
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-sustaining enzyme nanocapsules perform on-site chemical reactions†
Marina
Machtakova
,
Shen
Han
 ,
Yeliz
Yangazoglu
,
Ingo
Lieberwirth
,
Héloïse
Thérien-Aubin
* and
Katharina
Landfester
*
,
Yeliz
Yangazoglu
,
Ingo
Lieberwirth
,
Héloïse
Thérien-Aubin
* and
Katharina
Landfester
*
Max Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: therien@mpip-mainz.mpg.de; landfest@mpip-mainz.mpg.de
First published on 8th February 2021
Abstract
Nanoreactors offer a great platform for the onsite generation of functional products. However, the production of the desired compound is often limited by either the availability of the reagents or their diffusion across the nanoreactor shell. To overcome this issue, we synthesized self-sustaining nanoreactors carrying the required reagents with them. They are composed of active enzymes crosslinked as nanocapsules and the inner core serves as a reservoir for reagents. Upon trigger, the enzymatic shell catalyzes the conversion of the encapsulated payload. This concept was demonstrated by the preparation of nanoreactors loaded with sensing molecules for the detection of glucose in biological media. More importantly, the system introduced here serves as an adaptable platform for biomedical applications, since the nanoreactors display good cellular uptake and high activity within cells. Consequently, they could act as nanofactories for the in situ generation of functional molecules.
Introduction
Enzyme-based nanoreactors act as small-scale mobile factories for the on-site production of new compounds and are increasingly finding applications for sensing, diagnostics and therapy.1–4 However, the use of such nanosystems is often hampered not only by the loss of enzymatic activity but also by the limited access of reagents to the catalytic enzyme located in the inner cavity of the reactor. To address those limitations, we prepared highly efficient nanocapsules with a shell only composed of active enzymes able to encapsulate the required reactants in the core. This leads to the formation of self-sustaining nanoreactors.The concept of the nanoreactor is inspired by Nature, where enzymes perform multiple reactions in confined micro/nano-environments, such as subcellular organelles.5,6 In such systems, enzymes catalyze reactions with excellent specificity and with conversion rates that outperform chemical catalysts by several orders of magnitude.7 Artificial nanoreactors mimic the cellular organization of enzymes and confine the biological catalysts in nanocompartents.8
The enzyme-based nanoreactors can perform a myriad of reactions and when designed with multiple complementary enzymes, even biomimetic cascade reactions can be implemented.9,10 Such nanosystems can find use in a wide range of applications where they are employed as artificial organelles,11,12 diagnostic devices13 or therapeutic “drug nanofactories”.14,15 The efficiency of this concept has been demonstrated in a variety of environment from free suspensions16 to in cellulo after the uptake of the nanoreactors by cells.17,18
One of the early examples of this concept is the use of enzymes encapsulated in liposomes to replicate natural cellular functions.16 More recently, similar approaches showed the design of colloid- or polymersomes preserving the biological activity of enzymes that are encapsulated within the polymersome aqueous interior9,19 or bound to the membrane.20–22 Other related studies showed the fabrication of enzyme–polymer nano-constructs (proteinosomes), comprising a shell of (multiple) polymer–enzyme conjugates for the performance of enzymatic cascades.23
However, with the nanoreactors developed so far, the enzyme is encapsulated in or on a carrier, and the shell of the nanoreactor shell constitutes an additional diffusion barrier slowing down the influx rate of reactants.24 In such case, complex membrane modifications are required to ensure the nanoreactor permeability.19,25 Furthermore, the use of any additional carrier material, such as lipids or polymers, limits the effective enzyme concentration in the system due to the volume occupied by the non-catalytic carrier. This leads to a decrease in the overall catalytic efficiency of those nanostructured enzyme systems.26,27 In addition, in multiple cases, the chemical reactant, which should be converted into the desired product, can only be difficultly included in such systems, and has to be provided externally. Furthermore, in complex environments such as in vivo conditions, the availability of the reactant is limited by diffusion and the sufficient co-localization of enzyme and substrate can be hindered. Those drawbacks hamper the efficiency and the yield of the catalytic reactions and hence, the impact of the nanoreactors.
To overcome those limitations, we designed a self-sustaining full-enzyme nanoreactor system, which can carry the required reactants in its inner core. The only structural elements of the nanoreactor shell are crosslinked enzymes, and the inner aqueous core is a reservoir for the reagents needed for the catalytic reaction. Such nanoreactors were designed to increase the efficient mass fraction of active enzyme by 50–1000 folds in comparison to conventional nanoreactors,28–30 to be almost entirely composed of biocompatible and biodegradable enzymes,31 and to not require for the diffusion of reagents through the nanoreactor shell (Fig. 1). Alternatively, such nanoreactors can also convert non-encapsulated substrates if necessary.
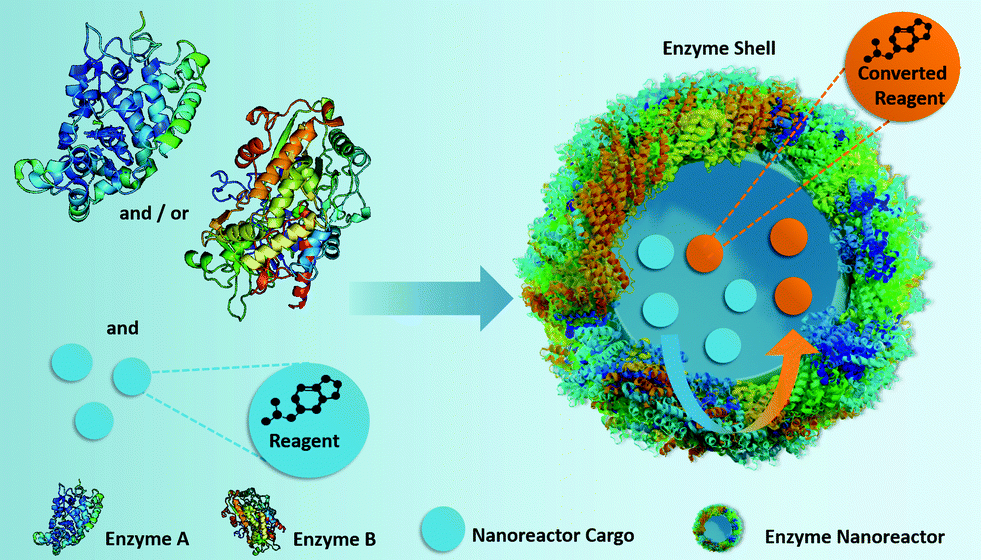 | ||
| Fig. 1 Fabrication of self-sustaining enzyme nanoreactors, which participate actively in both the transportation and the conversion of the reactants located in their inner core. | ||
Results and discussion
Preparation of enzyme nanoreactors
The formation of the enzyme nanoreactors proceeded through a stepwise process. First, an aqueous solution of the enzyme was used to prepare an inverse (water-in-oil) miniemulsion by a high-energy emulsification process. The preparation of the miniemulsion is a critical parameter for the size and size distribution of the resulting NCs. Typically, miniemulsions can be prepared by ultrasonication as a result of cavitation-induced droplet disruption.32–34 However, the high energy of the ultrasound waves and the long duration of their application are detrimental to sensitive biological molecules. The complex three-dimensional structure of the enzyme was disrupted by the use of ultrasound, resulting in a complete loss of enzymatic activity (Fig. S1†).To circumvent this crucial drawback, a milder emulsification process, microfluidization, was used. Microfluidization induces high mechanical stress on the droplets of the emulsion in a localized area for a very short time.35 The controlled collision of two microstreams in an interaction chamber produced a well-controlled emulsion and preserved a high degree of enzymatic activity (Fig. 2c and Fig. S1†).
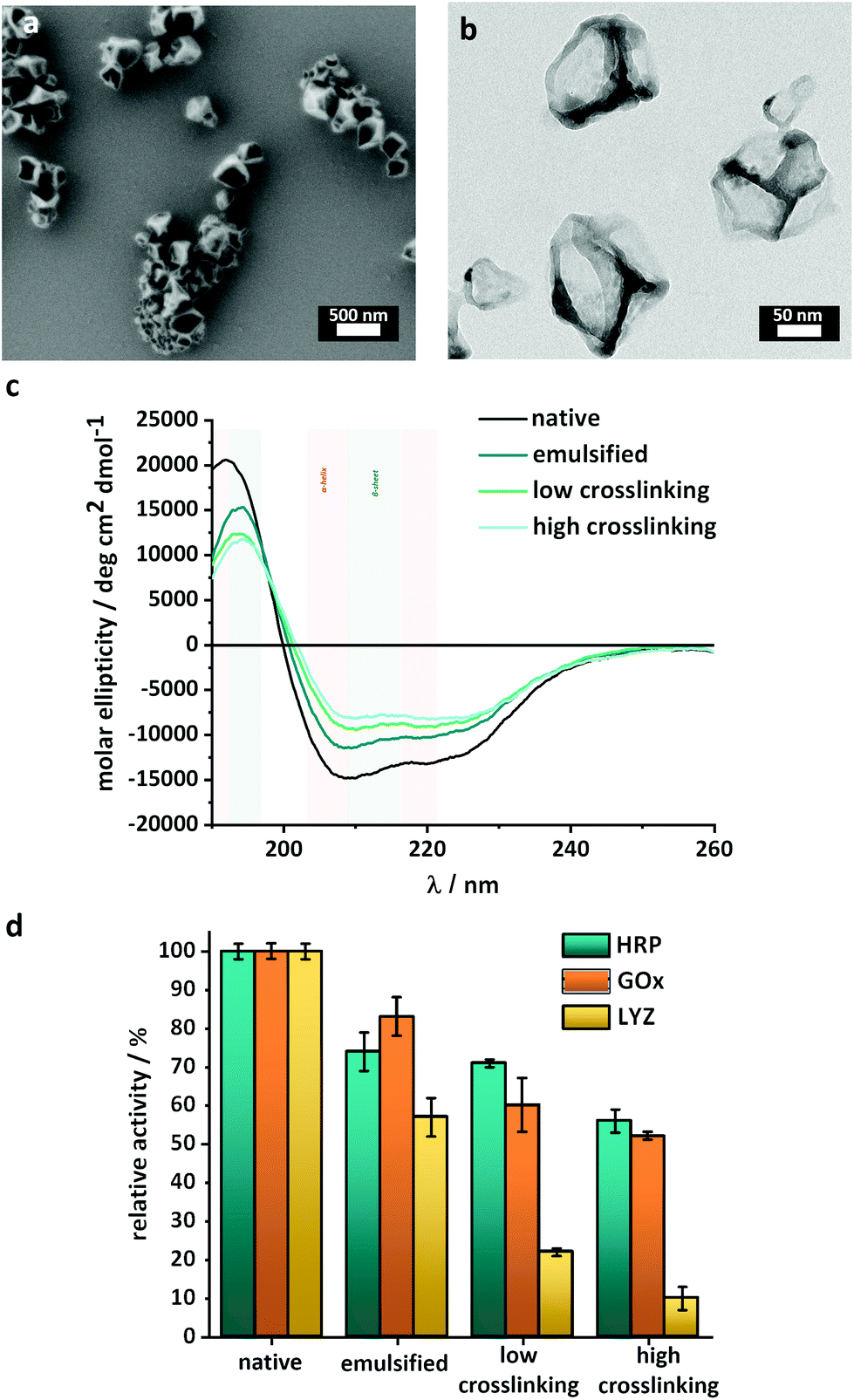 | ||
| Fig. 2 Characterization of the enzyme-nanoreactors. (a) SEM image of the HRP-nanoreactors, (b) TEM image of the HRP-nanoreactors, (c) CD-spectra of HRP after emulsification and crosslinking, (d) relative enzymatic activity of HRP, GOx and LYZ after emulsification and crosslinking. | ||
After the emulsification, the crosslinking of the enzyme molecules present at the interface of the aqueous droplets was carried out by the addition of the crosslinker 2,4-toluene diisocyanate (TDI) to the organic phase. The polyaddition between the isocyanate groups of the TDI and the accessible nucleophilic groups of the enzyme (Fig. S2†) resulted in the formation of a nanoreactor shell composed of crosslinked enzymes. Such interfacial crosslinking reaction occurring on inverse-miniemulsion droplets is an ideal technique to control the size of the resulting nanocapsules and to control the shell thickness.36
Furthermore, the shell of the resulting nanocapsules is semi-permeable, and while salts and small molecules can freely diffuse in and out of the inner core of the capsules,37,38 large macromolecules are trapped inside the core if added to the solution of capsule precursor before emulsification. The encapsulation of different payloads in such enzyme nanocapsules can potentially lead to the release of the degradation products of large macromolecular payloads such as polyprodrugs,14 or to the release of large payloads through the enzymatic degradation of the nanoreactor itself, which could occur under specific conditions.33
Finally, the nanoreactors were transferred to an aqueous buffer. Hollow enzyme NCs with mean diameters of 200–300 nm were obtained as confirmed by scanning electron microscopy (SEM) (Fig. 2a, Fig. S3 and S4†) and dynamic light scattering (DLS) (Fig. S5†). The low values of polydispersity index measured by DLS (<0.2) were indicative of the presence of individual and homogenous nanocapsules with a limited size distribution. Furthermore, electron microscopy (TEM) showed that the crosslinked enzymes formed a shell surrounding an empty core, and the thickness of this shell varied between 10 and 15 nm (Fig. 2b).
To demonstrate the versatility of this approach, the nanoreactors were prepared with different enzymes (Fig. S2†), namely horseradish peroxidase (HRP), glucose oxidase (GOx) and lysozyme (LYZ). Those enzymes were identified as prime candidates because HRP and GOx are robust enzymes, which can be used in clinical diagnostics, organic synthesis and food processing, and LYZ is a more delicate lytic enzyme able to degrade the cell wall of bacteria, making it of interest in medicine, cosmetics and in the food industry. Nanoreactors were formed with all the enzymes with the same control over size and size distribution (Fig. S5†). Such crosslinked enzyme or protein nanocapsules typically are biocompatible nanosystems, degradable by biological proteases, even after their crosslinking.33,39 The fraction of the enzymatic activity preserved during the synthesis was similar for HRP and GOx, but more limited for LYZ (Fig. 2c).
One of the critical factors influencing the catalytic activity of an enzyme is its folding and the resulting three-dimensional structure. External factors such as pH, temperature, pressure and chemical reactions can (irreversibly) alter the enzyme folding properties and decrease the catalytic activity. Circular dichroism spectroscopy (CD) was used to evaluate the secondary structure of the enzyme and qualitatively monitor changes in the structure after the different steps of the synthesis. The CD spectra (Fig. 2c and Fig. S6†) show the influence of each step of the nanoreactor formation on the 3D structure of the enzymes. The intensity of the peaks between 200 and 230 nm, typical of a mixture of α-helices and β-sheets, decreased after the emulsification and after the crosslinking of the enzyme. For the HRP nanoreactors, the results (Fig. 2b) indicated that a fraction of the native enzyme structure was either degraded or converted to random coils both after the emulsion and after the interfacial reaction. Similar results were obtained for the GOx and LYZ nanoreactors (Fig. S6†).
Enzymatic activity of the nanoreactors
In order to quantify the influence of the degradation observed by CD on the performance of the nanoreactors, enzymatic activity assays were used to measure the remaining biological activity. Fig. 2d shows that, in the case of both HRP and GOx, the most significant deactivation of the enzyme occurred during the emulsification process. In comparison, the reaction between the enzyme and the TDI only marginally influenced the enzymatic activity, in keeping with the loss of the 3D structure observed by CD.The nanoreactors synthesized with HRP and GOx both retained up to 70 ± 2% of their native activity. While HRP and GOx only showed a limited decrease of the activity during the crosslinking reaction, the synthesis of LYZ-nanoreactors was accompanied by a significant decrease in enzymatic activity. After the synthesis of LYZ nanoreactors, only ca. 22 ± 2% of the native enzymatic activity was maintained in agreement with the steep changes in the structure of LYZ observed by CD spectroscopy (Fig. S6b†). In the case of LYZ, the emulsification process also decreased the enzymatic activity. However, while GOx and HRP were relatively insensitive to the crosslinking reaction, LYZ was negatively impacted by the crosslinking reaction.
The result from the enzymatic activity assay demonstrated that the combination of a mild emulsification process with the covalent, but limited, crosslinking of the enzymes proceeding at room temperature and at physiological pH contributed to the preparation of enzymatically active, uniform, and reproducible full-enzyme-nanoreactors. A significant degree of enzymatic activity was preserved for the different enzymes used, chosen for their distinctive structural stability and sensitivity. Even in the case of LYZ, an enzyme both thermo- and mechano-sensitive, up to 22% of the native activity was preserved. The results provided a favorable assessment of the potential of our technique, where emulsification was followed by interfacial crosslinking, to synthesize nanoreactors from a range of different enzymes despite the partial loss of activity observed.
A reduction in the apparent enzymatic activity can be caused by a reduction in the number of available reaction sites where the substrates can bind to the enzyme or by a reduction of the reaction rate. The reaction kinetic of single-substrate enzymes, like those used here, can be described by the Michaelis–Menten model. The kinetic parameters obtained with the native enzyme were compared to those obtained with the emulsified and with the crosslinked enzyme-nanoreactors. The parameters Vmax (the maximum rate of substrate conversion) and Km (the Michaelis constant representing the substrate concentration at the half-maximal reaction rate) were used to describe the enzymatic reaction kinetic (eqn (S1) and (S2)†). To study the effect of the processing condition on the enzyme, the activity of each enzyme was measured using established protocols.
For the kinetic reaction profile of HRP, the conversion of a substrate (ABTS) was investigated for the native, emulsified and crosslinked enzyme (0.05 μM, based on the total amount of enzyme, both active and inactive) at different concentrations of hydrogen peroxide (between 1.0 and 2.5 mM) (Fig. 3). Both the emulsified and the crosslinked enzyme showed a moderate decrease in the initial rate of substrate conversion in comparison to the native enzyme (Fig. 3b). This slower initial reaction rate indicated a decrease in the catalytic rate constant. Moreover, the HRP nanoreactors reached a complete conversion of the substrate after a longer time (40 min for 1 mM H2O2 and 60 min for 2.5 mM H2O2) compared to the native HRP (20 min for 1 mM and 2.5 mM H2O2).
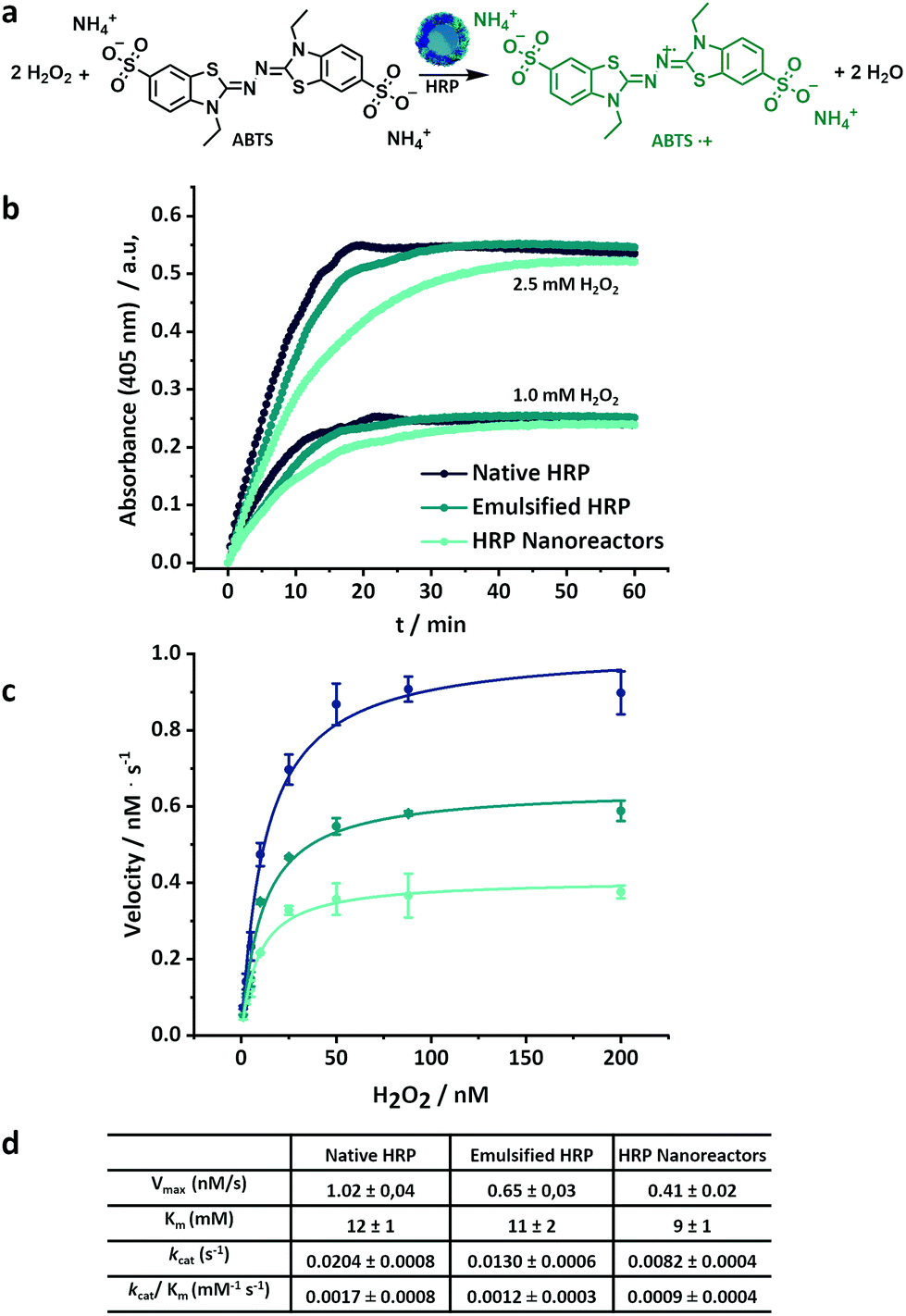 | ||
| Fig. 3 (a) Oxidation of ABTS in presence of hydrogen peroxide and horseradish peroxidase, (b) kinetic of the ABTS conversion in presence of native, emulsified and crosslinked HRP, (c) Michaelis–Menten plot of HRP-nanoreactors, lines are fits to eqn (S1),† (d) Kinetic parameters for native, emulsified and crosslinked HRP. | ||
Fig. 3c shows the Michaelis–Menten plots for the native, emulsified and crosslinked HRP and displays the relation between the substrate concentration (between 1.0 mM and 200 mM) and the reaction rate observed during the conversion. The resulting Km value of the HRP-nanoreactors was marginally lower than the Km of the native HRP (Fig. 3c and S7†), suggesting that the binding affinity of the substrate for the HRP might increase after the nanoreactor formation. The HRP nanoreactors showed a lower Vmax in comparison to the native enzyme. The decrease in Vmax was ascribed either to a reduction of the catalytic rate constant or to a decrease in the effective enzyme concentration. One possible explanation is that upon crosslinking, fewer active centers of the enzyme are accessible by the substrate, and thus, the reaction decelerates. Furthermore, the effective concentration of active enzyme decreases during the formation of the nanoreactors due to the loss of the enzyme structure (Fig. 2b).
To directly compare the catalytic efficiency of the different systems, the apparent enzymatic activity (kcat/Km) was calculated based on the total enzyme concentration. Approximately 71% and 53% of kcat/Km was retained for the emulsified and the crosslinked HRP in comparison to the native HRP. Similar experiments were carried out for glucose oxidase (Fig. S8†). The kcat/Km of the GOx systems was 66% (emulsified) and 29% (crosslinked) compared to native GOx.
Enzymatic activity of the nanoreactors in cells
The novel enzyme-based nanoreactors can be used for the performance of catalytic reactions inside living cells in biological and medical applications. Often, the intracellular activity of native enzymes is hampered by the instability and degradation of native proteins under physiological conditions and their poor cellular uptake.40 In contrast, the co-incubation of murine macrophages RAW264.7 with the enzyme nanoreactors resulted in the uptake of the particles by the cells (Fig. 4) by an endocytic pathway.41 To facilitate the observation of the HPR nanoreactors by confocal laser scanning fluorescence microscopy (CLSM), the reactors were labeled with a fluorescent dye, cyanine 5, during the synthesis. Furthermore, the HRP nanoreactors displayed no cytotoxicity towards the RAW264.7 cells (Fig. S9†). | ||
| Fig. 4 (a) DAB polymerization catalyzed by the HRP nanoreactors, (b) confocal fluorescence microscopy images of RAW264.7 cells incubated with HRP nanoreactors (red) for 24 h, before and 4 minutes after the addition of DAB. In the control experiment, the cells were exposed to DAB but were not co-incubated with the HRP nanoreactors. The scale bars are 50 μm. | ||
The nanoreactors can catalyze the production of new synthetic chemicals within the cellular compartment in order to influence the cell fate or cellular functions such as energy storage or cell differentiation. The production of synthetic material inside the cell is an important challenge, which could provide new possibilities to manipulate, track and control cellular behavior by the in situ generation of specific molecules.
One hurdle faced by the production of synthetic chemicals within the cell is the presence of a highly complex intracellular environment able to quench or prevent unconventional reactions.42 The enzyme nanoreactors overcome this hurdle by preserving their enzymatic activity in the cells. Furthermore, they can potentially outperform other systems by acting both as a catalyst for the reaction and as a transport module. Here, as a model reaction to visualize the enzymatic activity of the HRP nanoreactors in cells, the polymerization reaction of 3,3′-diaminobenzidine (DAB) was selected (Fig. 4a). While monomeric DAB is water-soluble and can freely cross the cellular membrane, polymerized DAB is water-insoluble and precipitated into large insoluble polymer clusters where the HRP activity is localized.43 After the co-incubation of the cells with the HRP nanoreactors (75 μg mL−1), the cells were washed. Then the cells were fixed with paraformaldehyde (PFA), to increase the permeability of the cell membrane44 to allow for the fast penetration of the DAB molecules into the cell, and finally exposed to a solution of DAB and hydrogen peroxide for 4 minutes.
During the reaction time of 4 minutes, the DAB was converted to poly(DAB) and formed black islands visible by confocal laser scanning fluorescence microscopy (CLSM) (Fig. 4c). The overlay of the different channels in the CLSM shows the colocalization of the poly(DAB) with the Cy5-loaded HRP-nanoreactors (Fig. 4b, red dots). When the HRP nanoreactors were not present in the cells, no polymerized DAB islands were observed (Fig. 4b, control). When the co-incubation period was increased, the amount of poly(DAB) formed during the 4 minutes of reaction increased as visualized by microscopy recorder after co-incubation periods of 1, 4, and 24 h (Fig. S10†). This was the result of the higher cellular uptake observed for longer co-incubation periods. The DAB polymerization within the cellular compartment demonstrated the ability to perform synthetic chemistry inside living cells using those new enzyme-nanoreactors. Here the HRP nanoreactors, which acted as a carrier for a fluorescent probe, efficiently crossed the cell membrane and then performed catalysis inside the cellular compartment. This approach can pave the way for the efficient control over synthetic intracellular chemistry and catalysis.
Enzymatic cascades in binary nanoreactors
A common characteristic of enzymatic reactions in biological systems is that multiple enzymes can be coupled in highly coordinated cascade reactions. Nature can control such cascades with the highest precision and efficiency by organizing the involved enzymes in a confined microenvironment or putting them in the same (cellular) compartments.45 The enhanced reaction rate of those reactions is thought to arise from the accelerated transport of intermediates between the active sites of the single enzymes in close vicinity.22 Inspired by the cellular organization of cooperating reaction partners, we designed binary enzyme nanoreactors by combining HRP and GOx in one nanoreactor. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of both enzymes was employed and hollow HRP-GOx-nanoreactors were obtained (Fig. S3†).
:1 ratio of both enzymes was employed and hollow HRP-GOx-nanoreactors were obtained (Fig. S3†).
During the formation of the binary nanoreactors, a decrease in the content of α-helix and β-sheet structures in the enzyme mixture was observed by CD spectroscopy in comparison to a native HRP/GOx mixture (Fig. S6c†). This result was in accordance with the decrease in the relative enzymatic activity of the enzyme mixture in comparison to the native mixture of HRP/GOx measured after the different steps of the HRP/GOx-nanoreactor formation (Fig. S11†). Respectively 83 ± 5% and 55 ± 4% of the original enzymatic activity was preserved after the emulsification of the enzyme mixture and after the crosslinking reaction. The model cascade reaction for the binary nanoreactors was composed of two conversion steps. First, GOx catalyzed the oxidation of β-D-glucose to D-glucono-δ-lactone and hydrogen peroxide. In the second step, HRP reacted with H2O2 to oxidize a substrate dye molecule (Fig. 5a).
 | ||
| Fig. 5 (a) Scheme of the GOX-HRP cascade reaction, (b) cascade reaction in a HRP/GOx nanoreactor, (c) cascade reaction in a mixture of HRP nanoreactors and GOx nanoreactors. (d) Substrate conversion and reaction rate of the mixed HRP/GOx nanoreactors and a mixture of single nanoreactors. | ||
The results show that the reaction rate in the nanoreactors made with a mixture of HRP and GOx outperformed a mixture of nanoreactors individually made with either HRP or GOx (Fig. 5b). The binary nanoreactors performed the same cascade reaction at a higher reaction rate than the mixture of nanoreactors. However, after extended reaction time, the total conversion of substrates was identical in both systems (Fig. S12†).
To investigate the causes for the acceleration of the reaction in the binary nanoreactors in comparison to the mixture of individual nanoreactors, the oxidation of the substrate by the HRP was triggered by the direct addition of hydrogen peroxide, rather than the cascade of the GOx/glucose. The addition of H2O2 resulted in the same reaction kinetic for both the binary nanoreactors and the mixture of the two individual nanoreactors (Fig. S13†). This result indicates that the co-immobilization of HRP and GOx in close vicinity accelerated the cascade and suggests that the main reason for the enhanced reaction rate was related to the transport of the hydrogen peroxide between the two enzymatic sites. The faster substrate turnover in the binary nanoreactor was likely influenced by the small interenzyme distances, which facilitate the fast transport of the intermediate (H2O2) between the two active sites while preventing the diffusion and dilution of the intermediate into the bulk of the solution. Those results are in good agreement with the reported enhanced cascade kinetics of enzyme reactions in micro/nano-confinements.10,46,47
Enzymatic conversion of encapsulated payload
Because the shell of the nanoreactors is semi-permeable,37,38 small hydrophilic molecules like glucose or peroxide can diffuse through the walls of the nanoreactors, and the catalytic reaction can be performed both on and in the nanoreactors, as observed in the previous cases. However, the method used here to synthesize the enzyme nanoreactors allows for the encapsulation of different reagents inside the hollow aqueous core,36,48 and macromolecules can be efficiently trapped inside the nanoreactor without leaking out (Fig. S14†).Thus, reagents can be encapsulated and carried together with the biocatalyst and can be converted by their carrier on-site. To demonstrate the ability of our system to perform reactions inside the aqueous core of the enzyme nanoreactors, polymer-functionalized luminol, a chemoluminescent reagent, was encapsulated in the HRP/GOx-nanoreactors.
Upon the addition of β-D-glucose to the luminol-containing binary-nanoreactos, the enzymatic cascade reaction was initiated. However, in the present case, the HRP converted the H2O2 to oxidize the luminol resulting in the emission of blue luminescence (Fig. 6a). Hence, the nanoreactors actually acted as an on-site nano-light bulb. When the luminol-derivative was encapsulated inside the hollow core, the reaction kinetics of the chemoluminescence was identical to the chemoluminescence generated by the addition of free luminol-derivative (Fig. 6b) to the external medium. Due to the semi-permeable nature of the shell, in the first case, the reaction could only occur within the confines of the nanoreactors, while in the second case, the reaction could only occur on the surface of the nanoreactor, given that the luminol-derivative cannot diffuse through the shell of the nanoreactor. Since the reaction kinetics was similar both in and on the surface of the nanoreactors, this highlighted the versatility of our nanoreactors, where the reaction rate was not influenced by the locus of the reaction.
 | ||
| Fig. 6 (a) Glucose activated on-site generation of chemoluminescence inside or on the surface of the HRP/GOx-nanoreactors, (b) Chemoluminescence of luminol loaded in the HRP/GOx nanoreactors (blue line) and outside of the nanoreactors (black line), inset: photograph of the visible chemoluminescence of HRP/GOx-luminol-nanoreactors upon the addition of H2O2, (c) Comparison of the quantification of glucose in biological media using the chemoluminescence of luminol in the binary HRP/GOx nanoreactors and by standard methods. | ||
Furthermore, the intensity of the light emitted by the nanoreactor increased with the addition of increasing concentration of glucose (Fig. S15a†). This makes the HRP/GOx nanoreactor containing luminol-derivatives an ideal candidate for glucose sensing in complex biological media. The HRP/GOx nanosensor containing luminol-derivatives showed a high sensitivity with a limit of detection (10σblank) for glucose of 0.4 μM. However, once the luminol reacted to emit light, it was transformed into aminophthalic acid. Consequently, each luminol molecule can only react once, and the catalysis can only be repeated until the encapsulated molecules were used up.
The glucose-responsive light emission from HRP/GOx-luminol nanoreactors was used to construct a simple, homogeneous and reagentless chemoluminescent glucose sensor and was used to quantify the glucose contained in real biological samples like serum and plasma. Despite the complex matrix of the biological media, it was possible to reliably quantify glucose by measuring the chemoluminescence emitted by the nanoreactors (Fig. 6). For the sensing experiment, to prevent the interference of the sample matrix, standard addition experiments were carried out (Fig. S15c†) with samples diluted by 100× (serum) or 1000× (plasma) times with PBS-buffer and five times with 0.1 M NaOH prior to the measurements. The quantification experiments showed glucose levels of 7.0 mM for human serum, 4.3 mM for animal (horse) serum and 40.2 mM for human plasma. The results obtained were in agreement with the glucose level measured by a commercial glucose assay or a commercial glucometer. No significant deviations were observed for the three methods, except for the glucometer, which failed to measure animal samples. The results confirm the potential of the self-sustaining enzyme nanoreactors for their use in detection and sensing.
Conclusions
We developed self-sustaining nanoreactors carrying the required reagents in their core. The enzymes were crosslinked as hollow nanoreactors without the use of any additional material. Their inner aqueous core was able to act as reservoir for the reactant and the enzymatic shell actively catalyzed the on-site conversion of the payload. We used three different enzymes for the synthesis of the nanoreactors, and preserved a high enzymatic activity after their formation. The nanoreactors were taken up by cells and were still able to perform their enzymatic catalysis within the cells. Furthermore, the combination of both horseradish peroxidase and glucose oxidase yielded nanoreactors performing an enzymatic cascade reactions. The encapsulation of luminol, a chemoluminescent reagent, allowed for the development of a precise sensor for the quantification of glucose in biological media. The results obtained prove the applicability of the system in complex biological environment.This approach is versatile and can be employed with different enzymes and reactants. Thus, it provides an adaptable platform for more complex biomedical applications, such as the development of in cellulo drug nanofactories.
Experimental
Synthesis of the enzyme nanoreactors
The enzyme nanoreactors were prepared by a polyaddition reaction in an inverse miniemulsion. In a typical experiment, the aqueous phase was prepared by dissolving 30 mg of enzyme and 1 mg of NaCl in 0.3 mL PBS-buffer (pH 7.0). When the nanoreactors were used to encapsulate luminol, 0.7 mg of dextran-luminol was added to the aqueous solution. For the encapsulation of Cy5, 0.3 mg of the dye was added to the aqueous solution. The organic continuous phase was prepared by dissolving 75 mg of polyglycerol polyricinoleate (PGPR) in 3 mL toluene. The aqueous and organic phases were combined and pre-emulsified with an ultraturrax (20000 rpm) for 60 seconds and homogenized by one cycle through a microfluidizer (LV1, microfluidic corporation) at an operating pressure of 10000 MPa in a 75 μm Y-shape interaction chamber. The emulsion was divided into three parts of 1 mL. A solution of 25 mg PGPR and 5 or 10 mg of crosslinking agent (TDI) in 0.5 mL of toluene was added dropwise to 1 mL of the miniemulsion over a period of 2 min. The reaction was stirred over 20 h at room temperature.
The synthesized enzyme-nanoreactors were purified by three cycles of centrifugation (30 min, 1200 RCF) followed by redispersion in fresh toluene for the removal of excess surfactant and unreacted crosslinker. To transfer the nanocapsules to an aqueous solution, 500 μL of the dispersion in toluene were added dropwise to a solution of Lutensol AT50 (0.1 wt%) in PBS buffer under sonication in an ultrasonic bath. The resulting dispersion was stirred in an open vial for 3 h to evaporate the toluene. The dispersion was purified using Amicon centrifugal filter (MWCO 300000, 2 times, 30 min) and redispersed in fresh PBS-buffer to remove the unreacted enzyme, excess of surfactant, and unencapsulated payload.
Determination of the enzyme concentration in the nanoreactors
The concentration of enzymes crosslinked as nanoreactors was quantified with a protein assay using bicinchoninic acid (BCA). Briefly, 100 mg of BCA, 200 mg of sodium carbonate, 16 mg of sodium tartrate and 95 mg of sodium hydrogen carbonate were dissolved in 10 mL of deionized water and the pH was adjusted to 11.3 by using 3.0 M NaOH. To this solution, 200 μL of 50 mg of CuSO4·5 H2O in 1 mL of deionized water were added, and 200 μL of this solution were mixed with 10 μL of protein standard (horseradish peroxidase) or the enzyme nanoreactor dispersion of unknown concentration and incubated at 60 °C for 30 min. The absorbance at 565 nm was recorded and the enzyme concentration was determined by comparison to the standard curve prepared with native HRP.Cell experiments
For the cell culture and co-incubation, first, RAW 264.7 cells (Mouse macrophage cell line, CLS; Eppelheim; Deutschland) were seeded in μ-Dish 35 mm Imaging Chamber (ibidi, Germany) with the concentration of 20000 cells per mL overnight. Then, HRP nanoreactors (Cy5-loaded) were added to the cells with a concentration of 75 μg mL−1 for 1, 4 or 24 hours and the cells were washed with fresh medium afterwards.
DAB staining
The cells were chemically fixed with 4% paraformaldehyde for 10 min and washed 3 times with PBS. Afterwards, The DAB/Cobalt and Urea Hydrogen Peroxide Tablets (Sigma-Aldrich, Germany) were taken from the freezer and were allowed to reach room temperature. Then the tablets were dropped into an appropriate container. The container was filled with 5 mL ultrapure water and vortexed until the tablets dissolved. (The DAB staining solution was used within 15 minutes.) The solution was added to the chemically fixed cells for 4 min. After staining the staining solution was removed and the cells were washed 2–3 times with PBS.Chemoluminescence measurements
In a typical measurement, 0.1 mL of a HRP/GOx-nanoreactors suspension in 0.1 M NaOH (0.6 mg mL−1) was loaded in a 96-well plate. Different glucose solutions (ranging from 2.5 mM to 1 μM) (0.1 mL) in 0.1 M NaOH were injected directly in the well and the chemoluminescence emission was detected for two minutes using a plate reader (M1000, Tecan).Glucose determination in serum and plasma
The horse serum was purchased from Gibco. Human serum and human plasma were obtained from human blood taken from healthy donors at the Department of Transfusion Medicine Mainz after physical examination and after obtaining written informed consent in accordance with the Declaration of Helsinki. The study was approved by the local ethics committee “Landesärztekammer Rheinland-Pfalz” (Bearbeitungsnummer: 837.439.12 (8540-F)). Written informed consent was obtained for any experimentation we carried out when using samples from human subjects. For the determination of the glucose concentration in plasma and serum, 10 μL of the sample was diluted 100 times with PBS-buffer for serum and 1000 times for plasma. Furthermore, the samples were diluted 5 times with 0.1 M NaOH. For the chemoluminescence measurements, 100 μL of the diluted serum or plasma samples were loaded in a 96-well plate. A suspension of HRP/GOx-nanoreactors in 0.1 M NaOH (0.6 mg mL−1, 100 μL) was injected directly in the well and the chemoluminescence emission was detected by the plate reader.The results were compared to the glucose level measured using a commercial glucose HK assay (Sigma-Aldrich). Additionally, a glucometer (Adia Diabetes Set) based on the electrochemical enzymatic detection of glucose was used. The concentration of glucose in plasma and serum was determined using the HK assay and the glucometer through a series of standard addition experiments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Else Kröner-Fresenius-Stiftung, the Max Planck Society and the MaxSynBio consortium are acknowledged for financial support. The authors thank Gunnar Glasser for recording the SEM images, Beate Müller for the HPLC analysis, Carole Champagnac for the cytotoxicity measurements, and Stefan Schuhmacher for help with graphical design. Open Access funding provided by the Max Planck Society.References
- K. Renggli, P. Baumann, K. Langowska, O. Onaca, N. Bruns and W. Meier, Adv. Funct. Mater., 2011, 21, 1241–1259 CrossRef CAS.
- X. Lian, Y. Huang, Y. Zhu, Y. Fang, R. Zhao, E. Joseph, J. Li, J.-P. Pellois and H.-C. Zhou, Angew. Chem., Int. Ed., 2018, 57, 5725–5730 CrossRef CAS.
- J. F. Mukerabigwi, Z. Ge and K. Kataoka, Chem. – Eur. J., 2018, 24, 15706–15724 CrossRef CAS.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS.
- F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS.
- A. Küchler, M. Yoshimoto, S. Luginbühl, F. Mavelli and P. Walde, Nat. Nanotechnol., 2016, 11, 409 CrossRef.
- K. Rina, P.-R. Javier, D. C.-N. Ruben and V.-D. Rafael, Nanotechnol. Rev., 2017, 6, 405–419 Search PubMed.
- W.-H. Chen, M. Vázquez-González, A. Zoabi, R. Abu-Reziq and I. Willner, Nat. Catal., 2018, 1, 689–695 CrossRef CAS.
- S. F. M. van Dongen, M. Nallani, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem. – Eur. J., 2009, 15, 1107–1114 CrossRef CAS.
- S.-M. Jo, F. R. Wurm and K. Landfester, ACS Appl. Mater. Interfaces, 2018, 10, 34230–34237 CrossRef CAS.
- B. Thingholm, P. Schattling, Y. Zhang and B. Städler, Small, 2016, 12, 1806–1814 CrossRef CAS.
- M. Marguet, C. Bonduelle and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 512–529 RSC.
- J. Liu, C. Cai, Y. Wang, Y. Liu, L. Huang, T. Tian, Y. Yao, J. Wei, R. Chen, K. Zhang, B. Liu and K. Qian, Adv. Sci., 2020, 7, 1903730 CrossRef CAS.
- W. Ke, J. Li, F. Mohammed, Y. Wang, K. Tou, X. Liu, P. Wen, H. Kinoh, Y. Anraku, H. Chen, K. Kataoka and Z. Ge, ACS Nano, 2019, 13, 2357–2369 CAS.
- S.-M. Jo, F. R. Wurm and K. Landfester, Nano Lett., 2020, 20, 526–533 CrossRef CAS.
- N. Wagner, M. Gutweiler, R. Pabst and K. Dose, Eur. J. Biochem., 1987, 165, 177–183 CrossRef CAS.
- S. F. M. van Dongen, W. P. R. Verdurmen, R. J. R. W. Peters, R. J. M. Nolte, R. Brock and J. C. M. van Hest, Angew. Chem., Int. Ed., 2010, 49, 7213–7216 CrossRef.
- M. Galliani, M. Santi, A. Del Grosso, A. Cecchettini, F. M. Santorelli, S. L. Hofmann, J.-Y. Lu, L. Angella, M. Cecchini and G. Signore, Bioconjugate Chem., 2018, 29, 2225–2231 CrossRef CAS.
- J. Kim and K. T. Kim, ACS Appl. Mater. Interfaces, 2020, 12, 23502–23513 CrossRef CAS.
- A. K. Johnson, A. M. Zawadzka, L. A. Deobald, R. L. Crawford and A. J. Paszczynski, J. Nanopart. Res., 2008, 10, 1009–1025 CrossRef CAS.
- T. Konno, J. Watanabe and K. Ishihara, Biomacromolecules, 2004, 5, 342–347 CrossRef CAS.
- J. N. Vranish, M. G. Ancona, E. Oh, K. Susumu, G. Lasarte Aragonés, J. C. Breger, S. A. Walper and I. L. Medintz, ACS Nano, 2018, 12, 7911–7926 CrossRef CAS.
- X. Huang, M. Li and S. Mann, Chem. Commun., 2014, 50, 6278–6280 RSC.
- S. Dutta, N. Kumari, S. Dubbu, S. W. Jang, A. Kumar, H. Ohtsu, J. Kim, S. H. Cho, M. Kawano and I. S. Lee, Angew. Chem., Int. Ed., 2020, 59, 3416–3422 CrossRef CAS.
- S. Varlas, J. C. Foster, P. G. Georgiou, R. Keogh, J. T. Husband, D. S. Williams and R. K. O'Reilly, Nanoscale, 2019, 11, 12643–12654 RSC.
- S. Talekar, A. Joshi, G. Joshi, P. Kamat, R. Haripurkar and S. Kambale, RCS Adv., 2013, 3, 12485–12511 CAS.
- L. Cao, L. v. Langen and R. A. Sheldon, Curr. Opin. Biotechnol., 2003, 14, 387–394 CrossRef CAS.
- L. D. Blackman, S. Varlas, M. C. Arno, A. Fayter, M. I. Gibson and R. K. O'Reilly, ACS Macro Lett., 2017, 6, 1263–1267 CrossRef CAS.
- D. Gräfe, J. Gaitzsch, D. Appelhans and B. Voit, Nanoscale, 2014, 6, 10752–10761 RSC.
- D. M. Vriezema, P. M. L. Garcia, N. Sancho Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2007, 46, 7378–7382 CrossRef CAS.
- G. Han, J.-T. Wang, X. Ji, L. Liu and H. Zhao, Bioconjugate Chem., 2017, 28, 636–641 CrossRef CAS.
- C. Tan, M. Arshadi, M. C. Lee, M. Godec, M. Azizi, B. Yan, H. Eskandarloo, T. W. Deisenroth, R. H. Darji, T. V. Pho and A. Abbaspourrad, ACS Nano, 2019, 13, 9016–9027 CrossRef CAS.
- K. Piradashvili, M. Fichter, K. Mohr, S. Gehring, F. R. Wurm and K. Landfester, Biomacromolecules, 2015, 16, 815–821 CrossRef CAS.
- A. Jafari, H. Sun, B. Sun, M. A. Mohamed, H. Cui and C. Cheng, Chem. Commun., 2019, 55, 1267–1270 RSC.
- M. S. Alkanawati, F. R. Wurm, H. Thérien-Aubin and K. Landfester, Macromol. Mater. Eng., 2018, 303, 1700505 CrossRef.
- K. Landfester and C. K. Weiss, Adv. Polym. Sci., 2010, 229, 1–49 CrossRef CAS.
- I. Schlegel, P. Renz, J. Simon, I. Lieberwirth, S. Pektor, N. Bausbacher, M. Miederer, V. Mailänder, R. Muñoz-Espí, D. Crespy and K. Landfester, Macromol. Biosci., 2018, 18, 1700387 CrossRef.
- S. Behzadi, C. Rosenauer, M. Kappl, K. Mohr, K. Landfester and D. Crespy, Nanoscale, 2016, 8, 12998–13005 RSC.
- K. Piradashvili, J. Simon, D. Paßlick, J. R. Höhner, V. Mailänder, F. R. Wurm and K. Landfester, Nanoscale Horiz., 2017, 2, 297–302 RSC.
- E. A. Prasetyanto, A. Bertucci, D. Septiadi, R. Corradini, P. Castro-Hartmann and L. De Cola, Angew. Chem., Int. Ed., 2016, 55, 3323–3327 CrossRef CAS.
- J. Dausend, A. Musyanovych, M. Dass, P. Walther, H. Schrezenmeier, K. Landfester and V. Mailänder, Macromol. Biosci., 2008, 8, 1135–1143 CrossRef CAS.
- J. Geng, W. Li, Y. Zhang, N. Thottappillil, J. Clavadetscher, A. Lilienkampf and M. Bradley, Nat. Chem., 2019, 11, 578–586 CrossRef CAS.
- G. Guizzunti and J. Seemann, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6590 CrossRef CAS.
- X. Yuan, N. Rietzschel, H. Kwon, A. B. W. Nuno, D. A. Hanna, J. D. Phillips, E. L. Raven, A. R. Reddi and I. Hamza, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E5144 CrossRef CAS.
- G. Begum, W. B. Goodwin, B. M. deGlee, K. H. Sandhage and N. Kröger, J. Mater. Chem. B, 2015, 3, 5232–5240 RSC.
- M. J. Männel, L. P. Kreuzer, C. Goldhahn, J. Schubert, M. J. Hartl and M. Chanana, ACS Catal., 2017, 7, 1664–1672 CrossRef.
- K. Vogele, J. List, F. C. Simmel and T. Pirzer, Langmuir, 2018, 34, 14780–14786 CrossRef CAS.
- B. Iyisan and K. Landfester, Macromol. Rapid Commun., 2019, 40, 1800577 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr08116g |
| This journal is © The Royal Society of Chemistry 2021 |