
An enzyme-mediated universal fluorescent biosensor template for pathogen detection based on a three-dimensional DNA walker and catalyzed hairpin assembly†
 *a
*a
Abstract
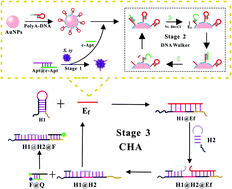
An enzyme-mediated universal fluorescent biosensor template for rapid detection of pathogens was developed based on the strategy of a three-dimensional (3D) DNA walker and catalyzed hairpin assembly (CHA) reaction. In the bacterial recognition step, a strand displacement reaction between bacteria and the double-stranded complex caused the release of the walker strand. The walker strand triggered the DNA walker to produce an enzyme fragment, and the DNA walker used gold nanoparticles (AuNPs) as the track to provide an excellent DNA ligand anchoring area. In the CHA step, the enzyme fragment induced the CHA cycle to yield fluorescence signals, which greatly enhanced the conversion ratio of trigger DNA and the sensitivity of the fluorescent biosensor. The effect of the distance and density of the DNA ligand was studied by adjusting the length of poly-adenine (PolyA), and was further explored by its reaction kinetics. By comparing the maximum reaction rate (Vmax), Michaelis constant (Km) and turnover number (Kcat), the optimized PolyA probe was assessed and identified. In this work, the optimized PolyA-DNA probe exhibited an outstanding sensitivity in Salmonella typhimurium (S. ty) detection, which is 11.9 times and 4.6 times higher than those of the SH-DNA and the MCH treated SH-DNA. Meanwhile, a detection limit of 28.1 CFU mL−1 was achieved in Escherichia coli (E. coli) detection. Furthermore, the biosensor achieved good selectivity and high repeatability with recoveries of 91%–115% for real sample detection. Considering these advantages, this template has great potential as a routine tool for pathogen detection and has wide applications in the field of global public health and food safety.
- This article is part of the themed collection: Editor’s Choice: Recent breakthroughs in nanobiotechnology
 Please wait while we load your content...
Please wait while we load your content...

