
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterconnection of organic–inorganic hybrid nano-building blocks towards thermally robust mesoporous structures†
Naoki
Tarutani
 *abc,
Riona
Sato
b,
Wataru
Yamazaki
b,
Kiyofumi
Katagiri
a,
Kei
Inumaru
a and
Takamasa
Ishigaki
bc
*abc,
Riona
Sato
b,
Wataru
Yamazaki
b,
Kiyofumi
Katagiri
a,
Kei
Inumaru
a and
Takamasa
Ishigaki
bc
aApplied Chemistry Program, Graduate School of Advanced Science and Engineering, Hiroshima University, 1-4-1 Kagamiyama, Higashi-Hiroshima, Hiroshima 739-8527, Japan. E-mail: n-tarutani@hiroshima-u.ac.jp
bDepartment of Chemical Science and Technology, Faculty of Bioscience and Applied Chemistry, Hosei University, 3-7-2 Kajino-cho, Koganei, Tokyo 184-8584, Japan
cResearch Center for Micro-Nano Technology, Hosei University, 3-11-15 Midori-cho, Koganei, Tokyo 184-0003, Japan
First published on 23rd June 2021
Abstract
The use of organic–inorganic hybrid nanoparticles will enable a control of the characteristics of both the nanoparticles and constructed fine structures. In this study, we report the synthesis of acrylate-intercalated layered manganese, cobalt, and nickel hydroxide nanoparticles and their assembly into ordered mesoporous structures. Polymerization of the intercalated acrylates takes place by means of a radical initiator. The formed organic network improved the thermal stability of the layered hydroxides, which results in thermally robust mesoporous structures. Additionally, it is found that the polymerization can be initiated and progressed at 200 °C without any initiators for the layered nickel hydroxide system. This allows for the scalable solid-state thermal polymerization of intercalated acrylates and the formation of thermally robust hierarchically ordered meso/macroporous powders as well as mesoporous films. The electrochemical characterization reveals that the thermally robust mesoporous films having regulated mesopores allow for the effective diffusion of molecules/solvent compared with the films having collapsed mesoporous structures.
Introduction
Organic–inorganic hybrid materials have been widely investigated from the fundamental to applied science fields. The advantage of such hybrid materials is the exploitation of properties derived from both the organic and inorganic moieties.1 In the field of nanomaterials, especially nanoparticles, the organic moieties play a significant role. During formation of nanoparticles, organic molecules are coordinated and/or adsorbed to stabilize the particles, which enables tuning of the particle diameter from the single-nanometer to tens-of-nanometer scales.2,3 Block copolymer ligands give nano-scale reaction space, which allows control size distribution4,5 and shape of nanoparticles.6,7 Solvent philicity control of the nanoparticles by surface molecules results in hydrophilic, lipophilic, and amphiphilic materials.8 Assembly control of inorganic nanoparticles by an organic modifier has been reported to produce dissipative structures in addition to thermodynamically stable structures.9–12The use of organic–inorganic hybrid nanoparticles as nano-building blocks (NBBs) and the control of their assembly show great potential for the design of fine structures to produce novel functionalities. Organic-molecules-adsorbed cubic nanoparticles have been assembled to form nanoparticle bilayers and show tandem catalytic properties.13 Superlattices of molecule-adsorbed nanoparticles have been investigated as optical,14 phonon-engineering,15 and lithium-ion storage materials,16 owing to the nano scale periodicity.
Porous structures, especially mesoporous structures, are some of the most promising fine structures to improve the functionalities of materials. Increasing the number of accessible active sites will maximize the ability to exploit the intrinsic properties of the crystals, and the porous structures will provide extrinsic properties such as a sieving effect17,18 and confinement effect.19,20 The first study on the synthesis of mesoporous materials reported the use of a molecular precursor,21 and subsequent studies have reported using NBBs.22–24 Sub-10 nm-sized nanoparticles/nanoclusters have been employed as precursor NBBs. However, the chemical composition of NBBs has been limited to metal oxides and precious metals because of the high thermal stability required to remove the block-copolymer templates.25,26 Despite the high functionalities, it has remained a challenge to synthesize crystalline mesoporous materials composed of thermally unstable phases under air atmosphere, such as hydroxides, nitrides, and sulfides, even with the NBB assembly approach.
Layered double hydroxides (LDHs) are organic–inorganic hybrid materials on the molecular level and are potential candidates as NBBs toward mesoporous materials. LDHs are composed of electrostatic bonds between the organic and inorganic moieties, which allows for a more versatile chemical composition with analogous layered structures. It is known that layered hydroxides tend to form coarse μm-sized crystals owing to rapid crystallization in the lateral direction.27 Crystallization control of LDHs to a size scale of sub-10 nm has been reported by Kuroda et al., who used tripodal ligands as stabilizers,28 and these were assembled into mesoporous structures.29 However, it has remained a challenge to synthesize mesoporous materials using LDH NBBs with a wide variety of chemical compositions.30 Additionally, our group reported another route to synthesize LDH NBBs using epoxides as alkalinizing agents,31 and these were assembled into ordered mesoporous structures.32 Highly ordered mesoporous structures were prepared using not only LDH NBBs but also layered single metal hydroxide NBBs having a wide range of chemical compositions.33,34
In this study, we focused on the organic moiety of the layered manganese, cobalt, and nickel hydroxide NBBs towards thermally robust mesoporous structures (Fig. 1). Acrylic acid was employed as an anionic species that was intercalated in the layered hydroxides. The layered metal hydroxide nanocrystals worked as NBBs and formed periodic mesostructures. Polymerization of the acrylates to form organic networks was achieved through post-treatment. The robustness of the mesoporous structures against thermal treatment was found to be improved after formation of the organic network. The thermally robust NBB network enabled design of hierarchically highly ordered meso/macroporous structures.
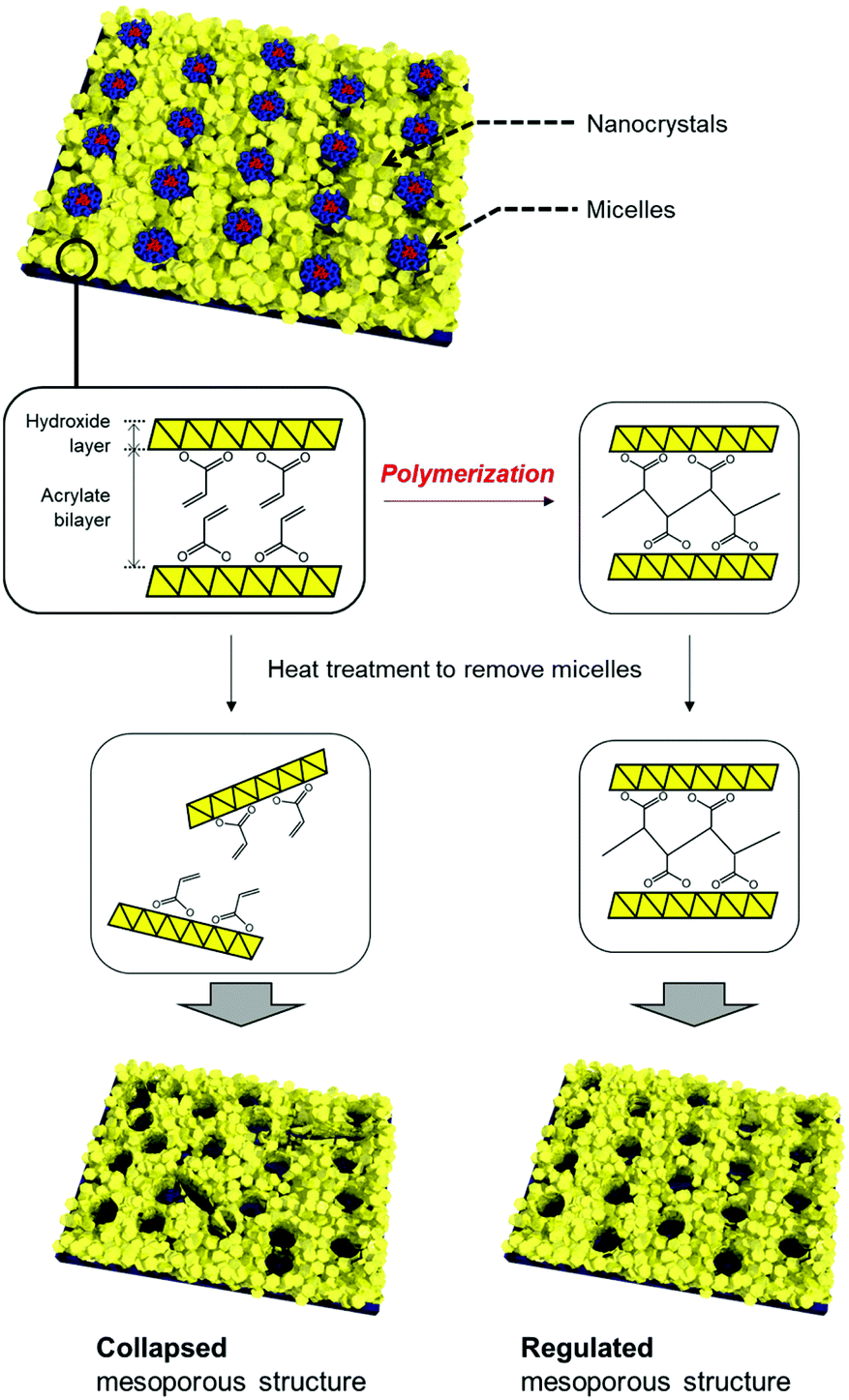 | ||
| Fig. 1 Schematic illustration of the synthetic strategy toward thermally robust mesoporous structures composed of layered metal hydroxide nanoparticles. | ||
Experimental
Chemicals
Manganese chloride tetrahydrate (MnCl2·4H2O, 99.0%), cobalt chloride hexahydrate (CoCl2·6H2O, 99.0%), nickel chloride hexahydrate (NiCl2·6H2O, 98.0%), acrylic acid (99%), ethanol (99.5%), propylene oxide (99.0%), Pluronic® F127 (F127), toluene (99.0%), 2,2′-azobis(isobutyronitrile) (AIBN, 98.0%), and polystyrene latex beads (10 wt% in H2O), deuterium oxide (D2O), deuterium chloride solution (35 wt% in D2O), ferrocene (95%), N,N-dimethylformamide (DMF, 99%), and tetrabutylammonium hexafluorophosphate (TBAPF6, 98%) were used as supplied. Acrylic acid, propylene oxide, F127, polystyrene latex beads, D2O, deuterium chloride solution, and TBAPF6 were purchased from Sigma-Aldrich Co. AIBN was purchased from Tokyo Chemical Industry Co., Ltd. Ferrocene and DMF were purchased from Nacalai tesque Inc. All other reagents were purchased from Wako Pure Chemicals Industries, Ltd. Ultrapure water with a resistivity of 18.2 × 106 Ω cm was used in all experiments.Thermally robust mesoporous layered metal hydroxide films through radical polymerization
MCl2·nH2O (M = Mn, Co, and Ni; n = 4–6), acrylic acid, and a specific amount of H2O were dissolved in 1.0 mL of ethanol (molar ratio of M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 1:6). Propylene oxide was added to the solution and stirred for 30 s. The amounts of metal salts, acrylic acid, and propylene oxide were 0.5, 2.0, and 7.5 mmol, respectively. The resultant homogenous solutions were left at room temperature (20–25 °C) for 6 h (M = Mn) or 60 min (M = Co and Ni). Ethanol (1 mL) with F127 (5.0 μmol) and H2O (3.0 mmol) was added to the solutions and stirred for 5 min. The obtained clear solutions were spin-coated on silicon substrates at 2000 rpm for 60 s. The dried films were immersed in a solution of AIBN dissolved in toluene (0.10 mol L−1, 8.0 mL) and heated at 65 °C for 20 h. Films were rinsed with toluene and heat treated at 250 °C for 6 h with a ramp rate of 1 °C min−1 under air atmosphere.
:H2O = 1:6). Propylene oxide was added to the solution and stirred for 30 s. The amounts of metal salts, acrylic acid, and propylene oxide were 0.5, 2.0, and 7.5 mmol, respectively. The resultant homogenous solutions were left at room temperature (20–25 °C) for 6 h (M = Mn) or 60 min (M = Co and Ni). Ethanol (1 mL) with F127 (5.0 μmol) and H2O (3.0 mmol) was added to the solutions and stirred for 5 min. The obtained clear solutions were spin-coated on silicon substrates at 2000 rpm for 60 s. The dried films were immersed in a solution of AIBN dissolved in toluene (0.10 mol L−1, 8.0 mL) and heated at 65 °C for 20 h. Films were rinsed with toluene and heat treated at 250 °C for 6 h with a ramp rate of 1 °C min−1 under air atmosphere.
Thermally robust mesoporous layered nickel hydroxide films through solid-state thermal polymerization
Spin-coated layered nickel hydroxide films were prepared according to the method written above. The as-dried films were heat treated in two steps, at 200 °C for 2 h and 250 °C for 6 h, in pre-heated ovens under air atmosphere. For a comparison, the as-dried films were heat treated in one step at 250 °C for 6 h in a pre-heated oven under air atmosphere.Solid-state thermal polymerization of acrylates intercalated in layered nickel hydroxide powders
NiCl2·6H2O (15 mmol) and acrylic acid (30 mmol) were dissolved in ethanol (30 mL). Propylene oxide (255 mmol) was added to the solution and stirred for 30 s. The resultant homogenous solutions were left at room temperature (20–25 °C) for 60 min. The obtained clear solutions were transferred to a glass Petri dish (φ 155 mm) and dried under atmospheric pressure for 14 h and vacuum condition for 1.5 h. The powders were heat treated at 200 °C for 2 h in pre-heated ovens under air atmosphere. For a comparison, as-dried powders were heat treated at 200 °C for 2 h under vacuum condition or at 150 °C for 20 h under air atmosphere.Thermally robust hierarchically ordered meso/macroporous materials through solid-state thermal polymerization
Layered nickel hydroxide powders were prepared according to the method written above. As-dried powders were redispersed in ethanol with a concentration of 100 g L−1. Redispersed colloids were mixed with ethanol including F127 (100 g L−1) at a volume ratio of 1:1 to prepare precursor solutions. Colloidal crystal templates were prepared by drying dilute polystyrene latex dispersed solutions (10 μL; volume ratio of reagent latex dispersed solution:H2O = 1:5) on silicon substrates at a temperature of 25 °C and relative humidity of 60 RH%. The colloidal crystal templates were heated to 100 °C on a hot plate. Prepared precursor solutions (2.0 μL) were injected into the colloidal crystal templates twice. Samples were heat treated at 200 °C for 2 h in a pre-heated oven under air atmosphere. Heat-treated samples were immersed in toluene and gently shaken for approximately 10 s and then dried at 100 °C. Dried samples were heat treated at 250 °C for 6 h with a ramp rate of 1 °C min−1 under air atmosphere.
Characterization
A field emission scanning electron microscope (SEM; S-8020, Hitachi, Japan) equipped with a transmittance SEM configuration was used to observe the meso-scale structures. The SEM was operated with a retarding mode of a low landing voltage of 1 kV (acceleration and deceleration voltage of 2.5 kV and 1.5 kV, respectively) to observe external surface structures without a structural change induced by an electron beam. An acceleration voltage of 30 kV was employed for the scanning transmission electron microscopy (STEM) mode observation. A transmission electron microscope (TEM; JEM-2100F, JEOL, Japan) operated with an acceleration voltage of 200 kV was employed to observe the NBBs that constructed the mesostructures. Powder X-ray diffraction (XRD) and small angle X-ray scattering (SAXS) measurements were used to characterize the crystal phases and meso-periodicities (Cu Kα radiation, SmartLab, Rigaku, Japan). The N2 adsorption–desorption technique was employed using a Belsorp-18 II instrument (MicrotracBEL Corp., Japan) to characterize the mesoporous structures. For measurement, samples were collected by scratching 20 films coated on glass substrates (76 mm × 26 mm). Samples were vacuum dried at 200 °C for 6 h prior to measurements. The specific surface area and the mesopore size distribution were estimated according to the Brunauer–Emmett–Teller (BET) method and the Barrett–Joyner–Halenda (BJH) method, respectively. Fourier transform infrared spectroscopy (IR) spectroscopy with an attenuated total reflectance attachment (FT/IR-6600, JASCO Corp., Japan) was used to characterize the chemical bond state of the samples. IR spectra were corrected considering the penetration depth of the evanescent wave. Raman spectroscopy (LabRAM HR Evolution, Horiba, Ltd, Japan) (532 nm laser of 25 mW) with a temperature control stage was also used to characterize the chemical bond states at a specific temperature under air atmosphere. Thermogravimetric-differential thermal analysis (TG-DTA; Thermo Plus Evo, Rigaku, Japan) was carried out to investigate the thermal behaviour at a ramp rate of 10 °C min−1 while continuously supplying air. 1H nuclear magnetic resonance (NMR) spectra were recorded using a Varian system500 spectrometer to characterize polymerization of acrylate species. The sample powders were dissolved in deuterium chloride-D2O solution.Electrochemical measurements
Electrochemical measurements were carried out in a conventional three-electrode cell configuration with 1.0 mol L−1 KOH aqueous solution as the electrolyte at room temperature. The electrolyte was N2 purged at 250 mL min−1 for 10 min before measurements. A platinum wire was used as counter electrode. Hg/HgO (filled with 1.0 mol L−1 KOH aqueous solution) and saturated calomel electrode (SCE) were used as the reference electrode. Sample films spin-coated on tin-doped indium oxide coated glass electrodes were used as the working electrode. The geometric area of the working electrode exposed to the electrolyte was 1 cm2. Double layer capacitances (Cdl) were determined by CV measurements within a potential window of −0.1–0.1 V vs. Hg/HgO in KOH solution with a scan rate of 40, 60, 80, and 100 mV s−1. Chronoamperometry measurements were performed to investigate electrolysis of ferrocene with an applied voltage of 0.5 V vs. SCE in DMF (25 mmol L−1 of ferrocene and 0.25 mol L−1 of PBAPF6) instead of KOH solution.Results and discussion
Radical polymerization of acrylates intercalated in layered metal hydroxides
As has been previously reported, the addition of propylene oxide to solutions dissolving metal salts induces an increasing solution pH.35 A carboxylic acid incorporated in the precursor solution showed the multiple roles of coordination, intercalation, and adsorption for the formation of stably dispersed carboxylate intercalated layered metal hydroxide NBBs (Metal = Mn, Fe, Co, Ni, and Cu).32,33 XRD patterns of the prepared samples were assigned as acrylate-intercalated layered metal hydroxides by considering the reported studies (Fig. S1†).34Fig. 2 shows SEM images of the heat-treated films prepared with/without AIBN treatment. Films prepared without the AIBN treatment step showed a mesoporous structure with numerous large cracks (Fig. 2a, c and e). On the other hand, well-regulated mesopores were observed for the films prepared with the AIBN treatment step (Fig. 2b, d and f). This clearly indicated that the AIBN treatment improved the thermal robustness of the mesostructures. | ||
| Fig. 2 SEM images of heat-treated films of (a) and (b) layered manganese hydroxides, (c) and (d) layered cobalt hydroxides, and (e) (f) layered nickel hydroxides. (a), (c) and (e) The films are AIBN treated before heat treatment. | ||
Fig. 3 shows the IR spectra of the films before and after AIBN treatment. The bands located at 1721, 1640, 1557, and 1431 cm−1 were assigned to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), ν(CC), ν(COO)as, and ν(COO)s, respectively. The bands of ν(CC) were broadened and weakened after AIBN treatment, which indicated free radicals attacked the CC bonds of the acrylate and initiated polymerization. The vibration differences between two ν(COO) bands, ΔCOO = ν(COO)as − ν(COO)s, provides information on the coordination environment of carboxy groups.36 The ΔCOO of acrylate-intercalated layered manganese, cobalt, and nickel hydroxides were 123, 123, and 129 cm−1, and increased to 187, 194, and 172 cm−1, respectively, after AIBN treatment. Considering that the ΔCOO of sodium acrylate (ionic bonding) is designated as 110–140 cm−1,37 the bond state of the acrylate species was changed from ionic to unidentate coordination during polymerization. As has been reported, the polymerization of acrylate enhanced the thermal stabilization of the layered structures of hydroxides.38,39 Therefore, polymerization of acrylate anions interconnected the hydroxide layers and NBBs, which led to an improvement of the thermal stability of the mesoporous structures.
O), ν(CC), ν(COO)as, and ν(COO)s, respectively. The bands of ν(CC) were broadened and weakened after AIBN treatment, which indicated free radicals attacked the CC bonds of the acrylate and initiated polymerization. The vibration differences between two ν(COO) bands, ΔCOO = ν(COO)as − ν(COO)s, provides information on the coordination environment of carboxy groups.36 The ΔCOO of acrylate-intercalated layered manganese, cobalt, and nickel hydroxides were 123, 123, and 129 cm−1, and increased to 187, 194, and 172 cm−1, respectively, after AIBN treatment. Considering that the ΔCOO of sodium acrylate (ionic bonding) is designated as 110–140 cm−1,37 the bond state of the acrylate species was changed from ionic to unidentate coordination during polymerization. As has been reported, the polymerization of acrylate enhanced the thermal stabilization of the layered structures of hydroxides.38,39 Therefore, polymerization of acrylate anions interconnected the hydroxide layers and NBBs, which led to an improvement of the thermal stability of the mesoporous structures.
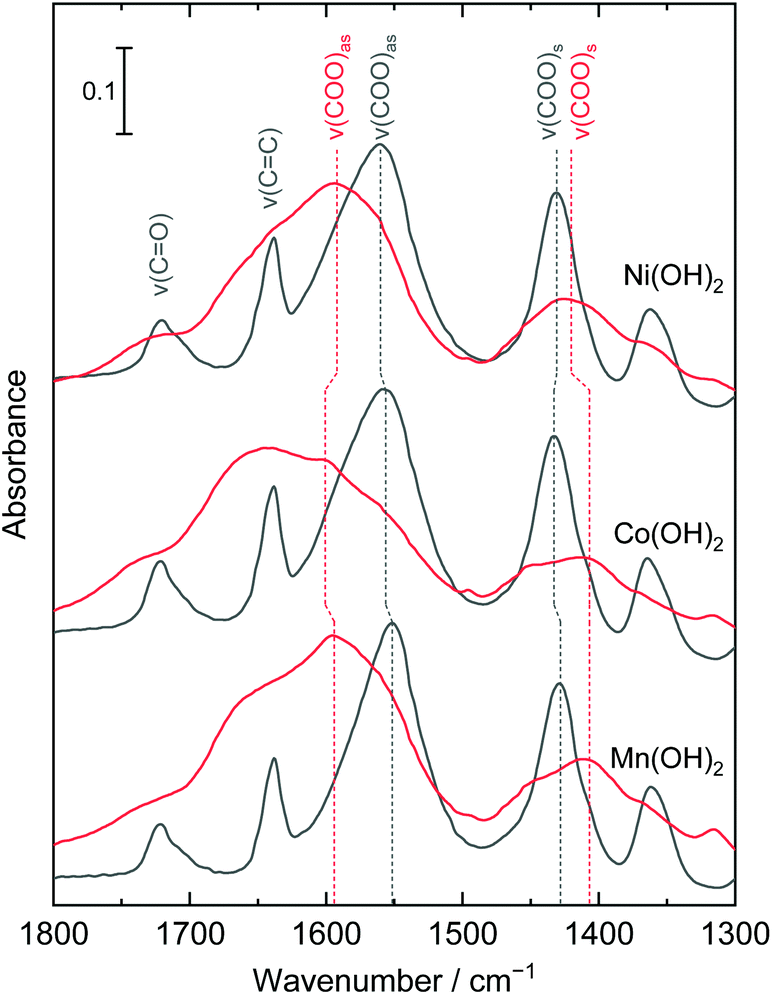 | ||
| Fig. 3 IR spectra of layered manganese, cobalt, and nickel hydroxide films before (black) and after (red) AIBN treatment. The bands are assigned considering previously reported studies.37,40–42 | ||
AIBN-initiated radical polymerization was a successful strategy to improve the thermal stability of mesoporous structures. However, polymerization of the organic moiety proceeded only in the case of films and not in the case of powders. Although polymerization needs diffusion of liquid/ions for the reaction to proceed, it may take a long time to penetrate the NBB interstices. As a result, the polymerization reaction was limited to the thin film scale, and was not applicable on the large bulk scale.
Solid-state thermal polymerization of acrylates intercalated in layered metal hydroxides
Acrylic acid is known to polymerize through a Michael addition reaction,43 which generates thermal energy and self-accelerates polymerization.44,45 It has been reported that alkali metal acrylates, alkali-earth metal acrylates, and transition metal acrylates show thermally induced solid-state polymerization.46,47 Considering these facts, the thermal behaviour of an acrylate-intercalated layered nickel hydroxide was investigated. Fig. 4 shows the TG-DTA curves of acrylate-intercalated layered nickel hydroxide powders. An exothermic event was detected at approximately 200 °C in the differential heat flow curve. An exothermic reaction at such a low temperature is similar to the thermal behaviour of transition metal acrylates47 and different from acrylate intercalated LDHs.38,39,48,49 The large amount of acrylate (acrylate/Ni ∼ 1.5 in molar ratio34) incorporated compared with conventional LDHs may change the thermal behaviour of materials. The exothermic reaction was not observed in the case of the powder after heat treatment at 200 °C for 2 h. This indicated that the reaction took place and completed within 2 h. Indeed, in situ Raman spectra indicated that the induction period of the thermal polymerization reaction was over the minute timescale (Fig. S2†).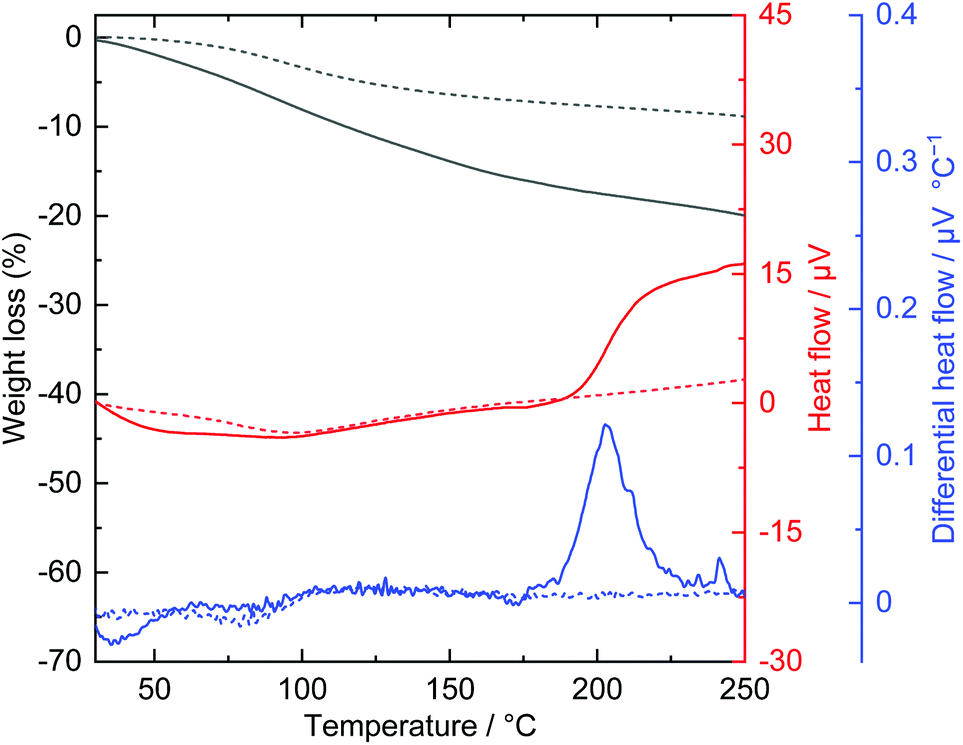 | ||
| Fig. 4 TG-DTA curves of as-dried (solid line) and heat treated at 200 °C (dot line) layered nickel hydroxide powders. | ||
The change of chemical bond condition was investigated using IR, Raman, and NMR spectroscopies (Fig. 5 and S3†). The part of detected band positions and assignments are listed in Table S1.† The IR spectra at a high frequency range of 3200–2700 cm−1 were obviously changed. The bands of ν(CH2)as, ν(CH2)s, and ν(CH) derived from the acrylate monomer disappeared and the band of ν(CH) derived from a polymeric species was detected. Although the Raman spectra showed the same features, the bands derived from the acrylate monomer species were still detected after heat treatment. In the lower frequency range of 1800–1050 cm−1, the ν(CC) bands derived from monomeric acrylate drastically decreased and the bands of τ(CH2) and ν(C–CH2) derived from polymeric species increased in both the IR and Raman spectra. These spectral changes indicated that polymerization of acrylate species took place during heat treatment at 200 °C. The ΔCOO were 121 cm−1 and 131 cm−1 before and after polymerization, which indicated that the ionic environment was identical during polymerization. The cumulative area ratios between the ν(CC) band and ν(COO)as band in the IR spectra, ACC/ACOO, decreased by an order of magnitude from 7.0 × 10–2 to 7.2 × 10−3 through heat treatment, which implied progress of polymerization reaction. The ACC/ACOO of the powders heat treated at 150 °C for 20 h under air atmosphere and at 200 °C for 2 h under vacuum condition were 4.8 × 10−2 and 6.2 × 10−2 (Fig. S4†). These results indicated the polymerization efficiently proceeded under an oxygen environment at a specific temperature. 1H NMR spectrum of powder treated at 200 °C showed signals of oligomeric and polymeric species (Fig. S3†). Considering cumulative signal area, 87% of monomers were reacted to form oligomers and polymers. Although degree of polymerization was deduced as low considering large signals of oligomers, it might be enough to interconnect each nanoparticle to improve robustness because nanoparticles close contact each other.
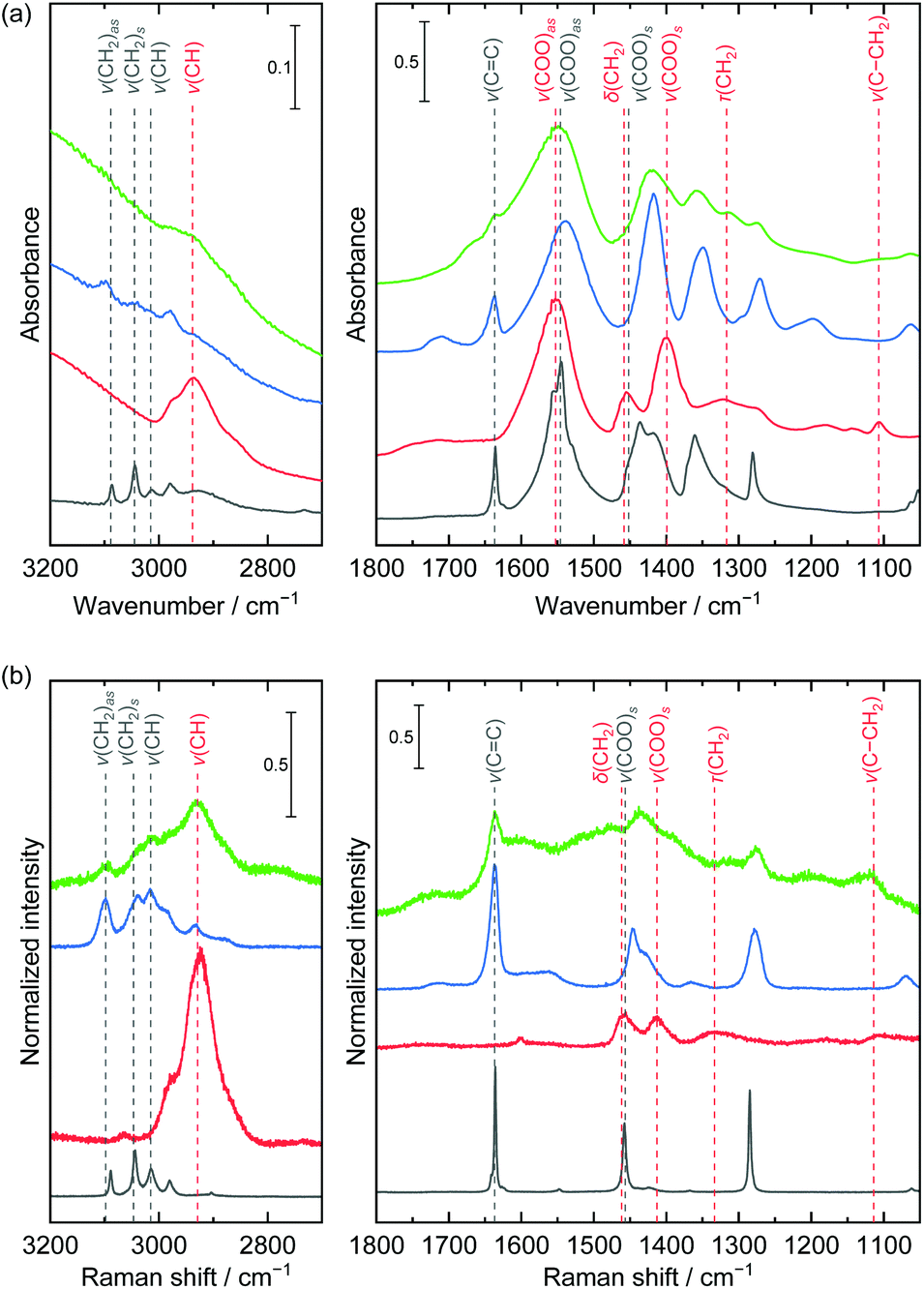 | ||
| Fig. 5 (a) IR and (b) Raman spectra of sodium acrylate (black), poly(acrylic acid sodium salt) (red), as-dried (blue) and heat treated at 200 °C (green) layered nickel hydroxide powders. The bands were assigned considering the previously reported studies.37,40–42 | ||
XRD patterns of the powders were identical before and after heat treatment at 200 and 250 °C (Fig. S5†). This indicated that the layered structures were thermally stable up to the decomposing temperature of the F127 micelles. The diffraction peaks assigned as 003 were shifted to a higher angle and the interlayer space decreased from 7.5 Å to 6.9 Å after heat treatment at 200 °C. Decrement of the interlayer space may correspond to the configuration change along with polymerization and/or elimination of the solvated species.50,51 This is the first report of solid-state thermal polymerization of anions included in layered metal hydroxides. Hypothetically, small-sized crystals (∼2 nm) enable the incorporation of relatively large amounts of anions and shorten the diffusion distance, which allows for the peculiar solid-state thermal polymerization.
Effect of solid-state thermal polymerization on robustness of mesoporous films
To investigate the effect of solid-state thermal polymerization on mesostructures, spin-coated films were employed. IR spectra of the films showed the same feature as the powder samples, where a band assigned as ν(CC) decreased by heat treatment at 200 °C (Fig. S6†). Fig. 6a shows the SEM image of the film heat treated in one step at 250 °C (not polymerized films). Mesopores with diameters of 6.5–10.5 nm were observed. In addition to the mesopores, nanometer-scale cracks were also observed over the entire area. The films heat treated in two steps at 200 °C and 250 °C (polymerized films) showed a regular mesopore size and orientation (Fig. 6b). TEM images revealed that the formed mesopores were surrounded by crystalline NBBs (Fig. 6c and d), which implied the formation of an ordered structure of NBBs and F127 micelles. Polymerized films showed monomodal size distributed mesopores (mode value of 7.6 nm) in the BJH pore size distribution, which agreed with the microscopy observations (average pore diameter of 8.5 nm) (Fig. S7†) and reported mesopore diameter templated using F127 and layered nickel hydroxide nanoparticles.52 Despite the different porous structures, the specific surface areas exhibited comparable values; not polymerized films were 131 m2 g−1 and polymerized films were 136 m2 g−1. Further analysis about mesopore area distribution indicated that polymerized film showed narrow pore area distribution and lower areal porosity (Fig. S8†), which implies robustness of the mesostructure. Although thermally robust mesoporous structures were successfully obtained in the case of layered nickel hydroxides, the layered manganese and cobalt hydroxide films showed collapsed mesostructures (Fig. S9†). Both of these hydroxides tended to oxidize at a lower temperature than the nickel hydroxides,51,53 which was proposed to lead to the collapse of the mesostructures before the polymerization reaction. Therefore, lower temperature polymerization process, such as AIBN-mediated process described above, is necessary for the layered manganese and cobalt hydroxide cases.
 | ||
| Fig. 6 (a) and (b) SEM and (c) and (d) TEM images of the films heat treated (a) in one step at 250 °C and (b)–(d) in two steps at 200 °C and 250 °C. Red arrows in (d) show mesopores. | ||
SAXS and XRD patterns of the films before and after solid-state polymerization and further heat treatment at 250 °C are shown in Fig. 7. The film before solid-state polymerization showed a peak at approximately 0.49 nm−1, which corresponded to a periodicity of 13 nm. The peak shifted to a larger scattering vector after solid-state polymerization and further heat treatment. Decreasing the periodicity means shrinking the matrix during heat treatment. The SAXS peak positions of the films prepared by one- or two-step heat treatment were comparable. The XRD pattern of the film obtained by two-step heat treatment was comparable to the pattern of the acrylate-intercalated layered nickel hydroxides. However, the pattern of the film prepared through a one-step heat treatment showed peaks assigned to NiO. Although the crystallite size of NiO (3.6 nm) was small enough to maintain mesoporous structures, dehydration may have led to the shrinkage of the matrixes and subsequent formation of cracks.54
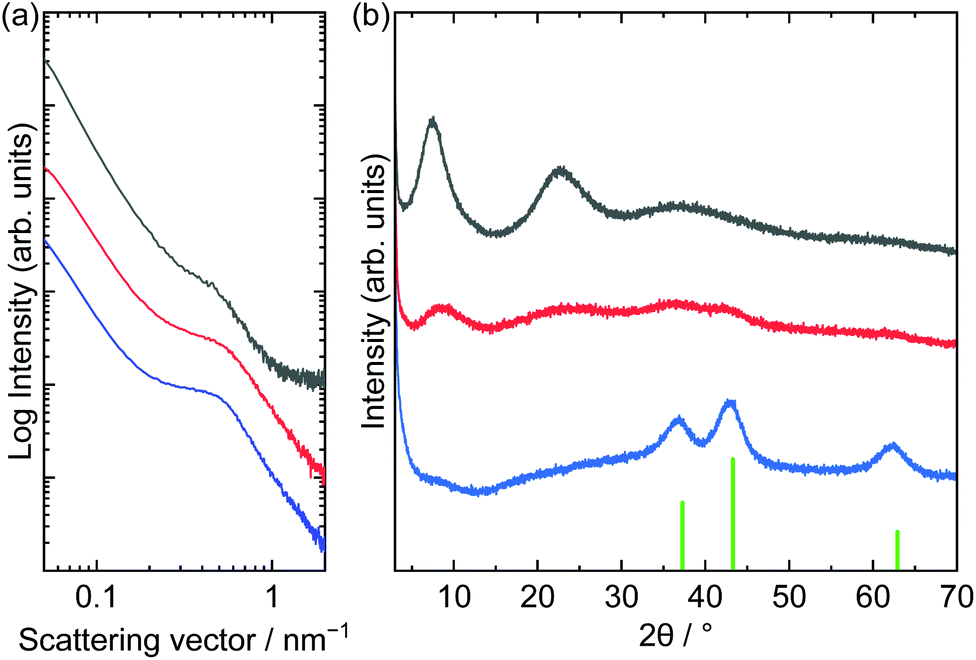 | ||
| Fig. 7 (a) SAXS and (b) XRD patterns of as-dried (black), two-step 200 °C and 250 °C heat treated (red) and one-step 200 °C heat treated (blue) films (green bar in (b): NiO (JCPDS: 00-047-1049)). | ||
In summary, thermally robust mesoporous structures composed of layered metal hydroxide nanoparticles were obtained by polymerization of intercalated/adsorbed acrylate species. Radical polymerization allowed low temperature reaction, which was applicable to all of layered manganese, cobalt, and nickel hydroxide systems. Although thermal polymerization route limited the applicable system as layered nickel hydroxide, it enabled to process large amount of sample which is difficult in the case of radical polymerization route.
Hierarchically ordered meso/macroporous materials
The dual templating approach, which uses micelle-based meso-templates and microparticle-based macro-templates, is a promising way to prepare hierarchically ordered porous materials. Polystyrene particles with measured diameters of 100, 240, and 720 nm formed hexagonal close-packed arrays by evaporating the suspensions (Fig. 8a and S10a, b†). The solution including NBBs and F127 was used to fill the interstices of polystyrene microparticles to form composites. The composites were thermally treated at 200 °C, immersed in toluene solution, and subsequently heat treated at 250 °C. The products showed a hexagonally ordered macroporous structure with a pore diameter of approximately 200 nm (Fig. 8b). TEM and STEM images revealed that the mesoporous structures with a pore dimeter of ∼7.5 nm were formed at the wall of the macroporous structures. Mesopores were found to be well aligned compared with the mesoporous structures discussed above. As has been reported, the alignment of micelles is affected by external factors such as the limitation of space.55 The thin macropore wall (thickness of 20–30 nm) may induced alignment of the F127 micelles and allow subsequent formation of hexagonally ordered mesoporous structures. Hierarchical porous materials with a macropore diameter of approximately 70 nm and 600 nm were also prepared through the same procedure (Fig. S10c–f†). | ||
| Fig. 8 (a) SEM images of the polystyrene latex beads template. (b) SEM, (c) TEM, and (d) STEM images of materials heat treated in two steps at 200 °C and 250 °C. | ||
Electrochemical characterization
Electrochemical measurement was employed to clarify the effect of polymerization of the NBBs. Solid-state thermally polymerized and not polymerized mesoporous layered nickel hydroxide films coated on tin-doped indium oxide coated glass were used as working electrodes. The Cdl were calculated as 2.53 mF cm−2 (solid-state polymerized film) and 2.36 mF cm−2 (not polymerized film) (Fig. S11†), which means electrochemical specific surface area was comparable. However, diffusion of solution/ion is expected to be limited in the case of not polymerized film because it included smaller pores due to collapsing the mesostructure. To investigate diffusion characteristics, electrolysis of ferrocene was performed. Chronoamperometry curves and Cottrell plots were shown in Fig. 9. Measured current was rapidly increased and gradually decreased with time. The currents were plotted against the time to the power of −1/2 considering Cottrell equation; i = −nFAD1/2c0π−1/2t−1/2, where i, n, F, A, D, c0, t are current, number of electrons, Faraday constant, electrode area, diffusion coefficient of redox species, initial concentration of redox species, and time, respectively. Here the slope is proportional to the diffusion coefficient. The slope, i.e. diffusion coefficient, of thermally polymerized film was about two times larger than the slope of not polymerized film. Considering that the small mesopores limit diffusion of solvent molecules due to strong interaction,19,20 it implies that the small pores produced as a result of collapsing the mesostructures retard diffusion of ferrocene/solvent molecules.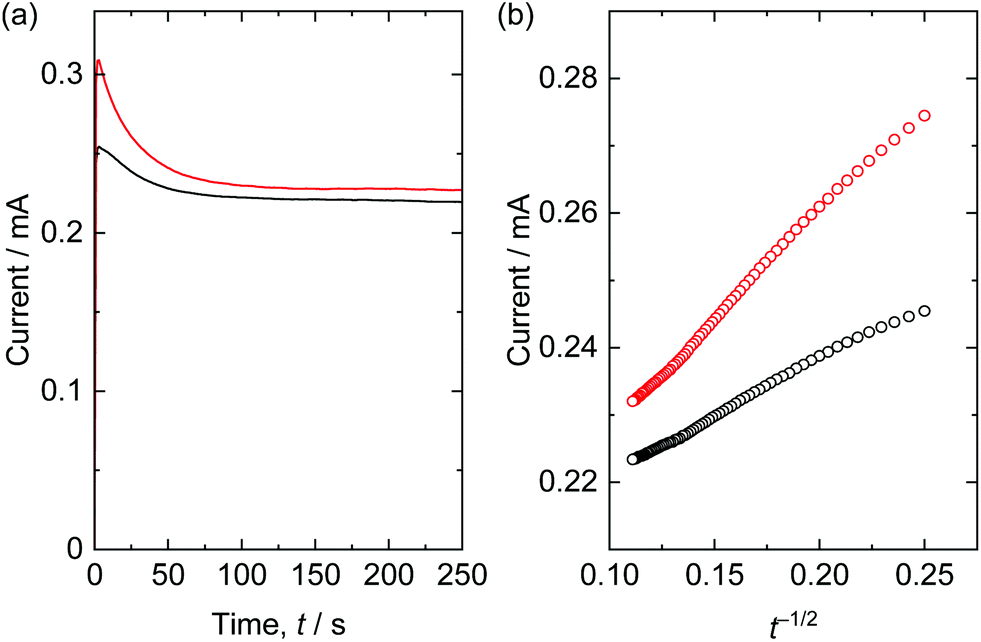 | ||
| Fig. 9 (a) Chronoamperometry curves and (b) Cottrell plots for electrolysis of ferrocene using synthesized working electrodes; mesoporous films prepared through one-step heat treatment (not polymerized, black) and two-step heat treatment (thermally polymerized, red). Applied voltage was 0.5 V vs. SCE. | ||
Conclusions
In this study, we demonstrated the preparation of thermally robust mesoporous layered hydroxide films by post-treatment. NBBs of acrylate-intercalated layered hydroxides of manganese, cobalt, and nickel were synthesized and assembled to form ordered mesostructures. Radical polymerization of the intercalated acrylates by an AIBN initiator improved the thermal stability of the hydroxides. A volume change of the hydroxide NBBs with heat treatment was suppressed and thermally robust mesoporous structures were obtained. In the case of layered nickel hydroxides, the polymerization of the intercalated acrylates successfully took place by simply heating at 200 °C without any initiator reagents. The solid-state thermal polymerization allowed the scalable reaction to progress, which led to the fabrication of hierarchically ordered meso/macroporous powders using micelle meso-templates and microparticle macro-templates. The electrochemical characterization revealed that molecules/solvent diffused effectively in the case of the regulated mesoporous structures compared with collapsed mesoporous structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present work is partially supported by JSPS KAKENHI Grant Number JP20K15368, JSPS Core-to-Core Program, MEXT Leading Initiative for Excellent Young Researchers, the Foundation for the Promotion of Ion Engineering, International Network on Polyoxometalate Science at Hiroshima University, and the Izumi Science and Technology Foundation (2019-J-112). We thank Prof. Riku Kawasaki (Hiroshima University) for helping NMR measurement and Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript.References
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS PubMed.
- L. T. Eunju, E. L. Kee, S. J. Jong and B. Y. Kyung, J. Am. Chem. Soc., 2008, 130, 6534–6543 CrossRef PubMed.
- N. R. Jana, Y. Chen and X. Peng, Chem. Mater., 2004, 16, 3931–3935 CrossRef CAS.
- X. Pang, L. Zhao, W. Han, X. Xin and Z. Lin, Nat. Nanotechnol., 2013, 8, 426–431 CrossRef CAS PubMed.
- X. Li, J. Iocozzia, Y. Chen, S. Zhao, X. Cui, W. Wang, H. Yu, S. Lin and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 2046–2070 CrossRef CAS PubMed.
- X. Pang, Y. He, J. Jung and Z. Lin, Science, 2016, 353, 1268–1272 CrossRef CAS PubMed.
- Y. Liu, J. Wang, M. Zhang, H. Li and Z. Lin, ACS Nano, 2020, 14, 12491–12521 CrossRef PubMed.
- D. M. Andala, S. H. R. Shin, H. Y. Lee and K. J. M. Bishop, ACS Nano, 2012, 6, 1044–1050 CrossRef CAS PubMed.
- F. Lu, K. G. Yager, Y. Zhang, H. Xin and O. Gang, Nat. Commun., 2015, 6, 1–10 Search PubMed.
- M. Rycenga, J. M. McLellan and Y. Xia, Adv. Mater., 2008, 20, 2416–2420 CrossRef CAS.
- R. Klajn, P. J. Wesson, K. J. M. Bishop and B. A. Grzybowski, Angew. Chem., Int. Ed., 2009, 48, 7035–7039 CrossRef CAS PubMed.
- H. Mamiya and B. Jeyadevan, Sci. Rep., 2011, 1, 157 CrossRef PubMed.
- Y. Yamada, C. K. Tsung, W. Huang, Z. Huo, S. E. Habas, T. Soejima, C. E. Aliaga, G. A. Somorjai and P. Yang, Nat. Chem., 2011, 3, 372–376 CrossRef CAS PubMed.
- B. Gao, G. Arya and A. R. Tao, Nat. Nanotechnol., 2012, 7, 433–437 CrossRef CAS PubMed.
- N. Yazdani, M. Jansen, D. Bozyigit, W. M. M. Lin, S. Volk, O. Yarema, M. Yarema, F. Juranyi, S. D. Huber and V. Wood, Nat. Commun., 2019, 10, 1–6 CrossRef CAS PubMed.
- Y. Jiao, D. Han, Y. Ding, X. Zhang, G. Guo, J. Hu, D. Yang and A. Dong, Nat. Commun., 2015, 6, 6420 CrossRef CAS PubMed.
- V. López-Puente, S. Abalde-Cela, P. C. Angelomé, R. A. Alvarez-Puebla and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2013, 4, 2715–2720 CrossRef.
- N. Vilà, E. André, R. Ciganda, J. Ruiz, D. Astruc and A. Walcarius, Chem. Mater., 2016, 28, 2511–2514 CrossRef.
- E. G. Solveyra, E. De La Llave, V. Molinero, G. J. A. A. Soler-Illia and D. A. Scherlis, J. Phys. Chem. C, 2013, 117, 3330–3342 CrossRef CAS.
- M. I. Velasco, M. B. Franzoni, E. A. Franceschini, E. G. Solveyra, D. Scherlis, R. H. Acosta and G. J. A. A. Soler-Illia, J. Phys. Chem. C, 2017, 121, 7533–7541 CrossRef CAS.
- T. Yanagisawa, T. Shimizu, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 1990, 63, 988–992 CrossRef CAS.
- G. J. A. A. Soler-Illia, E. Scolan, A. Louis, P.-A. Albouy and C. Sanchez, New J. Chem., 2001, 25, 156–165 RSC.
- M. S. Wong, E. S. Jeng and J. Y. Ying, Nano Lett., 2001, 1, 637–642 CrossRef CAS.
- Y. K. Hwang, Y.-U. Kwon and K.-C. Lee, Chem. Commun., 2001, 1738–1739 RSC.
- J. Y. Chane-Ching, F. Cobo, D. Aubert, H. G. Harvey, M. Airiau and A. Corma, Chem. – Eur. J., 2005, 11, 979–987 CrossRef CAS PubMed.
- S. C. Warren, L. C. Messina, L. S. Slaughter, M. Kamperman, Q. Zhou, S. M. Gruner, F. J. DiSalvo and U. Wiesner, Science, 2008, 320, 1748–1752 CrossRef CAS PubMed.
- G. J. A. A. Soler-Illia, M. Jobbágy, A. E. Regazzoni and M. A. Blesa, Chem. Mater., 1999, 11, 3140–3146 CrossRef.
- Y. Kuroda, Y. Miyamoto, M. Hibino, K. Yamaguchi and N. Mizuno, Chem. Mater., 2013, 25, 2291–2296 CrossRef CAS.
- Y. Oka, Y. Kuroda, T. Matsuno, K. Kamata, H. Wada, A. Shimojima and K. Kuroda, Chem. – Eur. J., 2017, 23, 9362–9368 CrossRef CAS.
- Y. Kuroda, T. Koichi, K. Muramatsu, K. Yamaguchi, N. Mizuno, A. Shimojima, H. Wada and K. Kuroda, Chem. – Eur. J., 2017, 23, 5023–5032 CrossRef CAS PubMed.
- Y. Tokudome, N. Tarutani, K. Nakanishi and M. Takahashi, J. Mater. Chem. A, 2013, 1, 7702–7708 RSC.
- N. Tarutani, Y. Tokudome, M. Jobbágy, F. A. Viva, G. J. A. A. Soler-Illia and M. Takahashi, Chem. Mater., 2016, 28, 5606–5610 CrossRef CAS.
- N. Tarutani, Y. Tokudome, M. Jobbágy, G. J. A. A. Soler-Illia and M. Takahashi, J. Sol-Gel Sci. Technol., 2019, 89, 216–224 CrossRef CAS.
- N. Tarutani, Y. Tokudome, M. Jobbágy, G. J. A. A. Soler-Illia, Q. Tang, M. Müller and M. Takahashi, Chem. Mater., 2019, 31, 322–330 CrossRef CAS.
- A. E. Gash, T. M. Tillotson, J. H. Satcher, J. F. Poco, L. W. Hrubesh and R. L. Simpson, Chem. Mater., 2001, 13, 999–1007 CrossRef CAS.
- G. B. Deacon, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- W. R. Feairheller and J. E. Katon, Spectrochim. Acta, Part A, 1967, 23, 2225–2232 CrossRef CAS.
- S. Rey, J. Mérida-Robles, K.-S. Han, L. Guerlou-Demourgues, C. Delmas and E. Duguet, Polym. Int., 1999, 48, 277–282 CrossRef CAS.
- A. Aguzzi, V. Ambrogi, U. Costantino and F. Marmottini, J. Phys. Chem. Solids, 2007, 68, 808–812 CrossRef CAS.
- A. Neppel, I. S. Butler and A. Eisenberg, Macromolecules, 1979, 12, 948–952 CrossRef CAS.
- J. R. Allan, J. G. Bonner, D. L. Gerrard and J. Birnie, Thermochim. Acta, 1991, 185, 295–302 CrossRef CAS.
- J. Dong, Y. Ozaki and K. Nakashima, Macromolecules, 1997, 30, 1111–1117 CrossRef CAS.
- L. B. Levy and J. D. Penrod, Plant/Oper. Prog., 1989, 8, 105–108 CrossRef CAS.
- M. Fujita, Y. Iizuka and A. Miyake, J. Therm. Anal. Calorim., 2017, 128, 1227–1233 CrossRef CAS.
- M. Fujita, Y. Izato, Y. Iizuka and A. Miyake, Process Saf. Environ. Prot., 2019, 129, 339–347 CrossRef CAS.
- W. Balcerowiak, J. Hetper, J. Beres and J. Olkowska, J. Therm. Anal., 1977, 11, 101–107 CrossRef CAS.
- A. Gronowski and Z. Wojtczak, J. Therm. Anal., 1983, 26, 233–244 CrossRef CAS.
- M. Tanaka, I. Y. Park, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 1989, 62, 3442–3445 CrossRef CAS.
- S. Shi, F. Guo, Y. Xia, Z. Su, X. Chen and M. Wei, J. Appl. Polym. Sci., 2011, 121, 1661–1668 CrossRef CAS.
- C. Vaysse, L. Guerlou-Demourgues, E. Duguet and C. Delmas, Inorg. Chem., 2003, 42, 4559–4567 CrossRef CAS PubMed.
- L. Poul, N. Jouini and F. Fievet, Chem. Mater., 2000, 12, 3123–3132 CrossRef CAS.
- N. Tarutani, K. Katagiri, K. Inumaru and T. Ishigaki, J. Phys. Chem. B, 2021, 125, 4883–4889 CrossRef CAS PubMed.
- S. Rouba, P. Rabu and M. Drillon, J. Solid State Chem., 1995, 118, 28–32 CrossRef CAS.
- T. E. Williams, D. Ushizima, C. Zhu, A. Anders, D. J. Milliron and B. A. Helms, Chem. Commun., 2017, 53, 4853–4856 RSC.
- K. Hirota, S. Hara, H. Wada, A. Shimojima and K. Kuroda, ACS Nano, 2019, 13, 2795–2803 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr08689d |
| This journal is © The Royal Society of Chemistry 2021 |