
Hypoxia-specific anti-RAGE exosomes for nose-to-brain delivery of anti-miR-181a oligonucleotide in an ischemic stroke model†
Minkyung
Kim
,
Youngki
Lee
and
Minhyung
Lee
 *
*
Department of Bioengineering, College of Engineering, Hanyang University, 222 Wangsimni-ro, Seondong-gu, Seoul 04763, Korea. E-mail: minhyung@hanyang.ac.kr; Fax: +82-2-2220-4454; Tel: +82-2-2220-0484
First published on 20th July 2021
Abstract
Ischemic stroke is caused by a reduction in blood flow to the brain due to narrowed cerebral arteries. Thrombolytic agents have been used to induce reperfusion of occluded cerebral arteries. However, brain damage continues to progress after reperfusion and induces ischemia-reperfusion (I/R) injury. The receptor for advanced glycation end-products (RAGE) is overexpressed in hypoxic cells of the ischemic brain. In this study, an exosome linked to RAGE-binding-peptide (RBP-Exo) was developed as a hypoxia-specific carrier for nose-to-brain delivery of anti-microRNA oligonucleotide (AMO). The RBP-Exos were less than 50 nm in size and had negative surface charge. In vitro studies showed that RBP-Exos delivered AMO181a to Neuro2A cells more efficiently than unmodified exosomes (Unmod-Exos). In addition, RAGE was downregulated by RBP-Exos, suggesting that the RBP moiety of the RBP-Exos reduced the RAGE-mediated signal pathway. MicroRNA-181a (miR-181a) is one of the upregulated miRNAs in the ischemic brain and its downregulation can reduce the damage to the ischemic brain. Cholesterol-modified AMO181a (AMO181a-chol) was loaded onto the RBP-Exo by hydrophobic interaction. The AMO181a-chol-loaded RBP-Exo (RBP-Exo/AMO181a-chol) was administered intranasally to a rat middle cerebral artery occlusion (MCAO) model. MiR-181a was knocked down and Bcl-2 was upregulated by intranasal delivery of RBP-Exo/AMO181a-chol. In addition, tumor necrosis factor-α (TNF-α) expression and apoptosis were reduced by RBP-Exo/AMO181a-chol. As a result, RBP-Exo/AMO181a-chol significantly suppressed infarct size compared with the controls. In conclusion, RBP-Exo was a hypoxia-specific carrier for nose-to-brain delivery of AMO181a-chol in an ischemic stroke model. Furthermore, the combined effects of RBP and AMO181a-chol exerted neuroprotective effects in the ischemic brain.
1. Introduction
Stroke has a high fatality rate. There are two types of strokes: ischemic strokes and hemorrhagic strokes. Ischemic stroke is due to narrowing of one or more cerebral arteries, which are occluded by emboli or thrombi. Thrombolytic agents such as recombinant tissue-plasminogen activator (rt-PA) have been used therapeutically to induce reperfusion of the infarcted areas of the brain.1 However, brain damage typically continues to progress after reperfusion. This post-reperfusion damage is called ischemia-reperfusion (I/R) injury, which is induced by inflammatory response, reactive oxygen species (ROS), and excitotoxicity. In particular, activation of pro-inflammatory cascades following transient cerebral ischemia results in severe inflammation-mediated damage of the brain.2,3To protect brain cells from I/R injury, various approaches have been investigated. Gene therapy has been studied for decades as one of the therapeutic options.4–6 In gene therapy, therapeutic nucleic acids are delivered into the brain using local or systemic injection. Two approaches to gene therapy have been studied for ischemic stroke therapy. A knock-in approach has been developed to induce therapeutic protein expression in the ischemic brain through gene delivery. The knock-in approach has shown positive effects in the treatment of ischemic brain injury in animal models. For instance, delivery of the heme oxygenase-1 (HO-1) gene reduced cell death in the ischemic brain and decreased the infarct volume.7–9 The other gene therapy approach is a knock-down approach using antisense oligonucleotides or small interfering RNAs. For example, local injection of high-mobility-group-box-1 (HMGB1) siRNA into the brain reduced infarct volume in animal models of ischemic strokes.10 Similarly, systemic administration of HMGB siRNA showed therapeutic effects in animal models.6
Although gene therapy appears promising, delivery into the brain remains a barrier to its use in ischemic strokes. Local stereotaxic injection of therapeutic genes with or without delivery vectors can deliver the genes efficiently into the brain; however, the needle used for direct injection causes damage to the brain tissue.11 Intravenous (IV) administration may be less invasive than local injection; however, delivery efficiency into the brain across the brain–blood barrier (BBB) is extremely low.11 Recently, many studies have been performed to evaluate intranasal (IN) administration for gene delivery to the brain,11,12 given IN administration delivers therapeutic agents into the brain by way of the olfactory and trigeminal nerves, bypassing the BBB.11 In addition, IN administration is less invasive than stereotaxic injection and more efficient than systemic IV injection. The use of IN delivery may also avoid many of the side effects caused by systemic delivery of therapeutic genes. Many previous studies of intranasal delivery of therapeutic agents to the brain demonstrated positive effects in brain-related diseases.10,13–17 For example, IN administration of HMGB1 siRNA nanoparticles complexed with a cationic polymer delivered the siRNA into the brain efficiently and reduced inflammation in the brain.18 The nanoparticles, composed of siRNAs and polymeric carriers, passed across cell membranes more efficiently than naked siRNA, increasing gene delivery efficiency. These findings suggest that an appropriate delivery carrier may be useful for the IN delivery of nucleic acids into the brain.
One of the promising carriers for nucleic acids is an exosome. Exosomes are the smallest subtype of extracellular vesicles with a size of 30–100 nm.19 Exosomes are biocompatible and do not induce toxicity to cells. Due to these characteristics, exosomes have been studied as novel carriers of nucleic acids such as small interfering RNA (siRNA) and antisense oligonucleotides.20,21 Exosomes have been investigated as potential carriers of genes into various organs following intravenous injection; however, exosomes have not been studied as nucleic acid carriers for IN administration.
One of the key factors involved in the pathophysiology of I/R injury is the receptor for advanced glycation end-products (RAGE). The transmembrane RAGE is a multi-ligand receptor for damage-associated molecular patterns (DAMPs) derived from host cells and pathogen-associated molecular patterns (PAMPs) derived from microorganisms.22 The DAMP family of biomolecules includes high-mobility group box-1 (HMGB-1), S100 proteins, and β-amyloid peptide, while PAMPs include lipopolysaccharides (LPS). Binding of DAMPs and PAMPs to RAGE activates the RAGE-mediated signal pathway and translocation of nuclear factor-κB (NF-κB) into the nucleus to result in severe inflammatory responses.23 In addition, RAGE is induced by positive feedback regulation, with RAGE activation increasing its expression.24–27 Therefore, RAGE inhibitors can reduce the expression of RAGE by interruption of this positive feedback loop. Recently, a recombinant RAGE-antagonist peptide (RAGE-binding peptide, RBP) was produced based on the RAGE binding domain of wild-type HMGB-1 (wtHMGB-1).28,29 In one such study, stereotaxic injection of RBP produced neuroprotective effects by reducing RAGE-mediated inflammatory responses in the ischemic brain.4 In particular, RAGE was reported to be overexpressed in neurons and glial cells under ischemic conditions.30 Therefore, specific binding of RBP to RAGE may be more frequent in hypoxic cells of the ischemic brains than in normal cells. These findings also suggest that RBP may be a specific ligand for targeted delivery of therapeutic agents to hypoxic cells. Indeed, RBP was shown to act as a hypoxia-specific ligand for the delivery of plasmid DNA to the ischemic brain after stereotaxic injection.4 In the current study, we engineered RBP-linked exosomes for hypoxia-specific delivery to the ischemic brain after IN administration. For this purpose, RBP was expressed on the surface of exosomes by creating a fusion protein with Lamp2b, an exosome membrane protein, to produce RBP-linked exosomes (RBP-Exo, Fig. 1A).
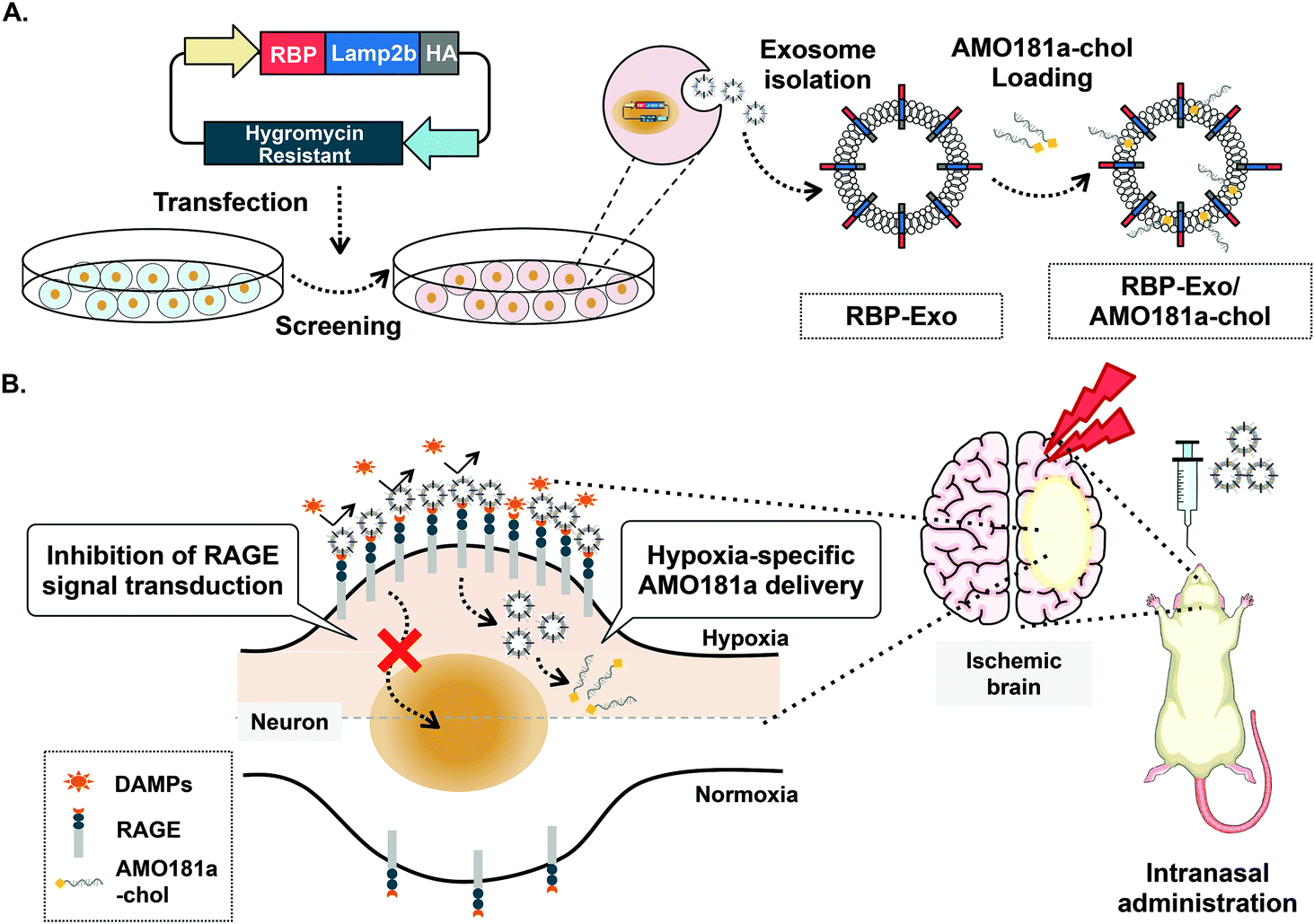 | ||
| Fig. 1 Schematic images. (A) Production of RBP-Exo. (B) Hypoxia-specific delivery of RBP-Exo/AMO181a-chol by intranasal administration. | ||
In this study, anti-microRNA oligonucleotide (AMO) was used as the test therapeutic nucleic acid for stroke therapy. MicroRNAs (miRNAs) are non-coding short RNA molecules of 18 to 25 nucleotides. MicroRNAs target the 3′-untranslated regions (3′-UTR) of specific mRNAs and regulate their translation. Various MiRNAs have been reported to play a key role in several brain diseases, likely due to up- or downregulation of key proteins.31,32 MicroRNA-181a (miR-181a) is one of the several miRNAs upregulated in the ischemic brain. Induction of miR-181a down-regulates the Bcl-2 anti-apoptotic protein and induces apoptosis. In previous studies, delivery of AMO against miR181a (AMO181a) increased Bcl-2 expression and attenuated ischemic neuronal damage.33–35 As a result, infarct volume was reduced by knockdown of miR181a. In this study, cholesterol-modified AMO181a (AMO181a-chol) was loaded into RBP-Exo, and the AMO181a-chol-loaded RBP-Exo (RBP-Exo/AMO181a-chol) was administered IN in a rat MCAO model of ischemic stroke. The anti-inflammatory effects of the RBP-Exo were evaluated using in vitro and in vivo studies (Fig. 1B). In addition, various assays were performed to demonstrate hypoxia-specific delivery of AMO181a and its therapeutic effects in the ischemic animal model. The results suggest that RBP-Exo may be useful as a hypoxia-specific carrier for nose-to-brain delivery of AMO181a.
2. Experimental section
2.1. Cell culture
Mouse neuroblastoma cells (Neuro2A) and HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) at 37 °C in a 5% CO2 incubator.2.2. Isolation of RBP-linked exosomes
The construction of pRBP-Lamp2b-HA has been previously reported.36 HA is a commonly used epitope tag, which does not interfere with the biodistribution of the recombinant protein. With the HA tag, the expression of recombinant RBP-Lamp2b-HA can be easily detected with the anti-HA antibody. Therefore, the HA cDNA was inserted the expression vector for the detection of RBP-Lamp2b-HA expression. To produce a stable cell line, 30 μg of pRBP-Lamp2b-HA was transfected into 2.5 × 106 cells of HEK293T with 150 μg of lipofectamine 2000 (Invitrogen, Carlsbad, CA). Then, the stable cells were screened with hygromycin at a 200 μg ml−1 concentration. The RBP-Lamp2b-expressing cells were cultured for 72 h in DMEM containing 10% exosome-depleted FBS. Since Lamp2b is an endosomal membrane protein, it is integrated into the exosome after translation in the cells. Due to the Lamp2b domain, Lamp2b fusion proteins such as RBP-Lamp2b-HA are integrated into the exosomes in the cells.6,37,38 Then, the Lamp2b integrated exosomes are excreted out of the cells. For isolation of the exosomes, the culture medium was collected. The harvested culture medium was centrifuged at 300g for 3 min and filtered through a Millex-GV 0.22 μm filter (Millipore, Molsheim, France) to remove large particles such as cell debris and apoptotic bodies. Then, an exoEasy Maxi kit (QIAGEN, Valencia, CA) was used to isolate exosomes according to the manufacturer's instructions. The concentration of purified exosomes was measured using the BCA protein assay (Invitrogen, Carlsbad, CA).2.3. Preparation of AMO181a-chol loaded exosomes
AMO against miRNA181a (AMO181a) and a negative control (scrambled AMO) with cholesterol modification were synthesized using Bioneer (Daejeon, Korea), and the sequences were as follows: AMO181a, 5′-mA.*.m.C.*.mU.mC.mA.mC.mC.mG.mA.mC.mA.mG.mC.mG.mU.mU.mG.mA.mA.mU.mG.*.mU.*.mU.-3′-cholesterol, and scrambled AMO181a, 5′-mA.*.mG.*.mU.mC.mA.mG.mC.mG.mA.mG.mA.mG.mC.mC.mU.mU.mG.mA.mU.mU.mG.*.mU.*.mU.-3′-Chl (mN = 2′-O-methyl nucleotide (N = A or C or G or U); * = phosphorothioate linkage). To load AMO181a into exosomes, the 3′-end of AMO181a was modified with cholesterol. The AMO181a-chol and exosomes were mixed at various weight ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:3, 1:5, 1:7, and 1:9) and incubated in phosphate-buffered saline (PBS) for 30 min at room temperature. The optimal AMO181a-chol-to-exosome ratio was established by measuring the uptake efficiency. The optimal weight ratio was 1:5. Therefore, the weight ratio was fixed at 1:5 for all experiments.
:1, 1:3, 1:5, 1:7, and 1:9) and incubated in phosphate-buffered saline (PBS) for 30 min at room temperature. The optimal AMO181a-chol-to-exosome ratio was established by measuring the uptake efficiency. The optimal weight ratio was 1:5. Therefore, the weight ratio was fixed at 1:5 for all experiments.
To measure the loading efficiency, Cy5-labeled AMO181a-chol (Cy5-AMO181a-chol) was synthesized using Bioneer (Daejeon, Korea). Cy5-AMO181a-chol and RBP-Exo were mixed and incubated for 30 min. The ratio of AMO181a-chol to RBP-Exo was 1:5 (w/w). Unloaded Cy5-AMO181a-chol was removed by ultracentrifugation at 100000g for 1 h. The supernatants were removed and the pellets were resuspended with PBS. The amount of Cy5-AMO181a-chol was quantified using a fluorometer (Molecular Devices, Sunnyvale, CA), with excitation at 650 nm and emission at 670 nm. The loading efficiency was calculated as follows: Loading efficiency (%) = (Amount of Cy5-AMO181a-chol after loading)/(Total amount of Cy5-AMO181a-chol) × 100.
2.4. Dynamic light scattering (DLS)
The exosomes were prepared with or without AMO181a-chol as described above. To measure the size and zeta potential, the exosomes were diluted in 1 mL of distilled water. Then, the sizes and zeta potentials of the exosomes were measured using the Zetasizer Nano ZS system (Malvern Instruments, Malvern, UK).2.5. Transmission electron microscope (TEM)
Exosomes were prepared with or without AMO181a-chol as described above. Mesh copper grids (Ted Pella, Redding, CA) were rinsed using a drop of distilled water. Then, the samples were placed on the grids and incubated for 30 min. The remaining sample droplets were absorbed into a tissue to remove excess particles. Finally, the samples were stained with 2% uranyl acetate for 10 min. The size and morphology of the exosomes were measured by transmission electron microscopy (JEM-2100F, JEOL, Tokyo, Japan).2.6. Western blot assay
Proteins contained in the exosomes were isolated using methanol/chloroform protein precipitation. 100 μg of the collected proteins were loaded into wells and separated by 10% SDS-PAGE. The proteins in the gel were transferred to a PVDF membrane, and immunoblotting was performed using a Pierce fast western blotting kit (Thermo Scientific, Rockford, IL) with anti-HA antibody (Cell Signaling, Danvers, MA), anti-GAPDH antibody (Cell signaling, Danvers, MA), and anti-CD63 antibody (Invitrogen, Carlsbad, CA).2.7. In vitro cellular uptake
Neuro2A cells were seeded in 12-well plates at a density of 1 × 105 cells per well and incubated for 24 h at 37 °C in a 5% CO2 incubator. Cy5-AMO181a-chol was synthesized chemically (Bioneer, Daejeon, Korea). Next, Cy5-AMO181a-chol-loaded Unmod-Exo (Unmod-Exo/AMO181a-chol) and Cy5-AMO181a-chol-loaded RBP-Exo (RBP-Exo/AMO181a-chol) were prepared. As a positive control, polyethylenimine (25 kDa, PEI25k)/AMO181a-chol complexes were prepared with Cy5-AMO181a-chol at a 1:1 weight ratio. The prepared complexes were added to the cells. The amount of Cy5-AMO181a-chol was fixed at 1 μg per well. The amount of exosomes was fixed at 5 μg per well. The cells were incubated for 4 h at 37 °C in serum-free DMEM, and then the medium was replaced with DMEM containing 10% FBS. After 20 h of additional incubation, the cells were harvested using trypsin–EDTA solution and transferred to micro-centrifuge tubes. The cells were centrifuged at 300g for 3 min and washed twice with cold FBS. Then, the cells were transferred to FACS tubes and analyzed by flow cytometry (BD FACS Calibur™, BD Biosciences Immunocytometry Systems, San Jose, CA).
2.8. Evaluation of RAGE after RBP-Exo treatment
Neuro2A cells were seeded at a density of 5 × 105 cells per well. To induce RAGE signaling, the cells were incubated for 24 h under hypoxic conditions (1% O2, 5% CO2 and 94% N2). Then, 5 μg of exosomes was treated into the RAGE-induced cells and the cells were incubated for an additional 4 h. After washing twice with PBS, the cells were incubated with anti-RAGE antibody (Abcam, Cambridge, UK) and Alexa 488 conjugated secondary antibody (Invitrogen, Carlsbad, CA) for 30 min, respectively. The cells were harvested using a cell scraper and the Alexa 488-positive cells were analyzed by flow cytometry using a BD FACS Calibur™ system (Becton & Dickinson and company, Bergen, NJ).2.9. Intranasal administration of exosomes in the animal model of ischemic brain injury
All animal experimental procedures were performed in accordance with the institutional guidelines of the Institutional Animal Care and Use Committees (IACUC). Animal experiments were approved by the IACUC of Hanyang University (accreditation number, 2019-0205A). Ten-week-old male Sprague Dawley (SD) rats (280–321 g) were anesthetized using 5% isoflurane in 70% N2O and 30% O2. The body temperature of the rats was maintained at 36.5 °C ± 0.5 °C during surgery using a heating pad (Harvard Apparatus, Holliston, MA, USA). The anesthetized rat was placed in the supine position, and the neck midline was incised to expose the right external carotid artery (ECA). Then, the right ECA was ligated using a thread. After fixing the common carotid artery (CCA) and internal carotid artery (ICA) sequentially, the exposed ECA was incised using micro-scissors. After that, 3.5 cm of 4-0 nylon suture was inserted into the ICA, and the CCA was occluded using a clip. After 1 h, the clip and inserted suture were removed for reperfusion. After an additional 1 h, the prepared AMO181a-chol-loaded exosomes and PEI25k/AMO181a-chol complexes were administered to the middle cerebral artery occlusion (MCAO) rats intranasally using a pressurized olfactory device (POD; Impel Neuropharma, Washington, USA). The amount of AMO181a was fixed at 15 μg per rat. The amount of the exosome was 75 μg per rat. Briefly, the catheter tube was filled with a 25 μL dose. Then, the tip of the POD was inserted into the nostril, and the loaded complexes were injected. The experimental animal groups include normal control, MCAO control, AMO181a-chol, scrambled AMO181a-chol, Unmod-Exo/AMO181a-chol, RBP-Exo/AMO181a-chol, and PEI25k/AMO181a-chol (n = 8).2.10. Real-time reverse transcription polymerase chain reaction (RT-PCR)
The RNA was extracted from brain tissues embedded in paraffin blocks using a miRNeasy PPFE kit (QIAGEN, Valencia, CA). Two 10-μm-thick sections under the bregma were deparaffinized, and RNA was extracted sequentially according to the kit manufacturer's instructions. Then, cDNA was synthesized using an iScript cDNA synthesis kit (BIO-RAD, Hercules, CA). Quantitative analyses of mRNA were performed using an Applied Biosystems 7500 real-time PCR system (Applied Biosystems, Foster City, CA) using a SensiFAST SYBR Lo-ROX kit (BIOLINE, Boston, MA). After initial heating at 50 °C for 120 s, 40 cycles of PCR amplification were performed, each consisting of three steps: denaturation at 95 °C for 15 s; annealing at 60 °C for 60 s; and extension at 60 °C for 15 s. The sequences of the GAPDH primers were as follows: forward primer 5′-GCCTTCCGTGTTCCTACC-3′ and reverse primer 5′-AGAGTGGGAGTTGCTGTTG-3′. The miRNA-181a primers were as follows: forward primer 5′-AACATTCAACGCTGTCGGTGAGT-3′ and reverse primer 5′-CTCCTTAGAATCTGTTTGCTCTCATA-3′.2.11. Immunohistochemistry
After cardiac perfusion, the rats were sacrificed. The harvested brains were fixed in 4% paraformaldehyde (PFA) overnight and embedded in paraffin. The paraffin-embedded brains were sectioned into 7 μm-thick slices that were placed on glass microscope slides. The brain slice centered on the bregma was used for immunohistochemistry. The samples were deparaffinized and hydrated using xylene and ethanol. Then, the samples were treated with blocking solution for 2 h at room temperature. After that, primary antibodies of rabbit anti-TNF-α antibody (1:100, Abcam, Cambridge, UK), rabbit anti-RAGE antibody (1:50, Abcam, Cambridge, UK), and rabbit anti-Bcl-2 antibody (1:50, Abcam, Cambridge, UK) were applied to the samples and incubated overnight at 4 °C. After washing twice with Tris-buffered saline (TBS) containing 0.025% Triton X-100 (TBST), goat-anti-rabbit Alexa-Fluor-488-conjugated antibody (Molecular Probes, Eugene, OR) was added to the samples, and the slides were incubated for 1 h in the dark. Finally, the samples were washed twice with TBST, and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). The stained samples were mounted and measured using an AxioScan.Z1 digital slide scanner (ZEISS, Oberkochen, Germany).
2.12. TUNEL assay
The paraffin-embedded brain samples were cut into 7 μm-thick slices and placed on glass microscope slides. Brain slices centered on the bregma were used for immunohistochemistry. The samples were deparaffinized and hydrated gradually. After that, the samples were immersed in PBS containing 3% Triton X-100 and fixed with 4% PFA. After washing, the fragmented DNA from apoptotic cells was labelled using an in situ BrdU-Red DNA fragmentation assay kit (Abcam, Cambridge, UK) according to the manufacturer's instructions. The labelled samples were mounted and analyzed using an AxioScan.Z1 digital slide scanner (ZEISS, Oberkochen, Germany).2.13. TTC staining
The brains of the MCAO model rats were harvested at 24 h after ischemia-reperfusion. The brain samples were sectioned into five 2 mm-thick slices from the side of forebrain. The 2 mm-thick slices were arranged in a row from the posterior part toward the anterior part of brain. The third slice was the sample under the bregma. The slices were incubated in a 2% 2,3,5-triphenyltetrazolium chloride (TTC, Sigma-Aldrich, St Louis, MO) solution at 37 °C for 10 min. The stained brain slices were fixed in 4% PFA at 4 °C. The infarct volume was measured using the Image J 1.42 software (NIH, Bethesda, MD, USA). The infarct volume was calculated as follows: Infarct volume (%) = (Infarct area × Thickness of each slice/Total area × Thickness of each slice) × 100. Five rats per group were subjected to TTC staining.2.14. Laser doppler analysis
The MCAO model was produced as described above. The monitoring point (4 mm lateral from bregma) was drilled to create a hole. To minimize heat damage due to drilling, saline (0.89% NaCl) was applied during hole formation. Then, the probe was placed in the hole, and the cerebral blood flow (CBF) signals were recorded using a BLF 21 laser Doppler flow meter (Transonic Systems Inc., Ithaca, NY).2.15. Statistical analysis
Statistical analysis of in vitro data was performed by ANOVA followed by a Newman–Keuls test. Statistical analysis of in vivo data was performed using the non-parametric Mann–Whitney U test to compare differences between groups. All data are presented as mean ± standard deviation, and results with p values less than 0.05 were considered statistically significant.3. Results and discussion
3.1. Isolation and characterization of exosomes
For hypoxia-specific delivery of AMO181a, exosomes exhibiting RBP were engineered using recombinant DNA technology. To create the exosomes exhibiting RBP, RBP cDNA was inserted into the Lamp2b expression plasmid at the 5′-end so that RBP was located at the N-terminus of Lamp2b in the fusion protein (Fig. 1A). Since the N-terminus of Lamp2b is located outside of the exosome, the RBP within the RBP-Lamp2b fusion protein is likewise located on the outside surface of the exosome membrane. Due to the direct linkage to Lamp2b, the RBP protein may have been located too close to the exosome membrane to interact with RAGE efficiently. A similar approach using T7-peptide-displaying exosomes for glioblastoma-targeted gene delivery was reported previously.38 The T7 peptide is a ligand for the transferrin receptor, which is overexpressed on glioblastoma cells, and may increase intracellular uptake of therapeutic genes into glioblastoma cells. In the cited study, T7 peptide was linked to Lamp2b directly, which could have led to the inability of the T7 peptide to interact effectively with the transferrin receptor as it had been located too close to the exosome membrane. However, T7 on the exosome surface was able to interact with its receptor and facilitated the delivery of nucleic acids into the glioblastoma cells. Given the T7 peptide was only seven-amino acid-residues (HAIYPRH) long, these findings indicate that direct linkage of a targeting ligand to Lamp2b may not interfere with its interaction with the corresponding receptor. Therefore, no linker peptide was introduced between RBP and Lamp2b in the current study. The HA tag was located at the 3′-end of Lamp2b for western blotting (Fig. 1A).Because RAGEs are overexpressed on hypoxic cells of the ischemic brain, interaction of RBP-Exos with RAGEs may facilitate gene delivery to hypoxic cells (Fig. 1B). Furthermore, interaction of RBP-Exo with RAGE can interfere with the interaction of DAMPs or PAMPs with RAGEs (Fig. 1B) and may result in downregulation of the inflammatory response in the ischemic brain.
To produce the RBP-Exos, pRBP-Lamp2b-HA was transfected into HEK293T cells. A stable RBP-Exo-producing cell line was established by screening transfected cells in the presence of hygromycin, since pRBP-Lamp2b-HA contained the hygromycin resistance gene. The RBP-Exos were isolated from the cell culture medium of the RBP-Lamp2b-expressing cells. Similarly, Unmod-Exos were isolated from the culture medium of normal HEK293T cells as a control. To confirm the presence of the RBP ligand on isolated exosomes, western blot assays were performed with isolated exosomes. The HA positive signal was detected in RBP-Exos but not in Unmod-Exos (ESI Fig. 1†). This result confirmed that the RBP-Lamp2b-HA fusion protein was expressed and located on the membrane of RBP-Exos.
AMO181a was loaded into the exosomes by simply mixing AMO181a-chol and the exosomes. Due to hydrophobic interactions, the cholesterol moiety was integrated into the exosome membrane. Unloaded AMO181a-chol was removed by ultracentrifugation. The loading efficiency was 24.75 ± 4.25%. In previous studies, therapeutic reagents were loaded into exosomes in various ways.17,39–42 In particular, electroporation has been used in many studies to load nucleic acids such as siRNAs and antisense oligonucleotides.6,37,38 However, the loading efficiency for electroporation did not exceed 10%. In contrast, hydrophobic drugs have been loaded into exosomes relatively efficiently through hydrophobic interactions.17,43–46 Similarly, cholesterol-modified siRNAs were used for efficient loading into exosomes.47,48 In the current study, AMO181a was modified with cholesterol, and the loading efficiency was greater than that of previous studies using electroporation. This result suggests that hydrophobically modified nucleic acids were efficiently loaded onto the exosomes. However, several points should be considered with regard to this method. First, the nucleic acid moiety of cholesterol-modified AMO181a may extend to the outside surface of exosomes and may be susceptible to enzymatic degradation. In the current study, AMO181a was modified at the 2′-methyl group, and phosphorothioate linkages were introduced in the first two and last two phosphodiester linkages. Therefore, enzymatic degradation was minimized, and the AMO181a on the outside surface of the exosomes was stable. Second, the size of exosomes may be increased by loading AMO181a-chol on the surface. The particle size was measured by dynamic light scattering in this study, and the AMO181a-chol-loaded exosomes showed slightly increased size compared with exosomes without AMO181a-chol (Fig. 2A). However, the size of all the exosomes was less than 50 nm, which was much smaller than when using a polyethylenimine (25 kDa, PEI25k)/AMO181a-chol complex (219.8 ± 3.6 nm; Fig. 2A). The PEI25k/AMO181a-chol complex was used as the control treatment in this study because PEI25k has been widely used as a carrier of plasmid DNA, siRNA, and antisense oligonucleotides.
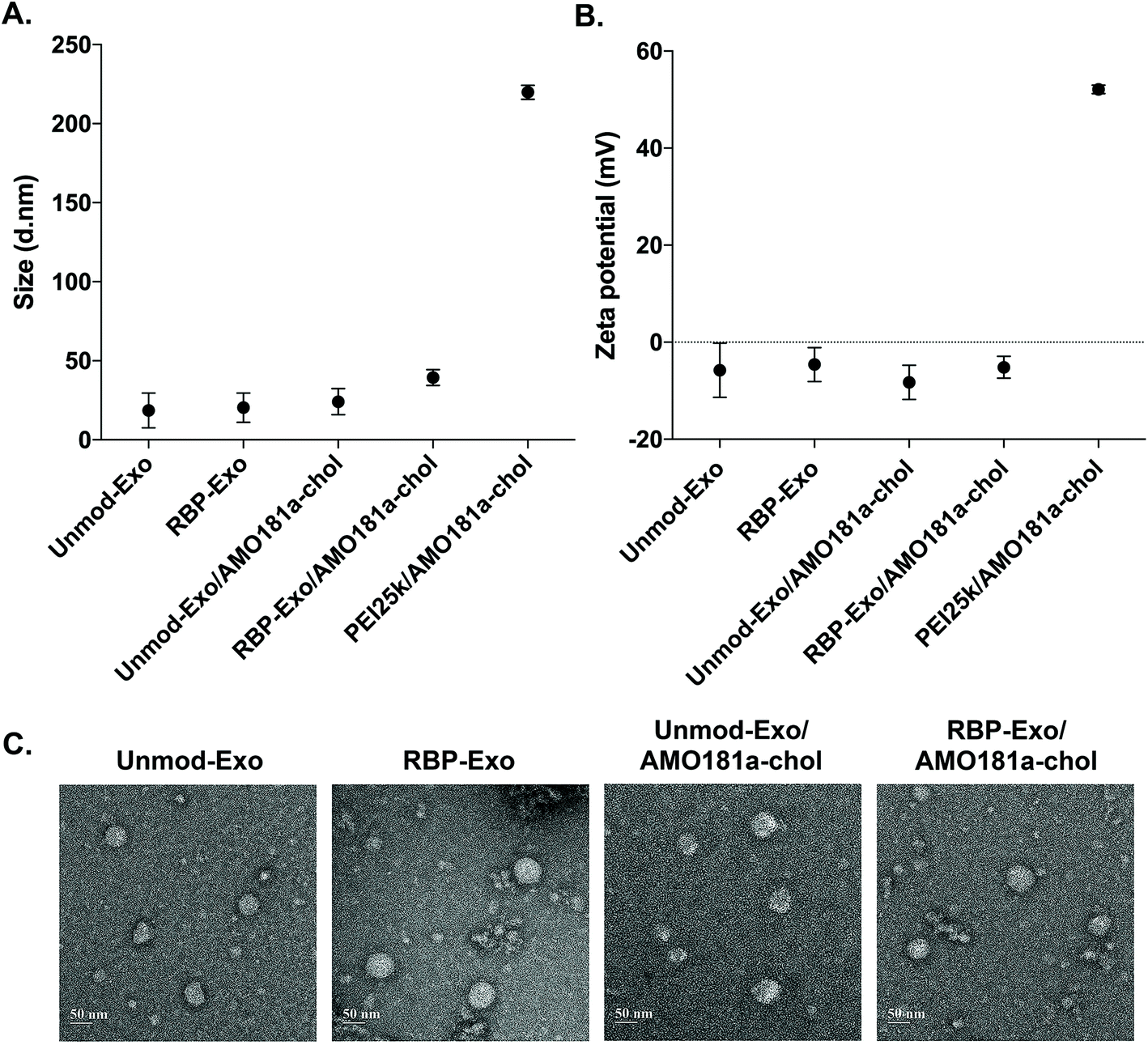 | ||
| Fig. 2 Physical characterization of RBP-Exo and Unmod-Exo. (A) Particle size and (B) zeta potential. The data are presented as the mean value ± standard deviation of triplicate experiments. (C) Transmission electron microscopy images. | ||
The zeta potentials of the exosomes also were measured (Fig. 2B). The results showed that the zeta potentials of the exosomes were around −0.8 to −11.8 mV. In contrast, the PEI25k/AMO18a complex had a strong positive surface charge of 52.1 ± 0.77 mV. Non-viral carriers such as PEI25k have been reported to induce cytotoxicity by aggregation on plasma membrane, due to a positive surface charge.49 The exosomes in this study may be free of cytotoxic effects due to the absence of a positive surface charge.
The TEM study showed that the exosomes had a spherical shape, and the loading of AMO181a-chol did not affect the morphological properties of the exosomes (Fig. 2C).
3.2. In vitro delivery of AMO181a-chol using RBP-Exo
The efficiency of in vitro delivery of the RBP-Exo to Neuro2A cells incubated under hypoxic conditions was evaluated using Cy5-AMO181a-chol. The cellular uptake of Cy5-AMO181a-chol was measured by flow cytometry and fluorescence microscopy. Both Cy5-AMO181a-chol-loaded Unmod-Exo (Unmod-Exo/AMO181a-chol) and PEI25k/Cy5-AMO181a-chol complexes were used as control treatments. Flow cytometry showed that the PEI25k/Cy5-AMO181a-chol complex had a greater delivery efficiency than any other treatment (Fig. 3A). However, this result may be relevant only to in vitro delivery assays. Since the PEI25k/Cy5-AMO181a-chol complex was larger in size and had a stronger positive surface charge than the exosomes, the PEI25k/Cy5-AMO181a-chol complex may not be suitable for in vivo AMO delivery: the strong positive surface charge may cause the PEI25k/Cy5-AMO181a-chol complex to interact with negatively charged plasma proteins to form aggregates. In addition, PEI25k is highly toxic to cells.50 The MTT assay results indicated that the PEI25k/AMO181a-chol complex was more cytotoxic than AMO181a-chol-loaded exosomes (ESI Fig. 2†). The flow cytometry results also indicated that RBP-Exo delivered Cy5-AMO181a-chol to cells more efficiently than Unmod-Exo (Fig. 3A). This finding may have been due to the RBP on the exosomes interacting more efficiently with overexpressed RAGE on Neuro2A cells cultured under hypoxic conditions.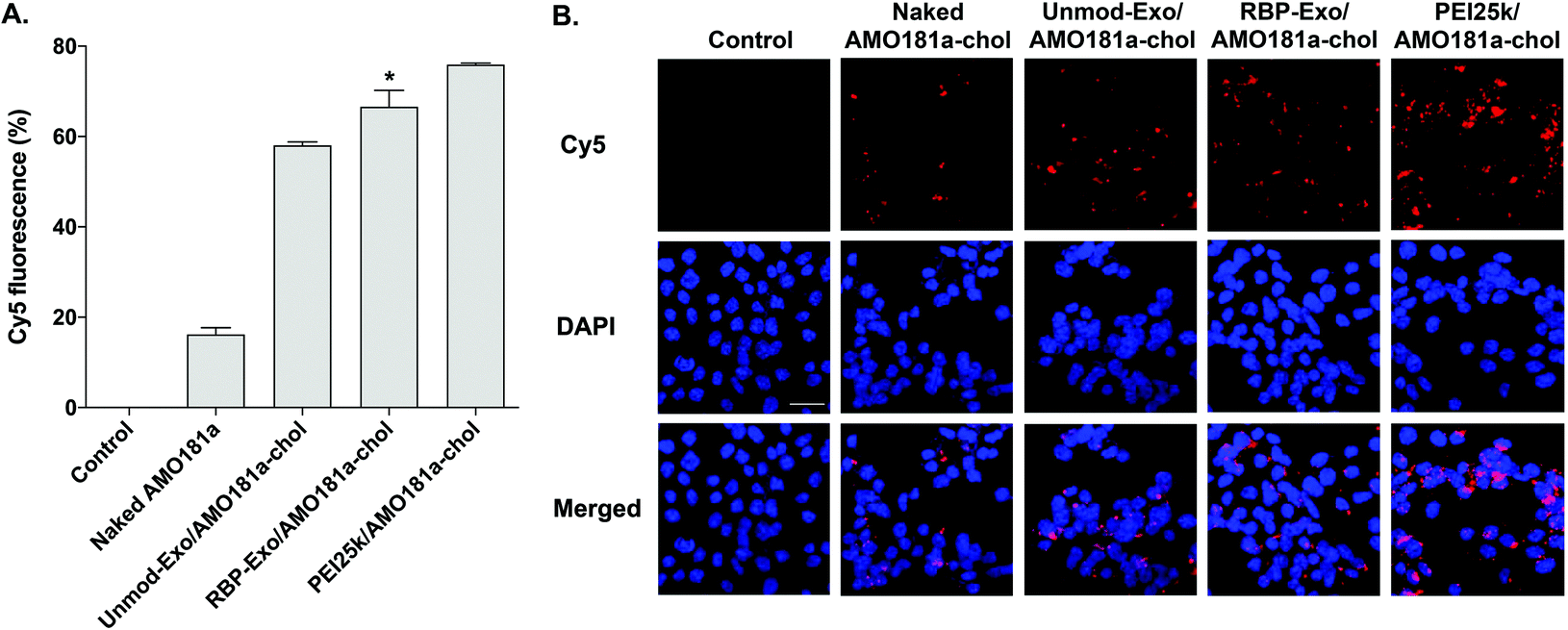 | ||
| Fig. 3 In vitro AMO181a delivery into Neuro2A cells. Cy5-labeled AMO181a-chol was loaded into RBP-Exo and Unmod-Exo. AMO181a delivery efficiency was measured by (A) flow cytometry and (B) fluorescence microscopy. The flow cytometry results are presented as the mean value ± standard deviation of triplicate experiments. The scale bar in (b) represents 25 μm. *p < 0.05 compared with the other groups. | ||
The fluorescence microscopy study also indicated that RBP-Exo/Cy5-AMO181a-chol had a greater delivery efficiency than Unmod-Exo/Cy5-AMO181a-chol or naked Cy5-AMO181a-chol (Fig. 3B). However, the PEI25k/Cy5-AMO181a-chol complex had the greatest delivery efficiency (Fig. 3B). These results supported the flow cytometry results (Fig. 3A).
3.3. In vitro anti-RAGE effect of RBP-Exos
The interaction between RAGE and DAMPs/PAMPs is antagonized by RBP binding to RAGE. As a result, RBP downregulates the RAGE signaling pathway. Similarly, the RBP moiety on RBP-Exos may potentially reduce RAGE expression on the plasma membrane of target cells, since RAGE expression is regulated by positive self-feedback of the RAGE signaling pathway.26 In this study, Neuro2A cells were cultured under hypoxic conditions to induce RAGE expression. To confirm the downregulation of RAGE by RBP-Exos, the cells were incubated with Unmod-Exos or RBP-Exos. Flow cytometry to detect anti-RAGE antibodies was performed to evaluate the anti-RAGE effect of RBP-Exos. The results showed that RAGE expression on the membrane of Neuro2A cells was induced under hypoxic conditions (Fig. 4). Incubation with RBP-Exos reduced the number of RAGE-positive cells, while Unmod-Exos did not produce this effect (Fig. 4). As described above, direct linkage of RBP and Lamp2b in RBP-Exos may have reduced the interaction between RBP and RAGE. However, this result confirmed that the RBP moiety of the RBP-Exo has anti-RAGE effects in target cells, suggesting that it interacts with RAGE on the cell surface.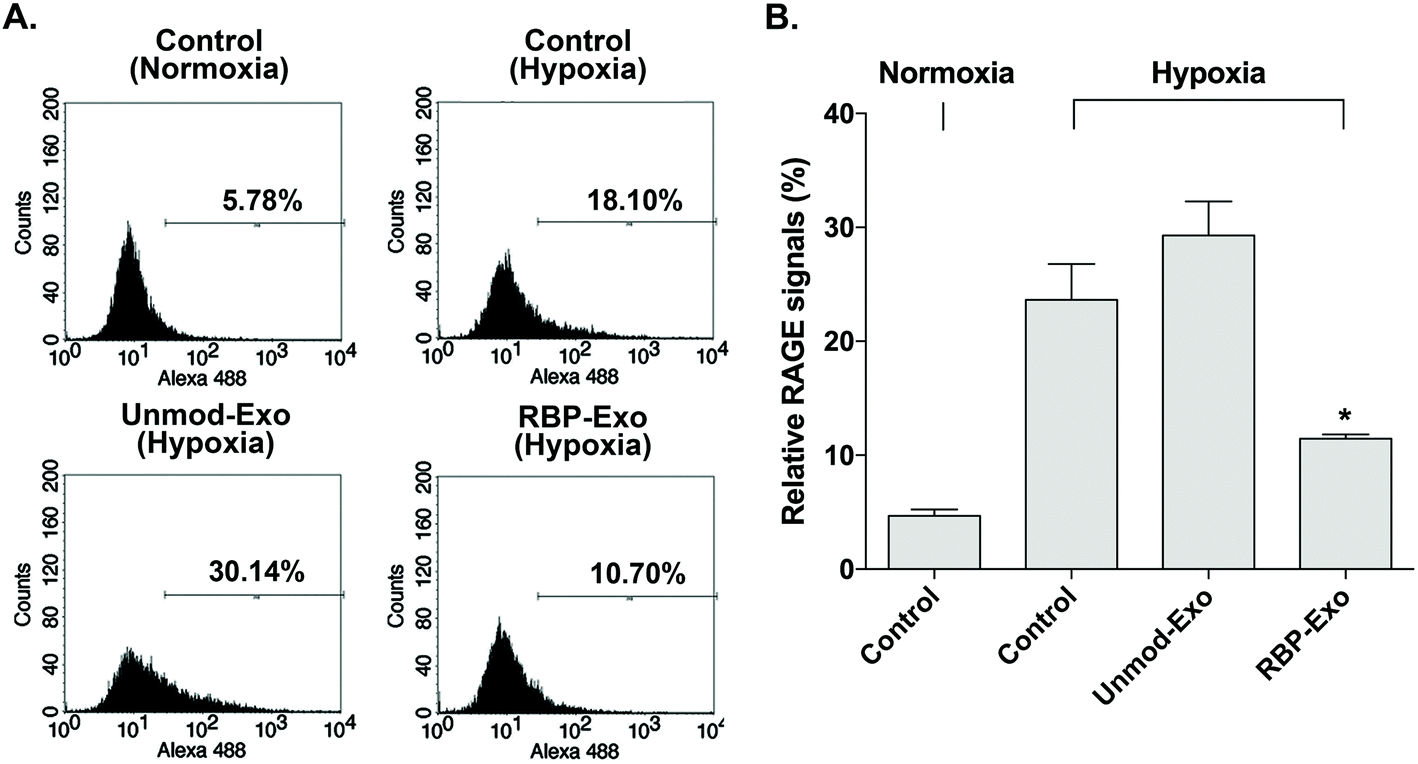 | ||
| Fig. 4 Downregulation of RAGE by RBP-Exo in vitro. The Neuro2A cells were incubated under hypoxic conditions to induce RAGE signaling. Then, RBP-Exo and Unmod-Exo were added to the RAGE-induced cells, and expression of RAGE was analyzed by flow cytometry. (A) Flow cytometry histograms, (B) quantitation of the flow cytometry results. The data are presented as the mean value ± standard deviation of triplicate experiments. *p < 0.05 compared with the other groups. | ||
3.4. Knockdown of miR-181a by intranasal delivery of RBP-Exo/AMO181a-chol
Ischemic stroke animal models have been developed by occluding the middle cerebral artery for 60 min. To ensure that the transient MCAO model was generated properly, cerebral blood flow (CBF) was measured using laser Doppler flowmetry (ESI Fig. 3†). After occluding the ICA, CBF was transiently reduced to around 42% and then recovered after reperfusion (ESI Fig. 3†). One hour after the 60 min occlusion, RBP-Exo/AMO181a-chol was administered into the ischemic stroke rats via intranasal instillation. After 24 h, the brains were harvested and analyzed using RT-PCR.In I/R injury, miR-181a is highly upregulated, causing deleterious effects such as additional inflammatory reactions and apoptosis.33,34 To confirm the knockdown of miR-181a by AMO181a-chol, real-time RT-PCR was performed with miR-181a-specific primers. Intranasal administration of RBP-Exo/AMO181a-chol significantly reduced the expression of miR-181a in the rats compared with the administration of RBP-Exo/scrambled AMO181a-chol (Fig. 5). This result suggests that the AMO181a-chol worked properly compared with scrambled AMO181a-chol. However, the difference between the effects of Unmod-Exo/AMO181a-chol and RBP-Exo/AMO181a-chol in terms of miR-181a inhibition was not statistically significant (Fig. 5).
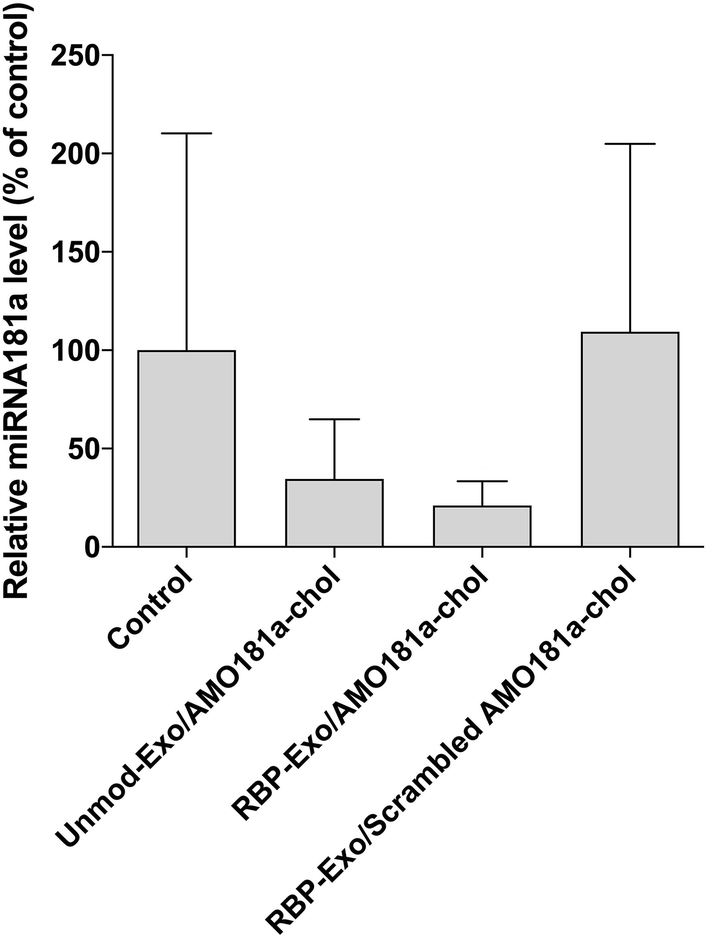 | ||
| Fig. 5 In vivo knock-down efficiency of miRNA-181a after intranasal delivery of RBP-Exo/AMO181a-chol. Real-time RT-PCR analysis was performed to measure miR-181a uptake in the ischemic brain of the stroke model rats after intranasal delivery of RBP-Exo/AMO181a-chol. The miR-181a content was normalized to GAPDH mRNA expression. The results are presented as the mean value ± standard deviation of septuplicate experiments. | ||
3.5. Effect of miR-181a knockdown on apoptosis by intranasal delivery of RBP-Exo/AMO181a-chol
One of the target proteins of miR-181a is Bcl-2, which is an anti-apoptotic protein.33,34 To evaluate Bcl-2 expression, the brain tissues were subjected to immunohistochemistry. The results showed that Bcl-2 expression level was reduced after a transient ischemic attack (Fig. 6). However, Bcl-2 level increased in the Unmod-Exo/AMO181a-chol and RBP-Exo/AMO181a-chol groups (Fig. 6). In particular, RBP-Exo/AMO181a-chol induced Bcl-2 more efficiently than Unmod-Exo/AMO181a-chol. Therefore, this result suggests that RBP-Exo delivers AMO181a-chol more efficiently than Unmod-Exo.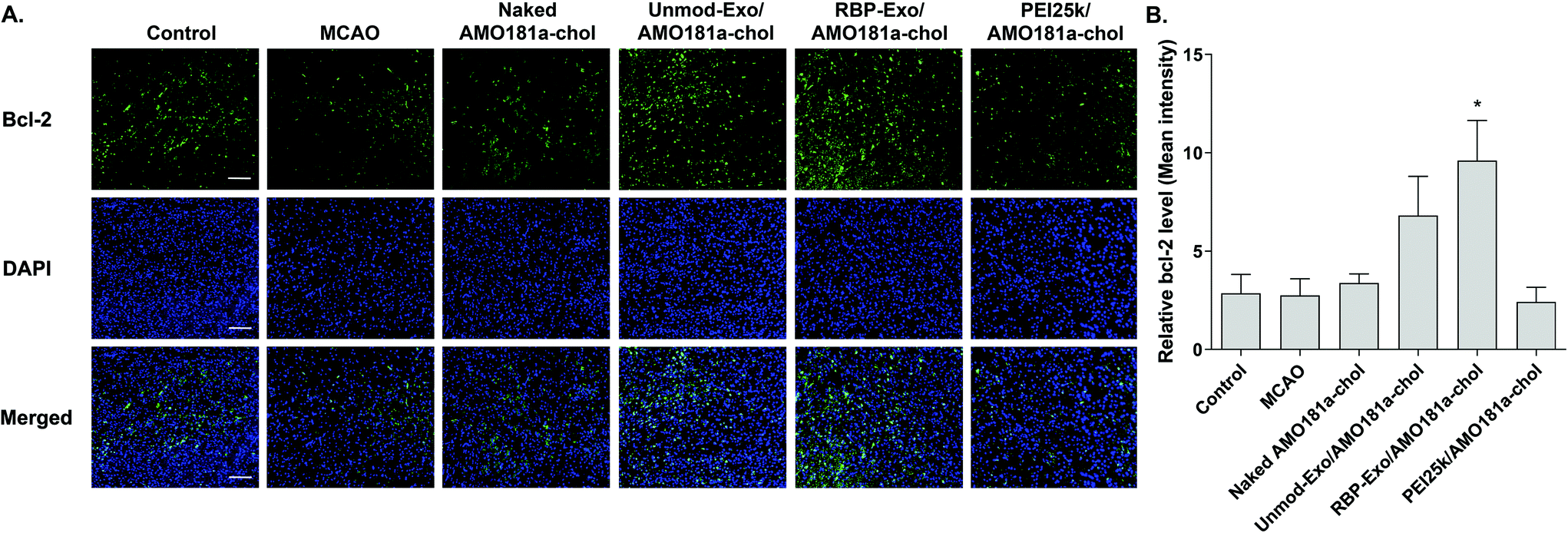 | ||
| Fig. 6 Bcl-2 expression in the ischemic brain after intranasal delivery of RBP-Exo/AMO181a-chol. Naked AMO181a-chol, Unmod-Exo/AMO181a-chol, RBP-Exo/AMO181a-chol, or PEI25k/AMO181a-chol was delivered into the ischemic brains by intranasal administration. After 24 h, the brains were harvested and immunostained with anti-Bcl-2 antibodies. The ischemic penumbra of the cerebral cortex was selected to evaluate the Bcl-2 expression level. Three sections per rat and three rats per group were evaluated for the Bcl-2 expression. (A) Fluorescence microscopy. The samples were observed by fluorescence microscopy. The scale bar represents 100 μm. (B) Quantitation of the Bcl-2 level. The quantitation of the Bcl-2 level was measured by ZEISS ZEN software. *p < 0.05 as compared with control, MCAO, naked AMO181a-chol, PEI25k/AMO181a-chol, but no statistical significance as compared with Unmod-Exo/AMO181a-chol. | ||
Apoptosis is modulated by miR-181a in the ischemic brain. Inhibition of miR-181a by AMO181a may reduce apoptosis in animal models of ischemic stroke. An in vivo TUNEL assay was performed to evaluate apoptosis in the harvested ischemic brains. The results indicated that apoptosis increased after transient MCAO (Fig. 7A and B); however, apoptosis was reduced after the IN administration of Unmod-Exo/AMO181a-chol or RBP-Exo/AMO181a-chol (Fig. 7A and B), with RBP-Exo/AMO181a-chol reducing apoptosis more than Unmod-Exo/AMO181a-chol (Fig. 7A and B).
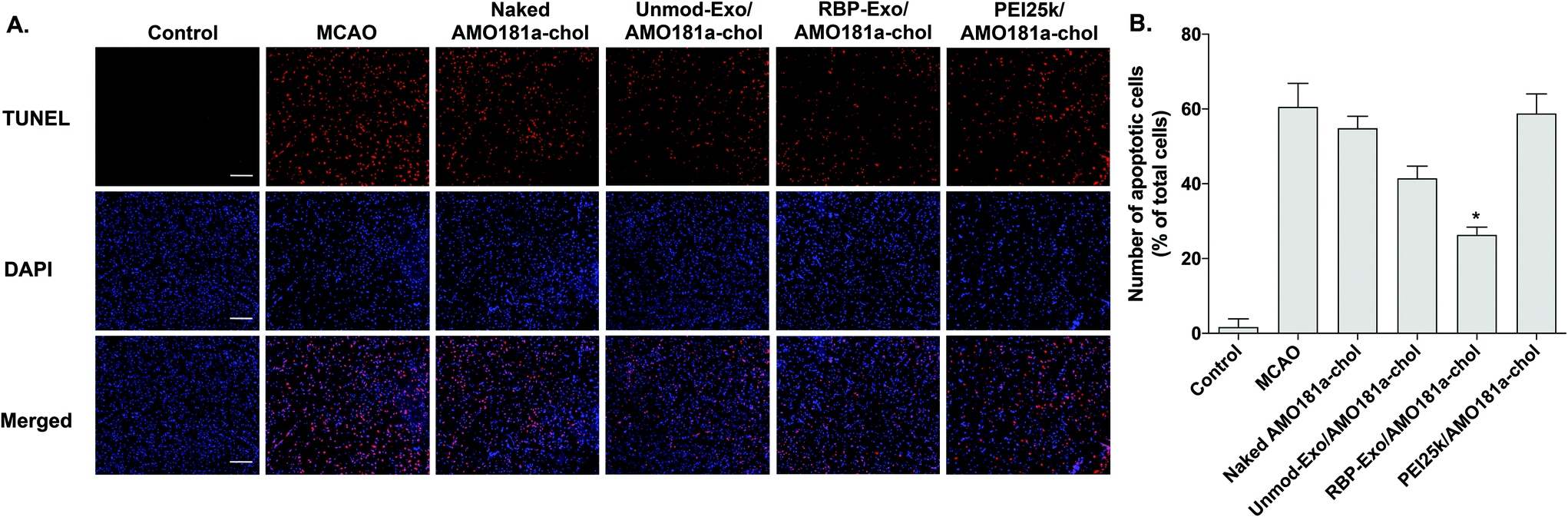 | ||
| Fig. 7 Apoptosis in the ischemic brain after intranasal delivery of RBP-Exo/AMO181a-chol. Naked AMO181a-chol, Unmod-Exo/AMO181a-chol, RBP-Exo/AMO181a-chol, or PEI25k/AMO181a-chol was delivered into the ischemic brains by intranasal administration. After 24 h, the brains were harvested and stained with anti-BrdU antibody. Three sections per rat and three rats per group were evaluated for TUNEL assay. (A) Fluorescence microscopy. The TUNEL-positive cells were analyzed by fluorescence microscopy. The scale bar represents 100 μm. (B) Quantification of apoptotic cells. The number of apoptotic cells was counted using the ImageJ software. The data are presented as the mean value ± standard deviation of triplicate experiments. *p < 0.05 compared with the other groups. | ||
3.6. Anti-RAGE effect on RAGE expression by intranasal delivery of RBP-Exo/AMO181a-chol
One of the therapeutic effects of RBP-Exo/AMO181a-chol was the suppression of RAGE. To evaluate the anti-RAGE effect, RAGE was immunostained in the harvested brains. The results showed that RAGE was overexpressed in the transient MCAO model. However, RAGE expression was reduced after the IN administration of naked AMO181a-chol, Unmod-Exo/AMO181a-chol, RBP-Exo/AMO181a-chol, or PEI25k/AMO181a-chol (Fig. 8). The reduction of RAGE expression in all treated animals may have been due in part to the anti-inflammatory effects of AMO181a; however, RAGE expression was further reduced in the samples from rats administered RBP-Exo/AMO181a-chol (Fig. 8). This finding suggests that the RBP moiety of RBP-Exo may interact with RAGE and reduce its expression on the surface of the plasma membrane.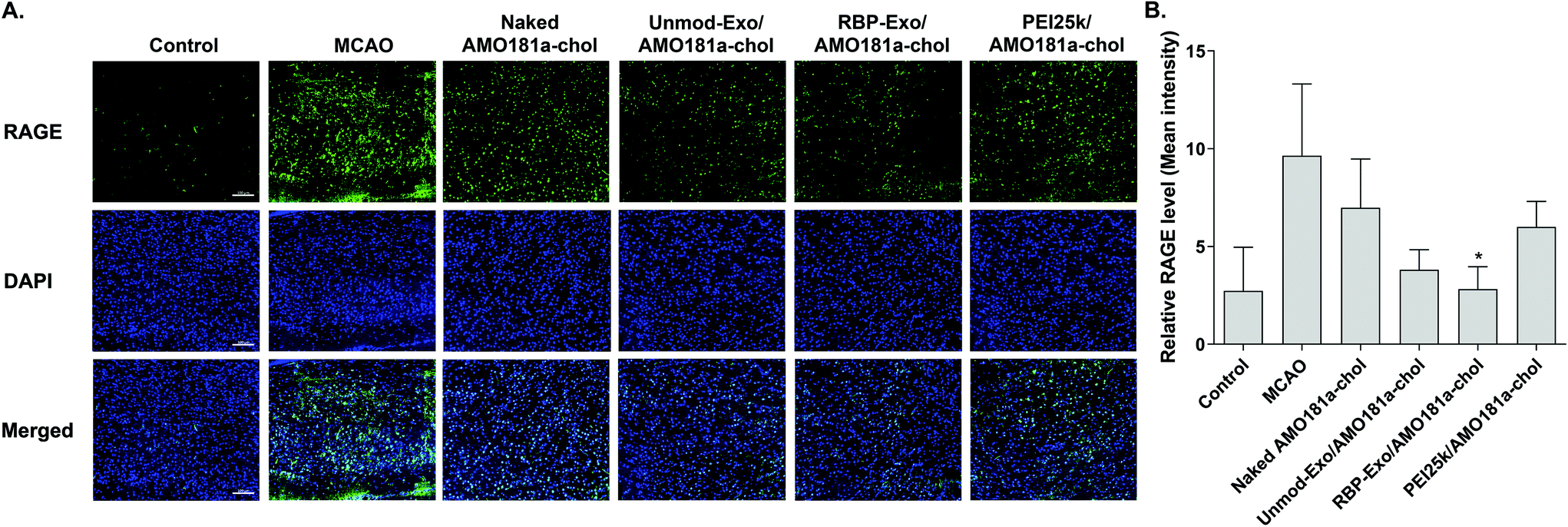 | ||
| Fig. 8 Downregulation of RAGE after intranasal delivery of RBP-Exo/AMO181a-chol. Naked AMO181a-chol, Unmod-Exo/AMO181a-chol, RBP-Exo/AMO181a-chol, or PEI25k/AMO181a-chol was delivered into the ischemic brains by intranasal administration. (A) Immunostaining of rage. After 24 h, the brains were harvested and immunostained with anti-RAGE antibodies. The ischemic penumbra of the cerebral cortex was selected to evaluate the RAGE expression level. Three sections per rat and three rats per group were evaluated for the RAGE expression. The scale bar represents 100 μm. (B) Quantitation of the RAGE level. The quantitation of the RAGE level was measured by the Zeiss ZEN software. *p < 0.05 as compared with MCAO, naked AMO181a-chol, and PEI25k/AMO181a-chol, but no statistical significance as compared with the other groups. | ||
The RAGE-mediated signal pathway eventually activates NF-κB and induces inflammation. Given the RAGE downregulation by RBP-Exo/AMO181a-chol, the expression of pro-inflammatory cytokines also may have been affected. Tumor necrosis factor-α (TNF-α) is a major pro-inflammatory factor and is released from neurons and glial cells.51 To evaluate the anti-inflammatory effects of RBP-Exo/AMO181a-chol, TNF-α expression in the rat brains was assessed by immunohistochemistry. The results demonstrated that RBP-Exo/AMO181a-chol effectively reduced TNF-α expression compared with the other treatments (Fig. 9). The infarct volume was evaluated by TTC staining of the harvested brains. The results showed that RBP-Exo/AMO181a-chol effectively reduced infarct volume compared with the other treatments (Fig. 10).
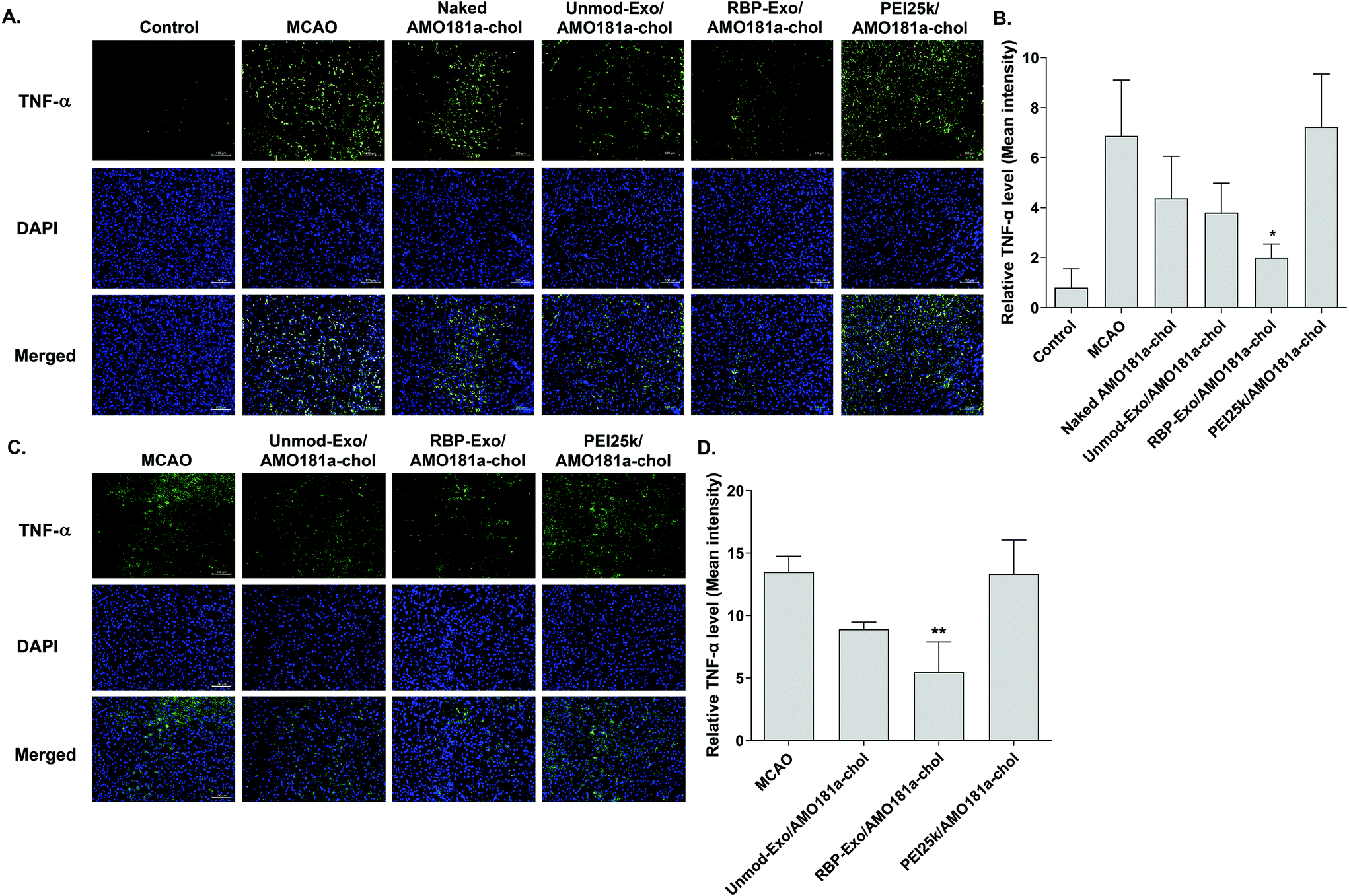 | ||
| Fig. 9 Downregulation of TNF-α after intranasal delivery of RBP-Exo/AMO181a-chol. Naked AMO181a-chol, Unmod-Exo/AMO181a-chol, RBP-Exo/AMO181a-chol, or PEI25k/AMO181a-chol was delivered into the ischemic brains by intranasal administration. After 24 h or 3 days, the brains were harvested and immunostained with anti-TNF-α antibody. The TNF-α-positive cells were analyzed by fluorescence microscopy. Three sections per rat and three rats per group was evaluated for TNF-α immunostaining. The ischemic penumbra of the cerebral cortex was selected to evaluate the TNF-α expression level. The scale bar represents 100 μm. The quantitation of the TNF-α level was measured by ZEISS ZEN software. *p < 0.05 as compared with the other groups. (A) Immunostaining with anti-TNF-α antibody (24 h post-injection), (B) quantitation of the TNF-α level (24 h post-injection, n = 3), (C) immunostaining with anti-TNF-α antibody (3 days post-injection), (D) quantitation of the TNF-α level (3 days post-injection, n = 3). *p < 0.05 as compared with the other groups. **p < 0.01 as compared with MCAO and PEI25k/AMO181a-chol, but no statistical significance as compared with Unmod-Exo/AMO181a-chol. | ||
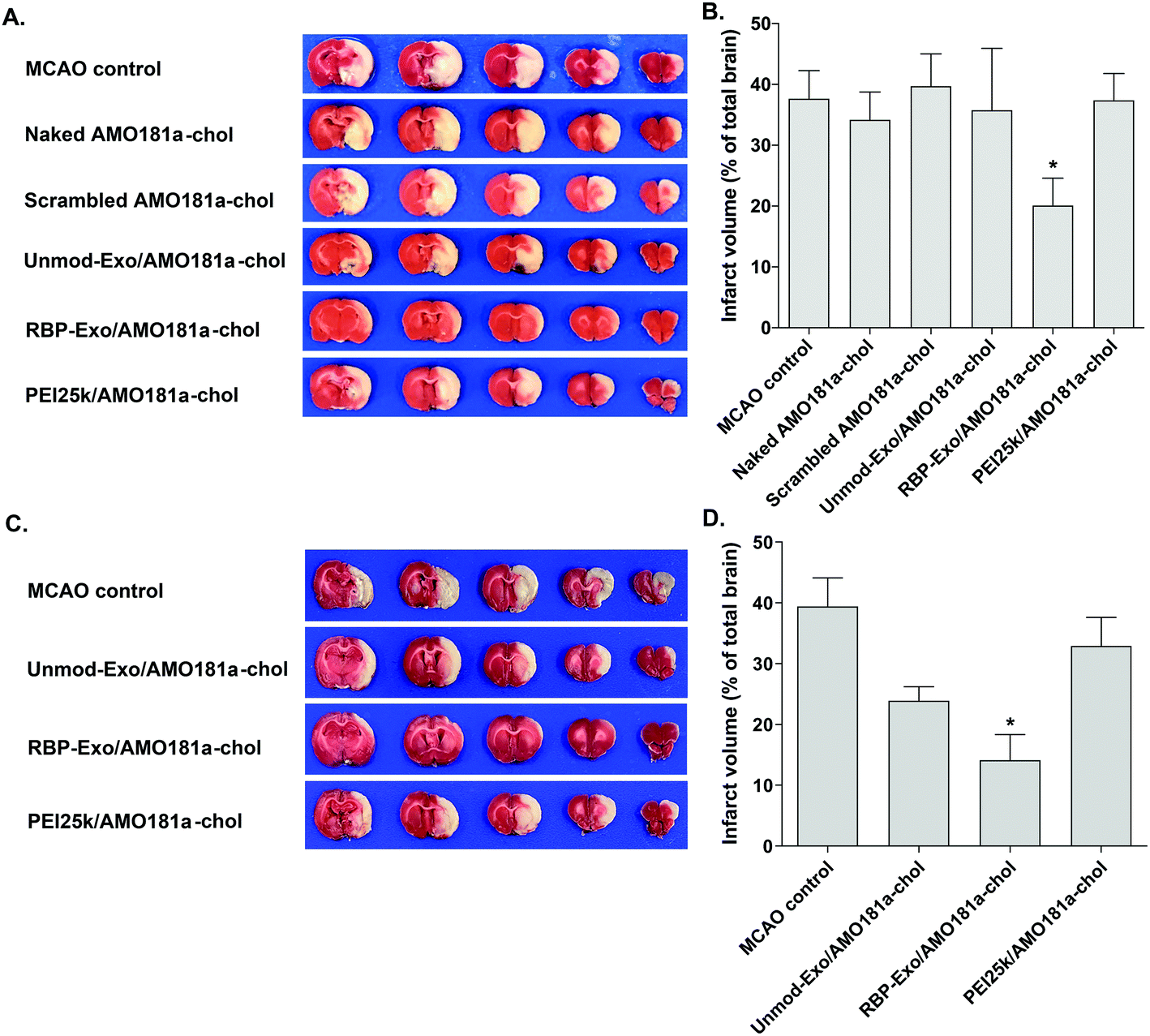 | ||
| Fig. 10 Infarct volume after intranasal delivery of RBP-Exo/AMO181a-chol. Naked AMO181a-chol, Unmod-Exo/AMO181a-chol, RBP-Exo/AMO181a-chol, or PEI25k/AMO181a-chol was delivered into the ischemic brains by intranasal administration. After 24 h or 3 days, the brains were harvested and stained with TTC solution. The infarct volume was measured by the ImageJ software. *p < 0.05 compared with the other groups. (A) TTC staining (24 h post-injection), (B) quantification of infarct volume from TTC staining results (24 h post-injection), (C) TTC staining (3 days post-injection), (D) quantification of infarct volume from TTC staining results (3 days post-injection). | ||
In summary, from the in vivo results we state that RBP-Exo/AMO181a-chol had a tendency to reduce miR-181a, RAGE, and TNF-α, compared with Unmod-Exo/AMO181a-chol. In addition, RBP-Exo/AMO181a-chol increased the Bcl2 expression and decreased apoptosis and infarct volume.
4. Conclusions
In this study, RBP-Exo was produced by genetic engineering and evaluated as a hypoxia-specific carrier for intranasal delivery of AMO181a-chol to the ischemic brain. Intranasal administration of RBP-Exo/AMO181a-chol delivered AMO181a into the ischemic brain more efficiently than Unmod-Exo/AMO181a-chol. These results suggest that the RBP moiety of RBP-Exo facilitates the delivery of AMO181a in ischemic tissues. The administration of RBP-Exo/AMO181a-chol reduced RAGE signaling, inflammatory cytokines, apoptotic cells, and infarct volume more efficiently than naked AMO181a-chol or Unmod-Exo/AMO181a-chol. Taken together, the results indicate that RBP-Exo is an efficient carrier for the intranasal delivery of AMO181a for ischemic stroke therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Individual Basic Science & Engineering Research Program (2019R1A2C1089560) through the National Research Foundation funded by the Ministry of Science and ICT in Korea.References
- NINDS Stroke Study Group, N. Engl. J. Med., 1995, 333, 1581–1587 CrossRef.
- T. Dalkara and E. M. Arsava, J. Cereb. Blood Flow Metab., 2012, 32, 2091–2099 CrossRef CAS PubMed.
- M. S. Sun, H. Jin, X. Sun, S. Huang, F. L. Zhang, Z. N. Guo and Y. Yang, Oxid. Med. Cell. Longevity, 2018, 2018, 3804979 Search PubMed.
- J. Oh, J. Lee, C. Piao, J. H. Jeong and M. Lee, Biomater. Sci., 2019, 7, 2174–2190 RSC.
- H. Hyun, Y.-W. Won, K.-M. Kim, J. Lee, M. Lee and Y.-H. Kim, Biomaterials, 2010, 31, 9128–9134 CrossRef CAS.
- M. Kim, G. Kim, D. W. Hwang and M. Lee, J. Biomed. Nanotechnol., 2019, 15, 2401–2412 CrossRef CAS PubMed.
- J. Lee, H. Hyun, J. Kim, J. H. Ryu, H. A. Kim, J. H. Park and M. Lee, J. Controlled Release, 2012, 158, 131–138 CrossRef CAS PubMed.
- H. Hyun, J. Lee, D. W. Hwang, S. Kim, D. K. Hyun, J. S. Choi, J. K. Lee and M. Lee, Biomaterials, 2011, 32, 306–315 CrossRef CAS PubMed.
- H. Hyun, Y. W. Won, K. M. Kim, J. Lee, M. Lee and Y. H. Kim, Biomaterials, 2010, 31, 9128–9134 CrossRef CAS PubMed.
- I.-D. Kim, J.-H. Shin, S.-W. Kim, S. Choi, J. Ahn, P.-L. Han, J.-S. Park and J.-K. Lee, Mol. Ther., 2012, 20, 829–839 CrossRef CAS PubMed.
- T. Rhim, D. Y. Lee and M. Lee, Pharm. Res., 2013, 30, 2429–2444 CrossRef CAS PubMed.
- J. J. Lochhead and R. G. Thorne, Adv. Drug Delivery Rev., 2012, 64, 614–628 CrossRef CAS PubMed.
- M. Van Woensel, N. Wauthoz, R. Rosiere, V. Mathieu, R. Kiss, F. Lefranc, B. Steelant, E. Dilissen, S. W. Van Gool, T. Mathivet, H. Gerhardt, K. Amighi and S. De Vleeschouwer, J. Controlled Release, 2016, 227, 71–81 CrossRef CAS PubMed.
- S. Yadav, S. K. Gandham, R. Panicucci and M. M. Amiji, Nanomedicine, 2016, 12, 987–1002 CrossRef CAS.
- I. Ullah, K. Chung, J. Oh, J. Beloor, S. Bae, S. C. Lee, M. Lee, P. Kumar and S. K. Lee, Sci. Rep., 2018, 8, 15041 CrossRef PubMed.
- E. Samaridou, H. Walgrave, E. Salta, D. M. Alvarez, V. Castro-Lopez, M. Loza and M. J. Alonso, Biomaterials, 2020, 230, 119657 CrossRef CAS.
- X. Zhuang, X. Xiang, W. Grizzle, D. Sun, S. Zhang, R. C. Axtell, S. Ju, J. Mu, L. Zhang, L. Steinman, D. Miller and H. G. Zhang, Mol. Ther., 2011, 19, 1769–1779 CrossRef CAS PubMed.
- I. D. Kim, J. H. Shin, S. W. Kim, S. Choi, J. Ahn, P. L. Han, J. S. Park and J. K. Lee, Mol. Ther., 2012, 20, 829–839 CrossRef CAS PubMed.
- C. Théry, L. Zitvogel and S. Amigorena, Nat. Rev. Immunol., 2002, 2, 569–579 CrossRef PubMed.
- M.-C. Didiot, L. M. Hall, A. H. Coles, R. A. Haraszti, B. M. Godinho, K. Chase, E. Sapp, S. Ly, J. F. Alterman and M. R. Hassler, Mol. Ther., 2016, 24, 1836–1847 CrossRef CAS PubMed.
- J. G. van den Boorn, M. Schlee, C. Coch and G. Hartmann, Nat. Biotechnol., 2011, 29, 325–326 CrossRef CAS PubMed.
- G. Fritz, Trends Biochem. Sci., 2011, 36, 625–632 CrossRef CAS PubMed.
- J. C. Tobon-Velasco, E. Cuevas and M. A. Torres-Ramos, CNS Neurol. Disord.: Drug Targets, 2014, 13, 1615–1626 CrossRef CAS.
- T. Kamide, Y. Kitao, T. Takeichi, A. Okada, H. Mohri, A. M. Schmidt, T. Kawano, S. Munesue, Y. Yamamoto and H. Yamamoto, Neurochem. Int., 2012, 60, 220–228 CrossRef CAS PubMed.
- P. Pichiule, J. C. Chavez, A. M. Schmidt and S. J. Vannucci, J. Biol. Chem., 2007, 282, 36330–36340 CrossRef CAS PubMed.
- A. Bierhaus, P. M. Humpert, M. Morcos, T. Wendt, T. Chavakis, B. Arnold, D. M. Stern and P. P. Nawroth, J. Mol. Med., 2005, 83, 876–886 CrossRef CAS PubMed.
- S. Muhammad, W. Barakat, S. Stoyanov, S. Murikinati, H. Yang, K. J. Tracey, M. Bendszus, G. Rossetti, P. P. Nawroth and A. Bierhaus, J. Neurosci., 2008, 28, 12023–12031 CrossRef CAS.
- S. Lee, C. Piao, G. Kim, J. Y. Kim, E. Choi and M. Lee, Eur. J. Pharm. Sci., 2018, 114, 275–284 CrossRef CAS PubMed.
- E. Choi, J. Oh, D. Lee, J. Lee, X. Tan, M. Kim, G. Kim, C. Piao and M. Lee, J. Controlled Release, 2018, 279, 40–52 CrossRef CAS PubMed.
- B. G. Hassid, M. N. Nair, A. F. Ducruet, M. L. Otten, R. J. Komotar, D. J. Pinsky, A. M. Schmidt, S. F. Yan and E. S. Connolly, J. Clin. Neurosci., 2009, 16, 302–306 CrossRef CAS PubMed.
- O. G. Bhalala, M. Srikanth and J. A. Kessler, Nat. Rev. Neurol., 2013, 9, 328–339 CrossRef CAS PubMed.
- S. E. Khoshnam, W. Winlow, Y. Farbood, H. F. Moghaddam and M. Farzaneh, J. Stroke, 2017, 19, 166–187 CrossRef PubMed.
- J. M. Moon, L. Xu and R. G. Giffard, J. Cereb. Blood Flow Metab., 2013, 33, 1976–1982 CrossRef CAS PubMed.
- L. J. Xu, Y. B. Ouyang, X. Xiong, C. M. Stary and R. G. Giffard, Exp. Neurol., 2015, 264, 1–7 CrossRef CAS PubMed.
- Y. B. Ouyang, Y. Lu, S. Yue, L. J. Xu, X. X. Xiong, R. E. White, X. Sun and R. G. Giffard, Neurobiol. Dis., 2012, 45, 555–563 CrossRef CAS PubMed.
- G. Kim, Y. Lee, J. Ha, S. Han and M. Lee, J. Controlled Release, 2021, 330, 684–695 CrossRef CAS PubMed.
- L. Alvarez-Erviti, Y. Q. Seow, H. F. Yin, C. Betts, S. Lakhal and M. J. A. Wood, Nat. Biotechnol., 2011, 29, 341–345 CrossRef CAS PubMed.
- G. Kim, M. Kim, Y. Lee, J. W. Byun, D. W. Hwang and M. Lee, J. Controlled Release, 2020, 317, 273–281 CrossRef CAS PubMed.
- D. Ha, N. N. Yang and V. Nadithe, Acta Pharm. Sin. B, 2016, 6, 287–296 CrossRef PubMed.
- T. Yong, X. Zhang, N. Bie, H. Zhang, X. Zhang, F. Li, A. Hakeem, J. Hu, L. Gan, H. A. Santos and X. Yang, Nat. Commun., 2019, 10, 3838 CrossRef PubMed.
- M. Khongkow, T. Yata, S. Boonrungsiman, U. R. Ruktanonchai, D. Graham and K. Namdee, Sci. Rep., 2019, 9, 8278 CrossRef PubMed.
- R. Kojima, D. Bojar, G. Rizzi, G. C. Hamri, M. D. El-Baba, P. Saxena, S. Auslander, K. R. Tan and M. Fussenegger, Nat. Commun., 2018, 9, 1305 CrossRef PubMed.
- Q. Zhan, K. Yi, H. Qi, S. Li, X. Li, Q. Wang, Y. Wang, C. Liu, M. Qiu, X. Yuan, J. Zhao, X. Hou and C. Kang, Theranostics, 2020, 10, 7889–7905 CrossRef CAS PubMed.
- W. J. Goh, C. K. Lee, S. Zou, E. C. Woon, B. Czarny and G. Pastorin, Int. J. Nanomed., 2017, 12, 2759–2767 CrossRef CAS PubMed.
- M. Vashisht, P. Rani, S. K. Onteru and D. Singh, Appl. Biochem. Biotechnol., 2017, 183, 993–1007 CrossRef CAS PubMed.
- C. Gong, J. Tian, Z. Wang, Y. Gao, X. Wu, X. Ding, L. Qiang, G. Li, Z. Han, Y. Yuan and S. Gao, J. Nanobiotechnol., 2019, 17, 93 CrossRef PubMed.
- R. A. Haraszti, R. Miller, M. C. Didiot, A. Biscans, J. F. Alterman, M. R. Hassler, L. Roux, D. Echeverria, E. Sapp, M. DiFiglia, N. Aronin and A. Khvorova, Mol. Ther., 2018, 26, 1973–1982 CrossRef CAS PubMed.
- M. C. Didiot, L. M. Hall, A. H. Coles, R. A. Haraszti, B. M. Godinho, K. Chase, E. Sapp, S. Ly, J. F. Alterman, M. R. Hassler, D. Echeverria, L. Raj, D. V. Morrissey, M. DiFiglia, N. Aronin and A. Khvorova, Mol. Ther., 2016, 24, 1836–1847 CrossRef CAS PubMed.
- D. Fischer, T. Bieber, Y. Li, H. P. Elsasser and T. Kissel, Pharm. Res., 1999, 16, 1273–1279 CrossRef CAS PubMed.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Biomed. Mater. Res., 1999, 45, 268–275 CrossRef CAS.
- G. Feuerstein, X. Wang and F. C. Barone, Cell. Mol. Neurobiol., 1998, 18, 695–701 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr07516g |
| This journal is © The Royal Society of Chemistry 2021 |