
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredicting the bulk modulus of single-layer covalent organic frameworks with square-lattice topology from molecular building-block properties†
Antonios
Raptakis
 ab,
Arezoo
Dianat
a,
Alexander
Croy
*a and
Gianaurelio
Cuniberti
ac
ab,
Arezoo
Dianat
a,
Alexander
Croy
*a and
Gianaurelio
Cuniberti
ac
aInstitute for Materials Science and Max Bergmann Center of Biomaterials, TU Dresden, 01062 Dresden, Germany. E-mail: alexander.croy@tu-dresden.de
bMax Planck Institute for the Physics of Complex Systems, 01187 Dresden, Germany
cDresden Center for Computational Materials Science (DCMS), TU Dresden, 01062 Dresden, Germany
First published on 22nd December 2020
Abstract
Two-dimensional Covalent Organic Frameworks (2D COFs) have attracted a lot of interest because of their potential for a broad range of applications. Different combinations of their molecular building blocks can lead to new materials with different physical and chemical properties. In this study, the elasticity of different single-layer tetrabenzoporphyrin (H2-TBPor) and phthalocyanine (H2-Pc) based 2D COFs is numerically investigated using a density-functional based tight-binding approach. Specifically, we calculate the 2D bulk modulus and the equivalent spring constants of the respective molecular building-blocks. Using a spring network model we are able to predict the 2D bulk modulus based on the properties of the isolated molecules. This provides a path to optimize elastic properties of 2D COFs.
Covalent Organic Frameworks (COFs) offer the unique possibility of engineering their chemical and physical features via a lego-like principle, where, through different combinations of molecular building blocks, novel material properties can emerge.1–3 As a result, they have gained increasing interest over the past years,2–6 and the potential of single-layer or few-layer 2D COFs has been demonstrated in various applications, including energy storage,7,8 supercapacitors,9,10 charge transport,11 optoelectronics,12,13 chemical catalysis14 and sensorics.15
The underlying combinatorial variety of possible materials, enabled by the previously mentioned bottom-up lego-principle, imposes additional challenges to experimental investigations, so that extensive computational studies are becoming invaluable. For example, in silico methods have been used to study photocatalysis,16 carbon capture,17,18 hydrogen storage,19 gas storage,20 methane adsorption21 and drug adsorption22 in 2D COFs. An especially challenging issue in this context is revealing possible correlations between the chemical and physical properties of COFs and the ones of their molecular constituents. This requires, for example, the identification of appropriate sets of non-redundant molecular descriptors. Learning COF properties using such fragment-level fingerprints can help to reduce the computational efforts and allows for an extended sampling of configurations. Since the reticular synthesis approach poses a highly combinatorial problem, we also expect that Machine Learning based approaches will become increasingly relevant in the near future.23
Although mechanical properties play an important role for many applications of COFs, up to now only a few experimental studies addressing COF elasticity are available. Specifically, measurements of the Young modulus of wafer-scale single-layer porphyrin-based imine COFs24 and of thin films of dihydroxyterethaldehyde-(triazine-triyl)trianiline (DHTA-TTA) COFs25 have been reported. In contrast, calculations of elastic properties of 2D COFs are routinely done using a range of different methods.26–28
In this work, we address the relation between elastic properties of a set of linker molecules and those of the resulting organic frameworks. To this end, we use a density-functional based tight binding (DFTB) approach to compute the bulk modulus of the COFs and the equivalent harmonic spring constants of the molecular building blocks. Based on a simple spring-network model,29 we are able to relate these two quantities.
The molecular spring constants can, in principle, be obtained experimentally and there have been several studies on their direct measurement.30–34 For example, in the work of Xu et al.,33 the spring constants of two small molecules, 1,8-octanedithiol and 4,4′-bipyridine, were measured in a setup consisting of two gold electrodes and a molecular junction. Similarly, the contact stiffness of small molecule ligands, bio-inspired peptide and peptoid oligomers was estimated by using atomic force microscopy.34
The COFs studied in this work include tetrabenzoporphyrin (H2-TBPor) and phtalocyanine (H2-Pc) as molecular cores and different linker molecules. For instance, in the example shown in Fig. 1, the core molecule is H2-TBPor (shaded in yellow) and the green shaded area indicates the linker molecule (bis(catecholato)diboron). In the context of our calculations and in the analytical approach, we take porphyrin and porphyrazine as core molecules, instead of H2-TBPor and H2-Pc, and consider the additional phenyls as part of the linker molecules as shown in Fig. 2. The geometry of the linker plays an important role for the elastic properties of the synthesized material. As linker molecules we use the following molecular systems: imine (N-benzylideneaniline), azo (azobenzene) and benzalphenylhydrazine (benzaldehyde phenylhydrazone), bis(catecholato)diboron and N-phthalimidophthalimide ([2,2′-biisoindoline]-1,1′,3,3′-tetraone) consisting of functionalized pyrolles, anthracene and phenazine.4,5,35 The first three consist of linear chains and the latter two include fused benzene rings. The resulting COFs are denoted as linker-core-COF as given in the caption of Fig. 2, which shows the different linker and core molecules. Due to the chemistry of the linkage, all the considered COF crystals feature a simple square lattice topology.
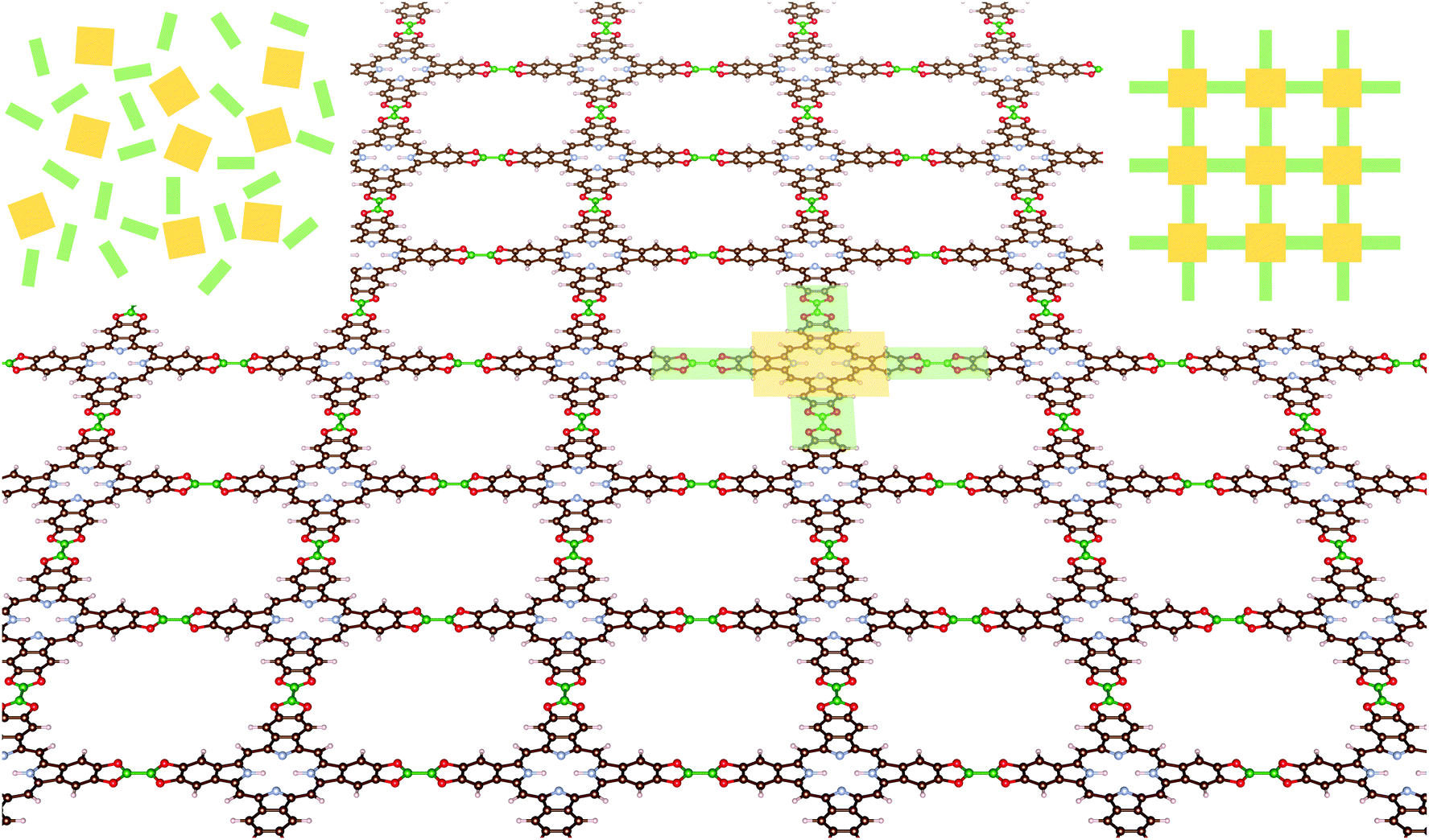 | ||
| Fig. 1 Schematic view of a CDB-TBPor-COF. The yellow and green areas indicate the core and linker molecule, respectively. Carbon atoms in the COF are colored brown, hydrogen grey, oxygen red, nitrogen light blue and boron green. | ||
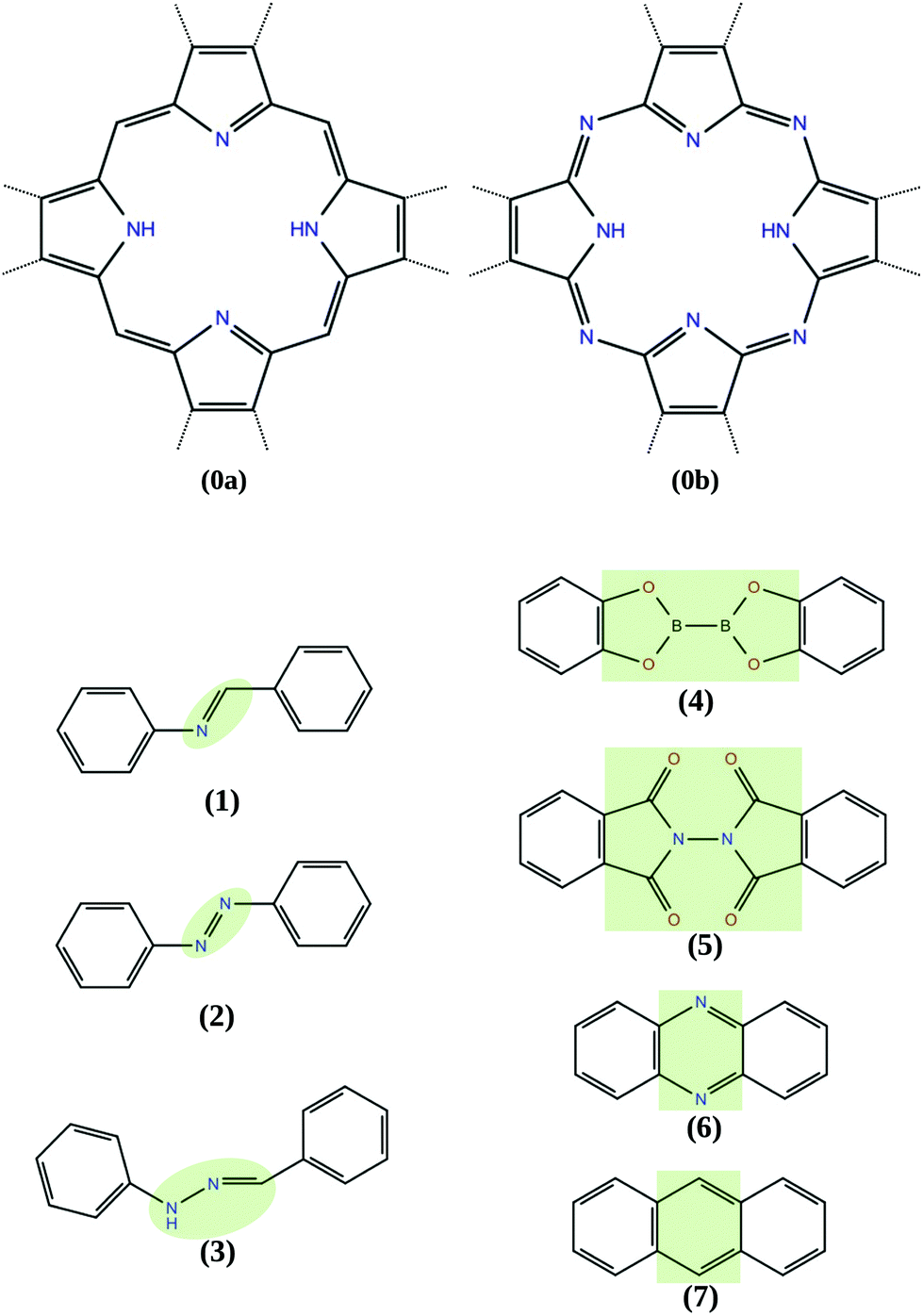 | ||
| Fig. 2 Core and linker molecules: (0a) porphyrin, (0b) porphyrazine, (1) imine (imine-TBPor-COF), (2) azo (azo-TBPor-COF), (3) benzalphenylhydrazine (BPH-TBPor-COF), (4) bis(catecholato)diboron (CDB-TBPor-COF), (5) N-phthalimidophthalimide (PP-TBPor-COF), (6) phenazine (phenazine-TBPor-COF), (7) anthracene (anthracene-TBPor-COF). | ||
This manuscript is structured as follows. In section 1 we review the concept of the equivalent spring constant of a molecule and use it to derive a model for the elastic properties of COFs with a square lattice topology. The computational methods are summarized in section 2. Then, we present and discuss the results of our study: the equivalent spring constants of the different linker and core molecules, the bulk moduli of the respective COFs and the possibility to tune the elastic properties by modifying the linker elasticity. Conclusions are given in section 4.
1 Equivalent molecular spring constants and 2D bulk moduli
1.1 Equivalent molecular spring constants
An equivalent spring constant can, in general, be associated with a complex system of connected springs. In particular, the combination of Hookean springs in series or parallel behaves like a single Hookean spring.36 In the context of molecules or molecular fragments, the equivalent molecular spring constant comprises the collective response of all chemical bonds to stretching or compression of the molecule. Depending on the chemical composition and molecular topology, the resulting behavior can approximately be Hookean and thus, the response of the molecule is considered as that of an effective Hookean spring. If the molecule consists of several fragments which are fused together, the equivalent molecular spring constant is approximately given in terms of those of the fragments.37,38 As an example, anthracene (see Fig. 2 (7)) consists of three fused benzene rings. If we assume each ring to be described by an effective spring with spring constant k, then the equivalent spring constant of the molecule consisting of n rings is obtained by simply adding the inverse spring constants36k(n) = k/n.1.2 Two dimensional bulk modulus
The bulk modulus is one of the two elastic constants of an isotropic material,39 and it quantifies the relative change of volume upon an applied pressure. For a two-dimensional material, the bulk modulus can be redefined according to the change in dimension as the relative change of area instead of volume and is obtained as: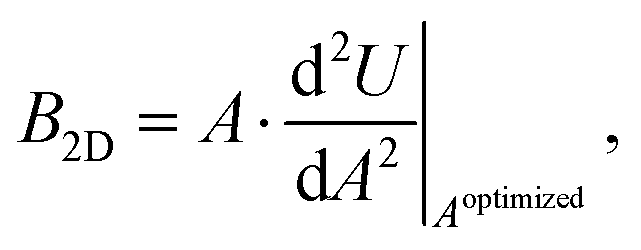 | (1) |
More specifically, we consider a square lattice with lattice constant a and area A(a) = a2. Since there are two identical bonds connecting neighboring sites in each unit cell, we can consider their respective contribution to the internal energy to be the same, i.e. U = Φx + Φy = 2Φ. Then, eqn (1) becomes
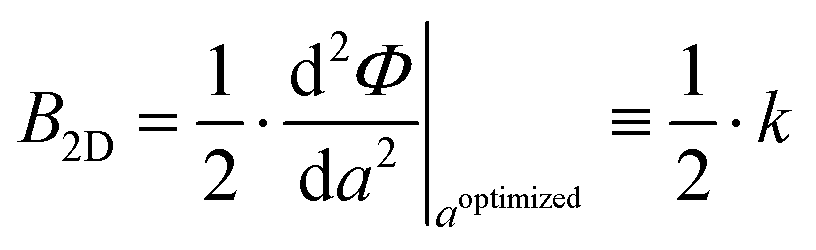 | (2) |
In two-dimensional porphyrin-based COFs with square lattice topology, the lattice sites are occupied by the core molecules which are in turn inter-connected by linker molecules. In Fig. 1, such a representative COF-structure is shown. Bi-axially stretching this 2D structure, will change the position of atoms depending on their local environment. If the building blocks are described by effective springs, then on each axis of the unit cell there are two springs in series, one corresponds to the core and one to the linker molecule. If we consider that each building block can be described as a linear spring, then according to the previous section the equivalent spring constant of the fused molecule can be written as
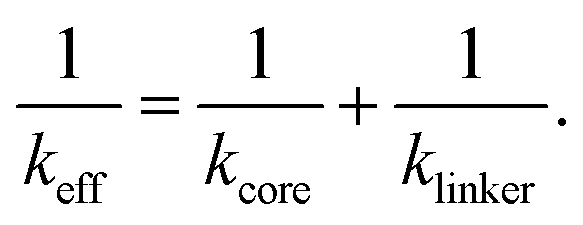 | (3) |
Combining eqn (2) and (3) then gives
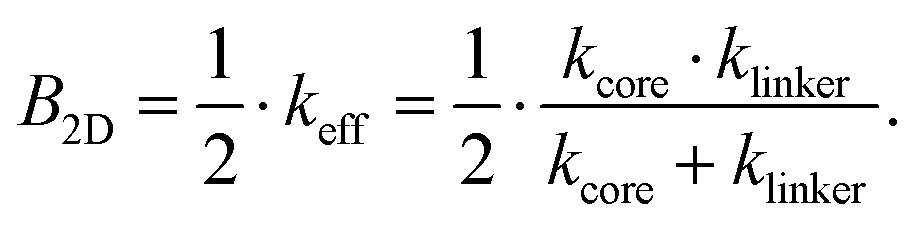 | (4) |
Eqn (4) is one of the central results of this work. It allows to calculate the bulk modulus of a single-layer 2D COF with square lattice topology only from the knowledge of the equivalent spring constants of the building blocks.
2 Computational methods
In this work, we mainly use the DFTB method to compute the mechanical properties of various COF structures. DFTB methods are much faster than ab initio and Density Functional Theory (DFT) methods and are, therefore, particularly attractive in applications to large systems and for extensive sampling of the configuration space.All calculations were performed using the self-consistent charge extension (SCC) method,40,41 implemented in the DFTB+ code (version 17.1). In order to better gauge the effect of the parametrization of the Hamiltonian matrix elements on our results, different Slater–Koster sets were used: 3ob-3-1 and mio-1-1,42 pbc-0-3,43 ob2-1-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44 and matsci-0-3.45 Those parametrizations have been optimized for different target systems. For example, mio-1-1 was designed for materials and biological systems and includes a second-order expansion of the energy, giving results comparable to DFT-Generalized Gradient Approximation (GGA). The 3ob-3-1 parametrization has been developed particularly for DFTB3.46 Compared to mio-1-1, it firstly improves non-covalent bonds by optimizing bond distances and energies and secondly, it reduces the overbinding energy.42 For organic and biological molecules, ob2-1-1 has been designed with a focus on long-range interactions. It has a very good performance for thermochemistry, geometries, and vibrational frequencies.44 The pbc-0-3 parametrization gives reasonable results for solid-state systems and iron clusters.43,47 Finally, matsci-0-3 is based on semi-relativistic calculations with self-consistent charge corrections,45 and gives results in quantitative agreement with the GGA functional of Perdew–Burke–Ernzerhof (PBE).48
44 and matsci-0-3.45 Those parametrizations have been optimized for different target systems. For example, mio-1-1 was designed for materials and biological systems and includes a second-order expansion of the energy, giving results comparable to DFT-Generalized Gradient Approximation (GGA). The 3ob-3-1 parametrization has been developed particularly for DFTB3.46 Compared to mio-1-1, it firstly improves non-covalent bonds by optimizing bond distances and energies and secondly, it reduces the overbinding energy.42 For organic and biological molecules, ob2-1-1 has been designed with a focus on long-range interactions. It has a very good performance for thermochemistry, geometries, and vibrational frequencies.44 The pbc-0-3 parametrization gives reasonable results for solid-state systems and iron clusters.43,47 Finally, matsci-0-3 is based on semi-relativistic calculations with self-consistent charge corrections,45 and gives results in quantitative agreement with the GGA functional of Perdew–Burke–Ernzerhof (PBE).48
For all calculations of COFs, periodic boundary conditions were used. The single layers of the square lattice were optimized using the SCC method with 30 Å distance between the layers to avoid interactions between them. We applied (7,7,1) k-points according to Monkhorst and Pack.49 The strain was changing the area of the unit cell around the equilibrium state in steps of 0.2%, while the z-direction of the lattice vector was always remaining the same.
To establish the applicability of DFTB for the computation of COF properties, we also carried out benchmark calculations for the bulk modulus using DFT based on the open source implementation of the Grid-based Projected Augmented Wave-method50 (GPAW51) code within the local density approximation (LDA) as well as GGA-PBE. The representation of the wave-function was implemented through plane-waves using a cut-off energy of 400 eV. For the COFs, we used (5,5,1) k-points. The linker molecules were put in the middle of the simulation box, keeping a vacuum of 15 Å in all directions. For the core molecule, the vacuum was 20 Å. The geometry optimization was achieved using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) optimizer.
3 Results and discussion
The equilibrium lattice constant of the COFs was found by using the equation of state52 as introduced by Murnaghan53,59 and implemented in ASE.54 This approach also yields the 3D bulk modulus B3D from the volume dependence of the total energy. To obtain the 2D bulk modulus we multiplied B3D by the height of the unit cell.For the calculations of the molecular spring constant of the linker molecules, we first optimize the molecular geometry. Then, applying a constraint to the outermost carbon atoms, the length of the molecule is changed according to the applied strain in steps of 0.2%. We compute the total energy for each length, which can be fitted by a polynomial
| Elinker(L) = c0 + c1·L + c2·L2 + c3·L3. | (5) |
The spring constant is then calculated from the second derivative of the energy around the minimum as
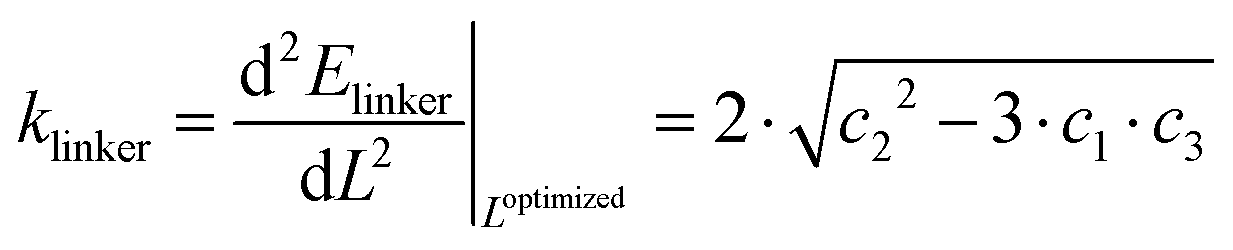 | (6) |
The concept for the estimation of the core spring-constant is similar as above. However, the length dependence is substituted by the change of area A:
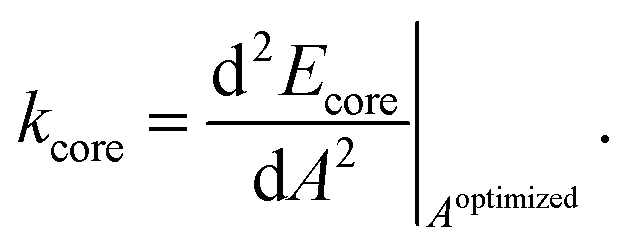 | (7) |
The underlying isotropic contraction or expansion of the molecule mimics more closely the behavior of the core in the COF.
3.1 Equivalent molecular spring constants
Fig. 3(a) shows the strain dependence of the energy for several linker molecules obtained from DFTB calculations with the matsci-0-3 parametrization. The energy at zero strain is shifted to zero for convenience. In all cases, a nearly parabolic behavior of the energy is observed and the curvature is used to extract the equivalent spring constant according to eqn (6) for the linker molecules and according to eqn (7) for the core molecules. The resulting values are summarized in Table 1. Of all the linker molecules, benzalphenylhydrazine has the smallest spring constant, while anthracene and phenazine have spring constants which are an order of magnitude larger. This difference in behavior is related to the chemical structure which can be inferred from Fig. 2. | ||
| Fig. 3 (a) Relative binding energy per atom as function of the strain for several molecules using matsci-0-3. (b) Spring constants using different Slater Koster parametrizations for selected molecules: anthracene, phenazine, N-phthalimidophthalimide and porphyrin (from left to right). | ||
| Molecule | Spring constant (N m−1) |
|---|---|
| Benzalphenylhydrazine | 35.2 |
| Imine | 117.0 |
| Azo | 123.4 |
| Bis(catecholato)diboron | 145.8 |
| N-Phthalimidophthalimide | 147.4 |
| Anthracene | 221.1 |
| Phenazine | 246.8 |
| Porphyrin | 163.8 |
| Porphyrazine | 195.4 |
It is interesting to compare the results for the spring constants for different Slater–Koster parametrizations. This is shown in Fig. 3(b) and summarized in Table 1 (ESI†) for phenazine, anthracence, N-phthalimidophthalimide and porphyrin. One can see that all parametrizations yield similar values, which are also consistent with the results from DFT. Since the different parametrizations are obtained by best fits to different target systems, the observed differences are reasonable. The ob2-1-1 parametrization systematically gives larger spring constants compared to the other ones and pbc-0-3 yields a drastically smaller value for N-phthalimidophthalide.
Next, we analyze the influence of the strain on different parts of the molecules. Fig. 4 show the relative change of specific bond lengths as a function of the applied strain for phenazine, anthracene and azo. Phenazine and anthraceneare molecules with similar structure and a planar arrangement of the atoms. Azo is planar as well, with two benzene rings being connected by a linear chain which consists of a N–N double bond. Fig. 4(a) shows that there is no change of the bond length between those nitrogens (orange line), and only a negligible change of the bond length between nitrogen and the ring (red line). The largest percentage of strain is absorbed by the outer rings (green line).
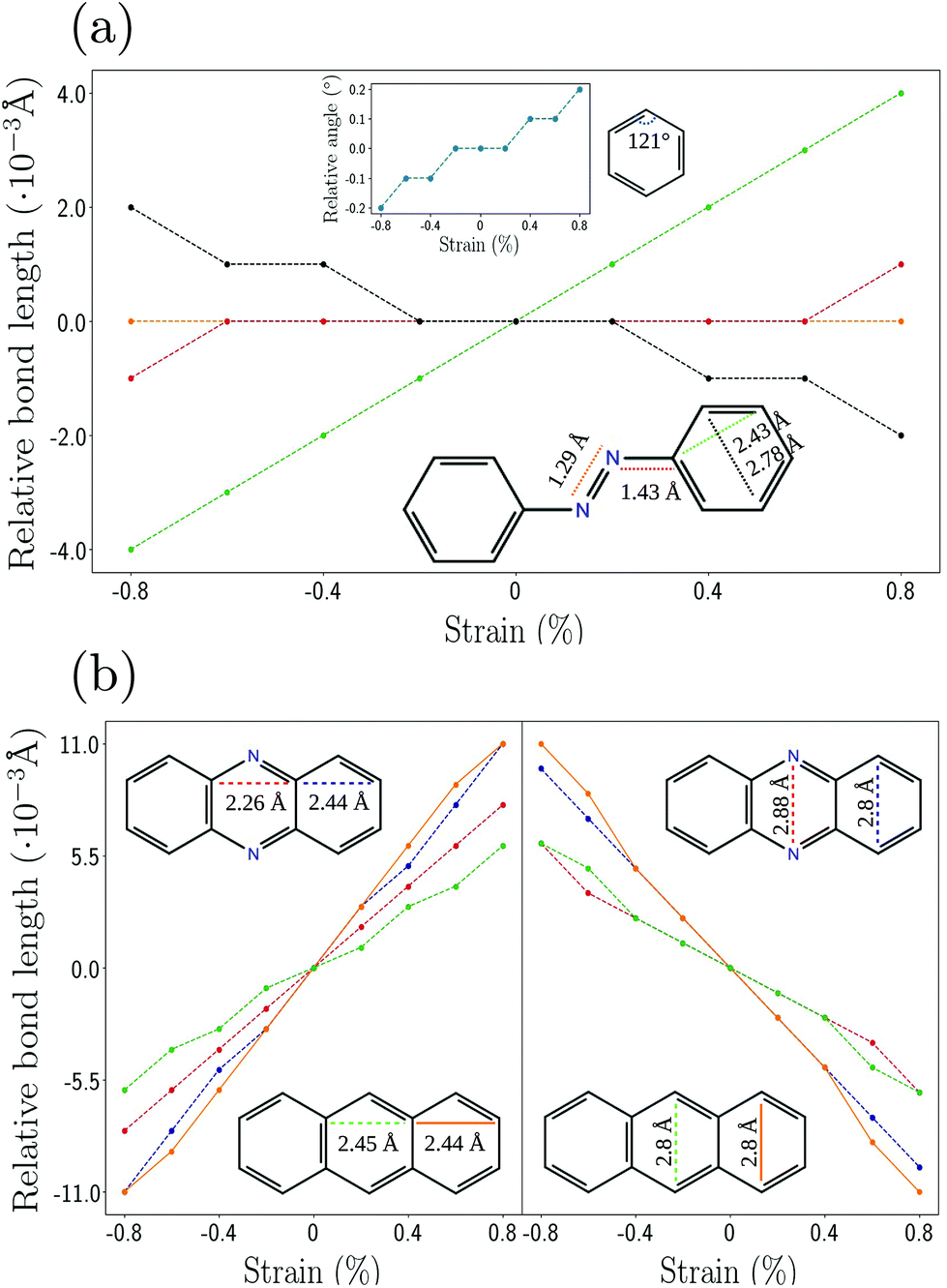 | ||
| Fig. 4 Strain analysis of specific distances and bond-lengths for the linker molecules of (a) azo and (b) phenazine and anthracene using DFTB+ and matsci-0-3. | ||
In contrast to this behavior, for anthracene and phenazine the width of all rings shows a similar increase upon applying strain. This is shown in Fig. 4(b). The width of the inner rings is changing with a slightly different rate than that of the edge ring, which implies that the strain is not transferred equally along the chain. This behavior might relate to the applied constraints, which keep the bond length between the constrained atoms fixed. We also observe a reduction of the height of the respective rings as the strain is increased. This effect is akin to the Poisson effect in the elasticity of solids.55,56
3.2 Bulk modulus
Fig. 5(a) shows the strain dependencies of the total energies for different COFs with TBPor cores and different linker molecules using matsci-0-3. The bulk moduli are extracted from the curvature at zero strain as described earlier in this section. The comparison with other parametrizations, shown in Fig. 5(b), again yields an overall quantitative agreement for the bulk moduli with the exception of BPH-TBPor-COF. The values of different parametrization are summarized in Table 2 (ESI†).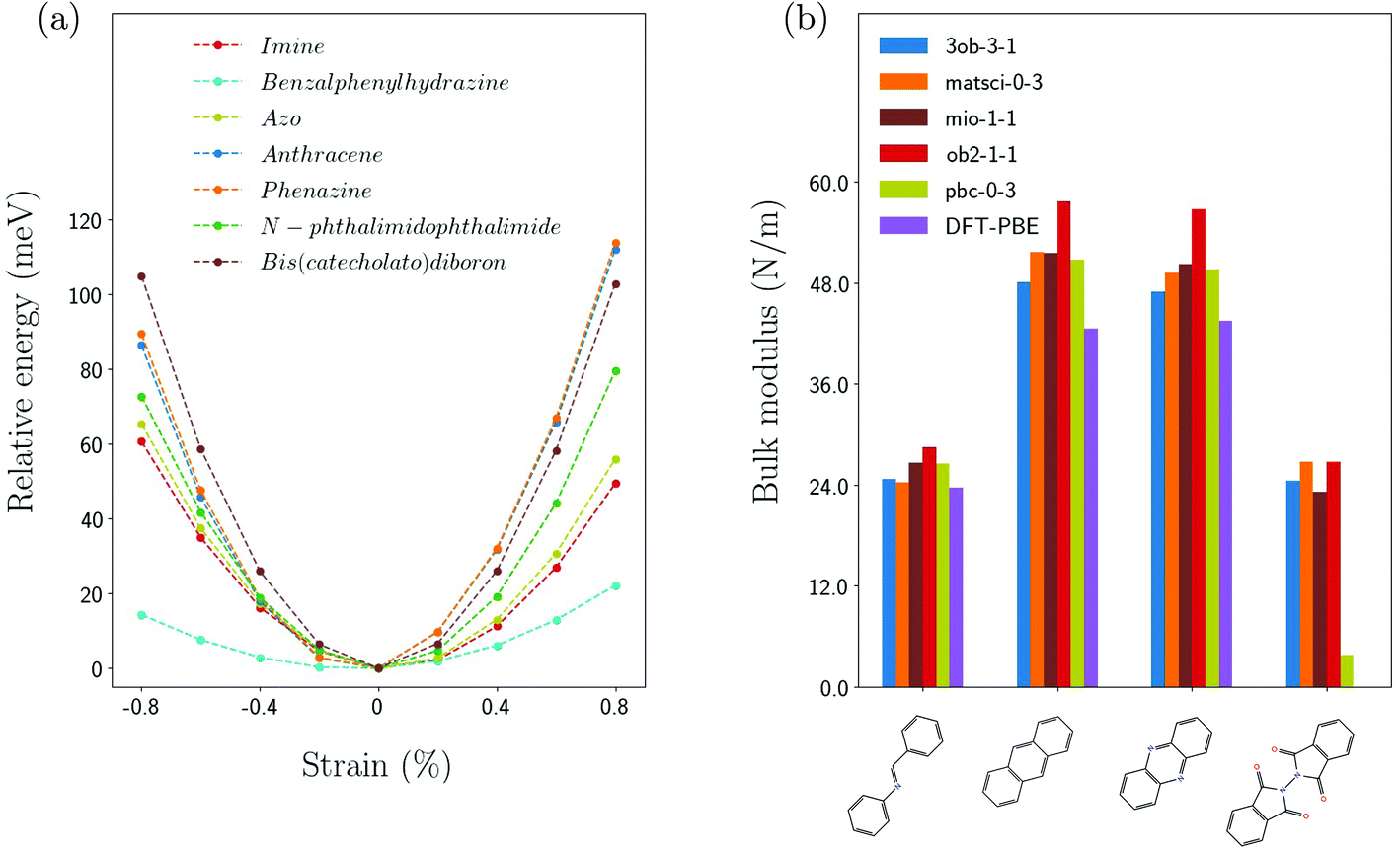 | ||
| Fig. 5 (a) Relative binding energy as function of strain for different TBPor-COFs using matsci-0-3. (b) Bulk modulus using different Slater–Koster parametrizations for selected linker molecules: imine, anthracene, phenazine and N-phthalimidophthalimide (from left to right). | ||
In Table 2 the calculated bulk moduli are compared with the analytical formula given by eqn (4) and using the equivalent spring constants from Table 1. While the bulk moduli of the COFs are much smaller than that of graphene (∼210 N m−1), they are comparable to other inorganic 2D crystals, such as silicene (∼45 N m−1) or InSe (∼35 N m−1).57,58 The analytical approach is in close agreement with the calculations for the linker molecules of bis(catecholato)diboron, anthracene and phenazine, which are planar. The results for linear chains have, however, relatively large deviations from the DFTB calculations, especially for benzalphenylhydrazine. The analytical approach for PP-TBPor-COF gives a relatively large deviation as well. Here, the constraints used for calculating the spring constant prevent a change of the torsional angle, which leads to an additional stiffening of the molecule. Due to the repulsion of the oxygen atoms a non-planar configuration of the linker molecule is preferred. In the COF, the linkage to the core leads to a more planar geometry. Moreover, by stretching the COF, the dihedral angle of the pyrroles in the linker molecules is changing, which in turn allows them to lie in the same plane as the core.
| Linker molecule | Bulk modulus (N m−1) | |
|---|---|---|
| Analytical approach | Calculation | |
| Benzalphenylhydrazine | 14.5 | 7.4 |
| Imine | 35.2 | 27.3 |
| Azo | 34.1 | 24.3 |
| Bis(catecholato)diboron | 38.8 | 35.3 |
| N-Phthalimidophthalimide | 34.1 | 26.7 |
| Anthracene | 47.0 | 49.2 |
| Phenazine | 49.2 | 51.6 |
In accordance with the discussion in the previous subsection, we find a very good agreement of the calculations with the analytical model for those linker molecules where the strain is evenly distributed along the molecule (anthracene, phenazine, bis(catecholato)diboron and N-phthalimidophthalimide). The molecules involving a linear chain (or single bonds) are seen to be too flexible to be described by a single effective spring. In the following we will focus only on the former set of molecules.
3.3 Manipulation of elastic properties
To further investigate the correlation between the bulk moduli of the COFs with the equivalent spring constants of the linker molecules, we added few benzene rings to bis(catecholato)diboron, phenazine, anthracene and N-phthalimidophthalimide. Additionally, we change the core to phtalocyanine. Table 3 (ESI†) summarizes the resulting equivalent spring constants and the respective bulk moduli of the COFs so built.The lower part of Fig. 6 shows the dependence of the equivalent spring constant on the number of inserted benzene rings. In accordance with the discussion in section 1 the spring constant of the molecule reducesas the number of benzene rings increases since those act as springs in series. A more quantitative analysis based on the combination of the spring constants of molecular fragments yield results in good agreement with the calculations for phenazine, anthracene and bis(catecholato)diboron (see ESI†).
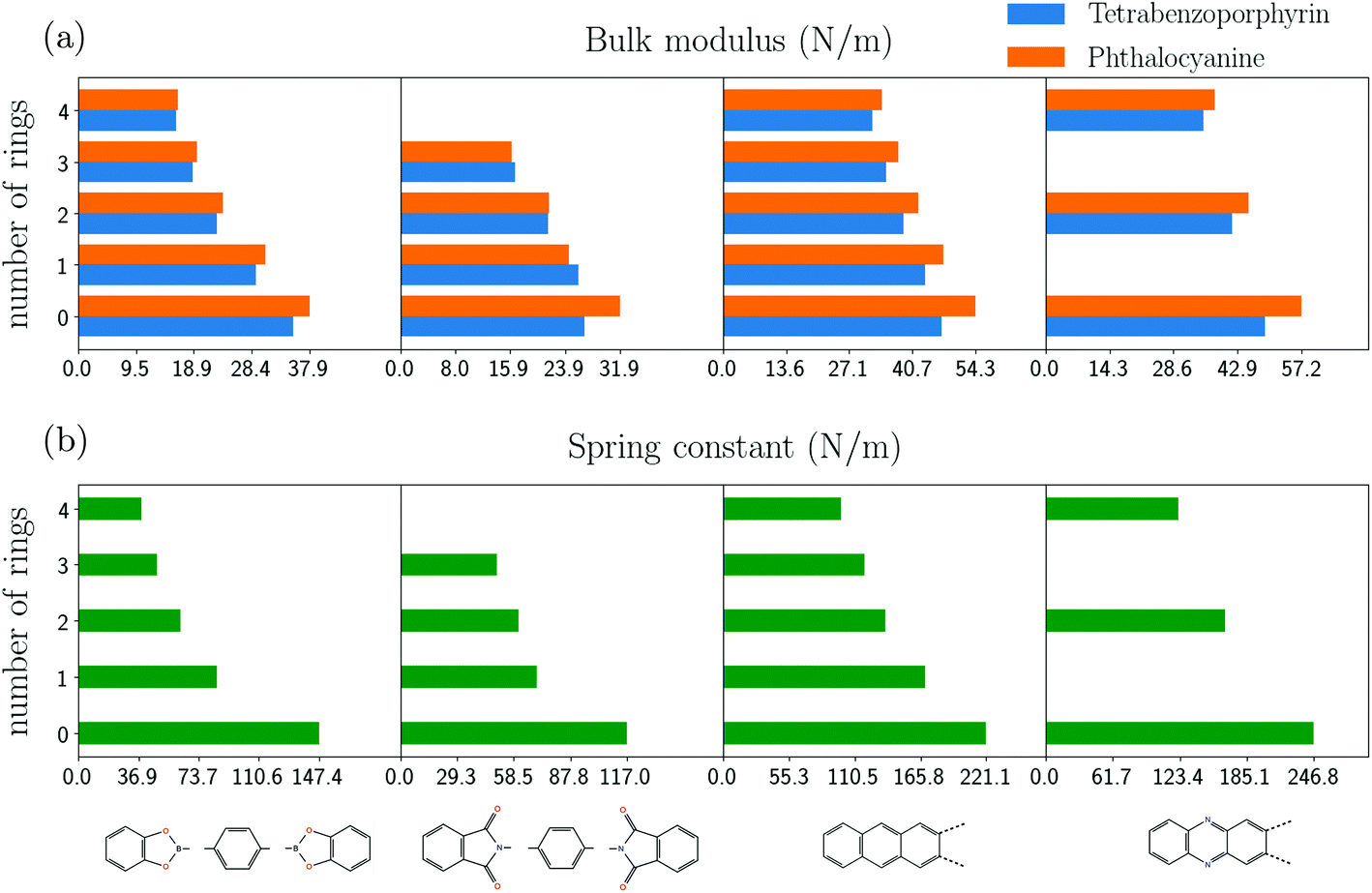 | ||
| Fig. 6 Comparison between (a) bulk moduli of COFs by changing the molecular core (blue denotes tetrabenzoporphyrin and orange denotes phthalocyanine) and (b) spring constant of molecular linkers with added phenyl-rings as shown on the bottom of the figure. Each column of bar-plots of (a) and (b) corresponds to the respective linker with phenyl-rings added as given. The structures from left to right are (COF(molecular linker)): CDB-COF(bis(catecholato)diboron), PP-COF(N-phthalimidophthalimide), anthracene-COF(anthracene), phenazine-COF(phenazine). All results are obtained with DFTB+ and matsci-0-3. | ||
For N-phthalimidophthalimide, the intrinsic torsional feature due to the presence of the opposing oxygens is replaced by a dihedral angle between the extra rings and the rest molecule. Moreover, these COFs become mostly planar, except for the added rings which are twisted with respect to each other. This effect is not accounted for in the Hookean spring model.
In the upper part of Fig. 6, the respective results of the bulk moduli for the COFs are shown. Similar to the spring constants, the bulk moduli are seen to become smaller as more benzene rings are added to the linker molecules. The comparison with the analytical formula (ESI†) yields an excellent agreement with the calculated values, with an exception for PP-COFs.
Additionally, we change the molecular core and replace H2-porphyrin (H2-TBPor-COF) by H2-porphyrazine (H2-Pc-COF). Phthalocyanine consists of the core of porphyrazine, which can be considered as functionalized porphyrazine. Firstly, the calculation of thespring constant shows that porphyrazine is stiffer than porphyrin. The calculated value of the spring constant for porphyrin is 163.8 N m−1, while for porphyrazine it is 195.4 N m−1. This reflects to the results of Table 3 in the ESI,† which shows that some COFs (with CDB, anthracene and phenazine linkers) with porphyrazine as core are stiffer. For the other COFs, however, the bulk moduli are very similar to the ones of TBPor-COFs, which indicates that the linker molecules determine the elastic behavior in those cases.
Motivated by the findings above, we show in Fig. 7 the bulk moduli of different COFs vs. the equivalent spring constants of the respective linker molecules. One can see a clear correlation between the spring constants and bulk moduli. As indicated above, the analytical model and eqn (4) yield results in excellent agreement with the calculations. A separate fit of eqn (4) to the calculated data gives kcore = 175.4 N m−1 for porphyrin and kcore = 209.4 N m−1 for porphyrazine, very close to the values found above (see Table 1).
 | ||
| Fig. 7 Correlation between spring constant and bulk modulus for the different cores. The fit according to eqn (4) gives values of kcore very close to the calculation for the core alone as listed in Table 1. The size of the circles for the structures is related to the number of the added benzene rings, while the color corresponds to the selected linker molecule as shown in the legend. | ||
In Fig. 7, there is one outlier for each core, which corresponds to the N-phthalimidophthalimide linker (smaller-sized circle). The spring constant of this linker has been overestimated, as explained in section 2. When rings are added into the linker, N-phthalimide partly lies on the same plane, while the rings tilt out of the plane. The corresponding COF structures are planar except for the added rings, which are tilted out of plane like in thecase of the isolated linker molecule.
Nevertheless, the fitting of eqn (4) verifies that we can predict the bulk modulus for planar 2D COFs which do not have any linear chain within the linker molecule.
4 Conclusions
In summary, we calculated the spring constants of several molecules, which can be used as linker molecules for building up COFs, using DFTB and different Slater Koster parametrizations. Using tetrabenzoporphyrin (H2-porphyrin based) and phthalocyanine (H2-porphyrazine based) as cores leads to monolayer crystals with a square lattice structure. The spring constant and thus the stiffness of the molecule were shown to be closely related to their structure. By adding benzene rings in selected linker molecules, we verified that the equivalent spring constants become smaller as more rings are added.Turning to the COFs, we calculated their bulk modulus from the strain dependence of the total energy. We found that the bulk modulus is correlated with the spring constant of the respective linker molecule. Modelling those linker molecules as Hookean springs we derived a simple analytical formula, given by eqn (4), to relate the bulk modulus to the corresponding spring constant. This analytical approach was shown to work very well for a set of linker molecules and explains the observed correlation.
Our results explicitly demonstrate the correlation between the properties of the molecular building blocks and the elastic properties of the resulting COFs. More generally, it is expected that such correlations also exist between other properties of the linker molecules and the COFs. Finding the relevant set of molecular descriptors is the foremost challenge for achieving a rational design of covalent organic frameworks.
Credit authorship contribution statement
Antonios Raptakis: Investigation, methodology, visualization, Writing – original draft, writing – review & editing. Arezoo Dianat: Supervision, methodology, writing – review & editing. Alexander Croy: Supervision, conceptualization, methodology, writing – review & editing. Gianaurelio Cuniberti: Funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R. thanks the International Max Planck Research School “Many Particle Systems in Structured Environment” and the Chair of Materials Science and Nanotechnology in the Faculty of Mechanical Engineering of TU Dresden for financial support; A. Mendez, H.-H. Lin, D. Bodesheim, S. Kampmann for helpful discussions; A. Ciesielski for the proposed linker molecules and L. Cusin for the abbreviations. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 813036. We also acknowledge the Center for Information Services and High Performance Computing (ZIH) at TU Dresden for computational resources.References
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453–465 CrossRef CAS.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef.
- S. Kim and H. C. Choi, ACS Omega, 2020, 5, 948–958 CrossRef CAS.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- X. Li, P. Yadav and K. P. Loh, Chem. Soc. Rev., 2020, 49, 4835–4866 RSC.
- D. Rodríguez-San-Miguel, C. Montoro and F. Zamora, Chem. Soc. Rev., 2020, 49, 2291–2302 RSC.
- F. Xu, S. Yang, X. Chen, Q. Liu, H. Li, H. Wang, B. Wei and D. Jiang, Chem. Sci., 2019, 10, 6001–6006 RSC.
- Z. Li, X. Feng, Y. Zou, Y. Zhang, H. Xia, X. Liu and Y. Mu, Chem. Commun., 2014, 50, 13825–13828 RSC.
- H. Peng, J. Raya, F. Richard, W. Baaziz, O. Ersen, A. Ciesielski and P. Samorì, Angew. Chem., 2020, 59, 2–10 CrossRef.
- M. Wang, H. Guo, R. Xue, Q. Li, H. Liu, N. Wu, W. Yao and W. Yang, ChemElectroChem, 2019, 6, 2984–2997 CrossRef CAS.
- X. Feng, L. Liu, Y. Honsho, A. Saeki, S. Seki, S. Irle, Y. Dong, A. Nagai and D. Jiang, Angew. Chem., Int. Ed., 2012, 51, 2618–2622 CrossRef CAS.
- A. K. Mandal, J. Mahmood and J.-B. Baek, ChemNanoMat, 2017, 3, 373–391 CrossRef CAS.
- M. Wang, M. Ballabio, M. Wang, H.-H. Lin, B. P. Biswal, X. Han, S. Paasch, E. Brunner, P. Liu, M. Chen, M. Bonn, T. Heine, S. Zhou, E. Cánovas, R. Dong and X. Feng, J. Am. Chem. Soc., 2019, 141, 16810–16816 CrossRef CAS.
- H. Hu, Q. Yan, R. Ge and Y. Gao, Chin. J. Catal., 2018, 39, 1167–1179 CrossRef CAS.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang, R. Deng, Y. Fu, L. Li, W. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC.
- Y. Wan, L. Wang, H. Xu, X. Wu and J. Yang, J. Am. Chem. Soc., 2020, 142, 4508–4516 CrossRef CAS.
- D. Ongari, A. V. Yakutovich, L. Talirz and B. Smit, ACS Cent. Sci., 2019, 5, 1663–1675 CrossRef CAS.
- K. S. Deeg, D. Damasceno Borges, D. Ongari, N. Rampal, L. Talirz, A. V. Yakutovich, J. M. Huck and B. Smit, ACS Appl. Mater. Interfaces, 2020, 12, 21559–21568 CrossRef CAS.
- M. Suri, M. Dornfeld and E. Ganz, J. Chem. Phys., 2009, 131, 174703 CrossRef.
- A. Sharma, A. Malani, N. V. Medhekar and R. Babarao, CrystEngComm, 2017, 19, 6950–6963 RSC.
- S. Keskin, J. Phys. Chem. C, 2012, 116, 1772–1779 CrossRef CAS.
- H. Hashemzadeh and H. Raissi, J. Phys. D: Appl. Phys., 2018, 51, 345401 CrossRef.
- J. Schmidt, M. R. G. Marques, S. Botti and M. A. L. Marques, npj Comput. Mater., 2019, 5, 2057–3960 Search PubMed.
- H. Sahabudeen, H. Qi, B. A. Glatz, D. Tranca, R. Dong, Y. Hou, T. Zhang, C. Kuttner, T. Lehnert, G. Seifert, U. Kaiser, A. Fery, Z. Zheng and X. Feng, Nat. Commun., 2016, 7, 13461 CrossRef CAS.
- Q. Hao, C. Zhao, B. Sun, C. Lu, J. Liu, M. Liu, L.-J. Wan and D. Wang, J. Am. Chem. Soc., 2018, 140, 12152–12158 CrossRef CAS.
- W. Zhou, H. Wu and T. Yildirim, Chem. Phys. Lett., 2010, 499, 103–107 CrossRef CAS.
- L. N. Duong, V. N. Tuoc and N. T. Thao, J. Phys.: Conf. Ser., 2019, 1274, 012010 CrossRef.
- O. G. Ziogos, I. Blanco and J. Blumberger, J. Chem. Phys., 2020, 153, 044702 CrossRef CAS.
- M. Ostoja-Starzewski, Appl. Mech. Rev., 2002, 55, 35–60 CrossRef.
- D. Ebeling, Q. Zhong, S. Ahles, L. Chi, H. A. Wegner and A. Schirmeisen, Appl. Phys. Lett., 2017, 110, 183102 CrossRef.
- D. R. Rotake and A. D. Darji, 2018 IEEE SENSORS, 2018, pp. 1–4 Search PubMed.
- B. Mehrafrooz and A. Shamloo, J. Theor. Biol., 2018, 448, 94–103 CrossRef CAS.
- B. Xu, X. Xiao and N. J. Tao, J. Am. Chem. Soc., 2003, 125, 16164–16165 CrossRef CAS.
- N. C. Miller, H. M. Grimm, W. S. Horne and G. R. Hutchison, Nanoscale Adv., 2019, 1, 4834–4843 RSC.
- E. Vitaku, C. N. Gannett, K. L. Carpenter, L. Shen, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2020, 142, 16–20 CrossRef CAS.
- K. Symon, Mechanics, Addison-Wesley Publishing Company, 1971 Search PubMed.
- M. Radiom, M. Plinio and T. A. Wesolowski, J. Mol. Model., 2018, 24, 24–36 CrossRef.
- R. Ramachandran, H. B. Li, W.-Y. Lo, A. Neshchadin, L. Yu and J. Hihath, Nano Lett., 2018, 18, 6638–6644 CrossRef CAS.
- N. Ashcroft and N. Mermin, Solid State Physics, Holt, Rinehart and Winston, 1976 Search PubMed.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- M. Elstner, J. Phys. Chem. A, 2007, 111, 5614–5621 CrossRef CAS.
- M. Gaus, A. Goez and M. Elstner, J. Chem. Theory Comput., 2013, 9, 338–354 CrossRef CAS.
- C. Köhler, Z. Hajnal, P. Deák, T. Frauenheim and S. Suhai, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 085333 CrossRef.
- V. Q. Vuong, J. Akkarapattiakal Kuriappan, M. Kubillus, J. J. Kranz, T. Mast, T. A. Niehaus, S. Irle and M. Elstner, J. Chem. Theory Comput., 2018, 14, 115–125 CrossRef CAS.
- B. Lukose, A. Kuc, J. Frenzel and T. Heine, Beilstein J. Nanotechnol., 2010, 1, 3762 Search PubMed.
- M. Gaus, Q. Cui and M. Elstner, J. Chem. Theory Comput., 2011, 7, 931–948 CrossRef CAS.
- C. Köhler, G. Seifert and T. Frauenheim, Chem. Phys., 2005, 309, 23–31 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- https://wiki.fysik.dtu.dk/gpaw/ .
- A. B. Alchagirov, J. P. Perdew, J. C. Boettger, R. C. Albers and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 026103 CrossRef.
- C. L. Fu and K. M. Ho, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 5480–5486 CrossRef CAS.
- https://wiki.fysik.dtu.dk/ase/ .
- H. Jafari, S. Sepehri, M. R. H. Yazdi, M. M. Mashhadi and M. M. S. Fakhrabadi, Phys. Scr., 2020, 95, 085004 CrossRef CAS.
- S. Feng, Y. Yang, F. Guo, L. Su, X. Cheng, C. Yuan and K. Yang, J. Alloys Compd., 2020, 844, 156098 CrossRef CAS.
- D. Akinwande, C. J. Brennan, J. S. Bunch, P. Egberts, J. R. Felts, H. Gao, R. Huang, J.-S. Kim, T. Li, Y. Li, K. M. Liechti, N. Lu, H. S. Park, E. J. Reed, P. Wang, B. I. Yakobson, T. Zhang, Y.-W. Zhang, Y. Zhou and Y. Zhu, Extreme Mech. Lett., 2017, 13, 42–77 CrossRef.
- A. Croy, Journal of Physics: Materials, 2020, 3, 02LT03 Search PubMed.
- F. D. Murnaghan, PNAS, 1944, 30, 244–247 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/D0NR07666J |
| This journal is © The Royal Society of Chemistry 2021 |