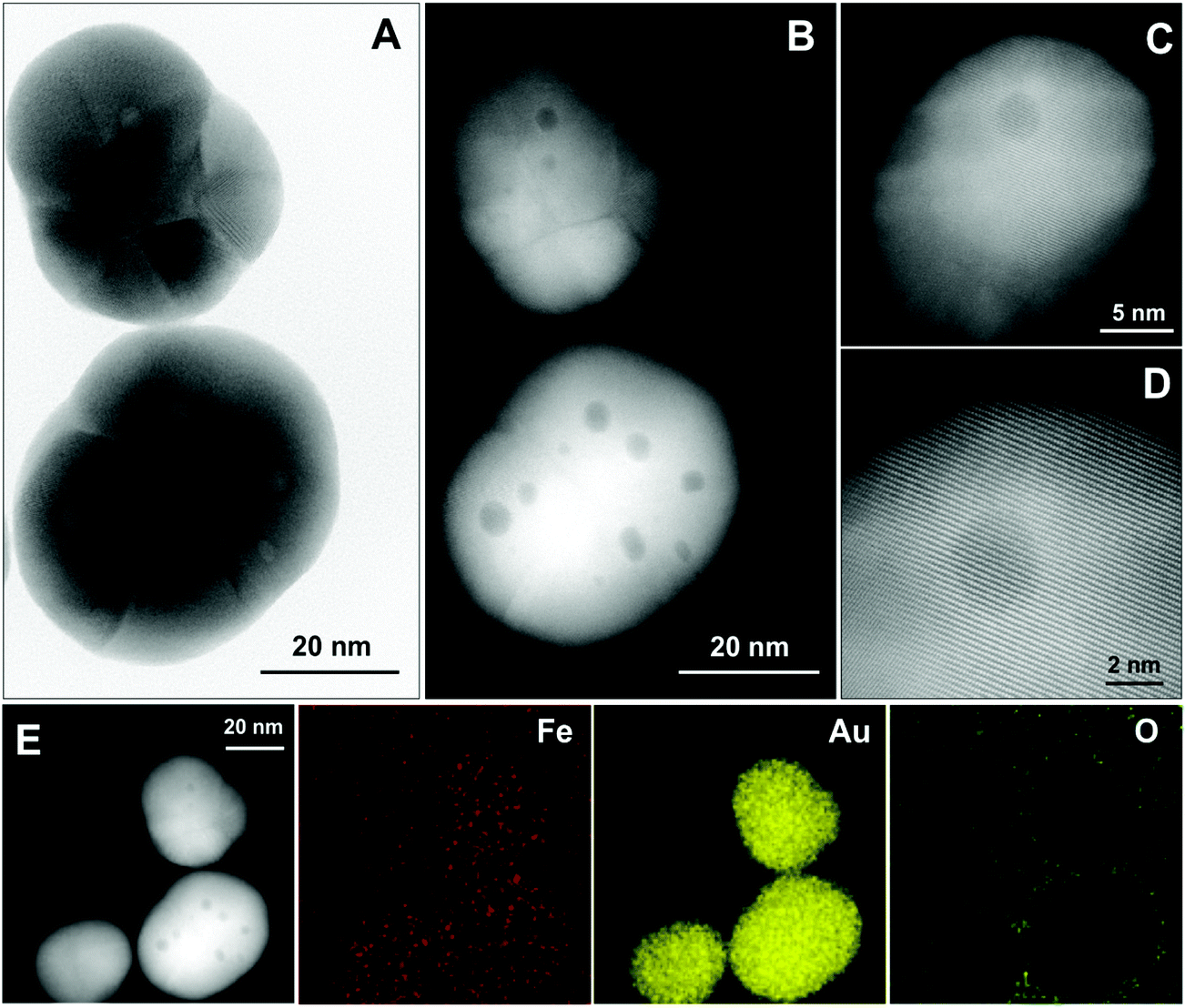 | ||
| Fig. 2 High-resolution TEM micrographs of AuFe NPs and EDX-elemental mapping. Bright-field (A) and dark-field (B) images of the same area. (C) HAADF-STEM image of another NP and the corresponding higher magnification image (D) showing continuous Au atomic columns at varying contrast. (E) STEM image and EDX elemental mapping of Fe, Au, and O in the NPs. | ||
Morphological, structural and compositional changes in the NPs upon annealing
Although STEM-EDX cannot resolve a different stoichiometry in the inclusions, we expect a somewhat higher Fe content in these regions of lower electron density (brightness). The formation of solid solution AuFe NPs is one possibility; however, such particles should be metastable because of the extremely low Fe solubility in Au at 25 °C (up to 1–2 mass%) according to the binary phase diagram.32 Such a transient state of AuFe NPs can be proved e.g. by stepwise annealing, tracking the structural and morphological changes. We annealed the NPs up to 700 °C in the TEM at low pressure (<5 × 10−7 mbar). Under such conditions, Fe is likely to be expelled from the Au host lattice46 finally forming Au/Fe Janus structures. Fig. 3 presents the morphology of one AuFe NP at 25 °C, then at 400, 500, 550, and 700 °C and finally at 25 °C again after exposure to air. Initially, the NP contains a visible inclusion of 8 nm diameter (Fig. 3a) that gets smaller upon heating up to 400–550 °C (Fig. 3b–d) and completely vanishes at 700 °C (Fig. 3e). This process is accompanied by the appearance of a strongly contrasted area on the NP surface at 400 °C (Fig. 3b) that expands with increasing temperature and finally forms a Janus structure with a low-contrast sector of ≈125° angle within a spherical Au NP (Fig. 3e). We attribute this phenomenon to the segregation of the Fe and Au phases while the latter recrystallizes. Note the healing of twin boundaries in Au during annealing. Remarkably, the morphology and crystalline structure of Au/Fe NPs do not change upon its exposure to air for one month after the annealing experiment (Fig. 3f).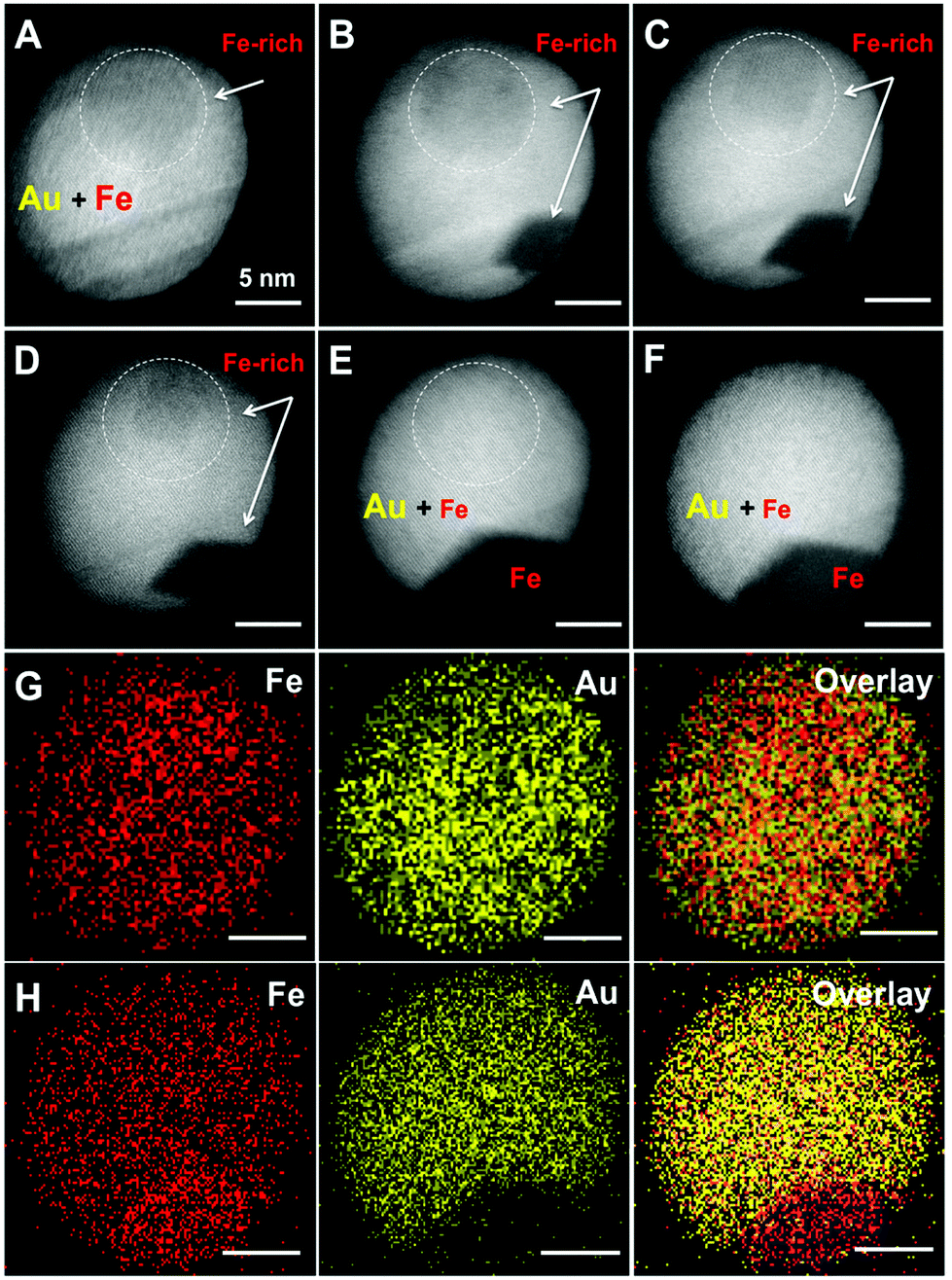 | ||
| Fig. 3 In situ annealing experiment in TEM. HAADF-STEM images of a AuFe NP on a SiN chip at 25 °C (A), 400 °C (B), 500 °C (C), 550 °C (D), and 700 °C (E) revealing the diffusion of Fe towards the surface of the hybrid NP. Panel (F) reflects the unchanged morphology at 25 °C after a one-month exposure to air demonstrating the resistance to atmospheric oxygen. The area shown by the white dotted circle is Fe-rich, having lower contrast that vanishes with increasing temperature. EDX-elemental mapping shows Fe and Au in the same NP before (G) and after annealing at 700 °C (H) proving the temperature induced phase segregation. The binning factors 4 and 2 in (G) and (H), respectively, were chosen for the best data visualization. The scale bar is 5 nm. | ||
The induced phase segregation of Fe and Au in the NPs at high temperatures is further verified by EDX-mapping of the NP (Fig. 3g and h). Before annealing (Fig. 3g), both, Fe and Au, are rather homogeneously distributed, although the tendency of Fe accumulation in the inclusion region of STEM image is confirmed in an element-specific manner. After annealing at 700 °C (Fig. 3h), the sector with low contrast in STEM consists of pure Fe. Nonetheless, residual Fe seems to rest within Au which cannot be unambiguously explained considering the amount of segregated Fe. Thus, either a significant amount of dissolved Fe is stable in the Au matrix at 700 °C or we may detect a Fe background signal from secondary and ternary X-ray photons excited in the steel chamber of the TEM.
Intrigued by the segregation of Fe and Au within the NP upon its transition to an equilibrium state, we investigated the NP's crystalline structure after annealing at 700 °C. Fig. 4 presents the HRTEM results with the corresponding fast Fourier transform (FFT) diagrams. Fig. 4a shows the contrast difference between the Au-rich (marked with a yellow square) and Fe-rich (marked with a red square) areas. FFT analysis (Fig. 4b–d) reveals an epitaxial relationship between the bcc Fe segment and the fcc Au host crystal. The epitaxial relation is Au (111) || Fe (110) and the fcc Au crystal is viewed along its [011] direction while the bcc Fe grain is oriented parallel to the [001] zone axis.
 | ||
| Fig. 4 Structural investigation of the Au/Fe NP on a SiN chip after annealing at 700 °C. Bright-field HRTEM image (A) and the corresponding FFT images (B and C) of the areas shown in (A) in yellow and red colors, respectively. The FFT pattern in (B) corresponds to the Au fcc structure (marked with yellow indices) and the NP is viewed along its [011] direction. The FFT pattern in (C) corresponds to the Fe bcc structure (marked with red indices) and the NP is viewed along its [001] direction. The FFT pattern in (D) corresponds to the Au/Fe interface area. The [111] and [110] crystallographic directions of Au and Fe align to each other. | ||
Magnetic characterization
The formation of AuFe solid solutions and phase separation in Au/Fe NPs by annealing were investigated by their field- and temperature-dependent magnetic responses. Fig. 5a presents the magnetic hysteresis loops at various temperatures of 5–300 K normalized to the total sample mass mtot = 15.43 mg. The overall shape of the hysteresis loops suggests the presence of several magnetic components. It is striking that the high field slopes change sign from paramagnetic at low temperatures to diamagnetic at high temperatures, which we ascribe to paramagnetic Fe atoms in the diamagnetic Au matrix as discussed below.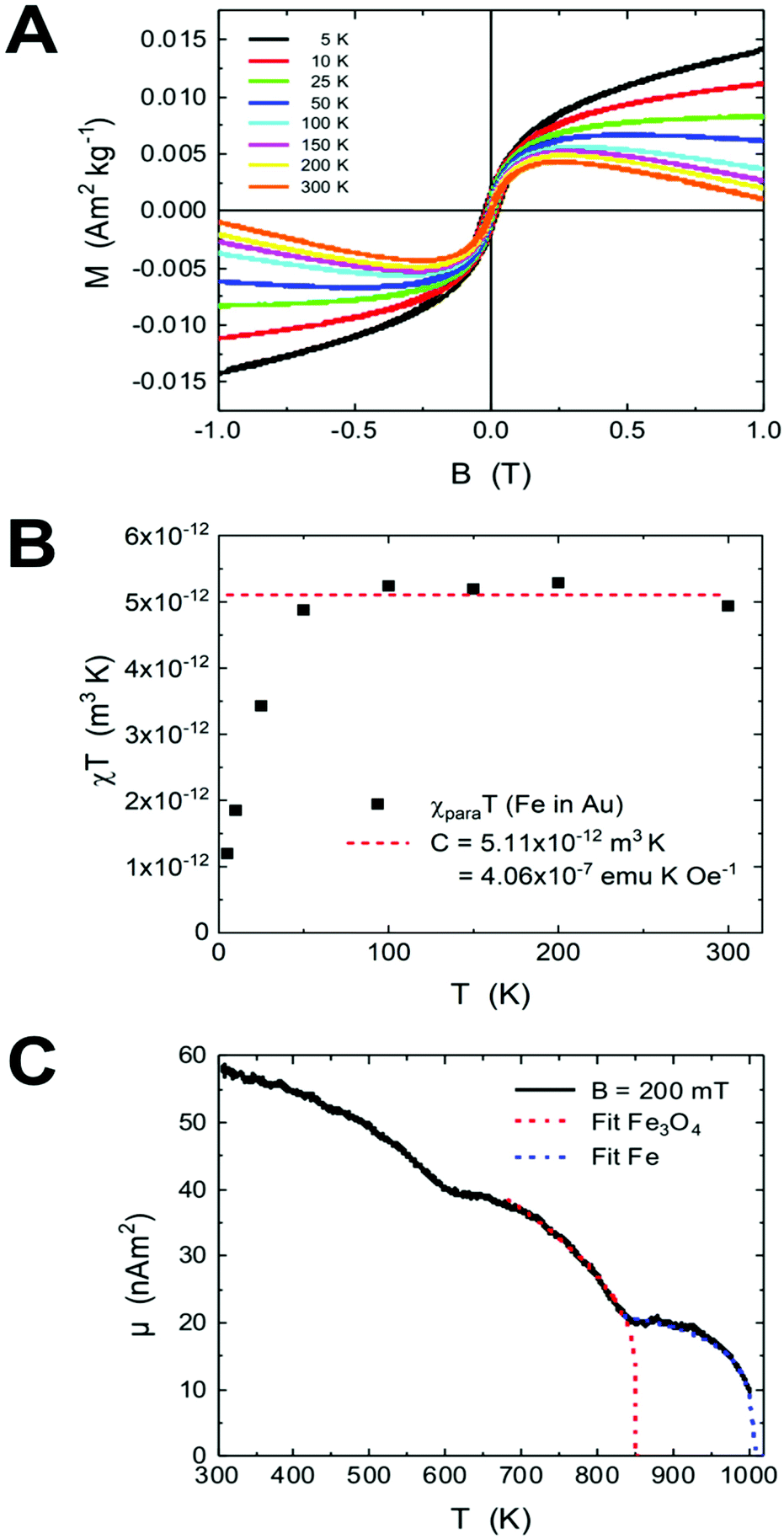 | ||
| Fig. 5 Magnetic characterization of AuFe NPs. (A) Hysteresis loops recorded at various temperatures between 5 K and 300 K. (B) χT (T) plot after subtraction of the Au diamagnetic component for the evaluation of the Curie constant; (C) high-temperature total magnetic moment of another sample at B = 200 mT. Two ordering temperatures are obtained as identified by the fits for Fe3O4 and Fe using mean field models while the dip at 600 K is due to diffusion. | ||
The hysteresis loops contain further a ferrimagnetic component originating from the residual uncovered Fe3O4 seeds present in the sample after purification (see the Experimental section). In Fig. S3,† the hysteresis loops of the Fe3O4 seed NPs (Fig. S3a†) and the hysteresis of AuFe NPs are plotted in a smaller range after the subtraction of the high-field slopes (Fig. S3b†). The coercive field μ0HC is 26 mT at 5 K, which is in agreement with the data previously reported in the literature,44 whereas the fitting of the coercive fields e.g. with Sharrock's equation is not applicable here. The ferrimagnetic component is very small (μFe3O4 = 1.02 × 10−7 A m2), which means that the diamagnetic Au prevails over the total sample mass. Considering the saturation magnetization of initial Fe3O4 seeds, we estimate mFe3O4 = (1.02 × 10−7 A m2)/(61.02 A m2 kg−1) = 1.67 μg (Fe3O4) or is equivalent to 1.21 μg (Fe).
The Fe oxide component is a side effect in the present experiments, which can be separated from Fe dissolved in the Au matrix. We get access to the paramagnetic contribution of Fe in Au by plotting the high-field magnetic susceptibility χ (slopes in Fig. 5a). This χ is, however, the sum of χpara (Fe) and χdia (Au). The paramagnetic Fe atoms can be described by the Langevin function (χ ∝ T−1), thus we plot χT (T) and subtract the linearly decreasing diamagnetic contribution of Au in this scaling (temperature-independent). After this, we obtain the susceptibility χFe in Au displayed in Fig. 5b. Below 50 K, χT is strongly decreasing which we ascribe to freezing of randomly oriented Fe magnetic moments in the Au matrix due to RKKY interaction. Above 50 K, χT can be approximated with a constant value. The Curie constant is given by
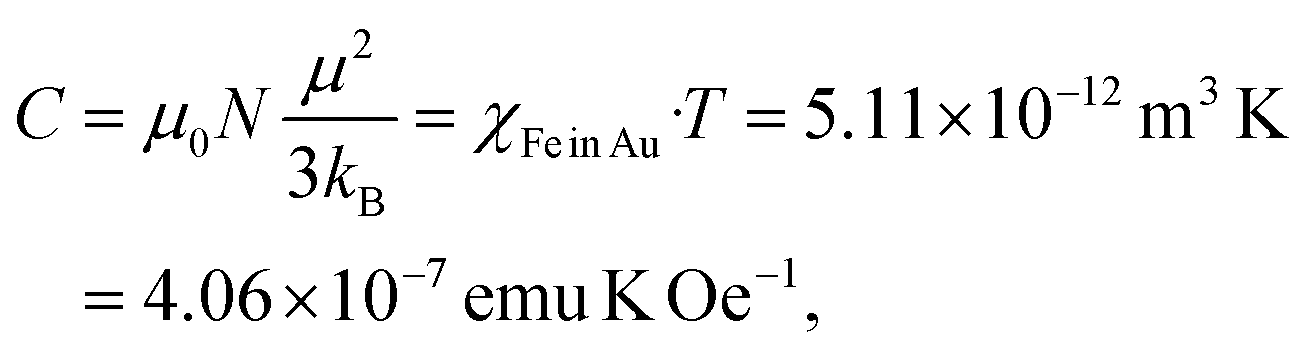 | (2) |
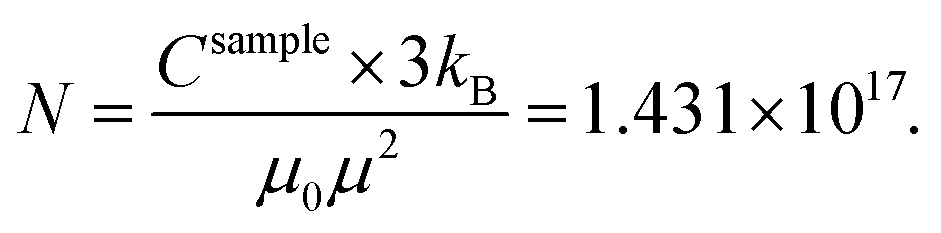 | (3) |
This leads to Fe mass mFe in Au = 20.16 μg dissolved in Au. Thus, the total sample mass mtot = mAu + mFe in Au + mFe3O4 = 15408 μg + 20.16 μg + 1.67 μg = 15.43 mg and about ≈0.14 Fe mass% or a Fe/Au atomic ratio of 0.5%. We estimated the error bars of the analysis to be in the order of 10–15%. In addition, the results proved that most Fe is dissolved in the Au matrix (20.16 μg) as compared to the residual Fe3O4 seeds (mFe = 1.21 μg).
Fig. 5c presents the high-temperature magnetization of the AuFe NPs upon annealing towards segregated Au/Fe NPs up to 1000 K at B = 200 mT. The two components (Fe atoms in Au and residual Fe3O4 seeds) show up in two magnetic ordering temperatures as indicated by the mean field model fits. Note that this measurement has been performed using the identical powder, but about 1 year after the low temperature measurements. During this period, significant Fe diffusion to the particle surface and progression of oxidation may lead to increased Fe3O4 content in the magnetic measurements. Overall, we observe 3 distinct regions, while the total magnetic moment of the sample decreases with increasing T. In the interval between 300 K and 850 K, the Fe3O4 signal decreases, which is overlaid by the segregation of Fe atoms in Au towards metallic Fe (cf. TEM annealing series) and explains the first dip in Fig. 5c. It should be noted that the sample powder was embedded in Zircar cement containing residual water from cement dispersion. Therefore, the oxidation of segregated Fe is possible upon heating, and the relative contributions of Fe in Au, segregated metallic Fe and Fe3O4 seeds change during the measurement. To fit the data in the 670–830 K interval, we use a mean field approach with the exponent δ = 0.5, which gives the Curie temperature TC (Fe3O4) = 850 K, and the remaining component has TC = 1006 K, pointing towards small metallic Fe segments in the Au/Fe NPs with slightly reduced Curie temperature TC as compared to bulk metallic Fe TC = 1043 K.50 Note that the maximum temperature of the device is 1000 K.
Biomedical applications of AuFe
As a first step towards the biomedical application of initial AuFe solid solution NPs synthesized in this work (Fig. S5†), we measured their plasmon resonance with the maximum at 533 nm in the UV-VIS spectrum (Fig. S5a†) and demonstrated that the viability of human prostate cancer cell lines LNCaP and PC-3 after 48 hours of incubation with NPs is above 90% and 85%, respectively, for the entire tested concentration range up to 150 μg mL−1 Au (Fig. S5b†).Discussion
In this work, we make use of the widely used method of iterative hydrogen tetrachloroaurate reduction in the presence of magnetite seeds aiming for core–shell iron oxide–gold NPs. Astonishingly and in contrast to previous reports, comprehensive characterization of the product reveals the successful synthesis of solid solution fcc AuFe NPs by employing the methods of colloidal chemistry at ambient temperature. In this discussion, we have summarized our data and compared the present results step-by-step with the data previously reported in the literature.According to low-resolution TEM (Fig. 1a and b) the size of NPs increases after the reduction of hydrogen tetrachloroaurate from 9 ± 2 nm to 32 ± 5 nm for Fe3O4 seeds and solid solution AuFe NPs, respectively. Thus, heterogeneous Au nucleation is successful although the final size of NPs is less than that reported for NPs prepared by an analogous Au coating procedure (50 ± 5 nm).26 The formation of a core-shell structure consisting of a Fe3O4 core surrounded by an Au shell, however, is not identified. This significant difference with respect to the work by Lyon et al.26 is likely explained by the different iron oxide seed treatment with strong electrostatic stabilization with perchloric acid, similar to the ones reported by Lo et al.38 In this case, NPs are strongly separated from neighbors and can facilitate the formation of one Au-containing particle per seed.
Diffraction data, XRD and electron diffraction, do not confirm the presence of a core–shell architecture. Both the SAED pattern (Fig. 1c) and the X-ray diffractogram (Fig. 1d) only reveal fcc Au in the NPs. This result has been reported by many other groups who investigated the core–shell structures and has been ascribed to a “heavy atom effect” from the compact Au shell overlapping signals of the Fe3O4 core.40,51,52 We agree with Luchini et al.51 pointing out that this phenomenon is not self-explaining and in fact different reasons shall be discussed: (a) iron oxide seeds degrade during the reduction of hydrogen tetrachloroaurate, (b) gold overlays the diffraction peaks of iron oxide or (c) iron oxide is completely shielded from X-rays. In favor of hypotheses (a) and (b), Lo et al.38 obtained XRD-patterns of core–shell NPs with diffraction peaks from both Fe3O4 and Au phases. However, uncoated Fe3O4 NPs were also observed in their TEM micrographs and it remains unclear whether the XRD has been measured for purified core–shell NPs or the as-obtained mixture containing Fe3O4 NPs. One can also see that the XRD diffraction peaks of Au do not significantly overlap with the corresponding Fe3O4 pattern (Fig. 1d), which again excludes hypothesis (b).
Since electrons in the TEM can pass the NPs and X-rays penetrate much deeper at Co-Kα energy, we also exclude (c) for our experiments. Nonetheless, hypothesis (c) is widely discussed in the literature. For example, authors claimed the growth of core–shell NPs although the diffractogram only shows gold peaks originating from a shell thickness as small as 0.7 nm.40 Another group52 questioned the presence of magnetite signals for a gold shell thickness of 1.2 nm. A more systematic study of this phenomenon is carried out in ref. 53 and 54. Some authors preliminarily covered the magnetic core with gold nuclei53 and subsequently coated further to a final Au thickness ranging from 5 to 15 nm. Their diffraction patterns show the peaks of the magnetic core at the stage of coating with nuclei. After complete coverage, however, these disappear. Another study of the step-by-step gold coating of Fe3O4 presents diffractograms with the characteristic peaks of gold and magnetite with the latter somewhat weakened in comparison with the uncoated state.54 At gold shell thicknesses of 0.5 nm, 1 nm, 5 nm, and 8 nm, however, the most intense Fe3O4 peak (311) is still visible.
The confirmation of a core–shell morphology based on XRD and bright-field TEM data is thus highly vulnerable and additional information is needed. We performed high-resolution electron microscopy (HRTEM), scanning transmission electron microscopy (STEM) in combination with the energy-dispersive X-ray spectroscopy (EDX) mapping (Fig. 2). Using a similar set of techniques, Wagener et al. and Tymoczko et al.35,55 have provided a solid proof of the core–shell Fe@Au NP structure. Besides this work, unambiguous proofs of a defined magnetite core surrounded by a gold shell are only provided in very few studies.56,57 Yet, those measurements were performed without the corresponding EDX analysis, so the localization and distribution of chemical elements remain unclear.
In the present work, we were able to detect the so-called inclusions (areas of different contrast with diameters up to 8 nm) inside the fcc Au matrix using HRTEM (Fig. 2a and b). For many NPs, these inclusions are also visible in low-resolution TEM images (Fig. S6†). Initially, we ascribed these inclusions to partially dissolved Fe3O4 cores, therefore suggesting a cluster–shell structure of our NPs. This, however, is disproved by higher magnification TEM (Fig. 2c and d) demonstrating that the crystallographic planes and atomic columns of Au are not distorted in regions with reduced contrast. One of the possible explanations of this contrast inside NPs could be the presence of nanosized pores. However, the production of nanoporous gold usually requires a template synthesis and sophisticated etching protocols.58–60 Recently, an elegant way of quick nucleation and growth of Au NPs in the stability region of cetyltrimethylammonium bromide has been suggested by Depciuch et al.61 which still involves elevated temperatures up to 60–90 °C, and in our case, the synthesis was carried out at room temperature. Therefore, the presence of nanosized pores in Au is unlikely. Another explanation of inner particle contrast is that the inclusions are regions of higher Fe content. If we consider these regions of reduced contrast as the only source of Fe in the NPs, then the 0.26 mass% (cf. ICP-MS data above) does not fit the mass estimate of Fe being only 0.02 mass% considering the average “core” size of 3.4 ± 1.0 nm and the average NP diameter of 32 ± 5 nm. This brings us to the conclusion that more Fe atoms are stored in the Au matrix than that suggested by TEM. On the other hand, the 0.26 mass% of Fe in the NPs roughly half of the 0.6 mass% calculated assuming the initial low-resolution TEM data: 9 nm iron oxide core, 11.5 nm Au shell thickness (derived from the total NP size) and Fe3O4 and Au densities of 5.2 g cm−3 and 19.3 g cm−3, respectively.62,63 This can be explained either by the prevalence of pure Au NPs, which is unlikely as mentioned above, or by the fact that a core–shell model is not (fully) applicable for our system.
More hints about NPs architecture can be obtained by EDX analysis. Strikingly, the elemental mapping at room temperature (Fig. 2e) shows the homogeneously spread signal of both iron and gold, without oxygen. It is worth mentioning that similar EDX mapping results were reported by other authors;51,64 however, no reasonable explanation has been given so far why the predominant accumulation of the Fe and O signals in the center of NPs is missing as expected for magnetite cores. The authors51 also admit that this could indicate another type of structure, different from a core–shell geometry. In most of the studies, the overall EDX spectrum showing Fe and Au signals (which also holds for our NPs, cf. Fig. S7†), is used as a proof of the Fe3O4 core.26,38,52,65 This approach is, however, questionable since it indicates no correlation between the morphology of NPs and the elemental distribution.
Thus, we assume in the following that all features described above – the absence of a defined magnetic core, the size of NPs, the inclusions and the homogeneous distribution of both Fe and Au within the hybrids and the lack of oxygen – point to an AuFe solid solution. These NPs could be formed during heterogeneous nucleation of Au NPs on the Fe ions forming a metal cluster.
To the best of our knowledge, room-temperature synthesis of such AuFe hybrid NPs has never been reported. The most common methods for the synthesis of solid solution AuFe NPs include gas phase condensation,33,34 laser ablation,35–37 and high-temperature decomposition of metal–organic precursors.66,67 Although the latter gives high NP yield and can be performed in both water and organic solvents, it still requires complex laser equipment and involves extremely high cooling rates to “freeze” the metastable solid solution state at high Fe content in an Au matrix. Nevertheless, this technique allows the programmed synthesis of either core–shell or solid solution AuFe NPs depending on the target composition and the particle diameter.35,46,55 In any case, the structure of NPs was confirmed by HRTEM and EDX measurements, unambiguously showing that the Fe signal is either concentrated in the core or uniformly distributed over the entire NP.36 In the present study, the correlation between HRTEM and EDX is not straightforward, most probably, due to the small size and number of inclusions and insufficient EDX sensitivity. Thus, we carried out an in situ annealing experiment up to 700 °C (Fig. 3) demonstrating the segregation of Fe from the Au matrix. The TEM results are representative for the sample batch as proven by VSM (Fig. 5). Notably, the final Au/Fe hybrids are stable for at least 1 year of air storage (Fig. S8†) and the formation of an fcc Au–bcc Fe epitaxy in a Janus particle is demonstrated (Fig. 4). To the best of our knowledge, this is the first report of such an epitaxy for Au/Fe Janus hybrids. A similar fcc/bcc orientation relationship was observed in the epitaxial growth of Fe on Au(111),68 diffusion bonding in steels,69 martensite transition in Fe70 or interphase precipitation in low-carbon steels.71 Very recently, bcc Fe cores in Fe@Au core–shell nanoparticles have been reported.72
Based on the final Au/Fe Janus NP geometry discovered by HRTEM after annealing, we sought to estimate the amount of Fe that can be dissolved in Au for solid solution NPs since the structural transformation can only lead to the migration of Fe atoms while their total quantity remains constant. Considering the Fe and Au volumetric densities of 7.9 g cm−3 and 19.3 g cm−3, respectively, and calculating from the Fe-rich projected area the volume of a spherical sector, we end up with 4 mass% Fe in this AuFe NP, which corresponds to the stoichiometry of Fe13Au87 in the initial solid solution NP, similar to what has been described by Amendola et al. for pulsed laser ablation in liquids.73 If more Fe could be expelled from Au at temperatures above 700 °C, this estimate of Fe content should be read as a lower limit. Thus, the amount of iron in gold for our NPs is at least twice that of the solubility in the equilibrium state.
The AuFe solid solution investigated by TEM is naturally restricted to a small number of NPs while magnetometry of mg amounts gives access to the mean values of the entire batch. From the magnetic response, we determined the Fe amount to be 0.14 mass% in the AuFe NPs (or a stoichiometry Fe0.5Au99.5), which differs from the ICP-MS result (0.26 mass% and reflects the mean stoichiometry of Fe0.9Au99.1) after HCl etching. Considering the very small amount of Fe, however, the deviation is acceptable. More interesting is the comparison with the 4 mass% estimated by the annealing experiment (Fig. 3). This shows that the choice of the NP for the TEM investigations is important. We have chosen this specific NP because the larger inclusion in the as deposited state also suggests a larger Fe content in this AuFe NP which in turn means that this result rather reflects the upper limit of Fe in solid solution.
Magnetometry revealed the expected T−1-dependence for the isolated Fe atoms in the Au matrix above T = 50 K further supporting the structural and morphological investigations. After HCl etching, however, a small amount of Fe oxides remains, which hindered the further detailed evaluation of the AuFe solid solution at low fields and temperatures. Importantly, magnetometry at high temperatures reconstructed the TEM annealing experiment for the entire batch. We extracted two magnetic ordering temperatures, for Fe3O4 (TN = 850 K) and for the segregated Fe in Au/Fe NPs (TC = 1006 K). The latter is reduced as compared to the volumetric value of 1043 K which we ascribed to the small size of segregated Fe segments in the Janus particles.
Towards the applications of the solid solution AuFe NPs, we obtained preliminary data of their optical properties (Fig. S5†), which proved the presence of the expected plasmon resonance peak at 533 nm. Remarkably, the Fe dissolved in the Au matrix does not alter the Au plasmon peak significantly at similar sizes.74–76 Further investigations of the optical properties of Au/Fe segregated NPs are certainly interesting but out-of-focus of the current work.
Besides magneto-optical and magneto-plasmonic applications the biomedical use of both, solid solution AuFe and segregated Au/Fe NPs, should be considered in the future. In the first step, we tested the toxicity to human cancer cell lines LNCaP and PC-3. The results indicate that the NPs are non-toxic to these cells after 48 hours of co-incubation. In order to optimize the large-scale production of Janus Au/Fe NPs with segregated iron and gold phases, it is conceivable to exchange the polymeric coating on AuFe NPs with a temperature-resistant shell such as silica using well-established protocols.77 Such shell would withstand the necessary annealing step for segregation. Another option would be the ex situ laser irradiation of the solid solution that could heat up the NPs to several hundred °C assisted by the plasmon resonance in the ns time regime. This would significantly speed up the Fe segregation process. Therefore, we believe that this new preparation route will allow for a variety of theranostic applications such as magnetic hyperthermia combined with photothermal therapy and dual-mode contrast agents for magnetic resonance imaging and computer tomography.
Conclusions
Room-temperature synthesis of solid solution fcc AuFe NPs is demonstrated. Upon annealing to 700 °C, the elements segregate to form Au/Fe Janus particles with ferromagnetic bcc Fe epitaxially matched to low index fcc Au planes. Based on our findings, previous reports using similar protocols may have to be revisited. AuFe solid solution and Au/Fe Janus particles are very interesting for biomedical applications and modelling the underlying physics. Since upscaling of the synthesis method seems achievable, this preparation route delivers new perspective materials for magnetoplasmonics and theranostics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. V. E. gratefully acknowledges the support from the Humboldt Research Fellowship for Postdoctoral Researchers provided by the Alexander von Humboldt Foundation and the support from the Add-on Fellowship for Interdisciplinary Life Science provided by the Joachim Herz Foundation. N. L. K. gratefully acknowledges the support from the Russian Foundation for Basic Research, grant number 18-29-09154.References
- S. A. Bansal, V. Kumar, J. Karimi, A. P. Singh and S. Kumar, Nanoscale Adv., 2020, 2, 3764–3787 RSC.
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256–2282 RSC.
- O. N. Metelkina, R. W. Lodge, P. G. Rudakovskaya, V. M. Gerasimov, C. H. Lucas, I. S. Grebennikov, I. V. Shchetinin, A. G. Savchenko, G. E. Pavlovskaya, G. A. Rance, M. del Carmen Gimenez-Lopez, A. N. Khlobystov and A. G. Majouga, J. Mater. Chem., 2017, 5, 2167–2174 CAS.
- S. Pshenichnikov, A. Omelyanchik, M. Efremova, M. Lunova, N. Gazatova, V. Malashchenko, O. Khaziakhmatova, L. Litvinova, N. Perov, L. Panina, D. Peddis, O. Lunov, V. Rodionova and K. Levada, J. Magn. Magn. Mater., 2021, 523, 167623 CrossRef CAS.
- Y. A. Nalench, I. V. Shchetinin, A. S. Skorikov, P. S. Mogilnikov, M. Farle, A. G. Savchenko, A. G. Majouga, M. A. Abakumov and U. Wiedwald, J. Mater. Chem. B, 2020, 8, 3886–3895 RSC.
- Q. Dong, H. Yang, C. Wan, D. Zheng, Z. Zhou, S. Xie, L. Xu, J. Du and F. Li, Nanoscale Res. Lett., 2019, 14, 235 CrossRef PubMed.
- M. V. Efremova, V. A. Naumenko, M. Spasova, A. S. Garanina, M. A. Abakumov, A. D. Blokhina, P. A. Melnikov, A. O. Prelovskaya, M. Heidelmann, Z.-A. Li, Z. Ma, I. V. Shchetinin, Y. I. Golovin, I. I. Kireev, A. G. Savchenko, V. P. Chekhonin, N. L. Klyachko, M. Farle, A. G. Majouga and U. Wiedwald, Sci. Rep., 2018, 8, 11295 CrossRef PubMed.
- R. A. Revia and M. Zhang, Mater. Today, 2016, 19, 157–168 CrossRef CAS PubMed.
- A. Tomitaka, H. Arami, A. Raymond, A. Yndart, A. Kaushik, R. D. Jayant, Y. Takemura, Y. Cai, M. Toborek and M. Nair, Nanoscale, 2017, 9, 764–773 RSC.
- E. Kozenkova, K. Levada, M. V. Efremova, A. Omelyanchik, Y. A. Nalench, A. S. Garanina, S. Pshenichnikov, D. G. Zhukov, O. Lunov, M. Lunova, I. Kozenkov, C. Innocenti, M. Albino, M. A. Abakumov, C. Sangregorio and V. Rodionova, Nanomaterials, 2020, 10, 1646 CrossRef CAS PubMed.
- H. Cai, K. Li, J. Li, S. Wen, Q. Chen, M. Shen, L. Zheng, G. Zhang and X. Shi, Small, 2015, 11, 4584–4593 CrossRef CAS PubMed.
- D. Maniglio, F. Benetti, L. Minati, J. Jovicich, A. Valentini, G. Speranza and C. Migliaresi, Nanotechnology, 2018, 29, 315101 CrossRef CAS PubMed.
- G. Brennan, N. D. Thorat, M. Pescio, S. Bergamino, J. Bauer, N. Liu, S. A. M. Tofail and C. Silien, Nanoscale, 2020, 12, 12632–12638 RSC.
- P. Guardia, S. Nitti, M. E. Materia, G. Pugliese, N. Yaacoub, J.-M. Greneche, C. Lefevre, L. Manna and T. Pellegrino, J. Mater. Chem. B, 2017, 5, 4587–4594 RSC.
- K. C.-F. Leung, S. Xuan, X. Zhu, D. Wang, C.-P. Chak, S.-F. Lee, W. K.-W. Ho and B. C.-T. Chung, Chem. Soc. Rev., 2012, 41, 1911–1928 RSC.
- A. Tomitaka, S. Ota, K. Nishimoto, H. Arami, Y. Takemura and M. Nair, Nanoscale, 2019, 11, 6489–6496 RSC.
- M. V. Efremova, M. M. Veselov, A. V. Barulin, S. L. Gribanovsky, I. M. Le-Deygen, I. V. Uporov, E. V. Kudryashova, M. Sokolsky-Papkov, A. G. Majouga, Y. I. Golovin, A. V. Kabanov and N. L. Klyachko, ACS Nano, 2018, 12, 3190–3199 CrossRef CAS PubMed.
- A. Majouga, M. Sokolsky-Papkov, A. Kuznetsov, D. Lebedev, M. Efremova, E. Beloglazkina, P. Rudakovskaya, M. Veselov, N. Zyk, Y. Golovin, N. Klyachko and A. Kabanov, Colloids Surf., B, 2015, 125, 104–109 CrossRef CAS PubMed.
- R. Henning, F. Liebig, C. Prietzel, B. Klemke and J. Koetz, Colloids Surf., A, 2020, 600, 124913 CrossRef CAS.
- P. G. Rudakovskaya, D. N. Lebedev, M. V. Efremova, E. K. Beloglazkina, N. V. Zyk, N. L. Klyachko, Y. I. Golovin, A. G. Savchenko and A. G. Mazhuga, Nanotechnol. Russ., 2016, 11, 144–152 CrossRef CAS.
- P. Quaresma, I. Osório, G. Dória, P. A. Carvalho, A. Pereira, J. Langer, J. P. Araújo, I. Pastoriza-Santos, L. M. Liz-Marzán, R. Franco, P. V. Baptista and E. Pereira, RSC Adv., 2013, 4, 3659–3667 Search PubMed.
- T. T. Nguyen, F. Mammeri and S. Ammar, Nanomaterials, 2018, 8, 149 CrossRef PubMed.
- S. V. Salihov, Y. A. Ivanenkov, S. P. Krechetov, M. S. Veselov, N. V. Sviridenkova, A. G. Savchenko, N. L. Klyachko, Y. I. Golovin, N. V. Chufarova, E. K. Beloglazkina and A. G. Majouga, J. Magn. Magn. Mater., 2015, 394, 173–178 CrossRef CAS.
- T. Härtling, T. Uhlig, A. Seidenstücker, N. C. Bigall, P. Olk, U. Wiedwald, L. Han, A. Eychmüller, A. Plettl, P. Ziemann and L. M. Eng, Appl. Phys. Lett., 2010, 96, 183111 CrossRef.
- T. Uhlig, U. Wiedwald, A. Seidenstücker, P. Ziemann and L. M. Eng, Nanotechnology, 2014, 25, 255501 CrossRef PubMed.
- J. L. Lyon, D. A. Fleming, M. B. Stone, P. Schiffer and M. E. Williams, Nano Lett., 2004, 4, 719–723 CrossRef CAS.
- J. Ren, F. Wang, G. Wei, Y. Yang, Y. Liu, M. Wei, Y. Huan, A. C. Larson and Z. Zhang, PLoS One, 2012, 7, e38350 CrossRef CAS PubMed.
- Y. Li, J. Liu, Y. Zhong, J. Zhang, Z. Wang, L. Wang, Y. An, M. Lin, Z. Gao and D. Zhang, Int. J. Nanomed., 2011, 6, 2805–2819 CrossRef CAS PubMed.
- X. Chao, F. Shi, Y.-Y. Zhao, K. Li, M.-L. Peng, C. Chen and Y.-L. Cui, Pharmazie, 2010, 65, 500–504 CAS.
- J. Lee, Y. Lee, J. K. Youn, H. B. Na, T. Yu, H. Kim, S.-M. Lee, Y.-M. Koo, J. H. Kwak, H. G. Park, H. N. Chang, M. Hwang, J.-G. Park, J. Kim and T. Hyeon, Small, 2008, 4, 143–152 CrossRef CAS PubMed.
- C. Hui, C. Shen, J. Tian, L. Bao, H. Ding, C. Li, Y. Tian, X. Shi and H.-J. Gao, Nanoscale, 2011, 3, 701–705 RSC.
- H. Fuse, N. Koshizaki, Y. Ishikawa and Z. Swiatkowska-Warkocka, Nanomaterials, 2019, 9, 198 CrossRef CAS PubMed.
- V. Velasco, D. Pohl, A. Surrey, A. Bonatto-Minella, A. Hernando, P. Crespo and B. Rellinghaus, Nanotechnology, 2014, 25, 215703 CrossRef CAS PubMed.
- Z. Fan and H. Zhang, Chem. Soc. Rev., 2016, 45, 63–82 RSC.
- P. Wagener, J. Jakobi, C. Rehbock, V. S. K. Chakravadhanula, C. Thede, U. Wiedwald, M. Bartsch, L. Kienle and S. Barcikowski, Sci. Rep., 2016, 6, 23352 CrossRef CAS PubMed.
- A. Tymoczko, M. Kamp, O. Prymak, C. Rehbock, J. Jakobi, U. Schürmann, L. Kienle and S. Barcikowski, Nanoscale, 2018, 10, 16434–16437 RSC.
- D. Zhang, B. Gökce and S. Barcikowski, Chem. Rev., 2017, 117, 3990–4103 CrossRef CAS PubMed.
- C. K. Lo, D. Xiao and M. M. F. Choi, J. Mater. Chem., 2007, 17, 2418–2427 RSC.
- L. Heinemann, G. R. Simpson, A. Boxall, T. Kottke, K. L. Relph, R. Vile, A. Melcher, R. Prestwich, K. J. Harrington, R. Morgan and H. S. Pandha, BMC Cancer, 2011, 11, 221 CrossRef CAS PubMed.
- L. Wang, J. Luo, Q. Fan, M. Suzuki, I. S. Suzuki, M. H. Engelhard, Y. Lin, N. Kim, J. Q. Wang and C.-J. Zhong, J. Phys. Chem. B, 2005, 109, 21593–21601 CrossRef CAS PubMed.
- H. Montazeri, A. Amani, H. R. Shahverdi, E. al D. Haratifar and A. R. Shahverdi, J. Nanostruct. Chem., 2013, 3, 25 CrossRef.
- F. Shi, W. Hui, Y. Cui and C. Chen, Nano, 2011, 06, 145–151 CrossRef CAS.
- S. V. Salikhov, A. G. Savchenko, I. S. Grebennikov and E. V. Yurtov, Bull. Russ. Acad. Sci.: Phys., 2015, 79, 1106–1112 CAS.
- M. V. Efremova, Y. A. Nalench, E. Myrovali, A. S. Garanina, I. S. Grebennikov, P. K. Gifer, M. A. Abakumov, M. Spasova, M. Angelakeris, A. G. Savchenko, M. Farle, N. L. Klyachko, A. G. Majouga and U. Wiedwald, Beilstein J. Nanotechnol., 2018, 9, 2684–2699 CrossRef CAS PubMed.
- H. S. C. O'Neill and W. A. Dollase, Phys. Chem. Miner., 1994, 20, 541–555 CrossRef.
- M. Kamp, A. Tymoczko, U. Schürmann, J. Jakobi, C. Rehbock, K. Rätzke, S. Barcikowski and L. Kienle, Cryst. Growth Des., 2018, 18, 5434–5440 CrossRef CAS.
- A. R. Kaufmann, S. T. Pan and J. R. Clark, Rev. Mod. Phys., 1945, 17, 87–92 CrossRef CAS.
- M. E. Gruner, J. Phys. D: Appl. Phys., 2010, 43, 474008 CrossRef.
- H. C. Herper, E. Hoffmann and P. Entel, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 3839–3848 CrossRef CAS.
- K. M. Krishnan, Fundamentals and Applications of Magnetic Materials, Oxford University Press, 2016 Search PubMed.
- A. Luchini, G. Vitiello, F. Rossi, O. Ruiz De Ballesteros, A. Radulescu, G. D'Errico, D. Montesarchio, C. de Julián Fernández and L. Paduano, Phys. Chem. Chem. Phys., 2015, 17, 6087–6097 RSC.
- I. Robinson, L. D. Tung, S. Maenosono, C. Wälti and N. T. K. Thanh, Nanoscale, 2010, 2, 2624–2630 RSC.
- H.-Y. Xie, R. Zhen, B. Wang, Y.-J. Feng, P. Chen and J. Hao, J. Phys. Chem. C, 2010, 114, 4825–4830 CrossRef CAS.
- M. Mandal, S. Kundu, S. K. Ghosh, S. Panigrahi, T. K. Sau, S. M. Yusuf and T. Pal, J. Colloid Interface Sci., 2005, 286, 187–194 CrossRef CAS PubMed.
- A. Tymoczko, M. Kamp, C. Rehbock, L. Kienle, E. Cattaruzza, S. Barcikowski and V. Amendola, Nanoscale Horiz., 2019, 4, 1326–1332 RSC.
- Y. Jin, C. Jia, S.-W. Huang, M. O'Donnell and X. Gao, Nat. Commun., 2010, 1, 41 CrossRef PubMed.
- X. Zhao, Y. Cai, T. Wang, Y. Shi and G. Jiang, Anal. Chem., 2008, 80, 9091–9096 CrossRef CAS PubMed.
- A. G. M. da Silva, T. S. Rodrigues, S. J. Haigh and P. H. C. Camargo, Chem. Commun., 2017, 53, 7135–7148 RSC.
- S. Pedireddy, H. K. Lee, W. W. Tjiu, I. Y. Phang, H. R. Tan, S. Q. Chua, C. Troadec and X. Y. Ling, Nat. Commun., 2014, 5, 4947 CrossRef CAS PubMed.
- G. W. Nyce, J. R. Hayes, A. V. Hamza and J. H. Satcher, Chem. Mater., 2007, 19, 344–346 CrossRef CAS.
- J. Depciuch, M. Stec, A. Maximenko, J. Baran and M. Parlinska-Wojtan, J. Mater. Sci., 2020, 55, 5257–5267 CrossRef CAS.
- T. L. Brown, H. E. LeMay, B. E. Bursten and B. E. Bursten, Chemistry: the central science, Prentice Hall Englewood Cliffs, NJ, 1994, vol. 8 Search PubMed.
- L. Blaney, Lehigh Rev., 2007, 15, 33 Search PubMed.
- I. Y. Goon, L. M. H. Lai, M. Lim, P. Munroe, J. J. Gooding and R. Amal, Chem. Mater., 2009, 21, 673–681 CrossRef CAS.
- T. T. Hien Pham, C. Cao and S. J. Sim, J. Magn. Magn. Mater., 2008, 320, 2049–2055 CrossRef CAS.
- H. L. Liu, J. H. Wu, J. H. Min and Y. K. Kim, J. Appl. Phys., 2008, 103, 07D529 CrossRef.
- I.-C. Chiang and D.-H. Chen, Adv. Funct. Mater., 2007, 17, 1311–1316 CrossRef CAS.
- H. F. Jurca, A. Damian, C. Gougaud, D. Thiaudière, R. Cortès, F. Maroun and P. Allongue, J. Phys. Chem. C, 2016, 120, 16080–16089 CrossRef CAS.
- Y. Gao, Z. Wang, Y. Liu, W. Li, C. Liu and H. Li, Metals, 2018, 8, 1012 CrossRef CAS.
- L. Sandoval, H. M. Urbassek and P. Entel, New J. Phys., 2009, 11, 103027 CrossRef.
- Y.-J. Zhang, G. Miyamoto, K. Shinbo and T. Furuhara, Scr. Mater., 2013, 69, 17–20 CrossRef CAS.
- M. Kamp, A. Tymoczko, R. Popescu, U. Schürmann, R. Nadarajah, B. Gökce, C. Rehbock, D. Gerthsen, S. Barcikowski and L. Kienle, Nanoscale Adv., 2020, 2, 3912–3920 RSC.
- V. Amendola, M. Meneghetti, O. M. Bakr, P. Riello, S. Polizzi, D. H. Anjum, S. Fiameni, P. Arosio, T. Orlando, C. de Julian Fernandez, F. Pineider, C. Sangregorio and A. Lascialfari, Nanoscale, 2013, 5, 5611–5619 RSC.
- D. L. Fedlheim and C. A. Foss, Metal Nanoparticles: Synthesis, Characterization, and Applications, CRC Press, 2001 Search PubMed.
- P. N. Njoki, I.-I. S. Lim, D. Mott, H.-Y. Park, B. Khan, S. Mishra, R. Sujakumar, J. Luo and C.-J. Zhong, J. Phys. Chem. C, 2007, 111, 14664–14669 CrossRef CAS.
- V. Amendola, S. Scaramuzza, S. Agnoli, S. Polizzi and M. Meneghetti, Nanoscale, 2014, 6, 1423–1433 RSC.
- L. M. Liz-Marzán, M. Giersig and P. Mulvaney, Langmuir, 1996, 12, 4329–4335 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1: Size distribution of Fe3O4 NPs and AuFe NPs; Fig. S2: Bright-field TEM image of AuFe NPs on a carbon-coated Cu grid before purification; Fig. S3: Magnetic characterization of Fe3O4 seeds and AuFe NPs; Fig. S4: Size distribution of inclusions (regions of a different contrast) in the TEM images of the AuFe NPs; Fig. S5: Properties of AuFe NPs for bio-applications; Fig. S6: TEM overview image indicating the inclusions. Fig. S7: Overview EDX spectrum of AuFe NPs. Fig. S8: HAADF-STEM of Au/Fe Janus particles 1 year after annealing. See DOI: 10.1039/d1nr00383f |
| ‡ Current address: Department of Nuclear Medicine, TUM School of Medicine, Technical University of Munich, 81675 Munich, Germany. E-mail: E-mail: mariia.efremova@tum.de |
| This journal is © The Royal Society of Chemistry 2021 |