
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLanthanide-porphyrin species as Kondo irreversible switches through tip-induced coordination chemistry†
B.
Cirera
*a,
J. M.
Gallego
 b,
J. I.
Martínez
b,
R.
Miranda
ac and
D.
Écija
*a
b,
J. I.
Martínez
b,
R.
Miranda
ac and
D.
Écija
*a
aIMDEA Nanoscience, Cantoblanco, Madrid, Spain. E-mail: borja.cirera@fhi-berlin.mpg.de; david.ecija@imdea.org
bInstituto de Ciencia de Materiales de Madrid (ICMM-CSIC), c/Sor Juana Inés de la Cruz 3, 28049 Madrid, Spain
cDepartamento de Física de la Materia Condensada, Universidad Autónoma de Madrid, Cantoblanco, Madrid, Spain
First published on 29th April 2021
Abstract
Metallosupramolecular chemical protocols are applied to in situ design dysprosium porphyrin complexes on Au(111) by sequential deposition of 2H-4FTPP species and Dy, resulting in the production of premetallated Dy-2H-4FTPP, partially metallated Dy-1H-4FTPP and fully metallated Dy-0H-4FTPP complexes, as determined by scanning tunneling microscopy (STM) and density functional theory (DFT) calculations. A zero bias resonance is found in the Dy-2H-4FTPP species which, upon study of its spatial distribution and behavior with temperature, is assigned to a Kondo resonance resulting from an unpaired spin in the molecular backbone, featuring a Kondo temperature (TK) of ≈ 21 K. Notably, the Kondo resonance can be switched off by removing one hydrogen atom of the macrocycle through tip-induced voltage pulses with submolecular precision. The species with this Kondo resonance can be laterally manipulated illustrating the potential to assemble artificial Kondo lattices. Our study demonstrates that the pre-metallation of macrocycles by lanthanides and their controlled manipulation is a novel strategy to engineer in situ tunable Kondo nanoarchitectures, enhancing the potential of coordination chemistry for spintronics.
The design of molecular systems on surfaces and the control over the inherent spin-electron interactions is crucial for the fundamental understanding of electronic transport and for the development of molecular electronics, spintronic devices and quantum computation. In the last two decades much attention has been devoted to study the Kondo phenomenon in interfacial nanosystems,1,2 including adsorbed magnetic atoms3–6 and molecules.7–19 The Kondo effect results from the many-body interaction between the spin of a localized magnetic impurity and the conduction electrons of the surrounding non-magnetic metal, giving rise to a resonance near the Fermi level for temperatures below the characteristic Kondo temperature (TK).1,20 Exploiting this effect in coordination complexes is particularly appealing owing to their potential for single-molecule spintronic applications. This phenomenon has been extensively investigated in surface-confined transition metal porphyrin macrocycles,21 which are tunable through removal of peripheral hydrogen atoms,7 tip-induced conformational changes,8 self-assembly19 or appropriate apical coordination to gas species.11,12 However, regarding lanthanides, only Tb-phthalocyanine has revealed a Kondo behavior when sublimated and deposited on Au(111).22 Thus, it is necessary to explore novel approaches to take advantage of the inherent magnetic properties of f-block elements, with the pre-metallation of macrocycles by lanthanides being a promising strategy.23
Here we report a low temperature scanning tunneling microscopy and spectroscopy study on the on-surface synthesis of 2D reticular porphyrin supramolecular nanoarchitectures and subsequent pre-, partial-, or fully-metallation by dysprosium atoms without altering the supramolecular design, giving rise to irreversible Kondo molecular switches. First, the exposure of a submonolayer coverage of 2H-4FTPP species to a beam of dysprosium affords the expression of: (i) pre-metallated Dy-2H-4FTPP (ii) Dy-1H-4FTPP and (iii) fully metallated Dy-4FTPP compounds. Importantly, the lanthanide ion is located below the molecular backbone on a metal “hollow” site, interacting simultaneously with the monomer and the substrate, as demonstrated by DFT calculations. A scanning tunneling spectroscopy inspection reveals that Dy-2H-4FTPP exhibits a zero bias peak close to Fermi level, which can be rationalized as a Kondo resonance in the molecular backbone. Both Dy-1H-4FTPP and Dy-0H-4FTPP species show no zero-bias resonances. Local tip-induced voltage pulses allow the controlled and irreversible transformation of the dysprosium compounds from Dy-2H-4FTPP to Dy-1H-4FTPP and from Dy-1H-4FTPP to Dy-0H-4FTPP species, respectively. Thus, the partial or full coordination with dysprosium of fluorinated porphyrins switches off irreversibly the Kondo resonance. Finally, the analysis of the evolution of the HWHM (half width at half maximum) of the Kondo resonance as a function of temperature shows a large Kondo temperature of TK ≈ 121 K. We envision our study will pave new avenues to engineer metallo-supramolecular nanoarchitectures of relevance for nanomagnetism, while enhancing the potential of coordination chemistry for spintronics.
Results and discussion
The deposition of a submonolayer coverage of 2H-4FTPP species on Au(111) held at room temperature (cf.Fig. 1a) gives rise to spatially extended closed-packed islands, highlighting the high mobility of the species. Each molecule reveals a two-fold symmetric appearance, characteristic of the nonplanar macrocycle deformation of the porphyrin family upon surface confinement (cf. inset Fig. 1a).21 The central depression is assigned to the free-base macrocycle, whereas the four terminal protrusions are attributed to the fluorine-aryl moieties.24 At positive bias voltage minor species (≈5%) with a larger apparent height can be detected (cf. Fig. SI1a and b†), distributed mainly in the frontiers between the hcp and the fcc regions of the herringbone reconstruction of Au(111). The four-lobed appearance is in good agreement with the simulation of premetallated Au-2H-4FTPP species, where an intact 2H-4FTPP is sitting on top of a gold adatom (cf. Fig. SI1c†). A similar result has been recently described in the 2H-TPP/Au(111)25 system, where an adatom can be observed underneath some species, resulting in the formation of Au-2H-TPP premetallated compounds. Moreover, in analogy with the reported Au-2H-TPP,25 we also observe the rigid shift of around 0.5 eV towards lower energy in STS between Au-2H-4FTPP and 2H-4FTPP species (see Fig. SI1d†).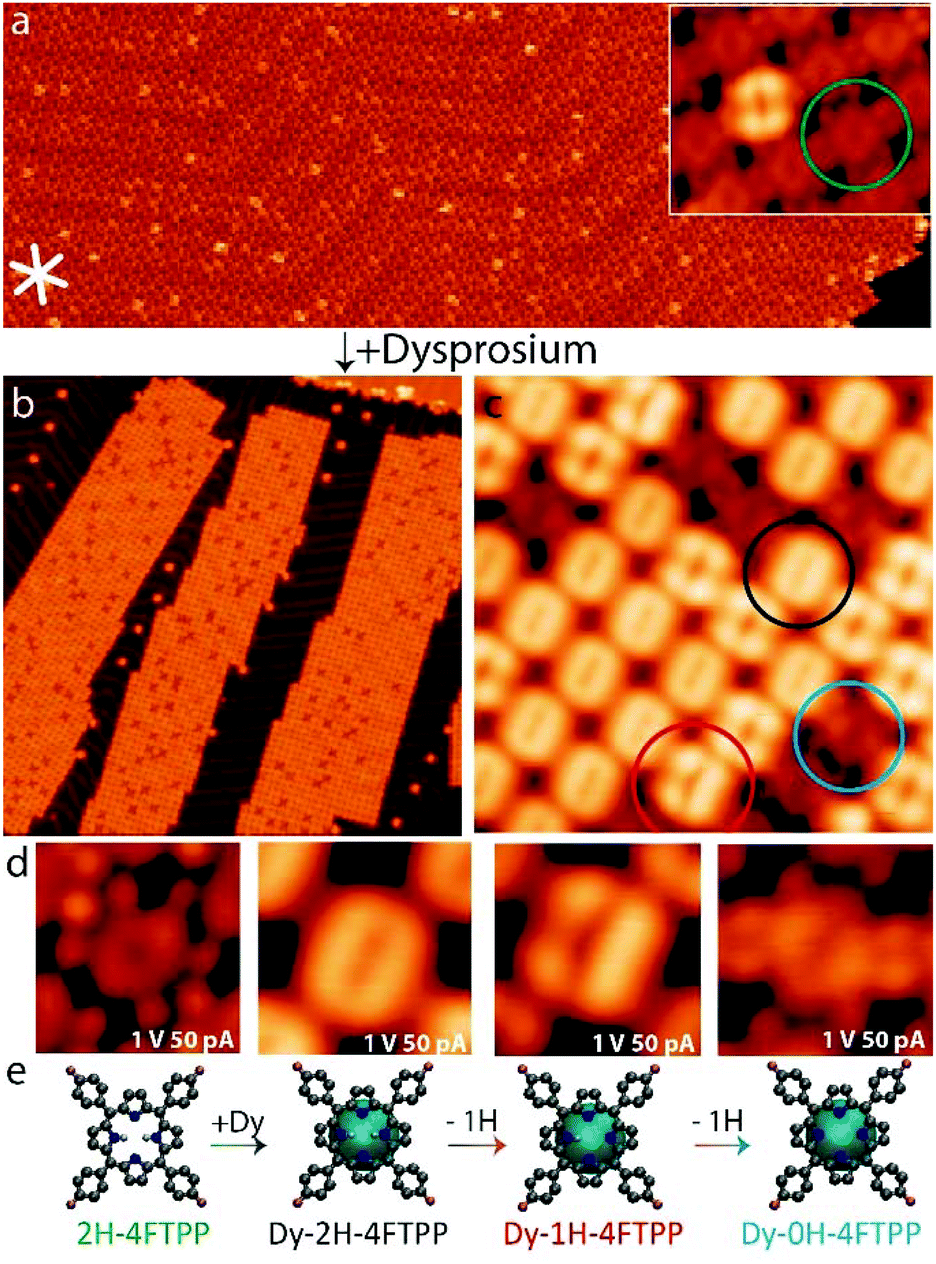 | ||
| Fig. 1 On-surface synthesis of dysprosium-porphyrinato derivatives by sequential deposition of 2H-4FTPP and dysprosium on Au(111). (a) Long-range image and zoomed-in (inset) self-assembly of pristine 2H-4FTPP on Au(111), with the minor Au-2H-4FTPP species showing a higher relative apparent height at positive Vb (width = 100 nm, Vb = 0.7 V, It = 100 pA; inset: width = 5.4 nm, Vb = 1.0 V, It = 250 pA). (b and c) Long range and close view images after deposition of Dy, where three new species, highlighted in black (Dy-2H), red (Dy-1H) and blue (Dy-0H), can be distinguished (Scanning parameters: b) (width = 100 nm, Vb = 1.0 V, It = 250 pA; (c) width = 10 nm, Vb = 1.0 V, It = 100 pA). (d and e) STM zoomed-in images and ball-and-stick models of the different species, identifying (from left to right) the initial intact species, the pre-metallated Dy-2H-4FTPP state, the intermediate Dy-1H-4FTPP and the final Dy-0H-4FTPP species (width = 2 nm, Vb = 1.0 V, It = 50 pA). All images have been acquired at 4 K. | ||
Next, we have inspected the tip-induced capabilities of the macrocycle towards tautomerization26–28 and deprotonation27 of the two protons in the inner macrocycle looking for the feasibility of subsequent metallation with lanthanides that will require partial or full deprotonation. As illustrated in Fig. SI2,† by stabilizing the tip at perturbative conditions (Vbias = 1.25 V and 2.9 nA) over one pyrrole of the macrocycle and opening the feedback loop, a clear telegraphic two-level graph is measured, each of the levels of conductance assigned to one tautomer of the 2H-4FTPP species. Such phenomenon can be also monitored topographically while scanning at perturbative conditions (Vbias ≥ 1.5 V) by detecting blurry lines over the macrocycles that correspond to the tautomeric movement of the inner protons (Fig. SI2f†). In addition, tip-induced voltage pulses at higher voltages over the macrocycle give rise to deprotonation of the inner protons (see Fig. SI3†), affording the in situ fabrication of 1H-4FTPP (pulse at Vbias = 2.1 V) and 0H-4FTPP (pulse at Vbias = 2.4 V) species on Au(111). Thus, as expected for a surface-confined tetrapyrrole, both tip-induced tautomerization and partial/full deprotonation of inner protons are allowed, which will be used in the following discussion as a fingerprint to identify intact 2H-4FTPP species.
Exposing a submonolayer of 2H-4FTPP (green circle) on Au(111) (Fig. 1a), held at room temperature, to a beam of Dy atoms gives rise to the successful synthesis of Dy-porphyrin species. Importantly, the molecular coverage was kept below the monolayer to avoid the formation of double-deckers.29,30 Large scale STM topographic images (Fig. 1b) show that the islands are smaller and exhibit an overall rectangular shape with rather straight edges. Remarkably, almost all molecules display now a greater apparent height, being “dark” species a minority, thus highlighting the role of the diffusion of Dy and interactions with the molecular adsorbates. However, the supramolecular self-assembly maintains the same unit cell, as shown in Fig. 1c. In addition to the remaining 2H-4FTPP and Au-2H-4FTPP compounds, three new species can be observed, encircled in black, red and blue, which can be better observed in the high resolution STM images in Fig. 1d.
At Vbias = 1 V (cf.Fig. 1d), Dy-2H-4FTPP (black) shows a doughnut-like shape with two pronounced lobes surrounded by four bright protrusions (better appreciated in Fig. 3), clearly different from the flower-like four-lobed shape of Au-2H-4FTPP, whereas Dy-1H-4FTPP (red) displays a characteristic asymmetric three-lobed appearance (cf.Fig. 1d and SI4†). Dy-0H-4FTPP (blue) shows a slightly higher apparent height of two of its central pyrroles when compared to intact species (green). Our rationalization of the Dy-porphyrinoids is determined by the excellent agreement between the simulated STM images and the experimental results. Fig. 2 shows the STM images simulated for the four species observed in the experiments, where it is apparent the difference in morphology between Dy-2H-4FTPP (black, a porphyrin physisorbed on top of a dysprosium atom) and Dy-1H-4FTPP (red, an intermediate coordination of the porphyrin to dysprosium by the loss of one proton), with two and three main lobes, respectively. Calculated STM images yield for both 2H-4FTPP and Dy-0H-4FTPP molecular species a central depression with two lobes (porphyrin macrocycle) surrounded by four protrusions (fluorine-aryl moieties) at the periphery in agreement with the experiment. At first sight, it might be surprising that intact 2H-4FTPP species and Dy-0H-4FTPP derivatives exhibit a similar topographic appearance. However, a closer inspection to experimental and theoretical results shows that Dy-0H-4FTPP display a slight higher contrast of the prominent lobes of the macrocycle at positive bias voltages compared to 2H-4FTPP, reflecting the change in conformation of the pyrroles to fully coordinate to the dysprosium adatom underneath (cf.Fig. 1d and SI5i†). Additionally, the absence of telegraphic noise (tautomerism) in Dy-0H-4FTPP species (using the same conditions described above for Dy-0H-4FTPP), the impossibility to deprotonate these species by vertical manipulation, as well a distinct electronic structure (to be described below), allow us to unambiguously discern between intact and fully metallated species (cf.Fig. 3).
 | ||
| Fig. 2 Theoretical rationalization of the partial and full metallation of 2H-4FTPP species by Dy on Au(111). (a and b) Side and top view of the relaxed structures of the four different species, highlighting the position of the Dy atom below the average molecular plane. (c) Keldish-Green theoretical STM images under constant-current regime at 1 V, in agreement with the experimentally observed appearance. | ||
 | ||
| Fig. 3 Electronic structure and tip-induced transformations of Dy-porphyrin derivatives on Au (111). (a) STS spectra recorded on top of the different species at 4 K (Feedback conditions for STS: It = 250 pA, Vb = −0.9 V, modulation: 10 mV, frequency = 773 Hz, integration time = 36 ms per point, sampling energy = 3 meV). (b) Model showing 9 monomers, five of them being intact (2H-4FTPP) and four of them Dy-2H-4FTPP species, to highlight the macrocycles in c-d containing dysprosium. (c) Topography and constant current dI/dV map at the approximate energy of the HOMO resonance (It = 225 pA, Vb = −0.8 V, width = 4.95 nm). (d) Topography and constant current dI/dV map at the approximate energy of the LUMO resonance, highlighting the tautomerization occurring while scanning in the macrocycle of the intact 2H-4FTPP species (It = 300 pA, Vb = 1.5 V, width = 5 nm). (e and f) Sequence showing that the Dy-species can be irreversibly transformed from Dy-2H-4FTPP into Dy-1H-4FTPP and subsequently into the final Dy-0H-4FTPP, as a result of the desorption of one inner proton (or two) and the subsequent bonding to the lanthanide ion underneath (All images: It = 150 pA, Vb = 1 V, width = 3.9 nm). | ||
DFT-optimized 2H-4FTPP, Dy-2H-4FTPP, Dy-1H-4FTPP and Dy-0H-4FTPP on Au(111) interfacial structures reveal further differences among them. The intact 2H-4FTPP/Au system is predicted to have the four fluorine-aryl moieties lying almost perpendicular to the metal surface, forming alternating dihedral angles of around 77° and 79° with the surface, the pyrrolic central part of the molecule lying on the metal rather flat at a distance of around 3.6 Å. The simulations indicate that for Dy-2H-4FTPP, Dy-1H-4FTPP and Dy-0H-4FTPP on Au(111), the lanthanide ion is always located underneath the molecular backbone on a metal three-fold “hollow” site at a perpendicular distance of around 2.4 Å from the Au surface, interacting simultaneously with the monomer and the substrate. Recent results on the direct visualization of the metallation of 2H-TPP by Cerium reveal that the lanthanide is adsorbed below the macrocycle plane.23 The Dy–Au distance barely varies independently on the molecular species adsorbed. It is important to mention that this adsorption arrangement is the most stable configuration energetically speaking after computing the relaxation of distinct starting-point structures. In all these cases, the dihedral angle of the fluorine-aryl moieties with respect to the metal surface reduces significantly from the 78° in 2H-4FTPP to 36°, 39° and 44°, respectively, mainly due to the increasing distortion of the pyrrolic central part of the molecules after interaction with the Dy atom, which also induces an increase of the perpendicular distance between the molecular center of mass and the Dy atom to 1.53, 1.46 and 1.38 Å for Dy-2H-4FTPP, Dy-1H-4FTPP and Dy-0H-4FTPP, respectively. For comparison, the binding energy of an isolated Dy atom adsorbed on a three-fold “hollow” Au site is 1.95 eV, which reduces around 20% when the molecular species are adsorbed on top. The binding energies of the 2H-4FTPP, 1H-4FTPP and 0H-4FTPP molecules attached to Dy/Au result in 2.65, 3.14 and 3.35 eV, respectively.
In all cases, calculations predict the Dy atom acting with an oxidation state ranging between +2.2 and +2.4. Hereby, it is worth to mention that Dy directly adsorbed on most noble metal surfaces has a 4f10 (i.e. Dy2+) configuration.31 On the other hand, we have experimentally shown that Gd and Dy, both from the family of lanthanides, upon coordination with molecular linkers on metal surfaces have a 4f9 (i.e. Gd+3and Dy+3) electronic structure,32,33 as typically encountered for metal–organic complexes based on lanthanides. In our previous study, coordinated Gd displays a +3 oxidation state when probed by XPS and a theoretical calculated charge of 2.1e−.30 Thus, we tentatively conclude that Dy also exhibits a +3 oxidation state after being adsorbed beneath the porphyrin macrocycle.
Next, we investigate the electronic structure of the species by means of scanning tunneling spectroscopy (STS), depicted in Fig. 3a. Intact species (green spectrum) shows two clear resonances at −0.9 and 1.6 eV (cf.Fig. 3a), which in analogy to other surface-confined tetrapyrrole derivatives are associated unambiguously to the HOMO and LUMO orbitals of the species, resulting in a gap of circa 2.5 eV, very similar to the results measured on 2H-TPP on Au(111),25 showing the very minor role of the fluorine substituents in the resulting electronic structure.24 Dy-2H-4FTPP is featureless in the HOMO region, shows a tiny resonance at 40 meV and suffers a transformation into Dy-1H-4FTPP for voltages above 1.4 eV. Dy-1H-4FTPP shows a prominent feature at 0.75 eV. Finally, the Dy-0H-4FTPP species display two clear resonances at −0.9 and 1.7 eV, tentatively assigned to the HOMO and LUMO orbitals of the molecule, respectively, which gives rise to an electronic gap of 2.6 eV. The upshift of the LUMO orbitals with respect to the Fermi level from Dy-2H-4FTPP to Dy-1H-4FTPP and from Dy-1H-4FTPP to Dy-0H-4FTPP is qualitatively well captured by our DFT simulations (cf. Fig. SI5†).
Fig. 3b shows schematically four Dy-0H-4FTPP and five 2H-4FTPP monomers, corresponding to the scenario depicted in Fig. 3c and d. By acquiring topographic STM images, together with dI/dV maps, at the approximate energies of the frontier orbitals (i.e. −0.8 and +1.5 V), both species can be unambiguously distinguished: (i) The HOMO orbital shows clearly different apparent intensities in the dI/dV map (Fig. 3c), (ii) intact 2H-4FTPP species exhibit tautomerization (Fig. 3d) while scanning (noise lines highlighted with arrows), and (iii) the LUMO of Dy-0H-4FTPP has a two-fold symmetric appearance and larger depression around its center, as compared to 2H-4FTPP, that displays four lobes and a smaller central void (Fig. 3d, right panel).
The observed abrupt change upon surpassing a certain voltage in the signal of the Dy-1H-4FTPP and Dy-2H-4FTPP spectra (black and red curves in Fig. 3a), is associated with an in situ modification of the investigated species. Fig. 3e and f illustrate that tip-induced voltage pulses, at the approximate energies (1.4 and 1.6 eV, respectively) where the abrupt changes are observed, allow to transform sequentially Dy-2H-4FTPP into Dy-1H-4FTPP and Dy-0H-4FTPP species, as shown for Ce-TPP derivatives on Ag(111).23 Such transformation can be performed both on self-assembled islands and on isolated Dy-2H-4FTPP species, previously displaced by lateral manipulation (cf. Fig. SI6†), highlighting not only the propensity towards fully metallation under appropriate tunneling conditions, but also the weak Dy–Au bonding that allows the dysprosium derivatives to be translated along the surface, opening new avenues to design specific metallo-supramolecular complexes with outmost spatial control.
Differential conductance (dI/dV) spectra measured close to the Fermi level reveal a pronounced zero bias peak (ZBP) on Dy-2H-4FTPP derivatives (cf. black line in Fig. 4a). Such resonance is not present for any of the other species. The spectral shape of the zero bias peak is well described by a Fano function including temperature and instrumental broadening (cf. violet curve in Fig. 4a and ESI† for details), giving at 4.4 K a Γ (HWHM) value of 10.2 meV and a q (line-shape factor) value of 15. The excellent fit hints towards an origin of the low-bias peak as a Kondo resonance. The peak line-shape and the large q value indicate that the indirect tunneling channel through the magnetic impurity dominates.1,2 In order to confirm this hypothesis, Γ was tracked as a function of temperature (cf.Fig. 4b) and fitted according to the Nagaoka's model5 for Kondo systems within the strong coupling Fermi liquid theory (cf.Fig. 4c), resulting in a large TK (Kondo temperature) of 121 K, indicative of a strong coupling between the spin and the conduction electrons.
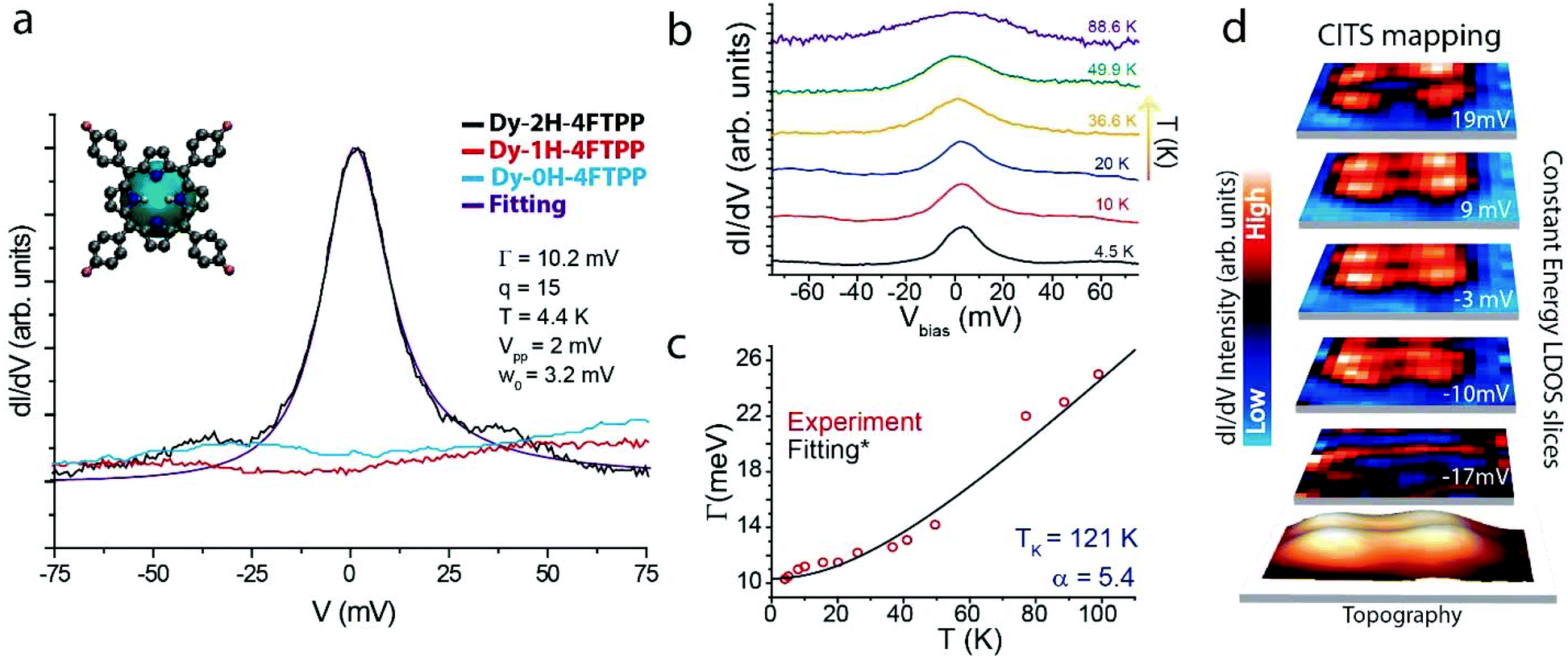 | ||
Fig. 4 Kondo resonance of Dy-2H-4FTPP on Au(111). (a) dI/dV spectra acquired close to the Fermi level for the three different Dy-porphyrin derivatives, where the initial Dy-2H-4FTPP species exhibits a strong resonance (Feedback conditions for STS: It = 250 pA, Vb = −0.1 V, modulation = 2 mV, frequency = 773 Hz, integration time = 300 ms per point, sampling energy = 0.5 meV). (b) dI/dV spectra recorded close to the Fermi level at different temperatures in order to track the evolution of Γ. (Feedback conditions for STS: It = 250 pA, Vb = −0.1 V, modulation: 2 mV, frequency = 773 Hz, integration time = 300 ms per point, sampling energy = 0.5 meV). (c) HWHM versus temperature fitted by  attributing the resonance to the Kondo effect. (d) CITS slices (25 × 25 pixels) at different energies (19, 9, −3, −10 and −17 mV from upper to bottom) in order to observe the spatial distribution of the Kondo resonance, highlighting its molecular shape. attributing the resonance to the Kondo effect. (d) CITS slices (25 × 25 pixels) at different energies (19, 9, −3, −10 and −17 mV from upper to bottom) in order to observe the spatial distribution of the Kondo resonance, highlighting its molecular shape. | ||
Fig. 4d reproduces LDOS slices at different energies close to the Fermi level extracted from a current-imaging-tunneling spectroscopy (CITS) map, where the spatial distribution of the Kondo resonance over the molecule can be visualized. The Kondo intensity shows a two-fold orbital shape with a clear depression over the center of the macrocycle, reflecting the topography of the orbital and, thus, anticipating the molecular origin of such a feature.2
As shown in Fig. 4a, the Kondo resonance can be irreversibly switched off by vertical manipulation transforming the Dy-2H-4FTPP complex to Dy-1H- or Dy-0H-4FTPP species, which do not show any feature close to the Fermi level. As mentioned before, Dy directly adsorbed on most noble metal surfaces has a 4f10 (i.e. Dy2+) configuration31 and a 4f9 (i.e. Dy+3) electronic structure upon coordination.33 The adsorption of the Dy atom underneath the macrocycle core gives rise to the decoupling of the molecular backbone from the Au substrate, while the bonding in Dy-2H-4FTPP permits the sufficient charge transfer between lanthanide and porphyrin to allow an unpaired spin to be delocalized in a molecular orbital of the porphyrin,13,15 as previously encountered for the related surface-confined porphyrin-lanthanide-porphyrin (double-decker) species.9,34 Hereby, the screening of this unpaired spin by the neighboring conducting electrons of the substrate gives rise to the Kondo resonance. Thus, the Kondo phenomenon in Dy-2H-4FTPP species is assigned to a purely molecular origin, i.e. the unpaired spin in the in situ synthesized lanthanide-macrocyclic compound,9 with a large Kondo temperature of 121 K. This value is close to the reported value for a Fe-polyphenyl complex on Cu(111), which also shows a Kondo peak,18 and where the Fe atom lies beneath the phenylene moiety, in analogy to our Dy-2H-4FTPP derivative. The controlled tip-induced elimination of hydrogen atoms from the macrocycle induces a stronger bond of the pyrrole unit with the lanthanide underneath, quenching the unpaired spin from the molecular orbital and removing the Kondo resonance.
Our experimental rationalization is qualitatively corroborated by spin-polarized density functional theory. As illustrated in Fig. SI5† (cf. ESI† for full discussion) the net integration of the computed spin-density, obtained as the difference between the spin-up and spin-down projected density of states, up to the Fermi level for the three (0H/1H/2H)-4FTPP species on Au(111) yields values of 0.02, 0.14 and 0.64 spin-down electrons from the 0H-4FTPP to the 2H-4FTPP cases. Thus, the highest value of electronic unpairing (0.64 e−) scattered by the Dy atom towards the molecule is obtained for the 2H-4FTPP species (considering the 0.85 e− transferred from the substrate to the molecule for this interface), which points it up as the best potential molecular candidate to yield a Kondo resonance from the theoretical point of view, agreeing with the experimental observation.
Finally, it is worth to point out that when individual local spins are arranged to form a spatially ordered lattice interesting many-body behavior may appear as a result of the interplay between Kondo screening and coupling among neighbor spins, including coherent non-local Kondo screening below a given temperature (different from individual Kondo temperature) controlled by intersite coupling,35 spatially periodic enhancement of the Kondo resonance36 or long range magnetic order.13,37 To this end, we found out that the Dy-2H-4FTPP species could be laterally manipulated with the STM tip (as illustrated in Fig. SI6†). This could contribute to form ordered lattices of Kondo scatterers, anticipating fascinating opportunities for in situ designing tailored Kondo systems with high Kondo temperatures obtained by embedding lanthanide elements in appropriate molecular nanoarchitectures.
Conclusions
In summary, we have shown the in situ synthesis of dysprosium porphyrin derivatives on Au(111) by the sequential deposition of a porphyrin archetype (2H-4FTPP) and dysprosium. Comparing experimental imaging with DFT calculated STM images, we could identify three distinct dysprosium complexes: premetallated Dy-2H-4FTPP, partially metallated Dy-1H-4FTPP and fully metallated Dy-0H-4FTPP. Tip-induced voltage pulses results in the sequential transformation of Dy-2H-4FTPP to Dy-1H-4FTPP and Dy-0H-4FTPP. Remarkably, only the Dy-2H-4FTPP species show a zero bias peak, which is assigned to the Kondo effect of an unpaired spin delocalized in a molecular orbital of the macrocycle, with a Kondo temperature of 121 K. Such a resonance can be switched off by removing the hydrogen atoms from Dy-2H-4FTPP to the other dysprosium derivatives. In addition, all dysprosium complexes can be laterally translated along the surface by the STM tip. Thus, our study paves new avenues towards the design of atomistic-tailored Kondo lattices, demonstrating the potential of pre-metallation of macrocycles for nanomagnetism, while expanding the knowledge at the nanoscale of metallo-supramolecular systems incorporating both lanthanides and porphyrins.Experimental section
The experiments were performed in a custom designed ultra-high vacuum system that hosts a low-temperature Omicron scanning tunneling microscope, where the base pressure was below 5 × 10−10 mbar. All STM images were taken in constant-current mode with electrochemically etched tungsten tips, applying a bias (Vb) to the sample and at a temperature of ∼4 K. The Au(111) substrate was prepared by standard cycles of Ar + sputtering (800 eV) and subsequent annealing to 723 K for 10 minutes. 2H-4FTPP species are commercial from Sigma Aldrich and were deposited by organic molecular-beam epitaxy (OMBE) from a quartz crucible held at 373 K onto a clean Au(111) at room temperature. Dy atoms were evaporated by electron beam evaporation from an outgassed Dy rod, with the sample held at room temperature at a distance of 12.5 cm, and with typical deposition times of 45 seconds at 5.5 mA of emission. Next, samples were transferred to the STM stage held at 4 K.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the H2020 European Research Council research and innovation programme (grant agreement No 766555). This work has been financed by the Spanish Ministerio de Ciencia, Innovación y Universidades (project PGC 2018-098613-B-C21, PGC2018-098613-B-C21, SpOrQuMat) and the Comunidad de Madrid (Project S2018/NMT-4321, NanoMagCost). IMDEA Nanociencia acknowledges support from the ‘Severo Ochoa’ Programme for Centres of Excellence in R&D (MINECO, Grant SEV-2016-0686).References
- M. Ternes, A. J. Heinrich and W.-D. Schneider, J. Phys.: Condens. Matter, 2008, 21, 053001 CrossRef PubMed.
- T. Komeda, Surf. Sci., 2014, 630, 343–355 CrossRef CAS.
- V. Madhavan, W. Chen, T. Jamneala, M. F. Crommie and N. S. Wingreen, Science, 1998, 280, 567 CrossRef CAS PubMed.
- J. Li, W.-D. Schneider, R. Berndt and B. Delley, Phys. Rev. Lett., 1998, 80, 2893–2896 CrossRef CAS.
- K. Nagaoka, T. Jamneala, M. Grobis and M. F. Crommie, Phys. Rev. Lett., 2002, 88, 077205 CrossRef CAS PubMed.
- A. F. Otte, M. Ternes, K. von Bergmann, S. Loth, H. Brune, C. P. Lutz, C. F. Hirjibehedin and A. J. Heinrich, Nat. Phys., 2008, 4, 847 Search PubMed.
- A. Zhao, Q. Li, L. Chen, H. Xiang, W. Wang, S. Pan, B. Wang, X. Xiao, J. Yang, J. G. Hou and Q. Zhu, Science, 2005, 309, 1542 CrossRef CAS PubMed.
- V. Iancu, A. Deshpande and S.-W. Hla, Nano Lett., 2006, 6, 820–823 CrossRef CAS PubMed.
- T. Komeda, H. Isshiki, J. Liu, Y.-F. Zhang, N. Lorente, K. Katoh, B. K. Breedlove and M. Yamashita, Nat. Commun., 2011, 2, 217 CrossRef PubMed.
- N. Tsukahara, S. Shiraki, S. Itou, N. Ohta, N. Takagi and M. Kawai, Phys. Rev. Lett., 2011, 106, 187201 CrossRef PubMed.
- A. Stróżecka, M. Soriano, J. I. Pascual and J. J. Palacios, Phys. Rev. Lett., 2012, 109, 147202 CrossRef PubMed.
- H. Kim, Y. H. Chang, S.-H. Lee, Y.-H. Kim and S.-J. Kahng, ACS Nano, 2013, 7, 9312–9317 CrossRef CAS PubMed.
- M. Garnica, D. Stradi, S. Barja, F. Calleja, C. Díaz, M. Alcamí, N. Martín, A. L. Vázquez de Parga, F. Martín and R. Miranda, Nat. Phys., 2013, 9, 368 Search PubMed.
- M. Garnica, D. Stradi, F. Calleja, S. Barja, C. Díaz, M. Alcamí, A. Arnau, A. L. Vázquez de Parga, F. Martín and R. Miranda, Nano Lett., 2014, 14, 4560–4567 CrossRef CAS PubMed.
- M. Garnica, F. Calleja, A. L. Vázquez de Parga and R. Miranda, Surf. Sci., 2014, 630, 356–360 CrossRef CAS.
- Q. Zhang, G. Kuang, R. Pang, X. Shi and N. Lin, ACS Nano, 2015, 9, 12521–12528 CrossRef CAS PubMed.
- S. Karan, N. Li, Y. Zhang, Y. He, I. P. Hong, H. Song, J.-T. Lü, Y. Wang, L. Peng, K. Wu, G. S. Michelitsch, R. J. Maurer, K. Diller, K. Reuter, A. Weismann and R. Berndt, Phys. Rev. Lett., 2016, 116, 027201 CrossRef PubMed.
- G. E. Pacchioni, M. Pivetta, L. Gragnaniello, F. Donati, G. Autès, O. V. Yazyev, S. Rusponi and H. Brune, ACS Nano, 2017, 11, 2675–2681 CrossRef CAS PubMed.
- V. Iancu, A. Deshpande and S.-W. Hla, Phys. Rev. Lett., 2006, 97, 266603 CrossRef PubMed.
- J. Kondo, Prog. Theor. Phys., 1964, 32, 37–49 CrossRef CAS.
- W. Auwärter, D. Ecija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef PubMed.
- K. Katoh, Y. Yoshida, M. Yamashita, H. Miyasaka, B. K. Breedlove, T. Kajiwara, S. Takaishi, N. Ishikawa, H. Isshiki, Y. F. Zhang, T. Komeda, M. Yamagishi and J. Takeya, J. Am. Chem. Soc., 2009, 131, 9967–9976 CrossRef CAS PubMed.
- F. Bischoff, K. Seufert, W. Auwärter, A. P. Seitsonen, D. Heim and J. V. Barth, J. Phys. Chem. C, 2018, 122, 5083–5092 CrossRef CAS.
- B. Cirera, B. de la Torre, D. Moreno, M. Ondráček, R. Zbořil, R. Miranda, P. Jelínek and D. Écija, Chem. Mater., 2019, 31, 3248–3256 CrossRef CAS.
- J. Mielke, F. Hanke, M. V. Peters, S. Hecht, M. Persson and L. Grill, J. Am. Chem. Soc., 2015, 137, 1844–1849 CrossRef CAS PubMed.
- P. Liljeroth, J. Repp and G. Meyer, Science, 2007, 317, 1203–1206 CrossRef CAS PubMed.
- W. Auwärter, K. Seufert, F. Bischoff, D. Ecija, S. Vijayaraghavan, S. Joshi, F. Klappenberger, N. Samudrala and J. V. Barth, Nat. Nanotechnol., 2012, 7, 41–46 CrossRef PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Nat. Chem., 2014, 6, 41–46 CrossRef CAS PubMed.
- D. Écija, W. Auwärter, S. Vijayaraghavan, K. Seufert, F. Bischoff, K. Tashiro and J. V. Barth, Angew. Chem., Int. Ed., 2011, 50, 3872–3877 CrossRef PubMed.
- K. Diller, A. Singha, M. Pivetta, C. Wäckerlin, R. Hellwig, A. Verdini, A. Cossaro, L. Floreano, E. Vélez-Fort, J. Dreiser, S. Rusponi and H. Brune, RSC Adv., 2019, 9, 34421–34429 RSC.
- A. Singha, R. Baltic, F. Donati, C. Wäckerlin, J. Dreiser, L. Persichetti, S. Stepanow, P. Gambardella, S. Rusponi and H. Brune, Phys. Rev. B, 2017, 96, 224418 CrossRef.
- J. I. Urgel, B. Cirera, Y. Wang, W. Auwärter, R. Otero, J. M. Gallego, M. Alcamí, S. Klyatskaya, M. Ruben, F. Martin, R. Miranda, D. Ecija and J. V. Barth, Small, 2015, 47, 6358–6364 CrossRef PubMed.
- B. Cirera, L. Dordevic, R. Otero, J. M. Gallego, D. Bonifazi, R. Miranda and D. Ecija, Chem. Commun., 2016, 52, 11227–11230 RSC.
- T. Komeda, H. Isshiki, J. Liu, K. Katoh, M. Shirakata, B. K. Breedlove and M. Yamashita, ACS Nano, 2013, 7, 1092–1099 CrossRef CAS PubMed.
- Y.-f. Yang, Z. Fisk, H.-O. Lee, J. D. Thompson and D. Pines, Nature, 2008, 454, 611–613 CrossRef CAS PubMed.
- Y. Jiang, Y. N. Zhang, J. X. Cao, R. Q. Wu and W. Ho, Science, 2011, 333, 324 CrossRef CAS PubMed.
- J. Girovsky, J. Nowakowski, M. E. Ali, M. Baljozovic, H. R. Rossmann, T. Nijs, E. A. Aeby, S. Nowakowska, D. Siewert, G. Srivastava, C. Wäckerlin, J. Dreiser, S. Decurtins, S.-X. Liu, P. M. Oppeneer, T. A. Jung and N. Ballav, Nat. Commun., 2017, 8, 15388 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr08992c |
| This journal is © The Royal Society of Chemistry 2021 |