
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactive molecular dynamics simulations of hydration shells surrounding spherical TiO2 nanoparticles: implications for proton-transfer reactions†
Federico A.
Soria
 and
Cristiana
Di Valentin
*
and
Cristiana
Di Valentin
*
Dipartimento di Scienza dei Materiali, Università di Milano Bicocca, via R. Cozzi 55, 20125 Milano, Italy. E-mail: cristiana.divalentin@unimib.it
First published on 12th February 2021
Abstract
In many potential applications, nanoparticles are typically in an aqueous medium. This has strong influence on the stability, optical properties and reactivity, in particular for their functionalization. Therefore, the understanding of the chemistry at the interface between the solvent and the nanoparticle is of utmost importance. In this work, we present a comparative ReaxFF reactive molecular dynamics investigation on spherical TiO2 nanoparticles (NSs) of realistic size, with diameters from 2.2 to 4.4 nm, immersed in a large drop of bulk water. After force field validation for its use for a curved anatase TiO2 surface/water interface, we performed several simulations of the TiO2 nanoparticles of increasing size in a water drop. We found that water can be adsorbed jointly in a molecular and dissociative way on the surface. A Langmuir isotherm indicating an adsorption/desorption mechanism of water on the NS is observed. Regarding the dissociative adsorption, atomistic details reveal two different mechanisms, depending on the water concentration around the NS. At low coverage, the first mechanism involves direct dissociation of a single water molecule, whereas, at higher water coverage, the second mechanism is a proton transfer reaction involving two water molecules, also known as Grotthuss-like mechanism. Thermal annealing simulations show that several water molecules remain on the surface in agreement with the experimental reports. The capacity of adsorption is higher for the 2.2 and 3.0 nm NSs than for the 4.4 nm NS. Finally, a comparative investigation with flat surfaces indicates that NSs present a higher water adsorption capacity (undissociated and dissociated) than flat surfaces, which can be rationalized considering that NSs present many more low-coordinated Ti atoms available for water adsorption.
1. Introduction
Titanium dioxide (TiO2) is one of most technologically relevant transition metal oxides as it is used in many applied fields. It offers wide-ranging properties for fundamental science and industrial applications, including photocatalysis,1–3 solar cell energy conversion,4–7 photoelectrochemistry,8,9 and more recently, photo-nanomedicine.10Because most of these applications take place in an aqueous environment, understanding the reactivity of TiO2 surfaces with water is of great importance. For this reason, several first-principles studies have investigated the detailed atomistic description of the water chemistry and multilayer dynamical structures on low-index flat surfaces.11–17 Also, several recent experimental studies have been carried out to determine the interaction at the interface between water and flat TiO2 surfaces.18–23 However, in practical applications, TiO2 is mostly used in the form of nanoparticles, which may partially dissolve and become round- or potato-shape in a diluted aqueous environment. Curved nanoparticles expose a larger number of undercoordinated Ti sites and are characterized by an enhanced surface tension. Both these features are expected to largely affect the surface interaction with the surrounding water environment.
Water interaction with curved TiO2 surfaces is of utmost importance because water can compete with any other chemical species for the adsorption on surface sites as a donor on low-coordinated Ti atoms and as an H-bonding species on low-coordinated O atoms.24,25 Also, dissociation of water on the surface sites or defects leads to the formation of OH species, which are crucial functional groups for surface chemistry, functionalization and molecular anchoring.26 Additionally, mono- or multi-layered water adsorption may alter the surface electronic properties and work function, as well as the surface energy leading to different reconstructions than in ultra-high vacuum conditions.27 Recently, it was even reported to affect the trapping dynamics of photogenerated charge carriers in TiO2.28,29
Modeling curved systems are computationally more expensive than flat ones because a realistic size of the nanosystem must be considered not to exaggerate the curvature and not to induce an excessive strain. For a diameter size of at least 2 nm, more than 600 atoms are needed. The addition of water multilayers around the nanosphere easily sums up to 2000–2500 atoms. On top of this, the description of a bulk of water surrounding the hydrated nanoparticles clearly leads to an extremely large model.26 Therefore, on the one hand, there is a need for models of several thousands of atoms and, on the other hand, there is a requirement of reliable accuracy in the description of the complex physics and chemistry of water,30 especially when in contact with spherical-shaped TiO2 nanoparticles.
First-principles calculations provide a very detailed and accurate description of metal–oxide/water interfaces.31 However, their substantial computational costs restrict the system size and time scale that can be probed, thus limiting their applicability in the study of solid surfaces in an aqueous environment. On the other hand, empirical force-field methods are faster and can simulate larger systems but cannot describe chemical reactions, such as proton transfers or dissociative adsorption on surfaces.32,33
With this scenario, there are two possible schemes to simulate the chemistry of realistic nanoparticles in a water environment: one of them is the QM/MM, as was shown previously by some of us,26 whereas the other possibility is the use of ReaxFF reactive molecular dynamics (MD).34,35 ReaxFF enables the modeling of bond formation and breaking on the basis of a bond-order formalism. It can simulate chemical reactions and, thus, allows for a dissociable model of water, enabling a reasonable description of proton-exchange dynamics and H-bond configurations at the interface.36 Therefore, ReaxFF acts as a bridge between quantum mechanics (QM) methods, such as those based on density functional theory (DFT) and empirical force-field methods, by being able to handle large simulations (in model size and time scale), while maintaining precision and reactivity that approximate that of QM-based methods. ReaxFF not only can describe the atomistic details of the chemical reaction but also can give other information, such as Arrhenius parameters,37 diffusion coefficients,38 Langmuir adsorption isotherms,39 among others. ReaxFF has been parametrized40 and used41–44 to describe the interface between flat TiO2 surfaces and water interface. ReaxFF has also been validated against DFT calculations to study the mechanism of diffusion of atomic H and of O vacancies on the flat and spherical TiO2 surfaces.45
In this work, we present a detailed study of the water surface reactivity and of the hydration shell of the spherical TiO2 nanoparticles (TiO2 NS). First, we assess the validity of the selected force field,40 which was parametrized for bulk (anatase, rutile and brookite), different crystal surfaces and faceted NP for its use on curved TiO2 surface/water interfaces, using previous DFT (B3LYP) and DFTB (MATORG + HBD) calculations by some of us as reference data.26 Secondly, we present several simulations for a 2.2 nm NS with an increasing number of surrounding hydration layers. We can explain the reaction and adsorption mechanism at different water concentrations. Later, we move to more realistic NS of 3.0 and 4.4 nm diameters with different layers of water to investigate the effect of increasing size and reduced curvature. We observe a similar capacity of water adsorption for the three investigated NSs. However, after annealing at 500 K, the larger NS is found to be left with a reduced capacity compared to the smaller ones. Finally, we compare the water adsorption capacity of curved systems with that of flat anatase (101) TiO2 surfaces.
2. Computational details
2.1 Reactive force field for TiO2/H2O systems
The ReaxFF forcefield provides an accurate description of bond breaking and bond formation during MD simulations by employing a bond-order/bond-energy relationship, which was developed by van Duin, Goddard, and co-workers.34,35 In the ReaxFF reactive force field, the total (system) energy is given by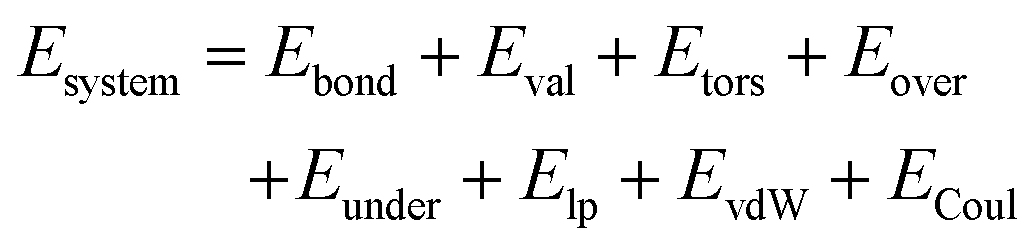 | (1) |
The terms in eqn (1) include bond energies (Ebond), valence angle energies (Eval), torsion-angle energies (Etors), the energy to penalize overcoordination of atoms (Eover), the energy to stabilize under-coordination of atoms (Eunder), lone-pair energies (Elp), and terms to handle nonbonded Coulomb (ECoul) and van der Waals (EvdW) interactions.
In this work, the Ti/O/H ReaxFF forcefield developed by Kim and coworkers was used. The developed force field parameters were fitted to a set of quantum mechanical calculations of TiO2/H2O systems and were found to predict the structures, energies, and equation of state for bulk TiO2 materials.40
The same force field was used by Raju and coworkers to study water adsorption and dissociation on anatase (101), (100), (112), and (001) and rutile (110) at 300.0 K (ref. 41) and the aggregation of anatase nanocrystals in vacuum and humid environments.42 In addition, other authors used the force field to compare the reactivity of water at the rutile (001), rutile (110), anatase (001), TiO2-B (100) and TiO2-B (001) surfaces.43 More recently, the same ReaxFF was used to investigate the structural and dynamical properties of anatase (101) and rutile (110) TiO2 interfaces with liquid bulk water. Based on this simulation, the authors reported a well-organized structure of water in the interface region within 6.5 Å from the surface and a spontaneous dissociation leading, rather controversially, to full coverage of O2c/Ob by H+ and partial coverage of Ti5c by OH−.44 Even the force field was used to describe the effect of high pressure H2 on the photocatalytic properties of a large spherical TiO2 nanoparticle (6 nm).45
2.2 Reactive molecular dynamics simulation setup
The ReaxFF-MD simulations were performed with the LAMMPS software package.46,47 MD simulations have been performed in the canonical (NVT) ensemble for 500 ps. A Velocity-Verlet algorithm was used with a 0.1 fs time step using the Berendsen thermostat and a temperature-damping constant of 100 fs to control the temperature of the entire system. The initial velocities are assigned according to the Boltzmann distribution. Each simulation was performed three times starting from different initial Boltzmann distributions in order to explore possible scenarios of the reaction process. In this way, we made about 50 different reactive simulations. System snapshots were saved every 0.01 ps during the simulations and they were used for the analysis of the evolution of chemical species and atom–atom radial distribution functions (RDFs).The spherical nanoparticles considered in this work are shown in Fig. 1. The initial structures have been directly carved from a bulk anatase TiO2 supercell. By different carving radii, we obtain nanoparticles of about 2.2 nm, 3.0 nm, and 4.4 nm diameter size, as described in detail in a previous work by some of us.48,49 The structures were kept stoichiometric and the too low-coordinated Ti and O atoms of the surface were saturated with dissociated H2O molecules: three-fold and some four-fold Ti atoms were coordinated to hydroxyl groups, whereas mono-coordinated O atoms were saturated with H atoms.
 | ||
| Fig. 1 Ball representation of the ReaxFF optimized structures, after simulated annealing, of the different nanospheres considered in this work. For each one, the stoichiometry and the approximate diameter are reported. Blue spheres represent the O atoms, grey spheres represent the Ti atoms and white small spheres represent the H atoms. | ||
The hydration shells around each nanosphere have been generated using the PACKMOL code.50 Spherical water shells were varied from 1 ML to bulk liquid water for the 2.2 nm NS, from 1 ML up to 6 ML for the 3 nm NS, and 6 ML for the 4.4 nm NS. In this way, we carried out simulations with a number of atoms varying between 1000 and 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500.
500.
For the simulations of water reactivity on the anatase (101) surface, we build a slab of 192 TiO2 units (three triatomic layers) placed into an orthorhombic supercell of dimensions of 30.4137 × 20.95596 × 59.00 Å. On each side of the slab, there are 32 Ti5c. Inside the periodic box, we put 1000 water molecules in order to get the bulk water density 1 g cm−3.
2.3 Simulation analysis
To perform the analysis of the behavior of the hydration shells around each nanosphere, we have calculated the g(d) distribution function by determining the MD-averaged number of water molecules within a distance d and d + Δd away from the nanosphere surface (Δd = 0.01 Å). The distance d is defined as the minimal distance between the O atom of the water molecule (Owater) and the superficial Ti atoms of the nanosphere (Tisup). The Tisup species not only include all the undercoordinated titanium atoms (Ti5c, Ti4c, Ti4c(OH), and Ti3c(OH)), but also fully coordinated Ti atoms (Ti6c_sup) when connected to at least one surface O2c.In parallel, to evaluate the structural properties of water on a flat surface, we have calculated the radial distribution function (RDF), g(r), between anatase (101) surface atoms (Ti5c or O2c) and water (O or H atoms). Also, for the flat surface, we have calculated the vertical distribution of the distances (ρ(Δz)) between the water molecules (O atoms) and the anatase (101) surface (Ti5c). For both analyses, we made use of the module implemented in Lammps software.46,47
Finally, to monitor the different molecular fragments produced during the ReaxFF-MD simulation, we have used the postprocessing tool (mol_fra.c) provided by Lammps software.46,47
3. Results and discussion
3.1 Force field (FF) validation for curved anatase TiO2 surface/water interface
In Fig. S2,† we present a comparison between ReaxFF and DFT (Fig. S2a†) and DFTB and DFT (Fig. S2b†) adsorption energies for both molecular and dissociated water molecules on different positions of the 2.2 nm NS. From the plots, it can observed a good agreement between ReaxFF and DFT for both dissociated and undissociated adsorption energies. On the other hand, DFTB calculations describe well the molecular adsorption but underestimate the dissociated adsorption energies which indicate a tend to favour molecular adsorption in comparison with DFT.26
In Fig. 2, we present the ReaxFF simulation of a NS (2.2 nm) in a water drop. For that, we plot the distance distribution function g(d) (as defined in the Computational details) for two different water drops around the NS as a function of the distance (d) in Fig. 2a. We consider a water drop of 6 ML, generated by 1000 molecules around the NS (∼1 nm of initial thick), and another one of a multilayer of 8958 water molecules (∼55 ML, about 4 nm thick, Fig. 2b). In Fig. 2c, we compare the g(d) of both water drop models with previous DFTB and DFTB/MM molecular dynamics calculations by some of us. The agreement is excellent with four distinct peaks centered at 1.9 Å, 2.3 Å, 3.9 Å, and 4.5 Å. The orange dashed lines are aligned to the center of the four peaks in the distribution. The first peak in the distribution is due to the Ti–OH moieties, resulting from the dissociated adsorption mode. These appear as green OHs in Fig. 2d. The second peak centered at 2.3 Å is due to molecularly coordinated water (orange H2O in Fig. 2d) to surface Ti atoms (Ti⋯OH2). The third peak is related to the water layer interacting through the H-bond to surface O atoms or to Ti–OH groups through the Hwater atoms and finally the peak at 4.5 Å is due to water molecules that are H-bonded to Ti–OH groups or coordinated to Ti atoms (Ti⋯OH2) through the Owater atom. Also, a similar distribution function was obtained for the H atoms of the water molecules and the O2c on the NS surface (Fig. S3†). Here, three main peaks are centered at 1.0, 1.6 and 3.0 Å, corresponding to the O–H bond, i.e. OH groups formed on O2c sites, H-bond between water molecules and O2c surface atoms, whereas the last peak indicates the long-distance interaction between H atoms of the second layer with the O2c surface atoms.
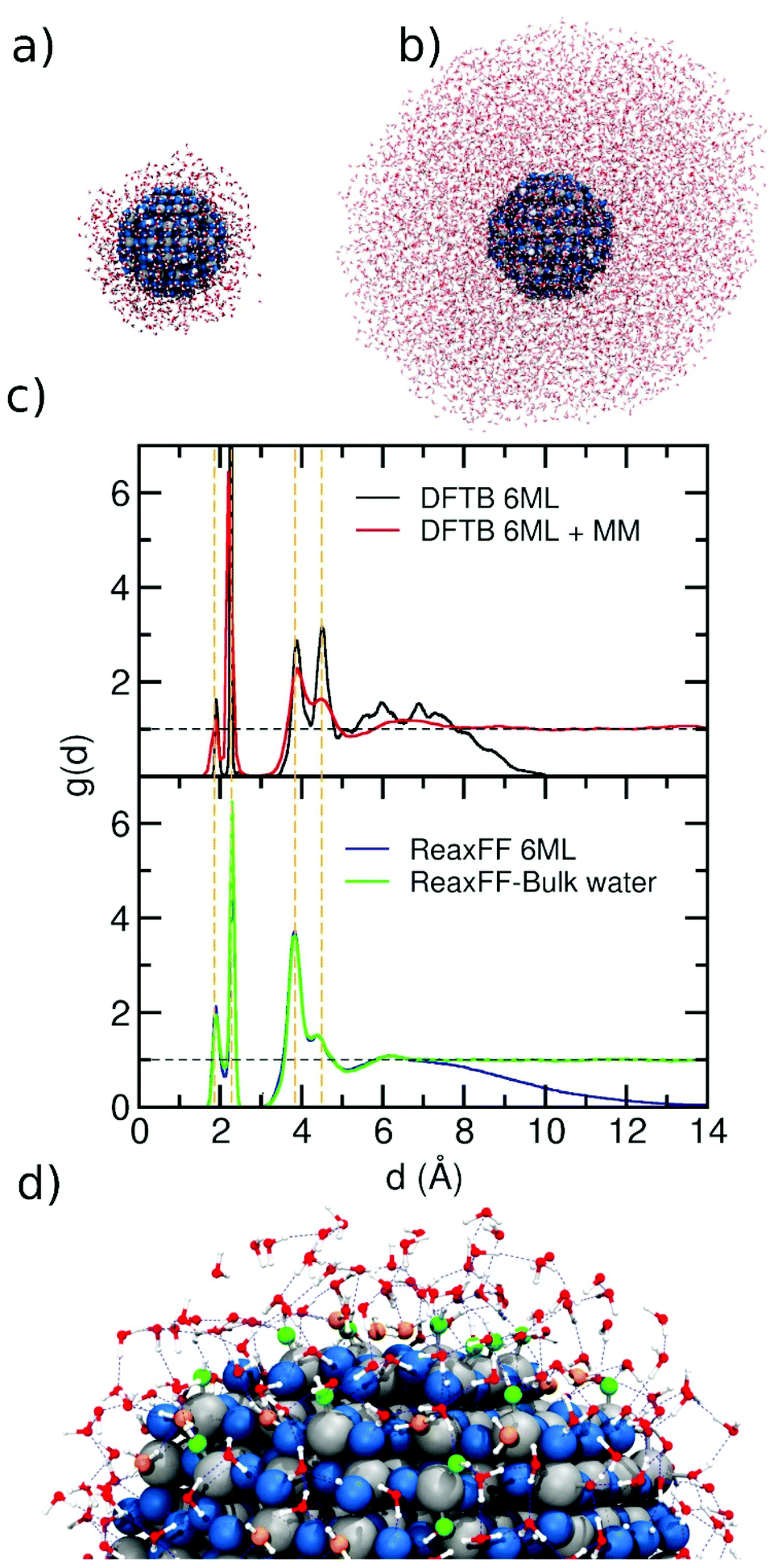 | ||
| Fig. 2 (a) and (b) Snapshot of the final structure from the ReaxFF molecular dynamics simulation of the NS model surrounded with a 6 ML and 55 ML of water, respectively. The overall stoichiometry is (TiO2)223·1000H2O and (TiO2)223·8952H2O in each case. (c) Distribution function g(d) as extracted from the DFTB molecular dynamics run26 (top) and from ReaxFF molecular dynamics simulations (bottom). A dashed horizontal black line is traced for g(d) = 1, which corresponds to bulk water density. For comparison, dashed vertical orange lines indicate the positions of the main peaks. (d) Zoomed-in image of a portion of the NS surrounded by water. Undissociated and dissociated water are marked in orange and green respectively. The blue dashed lines show the H-bond network. | ||
Comparing the g(d) distribution in Fig. 2c for 6 ML, we observed a more structured curve for DFTB (black line) than for ReaxFF (blue line). This may be rationalized by the fact that for DFTB, the simulation time is one order of magnitude less than for ReaxFF simulations (45 ps vs. 500 ps) which also explains the broader range of distance values that are reached during the ReaxFF-MD simulation.
Regarding the NS in a droplet of bulk water, the behavior of ReaxFF (green line) is very similar to that of DFTB/MM (red line) simulations. In the g(d) distribution from the ReaxFF simulation, we observe a tiny peak between 6 and 7 Å, similar to what the DFTB/MM feature, indicating that the force field provides a good description of the long-range interfacial effects. Finally, the comparison between 6 ML and bulk water with ReaxFF description, shows a very similar profile up to 7 Å, which suggests that a water model of 6 ML around the NS is sufficient to get a good description of NS in an aqueous environment.
The integral area of the peaks in the rdf plots provides the average coordination number of the atoms involved in the rdf distribution. These values for the peaks at 1.9 Å and 2.3 Å are 0.26 and 1.04 using DFTB and DFTB/MM, while using ReaxFF, the values are 0.42 and 0.83, respectively. The single values differ but their sums are very close, i.e. 1.29 and 1.25. The first peak is due to dissociatively adsorbed water, whereas the second peak is due to molecularly adsorbed water. The difference in the area of each single peak can be explained based on the results reported in Fig. S2,† where we correlate the adsorption energies of water on different sites of the 2.2 nm NP (dissociated or undissociated) with ReaxFF or DFTB against those obtained with DFT. Taking the DFT energies as the reference, we observe that DFTB can well reproduce the undissociated water adsorption but underestimates the dissociated adsorption energies, whereas ReaxFF describes both dissociated and undissociated water adsorption energies on the NP quite well. This explains why the value of the integral for the first peak indicates more dissociation with ReaxFF than with DFTB (or DFTB/MM).
Thus, we have shown that the force field, which was parameterized for bulk and for flat surfaces interacting with water, can be successfully used also for curved ones.
3.2 Water adsorption/dissociation at the curved surface of a spherical (2.2 nm) TiO2 nanoparticle
In this section, we investigated the adsorption mechanism of water on the 2.2 nm TiO2 NS. First, we will show a general mechanism which implies the formation of new OH groups on the NS decreasing of the number of undissociated H2O molecules around the NS. Then, we will focus on the atomistic details of the mechanism of Ti–OH formation.In Fig. 3, we first present the variation of the amount of certain relevant species on the surface as a function of the simulation time when the NS is decorated by 12 ML of water. The species considered are: (1) the undissociated water molecules around the NS (initial number is 1000, top panel), (2) the clean O2c sites on the NS surface (middle panel) and (3) the total OH groups on the NS (bottom panel), taking into account both the terminal OH (initial number is 20) and the bridging OH superficial groups. From the plot, we observe a decrease in the amount of undissociated water molecules correlated with a decrease in the amount of the clean O2c and with an increase in the total OH groups (terminal + bridging) present on the NS. The final number of (not dissociated) H2O is 955, with 45 molecules that have dissociated on the surface. The amount of clean O2c decreases from 124 to 79 units and total OH groups increase from 20 to 110 units. Thus, 90 new total OH groups are formed on the surface of which 45 are formed on the O2c sites (bridging OH groups) and 45 formed on the undercoordinated Ti surface atom (terminal OH groups). This is a clear evidence that dissociative adsorption is taking place on the surface at the same time as molecular adsorption (Fig. 2c, peak at 2.3 Å and Fig. 2d orange water molecules). This mixed dissociated/undissociated adsorption mode was observed experimentally by soft X-ray spectroscopies24,52 and by the electron yield in near-edge X-ray absorption fine structure (NEXAFS).24 In the next section, we will give details of the dissociation mechanism.
 | ||
| Fig. 3 Evolution of the amount of fragment/species as a function of time for a simulation of 6 ML of water around the NS system at 300.0 K. Top: entire water molecules around the NS. Middle: O2c sites on the NS. Bottom: total, bridging and terminal OH groups on the NS. | ||
As a next step, we varied the amount of water layers around the NS to investigate whether a different behavior is observed. The initial numbers of water molecules surrounding the NS were 150, 250, 500, 1000, 2000 and 8958, producing 1, 1.5, 3, 6, 12 and 55 ML of water, respectively. Then, in Fig. 4a, we report the variation of terminal OH groups is divided by the number of total Ti adsorption sites on the NS as a function of time for these six different initial amounts of water. An increase in the OH groups on the NS with time is always clearly observed. However, the shape of the curves for initial 150 and 250 water molecules is different from that for initial 500, 1000, 2000 and 8958 water molecules surrounding the NS. For the second set of data, we observe a more pronounced slope within the first 150 ps, which indicates a higher reaction rate. We rationalize this with the fact that for these water concentrations, the resulting aqueous environment is more condensate than for the first two sets. We evaluate the extent of condensation by counting the number of H-bonds, as normalized by the number of water molecules in the model. For the first two sets, we have less than 0.8 H-bonds per water molecule, whereas for the last four sets, more than 1.2 H-bonds per water molecule are computed. These results indicate that the reaction mechanism may depend on the water concentration around the NS. Similar conclusions were reached in a previous ab initio molecular dynamics study on small TiO2 (∼1 nm) nanoparticles.53 In the next section, we will discuss in detail different dissociation mechanisms depending on the different water concentrations.
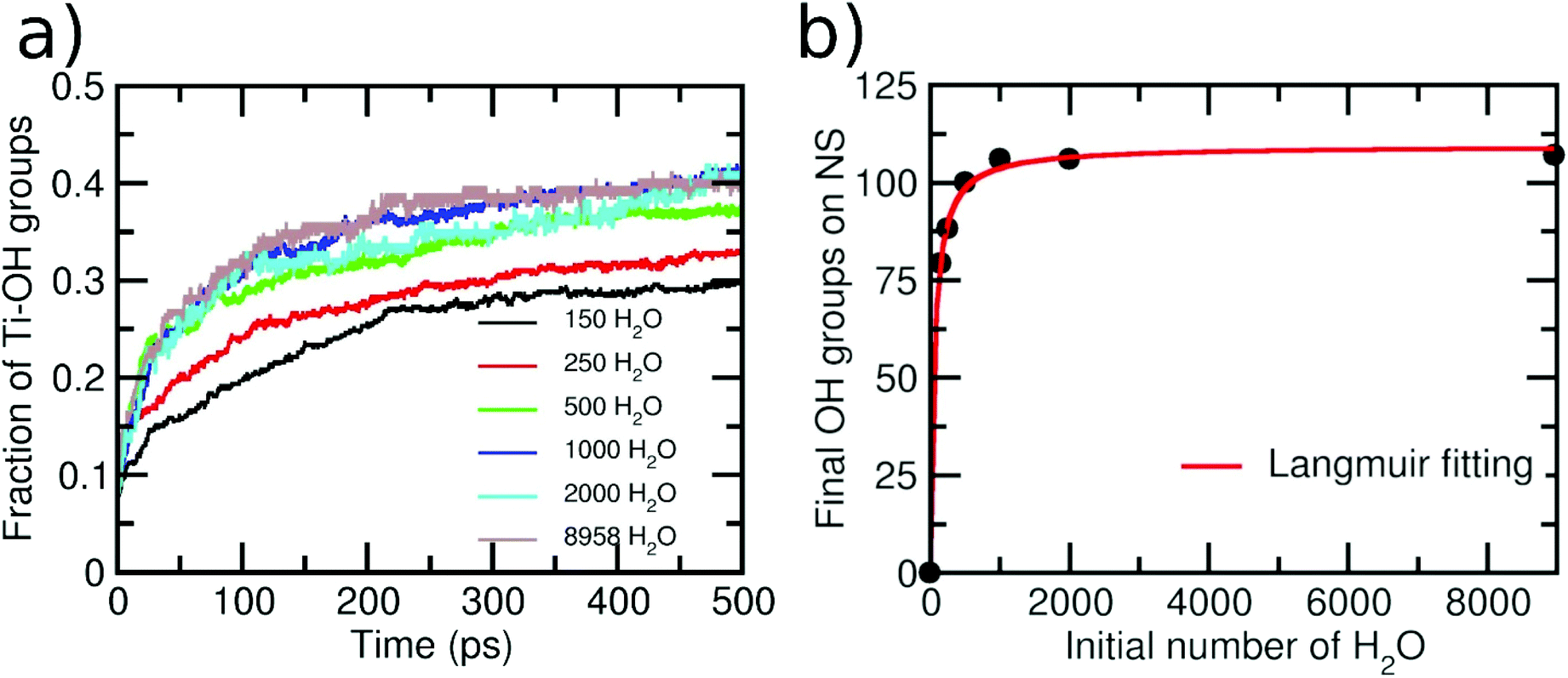 | ||
| Fig. 4 (a) Evolution of the amount of the terminal hydroxyl groups on the 2.2 nm NS normalized by the number of total Ti adsorption sites as a function of time for the simulations with different initial numbers of water molecules surrounding the NS. (b) Number of final hydroxyl groups on the NS after 500 ps of simulations as a function of the initial number of water molecules around the NS. The red line shows a fitting with a Langmuir isotherm. | ||
Finally, in Fig. 4b, we present a graph of the final number of OH groups (at the end of the simulation) on the NS surface as a function of the initial number of surrounding water molecules. Note that, for the case of 150 water molecules, the final number of OH groups is 80, giving rise to an extent of water dissociation on the surface is α = 0.29, which is close to the value that was computed at the DFT(HSE06) level of theory of α = 0.21.26 It can be seen that the number of OH groups becomes constant for a number of initial water molecules above 1000 (>6 ML). This result is in line with X-ray photoelectron spectroscopy experiments, which show that the Ti–OH species concentration is almost constant when a TiO2 nanoparticle is exposed to different degrees of humidity (RH).24 Our data can be nicely fit with a Langmuir isotherm, suggesting that there is an adsorption/desorption equilibrium in the system.54 This adsorption/desorption equilibrium is observed, at the atomic level, in terms of the protonation of some OH superficial groups on the NS (forming new water molecules) or in terms of the deprotonation of some adsorbed water molecules (forming OH groups on the NS surface).
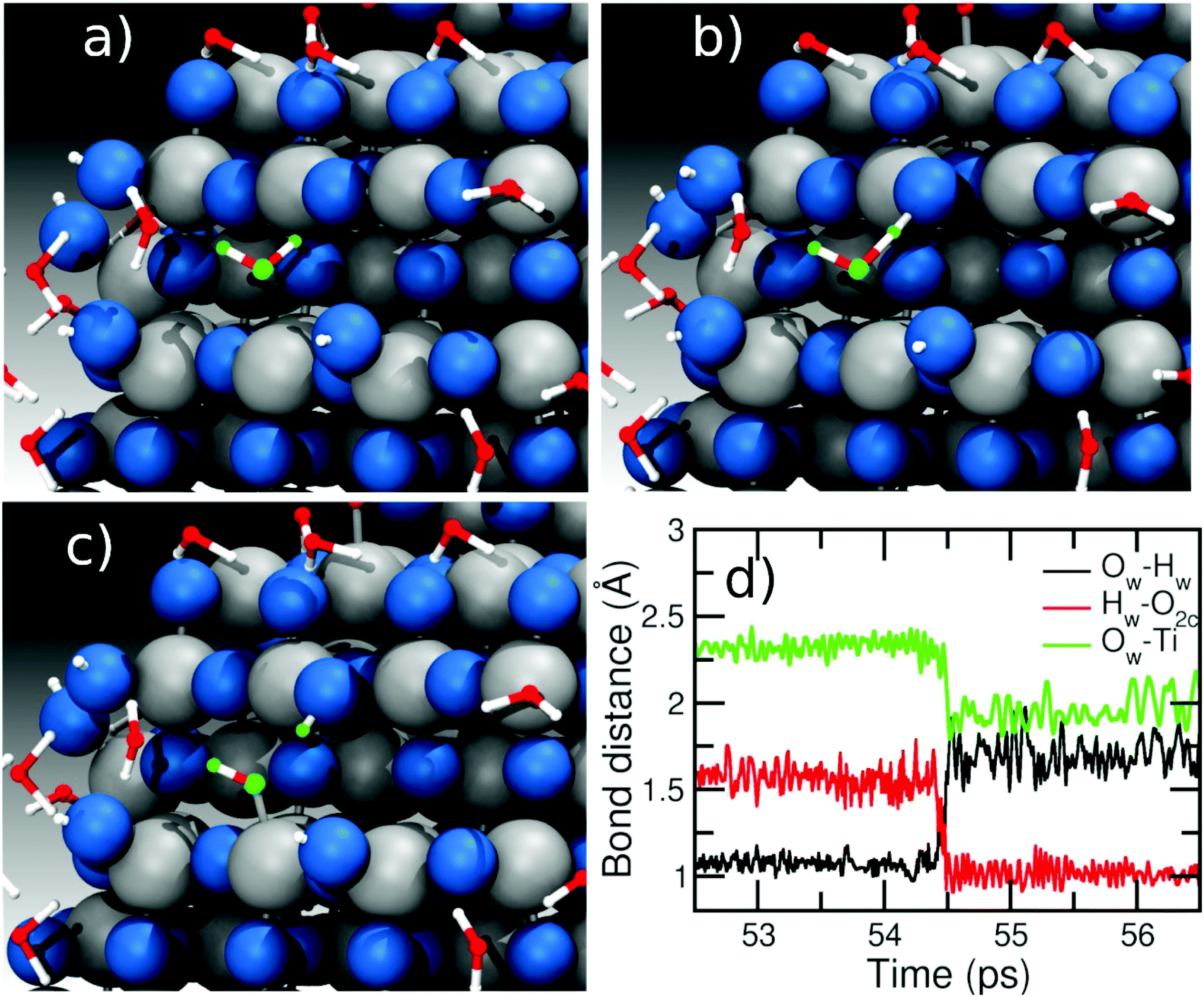 | ||
| Fig. 5 Details of the main mechanism observed for water dissociation on the NS surface at low coverage regimes involving one water molecule at 300 K. Panels (a), (b), and (c) show the evolution of the reaction of the water marked in green (see text). Panel (d) shows the evolution of the main bond distances during the reaction as a function of the time. | ||
In contrast, when the amount of water around the NS is larger (>500 molecules), we observe a different mechanism involving two water molecules, as described in Fig. 6. Panel (a) shows two water molecules (marked in orange and green) interacting with the surface. The ‘green’ water molecule is forming a H-bond of ∼1.5 Å with an O2c site (red line in the distance graph of Fig. 6d), while the ‘orange’ water is molecularly adsorbed on a Ti site (Ti–O bond is 2.3 Å, black line in Fig. 6d) and is H-bonded to the ‘green’ water with a H-bond distance of about 1.7 Å (blue line in Fig. 6d). From panel (b) to (c), two proton transfer reactions can be observed: the first one from the water molecules marked in green to the O2c site forming a bridging OH and the second one from the ‘orange’ to the ‘green’ water molecule, which form a terminal OH and a new water molecule. In this way, we observe that the molecule marked in green acts both as a donor of a proton towards an O2c site and as an acceptor of another proton from another water molecule (orange), which then binds to a Ti atom forming a terminal OH on the surface. In other words, the proton transfer proceeds through a Grotthuss-like mechanism,55 with a transient hydronium ion formed during the reaction. A similar mechanism was recently shown for water on an anatase (101) TiO2 surface using molecular dynamics with an ab initio-based deep neural network potential16 and also for different rutile faces by ab initio molecular dynamics (AIMD).56–58 We also observe reactions where several protons transfer take place to end up with two OH groups on the NS surface, as can be seen in Fig. S4 in the ESI.† Other authors have found that ReaxFF can correctly reproduce DFT reaction mechanisms on the TiO2 surface for other types of reactions.45
 | ||
| Fig. 6 Details of the main mechanism observed for water dissociation on the NS surface at high coverage regimes involving two water molecules at 300 K. Panels (a), (b), and (c) show the evolution of the reaction of the water molecules marked in green and orange (see text). The orange water acts as an acceptor and a donor of a proton. Panel (d) shows the evolution of the main bond distances during the reaction as a function of the time. | ||
The change in the mechanism going from low to high water coverage around the nanoparticle just discussed can also explain the different shapes in the curves of Fig. 4a. While for a higher number of water molecules around the NS (500 to 8958), the reaction of water dissociation is assisted by a second water molecule as a donor/acceptor moiety, at low water coverage (150 to 250 water molecules), the mechanism taking place just involves one water molecule whose dissociation is not assisted, changing the shape of the curve going from low to high water concentrations.
3.3 Electrostatic properties of the hydrated spherical TiO2 nanoparticle (2.2 nm)
In the previous section, we have observed some water chemistry on the surface of the spherical TiO2 nanoparticle, which we expect to cause charge redistribution at the TiO2/water interface and, consequently, variations in the resulting electric field and electrostatic potential. In this section, we will discuss these effects for the case of the nanoparticle of 2.2 nm diameter size.First, we have calculated the charge distribution profile (σ(r)) from the center of the NP across the water layers (shown in Fig. 7a), according to the H and O atomic distributions (Fig. S5†) and computed charge average, which are obtained from the simulations (it is important to note that in ReaxFF-MD calculations, the charges are not fixed but change during the simulation). In the plot of Fig. 7a, we observe an oscillation of the charge density in the region close to the surface up to 7 Å (18 Å from the center of the NP), but above that threshold, the charge density gets very close to zero, especially when we move towards the water bulk region. Similar charge oscillations in the region close to the surface have been previously observed for anatase flat surfaces, generating an electric double layer at the interface.59,60
 | ||
| Fig. 7 Calculated profiles of (a) charge distribution σ(r), (b) electric field E(r) and (c) electrostatic potential φ(r), for anatase 2.2 nm NP water interface. The distance is measured from the NP center. | ||
As a further step, we obtained the intrinsic electric field profile, E(r), through the integration according to the Poisson's equation:
 | (2) |
We performed a numerical integration from the middle of the bulk water region (rw) to the TiO2 surface with the boundary condition of E(rw) = 0. In Fig. 7b, we report the resulting electric field. In the region close to the surface, the electric field changes from −0.42 to 0.55 V Å−1. These high magnitudes evidence the charge changes occurring in the interface region because of the adsorption of water (dissociated and undissociated) on the TiO2 surface atoms.
Finally, we calculated the electrostatic potential, φ(r), through the integration of the electrostatic potential according to the following equation:
 | (3) |
The potential curve on the NP is shown Fig. 7c. The potential drop (surface potential) across the interface is found to be 0.47 V. Previous theoretical reports with the COMPASS potential and the SPC/E water model give values of 0.14, 0.28 and 0.62 V for water on anatase TiO2 (001), (100) and (101) surfaces, respectively.59 With ReaxFF, a value of 0.24 V for water on the anatase (101) surface was reported.44 Since we expect a strong dependence on the force field used to perform the calculations, only direct comparison of ReaxFF data is meaningful. Thus, comparing the value for the anatase (101) and for the NP, i.e., 0.24 vs. 0.47 V, we observe an increasing effect of the surface curvature on the potential drop. A recent experimental work reports a value of 0.18 V of surface potential for an amorphous TiO2 with 100 nm diameter immersed in water.61 The difference with respect to the computed value can be attributed to the different size and phase of the NP (100 nm vs. 2.2 nm and amorphous vs. anatase TiO2).
An interesting parameter to extract from the simulations is the ζ-potential, which is defined as the difference in the electrostatic potential at the shear plane (the “slipping plane”) between relatively immobile and mobile layers of water adjacent to the solid surface and can be measured experimentally, for example by electrophoresis.62 The unambiguous definition of the slipping plane (which separates surface-bound fluid from bulk) from simulations is nontrivial. Based on the distance distribution function (Fig. 2d), we observe a big effect of water on the surface up to distances of 7 Å. However, no sharp distinction between immobile and mobile layers can be rigorously made, except for the first adsorbed layers of water molecules. Beyond 7 Å from the surface (18 Å from the NP center), the oscillation of the electrostatic potential becomes close to 0 V in Fig. 7c. Experimentally, for anatase TiO2 NPs with a diameter below 20 nm, the ζ-potential at pH 7 is found to be around −20 mV (−0.02 V),62 which is within the oscillation range of our electrostatic potential plot. This prevents us from extracting a precise ζ-potential value from the plot.
Another clear fingerprint of a negative ζ-potential comes from the analysis of the total charge of the NP, which is found to be negative along all the simulation in water, as reported in Fig. S6,† in contrast to what is observed for the NP in vacuum, where the total charge is neutral along the whole simulation. These observations evidently agree with the experimentally observed negative ζ-potential value at pH 7 for TiO2 NPs.62
3.4 Increasing the size of the hydrated spherical TiO2 nanoparticles: from 2.2 nm to 3.0 and to 4.4 nm
In this section, we extended our analysis to more realistic NS models of diameter from 3.0 to 4.4 nm, which we surrounded by 1500 and 2500 water molecules, respectively, i.e. 6 ML of water (insets in Fig. 8). Based on the MD simulations data (500 ps), we extrapolated the distance distribution function g(d) for the two different NSs. In Fig. 8, we compare the g(d) distributions for the 2.2, 3.0 and 4.4 nm NSs. The behavior for the three models is very similar. Two peaks corresponding to dissociated (Ti–OH) and molecular water adsorption (Ti⋯OH2) are observed at 1.9 Å and 2.3 Å, respectively. Then, there is a third peak at 3.9 Å, related with the H-bond between the water molecules and surface O atoms or to Ti–OH groups. A tiny decrease in the peak at 4.5 Å (water molecules H-bonded to Ti–OH groups or Ti⋯OH2 through the Owater atom) is observed as the NS increases from 2.2 to 3.0 and to 4.4 nm. Finally, for all three NSs, at a distance between 6 and 7 Å, a small peak is observed before the density starts to slowly decrease. Therefore, we showed that the behavior of a water multilayer of a similar thickness (6ML) does not change with the increasing size of the NS from 2.2 to 4.4 nm.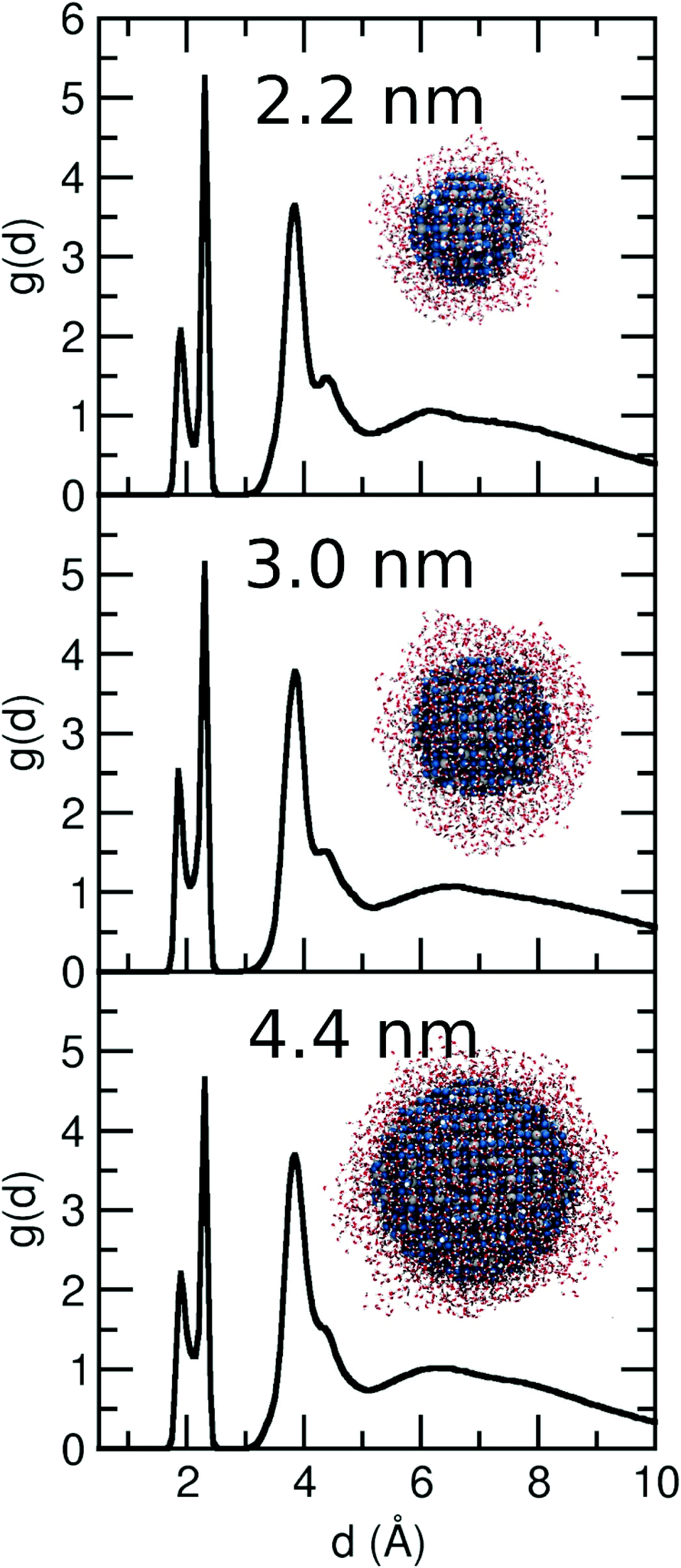 | ||
| Fig. 8 Distribution function g(d) as extracted from the ReaxFF molecular dynamics simulations for the 3.0 and 4.4 nm NS with 6 ML of water at 300 K. For comparison, we add the g(d) of the 2.2 nm NS shown in Fig. 2 for a 6 ML. The insets show the final structure after ReaxFF molecular dynamics simulation of 500 ps. The overall stoichiometry is (TiO2)399·1500H2O and (TiO2)1265·2400H2O in each case. | ||
In order to compare the capacity of water adsorption (molecular and dissociated) of each NS, we have integrated the first two peaks of the distribution function, which give an averaged number of molecules adsorbed by each undercoordinated Ti on the surface. The sum of the integrals for the 1st and 2nd peaks gives a value of 1.25, 1.35, and 1.20 for the 2.2, 3.0 and 4.4 nm NSs, respectively, suggesting a similar capacity of water adsorption of the three NSs.
In the case of the 3.0 nm NS, we also performed a comparative study of the mechanisms of water adsorption with respect to what is observed and discussed for the 2.2 nm NS in the previous section. Again, we have performed simulations varying the number of water molecules around the 3.0 nm NS. The amount of the terminal OH groups divide by the number of total Ti adsorption sites on the NS as a function of the time and the initial amount of water around the 3.0 nm NS is shown in Fig. 9a. An increasing number of total OH groups on the surface parallels a decreasing number of undissociated water molecules around the NS. As in the 2.2 nm NS, two different mechanisms emerge from the plot. Based on the shape of the curves, one mechanism is observed at low H2O concentrations (200 and 350 initial water molecules) and another at higher concentrations (750 and 1500 initial water molecules). As we explained before, the difference is due to the different reaction mechanism at different concentrations. While at low coverage, the main mechanism implies a single dissociation of a water molecule, at higher concentration, an assisted proton transfer reaction (involving at least two water molecules) is observed. For comparison with Fig. 4a, it can be seen that the fraction of Ti–OH groups on the surface at 500 ps for 3.0 nm NP is little lower to the 2.2 nm NP. Fig. 9b shows the final amount of the OH group as a function of initial water surrounding the 3.0 nm NS. Also, in this case, the data can be fitted with a Langmuir isotherm, which suggests an adsorption/desorption equilibrium in the system.54
 | ||
| Fig. 9 (a) Evolution of the amount of the terminal hydroxyl groups on the 3.0 nm NS normalized by the number of total Ti adsorption sites as a function of time for the simulations with different initial numbers of water molecules surrounding the NS. (b) Number of final hydroxyl groups on the NS after 500 ps of simulations as a function of the initial number of water molecules around the NS. The red line shows a fitting with a Langmuir isotherm. | ||
3.5 Water desorption under thermal annealing
In this section, we investigate the desorption process of the water multilayers around the NSs of different size. The goal of this part is assessing the ability of the ReaxFF to describe the desorption process of water from the NSs when the temperature of the whole system is increased. For that we have taken the final geometry of each NS surrounded by 6 ML of water as the starting point for the study of the desorption process at 500 K. We have run the simulation for 1 ns in order to desorb the largest possible portion of water molecules from the surface. Fig. 10 shows the final structure after 1 ns of simulation for each NS. Clearly, the resulting NSs are hydroxylated with some water molecules that are still undissociatively adsorbed after the thermal annealing. This is in agreement with previous experimental results by Matsumoto and coworkers, who have reported that water adsorbates on spherical TiO2 nanoparticles are very strongly bound to the surface and they cannot be completely eliminated even through an annealing process up to 700 K.28 We observed that these molecularly adsorbed molecules form two H-bonds with two different OH groups on the surface of the NS. This adsorption mode mostly takes place at the equatorial region of the NS. Also, we observed that the crystallinity of the NSs is kept after the annealing process. The final number of water molecules (undissociated and dissociated) is 128, 229 and 378 for the 2.2, 3.0 and 4.4 nm NS, respectively, i.e. an average of 1.06, 1.07 and 0.81 molecules per undercoordinated Ti on the surface, respectively. These values show a better water adsorption capacity of the smaller NSs after annealing, which is expected because in these NSs, the percentage of reactive sites on the surface is higher.49 For all the three NSs, we observe an increase in the diameter of about 0.4 nm, which is related with the water layer directly bonded to the surface. | ||
| Fig. 10 Ball representation of the ReaxFF structures after annealing for the three different nanospheres considered in this work. For each one, the stoichiometry and the approximate diameter are reported. Blue spheres represent the O atoms, grey spheres represent the Ti atoms and white small spheres represent the H atoms. | ||
3.6 Comparing curved vs. flat surfaces
Finally, we present a comparison of the reactivity of water on curved vs. flat surfaces. We consider the flat anatase (101) surface since it is the most stable anatase surface and has been extensively studied with respect to water reactivity both theoretically11–17,63 and experimentally.18–23,64 Much effort was devoted to the understanding of the reactivity of water on this surface, going from low coverage to the full monolayer to interfacing bulk water. A mixed molecular/dissociated water adsorption is the generally accepted mechanism on this surface. However, the degree of dissociation is still an open issue, since different types of experiments (from vacuum, few water monolayers, bulk water), or different surfaces (defect-free, reduced) or theoretical approaches (DFT, AIMD, DFTB, and ReaxFF) have been considered, leading to different results. Our goal in this part of the work is not to explain such differences but more simply to compare the capacity of water adsorption of this flat surface with respect to the curved surfaces of the NSs considered in the previous sections.The details of the model used to perform these calculations are reported in the Computational details above. Inside the periodic box, the bulk water density of 1 g cm−3 is reached by inserting 1000 water molecules. The final interface structure, after MD, is shown in Fig. 11a. After 500 ps of simulation at 300 K, we extracted the distribution ρ(Δz) of the vertical distances between the O atoms of the H2O molecules and the Ti5c plane of the surface, as shown the Fig. 11b. We observe four main peaks in the distribution (at 2.03 Å, ∼2.70 Å, ∼3.75 Å and a broad peak between 5 and 6.5 Å). For distances above 7 Å, a constant ρ(z) of 0.033 is observed, which is consistent with the bulk water density. A similar profile is observed on both sides of the slab (Fig. S7†). The position of peaks is comparable with that calculated for a trilayer of water on the same surface by Tilocca and Selloni12 with DFT MD simulations (∼2, ∼2.75, ∼4, and ∼5.5 Å) and by Fazio et al. with the DFTB method.17 This confirms a similar distribution of water on the surface as obtained by ReaxFF in comparison with the DFT-based method, in agreement with previous studies.41
 | ||
| Fig. 11 (a) Supercell model of the anatase (101) TiO2/water interface. (b) Vertical distribution of the distances between the water molecules (O atoms) and the surface (Ti5c). (c) Radial distribution function (g(r)) detecting interaction between anatase (101) surface atoms (Ti5c or O2c) and water (O or H atoms). | ||
In Fig. 11c, we present the radial distribution function for O of water (Ow) and T5c on the surface and the H of water (Hw) and O2c on the surface. For Ti5cOw, the first peak is observed between 1.8 Å and 2.5 Å, where three components centered at 1.92 Å, 2.16 Å, and 2.33 Å can be identified. A broader peak is observed between 3.5 Å and 4.8 Å in agreement with previous reports.14,16,44 For the O2cHw distribution, we observe a similar profile as the one for the three NSs above is observed, with three main peaks centered at 1.0, 1.6 and 3.0 Å, corresponding to the O–H bond, i.e. OH groups formed in O2c sites, the H-bond between water molecules and O2c surface atoms and the last peak indicating the long-distance interaction between H atoms of the second water layer with the surface O2c atoms. Regarding the first peak in the T5cOw distribution, a closer inspection shows that the first component at 1.92 Å corresponds to the Ti–OH bond, where the OH group forms only one H-bond to the surrounding water molecules. The peak component at 2.16 Å is also related to a Ti–OH moiety, but the OH group is forming two H-bonds with the water molecules of the second layer. Finally, a small component at 2.33 Å corresponds to Ti–OH2 indicating molecular adsorption. The formation of OH groups follows the same mechanism as that for the NS with a water molecule acting as an acceptor and a donor of a proton (Fig. 6). It is important to highlight an evident difference in the rdf plot of the Ti–Ow distribution, around the first peak, when comparing the curve for the anatase (101) slab and for the NP. We rationalize the difference in the rdf plots as due to the different types of surface Ti atoms present in the slab and NP models: while on the slab, only fivefold coordinated Ti5c atoms are present, on the NP surface, there are Ti5c, Ti4c, Ti3cOH and Ti4cOH. The rdf plot of the NP is the result of (dissociated or undissociated) water adsorption on all these types of superficial Ti atoms. The Ti4c and Ti3cOH can even bind up to two water molecules. Additionally, in general, the water adsorption energies on the NP are higher that on the slab. This produces a stronger interaction between the water molecules and the NP surface, causing a much slower water exchange between molecules at the interface and in the bulk, resulting in sharper peaks in the rdf around the NP than over the slab.
From the fragment analysis and the peaks of the rdf, we infer the formation of 102 total OH groups of which a fraction of 0.45 are terminal Ti–OH and the 0.55 are bridging OH (see Fig. S8†). This indicates that a degree 0.72 of the Ti5c is bonded to OH groups. These results are in agreement with previous ReaxFF calculations,44 but the extent of dissociation (0.72) is much higher in comparison with that obtained for the NPs (∼0.4) which is not expected due to the well-known higher reactivity of the curved surfaces with respect to flat ones. Previous DFT calculations15 and X-ray photoelectron spectroscopy experiments60 indicate that a mixed dissociated and undissociated water adsorption takes place, in line with what we found by ReaxFF calculations. However, the relative extent of dissociated water reported is 0.25. Based on a molecular dynamics study using an ab initio-based deep neural network potential, other authors observe only a 0.056 fraction of dissociated water after 25 ns of simulation time.16 We may conclude that the exact degree of water dissociation on the anatase (101) surface is still under debate, but seems to be lower than what we found here with ReaxFF. This discrepancy is probably due to a different evaluation of the relative energy gain associated with molecular/dissociated water adsorption on the anatase (101) surface, as detailed in Table S1.† Molecular adsorption is preferred in ReaxFF calculations in agreement with DFT ones; however, the difference with respect to the dissociated mode is smaller than that with DFT methods. A similar effect was observed previously, where ReaxFF overestimated the reactivity of H2 on the anatase (101) surface favoring the dissociative adsorption with respect to what was found with DFT.45
We wish to recall that for curved surfaces, due to their overall higher reactivity, a general better agreement between ReaxFF and DFT results has been reported and discussed above in sections 3.1.2 and 3.2.1. This shows that for the more reactive systems such as spherical TiO2 NP (or for instance, reduced flat TiO2 surfaces), the force field gives more satisfactory results than for systems with a lower reactivity. This is a valuable information for future investigations.
However, as we aforementioned, the goal here is the comparison of the overall capacity of water adsorption (molecular + dissociated) between flat and curved (NS) surfaces. The integral of the first peak in the T5cOw distribution for the anatase (101) model gives an average of water molecules adsorbed on undercoordinated Ti of 0.8. This value is lower than those calculated for the NSs, which are in the range between 1.20 and 1.35, indicating a higher water adsorption capacity of curved surfaces in comparison with the flat ones. This is probably because curved surfaces in NSs present more reactive low coordinated sites (Ti5c, Ti4c, Ti4c(OH), Ti3c(OH)) and defects, which can, in average, bind more water molecules, thus increasing the whole capacity of water adsorption.
4. Conclusions
In this work, we have presented a ReaxFF molecular dynamics investigation of realistic spherical anatase TiO2 nanoparticles (NSs) of 2.2, 3.0 and 4.4 nm of diameter immersed in different models of aqueous environments.First, we have focused the attention on the validation of the existing force field, which has been parametrized for TiO2 bulk and flat surfaces, for its application to curved surfaces and water interacting with the curved surfaces. For that we adsorbed an increasing number of water molecules going from a single isolated one up to water multilayers around the 2.2 nm NS, and compared the results with previous QM and QM/MM calculations.26 We found a very good agreement both for the molecular binding energy on the different sites of the NS and for the global behavior of multilayers surrounding the NS, which can be assessed by the position of the peaks in the distance distribution function g(d) of the water O atoms from the surface Ti atoms.
Then, we have analyzed the effect on the water adsorption mechanism of increasing the thickness of the water multilayers around both the 2.2 and the 3.0 nm NSs. We have observed that an increase in the number of OH species on the NS surface is accompanied by a decrease in the resulting total number of undissociated water molecules. This behavior, i.e. the final number of total OH species as a function of the initial water molecules, can be fitted with a Langmuir isotherm indicating an adsorption/desorption mechanism. A closer inspection of the simulations has proved that the formation of OH on the surfaces depends on the water concentration. At lower water coverage, the direct dissociation of one water molecule is observed, whereas at high water coverage, an assisted proton transfer mechanism governs the OH formation on the NS surface, in agreement with higher level calculations.16 The same conclusions hold for both the 2.2 and the 3.0 nm NSs.
The analysis of the charge density around the NP has indicated the formation of an electric layer in the interface NP/water with an oscillating profile of the electric potential up 7 Å from the NP surface. We have also observed that the originally neutral NP in a vacuum becomes negatively charged when immersed in water, which agrees with the experimentally reported negative ζ-potential at a pH of 7.
When changing the diameter size of the NSs from 2.2 to 3.0 to 4.4 nm, we did not observe a big difference in the capacity of water adsorption (which is in the range between 1.2 and 1.35 water molecules per undercoordinated surface Ti atom), considering both molecular and dissociated water.
We have also performed the thermal annealing of the nanoparticle/water systems (NS + 6ML) at 500 K for 1 ns, to simulate a desorption experiment. We have found that several water molecules (both dissociated and undissociated) remain bound to the NS surface, in agreement with what has been experimentally reported.28 The capacity of adsorption is higher for the 2.2 and 3.0 nm NSs (∼1 water molecule by undercoordinated Ti atom) than for the 4.4 nm NS (0.8 water molecule by undercoordinated Ti atom).
Finally, we have compared the behavior of the anatase NS curved surfaces with that of a flat anatase (101) surface. First, we have confirmed a similar water vertical distribution to that reported by previous ReaxFF41 and ab initio MD studies,14 and a radial distribution function in agreement with a previous ReaxFF simulation for water on the anatase (101) surface.44 Also, the percentage extent of dissociation, around 72%, is close to what was previously reported with ReaxFF;41,44 however, it is much higher than 5.6% that was recently obtained through a molecular dynamics study with an ab initio-based deep neural network potential,16 probably due to a different estimation of the relative adsorption energy for the molecular/dissociated adsorption modes. The capacity of water adsorption (undissociated + dissociated) of the flat anatase (101) surface is found to be lower than that registered in this work for the nanospheres. On average, the flat surface adsorbs 0.8 water molecules per Ti5c site, while for the NSs, this average is between 1.2 and 1.35. Clearly, defects, steps and more undercoordinated Ti atoms in the NS improve the water adsorption.
In conclusion, our present work with the ReaxFF method has allowed (1) the investigation of the water interface with TiO2 nanoparticles of realistic size (up to 4.4 nm), (2) the determination of the role played by the thickness of the water multilayer on the extent of dissociation at the curved surface, and (3) the identification of the mechanisms of dissociation at different water coverages. In particular, the ability of ReaxFF to correctly reproduce the reaction mechanisms, as observed before through higher level DFT-based calculations, is an extremely important result since it proves the reliability of this approximate and computational affordable method for the investigation of other chemical processes involving proton transfer reactions at the metal oxide surface in an aqueous environment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Prof. Annabella Selloni, Dr Gianluca Fazio and Dr Paulo Siani for fruitful discussions. The project has received funding from the European Research Council (ERC) under the European Union's HORIZON2020 research and innovation programme (ERC Grant Agreement No 647020).References
- M. T. Noman, M. A. Ashraf and A. Ali, Environ. Sci. Pollut. Res., 2019, 26, 3262–3291 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS.
- M. Bhogaita, S. Yadav, A. U. Bhanushali, A. A. Parsola and R. Pratibha Nalini, Mater. Today: Proc., 2016, 3, 2052–2061 Search PubMed.
- Y. Bai, I. Mora-Seró, F. De Angelis, J. Bisquert and P. Wang, Chem. Rev., 2014, 114, 10095–10130 CrossRef CAS.
- J. Bouclé and J. Ackermann, Polym. Int., 2012, 61, 355–373 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS.
- M. Kapilashrami, Y. Zhang, Y.-S. Liu, A. Hagfeldt and J. Guo, Chem. Rev., 2014, 114, 9662–9707 CrossRef CAS.
- S. Shen, J. Chen, M. Wang, X. Sheng, X. Chen, X. Feng and S. S. Mao, Prog. Mater. Sci., 2018, 98, 299–385 CrossRef CAS.
- T. Rajh, N. M. Dimitrijevic, M. Bissonnette, T. Koritarov and V. Konda, Chem. Rev., 2014, 114, 10177–10216 CrossRef CAS.
- A. Vittadini, A. Selloni, F. P. Rotzinger and M. Grätzel, Phys. Rev. Lett., 1998, 81, 2954–2957 CrossRef CAS.
- A. Tilocca and A. Selloni, J. Phys. Chem. B, 2004, 108, 4743–4751 CrossRef CAS.
- A. Tilocca and A. Selloni, Langmuir, 2004, 20, 8379–8384 CrossRef CAS.
- M. Sumita, C. Hu and Y. Tateyama, J. Phys. Chem. C, 2010, 114, 18529–18537 CrossRef CAS.
- R. Martinez-Casado, G. Mallia, N. M. Harrison and R. Pérez, J. Phys. Chem. C, 2018, 122, 20736–20744 CrossRef CAS.
- M. F. Calegari Andrade, H.-Y. Ko, L. Zhang, R. Car and A. Selloni, Chem. Sci., 2020, 11, 2335–2341 RSC.
- D. Selli, G. Fazio, G. Seifert and C. Di Valentin, J. Chem. Theory Comput., 2017, 13, 3862–3873 CrossRef CAS.
- M. J. Jackman, A. G. Thomas and C. Muryn, J. Phys. Chem. C, 2015, 119, 13682–13690 CrossRef CAS.
- I. M. Nadeem, G. T. Harrison, A. Wilson, C. L. Pang, J. Zegenhagen and G. Thornton, J. Phys. Chem. B, 2018, 122, 834–839 CrossRef.
- I. M. Nadeem, J. P. W. Treacy, S. Selcuk, X. Torrelles, H. Hussain, A. Wilson, D. C. Grinter, G. Cabailh, O. Bikondoa, C. Nicklin, A. Selloni, J. Zegenhagen, R. Lindsay and G. Thornton, J. Phys. Chem. Lett., 2018, 9, 3131–3136 CrossRef CAS.
- S. Hosseinpour, F. Tang, F. Wang, R. A. Livingstone, S. J. Schlegel, T. Ohto, M. Bonn, Y. Nagata and E. H. G. Backus, J. Phys. Chem. Lett., 2017, 8, 2195–2199 CrossRef CAS.
- M. F. Calegari Andrade, H.-Y. Ko, R. Car and A. Selloni, J. Phys. Chem. Lett., 2018, 9, 6716–6721 CrossRef CAS.
- W. Yuan, B. Zhu, X.-Y. Li, T. W. Hansen, Y. Ou, K. Fang, H. Yang, Z. Zhang, J. B. Wagner, Y. Gao and Y. Wang, Science, 2020, 367, 428 CrossRef CAS.
- F. Orlando, L. Artiglia, H. Yang, X. Kong, K. Roy, A. Waldner, S. Chen, T. Bartels-Rausch and M. Ammann, J. Phys. Chem. Lett., 2019, 10, 7433–7438 CrossRef CAS.
- H. Ali, R. Seidel, A. Bergmann and B. Winter, J. Mater. Chem. A, 2019, 7, 6665–6675 RSC.
- G. Fazio, D. Selli, L. Ferraro, G. Seifert and C. Di Valentin, ACS Appl. Mater. Interfaces, 2018, 10, 29943–29953 CrossRef CAS.
- J. Balajka, U. Aschauer, S. F. L. Mertens, A. Selloni, M. Schmid and U. Diebold, J. Phys. Chem. C, 2017, 121, 26424–26431 CrossRef CAS.
- K. Shirai, T. Sugimoto, K. Watanabe, M. Haruta, H. Kurata and Y. Matsumoto, Nano Lett., 2016, 16, 1323–1327 CrossRef CAS.
- K. Shirai, G. Fazio, T. Sugimoto, D. Selli, L. Ferraro, K. Watanabe, M. Haruta, B. Ohtani, H. Kurata, C. Di Valentin and Y. Matsumoto, J. Am. Chem. Soc., 2018, 140, 1415–1422 CrossRef CAS.
- A. Nilsson and L. G. M. Pettersson, Nat. Commun., 2015, 6, 8998 CrossRef CAS.
- F. De Angelis, C. Di Valentin, S. Fantacci, A. Vittadini and A. Selloni, Chem. Rev., 2014, 114, 9708–9753 CrossRef CAS.
- V. N. Koparde and P. T. Cummings, J. Phys. Chem. C, 2007, 111, 6920–6926 CrossRef CAS.
- R. S. Kavathekar, P. Dev, N. J. English and J. M. D. Macelroy, Mol. Phys., 2011, 109, 1649–1656 CrossRef CAS.
- A. C. T. Van Duin, S. Dasgupta, F. Lorant and W. A. Goddard, J. Phys. Chem. A, 2001, 105, 9396–9409 CrossRef CAS.
- K. Chenoweth, A. C. T. Van Duin and W. A. Goddard, J. Phys. Chem. A, 2008, 112, 1040–1053 CrossRef CAS.
- T. P. Senftle, S. Hong, M. M. Islam, S. B. Kylasa, Y. Zheng, Y. K. Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik, H. M. Aktulga, T. Verstraelen, A. Grama and A. C. T. Van Duin, npj Comput. Mater., 2016, 2, 15011 CrossRef CAS.
- F. A. Soria, W. Zhang, P. A. Paredes-Olivera, A. C. T. Van Duin and E. M. Patrito, J. Phys. Chem. C, 2018, 122, 23515–23527 CrossRef CAS.
- M. M. Islam, A. Ostadhossein, O. Borodin, A. T. Yeates, W. W. Tipton, R. G. Hennig, N. Kumar and A. C. T. Van Duin, Phys. Chem. Chem. Phys., 2015, 17, 3383–3393 RSC.
- M. Y. Sengul, C. A. Randall and A. C. T. Van Duin, ACS Appl. Mater. Interfaces, 2018, 10, 37717–37724 CrossRef CAS.
- S.-Y. Kim, N. Kumar, P. Persson, J. Sofo, A. C. T. Van Duin and J. D. Kubicki, Langmuir, 2013, 29, 7838–7846 CrossRef CAS.
- M. Raju, S.-Y. Kim, A. C. T. Van Duin and K. A. Fichthorn, J. Phys. Chem. C, 2013, 117, 10558–10572 CrossRef CAS.
- M. Raju, A. C. T. Van Duin and K. A. Fichthorn, Nano Lett., 2014, 14, 1836–1842 CrossRef CAS.
- L. Huang, K. E. Gubbins, L. Li and X. Lu, Langmuir, 2014, 30, 14832–14840 CrossRef CAS.
- Z. Futera and N. J. English, J. Phys. Chem. C, 2017, 121, 6701–6711 CrossRef CAS.
- S. Selcuk, X. Zhao and A. Selloni, Nat. Mater., 2018, 17, 923–928 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- H. M. Aktulga, J. C. Fogarty, S. A. Pandit and A. Y. Grama, Parallel Comput., 2012, 38, 245–259 CrossRef.
- G. Fazio, L. Ferrighi and C. Di Valentin, J. Phys. Chem. C, 2015, 119, 20735–20746 CrossRef CAS.
- D. Selli, G. Fazio and C. Di Valentin, J. Chem. Phys., 2017, 147, 164701 CrossRef.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef.
- S. Monti, J. Jose, A. Sahajan, N. Kalarikkal and S. Thomas, Phys. Chem. Chem. Phys., 2019, 21, 13099–13108 RSC.
- S. Benkoula, O. Sublemontier, M. Patanen, C. Nicolas, F. Sirotti, A. Naitabdi, F. Gaie-Levrel, E. Antonsson, D. Aureau, F.-X. Ouf, S.-I. Wada, A. Etcheberry, K. Ueda and C. Miron, Sci. Rep., 2015, 5, 15088 CrossRef CAS.
- E. G. Brandt, L. Agosta and A. P. Lyubartsev, Nanoscale, 2016, 8, 13385–13398 RSC.
- R. I. Masel, Principles of Adsorption and Reaction on Solid Surfaces, Wiley, 1996 Search PubMed.
- D. Marx, ChemPhysChem, 2006, 7, 1848–1870 CrossRef CAS.
- T. Zheng, C. Wu, M. Chen, Y. Zhang and P. T. Cummings, J. Chem. Phys., 2016, 145, 044702 CrossRef.
- L. Agosta, E. G. Brandt and A. P. Lyubartsev, J. Chem. Phys., 2017, 147, 024704 CrossRef.
- Z.-P. H. Hui-Li Wang and H. Li, Front. Phys., 2018, 13, 138107 CrossRef.
- L. Sang, Y. Zhang, J. Wang, Y. Zhaoa and Y.-t. Chen, Phys. Chem. Chem. Phys., 2016, 18, 15427 RSC.
- L. Lei, L. Sang, Y. Zhang and Y. Gao, ACS Omega, 2020, 5, 3522–3532 CrossRef CAS.
- M. Bischof, D. Biriukov, M. Předota, S. Roke and A. Marchioro, J. Phys. Chem. C, 2020, 124, 10961–10974 CrossRef.
- K. Suttiponparnit, J. Jiang, M. Sahu, S. Suvachittanont, T. Charinpanitkul and P. Biswas, Nanoscale Res. Lett., 2011, 6, 27 Search PubMed.
- C. E. Patrick and F. Giustino, Phys. Rev. Appl., 2014, 2, 014001 CrossRef.
- L. E. Walle, A. Borg, E. M. J. Johansson, S. Plogmaker, H. Rensmo, P. Uvdal and A. Sandell, J. Phys. Chem. C, 2011, 115, 9545–9550 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr07503e |
| This journal is © The Royal Society of Chemistry 2021 |