
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural dependence of electrosynthesized cobalt phosphide/black phosphorus pre-catalyst for oxygen evolution in alkaline media†
He
Xiao
 a,
Xiaoling
Du
a,
Man
Zhao
*a,
Ya
Li
a,
Tianjun
Hu
a,
Haishun
Wu
a,
Jianfeng
Jia
*a and
Nianjun
Yang
*b
a,
Xiaoling
Du
a,
Man
Zhao
*a,
Ya
Li
a,
Tianjun
Hu
a,
Haishun
Wu
a,
Jianfeng
Jia
*a and
Nianjun
Yang
*b
aKey Laboratory of Magnetic Molecules & Magnetic Information Materials, Ministry of Education, The School of Chemical and Material Science, Shanxi Normal University, Linfen 041004, China. E-mail: zm03030225@sxnu.edu.cn; jiajf@dns.sxnu.edu.cn
bInstitute of Materials Engineering, University of Siegen, Siegen 57076, Germany. E-mail: nianjun.yang@uni-siegen.de
First published on 29th March 2021
Abstract
The integration of black phosphorus (BP) with metal phosphides is known to produce high-performance electrocatalysts for oxygen evolution reduction (OER), although increased stability and prevention of the degradation of their lone pairs would be desirable improvements. In this work, cobalt phosphide (CoP)/BP heterostructures were electrochemically synthesized with a two-electrode system, where cobalt ions were generated in situ at a Co anode, and non-aggregated BP nanosheets (NSs) were exfoliated from the bulky BP cathode. With an electrolysis voltage of 30 V, the CoP/BP heterostructure exhibited a superior and stable OER performance (e.g., an overpotential of 300 mV at 10 mA cm−2, which is 41 mV lower than that obtained with a RuO2 catalyst). The CoOx formed in situ during the OER catalysis and remaining CoP synergistically contributed to the enhanced OER performance. The present strategy provides a new electrosynthetic method to prepare stable BP electrocatalysts and also further expands their electrochemical applications.
Introduction
Electrochemical water splitting is a promising approach to produce clean energy sources such as hydrogen, and it is a technique that is expected to tremendously alleviate the energy crisis.1–10 However, it is inevitably restricted by the oxygen evolution reaction (OER), a stepwise four-electron or a two-step-two-electron transfer process. During the OER process, the breaking of an O–H bond and O–O formation occur. In other words, high energy barriers need to be overwhelmed, and a large amount of applied energy is lost.11,12 To reduce such energy barriers for both processes, various noble metal oxides such as IrO2 and RuO2 have been utilized as OER catalysts. Unfortunately, their scarcity and high cost hinder their production on a large scale as well as their practical applications.13 Science and industry would greatly benefit if alternative OER electrocatalysts could be synthesized and if expensive noble catalysts could be replaced.Layered black phosphorus (BP) is a potential OER electrocatalyst whose effectiveness has recently been demonstrated. It is a two-dimensional (2D) material that features a tunable direct band gap, unique anisotropic properties, strong optical absorption, and high carrier mobility.14–21 Unfortunately, the active lone pairs of BP degrade, and thus, the stability of BP catalysts is quite poor during the OER process.15,22 To increase the stability of BP catalysts for the OER, passivation is required for exposed lone pairs. For example, the coupling of BP with a metal element (e.g., Co) or its derivations is a promising approach. Because the Fermi level of BP is higher than that of Co or its derivations, electron migration occurs from BP to Co or its derivations.15,22
Once defect-rich BP such as a BP nanosheet (NS) is utilized, the active lone pairs preferentially transfer over its entire surface. In this way, the degradation of the BP catalyst is efficiently hindered or the stability of the BP electrocatalyst is effectively increased. Moreover, the integration of a metal element (e.g., Co) or its derivations with BP NSs leads to the generation of a large number of active sites and an increase in the electrical conductivity of BP, eventually resulting in the production of high-performance OER catalysts.15,22,23 Cobalt phosphide (CoP) is also a promising candidate as an OER electrocatalyst because of a variety of valences of this interstitial compound, and thus, it exhibits metallic alloy properties and an extremely high electrical conductivity.24–27
Among the reported catalysts, wet chemical routes are generally employed to synthesize cobalt nanoparticles or CoP on exfoliated BP. The drawbacks of these wet chemical routes are quite obvious, such as a long reaction time and a multistep process. Thus, fine control during the synthesis of these catalysts is impossible. The obtained catalysts are not pure in most cases, and the used cobalt slats are expensive.
In this study, we attempted to develop a facile approach to prepare stable and pure OER catalysts from cobalt compounds and BP NSs. With respect to BP NSs, they are electrochemically exfoliated from bulky BP in solution.28 The thicknesses and exfoliation efficiencies of these BP NSs are finely tuned through the adjustment of applied anodic or cathodic potentials during an electrochemical exfoliation process.28,29 In the proposed two-electrode system, cobalt metal is applied as the anode. When the applied voltage exceeds the dissolution onset potential of this cobalt anode, cobalt metal dissolves as cobalt ions into solution.30 Then, further reaction between metal ions and exfoliated BP NPs occurs, and consequently, the composites (in most cases, so-named as heterojunctions) of cobalt ions and BP NSs are synthesized. To the best of our knowledge, such a two-step approach to electrosynthesize cobalt phosphide/black phosphorus (CoP/BP) heterojunctions has not been previously reported.
Herein, we report the details regarding the electrosynthesis of CoP/BP heterostructures and further refine their OER performance. Of special interest, the dependence of the OER activity on structure was investigated to further examine these CoP/BP heterostructures.
Experimental
Chemicals and materials
Cobalt wire was purchased from Runde Metal Material Company. Bulk black phosphorous was purchased from Kunming Black Phosphorous Company. Tetrabutylammonium tetrafluoroborate (TBABF4, 98%, powder) and proprylene carbonate (PC, ≥99.9%) were purchased from Aladdin. Anhydrous ethanol (99.5%) was purchased from Woke Company. Other chemicals were analytical grade and used as received without further purification.Preparation of CoP/BP heterojunctions
The steps for electrosynthesis of the CoP/BP heterojunctions are schematically shown in Fig. 1. A two-electrode system was developed. In the first step, 40 mg of black phosphorous was added to a porous plastic tube that was sealed, and a platinum plate was inserted to serve as the cathode. A cobalt wire was used as the anode. The electrolyte used was 0.1 M tetrabutylammonium tetrafluoroborate (TBABF4) dissolved in proprylene carbonate (PC) solvent. Unlike acidic or alkali ions, organic tetrabutylammonium cations (TBA+) with large diameters are more beneficial for exfoliating the bulky BP into nanosheets. At the same time, the organic electrolyte solution can reduce the oxidation and degradation of black phosphorus, while air or water can promote oxidation and degradation. Therefore, in this work, an organic electrolyte solution (0.1 M TBABF4 in PC solvent) was chosen to exfoliate black phosphorus.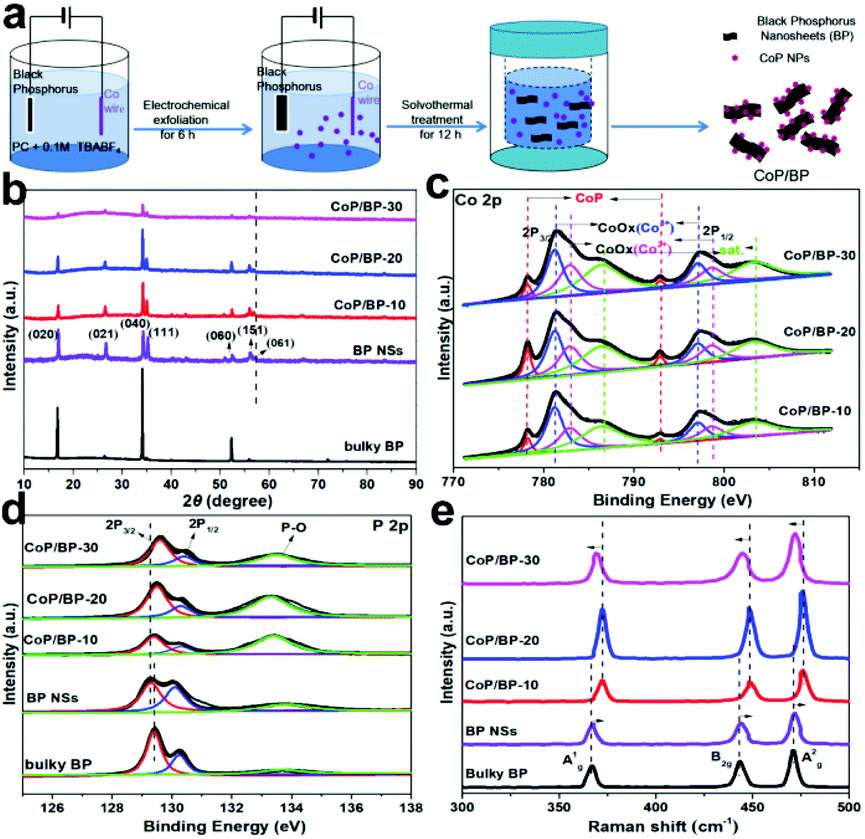 | ||
| Fig. 1 (a) Schematic plots of electrosynthesis of the CoP/BP heterojunctions; (b) XRD patterns of bulky BP, BP NPs electrosynthesized at 30 V, the CoP/BP-30 heterojunction, the CoP/BP-20 heterojunction, and the CoP/BP-10 heterojunction; (c) Co 2p XPS spectra of the CoP/BP-30, CoP/BP-20, and CoP/BP-10 heterojunctions; (d) P 2p XPS spectra and (e) Raman spectra of bulky BP, BP NPs electrosynthesized at 30 V, the CoP/BP-30 heterojunction, the CoP/BP-20 heterojunction, and the CoP/BP-10 heterojunction. | ||
A voltage of 10, 20, and 30 V was applied for 6 h to produce the CoP/BP-10, CoP/BP-20, and CoP/BP-30 heterojunctions, respectively. In the second step, the mixture was stirred for 12 h and then transferred to a 100 ml Teflon cup. Inside a stainless steel autoclave, Ar gas (0.5 h) was continuously flowed through the Teflon cup until the air was completely removed. Subsequently, the solution was treated at 160 °C for 12 h. During this process, the solution was stirred with a magnetic bar at a speed of 500 rpm. The resultant precipitates were filtered, washed three times with PC solution and twice with ethanol, and then dried at 80 °C for 8 h in a vacuum oven.
Characterization
X-ray diffraction (XRD) analysis was conducted on an X-ray diffractometer (Ultima IV-185) with Cu Kα radiation. Raman spectra were recorded on a Jobin Yvon LabRAM HR800 micro-Raman spectrometer. X-ray photoelectron spectroscopy (XPS, Thermo Fisher Scientific) was performed using an Al Kα X-ray source. Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images were collected on the FEI Tecnai F20 and JSM-7001F, respectively. The chemical composition (Co) of the obtained samples was measured by inductively coupled plasma optical emission spectrometry (ICP-OES).Electrochemical measurements
All the electrochemical tests were conducted at room temperature on a CHI 660E electrochemical workstation (CH Instruments, Inc., Shanghai) using a standard three-electrode system. A glassy carbon electrode (GCE, 3 mm in diameter), Hg/HgO, and a graphite rod were used as working electrode, reference electrode, and counter electrode, respectively. As-synthesized materials (5 μl ink) were coated drop by drop on a GCE. Such an ink was prepared by dispersing 5 mg material and 20 µL Nafion solution in 1 ml of water–ethanol solution with a volume ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The OER performance of these electrodes was tested in O2-saturated 1 M KOH solution. The linear sweep voltammograms (LSVs) were recorded at a scan rate of 5 mV s−1. The stability measurements were carried out by cyclic voltametric scanning at 2000 cycles in the potential range of 0 to 1.53 V with a scan rate of 50 mV s−1 and chronopotentiometric curve at 10 mA cm−2. The potential shown in this study refers to a revisable hydrogen electrode (RHE) according to the following equation of ERHE = EHg/HgO + 0.059 pH + EθHg/HgO, where ERHE is the converted potential vs. RHE and EHg/HgO is the experimental potential measured against the Hg/HgO reference electrode.
:1. The OER performance of these electrodes was tested in O2-saturated 1 M KOH solution. The linear sweep voltammograms (LSVs) were recorded at a scan rate of 5 mV s−1. The stability measurements were carried out by cyclic voltametric scanning at 2000 cycles in the potential range of 0 to 1.53 V with a scan rate of 50 mV s−1 and chronopotentiometric curve at 10 mA cm−2. The potential shown in this study refers to a revisable hydrogen electrode (RHE) according to the following equation of ERHE = EHg/HgO + 0.059 pH + EθHg/HgO, where ERHE is the converted potential vs. RHE and EHg/HgO is the experimental potential measured against the Hg/HgO reference electrode.
Results and discussion
Different from those previously reported, a two-step electrochemical process was developed in this study to synthesize the CoP/BP heterostructures (Fig. 1a). Here, cobalt ions were generated from a Co anode in situ, instead of using expensive cobalt salts dissolved in solution. In greater detail, electrochemical exfoliation of a bulky BP cathode and the generation of Co ions from a Co anode are simultaneously realized in the first step. During the electrolysis process, bulky BP is exfoliated into BP NSs. Defects are inevitably generated on these BP NSs, which result in unsaturated P sites with superior properties including smaller coordination number, additional lone electrons, and higher reducing activity.23The Co ions were dissolved into the electrolyte solution from the anodic Co wire. Because the defect occupation energy of Co is lower than the surface adsorption energy,31 Co ions in the electrolyte can be easily reduced to Co atoms on the defect sites in the subsequent solvothermal process.32 Then, the newly generated Co atoms can bond with the adjacent defective P atoms and form CoP. In addition to these defective P sites, the intrinsic P sites with lone electrons also contribute to the same influence. To vary the defective sites and the amount of Co ions during the fabrication of the CoP/BP heterostructures, the applied voltage in our two-electrode system can be varied as required. Throughout this article, the terms CoP/BP-30, CoP/BP-20, and CoP/BP-10 denote the CoP/BP heterojunctions synthesized under an electrolytic voltage of 30, 20, and 10 V, respectively.
These electrosynthesized BP NSs were initially characterized using X-ray diffraction (XRD) analysis. As control experiments, the XRD spectrum of bulky BP was also recorded. In both XRD spectra (Fig. 1b), patterns appear at 16.9°, 26.4°, 34.1°, 35.0°, 52.3°, 55.8°, and 56.5°, which can be assigned to the (020), (021), (040), (111), (060), and (061) planes of BP (PDF#65-2491), respectively. The relatively sharp peaks of BP NSs reveal the fine crystal quality even after exfoliation. Moreover, the CoP/BP-30, CoP/BP-20, and CoP/BP-10 heterojunctions show the same characteristic peaks as those of BP. Namely, these CoP/BP heterojunctions remain as crystalline phases of BP. The slight shift of the (061) peak on these CoP/BP heterojunctions to a small angle results from an enlarged layer-to-layer spacing as well as the doping of this Co species.33 The XRD spectra (Fig. 1b) show that the height of the characteristic peaks over CoP/BP-30 is lower than that of CoP/BP-20 and CoP/BP-10, indicating that the crystallinity of CoP and BP over CoP/BP-30 decreases in comparison with CoP/BP-20 and CoP/BP-10. This also can be concluded from the normalized XRD spectra (Fig. S1†) based on the (040) peak of BP.
The existence of Co, P, and O elements as well as the amount of Co in the CoP/BP heterojunctions have been confirmed by use of X-ray photoelectron spectroscopy (XPS).34 A comparison of their XPS survey spectra with those of BP NSs and bulky BP (Fig. S2†) clearly indicates the existence of elemental Co in the CoP/BP heterojunctions. The highest amount of Co was found in the CoP/BP-30 heterojunction. In order to further estimate the quantity of Co species over these CoP/BP heterostructures, the as-obtained inductively coupled plasma optical emission spectrometry (ICP-OES) results were combined with the XPS results. The loading amount of Co species over these CoP/BP heterostructures increases as the electrolytic voltage increases (Table S1†), with 15.96 wt%, 18.76 wt%, and 22.83 wt% for the CoP/BP-10, CoP/BP-20, and CoP/BP-30 heterojunctions, respectively. It is expected that the CoP/BP-30 heterojunction will exhibit superior catalytic performance in comparison to other heterojunctions, due to its largest number of active Co species.
The high-resolution Co 2p XPS spectra of three CoP/BP heterostructures were further recorded (Fig. 1c). Two peaks located at 778.2 and 792.9 eV correspond to Co 2p in CoP.35 Two peaks appearing at 781.5 and 798 eV are related to the inevitably oxidized CoOx (Co3+), while those at 782.9 and 798.6 eV result from oxidized CoOx (Co2+). These results clearly confirm the generation of CoP on the BP NSs. The high-resolution P 2p XPS spectra of bulky BP, BP NSs, and three CoP/BP heterostructures were also recorded (Fig. 1d). In the XPS spectrum of bulky BP, three peaks located at 129.6, 130.5, and 133.9 eV correspond to P 2p3/2, P 2p1/2, and P–O, respectively. For the BP NSs, their peaks shift to lower binding energies when compared with those of bulky BP. This confirms an electron-rich state of the P atom in BP NSs,33 which originates from the exposure of a larger number of defective edge-sites during the electrosynthesis (namely, electrochemical exfoliation) of BP NSs. Additionally, the binding energies of P 2p in three CoP/BP heterojunctions are positively shifted when compared with that of the BP NSs. This phenomenon indicates the strong electron interaction between CoP and BP. This suggests that these formed CoP/BP samples are heterostructures, not simple mixtures. Electrons can migrate from BP NSs to Co species, and because such electron transformation effectively suppresses the deterioration of the BP NSs,15 these electrosynthesized CoP/BP heterostructures are expected to provide excellent stability during the OER process.
The Raman spectra of bulky BP, BP NSs, and three CoP/BP heterostructures were recorded (Fig. 1e). In the Raman spectrum of bulky BP, three vibrational bands appear at approximately 366, 443, and 471 cm−1. They correspond to the out-of-plane A1g mode, in-plane B2g, and A2g vibration modes of BP, respectively.29,36 Compared with those in the Raman spectrum of bulky BP, the A1g, B2g, and A2g peaks of electrosynthesized BP NSs were positively shifted, which is an indication of successful exfoliation of bulky BP into thin BP NSs.28,33 In comparison with BP NSs, the CoP/BP heterostructures exhibited even further positive shifts for the A1g, B2g, and A2g characteristic peaks. The applied solvothermal process is thus also beneficial for the exfoliation of bulky BP into BP NSs. Note that the characteristic peaks of the CoP/BP heterostructures are negatively shifted once the Co content is enhanced. This is attributed to inhibited vibrations, derived from the electron transfer between Co species and BP.15
As a case study, the morphology and nanostructures of the CoP/BP-30 heterojunction were uncovered by means of transmission electron microscopy (TEM), high-resolution TEM (HRTEM), and scanning TEM (STEM). From one representative TEM image of the CoP/BP-30 heterojunction (Fig. 2a), one can see that the CoP/BP-30 heterojunction adequately retains the typical lamellar structure of the BP NSs. No structural damage was found (Fig. S3†). Numerous small amorphous aggregates are visible on the surface of the BP NSs (Fig. 2b). This leads to disappearance of the fine characteristic peaks of CoP or CoOx in the XRD spectrum of the CoP/BP-30 heterojunction. In the HRTEM image of the CoP/BP-30 heterojunction (Fig. 2c), the lattice spacing of 0.26 nm is indexed to the (040) plane of a BP crystal. Additionally, the lattice fringe spacings of 0.24 and 0.28 nm are well matched with the (102) and (002) planes of the CoP. This indicates that the generated CoP is uniformly grown on the surface of the BP NSs, but not mixed with the BP NSs. The elemental mapping of the CoP/BP-30 heterojunction was further conducted using STEM. Over the scanned area (Fig. 2d), homogeneous spatial distributions of phosphorus (Fig. 2e), cobalt (Fig. 2f), and oxygen (Fig. 2g) are seen. Consequently, CoP is generated and uniformly distributed over the entire surface of the BP NSs.
 | ||
| Fig. 2 (a, b) TEM, (c) HRTEM, and (d) STEM images of the CoP/BP-30 heterojunction; (e) EDS mapping of the elements of P, (f) Co and (g) O in the CoP/BP-30 heterojunction. | ||
As discussed, CoP in the CoP/BP heterostructure occupies the initial defects of BP NSs or forms bonds with lone pairs. It thus improves the electrical conductivity and/or increases the numbers of active electrocatalytic sites on the surface of BP NSs. Therefore, the CoP/BP heterostructure is expected to be a stable and excellent OER pre-catalyst.23 The OER activities of the CoP/BP heterostructures were evaluated in 1.0 M KOH. As control experiments, the OER performance of bulky BP and BP NSs was also investigated. In all these cases, the loading amount of these catalysts was exactly the same. Fig. 3a shows the related linear sweep voltammograms (LSVs) and Fig. S4† compares the corresponding overpotential values at a current density of 10 mA cm−2 for these catalysts. Clearly, the CoP/BP-30 heterojunction/catalyst exhibits superior OER catalytic activity among these used catalysts. Its overpotential at a current density of 10 mA cm−2 is 300 mV (without the iR compensation), which is comparable to that of a commercial RuO2 catalyst (341 mV), and much lower than that of the CoP/BP-20 heterojunction (350 mV), the CoP/BP-10 heterojunction (356 mV), BP NSs electrosynthesized at 30 V (435 mV), and bulky BP (about 520 mV). This lowest OER overpotential on the CoP/BP-30 heterojunction stems from the Co species integrated with BP NSs in that Co species play the dominant role in the OER performance of the CoP/BP heterostructures. Moreover, the CoP/BP-30 heterojunction featured the highest amount of CoP species, resulting in the strongest OER performance among the studied catalysts in this work.
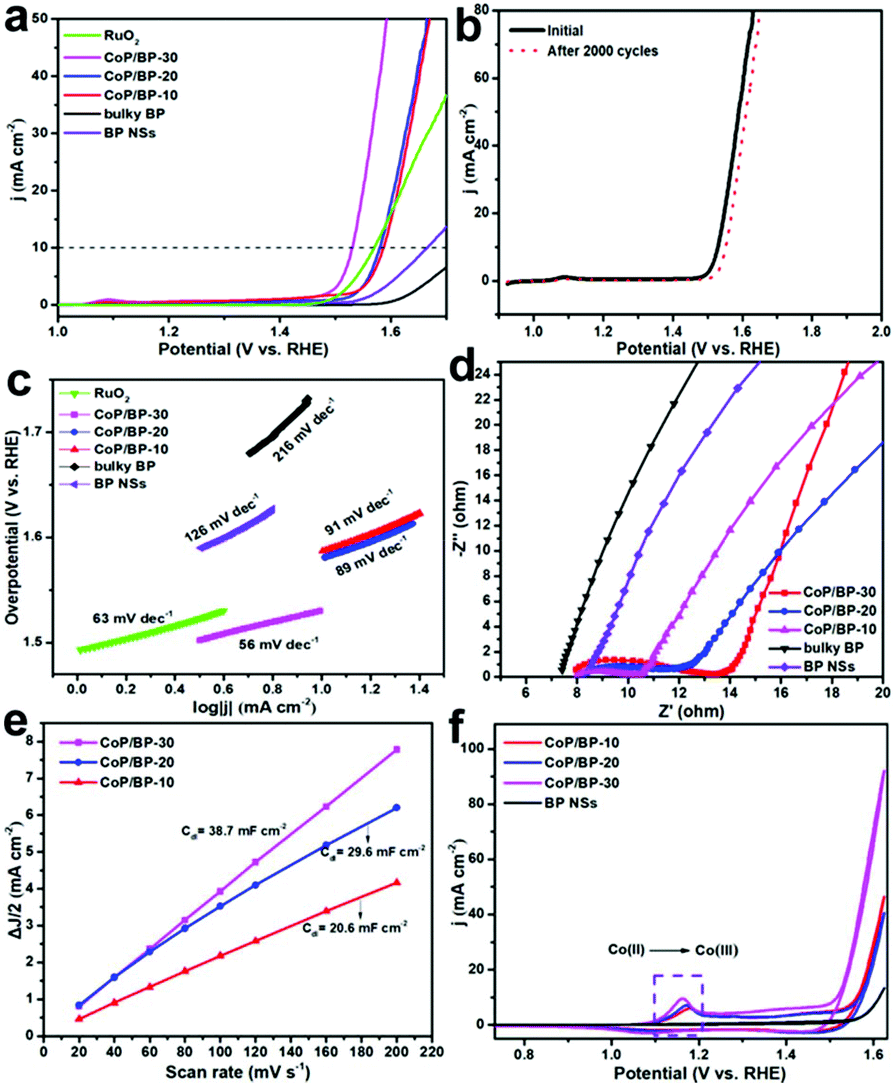 | ||
| Fig. 3 (a) The OER polarization curves of a RuO2 catalyst, bulky BP, BP NSs electrosynthesized at 30 V, the CoP/BP-30 heterojunction, the CoP/BP-20 heterojunction, and the CoP/BP-10 heterojunction in 1 M KOH solution. (b) A comparison of LSVs of the CoP/BP-30 heterojunction before and after 2000 OER cycles. (c) Tafel plots and (d) Nyquist plots of a RuO2 catalyst, bulky BP, BP NSs electrosynthesized at 30 V, the CoP/BP-30 heterojunction, the CoP/BP-20 heterojunction, and the CoP/BP-10 heterojunction for OER. (e) The Cdl values of the CoP/BP-30, CoP/BP-20, and CoP/BP-10 heterojunctions. (f) The CVs of BP NSs electrosynthesized at 30 V as well as the CoP/BP-30, CoP/BP-20, and CoP/BP-10 heterojunctions at a scan rate of 10 mV s−1. | ||
Mixed CoP/BP samples were also prepared by combining the commercial CoP with the obtained BP nanosheets based on the ratio over CoP/BP heterostructures tested by ICP-OES for comparison. The results (Fig. S5†) suggest that the CoP/BP-30 exhibits much higher OER performance than the CoP/BP mixture, indicating that the interaction between CoP and BP in the CoP/BP heterostructure also contributes to the enhanced OER performance. To more closely compare the active sites of the materials, the specific activity was calculated. CoP/BP-30 exhibited much higher specific activity compared with CoP/BP-20 and CoP/BP-10 at an overpotential of 300 mV (Fig. S6†), suggesting that its high intrinsic activity was attributable to a greater number of Co species-doped heterostructures at high voltage. The calculated TOF of the CoP/BP-30 is 0.02016 s−1, which is comparable with the same class of materials.37–39 In addition, the stability of the CoP/BP-30 heterojunction during the OER process was tested. The recorded LSVs remained almost unchanged even after 2000 cycling scans (Fig. 3b), and the chronopotentiometric curve showed a negligible slight decay after a testing period time of 9 h at 10 mA cm−2 (Fig. S7†), indicating excellent stability of the Co/BP heterojunction for the OER.
To obtain insight into the rate-determining step during the OER process under these conditions, Tafel analysis was conducted.15 It is known that in alkaline solutions, oxygen evolution undergoes a multistep reaction process to convert OH− to O2.11,12 If the first electron transfer process of OER is the rate-determining step, the corresponding Tafel slope is 120 mV dec−1. If the second chemical reaction process is the rate-limiting step, the relevant Tafel slope is 60 mV dec−1. Differently, if the electron–proton reaction is the rate-limiting step, the Tafel slope is 40 mV dec−1. In our case, the estimated Tafel slope of BP NSs is 126 mV dec−1 (Fig. 3c). This value is similar to 120 mV dec−1, suggesting that the electron transport process is the rate-determining step for the OER on the BP NSs. In contrast, the CoP/BP-30 heterojunction displays a Tafel slope of 56 mV dec−1. In other words, a chemical reaction step determines the OER rate on the CoP/BP-30 heterojunction, instead of an electron transfer step. The variation in rate-limiting steps reveals that sufficient electrons aggregate on the CoP/BP heterojunctions.
To further support such a statement, the Nyquist plots of used OER catalysts were recorded (Fig. 3d). Three CoP/BP heterojunctions exhibited a smaller radius in the semicircles than bulky BP and BP NSs. Their smaller charge transfer resistances prove that the introduction of CoP into BP increases the conductivities of CoP/BP heterojunctions. Moreover, the reaction rate of the obtained CoP/BP becomes faster as the electrolytic voltage increases. This indicates that the reaction rate is fastest on the CoP/BP-30 heterojunction, and also that a higher amount of CoP regulates the electronic properties of the CoP/BP heterojunctions. Furthermore, the electrochemically active surface area (ECSA) of the used catalysts (Fig. 3e) was determined with the assistance of their electrochemical double-layer capacitances (Cdl) (Fig. S8†).
In 1.0 M KOH solution, only capacitive currents were observed for all heterojunctions. The differences in current density at an overpotential of 0.396 V were plotted against the scan rates. The as-obtained linear regression enabled us to estimate the Cdl values of these heterojunctions and determine their ECSAs using the equation ECSA = Cdl/Cs, where Cs was assumed to be the same for all heterojunctions. The CoP/BP-30 heterojunction features the highest ECSA among the used catalysts in this study, and because it exposes active sites, it is definitely beneficial for the OER.
To further quantify the enhanced OER activity of the CoP/BP-30 heterojunction, the faradaic active sites (FASs) on the surface of the CoP/BP-30 heterojunction were calculated. The differences from the electrochemically active surface area (ECSA) based on the specific capacitance reflect the adsorption capacity of OH* or H2O*, and the faradaic active sites (FASs) reveal the statistical contribution of the active sites. However, the statistical contribution of the FASs is quite informative for evaluating the OER activity of an electrocatalyst.11 The faradaic contribution can serve as the active sites for water oxidation, which usually results from the reconstruction of catalytic interfaces.11 It has been verified that divalent Co2+ can evolve into active intermediate CoOOH species during the OER process, which also serve as the active sites for water oxidation.40 The intermediate CoOOH species dominates the related OER activity. In our case, divalent Co2+ is derived from the CoP and CoO in the CoP/BP heterojunction, as indicated by its Co 2p XPS spectrum.
The quantity of FASs of the used catalysts was calculated based on the redox process of a Co2+/Co3+ couple. In this way, the OER activity of the used CoP/BP heterojunctions is able to be revealed.41 In the cyclic voltammograms (CVs) of the CoP/BP heterojunctions (Fig. 3f), a pair of well-defined redox peaks is clearly seen in the potential range of 1.0 to 1.3 V (vs. the reversible hydrogen electrode, RHE), originating from the reversible faradaic reaction of Co3++ e− ↔ Co2+. Such waves do not exist in the CV of BP NSs. Therefore, the CoP/BP heterojunctions possess a greater number of FASs. The quantities of FASs for three CoP/BP heterojunctions were then precisely calculated according to the previously reported method.11 The charge density under the redox peak (q*, Table S2†) of the CoP/BP-30 heterojunction is 2423.2 mC cm−2 mg−1, which is higher than that of the CoP/BP-20 heterojunction (1675.9 mC cm−2 mg−1) and the CoP/BP-10 heterojunction (1489.0 mC cm−2 mg−1). Therefore, the CoP/BP-30 heterojunction exhibits superior OER activity among these used OER catalysts.
It has been reported that the electrochemical OER activity of transition metal phosphides (e.g., CoP) originates from the generation of a thin layer of oxides/oxyhydroxides (e.g., cobalt oxides) on the surface of the phosphides during the OER process. The formed oxides/oxyhydroxides provide additional catalytically active sites for the OER, while the remaining metal phosphides (e.g., CoP) facilitate the electron transfer for the OER. Furthermore, the synergistic effect between the CoP and its oxidized species promotes the OER kinetics.13 As a case study, the XPS spectra and TEM image of the CoP/BP-30 heterojunction after activation were obtained (named as CoP/BP-30R). In the high-resolution XPS spectra for Co 2p, P 2p, and O 1s (Fig. S9a and 9b†), the CoP peak disappears and the intensity of the P–O peak increases, validating the occurrence of the oxidation of cobalt phosphides into cobalt oxides/oxyhydroxides. This was further verified by its O 1s XPS spectrum (Fig. S9c†). Two peaks appeared at 531.2 and 532.6 eV, corresponding to metal–oxide and defect sites, respectively. A new peak located at 535.3 eV was assigned to hydroxyl species. Consequently, additional absorbed oxygen species are formed over the surface of the CoP/BP-30R heterojunction, consistent with the behavior of CoOx.35
The TEM image of the CoP/BP-30R heterojunction (Fig. 4a) reveals no variation of its morphology after activation. However, in its HRTEM image (Fig. 4b), lattice fringes appear that are different from the ones seen before activation. The d-spacings are 0.24 and 0.29 nm, resulting from the (101) of CoO and (220) of Co3O4, respectively. Namely, during the OER process, CoOx species are produced. Some corrugated morphologies of CoP/BP-30R after activation are also observed (Fig. 4c and d) in comparison with CoP/BP-30, and are probably due to the lattice dislocation of Co species. Because lattice dislocations can act as lattice defects,42–44 they actually serve as the active sites of the CoP/BP-30 heterojunction for the OER. The elemental mapping images of the scanned area (Fig. 4e) over CoP/BP-30R reveal the homogeneous spatial distributions of phosphorus (Fig. 4f), cobalt (Fig. 4g), and oxygen (Fig. 4h). These results further indicate that the generated CoOx species are highly dispersed over the BP NSs.
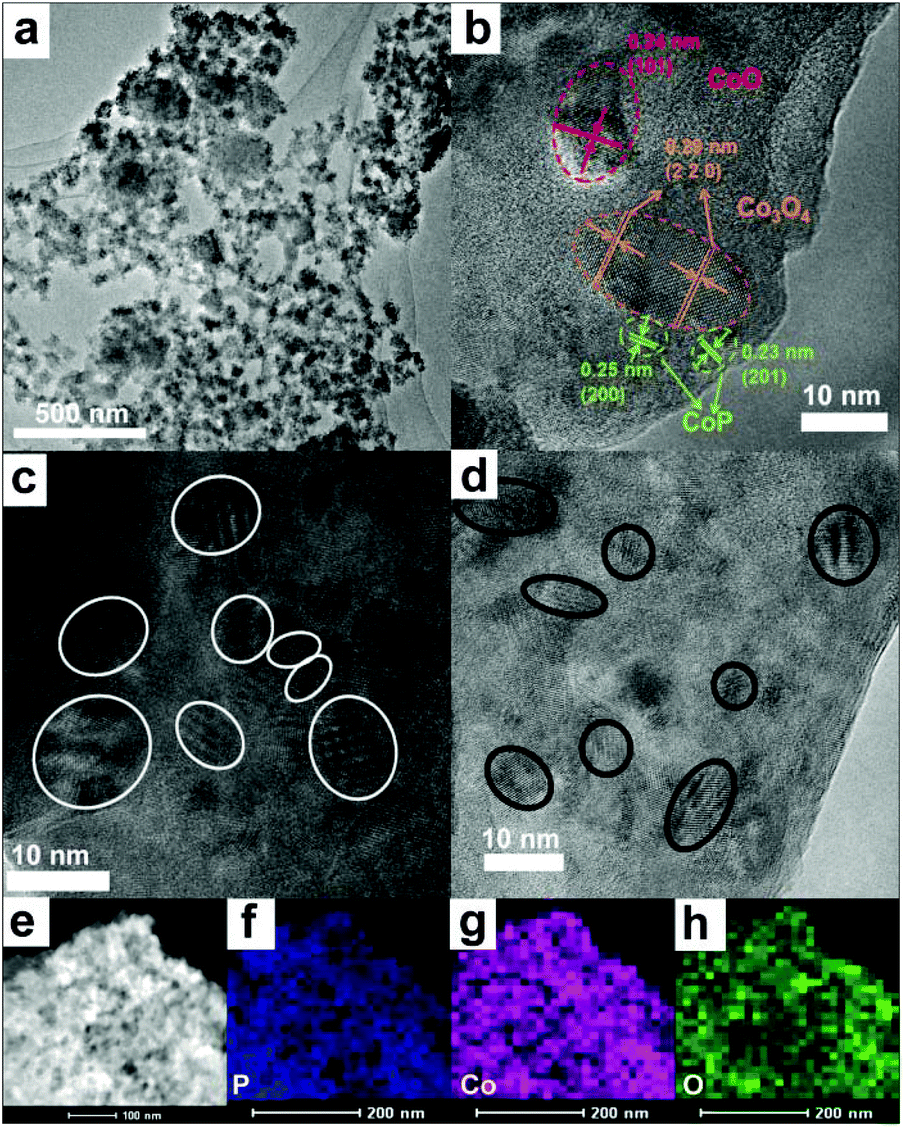 | ||
| Fig. 4 (a) TEM, (b, c, d) HRTEM, and (e) STEM images of the CoP/BP-30R heterojunction after activation. (f–h) EDS mappings of the elements of (f) P, (g) Co, and (h) O in the CoP/BP-30R heterojunction after activation. | ||
We also examined the microstructure and composition of the catalyst after the long-term chronopotentiometric measurement, and compared them with those after activation. The TEM and XPS (Fig. S10 and S11†) results of the catalyst after OER electrolysis for 9 h show almost the same morphology and chemical states as those obtained after the OER activation process. Therefore, we concluded that notwithstanding the compositional change, excellent performance was achieved by the OER during the entire long-term stability test.
Based on the above results, the structure–activity dependence of these CoP/BP heterostructures was analyzed. Fig. 5a summarizes the variation of the content of CoP on these heterojunctions, their ECSAs, and FASs, as well as the overpotential for the OER on these heterojunctions as a function of applied voltage during the course of electrosynthesis. The total content of CoP increased with the enhancement of electrolytic voltage. The CoP/BP-30 with the highest content of CoP showed the lowest overpotential and most optimal OER performance.
 | ||
| Fig. 5 (a) Relationship between OER activity and the content of CoP on the CoP/BP heterojunctions electrosynthesized with different electrolytic voltage. (b) Illustration of the OER reaction on the CoP/BP heterojunctions. | ||
The reasons for this are summarized as follows. First, the electron transfer from BP NSs to Co species via this engineered electronic interface not only enhances the OER performance of the CoP/BP heterostructure (smaller Tafel slope and smaller charge transfer resistance), but it also increases the stability through effectively suppressing the degradation of BP NSs caused by active lone pairs. Moreover, the divalent Co2+ species derived from CoP evolved into active CoOx species, which serve as active sites for the OER. The synergistic effect between the active oxidized species and remaining neighboring CoP further promotes the OER kinetics. The formed CoP nanoparticles also prevent the aggregation of BP nanosheets. A higher content of CoP is beneficial to achieve a larger ECSA. It is understandable that the OER activity of the CoP/BP heterostructure is positively related to the quantity of ECSA or FAS produced by active CoP sites. The OER reaction process is vividly illustrated in Fig. 5b.
Conclusions
A facile electrolysis-solvothermal method has been established to electrosynthesize CoP/BP heterostructures. This approach reduces the oxidation-induced degradation of BP nanosheets that occurs due to the reduction of the BP cathode. Simultaneously, cobalt phosphides are formed on the surface of BP nanosheets and act as the active sites for the OER. The electronic interface engineered by the growth of CoP on the BP nanosheets effectively suppresses the degradation of BP NSs that is caused by active lone pairs and greatly increases the stability of these heterojunctions. With a voltage of 30 V during such a process, the obtained CoP/BP-30 pre-catalyst exhibits excellent OER catalytic activity and stability. Such a method is thus promising for the large-scale production of BP-based electrocatalysts for high-performance OER processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (21571119), the Program for New Century Excellent Talents in University (NCET-12-1035), the Applied Basic Research Project of Shanxi Province (No. 201901D211393, 201901D211398), the Scientific and Technological Innovation Programs of the Higher Education Institution in Shanxi (2019L0466), the Foundation of the State Key Laboratory of Coal Conversion (J19-20-605), Shanxi Normal University School Fund (ZR1707), and 1331 Engineering of Shanxi Province.References
- Y. Hua, Q. Xu, Y. Hu, H. Jiang and C. Li, J. Energy Chem., 2019, 37, 1–6 CrossRef.
- J. Lin, P. Wang, H. Wang, C. Li, X. Si, J. Qi, J. Cao, Z. Zhong, W. Fei and J. Feng, Adv. Sci., 2019, 6, 1900246 CrossRef PubMed.
- C. Ding, J. Shi, Z. Wang and C. Li, ACS Catal., 2017, 7, 675–688 CrossRef CAS.
- T. Van Tam, S. G. Kang, M. H. Kim, S. G. Lee, S. H. Hur, J. S. Chung and W. M. Choi, Adv. Energy Mater., 2019, 1900945, 1900945 Search PubMed.
- Z. Yuan, J. Li, M. Yang, Z. Fang, J. Jian, D. Yu, X. Chen and L. Dai, J. Am. Chem. Soc., 2019, 141, 4972–4979 CrossRef CAS.
- L. Dai, Z. N. Chen, L. Li, P. Yin, Z. Liu and H. Zhang, Adv. Mater., 2020, 32, 1906915 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J. Tian, J. Chen, J. Liu, Q. Tian and P. Chen, Nano Energy, 2018, 48, 284–291 CrossRef CAS.
- M. Z. Rahman, C. W. Kwong, K. Davey and S. Z. Qiao, Energy Environ. Sci., 2016, 9, 709–728 RSC.
- X. Fan, Y. Liu, S. Chen, J. Shi, J. Wang, A. Fan, W. Zan, S. Li, W. A. Goddard and X.-M. Zhang, Nat. Commun., 2018, 9, 1809 CrossRef PubMed.
- G. Zhang, J. Yang, H. Wang, H. Chen, J. Yang and F. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 16159–16167 CrossRef CAS PubMed.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 CrossRef PubMed.
- Z. Liang, W. Zhou, S. Gao, R. Zhao, H. Zhang, Y. Tang, J. Cheng, T. Qiu, B. Zhu, C. Qu, W. Guo, Q. Wang and R. Zou, Small, 2020, 16, 190575 Search PubMed.
- Y. Zhang, Y. Zheng, K. Rui, H. H. Hng, K. Hippalgaonkar, J. Xu, W. Sun, J. Zhu, Q. Yan and W. Huang, Small, 2017, 13, 1700661 CrossRef PubMed.
- F. Shi, Z. Geng, K. Huang, Q. Liang, Y. Zhang, Y. Sun, J. Cao and S. Feng, Adv. Sci., 2018, 5, 1800575 CrossRef PubMed.
- V. Eswaraiah, Q. Zeng, Y. Long and Z. Liu, Small, 2016, 12, 3480–3502 CrossRef CAS PubMed.
- Q. Jiang, L. Xu, N. Chen, H. Zhang, L. Dai and S. Wang, Angew. Chem., Int. Ed., 2016, 55, 13849–13853 CrossRef CAS PubMed.
- P. Nakhanivej, X. Yu, S. K. Park, S. Kim, J. Y. Hong, H. J. Kim, W. Lee, J. Y. Hwang, J. E. Yang, C. Wolverton, J. Kong, M. Chhowalla and H. S. Park, Nat. Mater., 2019, 18, 156–162 CrossRef CAS PubMed.
- H. Liu, Y. Du, Y. Deng and P. D. Ye, Chem. Soc. Rev., 2015, 44, 2732–2743 RSC.
- W. Lei, G. Liu, J. Zhang and M. Liu, Chem. Soc. Rev., 2017, 46, 3492–3509 RSC.
- X. Ren, J. Zhou, X. Qi, Y. Liu, Z. Huang, Z. Li, Y. Ge, S. C. Dhanabalan, J. S. Ponraj, S. Wang, J. Zhong and H. Zhang, Adv. Energy Mater., 2017, 7, 1700396 CrossRef.
- F. Shi, K. Huang, Y. Wang, W. Zhang, L. Li, X. Wang and S. Feng, ACS Appl. Mater. Interfaces, 2019, 11, 17459–17466 CrossRef CAS PubMed.
- J. Wang, D. Liu, H. Huang, N. Yang, B. Yu, M. Wen, X. Wang, P. K. Chu and X. F. Yu, Angew. Chem., Int. Ed., 2018, 57, 2600–2604 CrossRef CAS PubMed.
- P. Ji, X. Luo, D. Chen, H. Jin, Z. Pu, W. Zeng, J. He, H. Bai, Y. Liao and S. Mu, ACS Sustainable Chem. Eng., 2020, 8, 17851–17859 CrossRef CAS.
- Z. Pu, T. Liu, I. S. Amiinu, R. Cheng, P. Wang, C. Zhang, P. Ji, W. Hu, J. Liu and S. Mu, Adv. Funct. Mater., 2020, 30, 2004009 CrossRef CAS.
- C. Lin, P. Wang, H. Jin, J. Zhao, D. Chen, S. Liu, C. Zhang and S. Mu, Dalton Trans., 2019, 48, 16555–16561 RSC.
- Y. Guo, P. Yuan, J. Zhang, H. Xia, F. Cheng, M. Zhou, J. Li, Y. Qiao, S. Mu and Q. Xu, Adv. Funct. Mater., 2018, 28, 1805641 CrossRef.
- A. Ambrosi, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2017, 56, 10443–10445 CrossRef CAS PubMed.
- H. Xiao, M. Zhao, J. Zhang, X. Ma, J. Zhang, T. Hu, T. Tang, J. Jia and H. Wu, Electrochem. Commun., 2018, 89, 10–13 CrossRef CAS.
- M. Tavakkoli, N. Holmberg, R. Kronberg, H. Jiang, J. Sainio, E. I. Kauppinen, T. Kallio and K. Laasonen, ACS Catal., 2017, 7, 3121–3130 CrossRef CAS.
- N. Suvansinpan, F. Hussain, G. Zhang, C. H. Chiu, Y. Cai and Y. W. Zhang, Nanotechnology, 2016, 27, 65708 CrossRef PubMed.
- X. Liu, J. D. Wood, K. S. Chen, E. Cho and M. C. Hersam, J. Phys. Chem. Lett., 2015, 6, 773–778 CrossRef CAS PubMed.
- D. Liu, J. Wang, J. Lu, C. Ma, H. Huang, Z. Wang, L. Wu, Q. Liu, S. Jin, P. K. Chu and X. F. Yu, Small Methods, 2019, 3, 1900083 CrossRef.
- H. Xiao, J. Zhang, X. Wang, Q. Zhang, H. Xie, Y. Han and Y. Tan, Catal. Sci. Technol., 2015, 5, 4081–4090 RSC.
- J. Wang, R. Gao, L. Zheng, Z. Chen, Z. Wu, L. Sun, Z. Hu and X. Liu, ACS Catal., 2018, 8, 8953–8960 CrossRef CAS.
- A. Favron, E. Gaufrès, F. Fossard, A. L. Phaneuf-Laheureux, N. Y. W. Tang, P. L. Lévesque, A. Loiseau, R. Leonelli, S. Francoeur and R. Martel, Nat. Mater., 2015, 14, 826–832 CrossRef CAS PubMed.
- H. Liu, Z. Liu, F. Wang and L. Feng, Chem. Eng. J., 2020, 397, 125507 CrossRef CAS.
- H. Liu, Z. Liu and L. Feng, Nanoscale, 2019, 11, 16017–16025 RSC.
- S. Wang, X. Yang, Z. Liu, D. Yang and L. Feng, Nanoscale, 2020, 12, 10827–10833 RSC.
- H. Y. Wang, S. F. Hung, H. Y. Chen, T. S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS PubMed.
- T. Audichon, E. Mayousse, T. W. Napporn, C. Morais, C. Comminges and K. B. Kokoh, Electrochim. Acta, 2014, 132, 284–291 CrossRef CAS.
- X. Li, X. Su, Y. Pei, J. Liu, X. Zheng, K. Tang, G. Guan and X. Hao, J. Mater. Chem. A, 2019, 7, 10745–10750 RSC.
- B. Zhang, X. Huang, Y. Zhang, G. Lu, L. Chou and Y. Bi, Angew. Chem., Int. Ed., 2020, 59, 18990–18995 CrossRef CAS PubMed.
- Y. Yang, H. Fei, G. Ruan, C. Xiang and J. M. Tour, ACS Nano, 2014, 8, 9518–9523 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr00062d |
| This journal is © The Royal Society of Chemistry 2021 |