
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering conductive protein films through nanoscale self-assembly and gold nanoparticles doping†
Sara H.
Mejias‡
 abc,
Elena
López-Martínez‡
c,
Maxence
Fernandez
c,
Pierre
Couleaud
ab,
Ana
Martin-Lasanta
ad,
David
Romera
ab,
Ana
Sanchez-Iglesias
c,
Santiago
Casado
a,
Manuel R.
Osorio
a,
Jose M.
Abad
ae,
M. Teresa
González
*a and
Aitziber L.
Cortajarena
*abcf
abc,
Elena
López-Martínez‡
c,
Maxence
Fernandez
c,
Pierre
Couleaud
ab,
Ana
Martin-Lasanta
ad,
David
Romera
ab,
Ana
Sanchez-Iglesias
c,
Santiago
Casado
a,
Manuel R.
Osorio
a,
Jose M.
Abad
ae,
M. Teresa
González
*a and
Aitziber L.
Cortajarena
*abcf
aIMDEA Nanociencia, Campus Universitario de Cantoblanco, C\Faraday, 9, 28049 Madrid, Spain. E-mail: teresa.gonzalez@imdea.org
bNanobiotechnology Unit (iMdea Nanociencia) associated with Centro Nacional de Biotecnología (CNB-CSIC), Campus Universitario de Cantoblanco, Madrid 28049, Spain
cCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramón 182, 20014, Donostia San Sebastián, Spain. E-mail: alcortajarena@cicbiomagune.es
dDepartamento de Química Orgánica, Universidad de Granada, Campus Fuentenueva s/n, E-18071 Granada, Spain
eDepartamento de Química Analítica y Análisis Instrumental, Facultad de Ciencias, Universidad Autónoma de Madrid, 28049 Madrid, Spain
fIkerbasque, Basque Foundation for Science, Mª Díaz de Haro 3, 48013 Bilbao, Spain
First published on 1st April 2021
Abstract
Protein-based materials are usually considered as insulators, although conductivity has been recently shown in proteins. This fact opens the door to develop new biocompatible conductive materials. While there are emerging efforts in this area, there is an open challenge related to the limited conductivity of protein-based systems. This work shows a novel approach to tune the charge transport properties of protein-based materials by using electron-dense AuNPs. Two strategies are combined in a unique way to generate the conductive solid films: (1) the controlled self-assembly of a protein building block; (2) the templating of AuNPs by the engineered building block. This bottom-up approach allows controlling the structure of the films and the distribution of the AuNPs within, leading to enhanced conductivity. This work illustrates a promising strategy for the development of effective hybrid protein-based bioelectrical materials.
Introduction
Protein-based materials, due to their structural flexibility and their chemical diversity,1 have brought new opportunities in a wide range of biotechnological and nanotechnological applications, such as tissue regeneration,2–4 catalysis,5–7 and plasmonics.8–10 For example, protein-based nanostructures can be formed with highly tunable properties, including dynamic assemblies.Moreover, protein-based functional hybrid structures are generated with the specific arrangement of active elements along the protein backbone.11–16 Recently, proteins have been proposed as candidates for the interfacing of biomolecules with electronic devices due to their intrinsic molecular conduction.17–19 Long-range electronic conductivity in protein-based materials arises from two mechanisms: (1) redox mediated via hopping electron transfer across redox centers, which are spaced closely enough for coherent overlap to occur,17,20,21 or (2) nonredox-mediated conduction with a band gap, that depends on the protein amino acid composition and the secondary structure of the protein scaffolds.17,22–25 Protonic conductivity occurs when protons are transported along the protein scaffold by charged amino acids, such as aspartic and glutamic acids.26–28 In addition, special charge transport occurs between redox active amino acids, such as tyrosine or tryptophan, where the charge transport happens by proton-coupled electron transfer simultaneously showing electronic- and protonic transport.23,29 These different types of transport can interplay depending on the composition of the material.30 The control of the charge transport mechanism in protein-based materials would allow tuning their conductive properties, necessary to generate bio-based conductive materials and devices. Several strategies have been reported to boost electronic or protonic charge transport in protein-based systems. For example, protonic charge transport in reflectin protein films has been tuned by varying the composition of the charge amino acids.26 The electronic transport has been favored by the introduction of redox active amino acids, such as tryptophan or tyrosine, or redox active compounds, such as naphthalene diimide (NDI) along the protein scaffold.14,31–35 Despite the increasing knowledge on transport mechanisms, charge transport through long distances is difficult to control and tune as it depends on the structure of the material and its chemical composition.17,18,30,36
The current work describes a novel approach that combines protein design, biomolecular templating of gold nanoparticles (AuNPs), and protein self-assembly to enhance the conductive properties of protein-based films (Fig. 1). The consensus tetratricopeptide repeat protein (CTPR) used as protein scaffold is made of a 34 amino acid helix-turn-helix module.37,38 The fold of CTPRs is defined by a few conserved residues; hence, there is a lot of room for engineering non-conserved amino acids.37 The modules generally appear in tandem repeats, from 2 to 20, generating a rigid right-handed superhelical structure.37 CTPRs assemble forming a transparent ordered solid film through their intrinsic self-assembly properties of the structured units.37,39 As previously demonstrated, these self-assembly capabilities enable the fabrication of photoconductive and electroactive functional assemblies based on CTPRs.11,40 CTPR protein-based films have shown protonic transport due to their ionic nature and high content of charged amino acids, and as such have been proven to be interesting actuators that respond to humidity in the environment.41 In parallel works, the incorporation of AuNPs for electrical interfacing of redox enzymes, or the AuNP-assisted assembly of heme protein resulted in effective improvement of the long-range charge transfer efficiency.42–44 AuNPs have been also used to generate AuNPs-peptide conductive nanocomposites.45 The aforementioned works show the value of design strategies to improve conductivity in protein-based systems, and inspired the use of AuNPs as electron conductive elements in a CTPR-based macroscopic hybrid films.
 | ||
| Fig. 1 Conductive protein-based biomaterials. Top. Engineered repeat proteins as building blocks to fabricate self-assembled conductive films. Bottom. Protein-AuNPs hybrids as building blocks to fabricate AuNPs-doped self-assembled films with enhanced conductivity. | ||
Herein, a CTPR unit was first designed to template AuNPs with nanometer precision through an orthogonal conjugation strategy. Then, protein self-assembly after solvent evaporation guided the formation of both solid films of CTPRs and CTPR-AuNPs.39 Remarkably, the CTPR films already displayed conductivity in the hundred nS/m range, recordable at micrometer length scales and mainly attributed to protonic charge transport. The CTPR-AuNPs hybrid films showed an enhancement of four orders of magnitude in conductivity when compared to the CTPR-only films, and, presumably, different charge transport properties. The current results demonstrate a strategy to fabricate protein-based conductive films with enhanced conductivity by using highly conductive nano-elements, which paves the way for future application of hybrid protein-based systems in the field of bioelectronics.
Experimental
Materials
All chemical reagents were purchased from Aldrich and used without further purification. Ultrafiltration tubes used were Amicon Millipore with a regenerated cellulose membrane and a cut-off of 3 kDa. Cobalt NTA affinity resin used is ABT 6BCL-QHCo-100 (Agarose Bead Technology). Ultrapure reagent grade water (18.2 MΩ, Wasserlab at 25 °C) was used in all experiments. Gold(III) chloride hydrate (HAuCl4, ≥99%), sodium citrate tribasic dihydrate (≥98%), sodium borohydride (NaBH4, ≥96%) and mercapto poly (ethylene glycol) amine functionalized (HS-PEG2K-NH2) were purchased from Sigma-Aldrich-Merck. All glassware was washed with aqua regia, rinsed with Milli-Q water, and dried before use.Measurements
UV-Vis and fluorescence spectra were recorded on a Synergy H4 microplate reader (BioTek) using 96-well plates. Fast Protein Liquid Chromatography (FPLC) was performed on a GE Life Science ÄKTA prime plus apparatus with a Superdex 75 column. Gel reader apparatus used was a Syngene G:Box Chemi XR5. Matrix Assisted Laser Desorption/Ionization Time Of Flight mass spectrometry (MALDI-TOF) was done by the National Center of Biotechnology (CNB) proteomic service in Madrid on an AB Sciex ABi 4800 MALDI TOF/TOF mass spectrometer. High Resolution Transmission Electronic Microscopy (HR-TEM) measurements were done on a JEOL JEM 1400 Plus.Gel filtration chromatography
Gel filtration chromatography was performed using an AKTA prime plus Fast Protein Liquid Chromatography (FPLC) equipment (GE Healthcare). After Ni-NTA column purification, the dialyzed elution fractions were injected into a Superdex 75 HR 10/30 size exclusion chromatography column (GE Healthcare) and run at 0.5 mL min−1 in PBS buffer with a detection UV absorption at 280 nm. The samples were collected in 0.5 mL fractions and stored at 4 °C.High resolution transmission electron microscopy (HR-TEM)
HR-TEM measurements were conducted on JEOL JEM 1400 Plus microscope. The samples for HR-TEM were prepared by drop contact of the sample solution at 100 nM concentration of protein with a TEM grid and blotted to dry. The analysis of the images was performed using ImageJ software. The statistical analysis was performed using a Mann–Whitney non parametrical test with GraphPad Prism software.Scanning tunnel microscopy (STM)
STM images were obtained with a home-built Scanning Tunnelling Microscope designed for room-temperature experiments.46 All the images were recorded in ambient conditions using commercial gold substrates (Arrandee) cleaned prior sample deposition by flame-annealing. Freshly cut gold wires (99.99%) were used as tips. Samples were prepared by the drop casting technique from an aqueous solution of CTPR164Cys-AuNPs. For individual molecule imaging, we used 0.1–10 nM concentrations, while for layer formation, we used a 1 μM protein concentration. A drop of 200–300 μL of the solution was deposited over our 1 cm2 gold substrates, which were rinsed with water several times after assembly periods of 10 to 30 minutes. The gold surface was then dried under N2 flow. Images were recorded using bias voltage values between 0.1 V and 1.5 V and a setpoint current between 500 pA and 5 nA. The body of the protein could not be imaged in range of voltages studied reflecting its low electrical conductance. The typical apparent height of the AuNP was 0.4 nm, accounting for their expected poor coupling to the gold substrate.X-ray diffraction (XRD)
XRD was performed in a Panalytical X'Pert PRO diffractometer with Cu tube (lambda Kα = 1.54187 Å) operated at 45 kV, 40 mA, Ni beta filter, programmable divergence and anti-scatter slits working in fixed mode, and fast linear detector (X'Celerator) working in scanning mode.Gold nanoparticles synthesis
Gold nanoparticles (∼3 nm) were prepared by fast reduction of HAuCl4 (20 mL, 0.125 mM) with freshly prepared NaBH4 (0.3 mL, 10 mM) in the presence of sodium citrate (0.25 mM) under vigorous stirring.47 The solution color changed from yellow to reddish. After two minutes, the seed solution was aged at 27 °C for 30 min before use to promote the decomposition of sodium borohydride. Mercapto poly (ethylene glycol) amine functionalized with a molecular weight of 2000 g mol−1 was used for ligand exchange. An aqueous solution of HS-PEG2K-NH2 (0.5 mL, 2.9 mM) was added dropwise to as synthesized gold nanoparticles under vigorous stirring. The mixture was allowed to react for 2 h. PEG-modified gold nanoparticles were centrifuged using Millipore Amicon Centrifugal Filter Units (10 kDa) and finally dispersed in water.Protein design and purification
Based on consensus CTPR16 protein, four cysteine residues were introduced in a loop position 33 of the CTPR repeats 2, 6, 10, and 14 to form CTPR164Cys protein. The mutation was introduced in CTPR1 by quick-change site directed mutagenesis. The CTPR164Cys gene was generated from the CTPR1 wild type gene by sequential additions of CTPR1 wild type or mutated repeats, depending on the CTPR repetition number, and cloned into pPro-EX-HTa vector. The protein was expressed as His-tagged fusion and purified using standard affinity chromatography methods as previously described.48 The protein was dialyzed into PBS buffer (150 mM NaCl, 50 mM phosphate buffer pH 7.4) with 2 mM β-mercaptoethanol and stored frozen at −20 °C. The protein concentration was determined by UV-absorbance at 280 nm using the extinction coefficient calculated from the amino acid composition.49Modification of AuNPs and conjugation with CTPR164Cys protein
1 mL of amine-PEG AuNPs at 160 nM were incubated with 39 μL of freshly prepared 22.9 μM 4-(N-maleimidomethyl) cyclohexane-1-carboxylic acid 3-sulfo-N-hydroxysuccinimide ester dissolved in water (Sulfo-SMCC) in a microtube for 30 min at room temperature (final molar ratio Sulfo-SMCC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AuNPs = 5:1). The AuNPs concentration was calculated from their absorption at 510 nm using the following equation: ln ε = 3.32111 × ln D + 10.80505, where ε is the molar extinction coefficient (M−1 cm−1) and D is the nanoparticle core diameter in nm of the gold nanoparticles measured by TEM, according to Liu et al.50 Protein cysteines were reduced using 5 mM dithiothreitol (DTT) for 45 minutes and purified using an Illustra NAP-5 desalting column equilibrated with a solution of 150 mM NaCl, 50 mM phosphate buffer pH 8.0. Before conjugation, the excess Sulfo-SMCC was removed from the AuNPs suspension by using an ultrafiltration unit Amicon® Ultra – 0.5 mL with a 3000 Da molecular weight cutoff (MWCO) at 21000 g for 5 min and by washing 5 times with 150 mM NaCl, 50 mM phosphate buffer pH 8.0. Purified AuNPs-SMCC were then incubated with 7.6 μL of 5.9 μM freshly reduced CTPR164Cys at room temperature with 20 rpm spinning for 1 h (final molar ratio CTPR164Cys:AuNPs = 1:4). After nanoparticle conjugation, the reaction was blocked by the addition of 5 μL of 10 mM β-mercaptoethanol. After overnight incubation, CTPR164Cys-AuNPs conjugates were purified from the excess of unbound AuNPs and linker using protein His-Tag and Ni-NTA column. Free AuNPs were eluted in the flow-through while CTPR164Cys-AuNPs conjugate was eluted from the nickel column with a 300 mM imidazole buffer solution.
:AuNPs = 5:1). The AuNPs concentration was calculated from their absorption at 510 nm using the following equation: ln ε = 3.32111 × ln D + 10.80505, where ε is the molar extinction coefficient (M−1 cm−1) and D is the nanoparticle core diameter in nm of the gold nanoparticles measured by TEM, according to Liu et al.50 Protein cysteines were reduced using 5 mM dithiothreitol (DTT) for 45 minutes and purified using an Illustra NAP-5 desalting column equilibrated with a solution of 150 mM NaCl, 50 mM phosphate buffer pH 8.0. Before conjugation, the excess Sulfo-SMCC was removed from the AuNPs suspension by using an ultrafiltration unit Amicon® Ultra – 0.5 mL with a 3000 Da molecular weight cutoff (MWCO) at 21000 g for 5 min and by washing 5 times with 150 mM NaCl, 50 mM phosphate buffer pH 8.0. Purified AuNPs-SMCC were then incubated with 7.6 μL of 5.9 μM freshly reduced CTPR164Cys at room temperature with 20 rpm spinning for 1 h (final molar ratio CTPR164Cys:AuNPs = 1:4). After nanoparticle conjugation, the reaction was blocked by the addition of 5 μL of 10 mM β-mercaptoethanol. After overnight incubation, CTPR164Cys-AuNPs conjugates were purified from the excess of unbound AuNPs and linker using protein His-Tag and Ni-NTA column. Free AuNPs were eluted in the flow-through while CTPR164Cys-AuNPs conjugate was eluted from the nickel column with a 300 mM imidazole buffer solution.
Film formation
Protein solid ordered films were generated as previously described.51 CTPR164Cys protein alone and CTPR164Cys-AuNPs conjugates were diluted to 3% (w/v) protein concentration in 10 mM NaCl, 10 mM Na phosphate pH 7.0 buffer. The solutions were deposited on different surfaces, depending on the experiments to be performed. Quartz cuvette was used for circular dichroism (CD) analysis, glass surface for conductivity measurements and silicon wafer for X-ray diffraction (XRD) analysis. The drop volumes also vary between 10 to 30 μL. The solvent was evaporated at room temperature for 12 hours, resulting in solid thin films.Lithography of the electrodes
A pattern of interdigitated electrodes with a defined channel of 20 μm in length L and 11.8 mm in width W was fabricated through clean-room processes of “maskless” lithography (“Heidelberg DWL66fs” model) and thermal evaporation. A Si/SiO2 wafer was coated by 2 mm “AZ 1512HS” (MicroChemicals GmbH) positive resin and exposed to a λ = 405 nm laser that printed the designed electrode pattern. The wafer was then introduced in a AZ351B developer (1:4 developer:water) to remove the resin parts exposed to the laser. To ensure that all the resin is removed, the wafer was further exposed to plasma (50 W) for 30 s. The revealed sample was introduced in a thermal evaporator (Nanosphere, de Oxford Vaccum Science model) where a chromium 5–10 nm thick layer was first evaporated to increase the adhesion to the substrate, followed by a 50 nm thick gold layer. To finish, a lift-off process was performed, introducing the sample in acetone to remove all the resin in the sample and obtain the desired pattern shown in Fig. S1.†
Electrical conductivity measurements
To study the conductive properties of the CTPR164Cys and CTPR64Cys-AuNPs, we prepared thin films of the protein and the conjugate devices over silicon wafers with gold electrodes patterned as described in the previous section (Fig. S1†). Using a Keithley 4200-SCS, we recorded current versus voltage curves in the interval (−1 V, +1 V) for both the CTPR164Cys protein and the CTPR164Cys-AuNPs conjugate. Olympus standard silicon nitride probes of 0.05 N m−1 and 18 kHz (OMCL-RC800PSA) were employed to contact the electrodes. The aspect ratio (W/L) of the interdigitated electrodes is 590, which allowed us to obtain measurable currents even for low-conducting plain CTPR164Cys films, and to reduce the effect of edge currents between the electrodes. Due to the low conductance of the CTPR164Cys, we prepared thicker film of this protein than of the AuNP conjugate. We prepared 4–5 μm thick films for the protein, and 20–100 nm thick films for the conjugate. The thickness of the films was measured using a profilomer for CTPR16 films and an atomic force microscope (AFM) in jumping mode for the CTPR164Cys-AuNPs films.Results
CTPR proteins were identified as good candidates to generate protein-based conductive films, due to their ability to form ordered macroscopic materials through their self-assembly properties. CTPR16, a protein composed of sixteen CTPR repeats, was chosen as scaffold considering its high stability and its 16 nm length which makes it suitable for templating several nanoparticles per protein. Moreover, as it has been demonstrated for squid ring teeth proteins, the number of tandem repetitions significantly and systematically enhances bulk transport properties.52 Therefore, sixteen CTPR repeats appeared as an appropriate protein size for promoting conductivity. To ensure a homogeneous and controlled distribution of AuNPs over the CTPR film, AuNPs were precisely introduced on selected regions of the CTPR scaffold by covalent linkage. CTPR16 protein was engineered with 4 unique cysteine residues in the loop of the 2nd, 6th, 10th, and 14th repeats, by the mutation R33C for the selective conjugation of the AuNPs, leading to CTPR164Cys (Fig. 2A). Position 33 within the CTPR sequence was selected since it is a non-conserved position53 and therefore a non-structural position, and it is located in a solvent exposed loop that will facilitate the conjugation. Repeats 2nd 6th, 10th and 14th were selected to encode a CTPR-AuNP complex with optimal inter-particle distance with 2 nanoparticles per superhelical turn54 and alternate particles facing opposite sides of the superhelix (Fig. 2A and S2†). These mutations did not significantly affect the structure or the stability of the scaffold, as confirmed by circular dichroism (CD) (Fig. 2B). The synthesized AuNPs functionalized with amine-polyethylene glycol (PEG) coating had an average diameter of 3.1 ± 0.7 nm and their colloidal stability is maintained by the PEG monolayer (see Experimental for details). These AuNPs showed a localized surface plasmon resonance (LSPR) around 510 nm.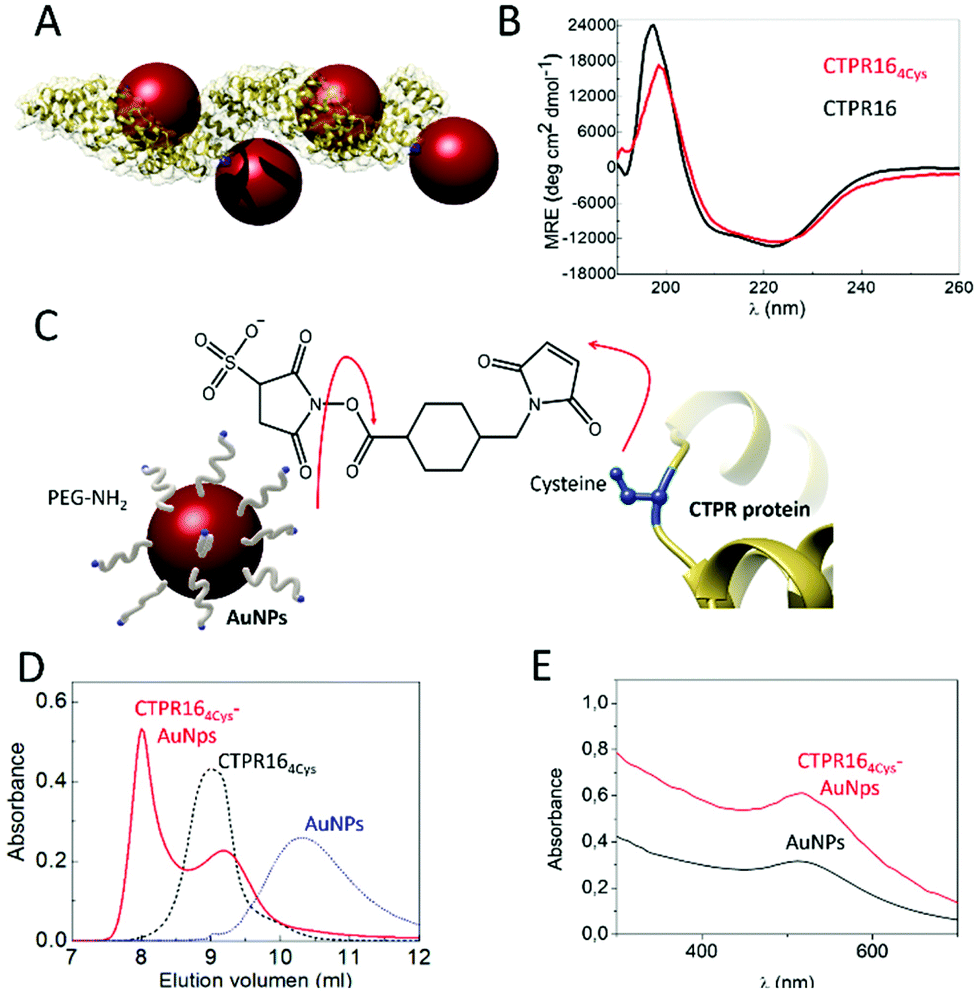 | ||
| Fig. 2 (A) Ribbon representation of the CTPR164Cys-AuNPs conjugated protein. Structural model is based on the structure of PDB ID: 2HYZ. The mutated cysteine residues are highlighted in blue. (B) CD spectra of CTPR164Cys (red line) compared with the original CTPR16 (black line). (C) Schematic representation of the conjugation strategy followed. The free amines from the PEGylated AuNPs and the thiol groups from the cysteines (shown in blue) of the CTPR164Cys protein are linked using sulfo-SMCC molecules. (D) Size exclusion chromatograms of AuNPs (blue dotted line), the CTPR164cys protein (black dotted line) and the CTPR164cys-AuNPs conjugates (red line). (E) UV-Visible spectra of the AuNPs with their characteristic LSPR peak at 520 nm (black line) and the CTPR164cys-AuNPs conjugate (red line). | ||
Amine-PEG AuNP were conjugated to CTPR164Cys proteins using sulfo-SMCC conjugation chemistry. As show in Fig. 2C, the NHS ester group of the sulfo-SMCC linker reacts with amine groups of the amine-PEG AuNPs while its maleimide group reacts orthogonally with the cysteines in the protein scaffold. After conjugation, the CTPR164Cys-AuNPs complexes were purified from unbound AuNPs by affinity chromatography. The eluate that exhibited a pink/purple color, characteristic of the AuNPs, was analyzed by size exclusion chromatography (Fig. 2D). The elution time for the CTPR164Cys-AuNPs was the shortest, which confirmed AuNPs incorporation to the CTPR164Cys that resulted in a larger hydrodynamic radius compared to CTPR164Cys, and to the AuNPs. Moreover, the UV-visible absorption spectrum of the CTPR164Cys-AuNPs conjugates showed the AuNPs LSPR peak at 510 nm (Fig. 2E).
The conjugation efficiency was analyzed by transmission electron microscopy (TEM) (Fig. 3A). TEM images were acquired and the distribution of the AuNPs showed the presence of spatially limited assemblies made of 3 or 4 particles, with a disposition in agreement with the arrangement encoded by the engineered CTPR164Cys scaffold (Fig. 3A, left panel). Such assembled nanostructures were not observed in the original amine-PEG functionalized AuNPs, for which mostly large aggregates of particles are observed (Fig. 3A, right panel). The CTPR164Cys-AuNPs conjugates also revealed some larger assemblies composed of more than 4 AuNPs per groups, which could be due to the linkage of two or more proteins through the AuNPs, as the activated AuNP-SMCC intermediates are not monovalent and could potentially be grafted by two or more proteins. However, the conjugates assemblies and the control-particle before conjugation show different behavior, with predominance of 1–4 assemblies in the conjugates, whereas predominance of large nonspecific aggregates in the control (Fig. S3†).
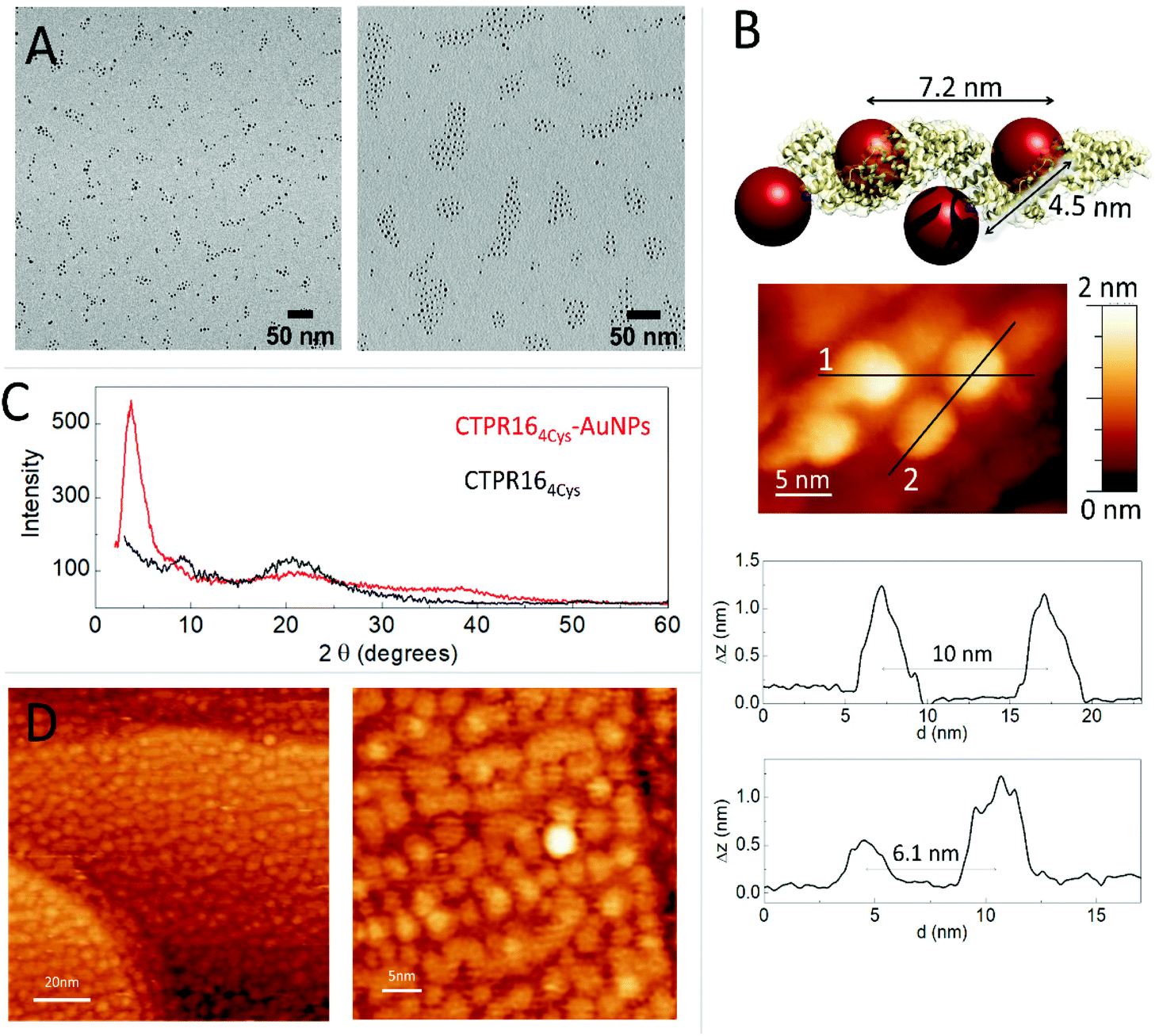 | ||
| Fig. 3 (A) Left: TEM micrograph of CTPR164Cys-AuNPs conjugates in discrete groups. Right: TEM micrograph of the PEGylated AuNPs in aggregates. (B) Model structure of CTPR164Cys-AuNPs conjugate based on the crystal structure of CTPR20 and the location of the cysteine mutations in the CTPR protein. A STM image of one CTPR164Cys-AuNPs conjugate is shown with the profile plots of lines 1 and 2 shown below (profile for line 1 on top, and profile for line2 on bottom). (C) Powder X-ray Diffraction (P-XRD) spectra of CTPR164Cys-AuNPs conjugate film in red and P-XRD spectra of the CTPR16 film in black. (D) STM images of the CTPR164cys-AuNPs conjugates deposited on a gold surface at different magnifications. | ||
The CTPR164Cys-AuNPs nanoscale structure was explored using scanning tunneling microscopy (STM) at bias voltages between 0.1 V and 1.5 V and at room temperature (Fig. 3B). Conjugated CTPR164Cys-AuNPs at 1 nM were deposited by drop casting over a freshly flame-annealed gold surface (see Experimental for details). Groups of four AuNPs disposed in good agreement with the disposition of the four cysteine residues in the designed protein were observed. According to image profiles, the distances between gold nanoparticles are slightly larger than expected from the model based on CTPR crystal structure, which can be explained by the flexibility of the CTPR spring-like backbone.55 Moreover, since STM images are based on the current measured between the tip and the sample, the obtained STM images already confirm the conductivity of the AuNPs attached to the scaffold protein.
CTPR164Cys and CTPR164Cys-AuNPs solid state assemblies were generated taking advantage of CTPR protein intrinsic interactions.39 A 300 μM protein solution was deposited into a planar surface and a transparent solid film was formed after drop drying. X-ray diffraction (XRD) was used to check internal order in the films (Fig. 2C). The XRD spectrum of the CTPR164Cys film showed two peaks at 2θ = 9.6° (001) and 20.0° (002) which could correspond to a lamellar packing of the protein with a periodical d-spacing of 9.1 Å, in agreement with previously described CTPR protein films.11 CTPR164Cys-AuNPs films also showed a peak at 2θ = 20.1°, related to the protein packing. In addition, these AuNPs-doped films exhibited two peaks which are not present in the pure protein film. A first peak at 2θ = 37.7° assigned to the crystal structure of AuNPs56 and a second peak at 2θ = 3.6° which corresponds to a periodical d-spacing of 2.3 nm. This spacing is attributed to the arrangement of gold nanoparticles within the solid film. It suggests a tighter packing of the AuNPs in the 3D film than the one observed by STM in a single molecule arrangement (Fig. 3B). To explore further the packing of the gold nanoparticles within the CTPR164Cys-AuNPs films, STM was performed (Fig. 3D). A 1 μM solution of CTPR164Cys-AuNPs conjugates (1000 times higher than the previous concentration for single molecule imaging) was deposited directly onto a gold surface. After solvent evaporation, a thin layer of CTPR164Cys-AuNPs was formed. STM images of the formed layer showed rows of AuNPs homogeneously distributed within the film. AuNPs are closer than expected from linear protein packing, as the proteins intercrossed minimizing AuNPs distances. The average inter-particle distance between AuNPs centers in the film is 4.2 ± 0.3 nm (Fig. 3D) with a nanoparticle gap of ∼ 0.6 nm. This smaller interparticle distance measured in the 2D film arrangement when compared with the inter-particle distance determined for individual CTPR164Cys-AuNPs (Fig. 3B), is in agreement with the tighter packing in the solid film previously determined by XRD (Fig. 3C). However, the inter-particle distance in film reported by XRD (2.3 nm) and by STM (4.2 nm) slightly differ since XRD reports on 3D arrangement and STM on 2D.
Charge transport properties of both CTPR16 protein and CTPR164Cys-AuNPs conjugate films were studied by measuring current–voltage (I–V) curves in the (−1 V, +1 V) interval (Fig. 4). The absolute value of the applied bias was limited to 1 V, which is smaller than thermoneutral voltage for water electrolysis.57 Films were formed onto Si/SiO2 wafers with interdigitated gold electrodes on top using different protein and conjugate batches to ensure reproducibility. The electrodes described a channel with a width W to length L ratio (W/L) of approximately 600, which minimized the effect of edge currents between the electrodes (see Experimental for details). At low applied voltage, (−0.3 V, 0.3 V) interval, films showed an ohmic behavior (constant G in Fig. 4B). In this range, protein films showed an average conductivity value of 140 ± 19 nS m−1, where current was transported through 20 μm of protein film according to the inter-distance length (L) between the electrodes. Conductivity was calculated using the equation σ = G × L/(t × W), where G = I/V is the conductance obtained from the I–V curve slope. The thickness of each film (t) was measured by atomic force microscopy (AFM) and W and L were defined by the electrode design (vide supra). In the case of the films doped with AuNPs, their conductivity increased by 4 orders of magnitude, with an average value of 1.37 ± 0.35 mS m−1 in the same applied voltage range. Measurements were performed across sets of independent batches of proteins and AuNP conjugates with high reproducibility of the current measurements when scaling the results by the film thickness (different curves in Fig. 4A). To better compare the profile of the curves, Fig. 4B shows the voltage dependence of the conductance normalized by its value in the low voltage regime (G/Glow). The G/Glow-V curves are symmetrical for both the CTPR164Cys and CTPR164Cys-AuNPs films. The conductance in the CTPR164Cys films showed a significant increase with the applied voltage, different from the behavior observed for CTPR164Cys-AuNPs films. Assuming that the edge resistivity is similar in the electrodes for all the films, these differences in the curve profile indicate differences in the main charge transport mechanism between the films with and without AuNPs. The normalized conductance of the CTPR164Cys films showed an exponential increase at absolute values of voltage ≥0.4 V. This non-linear profile is consistent with the presence of charge-carrier blocking at the electrical contacts, which is characteristic of protonic conductivity.27,41 This observation is in agreement with previously reported protonic charge transport for CTPR protein films.41 On the contrary, the CTPR164Cys-AuNPs films showed a quasi-linear increase in conductance for most of the explored voltage range (−1 V, 1 V), indicating a difference in their charge transport properties. The enhancement of the conductivity observed for the films with AuNPs is consistent with electron-like charge transport within the films. The inter-particle distance measured in the AuNP-doped films is compatible with hopping electron transfer between the AuNPs,58,59 in addition the described packing distance between consecutive layers within the protein films51,60 is also compatible with hopping electron transfer between layers. These effects would facilitate electron transport, thus increasing the conductivity along the CTPR164Cys-AuNPs films. This hypothesis is in agreement with the observations extracted from the G/Glowvs. V curves. A precise characterization of the charge transport mechanism through the protein films with and without AuNPs would require deeper studies combining several techniques and an analysis of the temperature and humidity dependence of the conductivity. However, the overall results shown here point to a mainly protonic charge transport in the CTPRs films and the emergence of electronic charge transport in the CTPR164Cys-AuNPs films.
 | ||
| Fig. 4 Transport properties of protein and protein-AuNPs thin films. (A) Current intensity(I) vs applied voltage(V) plot obtained for different CTPR16 films in black and CTPR164Cys-AuNPs films in red. The Y axis shows the intensity scaled by the film thickness (t) in logarithmic scale. (B) Normalized conductance (G/Glow) vs. applied voltage(V) for CTPR16 films in black and CTPR164Cys-AuNPs film in red obtained from the curves of part A. | ||
Conclusions
This work showed for the first time that gold nanoparticles, as an added electronic component, can be used to enhance the conductivity of protein-based films composed of engineered proteins. The main assets of this approach exploit (i) the rigid superhelical scaffold of CTPR proteins, (ii) their self-assembly properties that makes it possible to form protein films and (iii) the engineering of such proteins to form nanobioconjugates, enabling a strategy for controlled nanoparticles templating.Our study shows that a rigid superhelical scaffold, i.e. the engineered CTPR protein, can be used to regularly space up to 4 AuNPs per protein. Solid protein films were then formed, based on the self-assembly properties of CTPR proteins. Such films formed with pure protein or CTPR164Cys-AuNP conjugates exhibit long-range conductivity. CTPR164Cys-AuNPs films display a conductivity value of 1.37 ± 0.35 mS m−1, four orders of magnitude larger than the one measured for protein films. Furthermore, AuNPs seem to promote a change in the charge transport properties, as reflected by differences in the I–V curve profiles. The results presented are consistent with different charge transfer mechanisms: a mainly protonic mechanism for the CTPR films and an additional electronic component for the AuNPs-doped films. The latter favored by the short AuNP inter-particle gap observed in the CTPR164Cys-AuNPs films (∼0.6 nm), which is suitable for hopping electron transfer between AuNPs within the films. Although the conductive mechanisms along CTPR and CTPR164Cys-AuNPs films need further studies, our work opens the way to new designs and understandings of long-range conductivity through protein, which is both fundamentally interesting and potentially significant for the development of bioelectronics materials. We also demonstrate a generic approach for grafting any kind of nanoparticle on the scaffold of the proteins without altering its structure. Given the modular nature and the geometry of CTPR proteins, other inorganic nanomaterials could be arranged with adjustable inter-particle distances, making our approach a versatile tool for other applications such as optical coupling.
Author contributions
Conceptualization, A.L.C, and S.H.M.; Methodology, S.H.M., E.L-M, P.C., A.M-L., D.R., M.F., A.S-I, S.C., M.R.O, J.M.A, M.T.G.; Formal analysis, S.H.M., M.F., S.C., J.M.A, M.T.G., and A.L.C; Investigation: protein–nanoparticle conjugation and characterization, S.H.M., E.L-M, P.C., D.R., and M.F.; Investigation: nanoparticle synthesis, J.M.A., A.S-I.; Investigation: STM studies, A.M-L., and M.T.G.; Investigation: conductivity studies, S.H.M., S.C., M.R.O, and, M.T.G; Writing—original draft preparation, S.H.M., E-L.M, J.M.A., M.T.G., and A.L.C.; Funding acquisition, J.M.A., M.T.G., and A.L.C. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the European Research Council ERC-CoG-648071-ProNANO, ERC-PoC-841063-NIMM, Agencia Estatal de Investigación, Spain (PID2019-111649RB-I00; and MAT2017-88693-R), and the Basque Government (Elkartek KK-2017/00008), E.L-M thanks the Spanish Ministry of Science and Innovation for the FPI grant (BES-2017-079646). This work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency – Grant No. MDM-2017-0720 (CIC biomaGUNE) and SEV-2016-0686 (IMDEA Nanociencia).Notes and references
- J. Clarke and L. Regan, Curr. Opin. Struct. Biol., 2010, 20, 480–481 CrossRef CAS PubMed.
- S. Gomes, I. B. Leonor, J. F. Mano, R. L. Reis and D. L. Kaplan, Prog. Polym. Sci., 2012, 37, 1–17 CrossRef CAS PubMed.
- L. Quintanilla-Sierra, C. García-Arévalo and J. C. Rodriguez-Cabello, Mater. Today Bio., 2019, 2, 100007 CrossRef CAS PubMed.
- J. Teßmar, F. Brandl and A. Göpferich, in Fundamentals of Tissue Engineering and Regenerative Medicine, ed. U. Meyer, J. Handschel, H. P. Wiesmann and T. Meyer, Springer, Berlin, Heidelberg, 2009, pp. 495–517 Search PubMed.
- S. B. J. Kan, X. Huang, Y. Gumulya, K. Chen and F. H. Arnold, Nature, 2017, 552, 132–136 CrossRef CAS PubMed.
- D. W. Watkins, J. M. X. Jenkins, K. J. Grayson, N. Wood, J. W. Steventon, K. K. Le Vay, M. I. Goodwin, A. S. Mullen, H. J. Bailey, M. P. Crump, F. MacMillan, A. J. Mulholland, G. Cameron, R. B. Sessions, S. Mann and J. L. R. Anderson, Nat. Commun., 2017, 8, 358 CrossRef PubMed.
- A. E. Donnelly, G. S. Murphy, K. M. Digianantonio and M. H. Hecht, Nat. Chem. Biol., 2018, 14, 253–255 CrossRef CAS PubMed.
- D. Etezadi, J. B. Warner Iv, F. S. Ruggeri, G. Dietler, H. A. Lashuel and H. Altug, Light: Sci. Appl., 2017, 6, e17029–e17029 CrossRef CAS PubMed.
- E. Ferreira de Macedo, D. M. Ducatti Formaggio, N. Salles Santos and D. Batista Tada, Sensors, 2017, 17, 2765 CrossRef PubMed.
- S. H. Mejias, P. Couleaud, S. Casado, D. Granados, M. A. Garcia, J. M. Abad and A. L. Cortajarena, Colloids Surf., B, 2016, 141, 93–101 CrossRef CAS PubMed.
- S. H. Mejías, J. López-Andarias, T. Sakurai, S. Yoneda, K. P. Erazo, S. Seki, C. Atienza, N. Martín and A. L. Cortajarena, Chem. Sci., 2016, 7, 4842–4847 RSC.
- P. Couleaud, S. Adan-Bermudez, A. Aires, S. H. Mejías, B. Sot, A. Somoza and A. L. Cortajarena, Biomacromolecules, 2015, 16, 3836–3844 CrossRef CAS PubMed.
- S. H. Mejias, A. Aires, P. Couleaud and A. L. Cortajarena, Adv. Exp. Med. Biol., 2016, 940, 61–81 CrossRef CAS PubMed.
- J. Zhang, K. Zhou, Y. Zhang, M. Du and Q. Wang, Adv. Mater., 2019, 31, 1901485 CrossRef PubMed.
- S. Behrens, W. Habicht, K. Wagner and E. Unger, Adv. Mater., 2006, 18, 284–289 CrossRef CAS.
- C.-L. Chen, P. Zhang and N. L. Rosi, J. Am. Chem. Soc., 2008, 130, 13555–13557 CrossRef CAS PubMed.
- N. L. Ing, M. Y. El-Naggar and A. I. Hochbaum, J. Phys. Chem. B, 2018, 122, 10403–10423 CrossRef CAS PubMed.
- I. Ron, I. Pecht, M. Sheves and D. Cahen, Acc. Chem. Res., 2010, 43, 945–953 CrossRef CAS PubMed.
- V. V. Ptushenko, Biochemistry, 2020, 85, 955–965 CAS.
- H. B. Gray and J. R. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3534–3539 CrossRef CAS PubMed.
- M. Cordes and B. Giese, Chem. Soc. Rev., 2009, 38, 892–901 RSC.
- J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2014, 136, 2930–2939 CrossRef CAS PubMed.
- H. B. Gray and J. R. Winkler, Q. Rev. Biophys., 2003, 36, 341–372 CrossRef CAS PubMed.
- S. S. Skourtis, I. A. Balabin, T. Kawatsu and D. N. Beratan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3552–3557 CrossRef CAS PubMed.
- J. Blumberger, Chem. Rev., 2015, 115, 11191–11238 CrossRef CAS PubMed.
- D. D. Ordinario, L. Phan, W. G. W. Iv, J.-M. Jocson, E. Karshalev, N. Hüsken and A. A. Gorodetsky, Nat. Chem., 2014, 6, 596–602 CrossRef CAS PubMed.
- L. Glasser, Chem. Rev., 1975, 75, 21–65 CrossRef CAS.
- O. Silberbush, M. Amit, S. Roy and N. Ashkenasy, Adv. Funct. Mater., 2017, 27, 1604624 CrossRef.
- S. Y. Reece and D. G. Nocera, Annu. Rev. Biochem., 2009, 78, 673–699 CrossRef CAS PubMed.
- M. Amit, S. Appel, R. Cohen, G. Cheng, I. W. Hamley and N. Ashkenasy, Adv. Funct. Mater., 2014, 24, 5873–5880 CrossRef CAS.
- P. Carloni, W. Andreoni and M. Parrinello, Phys. Rev. Lett., 1997, 79, 761–764 CrossRef CAS.
- R. A. Jishi, N. C. Braier, C. T. White and J. W. Mintmire, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, R16009–R16011 CrossRef CAS.
- J. S. Lee, I. Yoon, J. Kim, H. Ihee, B. Kim and C. B. Park, Angew. Chem., Int. Ed., 2011, 50, 1164–1167 CrossRef CAS PubMed.
- B. Akdim, R. Pachter and R. R. Naik, Appl. Phys. Lett., 2015, 106, 183707 CrossRef.
- C. C. Moser, S. E. Chobot, C. C. Page and P. L. Dutton, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 1032–1037 CrossRef CAS PubMed.
- I. Ron, L. Sepunaru, S. Itzhakov, T. Belenkova, N. Friedman, I. Pecht, M. Sheves and D. Cahen, J. Am. Chem. Soc., 2010, 132, 4131–4140 CrossRef CAS PubMed.
- L. D. D'Andrea and L. Regan, Trends Biochem. Sci., 2003, 28, 655–662 CrossRef PubMed.
- T. Kajander, A. L. Cortajarena, S. Mochrie and L. Regan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 800–811 CrossRef CAS PubMed.
- T. Z. Grove, L. Regan and A. L. Cortajarena, J. R. Soc., Interface, 2013, 10, 20130051 CrossRef PubMed.
- J. López–Andarias, S. H. Mejías, T. Sakurai, W. Matsuda, S. Seki, F. Feixas, S. Osuna, C. Atienza, N. Martín and A. L. Cortajarena, Adv. Funct. Mater., 2018, 28, 1704031 CrossRef.
- N. A. Carter and T. Z. Grove, J. Am. Chem. Soc., 2018, 140, 7144–7151 CrossRef CAS PubMed.
- J. M. Abad, M. Gass, A. Bleloch and D. J. Schiffrin, J. Am. Chem. Soc., 2009, 131, 10229–10236 CrossRef CAS PubMed.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS PubMed.
- P. S. Jensen, Q. Chi, F. B. Grumsen, J. M. Abad, A. Horsewell, D. J. Schiffrin and J. Ulstrup, J. Phys. Chem. C, 2007, 111, 6124–6132 CrossRef CAS.
- T. Guterman, N. L. Ing, S. Fleischer, P. Rehak, V. Basavalingappa, Y. Hunashal, R. Dongre, S. Raghothama, P. Král, T. Dvir, A. I. Hochbaum and E. Gazit, Adv. Mater., 2019, 31, 1807285 CrossRef PubMed.
- M. T. González, A. Díaz, E. Leary, R. García, M. Á. Herranz, G. Rubio-Bollinger, N. Martín and N. Agraït, J. Am. Chem. Soc., 2013, 135, 5420–5426 CrossRef PubMed.
- J. Pérez–Juste, L. M. Liz–Marzán, S. Carnie, D. Y. C. Chan and P. Mulvaney, Adv. Funct. Mater., 2004, 14, 571–579 CrossRef.
- T. Kajander, A. L. Cortajarena and L. Regan, Methods Mol. Biol., 2006, 340, 151–170 CAS.
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4, 2411–2423 CrossRef CAS PubMed.
- X. Liu, M. Atwater, J. Wang and Q. Huo, Colloids Surf., B, 2007, 58, 3–7 CrossRef CAS PubMed.
- T. Z. Grove, L. Regan and A. L. Cortajarena, J. R. Soc., Interface, 2013, 10, 20130051 CrossRef PubMed.
- A. Pena-Francesch, H. Jung, M. A. Hickner, M. Tyagi, B. D. Allen and M. C. Demirel, Chem. Mater., 2018, 30, 898–905 CrossRef CAS.
- E. R. G. Main, Y. Xiong, M. J. Cocco, L. D'Andrea and L. Regan, Structure, 2003, 11, 497–508 CrossRef CAS.
- T. Kajander, A. L. Cortajarena, E. R. G. Main, S. G. J. Mochrie and L. Regan, J. Am. Chem. Soc., 2005, 127, 10188–10190 CrossRef CAS PubMed.
- S. S. Cohen, I. Riven, A. L. Cortajarena, L. De Rosa, L. D. D'Andrea, L. Regan and G. Haran, J. Am. Chem. Soc., 2015, 137, 10367–10373 CrossRef CAS PubMed.
- Y. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- A. Ursua, L. M. Gandia and P. Sanchis, Proc. IEEE, 2012, 100, 410–426 CAS.
- M. Gilbert and B. Albinsson, Chem. Soc. Rev., 2015, 44, 845–862 RSC.
- C. Li, C. Xu, D. Cahen and Y. Jin, Sci. Rep., 2019, 9, 18336 CrossRef CAS PubMed.
- N. A. Carter and T. Z. Grove, Biomacromolecules, 2015, 16, 706–714 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Figure. See DOI: 10.1039/d1nr00238d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |