
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBayberry tannin directed assembly of a bifunctional graphene aerogel for simultaneous solar steam generation and marine uranium extraction†
Fangwu
Wu‡
ab,
Dan
Liu‡
cd,
Guangyong
Li
a,
Liqiang
Li
a,
Lifeng
Yan
 e,
Guo
Hong
*cd and
Xuetong
Zhang
*af
e,
Guo
Hong
*cd and
Xuetong
Zhang
*af
aSuzhou Institute of Nano-tech and Nano-bionics, Chinese Academy of Sciences, Suzhou, 215123, P.R. China. E-mail: xtzhang2013@sinano.ac.cn
bNano Science and Technology Institute, University of Science and Technology of China, Suzhou, 215123, P.R. China
cInstitute of Applied Physics and Materials Engineering, University of Macau, Avenida da Universidade, Taipa, Macau SAR, Macao. E-mail: ghong@um.edu.cn
dDepartment of Physics and Chemistry, Faculty of Science and Technology, University of Macau, Avenida da Universidade, Taipa, Macau SAR 999078, Macao
eCAS Key Laboratory of Soft Matter Chemistry, iChEM, and Department of Chemical Physics, University of Science and Technology of China, Hefei 230026, China
fDivision of Surgery & Interventional Science, University College London, London NW3 2PF, UK
First published on 10th February 2021
Abstract
Solar steam generation and marine uranium extraction are promising methods for obtaining sufficient fresh water and nuclear fuel from the ocean, respectively, for overcoming water and energy crises. In this work, a bayberry tannin (BT) directed assembly of a bifunctional graphene aerogel (GA) has been designed for simultaneous solar steam generation (1.80 kg m−2 h−1 with a high solar efficiency of 95.5%) and marine uranium extraction (230.10 mg g−1 within 6 h). BT molecules are uniformly decorated inside the typical porous channels of GA, which integrates the excellent uranium binding of BT and the efficient light-to-heat conversion of GA. It is found that the hydrophilic nature of BT can improve fluid infiltration in the GA matrix for solar steam generation while the steam generation induced transpiration can accelerate the adsorption of uranium ions for marine uranium extraction. The unique bifunctional ability of the BT-GA composite paves a new way to utilize the abundant resources in the ocean.
Introduction
The sustainable development of fresh water and energy resources is the most urgent issue for humanity in the 21st century.1,2 Even though 70% of our planet's surface is covered with water, fresh water contributes only about 2.5% to the total amount while most of it cannot be used directly.3 On the other hand, nuclear energy is the most promising solution for energy shortage, and can provide base-load electricity on a large scale without greenhouse gas emission. The primary nuclear fuel, uranium, mainly exists in ores, salts and seawater. Specifically, there are 4.5 billion tons of uranium resources in the ocean and only 27 million tons are stored in terrestrial ores and salts.4 Considering a large amount of water and uranium storage in the ocean, freshwater regeneration and uranium extraction are becoming more and more attractive.For these purposes, a variety of methods and functional materials have been developed inconsistently. For freshwater regeneration, evaporation-enabled water desalination and purification are rapidly developed because of the high purity. As conventional water evaporation methods are usually powered by fossil fuels that produce environmentally harmful by-products, solar steam generation (SSG) stands out by utilizing solar energy.5–7 Through focusing on the phase change process of water, materials and structures with efficient solar-to-heat conversion and high heat utilization have been developed.8 For uranium extraction, the immobilization of a specific chemical group on porous materials is the most promising method.9,10 Plenty of porous materials have been utilized, such as mesoporous silica,11,12 polymers,13,14 carbon nanomaterials,15 MOFs,16 and even MXene.17 Much progress has been made in the improvement of the performance of the SSG and uranium extraction. However, there are still two problems that should be addressed: firstly, in order to graft specific chemical groups on porous materials, the preparation of most functional materials requires a complex chemical synthesis process. Secondly, porous materials have been used in both SSG and uranium extraction, but the lack of materials which have both functions is still an issue that remains unsolved. Therefore, it is necessary and valuable to develop a low cost and simple method to fabricate bifunctional porous materials for SSG and marine uranium extraction simultaneously.
Bayberry tannin (BT) has been recently reported as an excellent candidate for uranium extraction, and can be directly extracted from the bark of Myrica esculenta by a low-cost and environment-friendly method. It shows good selectivity and considerable adsorption capacity toward uranium ions by forming a chelate with it.18 However, bayberry tannins cannot be directly used in seawater and due to their water-soluble nature, an additional water resistant substrate is always required.19–22 Graphene aerogel (GA) has an inherent interconnected porous structure, which is suitable as the substrate of bayberry tannin. Moreover, graphene has been extensively studied for its excellent photothermal conversion ability.23–26 Its aerogel structure provides a wicking channel for water supply, facilitates steam escape, and plays a role in thermal insulation,27,28 and is one of the most promising structures for SSG. Therefore, it can be inferred that by grafting bayberry tannin on graphene aerogel it is possible to achieve both SSG and uranium extraction functions. In addition, graphene aerogel normally requires additional hydrophilization treatment to make it more hydrophilic due to the hydrophobic properties of graphene.29,30 Bayberry tannin is supposed to solve this problem since it is hydrophilic.
Herein, a novel bifunctional graphene aerogel (BGA) is designed and fabricated by integrating bayberry tannin and graphene aerogel, which simultaneously realizes solar steam generation and marine uranium extraction for the first time (Scheme 1a). The BGA combines the excellent solar-thermal conversion property and the unique porous structure of graphene aerogel with the uranium extraction ability of BT. Furthermore, BT endows BGA with excellent hydrophilicity, which facilitates water transport during solar steam generation, and rGO behaves as an excellent carrier of BT to adsorb uranium from seawater. During the sol–gel step in the fabrication process, no additional reducing agent is required as BT has the ability to reduce graphene oxide. It is also found that there is a synergistic effect between solar steam generation and marine uranium extraction in BGA. The increase of temperature during solar steam generation accelerates the diffusion rate31 of UO22+, leading to a faster uranium adsorption rate. Therefore, the water evaporation rate of BGA is up to 1.84 kg m−2 h−1 under one sun, and the equilibrium adsorption capacity of uranium of BGA reaches 230.10 mg g−1 within 6 h at the same time, which provides a new strategy to utilize seawater and has great prospects in the forthcoming solar steam generation as well as marine uranium extraction.
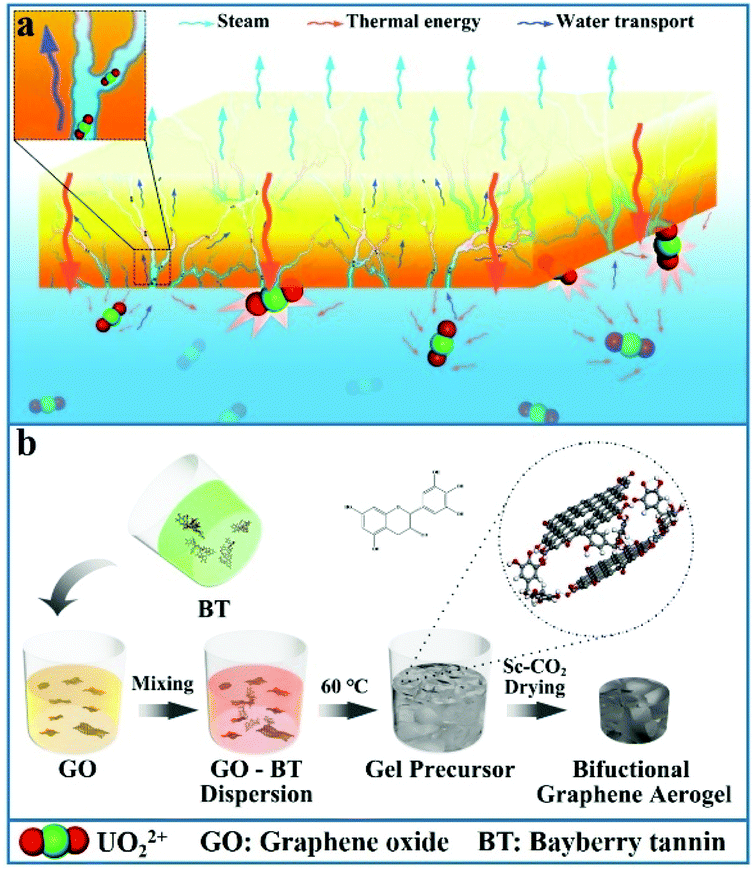 | ||
| Scheme 1 Schematic diagram of (a) simultaneous solar steam generation and uranium extraction based on the BGA and (b) the preparation processes of the BGA. | ||
Results and discussion
Fabrication of BGA
The synthetic pathway of BGA is shown in Scheme 1b. The colloidal dispersion of BT is firstly obtained through a simple dissolution process (Fig. S1†), which is stable because of its numerous hydrophilic groups. Then, the mixed dispersion of bayberry tannin and graphene oxide is prepared by mild stirring, meanwhile, the temperature of the mixed dispersion is maintained at 60 °C for 24 h to achieve complete sol–gel transition. During the sol–gel process, GO is slowly reduced by BT. After a hydrogen-bonded network is formed inside, the gel precursor of BGA is obtained. Finally, the light weight BGA is obtained by solvent replacement and supercritical CO2 drying (which is denoted as SC-CO2) (Fig. 1a). | ||
| Fig. 1 Optical images of the (a) BGA on Gypsophila paniculata and the (b) SEM image of the BGA (c) the strain–stress curve of BGA. (d) Nitrogen adsorption and desorption isotherms. The inset is the pore size distribution of the BGA fabricated with BT to GO feed mass ratios of 1/4, 1/2, 1, 2, 3 and 4. (e) The curves of storage modulus (G′) and loss modulus (G′′) with frequency for the BGA gel precursor and GO (f) The curves of storage modulus (G′) and loss modulus (G′′) with frequency for BGA gel precursor fabricated at different pH. | ||
The concentration of raw materials (BT and GO) and their mass feeding ratio are essential for the formation of the BGA gel precursor. Fig. S2† shows a series of optical images of BGA gel precursors fabricated with the mass feeding ratios of BT to GO from 1/8 to 4/1. Here, the concentration of BT is changed and the concentration of GO is maintained at 8 mg ml−1. For different ratios, the corresponding curves of storage modulus (G′) and loss modulus (G′′) with frequency under the shear force are shown in Fig. 2e and Fig. S3.† It is found that gel precursors with a stable shape can be obtained when the mass feeding ratio of BT to GO is higher than (or equal to) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. The dynamic rheological frequency scanning shows that the G′ value is larger than the G′′ value in all cases when the mass feeding ratio is higher than (or equal to) 1:4, indicating that the deformation of gel precursors is mainly elastic and a complete gel network has been formed.32 For the gel precursor obtained at a mass ratio of 1:8, its G′ value is smaller than the G′′ value when the frequency is higher than 36 Hz, indicating that the gel precursor mainly undergoes viscous deformation. Its rheological behaviour is similar to pure GO and the G′ value is smaller than the G′′ value when the frequency is greater than 22 Hz, which means that the gel network is not formed. Besides, with the increase of the mass feeding ratio, the G′ value increases and shows a peak at 3:1. When the feeding ratio is more than 3:1, the G′ value decreases slightly at high frequency, which may be ascribed to the adverse effect of excess BT on the stability of the gel network. The optical images of BGAs fabricated with different feeding mass ratios are illustrated in Fig. S4.† It can be intuitively seen that with the increase of the amount of BT, the shrinkage rate of BGA is becoming smaller, indicating that the anti-compression performance of BGA is becoming better. For the BGA fabricated with a BT concentration lower than 1 mg ml−1, the gel network is not stable enough. During the process of solvent replacement in anhydrous ethanol, part of the cross-linked network would be destroyed due to the elution of free BT, resulting in the collapse of the gel structure and a certain degree of contraction. When the BT concentration is higher than 1 mg ml−1, the gel network becomes stable enough to resist the deformation, and the structure of BGA fabricated at a mass feeding ratio of 3:1 is proved to be the most stable. The mechanical properties of BGA are shown in Fig. 1c, the BGA can maintain structural integrity even under 70% strain, and Young's modulus of BGA is up to 5.42 MPa. As shown in Fig. S5a,† the concentration of GO is also important for the formation of the BGA gel precursor. When the concentration of GO is lower than 4 mg ml−1, the gel network is not stable even if the BT concentration is up to 40 mg ml−1, which is further confirmed by corresponding dynamic rheological frequency scanning, as shown in Fig. S5b.†
:4. The dynamic rheological frequency scanning shows that the G′ value is larger than the G′′ value in all cases when the mass feeding ratio is higher than (or equal to) 1:4, indicating that the deformation of gel precursors is mainly elastic and a complete gel network has been formed.32 For the gel precursor obtained at a mass ratio of 1:8, its G′ value is smaller than the G′′ value when the frequency is higher than 36 Hz, indicating that the gel precursor mainly undergoes viscous deformation. Its rheological behaviour is similar to pure GO and the G′ value is smaller than the G′′ value when the frequency is greater than 22 Hz, which means that the gel network is not formed. Besides, with the increase of the mass feeding ratio, the G′ value increases and shows a peak at 3:1. When the feeding ratio is more than 3:1, the G′ value decreases slightly at high frequency, which may be ascribed to the adverse effect of excess BT on the stability of the gel network. The optical images of BGAs fabricated with different feeding mass ratios are illustrated in Fig. S4.† It can be intuitively seen that with the increase of the amount of BT, the shrinkage rate of BGA is becoming smaller, indicating that the anti-compression performance of BGA is becoming better. For the BGA fabricated with a BT concentration lower than 1 mg ml−1, the gel network is not stable enough. During the process of solvent replacement in anhydrous ethanol, part of the cross-linked network would be destroyed due to the elution of free BT, resulting in the collapse of the gel structure and a certain degree of contraction. When the BT concentration is higher than 1 mg ml−1, the gel network becomes stable enough to resist the deformation, and the structure of BGA fabricated at a mass feeding ratio of 3:1 is proved to be the most stable. The mechanical properties of BGA are shown in Fig. 1c, the BGA can maintain structural integrity even under 70% strain, and Young's modulus of BGA is up to 5.42 MPa. As shown in Fig. S5a,† the concentration of GO is also important for the formation of the BGA gel precursor. When the concentration of GO is lower than 4 mg ml−1, the gel network is not stable even if the BT concentration is up to 40 mg ml−1, which is further confirmed by corresponding dynamic rheological frequency scanning, as shown in Fig. S5b.†
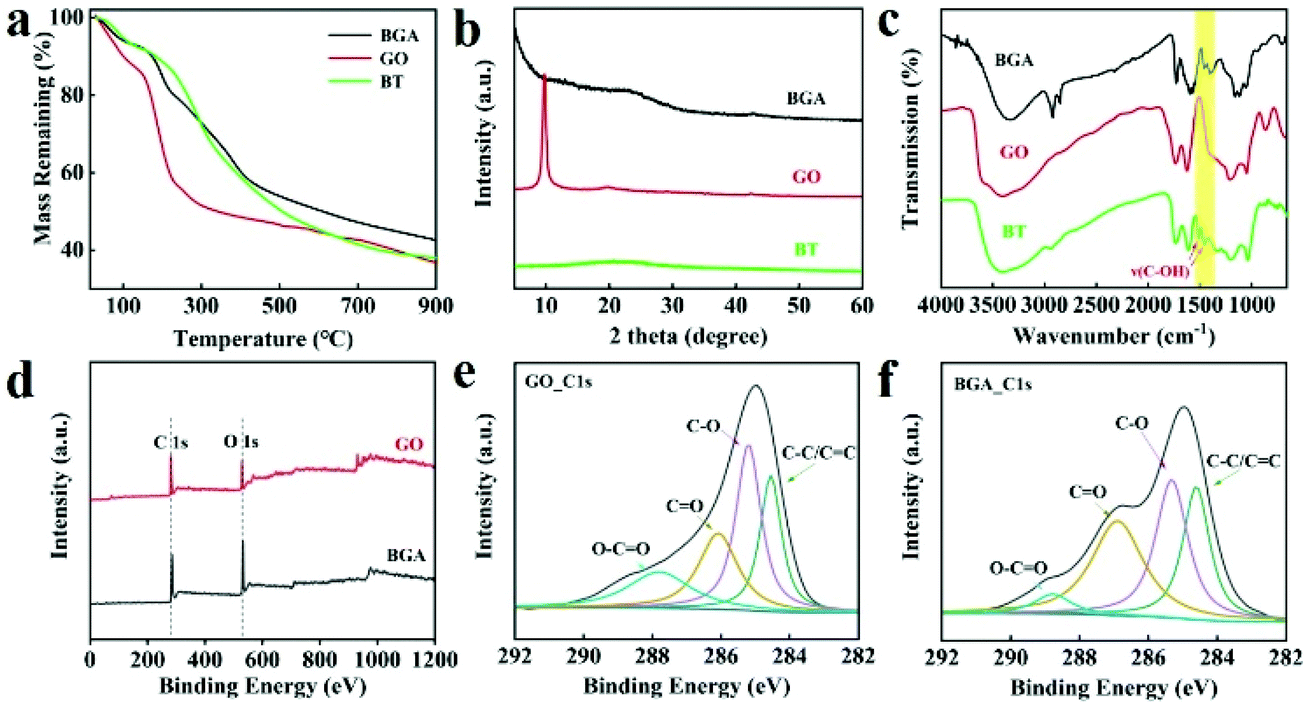 | ||
| Fig. 2 (a) TG curves of the BGA fabricated with different mass feed ratios of BT to GO. (b) XRD patterns of the BGA, GO, and BT. (c) IR spectra of the BGA, GO, and BT. (d) XPS spectra of the BGA and GO. (e) XPS C 1s spectra of GO. and (f) XPS C 1s spectra of BGA (unless otherwise specified, BGA in the figure refers to the aerogels obtained when the mass feed ratio of BT and GO is 3:1). | ||
The pH value of the mixed dispersion is also important for the formation of the BGA gel precursor. As seen from Fig. S6,† the gel cannot be formed when the pH is higher than 5. The G′ value is larger than the G′′ value when the pH is 8, but their difference is close to zero at 50 Hz. Moreover, the G′ and G′′ values of the gel precursor at pH = 8 are much smaller than that at pH = 2, indicating that the gel network is more stable at a low pH value. The reason is supposed to be that protons are dissociated from BT and GO under high pH, and then the number of hydrogen bonds between GO and BT sharply decreases, leading to the failure of gel formation. This phenomenon indicates that hydrogen bonding is necessary for the formation of BGA.
The microstructure of BGA is revealed by field emission scanning electron microscopy (FESEM). As shown in Fig. 1b and Fig. S7 and S8,† BGA has a large number of micropores, which is typical of an aerogel structure. Furthermore, there are no apparent BT aggregates observed, indicating that BTs are uniformly distributed inside BGA. The nitrogen adsorption and desorption test are used to analyse the pore structure of BGA. As illustrated in Fig. 1d, the nitrogen adsorption–desorption curve has a significant hysteresis loop, which is a typical type IV adsorption–desorption curve. The pore size distribution curve demonstrates that BGA has an open pore structure and consists of a large number of mesopores and some micropores. The BET specific surface area of BGA is concentrated in 300–400 m2 g−1, which is similar to most of the GAs reported (Table S1†). The pore structures of BGAs fabricated with different ratios of BT to GO have been analysed in Table S2.† The specific surface area of BGA decreases as the content of BT increases, but it is still very high. It should be noted that the specific surface area of BGA is lower than 300 m2 g−1 when the BT to GO mass feeding ratio is higher than 4.0. The possible reason is that fewer pores are formed and some of them are occupied by excess BTs. The rich pores and high specific surface area of BGA confirm its aerogel nature.
To obtain a deep understanding of the role of BT in the formation of BGA, thermogravimetric (TG) curves of BGA, GO, and BT are obtained, which are shown in Fig. 2a. It can be found that the BGA is more stable than GO and BT. And the residual mass of BGA is higher than those of GO and BT, indicating that GO has been reduced by BT. To further confirm the reduction of GO, the XRD spectra of GO and BGA are tested, which are shown in Fig. 2b. For GO, the diffraction peak at 2θ = 9.8° belongs to the (002) crystal plane, and the interplanar distance is about 0.90 nm. The peak is sharp with high intensity, indicating that most of the GO are high quality with fewer layers. For the BGA, the diffraction peak of the (002) plane shifts to 22.9°, which is close to the peak of natural graphite at about 26.2°. Compared with GO, the intensity and width of BGA's peak are lower and wider. The interplanar distance of the (002) plane in BGA is reduced to 0.39 nm, indicating that GO sheets are well assembled, and the oxygen-containing functional groups of GO are partially reduced.33 The oxygen-containing functional groups in GO before and after reduction were characterized by XPS, and the result is shown in Fig. 2d–f. The carbon–oxygen ratio increased from 1.03 to 1.36 after reduction, demonstrating that GO is reduced by BT during the preparation of BGA.34 The Raman spectra of BGA is also measured, which is shown in Fig. S9.† It can be seen that the average D/G ratio of BGA is higher than that of GO, indicating that GO is reduced by BT. Based on the chemical structure of BT, a possible reduction mechanism is proposed in Fig. S10.† The phenolic hydroxyl of BT reacts with the hydroxyl group and epoxy bond of GO, so some of the C–C bonds in graphene oxide are restored to the conjugated double bond.35,36
The characteristics of the functional groups in BGA are explored by infrared spectroscopy. As shown in Fig. 2c and Fig. S11,† BGA has a double peak at around 1500 cm−1, which corresponds to the stretching vibration peak of phenolic hydroxyl, the one that is originally [ν(C–OH)] and belongs to BT.17 The peak shifted to low wavenumber compared with BT, indicating that hydrogen bonds are formed between BTs and rGO.37,38 The hydrogen bond immobilizes BTs in BGA, which guarantees its function of uranium extraction. The immobilization of BTs in BGA is further confirmed by the Prussian blue method, which is a simple and effective method to detect the phenolic hydroxyl groups.39 Specifically, when BGA is added to the acidic Fe3+/[Fe(CN)6]3+ solution, the colour of the solution slowly changes from yellow to green and finally the colour becomes dark blue (Fig. S12†). The slow change of solution colour demonstrates that phenolic hydroxyl groups are not in a free state, which supports that there are hydrogen bonds between BTs and rGO inside BGA. Thus, BT not only reduces GO but also works as cross-linking agent during the formation of BGA.
As is well known, hydrophilicity is essential for materials used in solar steam generation.40 To explore the hydrophilicity of BGA, five kinds of graphene aerogels, including BGA, are prepared with the same batch of graphene oxide by using different reduction methods, which are hydrothermal, hydrazine hydrate, hydrogen iodide, ascorbic acid, and bayberry tannin, respectively. Their contact angles are shown in Fig. 3a. It can be seen that the contact angle of BGA is the smallest and close to 0°, indicating its strong hydrophilicity, which facilitates solar steam generation. The surface of BGA is hydrophilic because the immobilized BT has a number of oxygen-containing groups. The result further demonstrates that the BT binds with rGO inside BGA. To demonstrate the importance of hydrophilicity, the water transport performance of BGA and the other four graphene aerogels is tested, which is shown in Fig. 3b and Fig. S13.† The water transport rate (V, g min−1) is calculated by eqn (1):
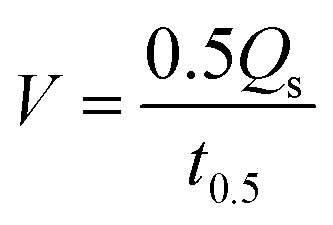 | (1) |
 | ||
| Fig. 3 (a) Contact angles of rGO films prepared by different reduction methods and (b) the variation of the water content of graphene aerogels prepared by different reduction methods. (c) UV-vis-NIR spectra of the BGA, GO, and BT films with 1 mm thickness. (d) Temperature–time curves of the top and bottom surface of the BGA and bulk water under one sun illumination. (e) Water evaporation rate of BGA and blank sample under different conditions, including darkness and one sun illumination. (f) Water evaporation rate and solar energy conversion efficiency of the BGA with different thicknesses. | ||
Solar steam generation performance of BGA
Solar steam generation (SSG) can be divided into two processes, namely photothermal conversion and thermal evaporation. In order to improve the performance of SSG, the absorber should possess high light absorption ability to ensure effective light-heat conversion, and the heat must be effectively utilized to evaporate water. To evaluate the SSG performance of BGA, the solar adsorption ability of BGA is firstly investigated by the UV-vis-NIR spectrum. As shown in Fig. 3c, the average absorptivity of BGA can reach 93.5% in the wavelength range from 200 to 2500 nm, which is higher than those of GO and BT. There are two reasons resulting in high light absorption of BGA. One reason is that the rGO inside BGA has continuous energy levels arising from the hybrid bonds, leading to broadband light absorption. The other reason is that the porous structure of BGA effectively decreases the interface reflection. The photothermal conversion of BGA is evaluated by monitoring the temperature of the upper and lower surface of BGA (the height is 5 mm) under one sun illumination. As shown in Fig. S14,† the temperature of the upper surface increases rapidly and reaches 46 °C in 60 s, while the temperature of the lower surface is only 36 °C in the same time. After 60 s, the temperature difference between the upper and lower surface becomes stable at about 10 °C. The high temperature of BGA after 60 s illumination indicates that BGA can efficiently convert solar light energy into thermal energy.41 The constant temperature difference between the upper and lower surface of BGA results from the low thermal conductivity of BGA. The same experiment has been conducted when BGA is placed in water. As shown in Fig. 3d, the temperature of the upper surface increases rapidly from 25 °C to 45 °C in 2 minutes and is maintained constant at 50 °C after 10 minutes. The temperature of the lower surface increases to 30 °C in 2 minutes and is maintained constant at 33 °C after 10 minutes. The temperature difference between the upper and lower surface is up to 17 °C, which is higher than that when BGA is placed in air. Meanwhile, the temperature of bulk water increases slowly, which is lower than 31 °C even after one hour. This phenomenon shows that there is almost no heat transfer from BGA to bulk water. The equilibrium temperature and heating rate in water are lower than the ones in air, demonstrating that the solar energy absorbed by BGA can be efficiently transferred to the water interface. This facilitates solar steam generation, as water evaporation mainly occurs at the interface between water and air.To evaluate the SSG performance of BGA, water evaporation rates of BGA and the blank sample under light or dark are measured, which is shown in Fig. 3e. For the blank sample, the water evaporation rate is 0.13 kg/(m2 h) in the dark and 0.36 kg m−2 h−1 in light, respectively. For BGA, the water evaporation rate is up to 0.38 kg m−2 h−1 in the dark and reaches 1.84 kg m−2 h−1 in light, which is one of the best performances among GA based materials (Table S3†). The water evaporation rates increase nearly three fold in the dark and five fold under one sun illumination compared with the blank sample. The reason is that BGA has rich interfaces with strong hydrophilicity, which provides a larger area for water evaporation and a higher water transport rate. The high rate of water evaporation of BGA demonstrates that it is suitable for solar steam generation. The performance of the BGA gel precursor is further measured to confirm the advantage of BGA, which is shown in Fig. S15.† The water evaporation rate of the BGA gel precursor is about 1.54 kg m−2 h−1 under one sun illumination, which is lower than that of BGA. The possible reason is that a considerable amount of bound water in the BGA gel precursor leads to a lower water transport rate. To investigate the effect of the drying method on SSG performance, the BGA fabricated by freeze-drying (this kind of BGA is denoted as F-BGA) is prepared. As shown in Fig. S16,† the water evaporation rate of F-BGA is 1.43 kg m−2 h−1, which is even lower than that of the BGA gel precursor. As seen in Fig. S17a and Table S2,† the BET specific surface area of F-BGA is 236.85 m2 g−1, which is much lower than that of BGA fabricated by SC-CO2, and the average pore size is 15.45 nm, which is larger than that of BGA fabricated by SC-CO2. It can be inferred that the growth of ice crystals during the process of freeze-drying is supposed to destroy a part of the channel structure and introduce some macropores (Fig. S17b†), leading to a decrease in the water evaporation rate.
The SSG performance is also affected by the thickness of BGA. BGAs with different thicknesses are prepared and their SSG performance is evaluated. The corresponding energy conversion efficiency is calculated by eqn (2) (see the Experimental section). As shown in Fig. 3f, the water evaporation rate is the highest when the thickness of the BGA is 5 mm. When it becomes thinner, the thermal insulation of BGA becomes worse. A large amount of heat is transferred to bulk water, resulting in energy loss and a lower water evaporation rate. When the thickness is more than 5 mm, the speed of water transport decreases due to the longer transport distance. The solar steam efficiency is evaluated using η = mhLV/Pin, in which m is the mass flux, hLV is the liquid vapor phase change enthalpy, and Pin is the received power density of the solar irradiations on the BGA surface. The result shows that solar energy conversion efficiency has the same trend as the water evaporation rate and reach 95.5% when the thickness is 5 mm. The heat from the environment could be ignored in our systems because the temperature of BGA is much higher than the temperature of the environment.42,43
As there are many ions in seawater, it is of considerable significance to explore the performance of BGA in high ion concentration water. The water evaporation rate of BGA in different concentrations of sodium chloride aqueous solution is measured. As shown in Fig. S18a,† even in 5% sodium chloride aqueous solution, the water evaporation rate of BGA is maintained at about 1.72 kg m−2 h−1 for 4 hours without an apparent decrease. The corresponding optical images of BGA at different times demonstrate that there are no salt crystals on the upper surface, indicating that the passage of water in BGA is not blocked at high ion concentrations (Fig. S18b†). As is well known, the highest salinity area in the world is the Red Sea, and its salinity is less than 5%. Therefore, BGA is suitable for solar steam generation in the marine environment.
Uranium extraction performance of BGA
BT exhibits a strong and specific binding with UO22+. The BT immobilized in BGA can absorb UO22+ by forming chelate bonds because the 4s/6d empty orbitals of UO22+ are able to accept free electron pairs of the phenolic oxygen atom (the inset of Fig. 4b). For the convenience of description, the BGA after the adsorption of UO22+ is defined as U-BGA. The infrared spectrum of U-BGA shows a peak at 931.76 cm−1 (Fig. S19†), which is attributed to the vibrational adsorption of [O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UO]2+. It can be found that the peak has an obvious red shift compared with that of UO2(NO3)2·6H2O (∼960 cm−1),44 which is supposed to be the effect of the chelating bond between the uranyl ion and BGA. After UO22+ adsorption, the pore structure of U-BGA is still maintained (Fig. S7d†), indicating that BGA has robust foam resistance. From the EDS mapping of U-BGA, it can be seen that uranium coexists with carbon and oxygen. Uranium is uniformly distributed inside BGA, demonstrating that UO22+ is uniformly absorbed by BGA. The result also confirms that bayberry tannins are uniformly distributed in BGA (Fig. 4a).
UO]2+. It can be found that the peak has an obvious red shift compared with that of UO2(NO3)2·6H2O (∼960 cm−1),44 which is supposed to be the effect of the chelating bond between the uranyl ion and BGA. After UO22+ adsorption, the pore structure of U-BGA is still maintained (Fig. S7d†), indicating that BGA has robust foam resistance. From the EDS mapping of U-BGA, it can be seen that uranium coexists with carbon and oxygen. Uranium is uniformly distributed inside BGA, demonstrating that UO22+ is uniformly absorbed by BGA. The result also confirms that bayberry tannins are uniformly distributed in BGA (Fig. 4a).
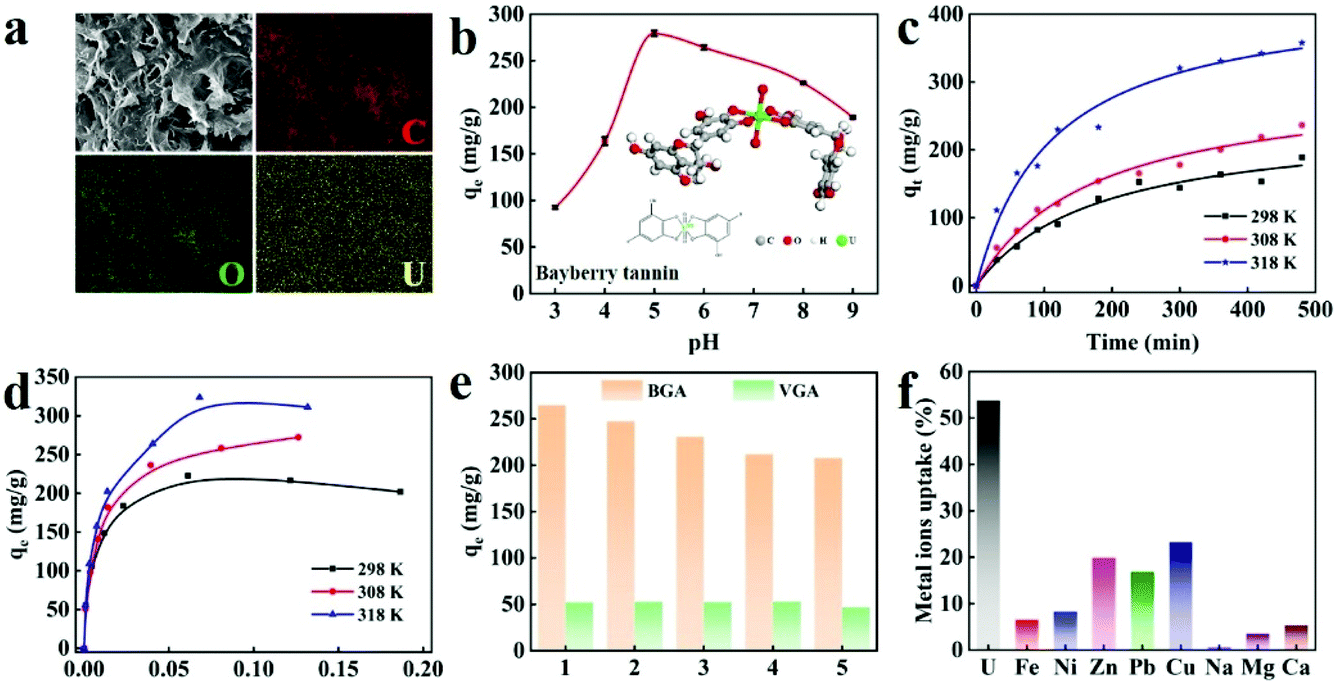 | ||
| Fig. 4 (a) The SEM image and EDS mapping of elements of U-BGA (the scale bar is 1 μm). (b) Effect of initial pH on the uranium adsorption capacity (initial uranium concentration: 0.25 mg mL−1, V m−1 ratio: 2000 mL g−1, temperature 298 K). The inset is the mechanism of uranium adsorption of BGA. (c) Uranium adsorption kinetics of BGA at different temperatures (initial uranium concentration: 0.25 mg mL−1, pH 5.0, V m−1 ratio: 2000 mL g−1). (d) Uranium adsorption isotherms of BGA at different temperatures (initial uranium concentration: 0.025–0.25 mg mL−1, pH 5.0, V m−1 ratio: 2000 mL g−1). (e) Cycle performance of BGA and VGA for uranium extraction at 298 K (initial uranium concentration: 0.25 mg mL−1, pH 5.0, V m−1 ratio: 2000 mL g−1). (f) The adsorption capacity of BGA towards different metal ions (the ratios of different metal ions are the same as that in natural seawater but the corresponding concentration is one hundred times that in natural seawater) (V m−1 ratio: 50000 mL g−1, temperature 298 K). | ||
The adsorption of metal ions is influenced by the initial pH of the solution. Thus, the adsorption of UO22+ by BGA at different pH is investigated (see formula (3) in the Experimental section). As shown in Fig. 4c, the UO22+ adsorption capacity of BGA increases rapidly with the increase of pH from 3 to 5 and decreases slightly under neutral and alkaline pH. The capacity of BGA is the highest when the pH is 5.0. The reason can be explained as follows. At lower pH values, the concentration of hydrogen ions is high. The adsorption sites, such as phenolic hydroxyl, are easily protonated, leading to electrostatic repulsion between the positively charged UO22+ and the protonated BGA. At higher pH values, UO22+ is converted into insoluble species (UO2(OH)2), which greatly decreases the adsorption capacity. Therefore, the following uranium adsorption experiments are conducted at pH = 5.0.
To investigate the UO22+ adsorption process of the BGA, the kinetic curves are measured at 298 K (Fig. S20†). It can be observed that the adsorption rate of UO22+ is very fast in the initial 300 minutes and the adsorption capacity of BGA reaches 279.10 mg g−1. The adsorption capacity is comparable with other typical uranium absorbents (Table S4†) and the adsorption rate is better than most of the uranium absorbents (Table S5†). The fast adsorption can be attributed to the unoccupied active chelating sites on the BGA in the initial stage. As the adsorption process continues, the number of active chelating sites gradually decreases until the adsorption reaches equilibrium and the adsorption rate decreases. In order to understand the mechanism of UO22+ adsorption, pseudo-first-order and pseudo-second-order models are employed for fitting the obtained kinetic data. The detailed fitting processes can be found in Fig. S20, S21 and Table S6.† For the BGA, the correlation coefficient of the pseudo-second-order kinetic model is higher, meaning that the UO22+ adsorption behaviour of BGA can be described by the pseudo-second-order kinetic model. Thus, the rate-limiting step is the chemical adsorption, that is, BT inside BGA exchanges electrons with uranium to form a coordination bond.45 Besides, the adsorption capacity of uranium increases with the increase in temperature. This indicates that high temperature promotes the thermal movement of uranyl ions and enhances the interaction between UO22+ and BGA.46
To understand the basic physicochemical behaviour during the adsorption process, the kinetic and adsorption isotherm curves at different temperatures are obtained (Fig. 4c and d), and are analysed using both Langmuir and Freundlich models (Fig. S22 and Table S7†). The experimental data are well consistent with the Langmuir model, explaining that the adsorption process is dominated by monolayer chemical adsorption.47 The cycling performance of BGA (shown in Fig. 4e) is also evaluated. The uranium adsorption capacity of BGA slightly decreases in the initial three cycles and then becomes stable. In comparison, the graphene aerogel prepared using vitamin C shows stable cycle performance with low capacity owing to physical adsorption. This further illustrates that the uranium adsorption of BGA is dominated by chemical adsorption, due to the specific interaction between BT and the uranium ion.
There are many competitive metal ions in seawater, which makes uranium extraction difficult. Thus, the ion adsorption selectivity is very important for uranium extraction from seawater. To determine the uranium ion selectivity of BGA, adsorption properties of Fe3+, Ca2+, Na+, and other coexisting ions in seawater are measured, and their competitive adsorptions with UO22+ are compared. For the convenience of measurement, the concentrations of UO22+ and other trace ions are one hundred times that in natural seawater (the detailed ion concentrations are listed in Table S8†). The results are shown in Fig. 4f. It can be seen that the extraction rate of UO22+ can still reach 54% even in the presence of various interfering ions. However, the single enrichment of uranium and vanadium is still a challenging problem due to the similar properties of uranium and vanadium.46,48 It is worth noting that although the content of Na+, Mg2+, and Ca2+ is much higher than that of UO22+, their extraction rate is very low. The results show that BGA possesses excellent selectivity for uranium, which is comparable with other typical materials.
The performance of BGA for simultaneous solar steam generation and marine uranium extraction
The simultaneous solar steam generation and uranium adsorption have been realized by BGA, as shown in Fig. 5a. It can be seen that the water evaporation rate is maintained at 1.80 kg m−2 h−1, and the adsorption capacity of UO22+ has reached 230.10 mg g−1 within 480 min at the same time. To explore the mutual effect between solar steam evaporation and uranium adsorption, a series of experiments have been conducted. Fig. 5b shows that the uranium adsorption curves of BGA under one sun illumination and in darkness. The adsorption rate under illumination is obviously higher than that in darkness due to higher temperature, indicating that the solar-thermal conversion of BGA is beneficial for uranium adsorption kinetics. As is well known, the uranium adsorption rate is greatly affected by the concentration and diffusion rate of uranium. Solar steam generation by BGA can not only increase the local concentration of uranium but also improve the diffusion rate of uranium, which greatly accelerates the adsorption of uranium. The photothermal effect of the BGA enhances the thermal motion of the uranyl ion near the adsorbent, which would increase the interaction between the uranium binding groups and the uranyl ions. The solar steam generation performance of BGA in uranium solution and deionized water is compared to confirm the effect of the uranium adsorption process, which is shown in Fig. 5d. The mass change of water in uranium solution is slightly lower than that in deionized water, indicating that solar steam generation is almost not affected by the adsorption process of UO22+.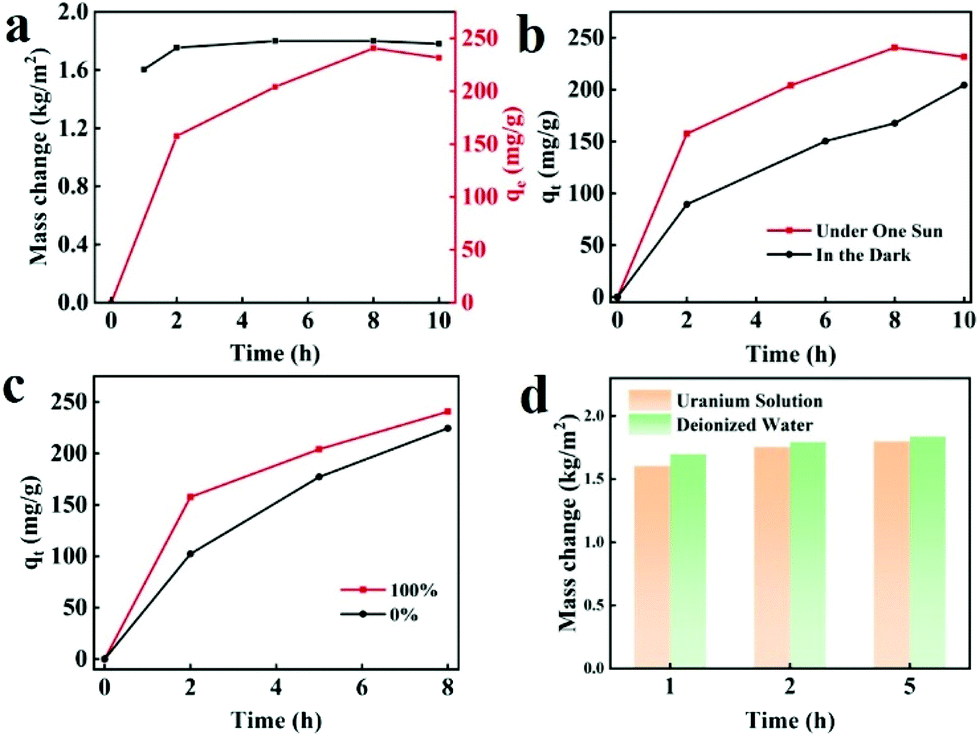 | ||
| Fig. 5 (a) The water evaporation rate and uranium adsorption kinetics of BGA. (b) Uranium adsorption kinetics of BGA under one sun illumination and in darkness. (c) Uranium adsorption kinetics of BGA with (100%) or without (0%) solar steam generation. (d) The water evaporation rate of BGA in deionized water and uranium solution. | ||
Conclusions
In summary, a novel bifunctional graphene aerogel has been designed and fabricated using bayberry tannin as both the reductant and cross-linker. The bayberry tannins are uniformly distributed on the highly porous structure of graphene aerogel, which improves the water affinity of BGA and endows BGA with the ability to absorb uranium. The BGA with a typical aerogel structure has high specific surface area (400 m2 g−1). The solar-thermal performance and uranium adsorption behaviours of BGA have been independently evaluated. The photothermal effect of BGA increases the temperature of the BGA–water interface and the steam induced transpiration, which enhances the thermal motion and diffusion rate of the uranyl ion near the adsorbent. The bifunction of BGA has been demonstrated. High-performance solar steam generation (up to 1.80 kg m−2 h−1) and uranium adsorption (up to 230.10 mg g−1 within 6 h) are simultaneously achieved, which provides a new solution for the crisis of water and energy.Experimental section
Materials
Bayberry tannin was provided by Prof. Xuepin Liao, Sichuan University, Chengdu, China. Graphite powder (1 μm) was purchased from Qingdao Tianheda Graphite Co., Ltd, Qingdao, China. Uranyl nitrate was purchased from Hubei Chushengwei Chemistry Co., Ltd, Wuhan, China. Chlorophosphonazo III was purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd, Shanghai, China. KMnO4, P2O5, K2S2O7, H2O2 (30%), HCl (37%), and H2SO4 were purchased from Sinopharm Chemical Reagent Co., Ltd, Shanghai, China. De-ionized water was used in all the experiments. All other reagents were used without further purification.Preparation of graphene oxide
Graphene oxide (GO) sheet was prepared from graphite powder by a modified Hummers method reported in our previous study.49Preparation of BGA
Bayberry tannin (BT) with a BT to GO mass ratio of 3:1 was added into a vial containing 4 mL 8.0 mg mL−1 GO dispersion. After continuous magnetic stirring, the resulting mixture was allowed to stand for 24 h in a 60 °C water bath. Following by washing at least four times with ethyl alcohol to replace water and supercritical drying with CO2 (40 °C, 10 MPa) for 12 h, the bifunctional graphene aerogel (BGA) was obtained.
Performance characterization of the BGA
Steam generation experiments were conducted at a temperature of 22 ± 2 °C and relative humidity of 50 ± 5% using a solar simulator (Newport 94043A) in which the light intensity was maintained at 1 kW m−2 (one sun illumination). The BGA with a diameter of 1.55 cm and thickness of 5 mm was floated on water in a cylindrical polystyrene container with an inner diameter of 2.7 cm (the exposed water surface was covered with PDMS) for solar steam generation. The mass change during water evaporation was measured using a lab balance (Quintix 224-1CN, 0.1 mg accuracy). All evaporation rates were measured under one sun illumination for 60 minutes after the light intensity stabilized. The corresponding energy conversion efficiency (η) was calculated according to the following formula: | (2) |
The adsorption experiments were performed in uranium (VI) solutions, which were prepared by dissolving uranyl nitrate in deionized water. 10 mg BGA was suspended in a 20 mL solution adjusted to pH 5.0 with the initial uranium(VI) concentration of 0.025–0.25 mg L−1, and adsorbed at constant ambient temperatures of 298 K, 308 K, and 318 K for 24 h. The change in the uranium concentration during the adsorption process was tracked using a UV-vis spectrophotometer (Agilent Cary5000, Palo Alto, CA, USA), and uranium adsorption capacity (qe, mg g−1) was calculated according to the following formula:
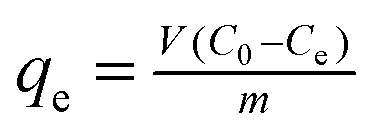 | (3) |
The bifunction verification experiments were conducted at a temperature of 22 ± 2 °C and relative humidity of 50 ± 5% using a solar simulator (Newport 94043A), in which the light intensity was maintained at 1.0 kW m−2 (one sun illumination). The BGA with the same size as used in solar steam generation investigation was floated on uranium (VI) solution (pH = 5.0, c = 0.25 mg L−1) in a cylindrical polystyrene container with the same size as mentioned above (the exposed water surface was covered with PDMS) for solar steam generation. The mass changed during water evaporation were measured by a lab balance (Quintix 224-1CN, 0.1 mg accuracy). All evaporation rates were measured under one sun illumination for 60 minutes after the light intensity stabilized.
The cycle performance of uranium adsorption of BGA has been determined using the following steps. The first step is the uranium adsorption on BGA. The BGA is placed in uranium solution at 298 K for 24 h. The second step is the elution of adsorbed uranium from BGA. The U-BGA is placed in 0.1 M HCl solution for 12 h at 298 K. The third step is freeze drying of BGA. The BGA is washed with deionized water to neutral pH and then dried by freeze drying. After this, the BGA is processed using the same steps.
Characterization
The morphology of the samples was examined by scanning electron microscopy (SEM) (Hitachi S-4800) with an acceleration voltage of 5–15 kV.The dynamic rheological measurements were recorded on a rotary rheometer (HAAKE RS6000) at 25 °C, and the storage modulus (G′) and loss modulus (G′′) of the gel precursors were measured at a frequency ranging from 0.1 to 100 Hz.
The pore size distribution and average pore diameter of the BGAs were analysed by the BJH nitrogen adsorption and desorption method (ASAP 2020, Micromeritics, USA). The surface areas of the BGAs were determined by the Brunauer–Emmett–Teller (BET) method, based on the amount of N2 adsorbed at pressures of 0.05 < P/P0 < 0.3
Thermal gravimetric analysis was carried out using a TG 209F1 Libra (NETZSCH) analyzer with a heating rate of 10 °C min−1 under a nitrogen atmosphere.
X-ray diffraction (XRD) patterns were recorded on a D8 Advanced spectrometer with a scanning rate of 0.05 s over an angular range of 5–60° (2θ).
IR spectra were studied with a Fourier transform infrared (FTIR) spectrophotometer (Nicolet 6700).
The contact angle was measured using an OCA 15EC, DataPhysics Instruments, GmbH.
The temperature–time curves were measured and recorded using a thermocouple and Keysight 34970 Data Acquisition system.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support from the Royal Society Newton Advanced Fellowship (NA170184), the National Key Research and Development Program of China (2016YFA0203301), the fund of the University of Macau (File No. SRG2017-00092-IAPME, MYRG2018-00079-IAPME, and MYRG2019-00115-IAPME), and the Science and Technology Development Fund, Macau SAR (File No. 081/2017/A2, 0059/2018/A2, 009/2017/AMJ, and 0092/2019/A2).Notes and references
- L. L. Zhu, C. F. Tan, M. M. Gao and G. W. Ho, Adv. Mater., 2015, 27, 7713 CrossRef CAS PubMed.
- C. W. Abney, R. T. Mayes, T. Saito and S. Dai, Chem. Rev., 2017, 117, 13935 CrossRef CAS PubMed.
- F. Gong, H. Li, W. B. Wang, J. G. Huang, D. W. Xia, J. X. Liao, M. Q. Wu and D. V. Papavassiliou, Nano Energy, 2019, 58, 322 CrossRef CAS.
- S. Das, A. K. Pandey, A. A. Athawale, M. Subramanianc, T. K. Seshagiri, P. K. Khanna and V. K. Manchanda, J. Hazard. Mater., 2011, 186, 2051 CrossRef CAS PubMed.
- M. Y. Tan, J. Wang, W. H. Song, J. H. Fang and X. T. Zhang, J. Mater. Chem. A, 2018, 7, 1244 RSC.
- B. Y. Gong, H. H. Yang, S. H. Wu, G. P. Xiong, J. H. Yan, K. F. Cen, Z. Bo and K. Ostrikov, Nano-Micro Lett., 2019, 11, 51 CrossRef PubMed.
- M. M. Gao, L. L. Zhu, C. K. Peh and G. W. Ho, Energy Environ. Sci., 2018, 12, 841 RSC.
- F. Zhao, Y. H. Guo, X. Y. Zhou, W. Shi and G. H. Yu, Nat. Rev. Mater., 2020, 5, 388 CrossRef.
- C. Abney, J. Gilhula, K. Lu and W. Lin, Adv. Mater., 2014, 26, 7993 CrossRef CAS PubMed.
- P. J. Lebed, J. D. Savoie, J. Florek, F. Bilodeau, D. Larivière and F. Kleitz, Chem. Mater., 2012, 24, 4166 CrossRef CAS.
- W. Zhang, G. Ye and J. Chen, J. Mater. Chem. A, 2013, 1, 12706 RSC.
- J. Li, J. Wang, W. Wang and X. T. Zhang, Molecules, 2019, 24, 1821 CrossRef PubMed.
- B. Yan, C. Ma, J. Gao, Y. H. Yuan and N. Wang, Adv. Mater., 2020, 32, 1906615 CrossRef CAS PubMed.
- J. L. Bahr and J. M. Tour, Chem. Mater., 2001, 13, 3823 CrossRef CAS.
- J. Górka, R. T. Mayes, L. Baggetto, G. M. Veith and S. Dai, J. Mater. Chem. A, 2012, 1, 3016 RSC.
- X. Min, W. Yang, Y. F. Hui, C. Y. Gao, S. Dang and Z. M. Sun, Chem. Commun., 2017, 53, 4199 RSC.
- I. Ihsanullah, Nano-Micro Lett., 2020, 12, 72 CrossRef CAS PubMed.
- V. U. Kavitha and B. Kandasubramanian, SN Appl. Sci., 2020, 2, 1081 CrossRef CAS.
- J. Meng;, X. Y. Lin, H. N. Li, Y. D. Zhang, J. Zhou, Y. Chen, R. Shang and X. G. Luo, RSC Adv., 2019, 9, 8091 RSC.
- J. Yu, X. G. Luo and B. Liu, J. Mater. Chem. A, 2018, 6, 15359 RSC.
- M. S. Alhumaimess, I. H. Alsohaimi, A. A. Alqadami, M. AliKhan, M. M. Kamel, O. Aldosari, M. R. Siddiqui and A. E. Hamedelniel, J. Mol. Liq., 2019, 281, 29 CrossRef CAS.
- W. Luo, G. Xiao, F. Tian, J. J. Richardson, Y. P. Wang, J. L. Guo, X. P. Liao and B. Shi, Energy Environ. Sci., 2019, 12, 607 RSC.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752 CrossRef CAS PubMed.
- P. Zhang, J. Li, L. Lv, Y. Zhao and L. T. Qu, ACS Nano, 2017, 11, 5087 CrossRef CAS PubMed.
- P. Zhang, F. Liu, Q. Liao, H. Z. Yao, H. Y. Geng, H. H. Cheng, C. Li and L. T. Qu, Angew. Chem., Int. Ed., 2018, 57, 16343 CrossRef CAS PubMed.
- M. Mohsenpour, S. Motahari, F. Tajabadi and M. Najaf, RSC Adv., 2020, 10, 41780 RSC.
- X. Hu, W. Xu, L. Zhou, X. Q. Li, L. Zhou, S. N. Zhu and J. Zhu, Adv. Mater., 2017, 29, 1606762 CrossRef PubMed.
- Y. Fu, G. Wang, X. Ming, X. G. Liu, B. F. Hou, T. Mei, J. H. Li, J. Y. Wang and X. B. Wang, Carbon, 2018, 130, 250 CrossRef CAS.
- X. Zhou, F. Zhao, Y. Guo, Y. Zhang and G. H. Yu, Energy Environ. Sci., 2018, 11, 1985 RSC.
- J. L. Yang, Y. S. Pang, W. X. Huang, S. K. Shaw, J. Schiffbauer, M. A. Pillers, X. Mu, S. R. Luo, T. Zhang, Y. J. Huang, G. X. Li, S. Ptasinska and M. Lieberman, ACS Nano, 2017, 11, 5510 CrossRef CAS PubMed.
- Y. Xie, C. L. Chen, X. M. Ren, X. X. Wang, H. Y. Wang and X. K. Wang, Prog. Mater. Sci., 2019, 103, 180 CrossRef CAS.
- T. Jiang, Z. Y. Sui, Q. S. Yang;, X. T. Zhang and B. H. Han, Soft Matter, 2015, 11, 3215 RSC.
- G. Y. Li, X. T. Zhang, J. Wang and J. H. Fang, J. Mater. Chem. A, 2016, 4, 17042 RSC.
- Z. Y. Sui, X. T. Zhang, Y. Lei and Y. J. Luo, Carbon, 2011, 49, 4314 CrossRef CAS.
- Y. D. Lei, Z. H. Tang, R. J. Liao and B. C. Guo, Green Chem., 2011, 13, 1655 RSC.
- O. Akhavan, M. Kalaee, Z. S. Alavi, S. M. A. Ghiasia and A. Esfandiar, Carbon, 2012, 50, 3015 CrossRef CAS.
- K. H. Thebo, X. T. Qian, Q. Zhang, L. Chen, H. M. Cheng and W. C. Ren, Nat. Commun., 2018, 9, 1486 CrossRef PubMed.
- Z. R. Yu, Z. X. Shi, H. J. Xu, X. D. Ma, M. Tian and J. Yin, Carbon, 2017, 114, 649 CrossRef CAS.
- C. Zhao, S. X. Xie, Y. Q. Pu, R. Zhang, F. Huang, A. J. Ragauskas and J. S. Yuan, Green Chem., 2016, 18, 1306 RSC.
- L. L. Zhu, M. M. Gao, C. K. N. Peh and G. W. Ho, Nano Energy, 2019, 57, 507 CrossRef CAS.
- H. Y. Ren, M. Tang, B. Guan, K. X. Wang, J. W. Yang, F. F. Wang, M. Z. Wang, J. Y. Shan, Z. L. Chen, D. Wei, H. L. Peng and Z. L. Liu, Adv. Mater., 2017, 29, 1702590 CrossRef PubMed.
- M. M. Gao, L. L. Zhu, C. K. Peh and G. W. Ho, Energy Environ. Sci., 2019, 12, 841 RSC.
- H. Q. Wang, C. Zhang, Z. H. Zhang, B. Zhou, J. Shen and A. Du, Adv. Funct. Mater., 2020, 29, 2005513 CrossRef.
- Q. Sun, B. Aguila, J. Perman, A. S. Ivanov, V. S. Bryantsev, L. D. Earl, C. W. Abney, L. Wojtas and S. Q. Ma, Nat. Commun., 2018, 9, 1644 CrossRef PubMed.
- Y. H. Yuan, S. L. Zhao, J. Wen, D. Wang, X. W. Guo, L. L. Xu, X. L. Wang and N. Wang, Adv. Funct. Mater., 2019, 29, 1805380 CrossRef.
- Y. Yuan, B. Niu, Q. Yu, X. Guo, Z. H. Guo, J. Wen, T. Liu, H. Q. Zhang and N. Wang, Angew. Chem., 2020, 132, 1236 CrossRef.
- B. Sun, B. Aguila, L. D. Earl, C. W. Abney, L. Wojtas, P. K. Thallapally and S. Q. Ma, Adv. Mater., 2018, 30, 1705479 CrossRef PubMed.
- D. Wang, J. A. Song, J. Wen, Y. H. Yuan, Z. L. Liu, S. Lin, H. Y. Wang, H. L. Wang, S. L. Zhao, X. M. Zhao, M. H. Fang, M. Lei, B. Li, N. Wang, X. L. Wang and H. Wu, Adv. Energy Mater., 2018, 8, 1802607 CrossRef.
- G. Y. Li, G. Hong, D. Dong, W. H. Song and X. T. Zhang, Adv. Mater., 2018, 30, 1801754 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Digital photos, the curves of storage modulus (G′) and loss modulus (G′′) with frequency, temperature–time curves under illumination, surface area and pore size distribution, more SEM images, Raman and FTIR spectra, diagram of the possible mechanism of GO reduction, water transport data, nitrogen adsorption and desorption isotherms, calculation and fitting details, uranium adsorption kinetics, etc. See DOI: 10.1039/d0nr08956g |
| ‡ Contributed equally |
| This journal is © The Royal Society of Chemistry 2021 |