
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-functional ionic porous organic framework for palladium scavenging and heterogeneous catalysis†
Shun-Shun
Qin
a,
Ze-Kun
Wang
a,
Lei
Hu
a,
Xing-Hao
Du
a,
Zheng
Wu
a,
Maria
Strømme
 b,
Qian-Feng
Zhang
a and
Chao
Xu
*ab
b,
Qian-Feng
Zhang
a and
Chao
Xu
*ab
aInstitute of Molecular Engineering and Applied Chemistry, Anhui University of Technology, Ma'anshan, 243002, P. R. China
bDepartment of Materials Science and Engineering, Uppsala University, The Ångström Laboratory, Box 35, 751 03 Uppsala, Sweden. E-mail: chao.xu@angstrom.uu.se; chao.xu0203@outlook.com
First published on 8th February 2021
Abstract
Porous organic frameworks (POFs) with predesigned structures and tunable porosities have been widely studied in adsorption and heterogeneous catalysis. Introducing ionic structure into the framework endows POFs with new functionalities that may extend their applications. Here, we report new applications for a guanidinium-based ionic POF (IPOF-Cl) in palladium scavenging and heterogeneous catalysis. Due to the ionic framework and the porous structure, the IPOF-Cl displays fast adsorption kinetics and high adsorption capacities (up to 754 mg g−1) of Na2PdCl4 in aqueous solutions via a chemisorption (ion exchange) process. Significantly, it shows excellent scavenging activity towards trace amount of [PdCl4]2− in aqueous solution. More importantly, the loaded [PdCl4]2− species on the IPOF substrate are further reduced into ultrafine Pd nanoparticles with size of ∼2–5 nm. The obtained IPOF-Pd(0) nanocomposite containing uniformly distributed Pd nanoparticles and hierarchical porous structure demonstrates high activity in catalyzing a range of Suzuki coupling reactions. This study provides new routes for the development of ionic porous organic materials for applications in metal scavenging and catalysis.
Introduction
Porous organic frameworks (POFs) are an emerging class of porous materials demonstrating unique characteristics of large specific surface areas, tunable porosities, and high physical and chemical stability.1–5 They are constructed by joining pure organic monomers via covalent bonds. From a synthetic point of view, numerous organic monomers and several organic reactions (e.g., condensation reactions, coupling reactions) can be applied in the synthesis of POFs.6,7 By judicious selection of the monomers and the reactions, various POFs with different building blocks and architectures have been successfully synthesized.8–12 In addition, the rich organic groups in POFs enable facile modification on the surface, offering an opportunity for the functionalization of POFs.13 In this context, the synthetic diversity and the principles of reticular chemistry allow the construction of novel POFs with predesigned structures and tailored properties for various applications.14–21Recently, a few examples of POFs consisting of ionic frameworks and counter-ions have been reported.22–26 Although the ionic POFs (IPOFs) usually have moderate surface areas due to the pore-blocking effect by the counter-ions, they demonstrate several unique advantages compared to their neutral analogs.27 For example, the ionic structure of IPOFs enables ion exchange which allows the pore size to be finely controlled by adjusting the size of counter-ions.28 Such IPOFs with controlled pore sizes demonstrated promising applications in size-selective adsorption and catalysis.25 In addition, the ionic interface in IPOFs triggers electrostatic interactions with guest molecules or ions that could enhance their performances in CO2 capture and in trapping ionic pollutants.26,29–32 Therefore, the design of IPOFs with tunable nanostructures, compositions and properties could further extend their applications in adsorption and catalysis.
Palladium catalysts (e.g., inorganic palladium salts, organopalladium, palladium nanoparticles) are widely used in a range of important organic transformations such as C–C coupling, hydrogenation, dehydrogenation.33 However, traditional homogeneous palladium catalysts usually suffer from significant recovery problems, which not only cause a waste of precious metal resource but also lead to heavy metal contamination in the products. In order to overcome these problems, it is highly desirable to develop efficient palladium scavengers and heterogeneous palladium catalysts.34–40 Given the ionic and porous structure, we envisioned that IPOFs may find applications in scavenging ionic palladium residues from solution via ion exchange and/or physical adsorption. In addition, immobilizing the palladium species in the porous channels of IPOFs offers a new route for the development of heterogeneous catalyst. Herein, we synthesize a dual functional IPOF-Cl composed of cationic guanidinium-based framework and chloride ions as charge-balance ions. The IPOF shows high adsorption uptake of tetrachloropalladate ions ([PdCl4]2−) in aqueous solutions. In addition, the adsorbed [PdCl4]2− ions in the porous channels are further reduced into ultrafine Pd nanoparticles (Scheme 1). As a result, the Pd nanoparticles immobilized IPOF shows high efficiency for catalytic conversion of a range of Suzuki coupling reactions.
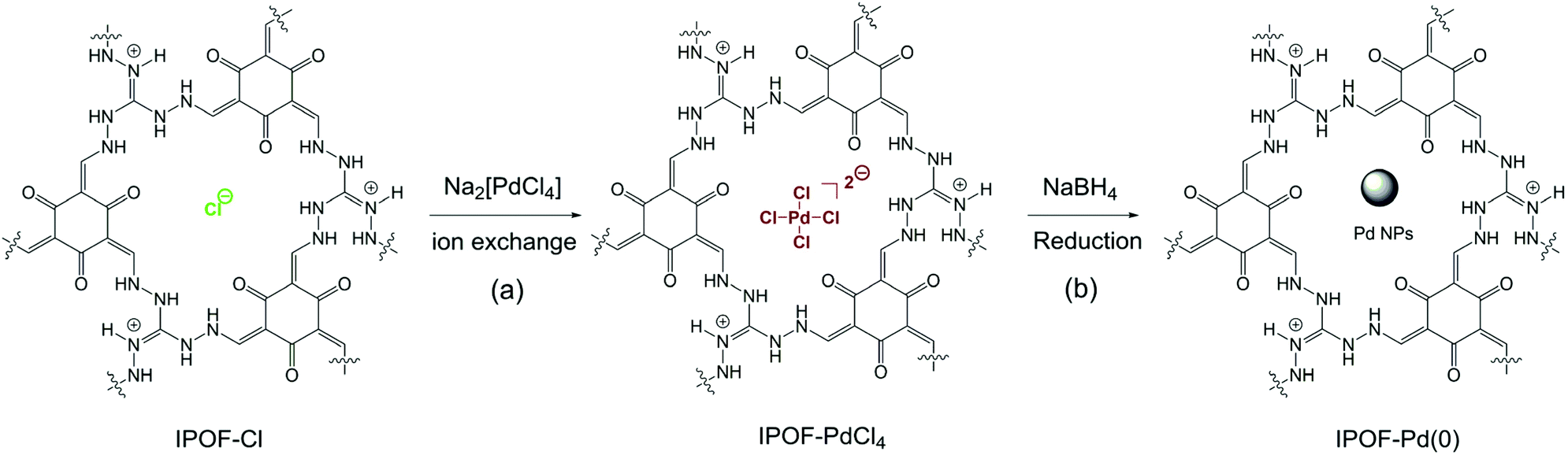 | ||
| Scheme 1 (a) Schematic reaction of ion exchange of IPOF-Cl with Na2PdCl4; (b) reduction of IPOF-PdCl4 by NaBH4 into IPOF-Pd(0). | ||
Results and discussion
IPOF-Cl was synthesized by the condensation reaction of triaminoguanidinium chloride (TGCl)24 with 1,3,5-triformylphloroglucinol (TFP, Shanghai Tensus Bio-tech) (Scheme S1†). TGCl is a hydrochloride salt of triaminoguanidine that contains three primary amine groups, one protonated tertiary amine and one counter-ion Cl−. The rich chemistry and the high reactivity of the amine groups make TGCl an ideal ionic monomer for the synthesis of IPOFs. The structure of IPOF-Cl was studied by infrared (IR), solid-state 13C nuclear magnetic resonance (NMR) spectroscopy, and powder X-ray diffraction (XRD). The strong bands at 1617 cm−1 and 1292 cm−1 displayed in the IR spectrum can be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–N stretching, respectively (Fig. 1a and S1†).41 The sharp peaks at 101 and 146 ppm observed in the 13C NMR spectrum can be attributed to the exocyclic CC carbons.19 The chemical shifts for carbons of guanidinium (CN) and keto (CO) are observed at 164 and 179 ppm, respectively.24 The peak detected at 192 ppm indicates the presence of unreacted aldehyde groups from the TFP monomer due to the reversible Schiff-base condensation reaction (Fig. 1b). Such unreacted aldehyde groups were also observed in various imine-linked porous organic polymers.42–44 The broad diffraction peaks observed for IPOF-Cl indicate relatively low crystallinity of the material compared with typical covalent organic frameworks.1,8 The diffraction peak at 2θ = ∼10.0 can tentatively be assigned to the 100 reflection. The intensive peak at 2θ = ∼26.8° suggests the existence of π–π stacking between the 2D layers in IPOF-Cl (Fig. 1c and S2†). The low crystallinity of IPOF-Cl could be explained by the fact that the repulsive interactions between the cationic layers hampered the formation of a highly ordered and closely stacked structure. In addition, N2 sorption measurements reveal that IPOF-Cl has a moderate surface area of 163 m2 g−1, which is lower than for many reported porous organic materials probably because the Cl ions partially block the porous channels. The significant adsorption uptake at high relative pressures and the existence of adsorption/desorption hysteresis suggest the formation of mesopores in IPOF-Cl. Pore size distribution analysis based on the adsorption isotherm indicates that IPOF-Cl contains micropores centered at 1.5 nm and mesopores with a broad distribution centered at ∼30 nm (Fig. 1d). These results are in agreement with those of reported results,17,24 which signifies the formation of guanidinium- and ketoenamine-linked IPOF-Cl with hierarchical porous structure.
C and C–N stretching, respectively (Fig. 1a and S1†).41 The sharp peaks at 101 and 146 ppm observed in the 13C NMR spectrum can be attributed to the exocyclic CC carbons.19 The chemical shifts for carbons of guanidinium (CN) and keto (CO) are observed at 164 and 179 ppm, respectively.24 The peak detected at 192 ppm indicates the presence of unreacted aldehyde groups from the TFP monomer due to the reversible Schiff-base condensation reaction (Fig. 1b). Such unreacted aldehyde groups were also observed in various imine-linked porous organic polymers.42–44 The broad diffraction peaks observed for IPOF-Cl indicate relatively low crystallinity of the material compared with typical covalent organic frameworks.1,8 The diffraction peak at 2θ = ∼10.0 can tentatively be assigned to the 100 reflection. The intensive peak at 2θ = ∼26.8° suggests the existence of π–π stacking between the 2D layers in IPOF-Cl (Fig. 1c and S2†). The low crystallinity of IPOF-Cl could be explained by the fact that the repulsive interactions between the cationic layers hampered the formation of a highly ordered and closely stacked structure. In addition, N2 sorption measurements reveal that IPOF-Cl has a moderate surface area of 163 m2 g−1, which is lower than for many reported porous organic materials probably because the Cl ions partially block the porous channels. The significant adsorption uptake at high relative pressures and the existence of adsorption/desorption hysteresis suggest the formation of mesopores in IPOF-Cl. Pore size distribution analysis based on the adsorption isotherm indicates that IPOF-Cl contains micropores centered at 1.5 nm and mesopores with a broad distribution centered at ∼30 nm (Fig. 1d). These results are in agreement with those of reported results,17,24 which signifies the formation of guanidinium- and ketoenamine-linked IPOF-Cl with hierarchical porous structure.
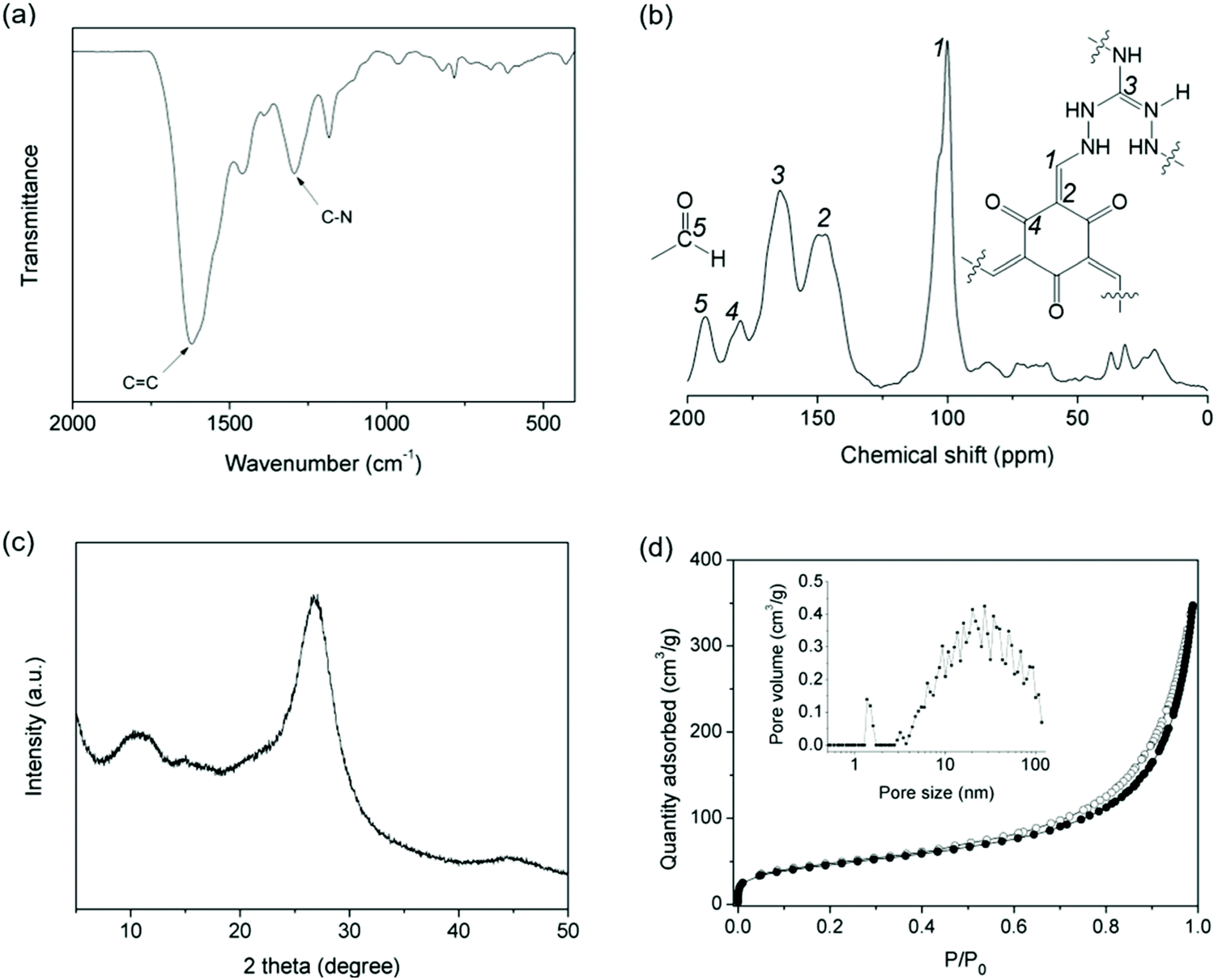 | ||
| Fig. 1 (a) Infrared spectrum, (b) solid state 13C nuclear magnetic resonance spectrum, (c) powder X-ray diffraction pattern of IPOF-Cl; (d) N2 adsorption and desorption isotherms of IPOF-Cl recorded at 77 K. The inset shows the pore size distribution of IPOF-Cl calculated from the adsorption branch using density functional theory model. | ||
As displayed in various ionic porous materials (e.g. zeolites,45 ionic porous organic polymers,46 ionic metal–organic frameworks47), the ionic structure could endow IPOF-Cl with ion exchange ability that can potentially be used as a scavenger for anionic metal complexes. In this context, we studied the capture capability of IPOF-Cl towards [PdCl4]2−, a widely used homogeneous catalyst in a range of organic reactions.48,49 Firstly, we studied the kinetic of [PdCl4]2− capture from an aqueous solution by using IPOF-Cl. Specifically, an aqueous solution of Na2PdCl4 (33 mL, 0.6 mg mL−1) was mixed and stirred with grinded IPOF-Cl (20 mg) at room temperature. The Pd content in the solution at different intervals was determined by Inductively Coupled Plasma-Atomic Emission Spectrometer (ICP-AES). Obviously, the concentration of Na2PdCl4 in the solution decreased rapidly and more than 75% of the uptake was reached within the initial 10 min. The capture kinetic curve fits well with the pseudo-second-order kinetic model, indicating that the capture process was dominated by an ion exchange (or chemisorption) process (Fig. 2a and S3†).50 In addition, the adsorption isotherm was recorded to study the effect of Na2PdCl4 concentration on the capture process. As shown in Fig. 2c and S4,† the adsorption isotherm fits well with the Langmuir adsorption model. The maximum capture capacity is calculated to be 852 mg g−1. The value is much higher than the theoretical capacity of 572 mg g−1 based on ion exchange, which can be attributed to a synergetic effect of ion exchange and physisorption of Na2PdCl4 in the IPOF. When it reached adsorption equilibration in the aqueous solution of Na2PdCl4 (0.8 mg mL−1), the adsorbent was collected and its porosity was characterized by N2 sorption measurement (Fig. S5†). Since [PdCl4]2− ions possess a larger molecular weight and size than the original counter-ions of Cl−, the replacement of Cl− with [PdCl4]2− in IPOF-Cl not only increases the bulk density and but also partially blocks the pores of the material. As a result, the surface area and the total pore volume of the sorbent are significantly reduced from 163 m2 g−1 and 0.352 cm3 g−1 for IPOF-Cl to 48 m2 g−1 and 0.109 cm3 g−1 for IPOF-PdCl4, respectively. The reduced porosity also demonstrates the efficient capturing ability of IPOF-Cl towards [PdCl4]2−.
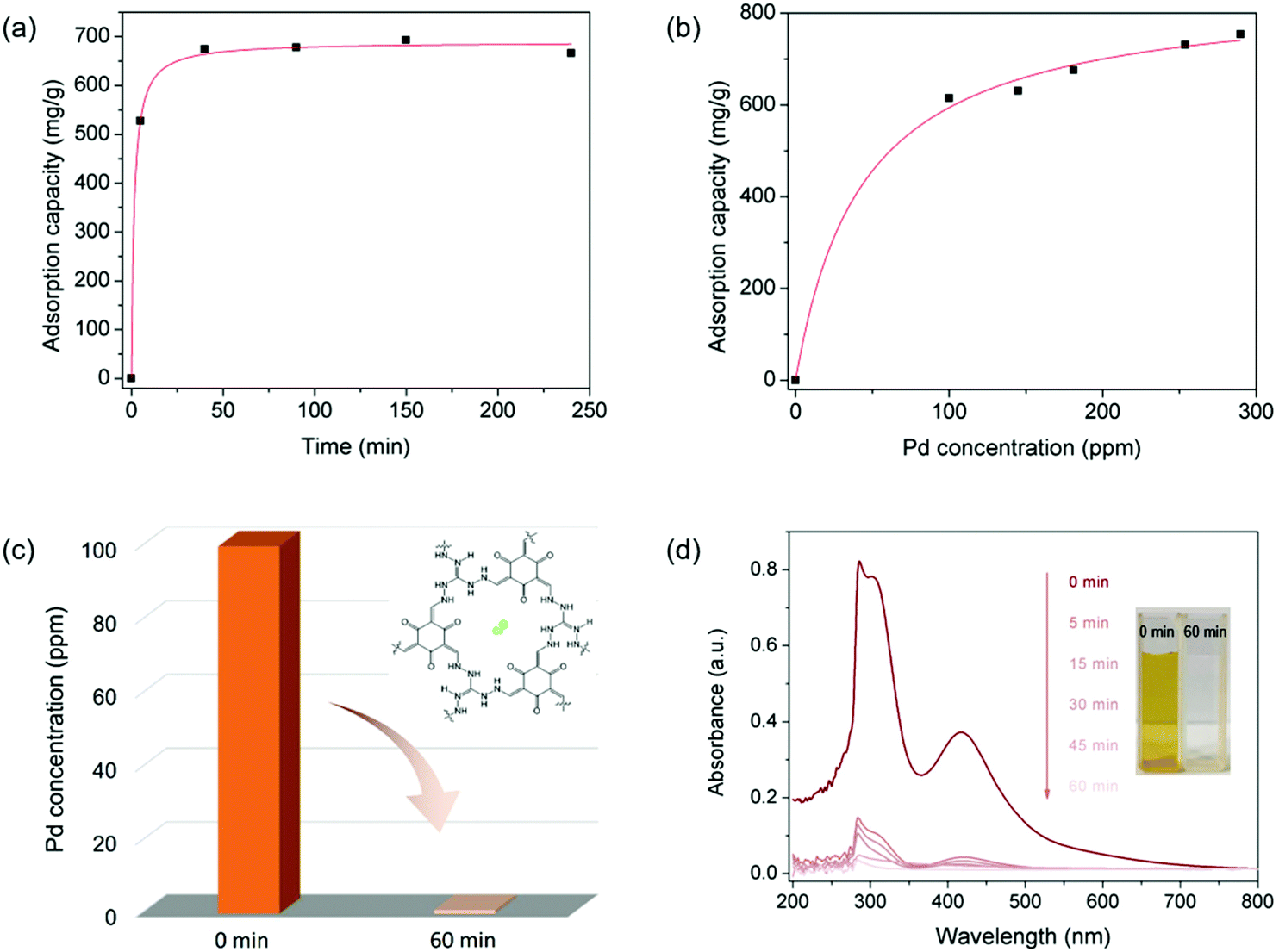 | ||
| Fig. 2 (a) Kinetic curve of Na2PdCl4 capture on IPOF-Cl; (b) Na2PdCl4 adsorption isotherm of IPOF; (c) scavenging performance of IPOF-Cl towards Na2PdCl4 in an aqueous solution; (d) UV–vis spectra of aqueous solution of Na2PdCl4 at different intervals during the scavenging experiment. The inset shows the optical image of the solution before and the after the scavenging experiment. The initial concentration of the solution was 100 ppm. | ||
Given the high uptake and the fast kinetic of the capture process, we anticipated that it can be used as an efficient scavenger for removing trace amount of Na2PdCl4 from aqueous solutions. Remarkably, the use of 10 mg of IPOF-Cl reduced the concentration of aqueous Na2PdCl4 solution (9 mL) from 100 ppm to <1 ppm in 60 min (Fig. 2c). In addition, the UV–vis spectra and the optical images of the solution clearly show the efficient removal of Na2PdCl4 in the solution by the use of IPOF-Cl (Fig. 2d).
The hierarchical porous structure of IPOF-Cl and its high loading capacity of [PdCl4]2− further inspired us to investigate the potential of IPOF-Cl in heterogeneous catalysis. After capturing [PdCl4]2− from the aqueous solution, the solid was collected by centrifugation and then thoroughly washed by water. The obtained complex of IPOF-PdCl4 was redispersed in water, followed by adding NaBH4 to reduce the Pd(II) species into Pd(0) nanoparticles that gave IPOF-Pd(0). The composition of IPOF-Pd(0) was examined by XRD analysis. The intensive diffraction peaks at 2θ = ∼40.1, 46.5 and 68.0° can be attributed to the 110, 200, and 220 reflections of Pd(0), respectively. The broad diffraction peak at 2θ = ∼26.8° suggests that the crystalline structure of the IPOF substrate mainly remained intact (Fig. 3a). In addition, no significant change is observed in the infrared spectra (Fig. S6 and S7†). These results demonstrate the high chemical and structural stability of the IPOF. Such high stability for ketoenamine-linked porous organic materials has also been observed in previous studies.41,51,52 The high-resolution X-ray photoelectron spectroscopy (XPS) spectrum of IPOF-Pd(0) shows two sets of peaks: 338.48 and 335.25 eV in the Pd 3d5/2 region and 343.6 and 340.5 eV in the Pd 3d3/2 region, indicating the existence of both Pd(II) and Pd(0) in the composite (Fig. 3b and S8†). The deconvolution analysis of the spectrum suggests that it contains 74 wt% of Pd(0) and 26 wt% of Pd(II) species, respectively. The surface Pd content in the composite is calculated to be 31.4 wt% from the XPS analysis, which is slightly higher than the bulk Pd content (25.4 wt%) derived from the thermogravimetric analysis of the composite in an air atmosphere (Fig. S9†). Transmission electron microscopy (TEM) images reveal the formation of ultrafine Pd nanoparticles with a narrow size distribution of ∼2–5 nm in the nanocomposites (Fig. 3c and d). The nanoparticles are uniformly dispersed in the POF substrate and no significant aggregation of the nanoparticles is observed. It appears that the templating effect of IPOF with ordered porous structure plays a key role in the formation of such narrowly distributed ultrafine Pd nanoparticles.52 In addition, the interactions between the Pd nanoparticles and the amine groups in the organic framework could also assist the stabilization of the nanoparticles.53,54 As expected, the surface area of the composites is reduced to 27 m2 g−1. Importantly, the pore size distribution analysis of the N2 adsorption isotherm confirms that the hierarchical porous structure of the IPOF substrate remained after loading the Pd nanoparticles (Fig. 3e–f).
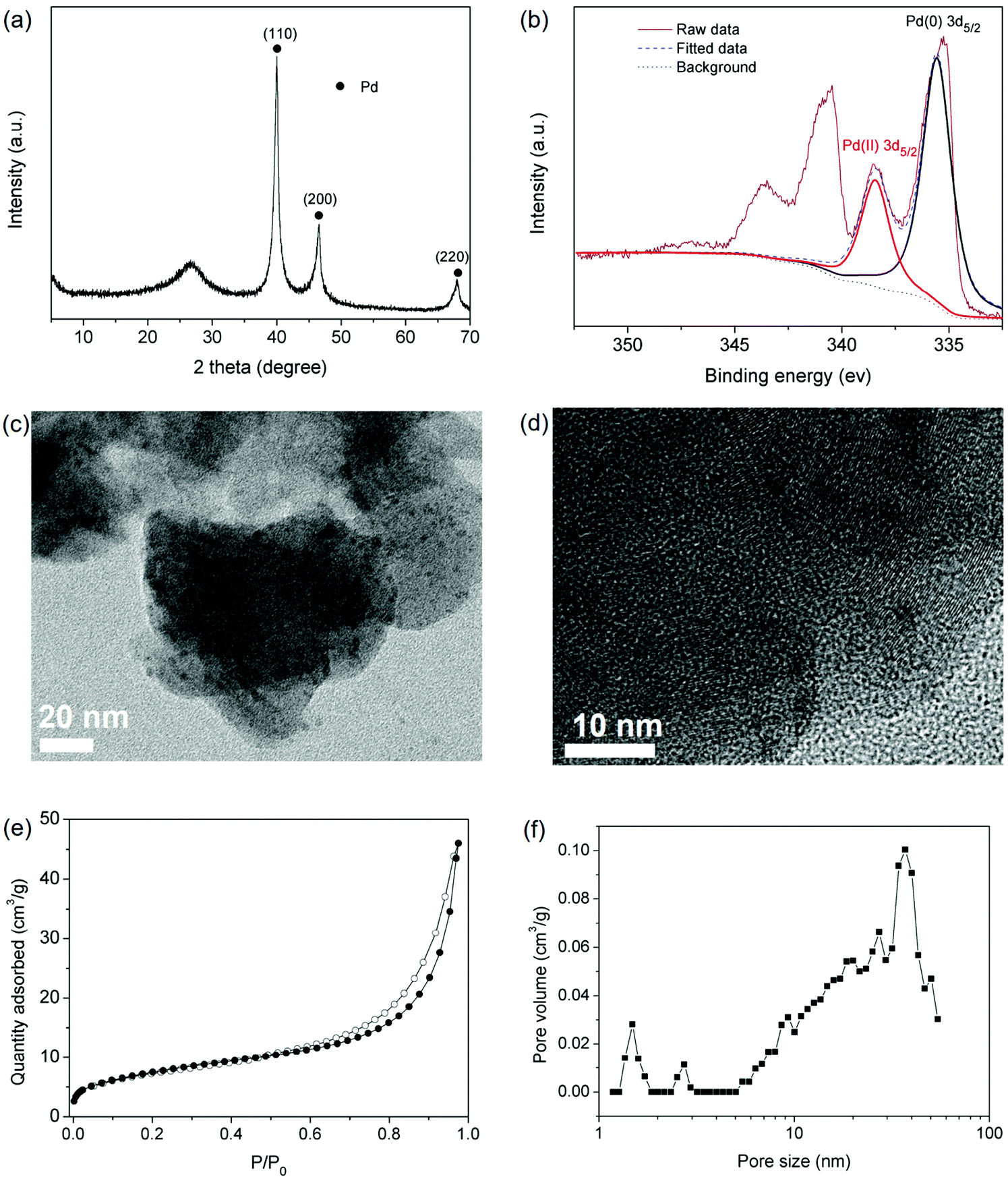 | ||
| Fig. 3 (a) Powder X-ray diffraction pattern of IPOF-Pd(0); (b) high-resolution Pd 3d X-ray photoelectron spectrum of IPOF-Pd(0) and its deconvoluted parts; (c) transmittance electron microscopy (TEM) and (d) high-resolution TEM image of IPOF-Pd(0); (e) N2 sorption isotherms of IPOF-Pd(0) recorded at 77 K; (f) pore size distribution of IPOF-Pd(0) calculated from the adsorption isotherm using density functional theory model. | ||
The catalytic activity of the IPOF-Pd(0) was evaluated in Suzuki coupling reactions of aryl halides and arylboronic acids in the presence of inorganic base. One of the advantages of the Suzuki coupling reaction is that the reaction can be conducted in various solvents including alcohol and water. Obviously, the high solubility of the arylboronic acid and the inorganic base in water could greatly promote the coupling reaction. More importantly, the use of water as the solvent provides a green and sustainable route for the organic synthesis. However, porous organic materials usually suffer from hydrolytic instability and/or hydrophobic nature preventing their applications in aqueous environments.55,56 Remarkably, the covalent nature and the ionic structure of the IPOF substrate endow high water stability and hydrophilicity of the catalyst, respectively, which greatly facilitate its catalytic application in aqueous solution. Therefore, we applied the catalytic reactions in a mixed solvent of water/ethanol (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 2:3). As expected, the catalyst showed high catalytic activity for a range of Suzuki coupling reactions (Table 1). The coupling reaction between iodobenzene and phenylboronic acid catalyzed by IPOF-Pd(0) (1 mol%) gave a high yield of 97% in 2 h (entry 1). The use of bromobenzene resulted in a slightly lower yield of 93% under the same condition (entry 2). Compared to iodobenzene and bromobenzene, chlorobenzene is an ideal starting material for various organic reactions because of its low cost. However, the low reactivity of chlorobenzene usually results in low yields largely limiting its practical uses in coupling reactions.57 Noteworthy, IPOF-Pd(0) showed high catalytic activity in the coupling reaction between phenylboronic acid and chlorobenzene. By increasing the catalyst amount to 2 mol% and prolonging the reaction time to 6 h, a relatively high yield of 83% can be achieved (entry 4). We also evaluated the scope of the reaction by varying the substituent group of the aryl halides and arylboronic acids. Having electron-withdrawing groups (–CF3, –F) or electron-donating groups (–CH3) on the organic substrates did not significantly affect the efficiency of the catalyst, which gave high yields of 82–95% (entries 5–11). A hot filtration experiment was also conducted and we found that no further conversion of the raw materials to the products was observed once the catalyst had been removed. This result suggested that no significant Pd leaching from the IPOF-Pd(0) occurred during the catalysis process due to the high physiochemical stability of the catalyst. In addition, the catalyst showed good recyclability and it can be recycled and used at least 5 times without significant loss of the catalytic activity (Fig. S10†). More importantly, the catalyst showed good chemical and structural stability during the catalytic conversions (Fig. S11†). The excellent catalytic performance of the IPOF-Pd(0) in Suzuki coupling reactions can be attributed to the synergetic results of (i) the uniform distribution of ultrafine Pd nanoparticles endowing high catalytic activity of the catalyst; (ii) the hierarchical porous structure of the IPOF substrate allowing for fast mass transport during the catalytic transformations; and (iii) the ionic structure and the resulted good water-dispersibility of the IPOF substrate increasing the compatibility between the starting materials and the catalyst.
:v = 2:3). As expected, the catalyst showed high catalytic activity for a range of Suzuki coupling reactions (Table 1). The coupling reaction between iodobenzene and phenylboronic acid catalyzed by IPOF-Pd(0) (1 mol%) gave a high yield of 97% in 2 h (entry 1). The use of bromobenzene resulted in a slightly lower yield of 93% under the same condition (entry 2). Compared to iodobenzene and bromobenzene, chlorobenzene is an ideal starting material for various organic reactions because of its low cost. However, the low reactivity of chlorobenzene usually results in low yields largely limiting its practical uses in coupling reactions.57 Noteworthy, IPOF-Pd(0) showed high catalytic activity in the coupling reaction between phenylboronic acid and chlorobenzene. By increasing the catalyst amount to 2 mol% and prolonging the reaction time to 6 h, a relatively high yield of 83% can be achieved (entry 4). We also evaluated the scope of the reaction by varying the substituent group of the aryl halides and arylboronic acids. Having electron-withdrawing groups (–CF3, –F) or electron-donating groups (–CH3) on the organic substrates did not significantly affect the efficiency of the catalyst, which gave high yields of 82–95% (entries 5–11). A hot filtration experiment was also conducted and we found that no further conversion of the raw materials to the products was observed once the catalyst had been removed. This result suggested that no significant Pd leaching from the IPOF-Pd(0) occurred during the catalysis process due to the high physiochemical stability of the catalyst. In addition, the catalyst showed good recyclability and it can be recycled and used at least 5 times without significant loss of the catalytic activity (Fig. S10†). More importantly, the catalyst showed good chemical and structural stability during the catalytic conversions (Fig. S11†). The excellent catalytic performance of the IPOF-Pd(0) in Suzuki coupling reactions can be attributed to the synergetic results of (i) the uniform distribution of ultrafine Pd nanoparticles endowing high catalytic activity of the catalyst; (ii) the hierarchical porous structure of the IPOF substrate allowing for fast mass transport during the catalytic transformations; and (iii) the ionic structure and the resulted good water-dispersibility of the IPOF substrate increasing the compatibility between the starting materials and the catalyst.
|
|
||||
|---|---|---|---|---|
| Entrya | Ar1-X | Ar2-B(OH)2 | Product | Yieldb (%) |
| a Reaction conditions: IPOF-Pd(0) (1 mol%), aryl halide (2.0 mmol, 1.0 equiv.), aryl boronic acid (2.4 mmol, 1.2 equiv.), K3PO4 (6.0 mmol, 3.0 equiv.), H2O (2.0 mL)/EtOH (3 mL), 85 °C, 2 h. b Yield of purified product after silica chromatography. c IPOF-Pd(0) (2 mol%), 6 h. | ||||
| 1 |
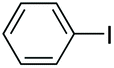
|
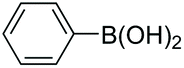
|
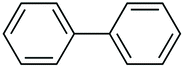
|
97 |
| 2 |
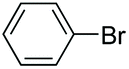
|
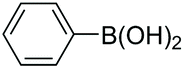
|
|
94 |
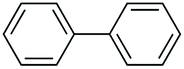
| 3 |
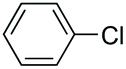
|
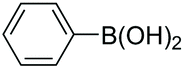
|
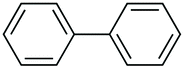
|
61 |
| 4c |
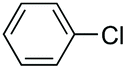
|
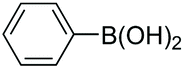
|
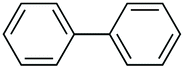
|
83 |
| 5 |
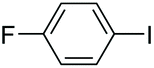
|
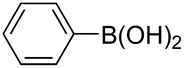
|
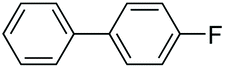
|
94 |
| 6 |
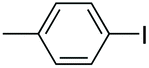
|
|
|
86 |
| 7 |
|
|
|
95 |
| 8 |
|
|
|
92 |
| 9 |
|
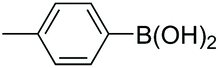
|
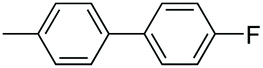
|
93 |
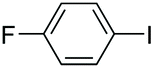
| 10 |
|
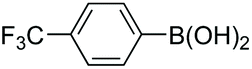
|
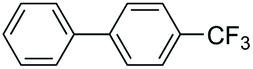
|
85 |
| 11 |
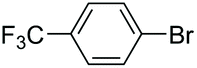
|
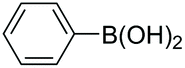
|
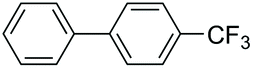
|
82 |
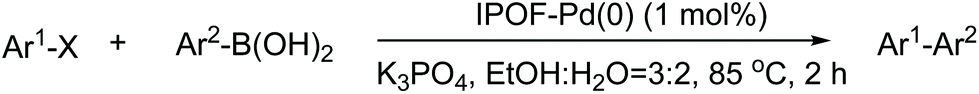
Conclusions
An ionic POF (IPOF-Cl) constructed by guanidinium and ketoenamine linkages has been studied for scavenging and heterogeneous catalysis applications. Owing to its ionic and hierarchical porous structure, IPOF-Cl showed high ion exchange capacity and excellent scavenging ability towards [PdCl4]2− in aqueous solutions. In addition, we have developed a new heterogeneous catalyst by reducing the captured [PdCl4]2− into Pd nanoparticles. Due to the synergetic effects of uniform distribution of Pd nanoparticles, high physiochemical stability, ionic structure, and hierarchical porous nature of the catalyst, the obtained nanocomposite of IPOF-Pd(0) demonstrated high catalytic activity and good recyclability in a series of Suzuki coupling reactions. We anticipate that the IPOF and its derived materials can be used in recovering precious metal ions, removing heavy metal pollutants, scavenging trace amount of metal ions, and immobilizing homogeneous catalysts that may find important practical applications in mining, water treatment, synthesis and handling of pharmaceuticals, industrial catalysis, and organic synthesis. Future studies may focus on the synthesis and functionalization of novel ionic porous organic materials for gas adsorption and separation, molecular sieving, and ion conduction applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Young Wanjiang Scholar Program and Åforsk research grant (19-493).References
- T. Q. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Y. Zhou, J. Niu, L. H. Li, Y. Y. Wang, J. Su, J. Li, X. G. Wang, W. D. Wang, W. Wang, J. L. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- Y. Li, W. Chen, G. Xing, D. Jiang and L. Chen, Chem. Soc. Rev., 2020, 49, 2852–2868 RSC.
- X. Zou, H. Ren and G. Zhu, Chem. Commun., 2013, 49, 3925–3936 RSC.
- T. Liu and G. Liu, Nat. Commun., 2020, 11, 4984 CrossRef CAS.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS.
- C. Xu and N. Hedin, Mater. Today, 2014, 17, 397–403 CrossRef CAS.
- S. Kandambeth, B. P. Biswal, H. D. Chaudhari, K. C. Rout, H. S. Kunjattu, S. Mitra, S. Karak, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, Adv. Mater., 2016, 29, 1603945 CrossRef.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673 CrossRef CAS.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS.
- A. Acharjya, P. Pachfule, J. Roeser, F.-J. Schmitt and A. Thomas, Angew. Chem., Int. Ed., 2019, 58, 14865–14870 CrossRef CAS.
- D. D. Medina, V. Werner, F. Auras, R. Tautz, M. Dogru, J. Schuster, S. Linke, M. Döblinger, J. Feldmann, P. Knochel and T. Bein, ACS Nano, 2014, 8, 4042–4052 CrossRef CAS.
- H. Ding, A. Mal and C. Wang, Mater. Chem. Front., 2020, 4, 113–127 RSC.
- S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Song, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS.
- Y.-N. Gong, W. Zhong, Y. Li, Y. Qiu, L. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 16723–16731 CrossRef CAS.
- Y. Qian, D. Li, Y. Han and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS.
- H. Yang, L. Yang, H. Wang, Z. Xu, Y. Zhao, Y. Luo, N. Nasir, Y. Song, H. Wu, F. Pan and Z. Jiang, Nat. Commun., 2019, 10, 2101 CrossRef.
- T. Banerjee, K. Gottschling, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CrossRef CAS.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS.
- X. Kong, S. Zhou, M. Strømme and C. Xu, Carbon, 2021, 171, 248–256 CrossRef CAS.
- X. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, C. Schlesiger, S. Praetz, J. Schmidt and A. Thomas, J. Am. Chem. Soc., 2019, 141, 6623–6630 CrossRef CAS.
- J. Roeser, D. Prill, M. J. Bojdys, P. Fayon, A. Trewin, A. N. Fitch, M. U. Schmidt and A. Thomas, Nat. Chem., 2017, 9, 977 CrossRef CAS.
- O. Yahiaoui, A. N. Fitch, F. Hoffmann, M. Fröba, A. Thomas and J. Roeser, J. Am. Chem. Soc., 2018, 140, 5330–5333 CrossRef CAS.
- S. Mitra, S. Kandambeth, B. P. Biswal, A. M. Khayum, C. K. Choudhury, M. Mehta, G. Kaur, S. Banerjee, A. Prabhune, S. Verma, S. Roy, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 2823–2828 CrossRef CAS.
- Z. Li, H. Li, X. Guan, J. Tang, Y. Yusran, Z. Li, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2017, 139, 17771–17774 CrossRef CAS.
- N. Huang, P. Wang, M. A. Addicoat, T. Heine and D. Jiang, Angew. Chem., Int. Ed., 2017, 56, 4982–4986 CrossRef CAS.
- C. Xu, G. Yu, J. Yuan, M. Strømme and N. Hedin, Mater. Today Adv., 2020, 6, 100052 CrossRef.
- H. Ma, B. Liu, B. Li, L. Zhang, Y.-G. Li, H.-Q. Tan, H.-Y. Zang and G. Zhu, J. Am. Chem. Soc., 2016, 138, 5897–5903 CrossRef CAS.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 CrossRef CAS.
- S. Jansone-Popova, A. Moinel, J. A. Schott, S. M. Mahurin, I. Popovs, G. M. Veith and B. A. Moyer, Environ. Sci. Technol., 2019, 53, 878–883 CrossRef CAS.
- H.-J. Da, C.-X. Yang and X.-P. Yan, Environ. Sci. Technol., 2019, 53, 5212–5220 CrossRef CAS.
- K. M. Gupta, K. Zhang and J. Jiang, Ind. Eng. Chem. Res., 2018, 57, 6477–6482 CrossRef CAS.
- X.-F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047–9050 CrossRef CAS.
- H. Ren, C. A. Strulson, G. Humphrey, R. Xiang, G. Li, D. R. Gauthier and K. M. Maloney, Green Chem., 2017, 19, 4002–4006 RSC.
- S. E. Hankari, A. E. Kadib, A. Finiels, A. Bouhaouss, J. J. E. Moreau, C. M. Crudden, D. Brunel and P. Hesemann, Chem. – Eur. J., 2011, 17, 8984–8994 CrossRef.
- C. Xu, S. Afewerki, C.-W. Tai, A. Córdova and N. Hedin, ChemistrySelect, 2016, 1, 5801–5804 CrossRef CAS.
- C. Xu, L. Deiana, S. Afewerki, C. Incerti-Pradillos, O. Córdova, P. Guo, A. Córdova and N. Hedin, Adv. Synth. Catal., 2015, 357, 2150–2156 CrossRef CAS.
- P. Zhang, Z. Weng, J. Guo and C. Wang, Chem. Mater., 2011, 23, 5243–5249 CrossRef CAS.
- A. Modak, J. Mondal, M. Sasidharan and A. Bhaumik, Green Chem., 2011, 13, 1317–1331 RSC.
- A. Modak, M. Pramanik, S. Inagaki and A. Bhaumik, J. Mater. Chem. A, 2014, 2, 11642–11650 RSC.
- K. Cui, W. Zhong, L. Li, Z. Zhuang, L. Li, J. Bi and Y. Yu, Small, 2019, 15, 1804419 Search PubMed.
- C. Xu, Z. Bacsik and N. Hedin, J. Mater. Chem. A, 2015, 3, 16229–16234 RSC.
- C. Xu and N. Hedin, Microporous Mesoporous Mater., 2016, 222, 80–86 CrossRef CAS.
- P. Pandey, A. P. Katsoulidis, I. Eryazici, Y. Wu, M. G. Kanatzidis and S. T. Nguyen, Chem. Mater., 2010, 22, 4974–4979 CrossRef CAS.
- S.-T. Yang, J. Kim and W.-S. Ahn, Microporous Mesoporous Mater., 2010, 135, 90–94 CrossRef CAS.
- Y. Su, Y. Wang, X. Li, X. Li and R. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 18904–18911 CrossRef CAS.
- X. Zhao, X. Bu, T. Wu, S.-T. Zheng, L. Wang and P. Feng, Nat. Commun., 2013, 4, 2344 CrossRef.
- A. Köllhofer and H. Plenio, Chem. – Eur. J., 2003, 9, 1416–1425 CrossRef.
- A. Köllhofer and H. Plenio, Adv. Synth. Catal., 2005, 347, 1295–1300 CrossRef.
- Y. S. Ho and G. McKay, Process Saf. Environ. Prot., 1998, 76, 332–340 CrossRef CAS.
- Z. D. Ding, Y. X. Wang, S. F. Xi, Y. Li, Z. Li, X. Ren and Z. G. Gu, Chem. – Eur. J., 2016, 22, 17029–17036 CrossRef CAS.
- S. Lu, Y. Hu, S. Wan, R. McCaffrey, Y. Jin, H. Gu and W. Zhang, J. Am. Chem. Soc., 2017, 139, 17082–17088 CrossRef CAS.
- D. Chen, W. Yang, L. Jiao, L. Li, S.-H. Yu and H.-L. Jiang, Adv. Mater., 2020, 32, 2000041 CrossRef CAS.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- L. M. Lanni, R. W. Tilford, M. Bharathy and J. J. Lavigne, J. Am. Chem. Soc., 2011, 133, 13975–13983 CrossRef CAS.
- A. Li, H.-X. Sun, D.-Z. Tan, W.-J. Fan, S.-H. Wen, X.-J. Qing, G.-X. Li, S.-Y. Li and W.-Q. Deng, Energy Environ. Sci., 2011, 4, 2062–2065 RSC.
- N. Candu, A. Dhakshinamoorthy, N. Apostol, C. Teodorescu, A. Corma, H. Garcia and V. I. Parvulescu, J. Catal., 2017, 352, 59–66 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr00172h |
| This journal is © The Royal Society of Chemistry 2021 |