
Anisotropic nanoporous morphology of ZnO-supported Co that enhances catalytic activity†
 ‡a
Adam A.
Corrao,
‡bc
Victoria
Petrova,
‡a
Taewoo
Kim,
d
Peter G.
Khalifah
*bc
and
Ping
Liu
*adf
‡a
Adam A.
Corrao,
‡bc
Victoria
Petrova,
‡a
Taewoo
Kim,
d
Peter G.
Khalifah
*bc
and
Ping
Liu
*adf
Abstract
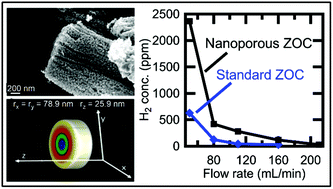
A novel conversion reaction synthesis (CRS) method is used to synthesize ZnO-supported Co nanoporous metal hybrid structures from a co-precipitated nanocomposite precursor of ZnO and Co3O4. After removal of Li2O with water, the resulting material consists of ZnO-supported Co nanoparticles that are interconnected to form anisotropic micro-particles. Additionally, individual ZnO nanoparticles have an anisotropic morphology, as revealed by synchrotron XRD analysis. Microscopy and surface area studies show these materials have an average pore size of 10–30 nm and specific surface areas up to 28 m2 g−1. The hybrid structure also has increased heat resistance compared to that of pure nanoporous metals; the Co phase within the ZnO–Co hybrid exhibits much less coarsening than the analogous nanoporous metal without ZnO at temperatures of 400 °C and above. These ZnO–Co hybrid materials were tested as heterogeneous catalysts for the steam reformation of ethanol at 400 °C. The nanoporous ZnO–Co hybrid material exhibits complete conversion of ethanol and high hydrogen selectivity, producing H2 with a molar yield of approximately 70%.
- This article is part of the themed collection: Editor’s Choice: Controlling anisotropy in nanomaterials
 Please wait while we load your content...
Please wait while we load your content...

