
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalable nanoprecipitation of niclosamide and in vivo demonstration of long-acting delivery after intramuscular injection†
James J.
Hobson
 ab,
Alison C.
Savage
ab,
Andrew B.
Dwyer
ab,
Catherine
Unsworth
ab,
Jonathan
Massam
ab,
Usman
Arshad
bc,
Henry
Pertinez
bc,
Helen
Box
bc,
Lee
Tatham
bc,
Rajith K. R.
Rajoli
bc,
Megan
Neary
bc,
Joanne
Sharp
bc,
Anthony
Valentijn
bc,
Christopher
David
bc,
Paul
Curley
bc,
Neill J.
Liptrott
bc,
Tom O.
McDonald
ab,
Andrew
Owen
*bcd and
Steve P.
Rannard
*abd
ab,
Alison C.
Savage
ab,
Andrew B.
Dwyer
ab,
Catherine
Unsworth
ab,
Jonathan
Massam
ab,
Usman
Arshad
bc,
Henry
Pertinez
bc,
Helen
Box
bc,
Lee
Tatham
bc,
Rajith K. R.
Rajoli
bc,
Megan
Neary
bc,
Joanne
Sharp
bc,
Anthony
Valentijn
bc,
Christopher
David
bc,
Paul
Curley
bc,
Neill J.
Liptrott
bc,
Tom O.
McDonald
ab,
Andrew
Owen
*bcd and
Steve P.
Rannard
*abd
aDepartment of Chemistry, University of Liverpool, Crown Street, L69 7ZD, UK. E-mail: srannard@liverpool.ac.uk
bCentre of Excellence in Long-acting Therapeutics (CELT), University of Liverpool, Liverpool, L7 3NY, UK
cDepartment of Pharmacology and Therapeutics, University of Liverpool, Liverpool, L7 3NY, UK
dMaterials Innovation Factory, University of Liverpool, Crown Street, L69 7ZD, UK
First published on 20th March 2021
Abstract
The control of COVID-19 across the world requires the formation of a range of interventions including vaccines to elicit an immune response and immunomodulatory or antiviral therapeutics. Here, we demonstrate the nanoparticle formulation of a highly insoluble drug compound, niclosamide, with known anti SARS-CoV-2 activity as a cheap and scalable long-acting injectable antiviral candidate.
Introduction
The global healthcare emergency created by the emergence and international transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has focussed industrial and academic researchers on interventions that can reduce the considerable societal, economic and health impacts. Vaccine development has been enabled by considerable funding and unprecedented international multidisciplinary collaboration; several clinical options are now available with additional candidates undergoing extensive evaluation.New therapeutic options have been slow to become established and the early focus of repurposing current medicines was based on the premise that the active pharmaceutical ingredients (APIs) contained within current clinical products would impact the circulating virus despite these options being formulated and optimised for numerous different indications.1 Early modelling studies showed that many APIs would suffer from the common issue of low exposures relative to in vitro targets; simply, oral dosing would not be able to generate the appropriate circulating concentrations required to match concentrations demonstrated to be required for antiviral activity. To date, only three APIs have approval for coronavirus disease 2019 (COVID-19) therapy, namely: dexamethasone (UK, Japan); favilavir (China, Italy, Russia); and remdesivir (US, Japan and Australia). In addition, emergency use authorisation in the US has been given for convalescent plasma, a remdesivir/baricitinib combination and bamlanivimab (monoclonal antibody).2 Very recent reports also suggest that benefits for severely ill patients may be achieved with tocilizumab and sarilumab.3
Repeated oral dosing requires patient populations to be highly adherent to dosing regimens to ensure clinical outcomes are achieved. Poor adherence, or non-adherence, can cause considerable complications including the emergence of drug resistance. Long-acting injectable (LAI) therapies have become a major focus for a range of indications after the considerable clinical success of therapies for indications ranging from schizophrenia and contraception to hormone therapies and alcohol dependence.4 The creation of LAI options has utilised several technologies and aqueous micro/nanodispersions have been shown to have numerous benefits over oil-based solutions of poorly water-soluble drugs which may lead to pain at the injection site that has been reported in some cases to persist for several months.
Niclosamide (NCL) has been shown to be highly active against SARS-CoV5 and SARS-CoV-26in vitro, with recent reports demonstrating an endocytic pH modification mechanism that inhibits viral entry. NCL has extremely low water solubility which is variably reported between 0.23–1.6 μg mL−1 under ambient conditions7,8 and subsequently has very poor bioavailability that is likely to prohibit direct repurposing for SARS-CoV-2;9 it is a cheap API that is typically used as an anthelminthic therapy but also antagonises TMEM16A in vitro and ex vivo leading to the suggestion of bronchodilation.10 Due to NCL also having known influenza and anticancer activity, numerous approaches, such as wet bead milling11 and high-pressure homogenisation,12 have been reported in attempts to provide aqueous formulations to improve systemic exposures. These have very recently also been focussed on SARS-CoV-2 therapy using nanomedicine approaches that include albumin nanoparticles,13 single-capillary electrospray processing,8 chitosan nanocarriers,14 biopolymer derived nanocarriers,15 nanoparticulate prodrugs16 and lipidic nanoparticles.17 There is a potential opportunity for LAI administration of NCL if injectable formulations with high drug concentrations can be achieved using scalable approaches. Additionally, the formation of formulations that may be dispersed at the point of need may overcome several supply chain issues experienced with vaccines and often seen with liquid injectable formulations.
Here, we demonstrate the use of nanoprecipitation to form redispersible solid drug nanoparticle (SDN) formulations that may be stored as solids, reconstituted with water and utilised as LAIs to provide extended drug exposure of 1 month. Importantly, SDNs are stabilised particles that consist entirely of the API and do not require nanocarrier encapsulation. As such, they do not suffer from limited drug loadings within the nanoparticle vehicle and offer a clinical option that may provide high injectable drug concentrations in water to minimise injection volume, Fig. 1; our recent studies have achieved >300 mg mL−1 for malaria prophylaxis candidates.18
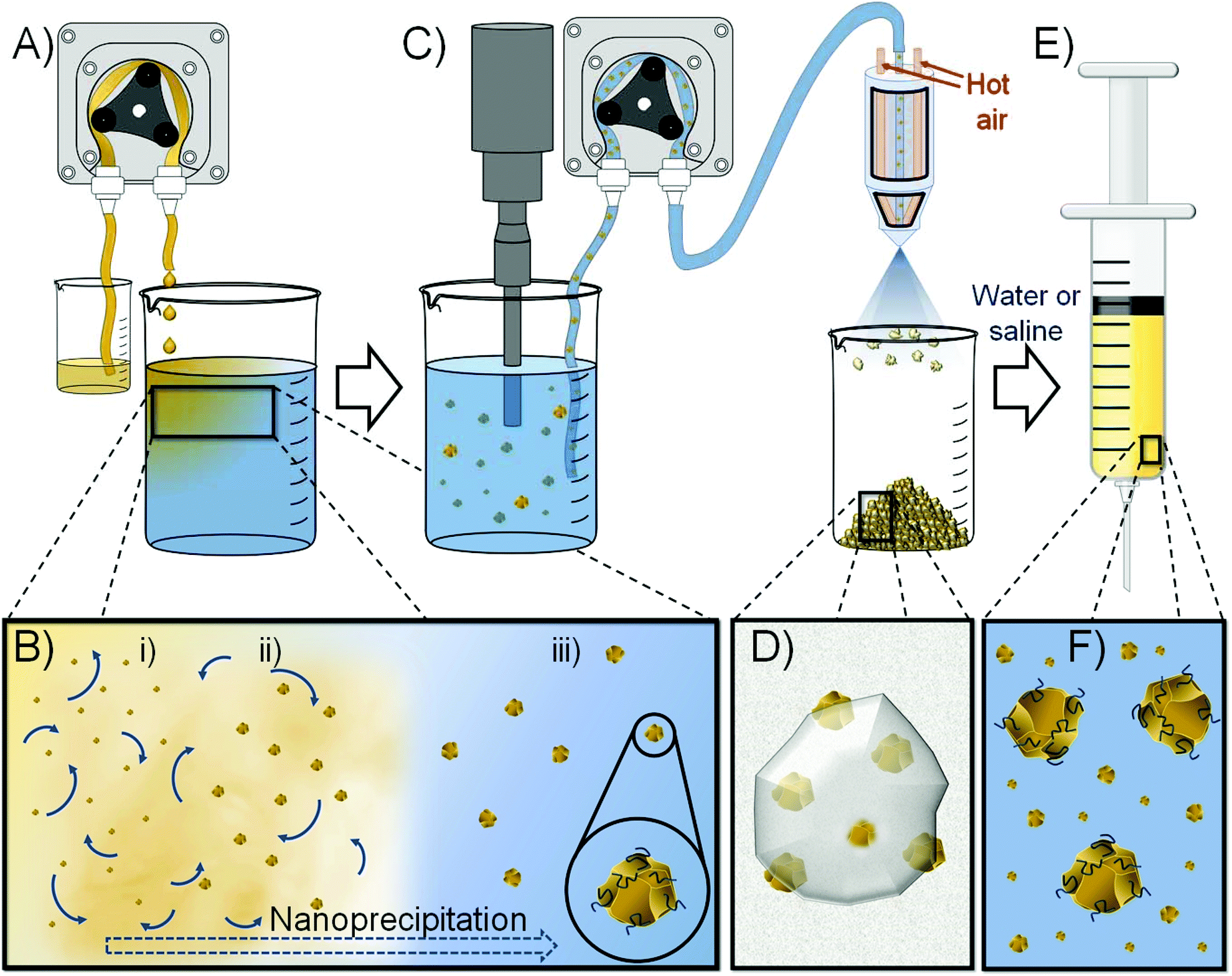 | ||
| Fig. 1 Schematic overview of the nanoprecipitation and spray drying process to form niclosamide (NCL) nanodispersions for long-acting injectable delivery. (A) Solution of NCL is pumped into an aqueous solution of stabilisers and sugars; (B) representation of the nucleation and growth of NCL nanoparticles as the good solvent is diluted by the aqueous bad solvent and particle growth is arrested by the stabilisers; (C) pumping of the nanoparticle dispersion after sonication into a spray dryer (spray dryer nozzle and beaker shown to simplify image); (D) spray dried powder contains water soluble excipients and NCL nanoparticles; (E and F) nanoparticles are redispersed in aqueous media for injection. | ||
Results and discussion
SDNs may be generated by a range of attrition-based manufacturing routes including nano-milling19 and high-pressure homogenisation,20 both of which are ‘top-down’ high energy processes that may damage APIs. To overcome the potential issues of ‘top-down’ processing, we have previously utilised ‘bottom-up’ approaches that manipulate the removal of organic solvents from solutions of API using techniques such as emulsion templated freeze drying (ETFD) or emulsion spray drying (ESD). These techniques have recently allowed the phase I human evaluation of orally dosed novel antiretroviral formulations21 (ClinicalTrials.gov Identifier: NCT02631473) and demonstrated the potential for LAI formulations for malaria prophylaxis.18NCL is particularly poorly soluble in a range of organic solvents, although detailed information was not readily available for many options typically employed in ‘bottom-up’ ETFD and ESD such as dichloromethane and chloroform. Solutions with appreciable concentrations are required for any scalable non-attrition process, therefore several organic solvents were screened and no single solvent was found that was able to produce concentrations at ≥50 mg mL−1 (ESI, Table S1†). Acetone was shown to produce homogeneous solutions at 10 mg mL−1 under ambient conditions and up to 50 mg mL−1 with heating to 65 °C; however, these solutions quickly phase separated when removed from heat.
Various combinations of the water miscible Class 3 solvents 2-butanone, ethanol and acetone provided relatively stable solutions of NCL at concentrations >50 mg mL−1 with heating. For example, a 2-butanone/ethanol (80/20 v/v) mixture was able to generate an NCL concentration of 75 mg mL−1 with heating to 60 °C, but maximum concentrations allowing further solution processing without considerable phase separation/crystallisation of 40, 60 and 65 mg mL−1 were only possible with ethanol/acetone, acetone/ethanol and 2-butanone/ethanol (80/20 v/v) mixtures, respectively. Acetone/ethanol was selected for evaluation as the 60 mg mL−1 solution offered superior stability after heating was removed.
The formation of new formulations with clinical relevance and the potential for translation to patient therapies is highly accelerated if stabilisers and excipients that have already been used in medicines are employed. The US Food and Drug Administration Center for Drug Evaluation and Research list of Inactive Ingredients was used to identify possible formulation ingredients for the formation of injectable nanoparticulate NCL dispersions; importantly, all candidate excipients also required significant solubility in the selected organic solvent combination to avoid precipitation during processing. A short-list of excipients used in current parenteral products with appreciable solubility was therefore generated, Table 1, and screening of nanoprecipitation processes was conducted on a 900 mg (NCL) scale.
Initial flash nanoprecipitation studies targeted an NCL drug content of 50% w/w relative to total solids. Rapid addition of a 60 mg mL−1 NCL solution (acetone/ethanol 80/20 v/v) with rapid stirring to an aqueous solution comprising a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w mixture of excipients led to a range of outcomes including the formation of several stable particle dispersions. The stable particle dispersions were transferred to a benchtop spray dryer and dried to yield a free-flowing powder with a 50/25/25 w/w ratio of NCL and the two excipients present within the aqueous non-solvent solution.
:1 w/w mixture of excipients led to a range of outcomes including the formation of several stable particle dispersions. The stable particle dispersions were transferred to a benchtop spray dryer and dried to yield a free-flowing powder with a 50/25/25 w/w ratio of NCL and the two excipients present within the aqueous non-solvent solution.
The dry powders were readily redispersed in saline or deionised water and analysed by dynamic light scattering (DLS) using a Malvern Panalytical ZetaSizer Ultra Photon Correlation Spectroscope. Hydrodynamic diameters (Dz) varied from 730 nm–4.3 μm with the smallest Dz values obtained from an excipient mixture comprising hydroxypropyl methylcellulose (HPMC; Mn = 10000 g mol−1) and sucrose. A 2-fold scale up (150 mL) of this nanoprecipitation was conducted as previously; however, a probe sonicator was utilised to ensure good mixing prior to spray drying and solutions/dispersions were controllably delivered using peristaltic pumping, Fig. 1. Scale-up may require the optimisation of mixing using options such as high shear mixer geometries.
Redispersion of the collected spray-dried powder from the scaled-up process into deionised water yielded a particle dispersion with Dz = 555 nm and dispersity (PDI) = 0.168, Fig. 2. Attempts to increase the drug content of the powder by repeating the process at a ratio of 60/20/20 w/w of NCL and the two excipients produced dispersions with slightly higher overall Dz values (960 nm) but a narrower distribution, Fig. 2, and excellent recovered yield (78%).
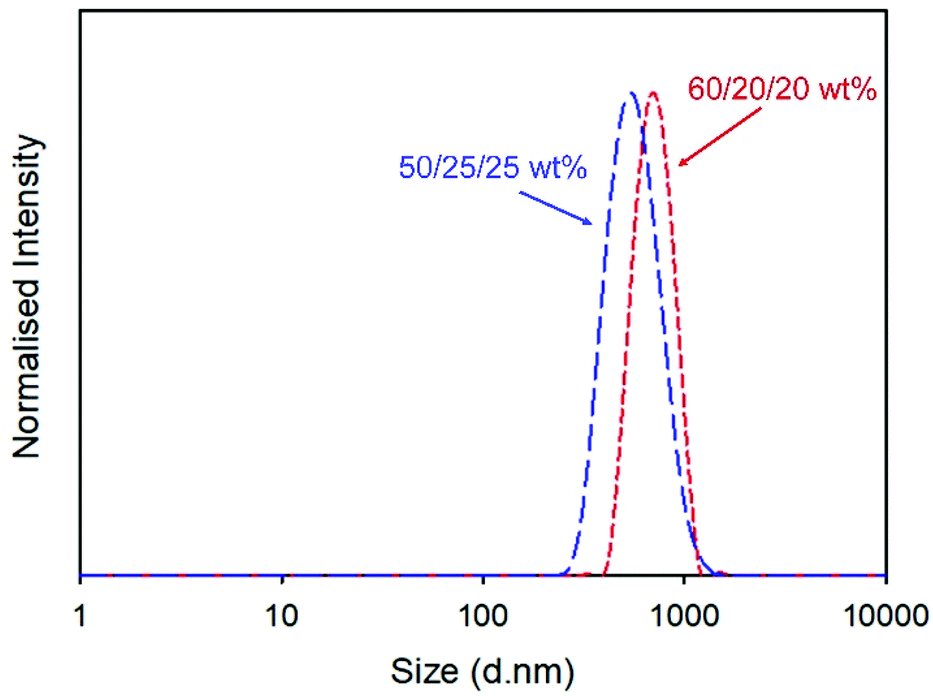 | ||
| Fig. 2 Distributions of redispersed NCL nanoprecipitates containing 50/25/25 (blue long dashed line) and 60/20/20 (red short dashed line) w/w formulations of NCL/HPMC/sucrose (1 mg mL−1) as determined by dynamic light scattering. Average of 3 repeated measurements shown. | ||
Confirmation of drug loading at 60 wt% relative to excipients in the recovered powders was achieved using 1H nuclear magnetic spectroscopy utilising a benzyl methacrylate (BzMA) internal standard at known concentration (5 mg mL−1) and utilising the empty region within the NCL spectrum between □ = 5.5–6.75 ppm; all components of the nanoprecipitated powder were soluble in deuterated dimethyl formamide and the non-overlapping aromatic resonance of NCL at approximately 8.75 ppm was compared to BzMA to establish the NCL concentration and subsequently the drug loading (ESI, Fig. S1†).
Nanoprecipitation has been widely studied for the formation of polymeric nanoparticles, either blank or drug containing, and also for the controlled formation of nanoparticles from organic compounds (including SDNs); the process has been commercialised at considerable scale and is used in the formation of beta-carotene particles for food, agriculture and beverage applications.32–34 The specific mechanism of nanoprecipitation has not been fully elucidated and models describing both nucleation-and-diffusion-limited-growth and diffusion-limited-cluster-cluster-aggregation have been described.35 In both cases, prevention of macroscopic particle formation is critical to maintain the formation of nanoparticles and polymers such as polysaccharides have been reported to deliver effective steric stabilisation.36–38 It is interesting to note that the observed Dz values increased with decreasing HPMC concentration as would be expected from a steric stabilisation mechanism. Our current understanding is that sucrose, as a water soluble hydrophilic and uncharged small molecule, is not specifically contributing to the stabilisation of the nanoprecipitation process but does act to separate the NCL nanoparticles in the final powder and allow redispersion.
The long-acting nature of NCL delivery from the nanoprecipitates was studied in vivo in Sprague Dawley rats. Pharmacokinetic (PK) studies of the drug were conducted over a 28-day period to establish the release kinetics from three different intramuscular (thigh) doses, namely 50, 100 and 200 mg kg−1. The spray-dried powder was redispersed in sterile water at NCL concentrations of 43.75, 87.5 and 175 mg mL−1 to enable different doses and the dispersions were administered using a 25-gauge needle (ESI, Fig. S2 and S3†). Each rat received two 0.2 mL injections and blood samples were taken via a tail vein bleed at intervals during the study. Plasma concentrations were determined using a validated liquid chromatography mass spectrometry method.39 Plasma PK data was fitted with an empirical biexponential model with first order input as a convenience function to provide a summary description of the PK profiles as a function of dose and give parameter estimates for the half-lives of the exponential phases, area under the curve (AUC) and apparent clearance. Model fitting was carried out via nonlinear regression in the R programming environment (v 4.0.3), Fig. 3.
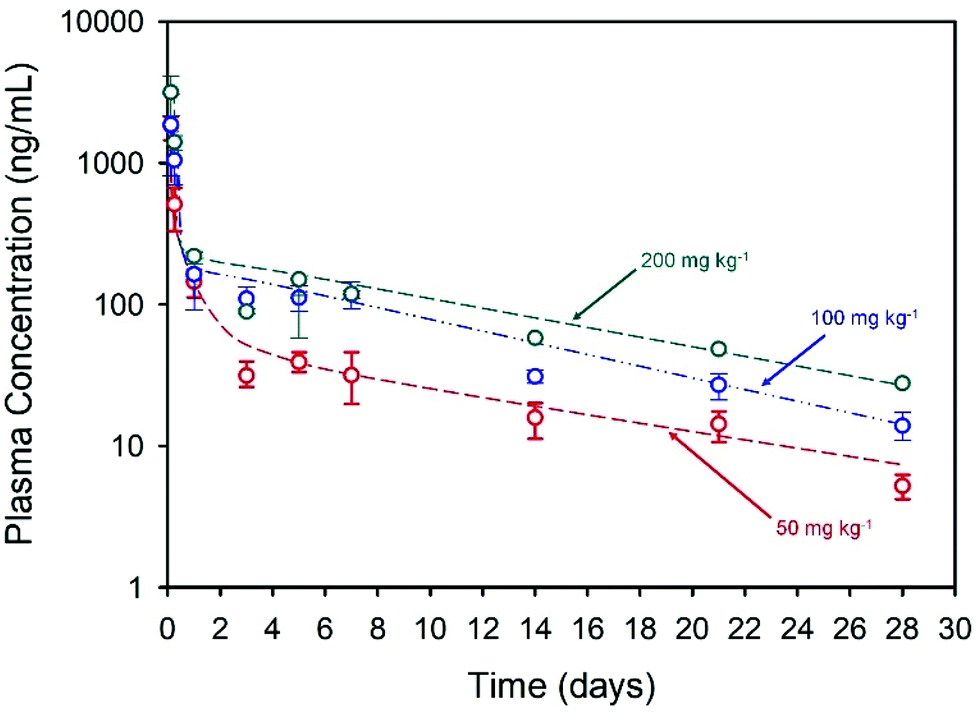 | ||
| Fig. 3 Pharmacokinetic profiles showing sustained NCL plasma concentrations over 28 days after a single intramuscular administration (rat model; n = 4) dosed at 50 mg kg−1 (red line), 100 mg kg−1 (blue line), and 200 mg kg−1 (green line). | ||
Within the first 24 hours following administration, mild issues with mobility were observed in three of the four rats in the high dose group and a decision was made to exclude these animals from further analysis. No other animals in any dosage group were observed to have impaired mobility after intramuscular injections, with weight increasing in all animals throughout the study. All of the injected doses exhibited rapid Cmax within 3 hours of administration with Cmax and AUC values increasing with dose, Table 2. Sustained plasma exposure of NCL was clearly achieved and the concentration-time profiles were adequately described by a biexponential model with first order input, likely reflecting the long-term drug release from the injection sites and resultant “flip-flop” kinetics rather than the intrinsic PK disposition of NCL; long-acting therapeutics benefit from the phenomenon of “flip-flop” kinetics which has been reported in numerous cases where extravascular drug administration may lead to an apparent rate of drug absorption being slower than the rate of drug elimination. This may be influenced by numerous factors including formulation and selection of administration site.40 No obvious local toxicity was observed during 28-day in vivo study; biopsies were not collected from the depot site for histological analysis during this early-stage demonstration.
| Dose (mg kg−1) | C max (ng mL−1) | T max (d) | Terminal T½ (d) | Apparent clearance (L h−1 kg−1) | AUC0-last (ng h mL−1) |
|---|---|---|---|---|---|
| 50 | 1408.6 | 2.5 | 10 | 1.5 | 28955 |
| 100 | 2041.3 | 2 | 8 | 1.7 | 55734 |
| 200 | 3125.3 | 3 | 9 | 2.4 | 74584 |
Owing to the purported immunomodulatory effects of NCL,3,41–44 samples collected at terminal bleed were assessed using the Luminex Rat Discovery 17-plex panel to establish cytokine/chemokine concentrations in the plasma of rats exposed to NCL compared with excipient-only controls (ESI, Fig. S4†). The one animal in the 200 mg kg−1 group was excluded from this analysis. Cytokine concentrations in animals exposed to NCL were generally higher than in controls. For example, IL-1β concentrations in control animals were 3.8 pg mL−1 compared to animals exposed to 50 mg kg−1 (5.6 pg mL−1) and 100 mg kg−1 (15.9 pg mL−1), respectively. However, the impact appeared to be cytokine- or chemokine-specific; for example, IL-2 concentration in rats exposed to 50 mg kg−1 niclosamide (1.29 pg mL−1) were higher than control rats (0.33 pg mL−1) but dose-dependent increases were not observed; this may suggest a degree of time-dependency or intricacy of the immunomodulatory effects on specific immune cell types and, as such, further studies will be required. When rat blood samples from animals treated with either excipients or niclosamide were exposed to the known inflammatory stimuli lipopolysaccharide (LPS) and phytohaemagglutinin (PHA), there appeared to be further immunomodulatory effects. The fold-difference between cytokine concentrations in unstimulated versus stimulated samples is summarised in Table 3.
| Dose and fold increase after stimulationa | |||
|---|---|---|---|
| Cytokine/Chemokine | Controlb | 50 mg kg−1 | 100 mg kg−1 |
| a Fold differences of unstimulated versus stimulated blood samples; bold and italic figures represent decreased values or no change. b Control sample contains excipients only. | |||
| CXCL12 | 3.8 | 4.1 | 3.7 |
| GM-CSF | 0.0 | 1.6 | 0.7 |
| IL-1β | 2.6 | 1.6 | 1.3 |
| IL-4 | 0.5 | 1.7 | 0.3 |
| IL-18 | 1.1 | 1.0 | 0.8 |
| TIMP-1 | 1.0 | 1.0 | 0.9 |
| VEGF | 1.5 | 1.3 | 1.2 |
| CXCL3 | 2.5 | 2.6 | 2.4 |
| ICAM-1 | 1.1 | 1.0 | 0.9 |
| IL-1α | 1.1 | 1.1 | 0.9 |
| IL-2 | 0.3 | 1.4 | 0.0 |
| IL-6 | 1.1 | 1.5 | 0.8 |
| L-selectin | 15.2 | 16.5 | 18.3 |
| TNFα | 8.4 | 8.9 | 5.3 |
Although there were higher concentrations of IL-1β in niclosamide exposed animals, and those concentrations increased upon stimulation, the degree of stimulation was markedly less in niclosamide exposed animals; a fold difference in control animals of 2.6, compared to a fold-difference in niclosamide groups of 1.6 and 1.3 in the 50 mg kg−1 and 100 mg kg−1 groups, respectively. Niclosamide appeared to inhibit the release of IL-1β, but L-selectin levels were markedly higher, following stimulation, in the 50 mg kg−1 (fold-difference 16.5) and 100 mg kg−1 (fold-difference 18.3) niclosamide groups than those in the control group (15.2), suggesting an enhancement of L-selectin release. Niclosamide enhanced the secretion of IL-2 in the 50 mg kg−1 group (fold-difference 1.4), but IL-2 responses were lower in the 100 mg kg−1 group. Additionally, niclosamide at higher concentrations appeared to inhibit the release of IL-6, following stimulation (100 mg kg−1, fold-difference 0.8) compared to the control group (fold-difference 1.1).
Cytokines and chemokines are vital mediators and regulators of the immune response to infection, but they can also exacerbate inflammation-related damage to tissues and organs thereby increasing inflammation in positive feedback manner. Therefore, a cautious approach should be taken with interpretation. A number of cytokines, particularly pro-inflammatory cytokines, have been associated with disease severity and poor prognosis (IL-1, IL-6, IL-12, IFNγ and TNFα)45 and NCL exerted several immunomodulatory effects that were evident 28 days after administration of the LAI. Some of these effects are consistent with an expected protective mechanism in SARS-CoV-2 infection but these observations are preliminary, and more work will be required to establish the benefit/risk.
Conclusions
The urgent need for interventions for COVID-19, and highly infectious variants of the SARS-CoV-2 virus, requires the reformulation of existing drug molecules using the multi-disciplinary tools available for advanced therapeutics. Therapies utilising small molecule drugs, including new active pharmaceuticals which will inevitably become available, will be essential for many years to come to compliment the vaccine for maximal population protection. The development of long-acting injectables that offer significant periods of protection and/or therapy, requires demonstration of the potential for the delivery of potent molecules using scalable approaches. Such approaches may offer a ‘test-and-treat’ option for symptomatic and asymptomatic patients and shorten infection and transmission timescales. The research presented here offers one such option and has targeted one of the most active drug molecules currently available; importantly, NCL is a generic compound and highly cost effective. The progression of this early work to preclinical studies using a Syrian Golden Hamster SARS-CoV-2 infection model is underway at much higher doses (700 mg kg−1 NCL) than those used in this current report (200 mg kg−1 NCL) to establish either prophylaxis, therapy (after infection) or both outcome options. Additionally, translation to a contract manufacturing and development organisation (CDMO) is in progress and early indications suggest that the process described here is highly scalable. Early studies have also shown that the formulations are stable under ambient conditions for several weeks, but regulatory-standard accelerated testing is required before depot site GLP toxicology can be conducted to support translation into early clinical testing.Author contributions
JJH, ACS, ABD, CU and JM all conducted the nanomaterials investigation; UA, HB, LT, MN, JS, AV, PC undertook the in vivo investigation and sample quantification; HP, RKRR conducted formal data analysis; CD undertook the immunological investigation; NJL, TOM, AO, SPR provided conceptualisation and supervision of the multidisciplinary programme.Conflicts of interest
Several of the co-authors are named inventors on patent filings aiming to facilitate rapid uptake and exploitation of the technology presented. AO and SR have received research funding from AstraZeneca and ViiV and consultancies from Gilead; AO has additionally received funding from Merck and Janssen and consultancies from ViiV and Merck not related to the current paper.Acknowledgements
The authors wish to thank UK Research and Innovation for an Engineering and Physical Sciences Council (EPSRC) Impact Acceleration Award funding (EPSRC IAA: COVID-19), EPSRC for funding via a Healthcare Impact Partnership Award (EP/R024804/1) and Unitaid for a supplement to Project LONGEVITY to enable CDMO engagement.References
- S. P. Rannard, T. O. McDonald and A. Owen, Br. J. Clin. Pharmacol., 2020 DOI:10.1111/bcp.14575.
- J. Craven, COVID-19 therapeutics tracker, https://www.raps.org/news-and-articles/news-articles/2020/3/covid-19-therapeutics-tracker, (accessed 16th January, 2021).
- A. C. Gordon, P. R. Mouncey, F. Al-Beidh, K. M. Rowan, A. D. Nichol, Y. M. Arabi, D. Annane, A. Beane, W. van Bentum-Puijk, L. R. Berry, Z. Bhimani, M. J. M. Bonten, C. A. Bradbury, F. M. Brunkhorst, A. Buzgau, A. C. Cheng, M. A. Detry, E. J. Duffy, L. J. Estcourt, M. Fitzgerald, H. Goossens, R. Haniffa, A. M. Higgins, T. E. Hills, C. M. Horvat, F. Lamontagne, P. R. Lawler, H. L. Leavis, K. M. Linstrum, E. Litton, E. Lorenzi, J. C. Marshall, F. B. Mayr, D. McAuley, A. McGlothlin, S. P. McGuinness, B. J. McVerry, S. K. Montgomery, S. C. Morpeth, S. Murthy, K. Orr, R. L. Parke, J. C. Parker, A. E. Patanwala, V. Pettilä, E. Rademaker, M. S. Santos, C. T. Saunders, C. W. Seymour, M. Shankar-Hari, W. I. Sligl, A. F. Turgeon, A. M. Turner, F. L. van de Veerdonk, R. Zarychanski, C. Green, R. J. Lewis, D. C. Angus, C. J. McArthur, S. Berry, S. A. Webb and L. P. G. Derde, medRxiv, 2021, DOI:10.1101/2021.01.07.21249390.
- A. Owen and S. Rannard, Adv. Drug Delivery Rev., 2016, 103, 144–156 CrossRef CAS PubMed.
- C.-J. Wu, J.-T. Jan, C.-M. Chen, H.-P. Hsieh, D.-R. Hwang, H.-W. Liu, C.-Y. Liu, H.-W. Huang, S.-C. Chen, C.-F. Hong, R.-K. Lin, Y.-S. Chao and J. T. A. Hsu, Antimicrob. Agents Chemother., 2004, 48, 2693–2696 CrossRef CAS PubMed.
- C. Prabhakara, R. Godbole, P. Sil, S. Jahnavi, T. S. van Zanten, D. Sheth, N. Subhash, A. Chandra, V. K. Nuthakki, T. P. Puthiyapurayil, R. Ahmed, A. H. Najar, S. M. Lingamallu, S. Das, B. Mahajan, P. Vemula, S. B. Bharate, P. P. Singh, R. Vishwakarma, A. Guha, V. Sundaramurthy and S. Mayor, bioRxiv, 2020, DOI:10.1101/2020.12.16.422529.
- B. Devarakonda, R. A. Hill, W. Liebenberg, M. Brits and M. M. de Villiers, Int. J. Pharm., 2005, 304, 193–209 CrossRef CAS PubMed.
- C. K. Lin, M. Y. Bai, T. M. Hu, Y. C. Wang, T. K. Chao, S. J. Weng, R. L. Huang, P. H. Su and H. C. Lai, Oncotarget, 2016, 7, 8993–9006 CrossRef PubMed.
- U. Arshad, H. Pertinez, H. Box, L. Tatham, R. K. R. Rajoli, P. Curley, M. Neary, J. Sharp, N. J. Liptrott, A. Valentijn, C. David, S. P. Rannard, P. M. O'Neill, G. Aljayyoussi, S. H. Pennington, S. A. Ward, A. Hill, D. J. Back, S. H. Khoo, P. G. Bray, G. A. Biagini and A. Owen, Clin. Pharmacol. Ther., 2020, 108, 775–790 CrossRef CAS PubMed.
- K. Miner, K. Labitzke, B. Liu, P. Wang, K. Henckels, K. Gaida, R. Elliott, J. J. Chen, L. Liu, A. Leith, E. Trueblood, K. Hensley, X.-Z. Xia, O. Homann, B. Bennett, M. Fiorino, J. Whoriskey, G. Yu, S. Escobar, M. Wong, T. L. Born, A. Budelsky, M. Comeau, D. Smith, J. Phillips, J. A. Johnston, J. G. McGivern, K. Weikl, D. Powers, K. Kunzelmann, D. Mohn, A. Hochheimer and J. K. Sullivan, Front. Pharmacol., 2019, 10, 51 CrossRef CAS PubMed.
- Y. Ye, X. Zhang, T. Zhang, H. Wang and B. Wu, Drug Dev. Ind. Pharm., 2015, 41, 1416 CrossRef CAS PubMed.
- Q. Fua, X. Jina, Z. Zhang and H. Lv, Int. J. Pharm., 2020, 584, 119432 CrossRef PubMed.
- B. Bhushan, P. Dubey, S. U. Kumar, A. Sachdev, I. Matai and P. Gopinath, RSC Adv., 2015, 5, 12078–12086 RSC.
- S. Naqvi, S. Mohiyuddin and P. Gopinath, R. Soc. Open Sci., 2017, 4, 170611 CrossRef PubMed.
- Y. Xie and Y. Yao, Int. J. Pharm., 2018, 535, 157–163 CrossRef CAS PubMed.
- G. B. Reddy, D. L. Kerr, I. Spasojevic, A. Tovmasyan, D. S. Hsu, B. E. Brigman, J. A. Somarelli, D. Needham and W. C. Eward, Mol. Cancer Ther., 2020, 19, 1448–1461 CrossRef CAS PubMed.
- G. Wang, H. Gaikwad, M. K. McCarthy, M. Gonzalez-Juarrero, Y. Li, M. Armstrong, N. Reisdorf, T. E. Morrison and D. Simberg, bioRxiv, 2020, DOI:10.1101/2020.12.18.423509.
- R. P. Bakshi, L. M. Tatham, A. C. Savage, A. K. Tripathi, G. Mlambo, M. M. Ippolito, E. Nenortas, S. P. Rannard, A. Owen and T. A. Shapiro, Nat. Commun., 2018, 9, 315 CrossRef PubMed.
- M. Li, M. Azad, R. Davé and E. Bilgili, Pharmaceutics, 2016, 8, 17 CrossRef PubMed.
- C. M. Keck and R. H. Müller, Eur. J. Pharm. Biopharm., 2006, 62, 3–16 CrossRef CAS PubMed.
- M. Giardiello, N. J. Liptrott, T. O. McDonald, D. Moss, M. Siccardi, P. Martin, D. Smith, R. Gurjar, S. P. Rannard and A. Owen, Nat. Commun., 2016, 7, 13184 CrossRef CAS PubMed.
- M. S. Malvankar-Mehta, A. Fu, Y. Subramanian and C. Hutnik, J. Ophthalmol., 2020, 2020, 7801093 Search PubMed.
- B. Shingleton, R. Rosenberg, R. Teixeira and M. O'Donoghue, J. Cataract Refractive Surg., 2007, 33, 1953–1957 CrossRef PubMed.
- X. Guan, R. Tang and G. H. Nancollas, J. Biomed. Mater. Res., Part A, 2004, 71, 488–496 Search PubMed.
- C. Kriegel, M. Festag, R. S. K. Kishore, D. Roethlisberger and G. Schmitt, Children, 2019, 7, 1 CrossRef PubMed.
- J. Moore, J. Am. Coll. Toxicol., 1984, 3, 1–82 Search PubMed.
- G. Dumortier, J. L. Grossiord, F. Agnely and J. C. Chaumeil, Pharm. Res., 2006, 23, 2709–2728 CrossRef CAS PubMed.
- S. V. Hemelryck, J. Dewulf, H. Niekus, M. van Heerden, B. Ingelse, R. Holm, E. Mannaert and P. Langguth, Int. J. Pharm.: X, 2019, 1, 100016 CAS.
- L. Yu, K. Li, X. Liu, C. Chen, Y. Bao, T. Ci, Q. Chen and J. Ding, J. Pharm. Sci., 2013, 102, 4140–4149 CrossRef CAS PubMed.
- B. M. Rayaprolu, J. J. Strawser and G. Anyarambhatla, Drug Dev. Ind. Pharm., 2018, 44, 1565–1571 CrossRef CAS PubMed.
- Y. Mehmood and U. Farooq, Open Sci. J. Pharm. Pharmacol., 2015, 3, 19–27 Search PubMed.
- F. Rocha, L. Yumi Sugahara, F. V. Leimann, S. M. de Oliveira, E. da Silva Brum, R. C. Calhelha, M. F. Barreiro, I. C. F. R. Ferreira, R. Porto Ineu and O. H. Gonçalves, Food Funct., 2018, 9, 3698–3706 RSC.
- H. T. Gordon and J. C. Bauernfeind, Crit. Rev. Food Sci. Nutr., 1982, 18, 59–97 CrossRef CAS PubMed.
- E. Kienzle, C. Kaden, P. P. Hoppe and B. Opitz, J. Anim. Physiol. Anim. Nutr., 2003, 87, 174–180 CrossRef CAS PubMed.
- E. Lepeltier, C. Bourgaux and P. Couvreur, Adv. Drug Delivery Rev., 2014, 71, 86–97 CrossRef CAS PubMed.
- R. Malviya, P. K. Sharma and S. K. Dubey, Mater. Sci. Eng., C, 2018, 91, 772–784 CrossRef CAS PubMed.
- C. Gavory, A. Durand, J. L. Six, C. Nouvel, E. Marie and M. Leonard, Carbohydr. Polym., 2011, 84, 133–140 CrossRef CAS.
- A. Chebil, M. Leonard, J. L. Six, C. Nouvel and A. Durand, Colloids Surf., B, 2018, 162, 351–361 CrossRef CAS PubMed.
- U. Arshad, H. Pertinez, H. Box, L. Tatham, R. Rajoli, M. Neary, J. Sharp, A. Valentijn, J. Hobson, C. Unsworth, A. Dwyer, A. Savage, T. O. McDonald, S. Rannard, P. Curley and A. Owen, bioRxiv, 2021, DOI:10.1101/2021.01.13.426426.
- J. A. Yáñez, C. M. Remsberg, C. L. Sayre, M. L. Forrest and N. M. Davies, Ther. Delivery, 2011, 2, 643–672 CrossRef PubMed.
- U. Thi Tran and T. Kitami, Commun. Biol., 2019, 2, 2 CrossRef PubMed.
- L. Liang, M. Huang, Y. Xiao, S. Zen, M. Lao, Y. Zou, M. Shi, X. Yang and H. Xu, Inflammation Res., 2015, 64, 225–233 CrossRef CAS PubMed.
- U. N. Shivaji, L. Jeffery, N. Batis, M. Iacucci and S. Ghosh, J. Crohns Colitis, 2019, 13, S134–S134 CrossRef.
- F. Morin, N. Kavian, C. Nicco, O. Cerles, C. Chéreau and F. Batteux, J. Immunol., 2016, 197, 3018–3028 CrossRef CAS PubMed.
- V. J. Costela-Ruiz, R. Illescas-Montes, J. M. Puerta-Puerta, C. Ruiz and L. Melguizo-Rodríguez, Cytokine Growth Factor Rev., 2020, 54, 62–75 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and analytical procedures, images and micrographs of injectable formulations, graphs from pharmacological evaluation. See DOI: 10.1039/d1nr00309g |
| This journal is © The Royal Society of Chemistry 2021 |