
Iron-based nanoparticles for MR imaging-guided ferroptosis in combination with photodynamic therapy to enhance cancer treatment†
Qifang
Chen‡
a,
Xianbin
Ma‡
b,
Li
Xie
a,
Wenjie
Chen
a,
Zhigang
Xu
 b,
Erqun
Song
a,
Xiaokang
Zhu
*a and
Yang
Song
*a
b,
Erqun
Song
a,
Xiaokang
Zhu
*a and
Yang
Song
*a
aKey Laboratory of Luminescence Analysis and Molecular Sensing (Southwest University), Ministry of Education, College of Pharmaceutical Sciences, Southwest University, Chongqing, 400715, China. E-mail: songyangwenrong@hotmail.com; zxk@swu.edu.cn; Fax: +86-23-68251225; Tel: +86-23-68250371
bSchool of Materials and Energy & Chongqing Engineering Research Center for Micro-Nano Biomedical Materials and Devices, Southwest University, Chongqing, 400715, China
First published on 19th February 2021
Abstract
Ferroptosis therapy, which applies ferroptotic inducers to produce lethal lipid peroxidation and induce the death of tumor cells, is regarded as a promising therapeutic strategy for cancer treatment. However, there is still a challenge regarding how to increase reactive oxygen species (ROS) accumulation in the tumor microenvironment (TME) to enhance antitumor efficacy. Herein, we designed a nanosystem coated with the FDA approved poly(lactic-co-glycolic acid) (PLGA) containing ferrous ferric oxide (Fe3O4) and chlorin E6 (Ce6) for synergistic ferroptosis-photodynamic anticancer therapy. The Fe3O4-PLGA-Ce6 nanosystem can dissociate in the acidic TME to release ferrous/ferric ions and Ce6. Then, the Fenton reaction between the released ferrous/ferric ions and intracellular excess hydrogen peroxide can occur to produce hydroxyl radicals (˙OH) and induce tumor cell ferroptosis. The released Ce6 can increase the generation and accumulation of ROS under laser irradiation to offer photodynamic therapy, which can boost ferroptosis in 4T1 cells. Moreover, magnetic monodisperse Fe3O4 loading provides excellent T2-weighted magnetic resonance imaging (MRI) properties. The Fe3O4-PLGA-Ce6 nanosystem possesses MRI ability and highly efficient tumor suppression with high biocompatibility in vivo due to the synergism of photodynamic and ferroptosis antitumor therapies.
Introduction
Breast cancer is the most prevalent type of malignancy and represents the leading cause of mortality among females worldwide.1 Triple-negative breast cancer (TNBC) is particularly aggressive and resistant to current therapies due to the lack of estrogen, progesterone and human epidermal growth factor 2 receptors.2,3 The mainstay of treatment for TNBC continues to be chemotherapy with innovative, multidrug combination systemic therapies, poly(ADP-ribose) polymerase (PARP) inhibitors or immunotherapy agents.4–6 However, optimal chemotherapy regimens have yet to be established for drug resistance. One of the promising methods of anti-TNBC treatment is photodynamic therapy (PDT), which is based on the adaptation of a photosensitizer to produce reactive oxygen species (ROS), such as singlet oxygen (1O2), superoxide radicals (O2˙−) or hydroxyl radicals (˙OH), under light excitation.7 The presence of ROS in tumor cells can cause peroxidation of cell membrane lipids or proteins and damage to nucleic acids, which leads to the instability of cells and consequently cell death.8 However, there are still deficiencies in PDT in the scope of localized treatment, limited light penetration through tissues and high oxygen consumption, which impede the PDT effects against deep, metastatic lesions under the hypoxic regions of the tumor microenvironment (TME).9–11 The application of combinational strategies with other therapeutic modalities, such as photothermal therapy (PTT), chemotherapy, immunotherapy and gene therapy, could improve the PDT effects at low dosages, thus overcoming the limitations of PDT.12–16Ferroptosis, a form of regulated cell death that is iron- and ROS-dependent, has been developed as a strategy for cancer therapy. Ferroptosis is different from apoptosis, necrosis and autophagy due to its dependence on iron and an imbalance in oxidation–reduction levels.17 Cellular ferroptosis can be reversed by the lipophilic antioxidant ferrostatin-1 (Fer-1) or the iron chelating agent deferoxamine (DFO) but not by apoptosis or autophagy inhibitors. Cysteine metabolism and glutathione peroxidase 4 (GPX4) inactivation play critical roles in ferroptosis initiation and lipid peroxidation (LPO) accumulation. Compared with other antitumor treatments, ferroptosis inducers bypass the drawbacks of apoptosis-mediated traditional therapeutics and thus supplement the current shortages in antitumor treatment. For example, ferroptosis inducers solve the reversal of resistance of first-line chemotherapy drugs mediated by apoptosis pathways.18 Ferroptosis inducers show good therapeutic effects on a variety of cancers, including liver cancer,19 kidney cancer,20 melanoma,21 breast cancer22 and pancreatic cancer.23 The emerging strategies of cancer therapy based on ferroptosis include the use of small molecules, nanomaterials and gene technologies.24,25 Among them, nanomaterials, especially iron-based nanoparticles (NPs), have attracted increasing attention due to their low adverse effects on normal tissues and prolonged circulating half-life in blood.26–28 Previous studies have demonstrated that iron-based NPs could release ferrous or ferric acids in acidic lysosomes and were further involved in the intracellular Fenton reaction to produce ROS and induce ferroptosis after the endocytosis of cells at the tumor site.29–33 With a deepening understanding of the biological process of ferroptosis, cancer therapeutic efficiency will be enhanced by simultaneously regulating multiple cell death pathways. To eradicate malignancies, ferroptosis of a nonapoptotic nature can also be combined with immunotherapy or chemical therapy.34,35
A common feature of PDT and ferroptosis is the generation and accumulation of lethal ROS in the TME, thus inducing the death of cancer cells. In addition, ferroptosis produces large amounts of oxygen through the Fenton reaction, which can alleviate hypoxia in the TME and improve the PDT effect.21,36 Therefore, it is appealing to combine PDT with ferroptosis therapy to improve the antitumor effects. To fulfill the combinational strategy, we designed a nanosystem by loading chlorin E6 (Ce6) and iron-based NPs (Fe3O4 NPs) with poly(lactic-co-glycolic acid) (PLGA) through hydrophobic interactions and chelation (Scheme 1). The formed Fe3O4-PLGA-Ce6 NPs may respond to the acidic TME through the degradable characteristics of PLGA to release ferric/ferrous ions and Ce6. We hypothesized that the released ferrous or ferric ions may react with excess intracellular H2O2 to trigger the Fenton reaction, resulting in intracellular ROS accumulation and LPO generation, which is required in ferroptosis.31,37,38 Ferroptosis always accompanies by depletion of glutathione (GSH), inactivation of GPX4 and seven members of the 12 transmembrane protein transporter solute carrier family 11 (SLC7A11), increase of Acyl-CoA synthetase long-chain family member 4 (ACSL4) and LPO in tumor cells, and eventually leads to the highly efficient tumor cell death. Under laser irradiation, the released Ce6 may generate singlet oxygen by the PDT effect and thus induce the death of tumor cells, which can boost ferroptosis.39 Furthermore, the endowed Fe3O4 is expected to allow visualization monitoring by magnetic resonance imaging (MRI) of tumor treatment mediated by PDT and ferroptosis therapy.40 MRI is a powerful medical diagnostic technique that can acquire three-dimensional (3D) tomographic images with excellent resolution for soft tissues.41,42 Iron-based NPs can be applied in T2-weighted MRI for good superparamagnetism.43 The ratio change of ferric to ferrous ions leads to responsive MRI, which could be employed to monitor the growth of tumors and the effectiveness of antitumor therapy.44,45 Our study provides a combination PDT-ferroptosis strategy to achieve antitumor effects by accumulating intracellular ROS through the utilization of iron-based NPs.
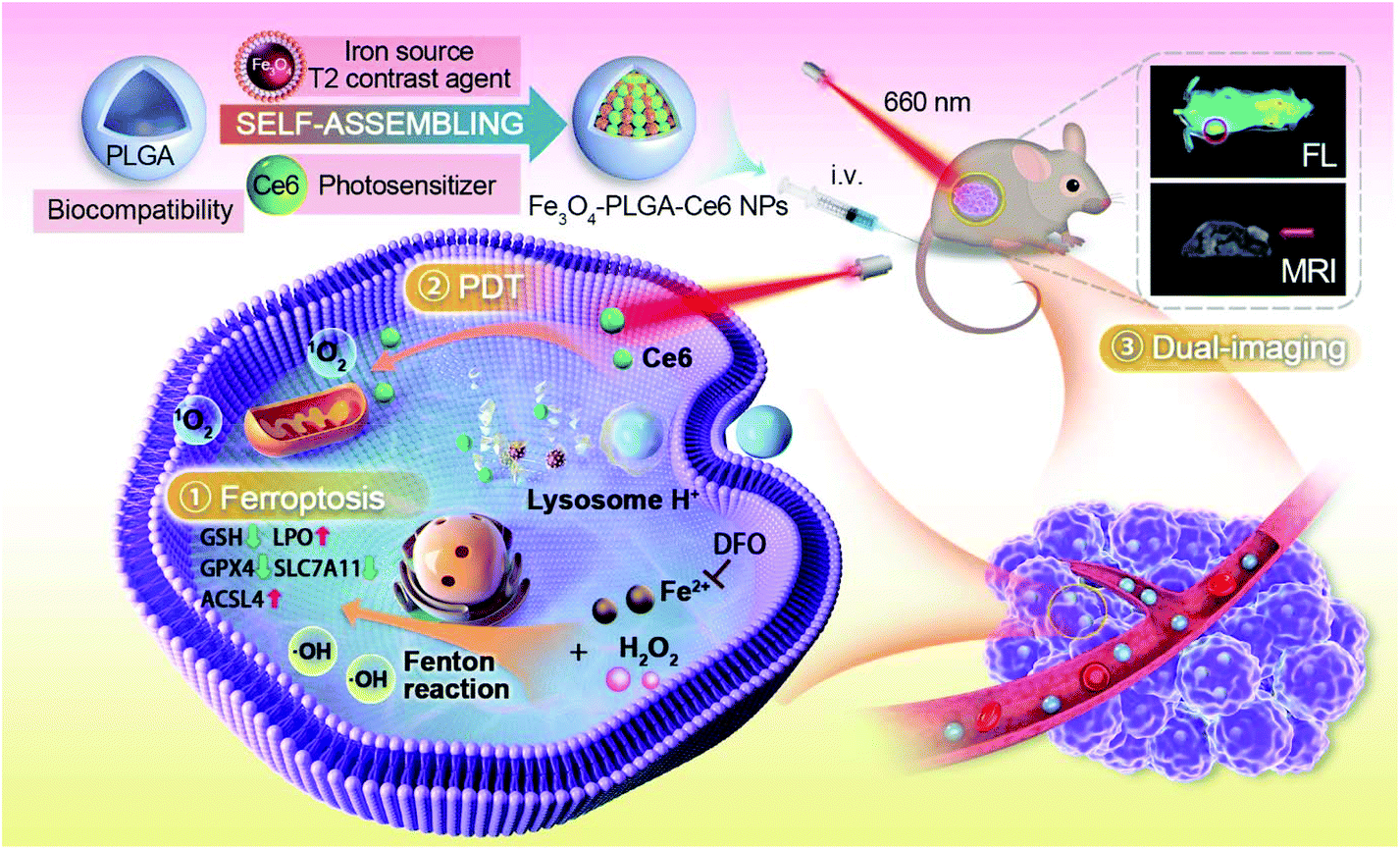 | ||
| Scheme 1 Schematic illustration of the construction of Fe3O4-PLGA-Ce6 nanoparticles and mechanistic study of ROS-mediated combination of PDT and MRI-guided ferroptosis. | ||
Materials and methods
Materials
Ferric chloride hexahydrate, iron powder, hexane, laurylamine, dimethyl sulfoxide, N,N′-dimethylformamide, 1,2-dichlorobenzene, oleic acid, tetramethyl ethylenediamine and citric acid were supplied by Aladdin Reagent Database Inc. (Shanghai, China). PLGA and Ce6 were acquired from Meilun Biotechnology Co., Ltd (Dalian, China). Cell Counting Kit-8 (CCK-8) was from Gen-View Scientific Inc. (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM) and terminal transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay kit were purchased from KeyGen Biotech Co., Ltd (Nanjing, China). The live/dead viability/cytotoxicity kit, the JC-1 assay kit, the annexin V-FITC/PI apoptosis analysis kit and 4′,6-diamidino-2-phenylindole (DAPI) were supplied by Wanlei Biotechnology Co., Ltd (Shenyang, China). Lysosome Blue and fluorescein diacetate/propidium iodide probe (FDA/PI) double-staining probes were supplied by Thermo Fisher Scientific (Waltham, MA, USA). Antibodies against GPX4, SLC7A11 and ACSL4 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Beta-actin (β-actin) was obtained from Proteintech Group Inc. (Wuhan, China). Real-time quantitative PCR (RT-qPCR) amplification primers for β-actin, GPX4, ACSL4 and SLC7A11 were synthesized by Sangon Biotech Co., Ltd (Shanghai, China). 2,7-Dichloro-fluorescein diacetate (DCFH-DA) was purchased from Sigma-Aldrich Co. LLC (Shanghai, China). ELISA kits were purchased from Preferred Biotechnology Co., Ltd (Shanghai, China). 2,2,6,6-Tetramethyl-4-piperidone (TEMP) and 5,5-dimethyl-1-pyrroline (DMPO) were purchased from Dojindo Co., Ltd (Shanghai, China). GSH, the lactate dehydrogenase (LDH) and malondialdehyde (MDA) kits were purchased from Jiancheng Bioengineering Institute (Nanjing, China).Preparation of citric acid (CA)-coated Fe3O4 and Fe3O4-PLGA-Ce6 NPs
First, oleic acid (OA)-Fe3O4 nanoparticles were synthesized according to a previous report with slight modification.46 Briefly, 1.15 g of FeCl3·6H2O (4 mmol) and 0.12 g of reduced iron powder (2 mmol) were added to a hexane solution containing 3.5 mL of oleic acid (12 mmol) and sonicated for 5 min at room temperature. Then, 8 mL of laurylamine (40 mmol) was added to the above solution. The resulting brown solution was transferred to a 100 mL Teflon-lined stainless-steel autoclave and heated to 200 °C for 3 h. After cooling to room temperature, the black-brown product was precipitated with 90 mL of ethanol three times and separated by a magnet. The particles were redispersed in hexane containing 0.2 mL of oleic acid and then centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. Finally, magnetic monodisperse Fe3O4 nanoparticles were obtained.
000 rpm for 10 min. Finally, magnetic monodisperse Fe3O4 nanoparticles were obtained.
Citric acid-coated Fe3O4 (CA-Fe3O4) nanoparticles were prepared as reported previously.47 OA-coated nanoparticles (60 mg) were dispersed in 15 mL of a 1,2-dichlorobenzene/N,N′-dimethylformamide solution (v/v, 50/50). Then, 0.05 g of citric acid was added to the above mixture and stirred electrically at 100 °C for 24 h. The resulting red-brown product was subsequently precipitated by the addition of ethyl ether (∼30 mL) and recovered with a magnet. The crude product was washed with acetone 3 times to remove free citric acid. The particles were redispersed in deionized water (DI water) water for future use.
Subsequently, 0.5 mL of CA-Fe3O4 nanoparticles (20 mg mL−1) and 1 mL of Ce6 (1 mg mL−1) were added to 1 mL of DMSO containing 10 mg of PLGA. The mixture stirred at room temperature for 24 h. Then, the mixture was transferred to a dialysis bag (MWCO: 3.5 KD) and purified for another 24 h. The product was stored away from light at 4 °C.
The morphologies of Fe3O4 and Fe3O4-PLGA-Ce6 NPs were examined via TEM (JEM-1200EX). The hydrated particle size and zeta potential of the Fe3O4-PLGA-Ce6 NPs were measured by DLS (ZEN3700). UV–vis absorbance spectra of Ce6 and the Fe3O4-PLGA-Ce6 NPs were acquired using a UV–vis absorption spectrophotometer (UV-2450). Fluorescence spectra of Ce6 and the Fe3O4-PLGA-Ce6 NPs were acquired with a fluorescence spectrophotometer (F-7000). The X-ray diffraction signal of the Fe3O4-PLGA-Ce6 NPs was obtained by an X-ray diffractometer (X'Pert3 Powder). The magnetic properties of the Fe3O4-PLGA-Ce6 NPs were measured by vibrating sample magnetometer (MPMS®SQUID-VSM).
Cell culture
The murine breast cancer 4T1 cell line was purchased from Army Medical University. Cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin at 37 °C with 5% CO2.Drug loading and release
The loading capacity (LC) and encapsulation efficiency (EE) of Ce6 and Fe3O4 into the Fe3O4-PLGA-Ce6 NPs were measured using UV-vis at 400 nm and AAS (TAS-990) in graphite furnace mode.To evaluate the release properties of Ce6 from the Fe3O4-PLGA-Ce6 NPs, 1 mL of sample solutions were loaded into dialysis bags (MWCO: 3.5 KD). Then, the dialysis bags were immersed in two different environments, PBS (pH 7.4) and cell culture medium (DMEM plus 10% FBS), and shaken at 37 °C at a moderate speed. One milliliter of PBS or cell culture medium outside the dialysis bag was measured by UV-vis at predetermined time points.
To evaluate iron release, Fe3O4-PLGA-Ce6 NPs solutions with different pH values were put into a dialysis bag (MWCO: 3.5 KD), which was immersed in PBS (pH 7.4) at 37 °C. At predetermined time intervals, 1 mL of PBS solution outside the dialysis bag was collected for the AAS test.
The LC and EE were calculated as follows: LC (%) = (weight of Ce6 or Fe3O4 encapsulated in nanoparticles)/(weight of nanoparticles) × 100%; EE (%) = (weight of Ce6 or Fe3O4 encapsulated in nanoparticles)/(total Ce6 or Fe3O4) × 100%.
1O2 generation by Fe3O4-PLGA-Ce6 NPs
1,3-Diphenylisobenzofuran (DPBF) was used as a probe to estimate the generation of singlet oxygen by the Fe3O4-PLGA-Ce6 NPs. Fifty microliters of DPBF (1.0 mg mL−1, dissolved in acetonitrile) was added to 1 mL of a Fe3O4-PLGA-Ce6 NPs dispersion (500 μg mL−1) in water solution.39 The absorption spectra of DPBF were measured and compared with the spectra without or with H2O2 (1 mM) at different times plus 660 nm irradiation at a power density of 0.5 W cm−2.Cellular uptake in vitro
One milliliter (5 × 104 cells) of 4T1 cells was seeded in 12-well plates and incubated for 24 h. Then, Fe3O4-PLGA-Ce6 NPs (at an equivalent dosage of 10 μg mL−1 Ce6) were added to the cell culture medium (DMEM plus 10% FBS) at different times (10 min, 20 min, 40 min, 60 min). After incubation at 37 °C, the cells were washed three times with PBS before flow cytometry (FCM) (BD FACS Melody™) analysis.Cytotoxicity assay
First, 100 μL (1 × 104 cells) of 4T1 cells were inoculated into 96-well plates for 24 h followed by the addition of different concentrations of Fe3O4, Ce6, and Fe3O4-PLGA-Ce6 NPs in the presence or absence of laser irradiation (660 nm, 0.5 W cm−2, 3 min) for 24 h. After washing with PBS three times to remove noninternalized nanoparticles or Ce6, the CCK-8 reagent (v/v: 10%) was added to the 96-well plate followed by incubation at 37 °C for another 1 h. Finally, the cell viability was determined with a microplate reader (ELX-800) at 450 nm. All data were obtained in quadruplicate. Relative cell viability was calculated as follows: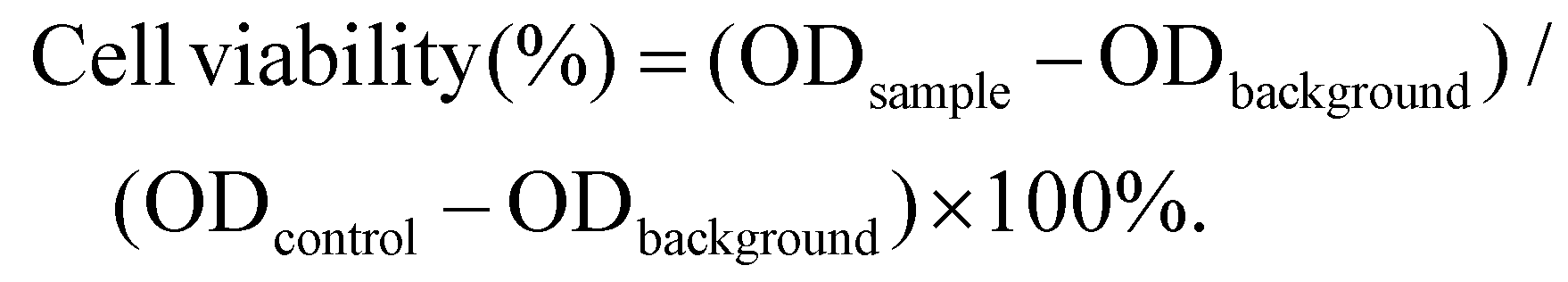
In order to intuitively observe the therapeutic effects of the Fe3O4-PLGA-Ce6 NPs on cancer cells, viable and dead cells were stained with FDA and PI after laser irradiation (660 nm, 0.5 W cm−2). Cell images were observed by the confocal laser scanning microscopy (N-SIM E).
Electron paramagnetic resonance/spin trapping (EPR)
EPR spectra were investigated using an electron paramagnetic resonance (Bruker EMX). The instrument settings were as follows: microwave attenuation of 15 dB, modulation of 1 G, sweep width of 100 G, and center of field at approximately 3434 G. TEMP (10 mM) was used as a spin trap to capture 1O2 and DMPO (150 mM) was used to trap ˙OH. For the determination of 1O2, 10 mM TEMP was mixed with a solution containing free Ce6 (10 μg mL−1) or Fe3O4-PLGA-Ce6 nanoparticles (containing 10 μg mL−1 Ce6) in PBS under a 660 nm laser for 3 min. For the detection of ˙OH, 5 mM H2O2 was added to the different PBS solutions (pH 7.4, pH 6.5, pH 5.0) containing Fe3O4-PLGA-Ce6 nanoparticles (containing 12.5 μg mL−1 Fe3O4) and DMPO (150 mM).Detection of the ROS and LPO levels
ROS generated in 4T1 cells was investigated by FCM using the DCFH-DA probe. 4T1 cells were treated with Ce6 (10 μg mL−1), Fe3O4 (12.5 μg mL−1), Fe3O4-PLGA-Ce6 NPs (containing Ce6 10 μg mL−1), Ce6 (10 μg mL−1) plus laser or Fe3O4-PLGA-Ce6 NPs (containing Ce6 10 μg mL−1) plus laser and coincubated at 37 °C for 6 h. After that, the medium was removed and the cells were washed with PBS. Cells were incubated at 37 °C for another 20 min with DCFH-DA after trypsinization. The DCF intensity was determined by FCM. Data were analyzed by FlowJo V10. The C11-BODIPY probe was used to detect the LPO level using the same procedure as the ROS assay. Data were analyzed by FlowJo V10.Mitochondrial membrane potential (ΔΨm, MMP) assay
The dye JC-1 was used to evaluate 4T1 cell MMP after the addition of Ce6, Fe3O4 and Fe3O4-PLGA-Ce6 NPs for 24 h followed by laser irradiation at 660 nm at 0.5 W cm−2 for 3 min. The cells were harvested after treatment with trypsin and stained with JC-1 for 15 min. Finally, the mixture was analyzed using FCM.LDH assay
Briefly, 4T1 cells were cultured in 48-well plates at a density of ∼1 × 104 cells per well. After 24 h, the cells were pretreated with different formulas at 10 μg mL−1 Ce6 for another 24 h. Ten microliters of supernatant was collected and used for LDH activity analysis, and then the absorbance at 450 nm was measured with a microplate reader (BioTek ELX800).GSH assay
Briefly, 4T1 cells at a concentration of 5 × 105 cells per well were seeded into 6-well plates. Cells were pretreated with different formulas at 10 μg mL−1 Ce6 and cultured for 24 h. Cell lysis solutions were harvested, and protein concentrations were determined. Total GSH levels were measured by a commercial GSH kit according to the manufacturer's instructions.Western blotting study
4T1 cells were seeded in a 6-well plate. After culture at 5% CO2 and 37 °C for 24 h, cells were treated with Ce6 (10 μg mL−1), Fe3O4 (12.5 μg mL−1), Fe3O4-PLGA-Ce6 NPs (containing 10 μg mL−1 Ce6), Ce6 (10 μg mL−1) plus laser and Fe3O4-PLGA-Ce6 NPs (containing 10 μg mL−1 Ce6) plus laser for another 24 h. The cell lysates were collected after centrifugation at 12000 rpm and 4 °C for 10 min. The cell lysates containing loading buffer were analyzed by electrophoresis on 12.5% denaturing polyacrylamide gels.
RNA extraction and RT-qPCR
First, total RNA was extracted with a total RNA Purification Kit (BioTek). The purified RNA (2.0 μg) was reverse-transcribed into cDNA. After that, the cDNA was used for DNA amplification for RT-qPCR analysis using the LightCycler 96 instrument protocol with FastStart Essential DNA Green Master. Subsequently, 40 cycles of PCR were carried out at 60 °C. Each sample was analyzed in three independent procedures. The relative GPX4, ACSL4 and SLC7A11 expression levels were obtained by the 2−ΔΔCt method and normalized to the internal control gene β-actin.MDA assay
Cells were treated with different formulas for 24 h. Cell lysis solutions were collected, and the protein concentration of each formula was determined. Immediately, MDA levels were measured by a commercial MDA kit according to the manufacturer's instructions.Lillie divalent iron staining
4T1 cells were incubated in confocal dishes at ∼1 × 104 cells per well. After 24 h of incubation, cells were treated with different formulas for an additional 24 h. After washing three times with PBS, cells were incubated with Lillie divalent iron staining reagent in the dark at room temperature for 40 min. After rinsing six times with PBS, the cells were examined under a reversed fluorescence microscope (Olympus IX71).Penetration study
Briefly, 10 mL of agarose solution (10 mg mL−1) containing 200 μL of TFA was dissolved using microwave heating. Then, 100 μL of the above solution was rapidly added to a 96-well plate for ultraviolet sterilization for 0.5 h. Finally, 4T1 cells (2 × 103 per well) were seeded into the precoated plate and incubated for 4 days to form similar spheroids. Afterward, the cancer cell spheroids were incubated for 1 or 4 h with Fe3O4-PLGA-Ce6 NPs (containing 10 μg mL−1 Ce6). The cells were washed with PBS and observed using CLSM via the Z-axis.In vivo real-time fluorescence imaging
4T1 tumor-bearing mice (n = 3) were subjected to in vivo real-time fluorescence imaging studies at the dose of 5.0 mg kg−1 Ce6. Fluorescent images were acquired at 6, 24, 36, 48 and 72 h after i.v. injection using an in vivo imaging system (NEWTON 7.0). After 72 h, the mice were sacrificed. The tumor and main organs, including the kidney, lung, spleen, liver and heart, were collected and subjected to ex vivo imaging.In vivo MRI
Before and after injection via tail vein of Fe3O4 and Fe3O4-PLGA-Ce6 NPs at a 6.25 mg kg−1 Fe3O4, subcutaneous tumors were observed using a clinical 0.5-T MRI system (MesoMR23-060H-I). Axial T2-weighted images of the subcutaneous tumors were obtained at 24, 48 and 72 h using the following parameters: TR/TE = 195/18.2 ms, average = 8, FOV = 100 mm, matrix = 256 × 256, and section thickness = 9.0 mm. After 72 h, all the mice were sacrificed. The tumor tissues were used to observe iron accumulation by fluorescence staining microscopy.In vivo antitumor study
BALB/c female mice (4 weeks, 16–20 g) were obtained from Teng Xin Technology. All animal experiments were conducted following the National Institutes of Health guidelines for the care and use of laboratory animals. To establish the tumor models, all animals were subcutaneously inoculated with 200 μL of saline loaded with 4T1 cells (5 × 106 per mL). After the tumor volume reached 80–100 mm3, all tumor-bearing BALB/c mice were randomly assigned to 6 groups (n = 6 for each group) as follows: PBS, Ce6, Fe3O4, Fe3O4-PLGA-Ce6 NPs, Ce6 plus laser (660 nm, 0.5 W cm−2, 8 min) and Fe3O4-PLGA-Ce6 NPs plus laser (660 nm, 0.5 W cm−2, 8 min). The dosage was fixed at an equivalent Ce6 concentration of 5.0 mg kg−1 and Fe3O4 concentration of 6.25 mg kg−1 every 3 days (days 0, 3, 6 and 9) for the 12-day treatment process. Body weights and tumor sizes were continuously monitored during the treatment course. Tumor volumes were calculated according to the following formula: tumor volume = (axial length) × (axial width)2 × 0.5.On the 12th day, all mice were sacrificed to collect the tumors and vital organs, which were thereafter weighed and photographed. Subsequently, tissue sections were formalin-fixed and paraffin-embedded for histological analysis. These sections were stained with hematoxylin–eosin (H&E), TUNEL, Ki67 and GPX4 for observation under a microscope.
Safety evaluation
For the hemolysis assay, peripheral blood was drawn from the orbital venous plexus of BALB/c female mice. Erythrocytes were centrifugally separated (3000 rpm, 5 min) and washed five times with PBS. The erythrocytes were diluted to 2% with PBS. Afterwards, different test samples were incubated with the 2% red blood cells (RBCs) at 37 °C for 1 h. Erythrocytes cultured in PBS served as a negative control. The Triton X-100-treated group was the positive control. Subsequently, the samples were centrifuged at 3000 rpm for 10 min. Finally, the supernatant was transferred to a quartz cuvette, and the absorbance at 540 nm was measured using a UV-vis spectrophotometer. The results of hemolysis were calculated by the following formula: Hemolysis rate (%) = [(sample absorbance − negative control absorbance)/(positive control absorbance − negative control absorbance)] × 100%.BALB/c female mice were used for routine blood analysis. Mice (n = 4, 18–22 g) were treated with different formulas (PBS, Fe3O4, Ce6, and Fe3O4-PLGA-Ce6 NPs) through the tail vein at the equivalent Ce6 concentration of 5.0 mg kg−1 and Fe3O4 concentration of 6.25 mg kg−1. After 1 or 7 days, blood was collected from the eye sockets to detect hematological indicators (WBC, RBC, MONs, RDW, PDW, MPV, MCV, HCT and PCT). The data were obtained with a hematology analyzer.
To further evaluate the safety of the different formulas in vivo, we collected peripheral blood serum from the orbital venous plexus of BALB/c mice to detect the levels of TNF-α and IL-1β by ELISA kits. Additionally, biochemical indexes, such as the serum levels of ALT, AST and CREA, were detected with an automated analyzer.
Statistical analysis
Statistical analysis was obtained by t-test analysis of variance. All data were shown as mean ± standard deviation (S.D.). All results were expressed as mean plus or minus the standard error.Results and discussion
Preparation and characterization of Fe3O4-PLGA-Ce6
CA-coated ferroferric oxide NPs (Fe3O4 NPs) ∼10 nm in size were obtained by surface modification (Fig. S1A†). The powder X-ray diffraction (XRD) patterns of the Fe3O4 nanoparticles in Fig. S1B† showed clear diffraction peaks, as previously reported.46 CA-coated Fe3O4, Ce6 and PLGA were then mixed, fabricating self-assembled Fe3O4-PLGA-Ce6 NPs through chelation and hydrophobic interactions. Fe3O4-PLGA-Ce6 NPs displayed a spherical morphology and uniform size distribution profile with an average diameter of 85 nm as observed by transmission electron microscopy (TEM) (Fig. 1A), which was smaller than the hydrodynamic diameter measured by dynamic light scattering (DLS) (Fig. 1B). These nanoparticles showed an optimal size with enhanced permeability and retention effects and pharmacokinetics.48 The zeta potential was −30.1 mV in PBS (pH 7.4), as shown in Fig. S2,† which can elongate the retention time in vivo.49 Fe3O4-PLGA-Ce6 NPs had a maximum absorption peak at approximately 405 nm according to the absorbance of Ce6, which indicated the successful loading of Ce6 (Fig. 1C). Additionally, Fe3O4-PLGA-Ce6 NPs had a slight redshift in the fluorescence spectrum compared with that of free Ce6, with a peak at 670 nm, which should result from hydrophobic interactions and chelating forces (Fig. 1D). The loading effectiveness of the nanosystem was then investigated. The loading contents of Ce6 and Fe3O4 were 22.1% and 25.5%, respectively (Table S1†). MRI is deemed to be a very powerful diagnostic technique that provides accurate anatomical information.50 To explore the potential applications of the nanoparticles as MRI contrast agents, the transverse relaxivity (r2) of the Fe3O4-PLGA-Ce6 NPs was determined. As shown in Fig. 1E, the saturation magnetization was calculated to be 60 emu g−1 by a superconducting quantum interference device vibrating sample magnetometer. The 1/T2 signal intensity was proportional to the increase in iron ion concentration, resulting in darkened images. A linear increase in the R2 (1/T2) values of the Fe3O4-PLGA-Ce6 NPs against iron concentrations reached 105.1 s−1 mM−1 (Fig. 1F), which is similar to that of commercial MRI contrast agents Feridex (93 s−1 mM−1) and Resovist (143 s−1 mM−1).51These results indicated the conceivable capacity of Fe3O4-PLGA-Ce6 NPs as a T2 MRI contrast agent for visualizing the drug delivery process in vivo.52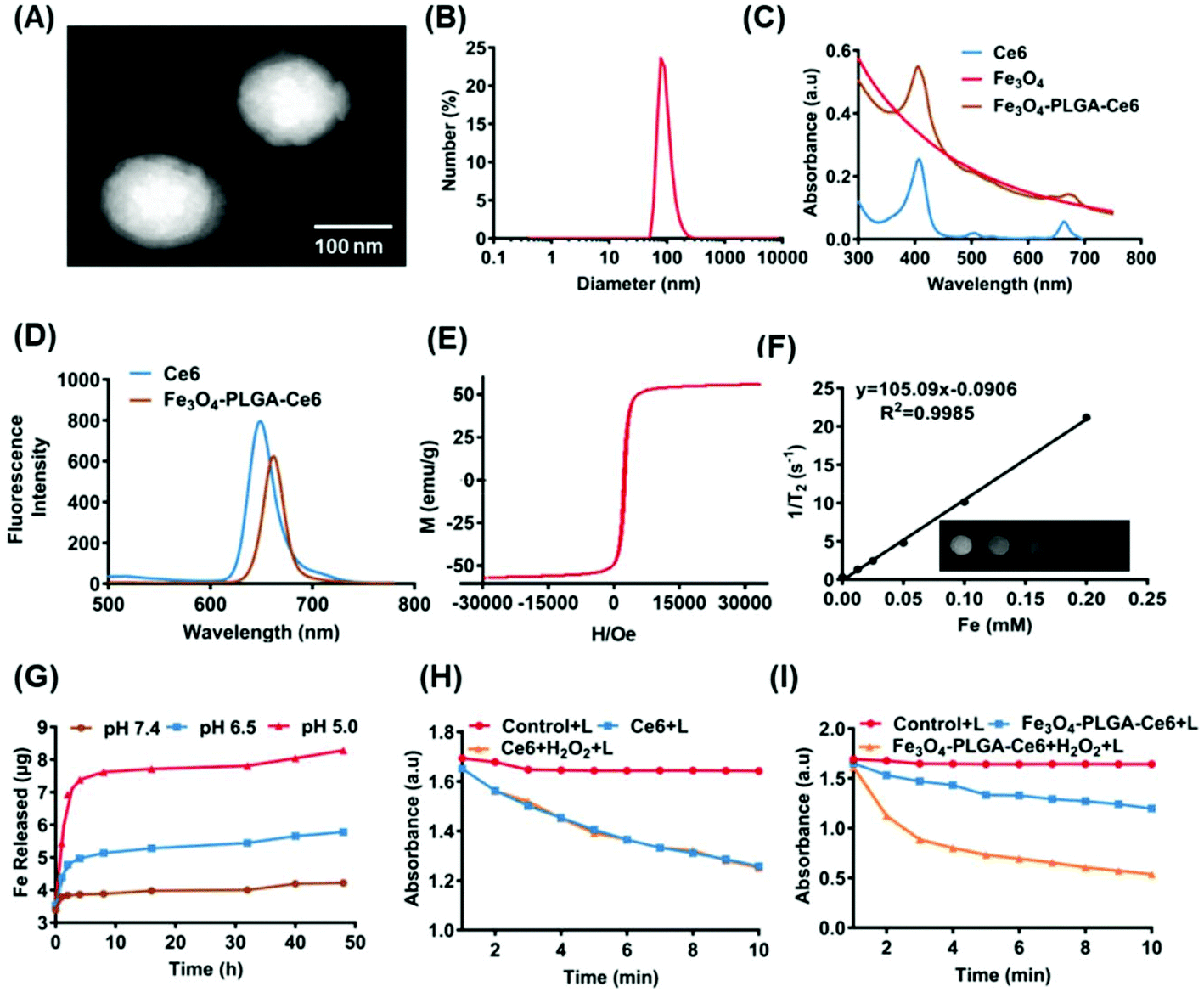 | ||
| Fig. 1 Characterization of Fe3O4-PLGA-Ce6 NPs. (A) TEM image (scale bars: 100 nm) of Fe3O4-PLGA-Ce6 NPs. (B) Hydrodynamic diameters of Fe3O4-PLGA-Ce6 NPs in water. (C) Absorption spectrum of Fe3O4, Ce6 and Fe3O4-PLGA-Ce6 NPs. (D) Fluorescent spectra of Ce6 and Fe3O4-PLGA-Ce6 NPs. (E) Magnetic hysteresis curve of Fe3O4-PLGA-Ce6 NPs. (F) T2 relaxation rate (1/T2, s−1) as a function of Fe concentration (mM) for Fe3O4-PLGA-Ce6 NPs. Inset: T2-weighted MRI image of Fe3O4-PLGA-Ce6 NPs. (G) Fe ions release in different pH value (pH 5.0, pH 6.5 and pH 7.4). (H) Decay curves of DPBF absorption at 410 nm in free Ce6 solution with or without H2O2 at different time under laser irradiation (660 nm, 0.5 W cm−2). L represents laser. (I) Decay curves of DPBF absorption at 410 nm in Fe3O4-PLGA-Ce6 solution with or without H2O2 at different time under laser irradiation (660 nm, 0.5 W cm−2). L represents laser. | ||
Subsequently, we examined the drug release profiles at different pH values, which imitated physiological (pH 7.4), tumor microenvironment (pH 6.5) and lysosomal fluid (pH 5.0) environments. As shown in Fig. 1G, more iron ions were released at low pH than at pH 7.4, suggesting a significant improvement in the disintegration of the nanoparticles in the acidic TME. Accordingly, we also conducted a Ce6 release experiment in phosphate-buffered saline (PBS) and cell culture medium. As shown in Fig. S3,† within the initial 8 h, Fe3O4-PLGA-Ce6 NPs released ∼18% of Ce6 in cell culture medium, which was the same release rate as that in PBS because of Ce6 diffusion. The Fe3O4-PLGA-Ce6 NPs were released faster in cell culture medium and to a greater extent than in PBS after 8 h. Finally, the Fe3O4-PLGA-Ce6 NPs released 98% of Ce6 in cell culture medium within 48 h. This result indicated that the release of Ce6 contributed to rival binding between Ce6 and the serum components in FBS, except for the diffusion after 8 h.
As 1O2 is one of the basic elements of PDT, we used DPBF as a probe to evaluate the ability of Fe3O4-PLGA-Ce6 NPs to generate singlet oxygen with or without H2O2 under laser irradiation. DPBF specifically reacts with singlet oxygen, resulting in a decrease in its characteristic absorbance peaks. As shown in Fig. 1H, compared with the DPBF plus laser irradiation group, the absorbance intensity of the Ce6 plus laser irradiation group gradually decreased, demonstrating the generation of 1O2. No significant difference in 1O2 generation was observed by treatment with free Ce6 in the presence or absence of H2O2, demonstrating that H2O2 cannot enhance 1O2 production under laser irradiation. However, there was an efficient decrease in the absorbance intensity of the Fe3O4-PLGA-Ce6 NPs plus laser irradiation group in the presence or absence of H2O2 compared to that of the DPBF plus laser irradiation group, suggesting the efficient generation of 1O2. Importantly, the absorbance intensity greatly decreased after the addition of H2O2 to Fe3O4-PLGA-Ce6 NPs, indicating that H2O2 can improve the production efficiency of 1O2 under laser irradiation, as shown in Fig. 1I. Therefore, Fe3O4-PLGA-Ce6 NPs can produce 1O2 under laser irradiation, indicating its PDT properties.
Cellular uptake and cytotoxicity
Fe3O4-PLGA-Ce6 NPs should be efficiently internalized by tumor cells after they arrive at the tumor site, which is highly interrelated with their subsequent lethal effects.53 As shown in Fig. 2A, 4T1 cells pretreated with Fe3O4-PLGA-Ce6 NPs exhibited obvious rightward fluorescence signal shifts as time increased as shown by FCM analysis. In Fig. 2B, we observed a rapid increase in the mean fluorescence intensity (MFI) with increasing incubation time, suggesting successful internalization into 4T1 cells. After internalization, almost all of the nanoparticles would be sequestered in cellular lysosomes. The pH in the lysosome is lower than that in the normal physiological environment (pH 7.4), which is conducive to the rapid dissociation of the PLGA-coated nanostructures. As shown by CLSM images (Fig. S4†), the red fluorescence of Ce6 from the Fe3O4-PLGA-Ce6 NPs overlapped well with the blue fluorescence from LysoTracker Blue after coculture, demonstrating the internalization of Fe3O4-PLGA-Ce6 in the lysosome after cellular endocytosis. Therapeutic drugs, including iron ions and Ce6, are expected to escape from lysosomes and localize to the cytoplasm to induce follow-up treatment effects. As shown in Fig. S5,† the red fluorescence of Ce6 from the Fe3O4-PLGA-Ce6 NPs was significantly disturbed, with green fluorescence from the lysosome tracker appearing at approximately 6 h, displaying a time-dependent lysosomal escape.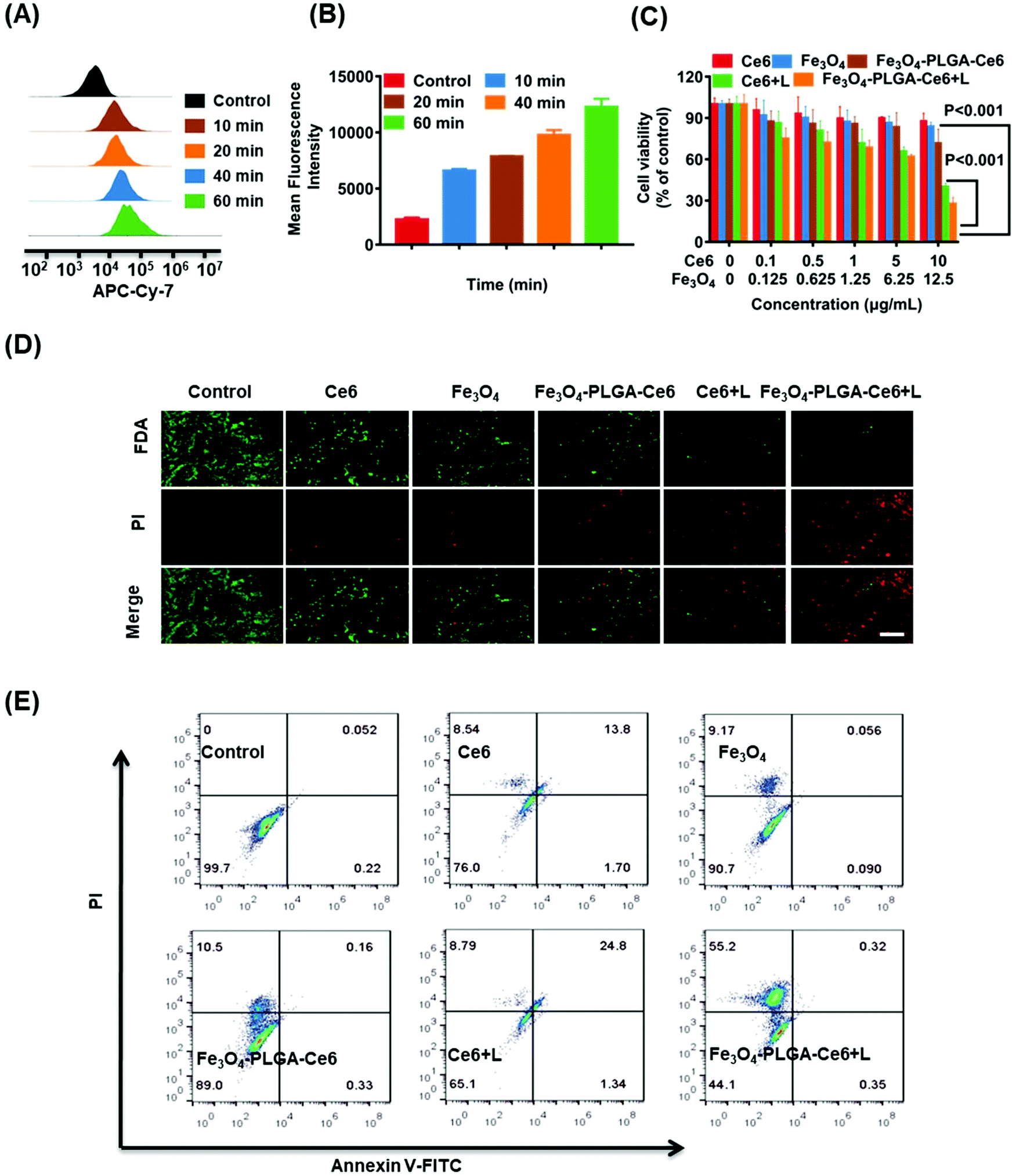 | ||
| Fig. 2 In vitro cellular uptake and cytotoxicity of Fe3O4-PLGA-Ce6 NPs. (A) FCM histogram profiles of 4T1 cells incubated with Fe3O4-PLGA-Ce6 NPs at different time. (B) Mean fluorescence intensity of Fe3O4-PLGA-Ce6 NPs. (C) Cell viabilities of 4T1 cells after the co-incubation with Ce6, Fe3O4, Fe3O4-PLGA-Ce6, Ce6 + L and Fe3O4-PLGA-Ce6 + L at different concentrations. (D) FDA/PI (live/dead) staining analysis of 4T1 cells after the co-incubation with different formulas for 24 h. (E) Annexin V-FITC/PI analysis of 4T1 cells treated with different formulas for 24 h by FCM. Drug dose: Ce6 10 μg mL−1, Fe3O4 12.5 μg mL−1, Fe3O4-PLGA-Ce6 NPs with 10 μg mL−1 Ce6, Ce6 + L 10 μg mL−1 and Fe3O4-PLGA-Ce6 + L with 10 μg mL−1 Ce6. L represents laser. The bar: 200 μm. | ||
Owing to their escape from lysosomes, cytotoxicity is expected to be induced once ferrous ions and Ce6 are released into the cytoplasm. Cell viability was measured using a CCK-8 assay in 4T1 cells following treatment with different formulas at 24 h in the absence or presence of laser irradiation. As shown in Fig. 2C, compared with the control, there was no significant cytotoxicity in the Ce6 and Fe3O4 treated groups. The viability of 4T1 cells cocultured with Ce6 under laser irradiation for 24 h decreased to 40%, demonstrating that Ce6 had a good phototoxic effect. Interestingly, the viability of 4T1 cells treated with the Fe3O4-PLGA-Ce6 NPs under laser irradiation was 25%, which was the lowest among all groups. These results indicated the synergistic cytotoxicity of the photochemical process and iron ions. It could therefore be inferred that the photochemical process promotes the release of iron ions. The released iron reacts with intracellular H2O2, triggers the Fenton reaction and then induces the generation of ROS in tumor cells, thus boosting photocytotoxicity. In addition, in Fig. 2D, an increased amount of red fluorescence was observed in the 4T1 cells treated with Fe3O4-PLGA-Ce6 plus laser irradiation compared with the other groups by FDA/PI probe staining, which was consistent with the cell cytotoxicity results. The quantified results were measured by a FITC-annexin V/PI double-staining assay after coincubation with different formulas using FCM (Fig. 2E). Free Ce6 could initiate 13.8% apoptosis, while the ratio of apoptotic cells increased to 24.8% due to the increase of 1O2 production after laser irradiation. Compared with the other formulas, Fe3O4-PLGA-Ce6 plus laser irradiation possessed the highest cell death rate of 55.2% in 4T1 cells, which could be attributed to the rapid release of Ce6 and Fe3O4. All of the results showed that the combination of laser irradiation and iron ions increased cytotoxicity in 4T1 tumor cells.
ROS-based cell death in vitro
Excessive ROS may destroy cellular redox homeostasis and trigger cell death. Therefore, upregulation of ROS levels can be a promising approach for cancer therapy. Photosensitizers under laser irradiation can generate 1O2, which can be assayed by EPR. TEMP, a typical spin-trapping agent, was used to capture the 1O2 produced by PDT. As illustrated in Fig. 3A, the typical three-line peak with equal intensities was observed in both of the Ce6/TEMP and Fe3O4-PLGA-Ce6/TEMP systems after laser irradiation for 5 min, indicating the generation of 1O2 by PDT. Moreover, Fe3O4 can react with intracellular H2O2 and then trigger the Fenton reaction, thus producing ˙OH. DMPO was applied to capture ˙OH at different pH values. The four-line peak of DMPO-˙OH in water was detected in the Fe3O4-PLGA-Ce6/H2O2/DMPO system, as shown in Fig. 3B. The baseline of Fe3O4-PLGA-Ce6/H2O2/DMPO showed a slight slope at pH 7.4 due to Fe3O4 loading, which can be verified by the EPR spectrum of the Fe3O4-PLGA-Ce6 NPs in Fig. S6.† The spectrum was composed of a broad and almost symmetric line at approximately 3432 G (g = 2.001) due to the characteristics of the superparamagnetic nanoparticles. Additionally, the intensity of the DMPO-˙OH peak at pH 7.4 was much weaker than that at pH 6.5 and pH 5.0. This phenomenon could be explained by the fact that an acidic environment favors the degradation of Fe3O4-PLGA-Ce6 and triggers the Fenton reaction. All of these observations verified that Fe3O4-PLGA-Ce6 could generate ROS through photochemical processes and Fenton reactions.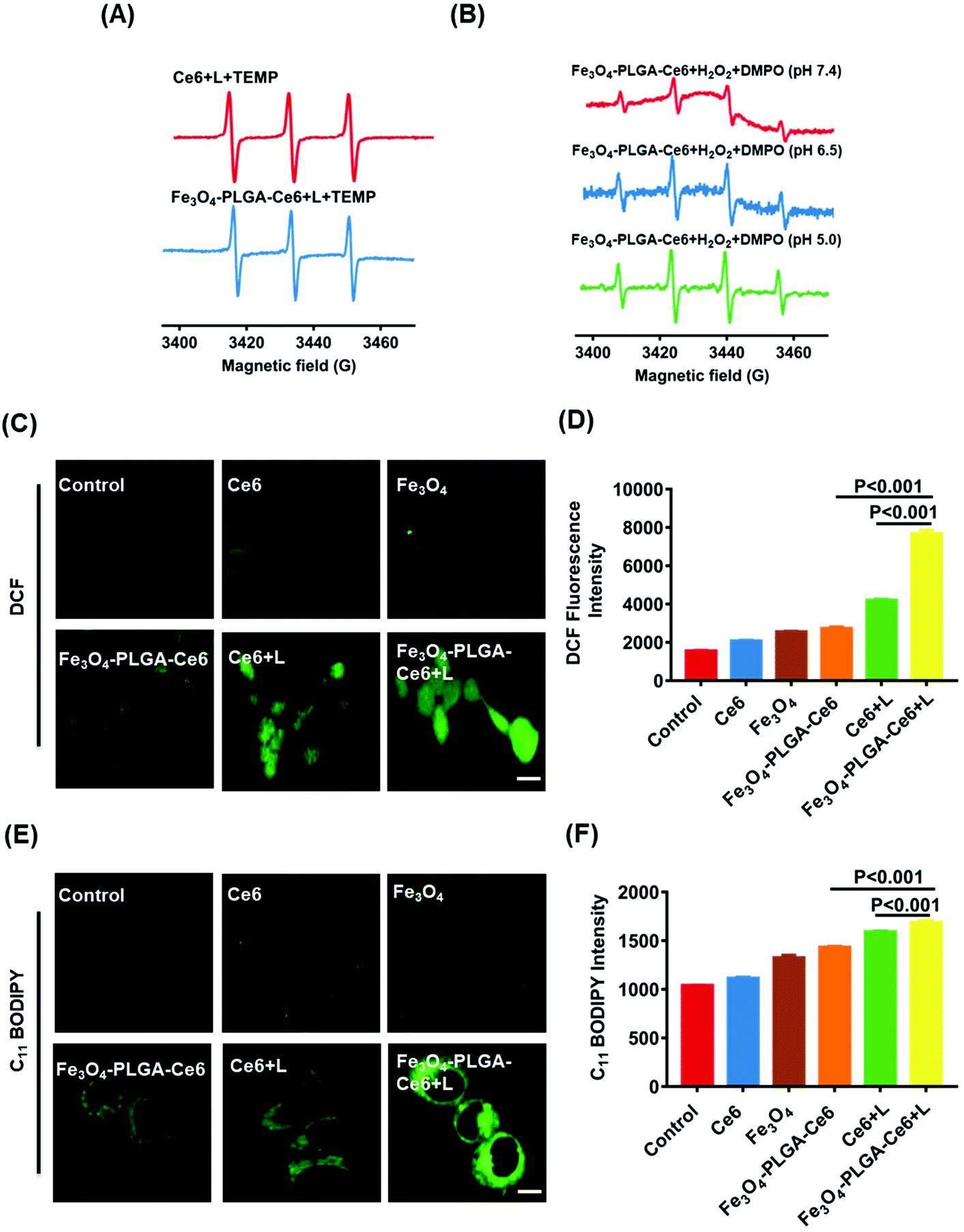 | ||
| Fig. 3 ROS-based cell death in vitro. (A) EPR spectra of singlet oxygen generated in the Ce6/TEMP/L and Fe3O4-PLGA-Ce6 NPs/TEMP/L systems. L represents laser. (B) The EPR spectra of Fe3O4-PLGA-Ce6 NPs and hydroxyl radical generated in the H2O2/Fe3O4-PLGA-Ce6 NPs/DMPO at pH 7.4, pH 6.5 and pH 5.0. (C) CLSM images of 4T1 cells stained by DCFH-DA probe after treated with different formulas for 6 h with or without laser irradiation (green indicated DCF fluorescence). (D) Quantitative DCF fluorescence intensity of 4T1 cells stained by DCFH-DA probe in different formulas for 6 h by FCM. (E) CLSM images of 4T1 cells stained by C11 BODIPY probe in different formulas for 6 h. The bar is 10 μm. (F) Quantitative fluorescence intensity of 4T1 cells stained by C11 BODIPY probe in different formulas for 6 h by FCM. Drug dose: Ce6 10 μg mL−1, Fe3O4 12.5 μg mL−1, Fe3O4-PLGA-Ce6 NPs with 10 μg mL−1 Ce6, Ce6 + L 10 μg mL−1 and Fe3O4-PLGA-Ce6 + L with 10 μg mL−1 Ce6. L represents laser. The bar: 10 μm. | ||
To gain insights into the mechanism underlying the anticancer action at the cellular level, we examined the intracellular generation of ROS from Fe3O4-PLGA-Ce6 using DCFH-DA as a fluorescent probe. As shown in Fig. 3C, strong green fluorescence was observed in Fe3O4-PLGA-Ce6 NPs plus laser irradiation-incubated 4T1 cells, which implied that this group contained the highest intracellular ROS concentration as a consequence of the synergism of PDT and the Fenton process. FCM analysis came to the same conclusion that the highest intracellular ROS level was observed in the Fe3O4-PLGA-Ce6 plus laser irradiation group (Fig. 3D). ROS are known to initiate LPO and then result in cell death.54 C11-BODIPY was selected as a ratiometric fluorescent indicator to evaluate LPO by monitoring its fluorescence intensity. As shown in Fig. 3E, the fluorescence intensity of 4T1 cells treated with Fe3O4-PLGA-Ce6 under laser irradiation significantly increased, clearly suggesting the capacity of Fe3O4-PLGA-Ce6 plus laser irradiation to activate LPO through the production of ROS by the synergism of PDT with the Fenton process. The FCM assessment in Fig. 3F also showed similar results, which implied that stimulation with PDT could improve the production of LPO. All of these results could be interpreted as the ROS generated by the Fenton reaction play vital roles in the exertion of the cytotoxic effects of the Fe3O4-PLGA-Ce6 NPs in vitro.
Endogenous ROS is mainly derived from the mitochondria. Thus, the anticancer mechanism may be related to mitochondrial dysfunction.55 Therefore, we used the JC-1 assay to MMP by FCM as shown in Fig. S7.† Compared to the other groups, the FITC/PI signal values (green/red) of 4T1 cells treated with the Fe3O4-PLGA-Ce6 NPs plus laser irradiation notably increased with decreasing MMP. These results demonstrated that Fe3O4-PLGA-Ce6 NPs under laser irradiation abnormally regulated the level of endogenous ROS by disrupting mitochondrial function, resulting in 4T1 cell death.
Mechanistic evaluation of ferroptosis
Based on the high ROS-mediated cell death and the presence of iron ions in the Fe3O4-PLGA-Ce6 NPs, we hypothesized that tumor cell death after treatment with Fe3O4-PLGA-Ce6 NPs is dependent on ROS and iron, which are characteristics of ferroptosis. Therefore, we applied two specific inhibitors to test the features of ferroptosis in Fe3O4-PLGA-Ce6 NPs treated 4T1 cells. Fer-1, a typical ferroptosis inhibitor, can reverse cell death by blocking LPO. As shown in Fig. 4A, Fer-1 could significantly reduce the 4T1 cytotoxicity induced by Fe3O4-PLGA-Ce6 plus laser irradiation. Similarly, the inhibition of cell viability was mitigated by DFO coincubation, suggesting that iron is involved in the process of cell death. The addition of DFO or Fer-1 also significantly decreased LDH level in tumor cells treated with Fe3O4-PLGA-Ce6 NPs plus laser irradiation, as shown in Fig. 4B. Reversal of the cell death by ferroptosis inhibitors and iron-chelating agents indicated the occurrence of ferroptosis. The depletion of GSH is generally regarded as a marker in ferroptosis. As shown in Fig. 4C, cells treated with Fe3O4-PLGA-Ce6 NPs plus laser exhibited a relatively lower level of GSH than the control group, while the depletion of GSH levels in the other groups was almost negligible. These results demonstrated that ferroptosis participated in cell death after treated with Fe3O4-PLGA-Ce6 NPs plus laser irradiation.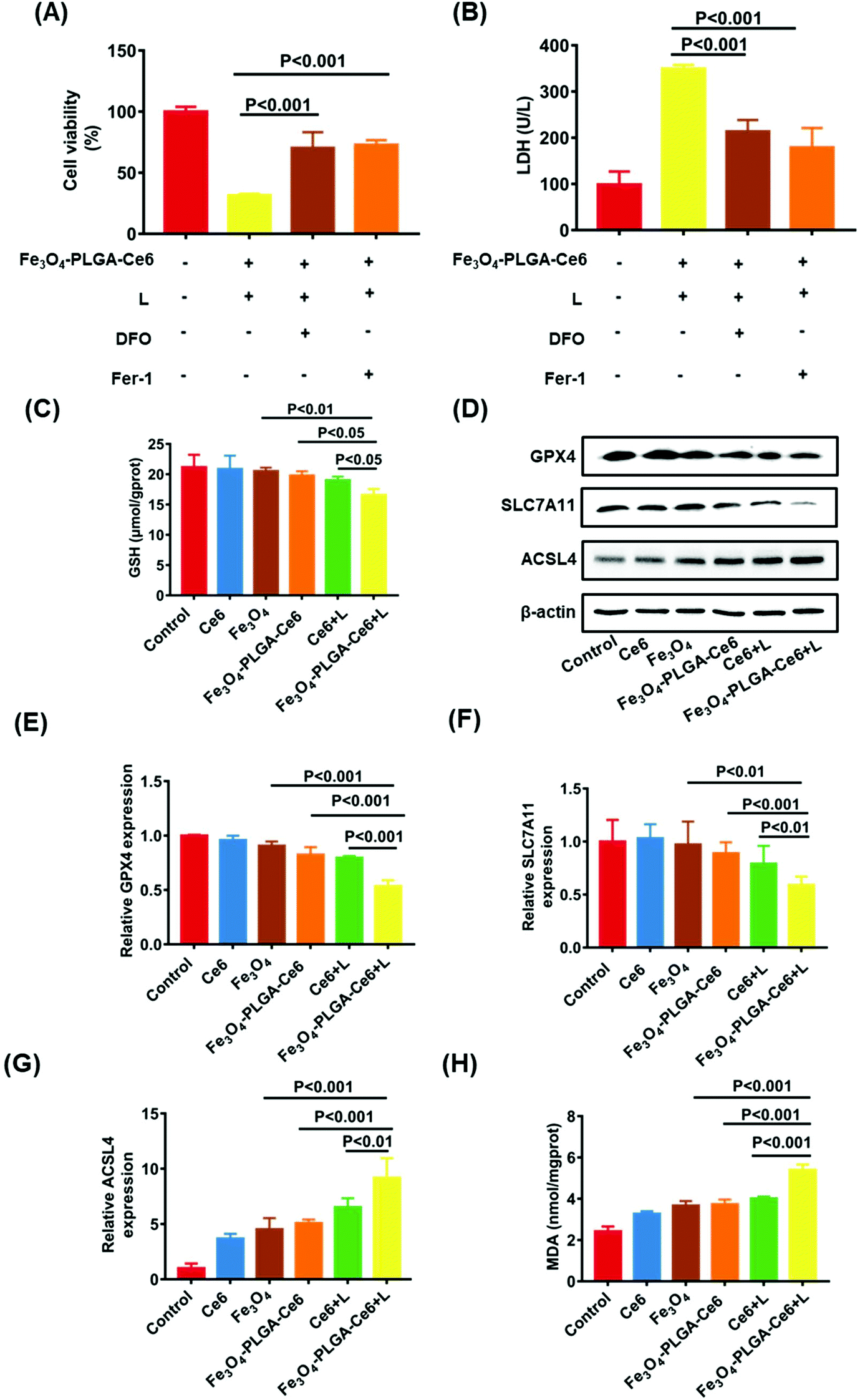 | ||
| Fig. 4 Mechanism evaluation of ferroptosis. (A) Cell viability after treated with DFO (100 μM), Fer-1 (5 μM) and Fe3O4-PLGA-Ce6 NPs with 10 μg mL−1 Ce6 under laser irradiation for 24 h. (B) LDH level of 4T1 cells after treated with DFO (100 μM), Fer-1 (5 μM) and Fe3O4-PLGA-Ce6 NPs with 10 μg mL−1 Ce6 under laser irradiation for 24 h. (C) Intracellular GSH level of 4T1 cells after treated with different formulas for 3 h. (D) Western blot analysis of GPX4, SLC7A11 and ACSL4 expression in 4T1 cells after the treatment with different formulas. (E–G) RT-qPCR analysis of GPX4, SLC7A11 and ACSL4 gene expression in 4T1 cells after the treatment with different formulas for 24 h. (H) Intracellular MDA level of 4T1 cells after the treatment with different formulas for 24 h. Drug dose: Ce6 10 μg mL−1, Fe3O4 12.5 μg mL−1, Fe3O4-PLGA-Ce6 NPs with 10 μg mL−1 Ce6, Ce6 + L 10 μg mL−1 and Fe3O4-PLGA-Ce6 + L with 10 μg mL−1 Ce6. L represents laser. | ||
GPX4, which is inactivated with the consumption or depletion of GSH, is the key regulator of ferroptosis.56,57 SLC7A11, which mediates cystine uptake and glutamate release, is essential for iron overload-mediated ferroptosis.58 ACSL4 induces the accumulation of lipid intermediates and can also be used as an indicator to predict ferroptosis.59 Therefore, we evaluated the protein levels of GPX4, SLC7A11 and ACSL4 after treatment with different formulas. As expected, the protein levels of GPX4 and SLC7A11 were slightly downregulated when 4T1 cells were treated with Fe3O4-PLGA-Ce6 NPs compared with control or free Ce6 group, as shown in Fig. 4D, indicating the occurrence of ferroptosis. Compared with Fe3O4-PLGA-Ce6 NPs treatment, the protein expression levels of GPX4 and SLC7A11 were significantly downregulated after treatment with Fe3O4-PLGA-Ce6 NPs plus laser irradiation, indicating that ferroptosis can be boosted by PDT. Moreover, the protein levels of ACSL4 were obviously upregulated in 4T1 cells treated with Fe3O4-PLGA-Ce6 NPs and Fe3O4 compared with control or free Ce6-treated cells, showing that the released iron ions participated in the Fenton reaction in 4T1 cells to induce ferroptosis (Fig. 4D). In addition, the protein levels of ACSL4 were the highest in 4T1 cells treated with Fe3O4-PLGA-Ce6 NPs plus laser irradiation among all formulas, again indicating that ferroptosis can be boosted by PDT. The results of the gene assay in Fig. 4E–G supported that Fe3O4-PLGA-Ce6 NPs induced ferroptosis, which can be enhanced by PDT in 4T1 cells.
Since the production and accumulation of LPO are related to iron concentration, we investigated Fe2+ release and accumulation of Fe3O4-PLGA-Ce6 NPs under laser irradiation. As shown in Fig. S8,† the released content of iron was the highest in the Fe3O4-PLGA-Ce6 plus laser irradiation group as detected by atomic absorption spectroscopy (AAS). In addition, we investigated ferrous ion accumulation in 4T1 cells with a Lillie ferrous staining assay (Fig. S9†). More light blue foci were observed in the Fe3O4-PLGA-Ce6 NPs group than in the Fe3O4 group, showing that the intracellular acidic environment was conducive to lysis of the Fe3O4-PLGA-Ce6 NPs. Importantly, more light blue foci were observed in the Fe3O4-PLGA-Ce6 plus laser irradiation group than in the Fe3O4-PLGA-Ce6 without laser irradiation group, which was evidence of an increase in ferrous ions. Thus, we deduced that the photochemical process promoted iron ion release and then induced ferroptosis. MDA levels were used to evaluate the lipid peroxidation level. As shown in Fig. 4H, MDA levels dramatically increased in the Fe3O4-PLGA-Ce6 NPs plus laser irradiation group compared to the Fe3O4-PLGA-Ce6 NPs group, suggesting that laser irradiation can promote the generation of LPO. Compared to the Ce6 plus laser irradiation group, the MDA level was dramatically increased in the Fe3O4-PLGA-Ce6 NPs plus laser irradiation group, suggesting that the release of iron ions contributes to the generation of LPO. Together, these results provide evidence for the occurrence of cell death by ferroptosis, which can be boosted by PDT.
Tumor-targeting ability and safety in vivo
Before the in vivo studies, we assessed tumor penetration ability. The penetration efficiency of the Fe3O4-PLGA-Ce6 NPs was recorded by CLSM Z-stack scanning at a scanning depth of 60 μm, as shown in Fig. S10.† The red fluorescence intensity in the multicellular spheroids (MCSs) increased with time and scanning depth, which showed the optimum penetration efficiency of Fe3O4-PLGA-Ce6 NPs. Generally, the excellent penetration ability of Fe3O4-PLGA-Ce6 NPs provides a high opportunity to enter tumor tissue in vivo.Encouraged by the ferroptosis and photodynamic synergistic effects in vitro, we evaluated the tumor-targeting and retention ability of Fe3O4-PLGA-Ce6 NPs in 4T1 tumor-bearing mice model. Ce6 loading into the Fe3O4-PLGA-Ce6 NPs endowed fluorescence imaging ability. Thus, free Ce6 and Fe3O4-PLGA-Ce6 NPs were prepared and injected into 4T1 tumor-bearing mice for observation at different times. As shown in Fig. 5A, the fluorescent signal from free Ce6 appeared in the tumor area until 6 h, reached its highest value at 48 h, and then decreased gradually due to in vivo metabolism. However, the fluorescence intensity of the Fe3O4-PLGA-Ce6 NPs significantly increased within 48 h in the tumor site and remained almost unchanged after 48 h, indicating the strong targeting and retention ability of the Fe3O4-PLGA-Ce6 NPs in the tumor site. The corresponding results were confirmed by the fluorescent images of the ex vivo tumors and major organs at 72 h post-injection, as shown in Fig. 5B. The fluorescence intensity in the Fe3O4-PLGA-Ce6 NPs group was stronger than that in the free Ce6 group, indicating the good retention ability of the Fe3O4-PLGA-Ce6 NPs. To further confirm the tumor-targeting and retention effects, the accumulation ability of free Ce6 and Fe3O4-PLGA-Ce6 NPs in ex vivo tumors was evaluated by slice staining (Fig. S11†). A substantial number of Fe3O4-PLGA-Ce6 NPs were observed to accumulate in the tumor site compared to free Ce6, indicating the better tumor targeting and retention ability of Fe3O4-PLGA-Ce6 NPs.
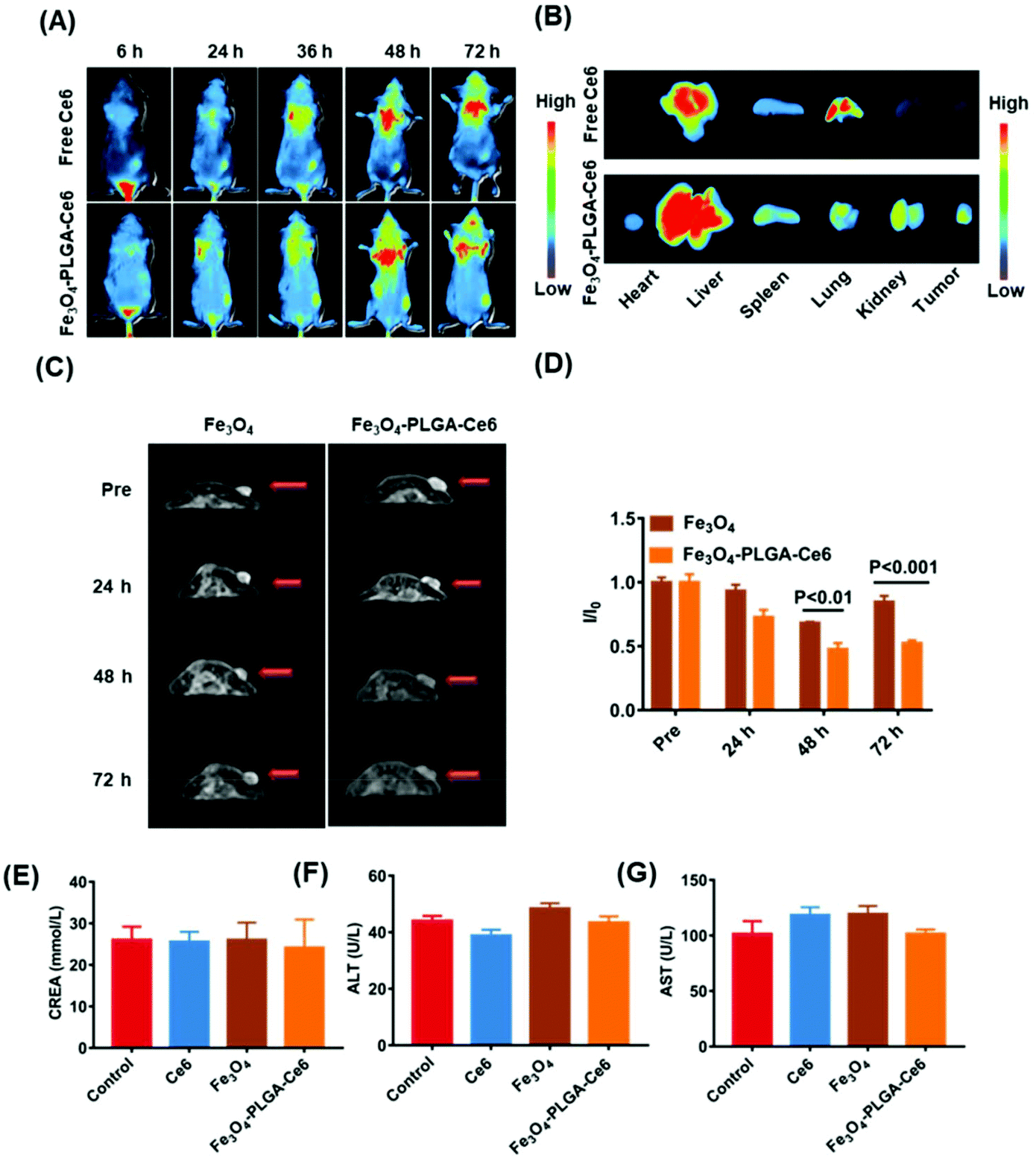 | ||
| Fig. 5 Tumor-targeting ability and safety in vivo. (A) In vivo fluorescence images of free Ce6 and Fe3O4-PLGA-Ce6 NPs in 4T1 tumor-bearing female mice. (B) Ex vivo fluorescence images of free Ce6 and Fe3O4-PLGA-Ce6 NPs distribution in tumor and main organs at 72 h post-injection. (C) In vivo T2-weighted MR image. (D) I/I0 signal intensity ratio level of tumor area in the T2-weighted MR imaging. (E–G) The CREA, ALT and AST level in healthy BALB/c mice after treated with different formulas. Drug dose: Ce6 5 mg kg−1, Fe3O4 6.25 mg kg−1, Fe3O4-PLGA-Ce6 NPs with 5 mg kg−1 Ce6. Data are given as means ± SD (n = 4). | ||
In vitro experiments showed that Fe3O4-PLGA-Ce6 NPs could be successfully used as a T2-weighed contrast agent due to Fe3O4 loading. Thus, we next evaluated the tumor-targeting performance of Fe3O4 and Fe3O4-PLGA-Ce6 NPs in vivo by MRI, as shown in Fig. 5C and D. The T2-weighted imaging signal intensity of the free Fe3O4 group decreased at 48 h in the tumor site and then gradually increased, likely due to iron clearance. After Fe3O4-PLGA-Ce6 NPs injection, the T2-weighted imaging signal intensity in the tumor area showed a better contrast effect than that in the Fe3O4 group at 48 h and remained excellent T2 contrast effect until 72 h, which again demonstrated the success of utilizing Fe3O4-PLGA-Ce6 NPs for tumor-targeted delivery. To further observe iron accumulation in the tumor site, ex vivo slice staining images are shown in Fig. S12.† More iron ions accumulated in the tumor tissue of the Fe3O4-PLGA-Ce6 NPs group than in the Fe3O4 group at 72 h. These results demonstrated that Fe3O4-PLGA-Ce6 NPs could be not only a promising MRI T2 contrast agent for solid tumors to guide the synergism of ferroptosis and PDT but also a successful tumor-targeting agent.
Next, we systemically evaluated the safety of the different formulas in vivo. First, the hemolytic activity of the Fe3O4-PLGA-Ce6 NPs was evaluated by a hemolysis assay. As shown in Fig. S13,† after coincubation with Fe3O4-PLGA-Ce6 NPs and RBCs, no significant hemolytic toxicity was observed in the photographs of the centrifuged RBC solutions or according to the hemolysis ratios, indicating the biosafety of the Fe3O4-PLGA-Ce6 NPs. In addition, we investigated the in vivo biocompatibility of Ce6, Fe3O4 and Fe3O4-PLGA-Ce6 NPs in BALB/c mice. After injection of different formulas, blood samples were taken on the first and seventh days and were used to evaluate the acute toxicity and chronic toxicity, respectively. Nine blood indexes, including white blood cell (WBC) count, RBC count, red cell distribution width (RDW), routine blood levels of hematocrit (HCT), mean platelet volume (MPV), monocytes (MONs), mean corpuscular volume (MCV), platelet volume distribution width (PDW), and thrombocytocrit (PCT), of the three groups were tested by a hematology analyzer. The nine indexes mentioned above showed no obvious abnormalities in the treatment groups compared with the control group on days 1 and 7 (Fig. S14†), indicating no significant adverse reactions in the hematochemistry of mice. No obvious fluctuation in mouse weights was observed during the 7-day evaluation of biocompatibility, as shown in Fig. S15.† From the in vivo biodistribution results, more aggregation was observed in the liver and kidney. Therefore, we chose two liver toxicity indicators and kidney indicators for serum biochemical parameter detection. As shown in Fig. 5E–G, the levels of creatinine (CREA), alanine aminotransferase (ALT) and aspartate aminotransferase (AST) also showed no significant abnormalities in the treatment groups compared with the control group. These results consistently suggested that certain doses of different formulas had good biological safety.
Ferroptosis and photodynamic synergistic efficacy in vivo
Having acquired admirable permeability and accumulation efficiency results for Fe3O4-PLGA-Ce6 NPs in vivo, the synergistic anticancer effects were further explored. As shown in Fig. 6A, 4T1 tumor-bearing mice were i.v. injected with Ce6, Fe3O4 and Fe3O4-PLGA-Ce6 (n = 6 mice per group) on day 0, 3, 6 and 9. At 24 h post injection, the tumors were irradiated with a 660 nm laser (0.5 W cm−2) for 8 min. During the entire 12-day treatment period, different formulas were injected 3 times through the tail vein. Tumor volumes were monitored after injection of the different formulas, as shown in Fig. 6B. The group intravenously injected with PBS was defined as the negative control. Without laser irradiation, the group treated with Fe3O4-PLGA-Ce6 NPs showed stronger tumor inhibition efficacy than the group treated with free Fe3O4 and Ce6 with regards to the accumulation and retention of nanoparticles. Ce6 plus laser irradiation also showed moderate antitumor ability because of the PDT efficacy. The antitumor effects of the Fe3O4-PLGA-Ce6 NPs plus laser irradiation group were the best, indicating the effectiveness of ferroptosis/PDT combination therapy. Tumor-bearing mice were then sacrificed for tumor harvest. The volumes of the tumors in the combination therapy group were the smallest, as shown in the photographs in Fig. 6C after 12 days of treatment. Correspondingly, the tumor inhibition rates were calculated for the Ce6 (19.8%), Fe3O4 (32.5%), Fe3O4-PLGA-Ce6 (53.5%), Ce6 plus laser (65.5%) and Fe3O4-PLGA-Ce6 plus laser groups (92.4%), as shown in Fig. 6D. As expected, the tumor inhibition rate was the highest after treatment with the Fe3O4-PLGA-Ce6 NPs plus laser, which was consistent with the in vitro results. All of these results indicated the effectiveness of ferroptosis/PDT combination therapy. During the treatment period, the body weights of the BALB/c mice treated with the different formulas had no appreciable change, implying the safety of the Fe3O4-PLGA-Ce6 NPs in vivo (Fig. 6E). In addition, the major organs (including the heart, liver, spleen, kidneys, and lungs) were also collected after 12 days of different treatments for H&E analysis. No obvious histopathological abnormalities were found in these tissue sections, suggesting negligible adverse toxicity of the Fe3O4-PLGA-Ce6 NPs (Fig. S16†).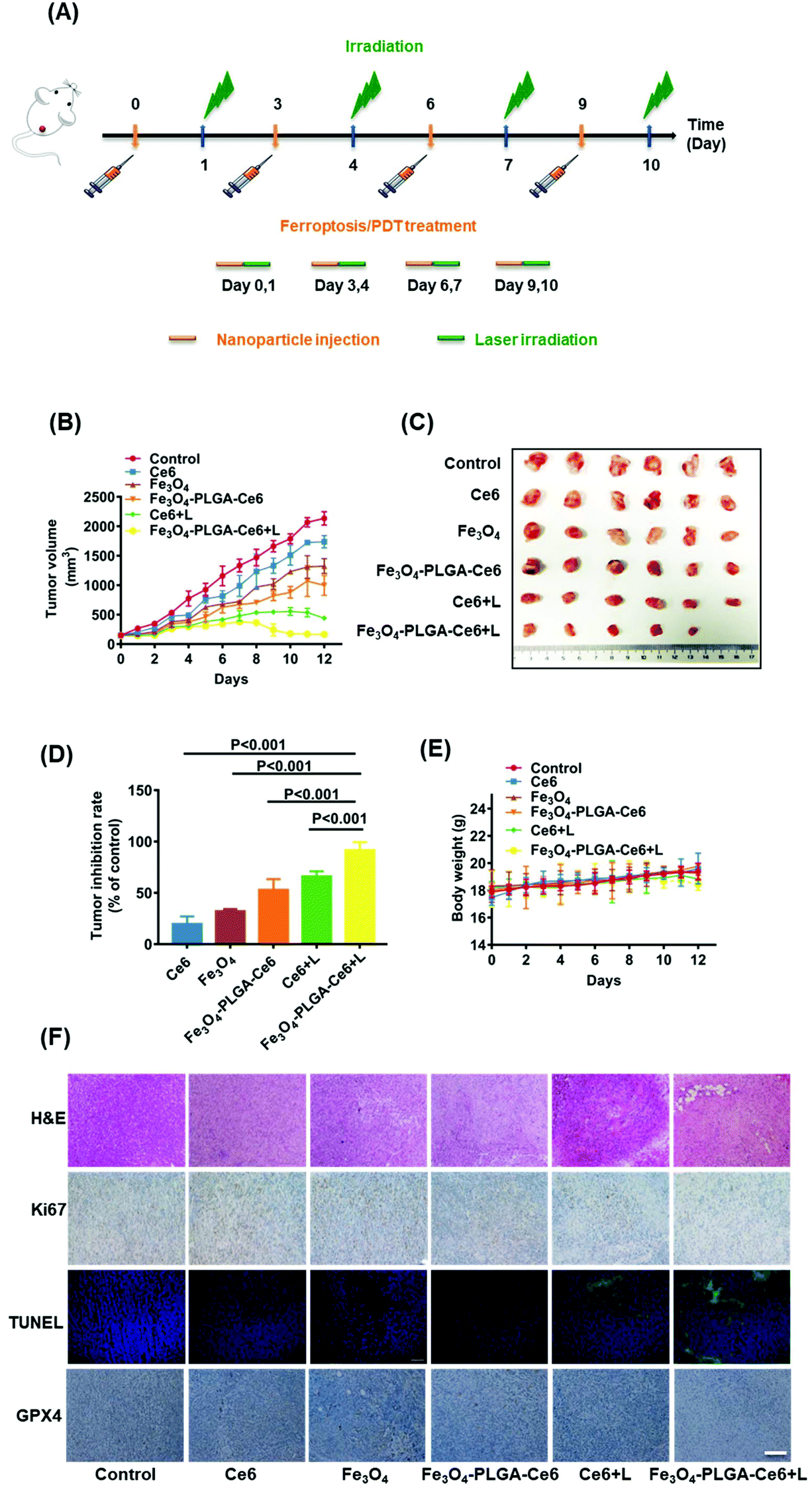 | ||
| Fig. 6 Ferroptosis and photodynamic synergistic efficacy in vivo. (A) Timeline of the ferroptosis/PDT treatment. 4T1 tumor-bearing mice were treated with three injections of Ce6, Fe3O4, or Fe3O4-PLGA-Ce6 on 0, 3, 6 and 9 day. Laser irradiation was performed on 1, 4, 7 and 10 day. (B) Tumor growth curves for 4T1 tumor-bearing female mice during 12 days treatment. (C) Photographs of harvested tumors from 4T1 tumor-bearing female mice after 12 days of different treatments. (D) Tumor inhibition rate of mice treated with different formulas. (E) Animal weight changes of different treatment during 12 days. (F) Histological observation of the tumor tissues with H&E, Ki67 staining, TUNEL and GPX4 staining from 12 days of different treatments. The bar: 150 μm. Drug dose: Ce6 5 mg kg−1, Fe3O4 6.25 mg kg−1, Fe3O4-PLGA-Ce6 NPs with 5 mg kg−1 Ce6, Ce6 + L 5 mg kg−1 and Fe3O4-PLGA-Ce6 NPs + L with 5 mg kg−1 Ce6. L represents laser. Data shown as means ± SD (n = 6). | ||
Subsequently, H&E staining images of the tumor tissue verified the superior therapeutic effects of the Fe3O4-PLGA-Ce6 NPs plus laser group (Fig. 6F). Almost all of the cancer cells maintained complete morphology in all groups without laser irradiation. In contrast, most tumor cells lost their cellular integrity after treatment with laser irradiation. To further understand the tumor inhibition mechanism of the Fe3O4-PLGA-Ce6 NPs plus laser group, we conducted immunohistochemistry analysis by Ki-67 and TUNEL assays. A similar tendency of tumor cell proliferation inhibition could be observed in Ki-67 and TUNEL expression levels. Moreover, the GPX4 expression level in the Fe3O4-PLGA-Ce6 NPs plus laser group was significantly lower than that of the other groups, which revealed that the ferroptosis was attributed to the therapeutic effects in vivo. These results strongly support the therapeutic effects of Fe3O4-PLGA-Ce6 NPs plus laser irradiation through the synergistic effects of ferroptosis and PDT.
Conclusions
In this study, we constructed self-assembled Fe3O4-PLGA-Ce6 NPs through chelation and hydrophobic interactions. These nanoparticles allowed fluorescence imaging and MRI guidance due to Ce6 and Fe3O4 loading, respectively. 4T1 cells treated with Fe3O4-PLGA-Ce6 NPs plus laser irradiation could significantly increase the intracellular ROS concentration and thus induce severe cell death. We concluded that the Fe3O4-PLGA-Ce6 NPs plus laser irradiation induced cell death that coincided with the features of ferroptosis in vitro, such as depletion of GSH, generation of LPO, reduction in the levels of GPX4 and SLC7A11 and an increase the level of ACSL4. The Ce6 and Fe3O4 cargo significantly induced synergistic PDT and ferroptosis because of the good permeability and retention effect of the formed nanoparticles after intravenous injection. Therefore, superior therapeutic effects with few abnormalities were achieved in a 4T1 tumor-bearing model. These satisfactory results suggested that the combination therapy strategy was feasible and demonstrated that Fe3O4-PLGA-Ce6 NPs could be a promising type of adjuvant nanomedicine for future clinical translation.Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Southwest University and experiments were approved by the Animal Ethics Committee of Southwest University.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by Fundamental Research Funds for the Central Universities (XDJK 2020C062) and National Natural Science Foundation of China (21976145 and 21602180).References
- H. Kostrhunova, J. Zajac, L. Markova, V. Brabec and J. Kasparkova, Angew. Chem., 2020, 59, 21157–21162 CrossRef CAS.
- S. J. Isakoff, Cancer J., 2010, 16, 53–61 CrossRef CAS.
- S. E. Santana-Krimskaya, M. A. Franco-Molina, D. G. Zarate-Trivino, H. Prado-Garcia, P. Zapata-Benavides, F. Torres-Del-Muro and C. Rodriguez-Padilla, Biomed. Pharmacother., 2020, 126, 110062 CrossRef CAS.
- Y. J. Ni, K. R. Schmidt, B. A. Werner, J. K. Koenig, I. H. Guldner, P. M. Schnepp, X. J. Tan, L. Jiang, M. Host, L. H. Sun, E. N. Howe, J. M. Wu, L. E. Littlepage, H. Nakshatri and S. Y. Zhang, Nat. Commun., 2019, 10, 2860 CrossRef.
- W. D. Foulkes, I. E. Smith and J. S. Reis, N. Engl. J. Med., 2010, 363, 1938–1948 CrossRef CAS.
- G. Bianchini, J. M. Balko, I. A. Mayer, M. E. Sanders and L. Gianni, Nat. Rev. Clin. Oncol., 2016, 13, 674–690 CrossRef CAS.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, CA Cancer J. Clin., 2011, 61, 250–281 CrossRef.
- J. Dang, H. He, D. Chen and L. Yin, Biomater. Sci., 2017, 5, 1500–1511 RSC.
- C. D. Phung, T. H. Tran, L. M. Pham, H. T. Nguyen, J. H. Jeong, C. S. Yong and J. O. Kim, J. Controlled Release, 2020, 324, 413–429 CrossRef CAS.
- M. Y. Peng, D. W. Zheng, S. B. Wang, S. X. Cheng and X. Z. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 13965–13975 CrossRef CAS.
- J. Xu, L. Xu, C. Wang, R. Yang, Q. Zhuang, X. Han, Z. Dong, W. Zhu, R. Peng and Z. Liu, ACS Nano, 2017, 11, 4463–4474 CrossRef CAS.
- Z. Dong, H. Gong, M. Gao, W. Zhu, X. Sun, L. Feng, T. Fu, Y. Li and Z. Liu, Theranostics, 2016, 6, 1031–1042 CrossRef CAS.
- K. Lu, C. He, N. Guo, C. Chan, K. Ni, R. R. Weichselbaum and W. Lin, J. Am. Chem. Soc., 2016, 138, 12502–12510 CrossRef CAS.
- D. W. Zheng, J. X. Fan, X. H. Liu, X. Dong, P. Pan, L. Xu and X. Z. Zhang, Adv. Mater., 2018, 30, e1801622 CrossRef.
- S. J. Dixon and B. R. Stockwell, Nat. Chem. Biol., 2014, 10, 9–17 CrossRef CAS.
- J. L. Roh, E. H. Kim, H. J. Jang, J. Y. Park and D. Shin, Cancer Lett., 2016, 381, 96–103 CrossRef CAS.
- X. Sun, Z. Ou, R. Chen, X. Niu, D. Chen, R. Kang and D. Tang, Hepatology, 2016, 63, 173–184 CrossRef CAS.
- M. J. Kerins, J. Milligan, J. A. Wohlschlegel and A. Ooi, Cancer Sci., 2018, 109, 2757–2766 CrossRef CAS.
- M. Luo, L. Wu, K. Zhang, H. Wang, T. Zhang, L. Gutierrez, D. O'Connell, P. Zhang, Y. Li, T. Gao, W. Ren and Y. Yang, Cell Death Differ., 2018, 25, 1457–1472 CrossRef CAS.
- S. Ma, E. E. Henson, Y. Chen and S. B. Gibson, Cell Death Dis., 2016, 7, e2307 CrossRef CAS.
- N. Eling, L. Reuter, J. Hazin, A. Hamacher-Brady and N. R. Brady, Oncoscience, 2015, 2, 517–532 CrossRef.
- Z. Y. Shen, J. B. Song, B. C. Yung, Z. J. Zhou, A. G. Wu and X. Y. Chen, Adv. Mater., 2018, 30, e1704007 CrossRef.
- C. Liang, X. L. Zhang, M. S. Yang and X. C. Dong, Adv. Mater., 2019, 31, e1904197 CrossRef.
- M. Sang, R. Luo, Y. Bai, J. Dou, Z. Zhang, F. Liu, F. Feng, J. Xu and W. Liu, Theranostics, 2019, 9, 6209–6223 CrossRef CAS.
- S. Zhao, X. Yu, Y. Qian, W. Chen and J. Shen, Theranostics, 2020, 10, 6278–6309 CrossRef CAS.
- M. Jaganjac, S. B. Sunjic and N. Zarkovic, Antioxidants, 2020, 9, 191 CrossRef CAS.
- P. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang, Z. Gu and J. Lin, Nano Lett., 2017, 17, 928–937 CrossRef CAS.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS.
- T. Liu, W. Liu, M. Zhang, W. Yu, F. Gao, C. Li, S. B. Wang, J. Feng and X. Z. Zhang, ACS Nano, 2018, 12, 12181–12192 CrossRef CAS.
- K. Jiang, Y. Chen, D. Zhao, J. Cheng, F. Mo, B. Ji, C. Gao, C. Zhang and J. Song, Nanoscale, 2020, 12, 18682–18691 RSC.
- J. Cheng, A. Kenaan, D. Zhao, D. Qi and J. Song, Nanomedicine, 2020, 30, 102286 CrossRef CAS.
- F. Zhang, F. Li, G. H. Lu, W. D. Nie, L. J. Zhang, Y. L. Lv, W. E. Bao, X. Y. Gao, W. Wei, K. Y. Pu and H. Y. Xie, ACS Nano, 2019, 13, 5662–5673 CrossRef CAS.
- W. M. Wang, M. Green, J. E. Choi, M. Gijon, P. D. Kennedy, J. K. Johnson, P. Liao, X. T. Lang, I. Kryczek, A. Sell, H. J. Xia, J. J. Zhou, G. P. Li, J. Li, W. Li, S. Wei, L. Vatan, H. J. Zhang, W. Szeliga, W. Gu, R. Liu, T. S. Lawrence, C. Lamb, Y. Tanno, M. Cieslik, E. Stone, G. Georgiou, T. A. Chan, A. Chinnaiyan and W. P. Zou, Nature, 2019, 569, 270–274 CrossRef CAS.
- B. W. Yang, Y. Chen and J. L. Shi, Chem. Rev., 2019, 119, 4881–4985 CrossRef CAS.
- Y. Zou and S. L. Schreiber, Cell Chem. Biol., 2020, 27, 463–471 CrossRef CAS.
- Z. Shen, T. Liu, Y. Li, J. Lau, Z. Yang, W. Fan, Z. Zhou, C. Shi, C. Ke, V. I. Bregadze, S. K. Mandal, Y. Liu, Z. Li, T. Xue, G. Zhu, J. Munasinghe, G. Niu, A. Wu and X. Chen, ACS Nano, 2018, 12, 11355–11365 CrossRef CAS.
- J. P. Wei, J. C. Li, D. Sun, Q. Li, J. Y. Ma, X. L. Chen, X. Zhu and N. F. Zheng, Adv. Funct. Mater., 2018, 28, 1706310 CrossRef.
- Z. Wei, P. Liang, J. Xie, C. Song, C. Tang, Y. Wang, X. Yin, Y. Cai, W. Han and X. Dong, Chem. Sci., 2019, 10, 2778–2784 RSC.
- H. Xiang, P. Dong, L. Pi, Z. Wang, T. Zhang, S. Zhang, C. Lu, Y. Pan, H. Yuan and H. Liang, J. Mater. Chem. B, 2020, 8, 1432–1444 RSC.
- G. Zhang, L. Zhang, Y. Si, Q. Li, J. Xiao, B. Wang, C. Liang, Z. Wu and G. Tian, Chem. Eng. J., 2020, 388, 124269 CrossRef CAS.
- P. Shou, Z. Yu, Y. Wu, Q. Feng, B. Zhou, J. Xing, C. Liu, J. Tu, O. U. Akakuru, Z. Ye, X. Zhang, Z. Lu, L. Zhang and A. Wu, Adv. Healthcare Mater., 2020, 9, e1900948 CrossRef.
- B. Yu, B. Choi, W. Li and D. H. Kim, Nat. Commun., 2020, 11, 3637 CrossRef CAS.
- W. Fei, D. Chen, H. Tang, C. Li, W. Zheng, F. Chen, Q. Song, Y. Zhao, Y. Zou and C. Zheng, Nanoscale, 2020, 12, 16738–16754 RSC.
- Y. C. Chang and D. H. Chen, J. Colloid Interface Sci., 2005, 283, 446–451 CrossRef CAS.
- M. Lattuada and T. A. Hatton, Langmuir, 2007, 23, 2158–2168 CrossRef CAS.
- S. K. Golombek, J. N. May, B. Theek, L. Appold, N. Drude, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2018, 130, 17–38 CrossRef CAS.
- H. Yu, J. M. Li, K. Deng, W. Zhou, C. X. Wang, Q. Wang, K. H. Li, H. Y. Zhao and S. W. Huang, Theranostics, 2019, 9, 7033–7050 CrossRef CAS.
- J. Li, X. Li, S. Gong, C. Zhang, C. Qian, H. Qiao and M. Sun, Nano Lett., 2020, 20, 4842–4849 CrossRef CAS.
- B. Li, T. Gong, N. Xu, F. Cui, B. Yuan, Q. Yuan, H. Sun, L. Wang and J. Liu, Small, 2020, 16, e2003969 CrossRef.
- F. Zhang, F. Li, G. H. Lu, W. Nie, L. Zhang, Y. Lv, W. Bao, X. Gao, W. Wei, K. Pu and H. Y. Xie, ACS Nano, 2019, 13, 5662–5673 CrossRef CAS.
- W. Bao, X. Liu, Y. Lv, G. H. Lu, F. Li, F. Zhang, B. Liu, D. Li, W. Wei and Y. Li, ACS Nano, 2019, 13, 260–273 CrossRef CAS.
- T. Xu, W. Ding, X. Ji, X. Ao, Y. Liu, W. Yu and J. Wang, J. Cell. Mol. Med., 2019, 23, 4900–4912 CrossRef.
- Z. Zou, H. Chang, H. Li and S. Wang, Apoptosis, 2017, 22, 1321–1335 CrossRef CAS.
- T. M. Seibt, B. Proneth and M. Conrad, Free Radicals Biol. Med., 2019, 133, 144–152 CrossRef CAS.
- S. Wang, F. Li, R. Qiao, X. Hu, H. Liao, L. Chen, J. Wu, H. Wu, M. Zhao, J. Liu, R. Chen, X. Ma, D. Kim, J. Sun, T. P. Davis, C. Chen, J. Tian, T. Hyeon and D. Ling, ACS Nano, 2018, 12, 12380–12392 CrossRef CAS.
- B. R. Stockwell, J. P. F. Angeli, H. Bayir, A. I. Bush, M. Conrad, S. J. Dixon, S. Fulda, S. Gascon, S. K. Hatzios, V. E. Kagan, K. Noel, X. J. Jiang, A. Linkermann, M. E. Murphy, M. Overholtzer, A. Oyagi, G. C. Pagnussat, J. Park, Q. Ran, C. S. Rosenfeld, K. Salnikow, D. L. Tang, F. M. Torti, S. V. Torti, S. Toyokuni, K. A. Woerpel and D. D. Zhang, Cell, 2017, 171, 273–285 CrossRef CAS.
- S. Doll, B. Proneth, Y. Y. Tyurina, E. Panzilius, S. Kobayashi, I. IngoId, M. Irmler, J. Beckers, M. Aichler, A. Walch, H. Prokisch, D. Trumbach, G. W. Mao, F. Qu, H. Bayir, J. Fullekrug, C. H. Scheel, W. Wurst, J. A. Schick, V. E. Kagan, J. P. F. Angeli and M. Conrad, Nat. Chem. Biol., 2017, 13, 91–98 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr08757b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |