
Hydrocarbon contamination in angström-scale channels†

Abstract
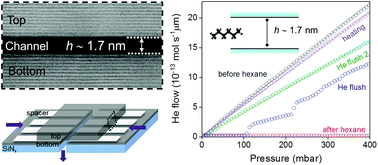
Nonspecific molecular adsorption such as airborne contamination occurs on most surfaces including those of 2D materials and alters their properties. While surface contamination is studied using a plethora of techniques, the effect of contamination on confined systems such as nanochannels/pores leading to their clogging is still lacking. We report a systematic investigation of hydrocarbon adsorption in angstrom (Å) slit channels of varying heights. Hexane is chosen to mimic the hydrocarbon contamination and the clogging of the Å-channels is evaluated via a helium gas flow measurement. The level of hexane adsorption, in other words, the degree of clogging depends on the size difference between the channels and hexane. A dynamic transition of the clogging and revival process is shown in sub-2 nm thin channels. Long-term storage and stability of our Å-channels are demonstrated here for up to three years, alleviating the contamination and unclogging the channels using thermal treatment. This study highlights the importance of the nanochannels’ stability and demonstrates the self-cleansing nature of sub-2 nm thin channels enabling a robust platform for molecular transport and separation studies. We provide a method to assess the cleanliness of nanoporous membranes, which is vital for the practical applications of nanofluidics in various fields such as molecular sensing, separation and power generation.
- This article is part of the themed collections: Celebrating the 200th Anniversary of the University of Manchester, Celebrating International Women’s Day: Women in Nanoscience and Nanoscale 2021 Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

