
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalently bonded surface functional groups on carbon nanotubes: from molecular modeling to practical applications†
Aleksandra
Benko
 *a,
Joanna
Duch
b,
Marta
Gajewska
c,
Mateusz
Marzec
c,
Andrzej
Bernasik
d,
Marek
Nocuń
a,
Witold
Piskorz
b and
Andrzej
Kotarba
*b
*a,
Joanna
Duch
b,
Marta
Gajewska
c,
Mateusz
Marzec
c,
Andrzej
Bernasik
d,
Marek
Nocuń
a,
Witold
Piskorz
b and
Andrzej
Kotarba
*b
aAGH University of Science and Technology, Faculty of Materials Science and Ceramics, 30 A. Mickiewicz Ave., 30-059 Krakow, Poland. E-mail: akbenko@gmail.com; Tel: +48 12 617 52 06
bFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387, Krakow, Poland. E-mail: kotarba@chemia.uj.edu.pl; Tel: +48 12 686 25 09
cAGH University of Science and Technology Academic Centre for Materials and Nanotechnology, 30 A. Mickiewicz Ave., 30-059 Krakow, Poland
dAGH University of Science and Technology, Faculty of Physics and Applied Computer Science, 30 A. Mickiewicz Ave., 30-059 Krakow, Poland
First published on 12th May 2021
Abstract
The aim of this work was to investigate how chemical functionalization affects the electronic properties of multi-walled carbon nanotubes, altering the electrophoretic deposition process: a method of choice for the fabrication of high quality, all-carbon nanotube (CNT) layers. Wet chemistry methods were applied to modify the surfaces of CNTs by insertion of various oxygen- and nitrogen-containing groups. Transmission electron microscopy revealed no significant changes in the material morphology, while X-ray photoelectron spectroscopy and Raman spectroscopy showed that changes in the chemical composition did not translate to the changes in the structure. Molecularly modelled optimized surface functional group geometries and electron density distributions allowed the calculation of the dipole moments (–COOH = 0.77; –OH = 1.65; –CON(CH3CH2)2 = 3.33; –CONH2 = 2.00; –NH2 = 0.78). Due to their polarity, the introduction of surface functional groups resulted in significant modifications of the electronic properties of CNTs, as elucidated by work function measurements via the Kelvin method and ultraviolet photoelectron spectroscopy. The work function changed from 4.6 eV (raw CNTs) to 4.94 eV for the –OH functionalized CNTs and 4.3 eV for the CNTs functionalized with –CON(CH3CH2), and was inversely proportional to the dipole moment values. Finally, using CNT dispersions, electrophoretic deposition was conducted, allowing the correlation of the work function of CNTs and the measured electrophoretic current with the impact on the deposits' qualities. Thus, a rational background for the development of carbon-based biomaterials was provided.
1. Introduction
Since raw carbon nanotubes (CNTs) are entirely hydrophobic, their applications as biomaterials require surface functionalization. Such modifications are one of the main strategies to improve the dispersibility in the solvent of choice. Additionally, the presence of functional groups enables anchoring of molecules for further modification, including attachment of bioactive compounds, recently extensively explored.1 Functionalization can either be covalent or non-covalent and the strategy applied depends mostly on the target application. For example, applications that benefit from the CNTs’ outstanding mechanical and electrical properties will most often require materials that have a low share of amorphous carbon in their structure. While non-covalent functionalization is more versatile and does not change the structure and length of CNTs, it has two major flaws: it is usually temporary (i.e. de-agglomeration can occur after prolonged usage) and it does not remove impurities, such as amorphous carbon or residual metallic catalysts. As a result, the quality of the obtained products in terms of biocompatibility is lower.2,3 Thus, for most applications, especially biomedical, covalent modification is the preferred method, even though it has been reported to alter the structure of CNTs.4–7 Among other strategies, oxidation is the simplest and the most often applied as it removes impurities, shortens the CNTs, and generates surface groups that are suitable sites for further modifications.8 Oxidized CNTs, typically fabricated via the protocol established by Smalley and his co-workers,9 have been proven to contain various types of functional groups: carboxy, hydroxy, carbonyl, etc.10–12 Usually, the group types and their surface concentration are tailored to meet specific needs, and the modification process is controlled in such a manner that it yields materials of the desired level of structural integrity. Importantly, by obtaining short CNTs that are easily dispersible in various solvents, one obtains materials that can be successfully used as polymer matrix modifiers that bond chemically to the polymer,13 and drug carriers14 that can be phagocytized15 and biodegraded16 inside the cells. Short CNTs are also perfect candidates to fabricate high quality, electrophoretically deposited layers on surfaces which can then be further used in numerous applications,17 such as biosensors or other electronic devices18,19 or as biomaterials.20,21 It has also been reported that the surface oxidation of CNTs is beneficial for further modifications, tailored to introduce different functional groups for biological applications.22Covalent modification of CNTs affects not only their impact on cells or their behavior in various solvents, but also dramatically alters their electronic properties, such as surface charge, its distribution, and electron-donor properties. These features indicate the material's electrochemical applicability and can also have a direct impact on the interactions with bacteria and cancer cells, participating in reactive oxygen species generation or affecting the oxidative stress generated in normal cells. This, in turn, is known to activate some important tissue protection, anti-tumor mechanisms.23 Recently, it has been reported that there is a direct correlation between bacteria adhesion to carbonaceous surfaces and the material's electron-donor properties, parameterized by the material's work function (Φ).5,24–26 By definition, Φ is the amount of energy that needs to be delivered to the material's surface to deprive it of an electron. One of the oldest methods to measure this parameter is the Kelvin probe which measures the potential difference between the surface and the working electrode, recalculated into Φ.27 It is worth noting that most of the research aiming to combine the material's biological performance with its surface electron-donor properties is purely phenomenological and there is no insight into how the functional groups affect the electrochemical properties of CNTs at the molecular level.
In our recent study,26 we have correlated the work function values with the biological performance of the electrophoretically deposited layers of carbon nanotubes. These layers were composed of CNTs with different types and amounts of functional groups and in our experiments, we have observed that these materials behaved differently during their handling (dispersability) and processing (EPD). A full understanding of the phenomena guiding this behavior is of great importance as it creates the possibility for optimizing the functionalization process to obtain the final product having desired properties. Hence, by understanding the effect functionalization has on the CNTs’ dipole moment and electron-donor properties, one can elucidate the interactions between the CNTs’ functional groups and the dispersants that are used to process them into the desired materials. Consequently, one can predict the material's behavior during its processing, control the process (mixing with the solvent, deposition), and fine-tune the final product's performance. This study aimed to identify how does the introduction of various types of covalently bonded functional groups affect the materials' properties, (such as surface composition, charge, and polarity), the resulting functional features, (i.e. dispersibility in the solvent of choice, ability to form stable dispersions), the electrophoretic deposition (EPD) process, and finally, the quality of the carbon-based biomaterials. Multi-walled carbon nanotubes (MWCNTs) were selected as a model because their shape and size have been proven to be more appealing for biomedical applications than their single-walled counterparts.15 Two types of oxidized CNTs were prepared in accordance with Smalley's protocol (with modifications),9 using two acidic mixtures of different oxidation strengths. From these, nitrogen modifications were derived. The purpose was to test how the electronegativity of the atom attached to the surface of CNTs can affect the material's properties and what are the implications in the processability of the CNTs.
2. Materials and methods
2.1. Materials
Multi-walled carbon nanotubes were purchased from NanoAmor company, USA (stock #1213NMGS).28 Concentrated sulfuric, nitric and hydrofluoric acids, ethanol, acetone, ammonia, and tetrahydrofuran (THF) were supplied by Avantor Performance Materials, Poland. Dicyclohexylcarbodiimide (DCC) and diethylamine (DEA) were purchased from Sigma Aldrich.2.2. CNT functionalization
In brief, methods employed for the CNT functionalization by wet chemistry methods are presented in Fig. 1. In the first step, chemical oxidation was performed by refluxing in a mixture of concentrated sulfuric and nitric acids in two different ratios: 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1.5:1. The ratios were altered to produce environments of different oxidation strengths that would yield carbon atoms of varying oxidation numbers: mainly +3 or mainly +2. The heating and washing steps were identical to those in our previous study.10 The as-received materials are denoted as HO (high oxidation state CNTs) and LO (low oxidation state CNTs).
:1 and 1.5:1. The ratios were altered to produce environments of different oxidation strengths that would yield carbon atoms of varying oxidation numbers: mainly +3 or mainly +2. The heating and washing steps were identical to those in our previous study.10 The as-received materials are denoted as HO (high oxidation state CNTs) and LO (low oxidation state CNTs).
 | ||
| Fig. 1 Schematic illustration of the applied chemical modifications of CNTs. | ||
For the obtainment of amide-modified CNTs, a method designed by Jeong et al.29 was employed, with small modifications. 100 mg of HO and LO were separately placed in 50 ml of THF and dispersed with the aid of a probe sonicator (Vibra Cell, VCX 130 from Sonics & Materials, Inc.) set to 30% of maximum power, operating in a constant mode for 2 minutes. After obtaining a homogenous suspension, 100 mg of DCC was added to each sample. The flasks were then placed on a magnetic stirrer and left for 5 hours to complete the reaction between DCC and oxygen functional groups (mainly carboxyl, but also hydroxyl, carbonyl, ester, aldehyde), occurring via deprotonation. Afterward, the as-obtained suspension was centrifuged (Sigma 3k30 centrifuge, 26 K RPM, 18 °C) and successively washed three times with THF to remove any unreacted compounds and reaction by-products. The as-received CNT slurry was resuspended in 20 ml of THF and 50 ml of ammonia (HNH and LNH) or DEA (HDEA) were added to the mixture. The flasks were placed on a magnetic stirrer and the mixture was left for 2 h to complete the reaction between the amine and the DCC-activated functional groups. The suspensions were successively centrifuged in THF, a THF:ethanol (1:1) mixture, and then ethanol to remove any substrate residues and reaction by-products, and left to dry in a vacuum drier (Vacucell 55 Comfort) at 40 °C.
2.3. Morphology, chemical composition, and structure of the CNTs
The morphology of the as-obtained CNTs was evaluated using a transmission electron microscope (TEM, FEI Tecnai TF20 X-TWIN (FEG)), operating at an accelerating voltage of 200 kV. The samples were prepared as follows: a small amount of each type of CNTs was dispersed in ethanol using an ultrasonic probe. The solutions were then drop-cast onto carbon coated copper TEM grids and left to dry under room conditions for approx. 30 min.The chemical composition of the materials was investigated by X-ray photoelectron spectroscopy (XPS, Vacuum Systems Workshop, Ltd, England) with the identification of distinctive chemical states of atoms revealed via deconvolution (XPS Peak 4.1 software). Full details of the experimental procedures can be found in our previous study.17 The results of the XPS study were further confirmed by performing thermogravimetric analysis (TGA, STA 449 F3 Jupiter, Netzsch), which can be used to reveal the overall amount of functional groups present in the sample. Approx. 5 mg of either HO or LO were separately placed inside the Al2O3 crucibles and heated up to 1360 °C under the protective flow of nitrogen. Further analysis of the samples’ chemical composition was performed through Fourier-transform infrared spectroscopy (FTIR, Tensor 27, Bruker), operating in the transmittance mode. A small amount of the sample was mixed with 200 mg of potassium bromide (KBr) and compressed into a pellet. A pellet made from solely KBr served as a background. Spectra were obtained in the spectral range of 400–4000 cm−1, with a 2 cm−1 resolution. An average of 128 scans was collected for each sample. Following the measurement, an atmospheric compensation, manual baseline correction and smoothing were done using the OPUS 7.2 software.
Micro-Raman spectroscopic measurements were carried out under ambient conditions using a Renishaw InVia dispersive spectrometer integrated with a Leica DMLM confocal microscope. Spectra were recorded with a laser line of 514.5 nm, a 50× magnification lens, and a resolution of 2 cm−1. The Raman scattered light was collected in the spectral range of 100–3000 cm−1 by applying exposure times of at least 15 s. The samples were investigated in the form of flattened powder. The Raman spectrum deconvolution for the determination of spectral parameters was performed with the Fityk 1.3.1 software program.
All of the graphs were prepared using Origin 2021 software.
2.4. Molecular modeling
Molecular modeling was performed with the use of an analogous model and at the same level of theory (DFT+D) as in our previous article.5 Briefly, the GGA PBE30,31 augmented with Grimme32 semiempirical dispersion approximation and the SVP basis set was used, as implemented in the Turbomole33 5.10 code. The nanotube segment (C280) has been cut off from the library-imported nanotube, yielding 22.23 Å diameter and 56 C atoms in the zigzag ring. The resultant segment with a length of 9.37 Å, and dangling bonds capped by 56 H atoms (C–H bond length equal to 1.14 Å), was subsequently optimized having the H positions fixed.2.5. Work function determination
The work function (Φ) of raw and functionalized MWCNTs was determined by the Kelvin probe technique. During the experiments, the contact potential difference (VCPD), which is a difference between Φ of the stainless-steel reference tip (Φref = 4.3 eV) and Φ of the investigated surface, was measured using a KP6500 probe (McAllister Technical Services). The experimental parameters such as the gradient of the peak-to-peak versus backing potential, amplitude, and vibration frequency were set to 0.1, 40 a.u., and 120 Hz, respectively. The individual VCPD was an average of 60 successively measured values, each being calculated based on 32 points. The flattened powder samples were investigated under ambient conditions. The applied procedure allows for high repeatability of the measurements with the experimental error smaller than 0.01 eV.To confirm the results obtained by the Kelvin probe technique, two materials having distinctively different Φ values were selected for further evaluation using ultraviolet photoelectron spectroscopy (UPS). Approx. 10 mg of HO and LNH were separately placed inside plastic test-tubes and suspended in 3 ml of the previously established EA solvent.17 The as-prepared suspensions were then transferred into a glove box. Samples were prepared by spin-coating (Osilla) onto gold-covered substrates, under an Ar atmosphere, and transferred with a transport vessel under Ar to the UHV system to avoid air-borne surface contamination deposition. The measurement times were kept as short as possible to avoid the possible UV degradation damage of the examined materials. UPS measurements were performed on a PHI VersaProbeII apparatus (ULVAC-PHI, Chigasaki, Japan) using the He I line (21.22 eV) from a UHV gas discharge lamp. The sample was set to an acceleration potential of −5 V leading to a much more pronounced secondary electron cut off (SE cut-off). The work function, measured as the difference between the photon energy and SE cut-off position, was then calculated. For each UPS spectrum, the emission features due to the secondary line excitations of the He–I gas discharge are subtracted. Since the actual relative intensities of the satellite excitations depend on the He discharge pressure, the secondary line sub-spectra were adjusted marginally in intensity to the measured UPS spectrum and subtracted incrementally starting with the highest photon energy satellite.
2.6. Impact of the CNTs’ physicochemical properties on the key process parameters of the EPD
To investigate the impact the surface modifications have on some of the functional features, solutions of all 5 types of the as-fabricated CNTs were prepared, separately, as follows. Approx. 35 mg of CNTs were placed in a plastic test-tube and the following solvents were added successively: 7.5 ml of EtOH, 2.5 ml of acetone, and 2 ml of H2O. Each addition was followed by 2 minutes of sonication using a probe sonicator. Thus, suspensions in the EA solvent, like the one established in our previous studies, were prepared.17 The stability of the suspensions (i.e. tendency to sediment through the formation of agglomerates or flocculation) over a given period of time was identified by visual inspection.Independently, the suspensions were evaluated for their applicability and behavior during the electrophoretic deposition process (EPD). The set-up was identical to that in our previous studies, with the DC voltage source (TTi EL561R) set to 30 V, the distance between electrodes fixed at 5 mm, a stainless steel plate as the counter electrode, and a titanium plate as the working electrode. A digital multimeter (Agilent 34405A), connected to a PC equipped with appropriate software (BenchVue), was used to record the current flowing through the set-up upon generation of the electric field. The following parameters were evaluated: (1) the sign of the electrode the particles were deposited on, indicative of the overall surface charge sign of CNTs;34 (2) the mean value of the current flowing through the circuit upon generation of an electric field, with the same voltage of 30 V applied, indicative of the suspension quality and an estimate of the CNT surface charge;34 (3) having established the overall surface charge sign and the value of the current flowing through the circuit upon generation of an electric field, the value of the applied voltages and the deposition times were altered to provide similar deposition kinetics for the evaluation of the deposits’ morphology.34 The macroscopic and microscopic observations to evaluate the quality of the deposits were made using a digital optical microscope (Keyence, VHX-5000) and a scanning electron microscope (SEM, Nova NanoSEM 200, manufactured by FEI Europe Company). SEM was operated under low vacuum conditions, and an ultra-high-resolution Helix detector was employed.
3. Results
3.1. Morphology, chemical composition, and the structure of the CNTs
The results of the TEM investigations of the CNT morphologies can be found in Fig. 2. Similar to our previous work,17 the CNTs used herein also have graphenic planes arranged along the axis at a certain angle, which appears to be irrespective of the chemical treatment, suggesting that this is an intrinsic quality of the starting material. In all the CNTs, bends in the structures are visible, characteristic of the presence of structural defects, which have been reported to facilitate the CNT buckling.35 The HO CNTs and their amidized derivatives HNH and HDEA appear to be shorter than the LO and LNH CNTs. This suggests that the chemical oxidation applied for the HO fabrication was more efficient in cutting the CNTs. The amidization slightly blurs the sharpness of the graphene planes as compared to the non-amidized materials. Since the process itself is conducted in a mild environment, which should not have altered the level of structural arrangement in HNH, LNH, and HDEA (this can be confirmed by the fact that in the Raman spectra of these samples, the ID/IG ratios are not increased (ESI Table 3†)), the sharpness reduction should rather be attributed to the presence of some residues enveloping these CNTs. These are most likely some DCC residues or a synthesis by-product, dicyclohexylurea. The presence of some organic residues with carbon in the sp2 hybridization is also supported by the XPS data (ESI Table 1†). One must take into account that the structural arrangement observed via HR-TEM gives information only about the local state of the material instead of providing a more global information about the sample's structure. Furthermore, the sharpness of the planes is random, depending on the CNT alignment with the electron beam; only those graphene planes that are aligned perpendicularly to the beam will be visible. This might also have contributed to slight blurring of the graphene planes in HNH, LNH and HDEA. | ||
| Fig. 2 TEM evaluation of the CNT morphology, presented with increasing magnifications. | ||
Deconvoluted XPS O 1s peaks of the tested samples, presented along with the summary of their overall atomic composition can be found in Fig. 3. Quantitative evaluation based on the deconvoluted O 1s peak is given in Table 1. A graphical representation of carbon and nitrogen deconvoluted peaks, along with the quantitative evaluation of their chemical states can be found in the ESI 1–6 and Tables 1 and 2.†
 | ||
| Fig. 3 Deconvoluted O 1s peaks, obtained through XPS analysis, presented along with the overall atomic composition. | ||
| B.E [ev] | Oxygen species | LO (at%) | HO (at%) | LNH (at%) | HNH (at%) | HDEA (at%) |
|---|---|---|---|---|---|---|
| 531.3–531.5 | N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
2.6 | 2.5 | |||
| 531.9 | C–OH, CO, CHO |
4.6 | 7.3 | |||
| 532.5–533.3 | C–O–C, COO*H, COO*R | 5.0 | 7.1 | 5.6 | 6.7 | 5.7 |
| 534.0–535.0 | CO*O, acid anhydride, CO*ON | 3.4 | 6.0 | 3.7 | 2.9 | |
| 535.7–536.7 | H2O, O2 | 1.9 | 2.9 | 1.3 | ||
| Total | 14.9 | 16.0 | 11.9 | 12.1 | 14.3 |
The overall amount of oxygen was 16% in HO and 15% in LO. The oxygen content in the oxidized samples was further confirmed by performing a thermogravimetric analysis: by heating the CNTs up to 1360 °C under a protective atmosphere. The total mass loss was 14.6% for HO and 10.6% for LO. After normalizing these values to the atomic masses of carbon and oxygen and subtracting an average mass loss of the unmodified CNTs (2%), values of 16.5% and 11.6% were obtained, respectively. The obtained results and the decomposition steps are in line with the XPS results (ESI 7†).36,37
A reaction with ammonia yielded materials with approx. 12% of oxygen and 2% of nitrogen (in both HO and LO), while with DEA 14% of oxygen and 2% of nitrogen were obtained, suggesting that in all cases a reaction between the CNTs and amine/ammonia took place, with a similar efficiency. Deconvolution of the C 1s and O 1s peaks revealed that in LO and HO there was approx. 30% of the CC bonds (sp2 carbon), indicating similar amounts of defects/functional groups in the CNT structure. In LO, another 30% of carbon atoms were connected via a single bond with oxygen and carbon atoms (including hydroxy and ether), while 18% were attributed to the presence of the double bonds between C and O. These results indicate that the majority of carbon atoms inside the LO CNTs had a coordination number equal to 2. Meanwhile, in the HO, nearly 30% of carbon atoms were found to be connected with oxygen via a double bond, suggesting a high share of carbon atoms with a coordination number of 3. Thus, as expected, the two chemical oxidation strategies yielded environments of different oxidation strengths, with a higher one found for a higher H2SO4:HNO3 ratio (as for HO). In both types of oxidized CNTs, a reaction with ammonia causes the formation of amine and amide functional groups, with a slightly higher share of the latter observed for the reaction with HO, which should be connected with an initially higher number of carboxyl species in these CNTs. In HDEA, no new C–N bonds were created, and the majority of nitrogen atoms were either connected via an amide bond or due to C–NH–C groups, some of which were from the diethylamine backbone. In all of the amidized samples, an increase in the amount of CC bonds was observed (from 30 to 40% in HNH and HDEA), and this effect was the highest for LNH (from 30 to more than 50%). This observation indicates the incomplete removal of DCC or the reaction by-products. Supposedly, there was some sort of chemical interaction between the LO and the DCC/by-product, resulting in their obstructed removal through washing off.38–51 As our recent study has proven, the EPD process of HNH and LNH yields layers with an amount of sp2 carbons similar to those of the HO and LO.26 This indicates that the EPD process may serve as a fractioning tool to remove the amidization by-products.
The chemical composition of the CNTs was further investigated using FTIR, operating in the transmittance mode. FTIR analysis (Fig. 4) of the CNTs fabricated in this study revealed that there is an appreciable amount of oxygen-based functional groups in both types of oxidized CNTs (HO and LO), which is indicated by the characteristic bands at 3436, 1700 and 1179 cm−1, attributed to OH and CO in carboxyl and C–O bonds, respectively. As expected, higher intensities of these bands are found in HO which was oxidized in an environment of higher oxidative strength. More importantly, in the HO sample, the OH and carboxyl-attributed bonds have higher intensities and there is a small shoulder at 1646 cm−1, suggesting the presence of another CO containing chemical species. All these observations suggest the presence of a higher amount of carbon atoms with an oxidation number of +3 in HO, as compared to LO. Finally, a small-intensity doublet at 2915 and 2843 cm−1 visible in the spectra of HO, HNH, LO, and LNH is indicative of a C–H stretching. Together with the presence of CO – attributed bonds, this indicates the presence of small amounts of aldehyde functional groups in the samples. Another high intensity bond, found in all the spectra at 1562 cm−1, should be related to the CC stretching vibrations in all samples and to the amide II (N–H) vibrational modes in HNH, LNH, and HDEA.52,53 In all cases, the DCC-activated reaction with ammonia had caused reduction in the intensity of bonds at 1700 and 1179 cm−1, indicating a reaction with the CO and C–O bonds, respectively. Reduction of the C–O bonds was more pronounced in the LO-LNH transition. This indicates that in the abundance of carboxyl functional groups, a reaction of amines with hydroxyl groups is less favored. However, in the state of carboxyl functional group deficiency, OH groups also become activated by DCC and react with the amines. In LNH, HNH and HDEA, altered intensities of the bonds between 1090 and 793 cm−1 should be connected with the presence of amide/amine chemical species, with higher amounts observed for the HO-derived sample. These observations suggest that, as expected, the amide formation reaction was more effective in CNTs with a higher share of carboxyl groups (HO).49,54 In general, HDEA reveals more complex shapes of the bonds below 1562 cm−1, indicative of the presence of higher amounts of IR-active bonds, possibly related to the coupled interactions between the C and O/H atoms.
 | ||
| Fig. 4 FTIR analysis of the as-fabricated CNTs. The spectra are offset for better clarity. | ||
The presented results show a good correlation with the XPS analysis (Fig. 3 and Table 1).
The structural changes of CNTs after the treatment have been monitored by micro-Raman spectroscopy. For all the investigated materials, the Raman spectra (Fig. 5) exhibited bands characteristic of multiwalled carbon nanotubes: D (1345 ± 1 cm−2), G (1571 ± 2 cm−2), D′ (1629 ± 3 cm−2), and 2D (2868 ± 2 cm−2). A table presenting the peak positions can be found in ESI Table 3.† Small differences in the peak positions can result from the tube bundling, variations in the defect density and inhomogeneity in the tube size. The G band originates from the in-plane stretching vibrations of sp2-hybridized carbon atoms. Both D and D′ bands are typically induced by the structural disorder coming from the lattice defects, amorphous carbon on the outer walls, and/or intercalated ions.55 The 2D band, the first overtone of the D mode, is usually associated with the degree of carbon nanotube crystallinity.56 One of the useful parameters, which allows evaluating the structural disorder of the nanotubes, is the ID/IG ratio (I is the integral surface area). Indeed, a decreasing ID/IG ratio denotes an increasing degree of graphitization.11,57 The calculated ID/IG ratios for the investigated carbon materials are presented in Fig. 5. It is reported that the ID/IG of the native CNTs is 1.0, whereas for the O- and N-functionalized materials the ratios are slightly higher indicating the reduction of the graphitic order, the introduction of functional groups, and the formation of defects on the surface during the treatment. In addition, the area of the 2D band decreased after the CNT modification confirming that the crystallinity of carbon nanotubes has been interrupted. Interestingly, in the HO and LO, the ID/IG is identical and equal to 1.7, indicating that the employed chemical treatment generated a similar amount of defects in the CNT structures. This result is in agreement with the XPS analysis, where the number of CC groups (sp2 hybridized carbon) was the same and equal to 30% for these two samples. Hence, it can be suggested that both of the applied acidic mixtures damage the CNT structure to a similar extent, but in the HO, higher amounts of oxygen and carbon atoms with higher oxidation states of +2 and +3 are being generated. Modification with ammonia leads to a decrease in the ID/IG ratio to approx. 1.3, irrespective of the sample. These results, combined with the C 1s XPS analysis (ESI Table 1†), indicate the presence of some DCC residues in the samples. As our previous article suggests, these residues are not deposited during the EPD process, indicating its ability to purify the CNTs by selective deposition. If the CNTs are to be used in the powdered form, a more thorough purification would be needed.
 | ||
| Fig. 5 Raman spectra of the untreated and wet chemistry modified CNTs with the marked characteristic bands (D, G, D′, 2D), presented along with the ID/IG ratio. | ||
3.2. Molecular modeling and work function
For the in-depth insight into the surface functionalities, the molecular modeling of the generated functional groups was performed. The collection of the calculated functional groups, selected based on the XPS results, is presented in Table 2. Together with the molecular geometry, the electron isodensity (with the superimposed electrostatic potential) is shown: color coding represents negative (blue), neutral (green), and positive (red) potentials. Having the optimized geometry and charge density, the dipole moments were evaluated, derived from Hirshfeld atomic charges (see column 4). Due to the different orientations of the surface dipoles, for the sake of comparison, their projection in the direction perpendicular to the carbon surface was determined. It is worth emphasizing that the values of dipole moments change in a broad range of 0.30–1.31 a.u., and their orientations differ qualitatively from the negative (−0.61 a.u.) to positive (1.10 a.u.). The obtained results clearly illustrate the strong impact of the introduced functionalities on the surface electronic properties and the possibility of their fine-tuning. This can be done by selecting the functional groups and adjusting their surface coverage. One of the most sensitive parameters to monitor the changes in the electronic surface state is the work function, as reported elsewhere.5,24–26| Functional groups | Model | Dipole moment/a.u. (component in the normal direction/a.u.) | |
|---|---|---|---|
| Formula | Structure | ||
| –CON(CH3CH2)2 |

|

|
1.31 (1.10) |
| –CONH2 |

|

|
0.79 (0.21) |
| –NH2 |

|

|
0.31 (0.07) |
| –COOH |
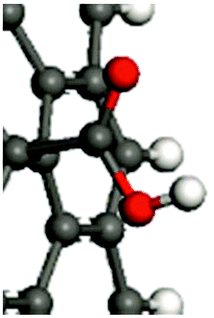
|
|
0.30 (−0.02) |
| –OH |
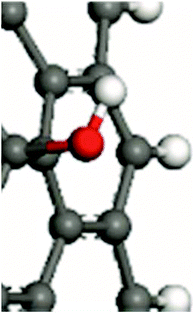
|

|
0.65 (−0.61) |
As expected, depending on the chemical nature of the introduced functionalities, the work function (Φ) of the CNTs (4.60 ± 0.01 eV) can be modified in a wide range. The surface oxygen species lead to the Φ increase, whereas the formation of nitrogen-containing groups decreases the Φ value. The dependence between electronic properties expressed as a work function, and the values of the dipole moments, represented by the component in a normal direction, are presented in Fig. 6, where the negative and positive influences are color-coded (grey shadowing represents the work function increase, connected with the oxidation, whereas the nude one stands for the work function reduction, created by an amide modification). It was observed that the most negative dipole moment was formed upon functionalization of the carbon surface with –OH groups and, as a consequence, the work function of the LO CNTs is the highest (4.94 ± 0.01 eV). The most positive dipole moment was calculated for –CON(CH3CH2)2 functional groups and the experimentally determined work function of the HDEA CNTs is the lowest one (4.31 ± 0.01 eV), whereas for the rest of the samples the WF values were as follows: HO = 4.84 ± 0.01 eV, HNH = 4.57 ± 0.01 eV, LNH = 4.35 ± 0.01 eV.
 | ||
| Fig. 6 Work function values of functionalized carbon nanotubes as a function of the formed surface dipole moments (x components). | ||
To confirm the work function values determined by the Kelvin probe technique, two CNTs having distinctively different Φ were selected for further evaluation using the UPS: HO and LNH. The resultant spectra are given in ESI 8.† The spectra were then subtracted from the photon energy value (21.22 eV), and from these, the work function was found to be the low kinetic energy cutoff (Fig. 7). The obtained values revealed very good agreement with the ones obtained with the Kelvin probe method, showing good accuracy of the latter. It is important to note that the samples for the UPS were prepared from liquids under a protective atmosphere, while for the Kelvin probe analysis the samples were prepared from powders under ambient conditions. Still, the results showed a good correlation, indicating that the samples are fairly insensitive towards the atmospheric contamination (or it proceeds to a similar extent regardless of the material type and the media it was prepared in (liquid vs. air)).
 | ||
| Fig. 7 Work function values obtained through the UPS analysis of the valence band shift. | ||
The experimentally measured changes in the work function are directly related to the theoretical surface dipoles and can be described by two models, developed by Helmholtz58 and Topping,59 respectively.
| ΔΦ = 4πeμN |
where ΔΦ is the measured work function change, e is elementary charge, μ is the dipole moment, N is the number of adsorbates per unit square, ε0 is the vacuum permittivity, and α is the polarizability.
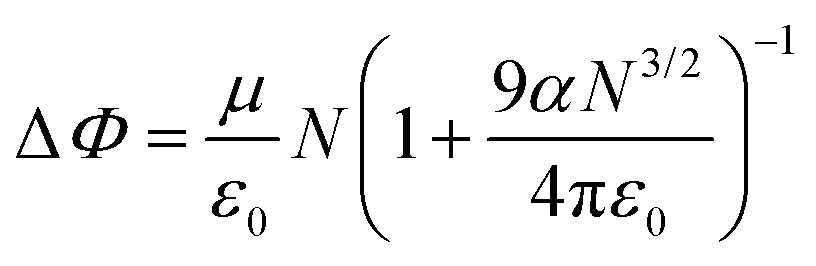
The Helmholtz approach assumes a low concentration of surface species and is narrowed to non-interacting dipoles. Depending on the dipole direction, a linear increase or decrease of the work function can be observed. At a higher surface coverage, according to Topping, mutual depolarization of dipoles of similar orientation takes place. As a result, the work function changes in a nonlinear way upon further introduction of surface adspecies.
The observed changes in the CNT surfaces lead to several important practical consequences. Increasing the polarity of the surface of the nanotubes allows for the facile preparation of stable CNT suspensions, which is an initial, indispensable step for the electrodeposition process. Another important implication is the generation of surface dipole–solvent molecule electrostatic interactions that influence the hydrodynamic radius and change the kinetics of the electrophoretic process.17 This in turn creates the possibility for the engineering of the deposited carbon film morphology and micro-hardness,60 essential for carbon-based biomaterials. Indeed, as reported in our previous study,26 the morphology of the EPD films influences both the adhesion of the cells and the bacteria.
3.3. Results of the impact of the CNTs’ physicochemical properties on the key process parameters of the EPD
The functional features of the as-modified CNTs were reflected by the tendency of the dispersions to sediment/agglomerate/flocculate and the performance of the EPD process. The EPD process was followed by the microscopic evaluation of the obtained deposits. The tendency of the suspensions to sediment/flocculate was evaluated across 3 months. At this time point, the solutions were visually inspected and the results are given in (ESI 9†). No obvious sedimentation was observed regardless of the CNT type. The suspensions were all black, with no clear border between the solvent and the CNTs visible. Thus, in all of the materials, there is a large population of CNTs for which interactions with the solvents can block spontaneous CNT–CNT agglomeration via van der Waals and/or π–π forces. Tilting the test tubes upside down and looking through them at the light source revealed that in all the suspensions, there was a minute portion of sedimented agglomerates visible, with the lowest amount observed for the HO. Upon vigorous shaking, this sediment disappeared in the HO but was still observed in all of the other suspensions. This observation suggests that the carboxyl functional groups are determinants for long-term suspension stability. Amide modification increases the particles’ susceptibility to interact with one another, most likely by reducing the overall surface charge, and diminishing the solvent–particle interactions.34 This effect is stronger for less polar functional groups such as DEA. OH modified CNTs (LO) and their amide-modified derivatives are found to have the highest tendency to agglomerate.During the EPD, all of the CNTs deposited readily on a positive electrode, indicating that the overall charge of the CNTs was negative, irrespective of the functional groups. However, deposition using a constant voltage of 30 V revealed differences in the average value of the current flow over a 15 s period, as presented in Fig. 8. Since the diluent, voltage, electrode, and the distance between them were identical in all the experiments, differences in the value of the current flow should be attributed solely to the character of the CNT surface functional groups, which are the main current carriers in this EPD process.34 Generally, higher currents were observed for the CNTs with a higher coordination number and a higher share of carboxyl-based species (HO, HNH, and HDEA) than those observed for their counterparts having the majority of carbon atoms with a coordination number of 2 and hydroxy-based groups, following the trend: HDEA > HNH > LNH > HO > LO. This is most likely due to the higher dissociation constant of the carboxyl functional groups, as compared to the hydroxyls. Amine modification increases the current flow, with the strongest impact observed for HDEA. Meanwhile, the lowest value was observed for the LO. To further explore this phenomenon, the mean current flow values were plotted against the values of the surface dipole moments, associated with the most abundant surface functional groups. It was found that a direct correlation existed: the lower the dipole moment, the lower the mean current flow value. It has been reported elsewhere that during the EPD process, the applied DC electrical field induces the polarization of individual nanotubes, leading to the generation of dipole moments.60,61 Additional influence on the charge of CNTs comes from the dipole moments formed upon functionalization (introduction of polar oxygen- and nitrogen-containing functional groups). As a consequence of the resultant surface charge, CNTs are aligned in the direction of the electric field, and the movement of CNTs towards the anode is improved. An inverse correlation was observed for the work function value: the higher the Φ value, the lower the current flow. The results show that the composite coating becomes coarser when the depositing current increases (Fig. 9).
 | ||
| Fig. 8 Current flow during 15 s deposition at an applied value of 30 V and mean current values during the functionalized carbon nanotube layer deposition as a function of the surface dipole moments (x component: normal to the CNT surface). | ||
 | ||
| Fig. 9 Results of the EPD deposition, based on the optimized parameters (time and voltage). The morphology of the deposits was investigated by SEM. | ||
Modification of the electronic structure, which can be quantified by the perpendicular component of the dipole moment, has an impact on the two experimentally measured quantities, namely the work function and the electrophoretic current density. Both of them can serve as suitable descriptors when tuning the parameters for the electrodeposition process. Carboxy functional groups yield materials that form stable CNT–solvent suspensions, having a moderate and nearly linear current flow during the deposition. Similarly stable deposition is reported for HNH. Taking into account the biological performance of carbon nanotube-based materials (cytocompatibility combined with antibacterial and anticancer properties), it can be inferred that for healthy mammalian cells, the preferential functional group is CONH2, which exhibits the second-highest dipole moment normal component.26
Next, the applied voltage was adjusted, so that the current flow during the deposition was similar regardless of the CNT type. The optimal range between 0.8 and 1.5 mA was chosen. The results of the current flows, recorded for at least 5 independent measurements, are presented as a mean ± standard deviation (Fig. 9). The time of deposition was altered based on the visual inspection of the obtained layers and the results from our previous studies.10,17 Interestingly, even though the current flow was the highest for HDEA, the time needed to deposit layers of visually similar qualities was longer, as compared to that for HNH, which had similar values of the current flow. This might be due to the fact that a very high conductivity of the suspension is known to reduce the particle mobility in the solvent.34 Jan Ma and Wen Cheng found that the deposit density was lower when a very high voltage was applied.62 While they did not measure the value of the current, it is known to increase with the increasing voltage. Thus, the HDEA films may also be affected by a very high electric field created in the circuit, possibly causing turbulences in the electrophoretic flux which disturbs the deposition.34
Following the deposition, the samples were subjected to microscopic observations using a digital, optical microscope (ESI 10†), and SEM (additional images of HO, LO, HNH, and LNH can also be found in our recent study).26 These revealed that the smoothest and the most homogenous layer was obtained using the HO CNTs, which is in agreement with our previous studies where high quality and uniform deposits were obtained for these CNTs.17 In this sample, CNTs adhere to one another along their sidewalls and no agglomerates are visible. Such alignment suggests good dispersion stability and the ability of the CNTs to organize and pack at the material's surface during the EPD. Hence, the deposition process was optimal. Meanwhile, in LO, LNH, and HNH deposits, places not covered with CNTs are visible and in the case of LO and LNH layers, agglomerates can be found, indicating that flocculation in the solvent had occurred prior to deposition. Similar to HO, LO CNTs also adhere to one another along their sidewalls, but the layer is visually less dense, with pores clearly visible. For DEA, the titanium coverage with CNTs is complete, but inhomogeneous distribution is apparent, especially at the sample's edges.
Thus, the presence of carboxyl groups seems to be the determining factor for obtaining stable, non-flocculating suspensions, and reduction of the number of these in favor of hydroxy species results in the formation of agglomerates. Amide modification is another factor decreasing the suspension stability and successive deposition; the presence of various functional groups with different surface charges may possibly increase the solution's tendency to agglomerate.
To sum up, the presented results clearly illustrate that the functional groups generated on the CNT surface have the determining effect on the electrophoretic deposition process. Differences in the size and polarity (dipole moments) affect the solvent ion–surface interactions and thus alter the CNT flux in the electrostatic field created during the EPD. As the kinetics of the process is affected, surfaces of different morphologies are obtained. Highly stable suspensions of HO, which have a low normal component of the dipole moments, are responsible for static current flow values during the EPD, creating uniform and highly packed layers (as reported in our previous study).17 Higher current flow values in the HNH and HDEA deposition (due to high values of the normal component of the dipole moments) create porous layers, while the tendency of LO and LNH suspensions to agglomerate (due to their higher lengths and smaller (i.e. consisting of fewer atoms), less polar functional groups) results in layers of higher amounts of microstructural features.
As reported in our recent studies,26 the morphology and the surface chemistry of the generated surfaces both have a remarkable influence on the biological response, i.e. the interaction with healthy cells (fibroblasts), cancer cells (melanoma), bacteria (methicillin-resistant Staphylococcus aureus (MRSA), and multidrug-resistant Escherichia coli (MDR-EC)). The combined approach involving theoretical modelling and experiments provides sensible guidelines for designing the electrophoretic process for the fabrication of carbon-based biosurfaces with adjustable properties, specifically tailored to meet the criteria of the target application.
4. Conclusions
In this work, the fundamental investigations on the chemical functionalization of carbon nanotubes (CNTs) were performed, interlacing experimental and theoretical approaches. The modifications were realized through wet chemistry methods which resulted in the generation of surface oxygen- and nitrogen-containing functional groups. The obtained materials were thoroughly characterized by several spectroscopic and microscopic methods, whereas the changes in electronic properties were quantified by work function measurements. Insight into the molecular nature of the introduced surface species was obtained via DFT modeling which allowed to establish the corresponding surface dipole moments based on the functional groups’ geometry and electron density distribution. The obtained experimental and theoretical results clearly revealed that the dipole moments and work function dramatically change upon functionalization. While the O-containing groups (–COOH, –OH) lead to a substantial work function increase from 4.6 eV (raw MWCNTs) up to 4.9 eV, the N-containing groups (–CON(CH3CH2)2, –CONH2, –NH2) lead to the work function decrease down to 4.3 eV. It was also found that the changes in the work function were correlated with the normal component of the generated surface dipoles, evaluated by molecular modeling (μ–COOH = 0.30; μ–OH = 0.65; μ–CON(CH3CH2)2 = 1.31; μ–CONH2 = 0.79; μ–NH2 = 0.31 a.u.). Increasing the polarity of the surface of the nanotubes and generating electrostatic interactions between the surface dipoles and solvent molecules affected the kinetics of the electrophoretic process. It was found that a direct correlation existed between the value of the current flow and the normal component of the surface dipole moments, with lower values of the latter observed to be favorable. A rational background for the development of carbon-based biomaterials was provided with the dipole moments and work function changes found to be suitable descriptors. Thus, the obtained results are found to have a practical aspect and can be utilized while designing the fabrication process of biomaterial surfaces. The results of the present study provide new guidance for the rationale planning of the CNT based material fabrication process. Fine tuning of the CNT surface electronic properties can be considered as a suitable tool for obtaining products that meet the criteria of specific applications.Experiments on the biological performance of CNTs in the free form are in progress in our laboratory. Right now, we are also investigating the separated effects of the surface chemistry and the morphology of all-CNT layers on the biological response of different cell types.
Author contributions
A. B.: conceptualization; methodology; validation; formal analysis; investigation: CNT functionalization, EPD, and SEM; data curation: EPD and XPS; visualization: TEM, XPS, EPD, and SEM; supervision; funding acquisition: 2017/24/C/ST8/00400; project administration; writing – the original draft; writing – review & editing; J. D.: methodology; validation; formal analysis; investigation: work function and Raman spectroscopy; data curation: work function and Raman spectroscopy; visualization: DFT, work function and Raman spectroscopy; writing – the original draft; writing – review & editing; M. M. and A. B.: investigation: UPS and proofreading of the revised draft; M. N.: investigation: XPS and proofreading of the original draft; M. G.: investigation: TEM and proofreading of the original draft; W. P.: methodology; validation; investigation; visualization: DFT; data curation: DFT; proofreading of the original draft; A. K.: conceptualization; methodology; supervision; funding acquisition: 2020/37/B/ST5/03451; project administration; writing – the original draft; writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Science Centre, Poland, under grants no. UMO-2017/24/C/ST8/00400 and 2020/37/B/ST5/03451.References
- N. K. Mehra and S. Palakurthi, Drug Discovery Today, 2016, 21, 585–597 CrossRef CAS PubMed.
- O. Vittorio, V. Raffa and A. Cuschieri, Nanomedicine, 2009, 5, 424–431 CrossRef CAS PubMed.
- C. P. Firme III and P. R. Bandaru, Nanomedicine, 2010, 6, 245–256 CrossRef PubMed.
- I. D. Rosca, F. Watari, M. Uo and T. Akasaka, Carbon, 2005, 43, 3124–3131 CrossRef CAS.
- J. Duch, M. Mazur, M. Golda-Cepa, J. Podobiński, W. Piskorz and A. Kotarba, Carbon, 2018, 137, 425–432 CrossRef CAS.
- M. T. Martinez, M. A. Callejas, A. M. Benito, M. Cochet, T. Seeger, A. Ansón, J. Schreiber, C. Gordon, C. Marhic, O. Chauvet, J. L. G. Fierro and W. K. Maser, Carbon, 2003, 41, 2247–2256 CrossRef CAS.
- J. Zhang, H. Zou, Q. Qing, Y. Yang, Q. Li, Z. Liu, X. Guo and Z. Du, J. Phys. Chem. B, 2003, 107, 3712–3718 CrossRef CAS.
- A. Moya, M. Hernando-Pérez, M. Pérez-Illana, C. San Martín, J. Gómez-Herrero, J. Alemán, R. Mas-Ballesté and P. J. de Pablo, Nanoscale, 2020, 12, 1128–1137 RSC.
- J. Liu, A. G. Rinzler, H. Dai, J. H. Hafner, R. K. Bradley, P. J. Boul, A. Lu, T. Iverson, K. Shelimov, C. B. Huffman, F. Rodriguez-Macias, Y.-S. Shon, T. R. Lee, D. T. Colbert and R. E. Smalley, Science, 1998, 280, 1253–1256 CrossRef CAS PubMed.
- A. Benko, A. Frączek-Szczypta, E. Menaszek, J. Wyrwa, M. Nocuń and M. Błażewicz, J. Mater. Sci.: Mater. Med., 2015, 26, 1–13 CrossRef CAS PubMed.
- A. Benko, A. Przekora, A. Wesełucha-Birczyńska, M. Nocuń, G. Ginalska and M. Błażewicz, Appl. Phys. A, 2016, 122, 1–13 CrossRef CAS.
- A. Przekora, A. Benko, M. Nocun, J. Wyrwa, M. Blazewicz and G. Ginalska, Mater. Sci. Eng., C, 2014, 45, 287–296 CrossRef CAS PubMed.
- C. Harito, D. V. Bavykin, B. Yuliarto, H. K. Dipojono and F. C. Walsh, Nanoscale, 2019, 11, 4653–4682 RSC.
- D. A. Scheinberg, M. R. McDevitt, T. Dao, J. J. Mulvey, E. Feinberg and S. Alidori, Adv. Drug Delivery Rev., 2013, 65, 2016–2022 CrossRef CAS PubMed.
- K. Kostarelos, Nat. Biotechnol., 2008, 26, 774–776 CrossRef CAS PubMed.
- G. P. Kotchey, Y. Zhao, V. E. Kagan and A. Star, Adv. Drug Delivery Rev., 2013, 65, 1921–1932 CrossRef CAS PubMed.
- A. Benko, M. Nocuń, K. Berent, M. Gajewska, Ł. Klita, J. Wyrwa and M. Błażewicz, Appl. Surf. Sci., 2017, 403, 206–217 CrossRef CAS.
- M. Pacios Pujadó, in Carbon Nanotubes as Platforms for Biosensors with Electrochemical and Electronic Transduction, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 1–78, DOI:10.1007/978-3-642-31421-6_1.
- J. Budhathoki-Uprety, J. Shah, J. A. Korsen, A. E. Wayne, T. V. Galassi, J. R. Cohen, J. D. Harvey, P. V. Jena, L. V. Ramanathan, E. A. Jaimes and D. A. Heller, Nat. Commun., 2019, 10, 3605 CrossRef PubMed.
- A. Benko, A. Wiecheć, B. Rajchel, E. Długoń and M. Błażewicz, Acta Phys. Pol., A, 2016, 129, 176–178 CrossRef.
- A. R. Boccaccini, F. Chicatun, J. Cho, O. Bretcanu, J. A. Roether, S. Novak and Q. Chen, Adv. Funct. Mater., 2007, 17, 2815–2822 CrossRef CAS.
- A. Battigelli, C. Ménard-Moyon, T. Da Ros, M. Prato and A. Bianco, Adv. Drug Delivery Rev., 2013, 65, 1899–1920 CrossRef CAS PubMed.
- A. Loboda, A. Jozkowicz and J. Dulak, Vasc. Pharmacol., 2015, 74, 11–22 CrossRef CAS PubMed.
- J. Duch, M. Golda-Cepa and A. Kotarba, Appl. Surf. Sci., 2019, 463, 1134–1140 CrossRef CAS.
- W. Pajerski, J. Duch, D. Ochonska, M. Golda-Cepa, M. Brzychczy-Wloch and A. Kotarba, Mater. Sci. Eng., C, 2020, 113, 110972 CrossRef CAS PubMed.
- A. Benko, D. Medina-Cruz, J. Duch, T. Popiela, S. Wilk, M. Bińczak, M. Nocuń, E. Menaszek, L. D. Geoffrion, G. Guisbiers, A. Kotarba and T. J. Webster, Mater. Sci. Eng., C, 2020, 111703, DOI:10.1016/j.msec.2020.111703.
- M. Rohwerder, in Encyclopedia of Interfacial Chemistry, ed. K. Wandelt, Elsevier, Oxford, 2018, pp. 414–422, DOI:10.1016/B978-0-12-409547-2.13405-5.
- I. Nanostructured & Amorphous Materials, Amorphous Products | Nanoscale Products - Short MWNT (95+%, OD 10–30 nm), http://www.nanoamor.com/inc/sdetail/4021.
- Y. N. Jeong, M. Y. Choi and H. C. Choi, Electrochim. Acta, 2012, 60, 78–84 CrossRef CAS.
- J. C. Slater, Phys. Rev., 1951, 81, 385–390 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, WIREs Comput. Mol. Sci., 2014, 4, 91–100 CrossRef CAS.
- L. Besra and M. Liu, Prog. Mater. Sci., 2007, 52, 1–61 CrossRef CAS.
- Y. Y. Zhang, Y. Xiang and C. M. Wang, J. Appl. Phys., 2009, 106, 113503 CrossRef.
- S. Liang, G. Li and R. Tian, J. Mater. Sci., 2016, 51, 3513–3524 CrossRef CAS.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS.
- J. T. Titantah and D. Lamoen, Diamond Relat. Mater., 2007, 16, 581–588 CrossRef CAS.
- K. Wepasnick, B. Smith, J. Bitter and D. Howard Fairbrother, Anal. Bioanal. Chem., 2010, 396, 1003–1014 CrossRef CAS PubMed.
- B. Lesiak, J. Zemek, P. Jiricek and L. Stobinski, Phys. Status Solidi B, 2009, 246, 2645–2649 CrossRef CAS.
- X. Ling, Y. Wei, L. Zou and S. Xu, Appl. Surf. Sci., 2013, 276, 159–166 CrossRef CAS.
- Y. Zhang, Y. Li and P. Zhang, J. Mater. Sci., 2014, 49, 3469–3477 CrossRef CAS.
- A. Pacuła, R. P. Socha, M. Zimowska, M. Ruggiero-Mikołajczyk, D. Mucha and P. Nowak, Appl. Clay Sci., 2013, 72, 163–174 CrossRef.
- M. Zhao, L. Meng, L. Ma, L. Ma, X. Yang, Y. Huang, J. E. Ryu, A. Shankar, T. Li, C. Yan and Z. Guo, Compos. Sci. Technol., 2018, 154, 28–36 CrossRef CAS.
- L. Hussein, RSC Adv., 2016, 6, 13088–13100 RSC.
- C. Morant, J. Andrey, P. Prieto, D. Mendiola, J. M. Sanz and E. Elizalde, Phys. Status Solidi A, 2006, 203, 1069–1075 CrossRef CAS.
- G. Gabriel, G. Sauthier, J. Fraxedas, M. Moreno-Mañas, M. T. Martínez, C. Miravitlles and J. Casabó, Carbon, 2006, 44, 1891–1897 CrossRef CAS.
- P. Song, Y. Shen, B. Du, Z. Guo and Z. Fang, Nanoscale, 2009, 1, 118–121 RSC.
- B. R. C. de Menezes, F. V. Ferreira, B. C. Silva, E. A. N. Simonetti, T. M. Bastos, L. S. Cividanes and G. P. Thim, J. Mater. Sci., 2018, 53, 14311–14327 CrossRef CAS.
- S. Kundu, W. Xia, W. Busser, M. Becker, D. A. Schmidt, M. Havenith and M. Muhler, Phys. Chem. Chem. Phys., 2010, 12, 4351–4359 RSC.
- A. Benko, M. Nocuń, M. Gajewska and M. Błażewicz, Polym. Degrad. Stab., 2019, 161, 260–276 CrossRef CAS.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- D. V. Rodriguez, Doctor of Philosophy, Macdonald Campus of McGill University, 2015.
- U. J. Kim, C. A. Furtado, X. Liu, G. Chen and P. C. Eklund, J. Am. Chem. Soc., 2005, 127, 15437–15445 CrossRef CAS PubMed.
- S. Osswald, M. Havel and Y. Gogotsi, J. Raman Spectrosc., 2007, 38, 728–736 CrossRef CAS.
- F. Pourfayaz, Y. Mortazavi, A.-A. Khodadadi, S. H. Jafari, S. Boroun and M. V. Naseh, Appl. Surf. Sci., 2014, 295, 66–70 CrossRef CAS.
- J.-P. Tessonnier, D. Rosenthal, T. W. Hansen, C. Hess, M. E. Schuster, R. Blume, F. Girgsdies, N. Pfänder, O. Timpe, D. S. Su and R. Schlögl, Carbon, 2009, 47, 1779–1798 CrossRef CAS.
- G. A. Somorjai and Y. Li, Introduction to Surface Chemistry and Catalysis, Wiley, New York, 2nd edn, 2010 Search PubMed.
- J. Brison, N. Mine, S. Poisseroux, B. Douhard, R. G. Vitchev and L. Houssiau, Surf. Sci., 2007, 601, 1467–1472 CrossRef CAS.
- J. Cho, K. Konopka, K. Rożniatowski, E. García-Lecina, M. S. P. Shaffer and A. R. Boccaccini, Carbon, 2009, 47, 58–67 CrossRef CAS.
- L. Li, W. Liu, F. Yang, W. Jiao, L. Hao and R. Wang, Compos. Sci. Technol., 2020, 187, 107946 CrossRef CAS.
- J. Ma and W. Cheng, Mater. Lett., 2002, 56, 721–727 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr09057c |
| This journal is © The Royal Society of Chemistry 2021 |