
Advanced nitric oxide donors: chemical structure of NO drugs, NO nanomedicines and biomedical applications
Yueqi
Yang
ab,
Zhangjian
Huang
*a and
Li-Li
Li
 *b
*b
aState Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, Nanjing 210009, P. R. China. E-mail: zhangjianhuang@cpu.edu.cn
bLaboratory for Biomedical Effects of Nanomaterials and Nanosafety, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology (NCNST), No. 11 Beiyitiao, Zhongguancun, Beijing, 100190, P. R. China. E-mail: lill@nanoctr.cn
First published on 8th December 2020
Abstract
Nitric oxide (NO), as an endogenous diatomic molecule, plays a key regulatory role in many physiological and pathological processes. This diatomic free radical has been shown to affect different physiological and cellular functions and participates in many regulatory functions ranging from changing the cardiovascular system to regulating neuronal functions. Thus, NO gas therapy as an emerging and promising treatment method has attracted increasing attention in the treatment of various pathological diseases. As is known, the physiological and pathological regulation of NO depends mainly on its location, exposure time and released dosage. However, NO gas lacks effective accumulation and controlled long-term gas releasing capacity at specific sites, resulting in limited therapeutic efficacy and potential side effects. Thus, researchers have developed various NO donors, but eventually found that it is still difficult to control the long-term release of NO. Inspired by the self-assembly properties of nanomaterials, researchers have realized that nanomaterials can be used to support NO donors to form nanomedicine to achieve spatial and temporal controlled release of NO. In this review, according to the history of the medicinal development of NO, we first summarize the chemical design of NO donors, NO prodrugs, and NO-conjugated drugs. Then, NO nanomedicines formed by various nanomaterials and NO donors depending on nanotechnology are highlighted. Finally, the biomedical applications of NO nanomedicine with optimized properties are summarized.
1. Introduction
The discovery of nitric oxide (NO) in mammals in the 1980s was a major advance in the history of life sciences. Since then, among many signal molecules, NO as a type of colorless, odorless gasotransmitter and a highly active free radical molecule has attracted increasing attention. NO can be synthesized via three different NO synthases (NOS) in the body, including neuronal NO synthase (nNOS) expressed in neuronal cells, endothelial NO synthase (eNOS) expressed in endothelial cells and inducible NO synthase (iNOS). The low levels of NO produced by nNOS and eNOS directly interact with specific molecules, such as metals, lipid free radicals and DNA free radicals to regulate physiological function. In the presence of iNOS, the production of NO is much greater and indirect effects such as nitrosation, nitration, and oxidation reactions occur.1 In summary, NO can inhibit enzyme function, but it also has antioxidant properties and the ability to protect cells from cytokine-induced damage and apoptosis. Moreover, NO can react with other free radicals (such as superoxide and lipid-derived free radicals) to form products such as ONOO−, which can further react with other biological targets. Therefore, this diatomic free radical has been shown to affect different physiological and cellular functions and to participate in many regulatory functions ranging from changing the cardiovascular system to regulating neuronal function.2,3 With further research, it has been found that NO plays an important role in various pathophysiological diseases, such as arthritis, atherosclerosis, cancer, diabetes, various degenerative neuronal diseases, stroke and myocardial infarction. In summary, NO is an extremely important endogenous mammalian bioregulator in the human body, and it plays an extremely important physiological role in the biological system.4,5The duration of NO exposure time and the kinetic behavior of NO are key determinants in its biological applications. However, therapeutic NO gases have an extremely short half-life, aimlessly diffuse everywhere, and are hard to effectively accumulate in target tissues, leading to limited NO gas therapy efficacy.6 Consequently, problems with spatial and temporal NO generation can lead to NO failing to achieve the desired therapeutic effect and may even cause adverse effects due to a large amount of enrichment in a specific space.2 Thus, to solve these problems, researchers have developed many NO donors and prodrugs. For example, O2-derived diazeniumdiolates can be activated in response to specific conditions. However, the emergence of these small chemical compounds still cannot solve the long-term sustained release of NO.
The size of nanomaterials on the nanoscale is similar to the size of basic biological molecules such as DNA.7 Nanomedicine therapy usually consists of therapeutic entities such as small molecule drugs, gas drugs, peptides, proteins, and nucleic acids, and then components that are assembled with the therapeutic entities such as lipids and polymers to form nanomedicines. Nanomedicines have the characteristics of small particles, large specific surface area, high surface reactivity, many active centers and strong adsorption capacity. Their applications range from drug delivery to biomedical imaging.8,9 Currently, due to the rapid development of science, about 20 drugs based on nanomedicines have been developed.10 The self-assembly of nanomaterials can realize the encapsulation, covalent attachment, adsorption and co-assembly of NO donors. Consequently, the nanomaterial can reach the target site as a carrier of NO, achieving the controlled and sustained release of NO, extending the half-life of NO gas drugs, and reducing the harmful side effects of NO on normal tissues.11 Furthermore, the use of nano-platforms can easily achieve synergistic treatment using NO and other strategies (such as traditional chemical drugs and photothermal therapy). Because of these excellent properties of NO nanomedicine, it is widely used in various physiological and pathological diseases, including cancer, infection, hyperglycemia, glaucoma, and vasodilation.
In this review, the chemistry section mainly introduces common NO donors, NO prodrugs, and antibodies or peptides conjugated with NO drugs. In the materials section, the different ways of achieving NO donor loading with different nanomaterials are mainly introduced. Finally, the common biomedical applications of NO nanomedicine are summarized.
2. NO drugs
2.1 NO donors
Organic nitrates are the earliest discovered NO donor drugs. They are classic vasodilators, including organic nitrates, organic nitrites, and nitrite thiol esters, among which organic nitrates are the most studied. Early organic nitrate drugs include nitroglycerin, erythrityltetranitrate, and isosorbidedinitrate. These drugs are quickly absorbed through the oral cavity and have apparent anti-angina pectoris effects (Fig. 1).12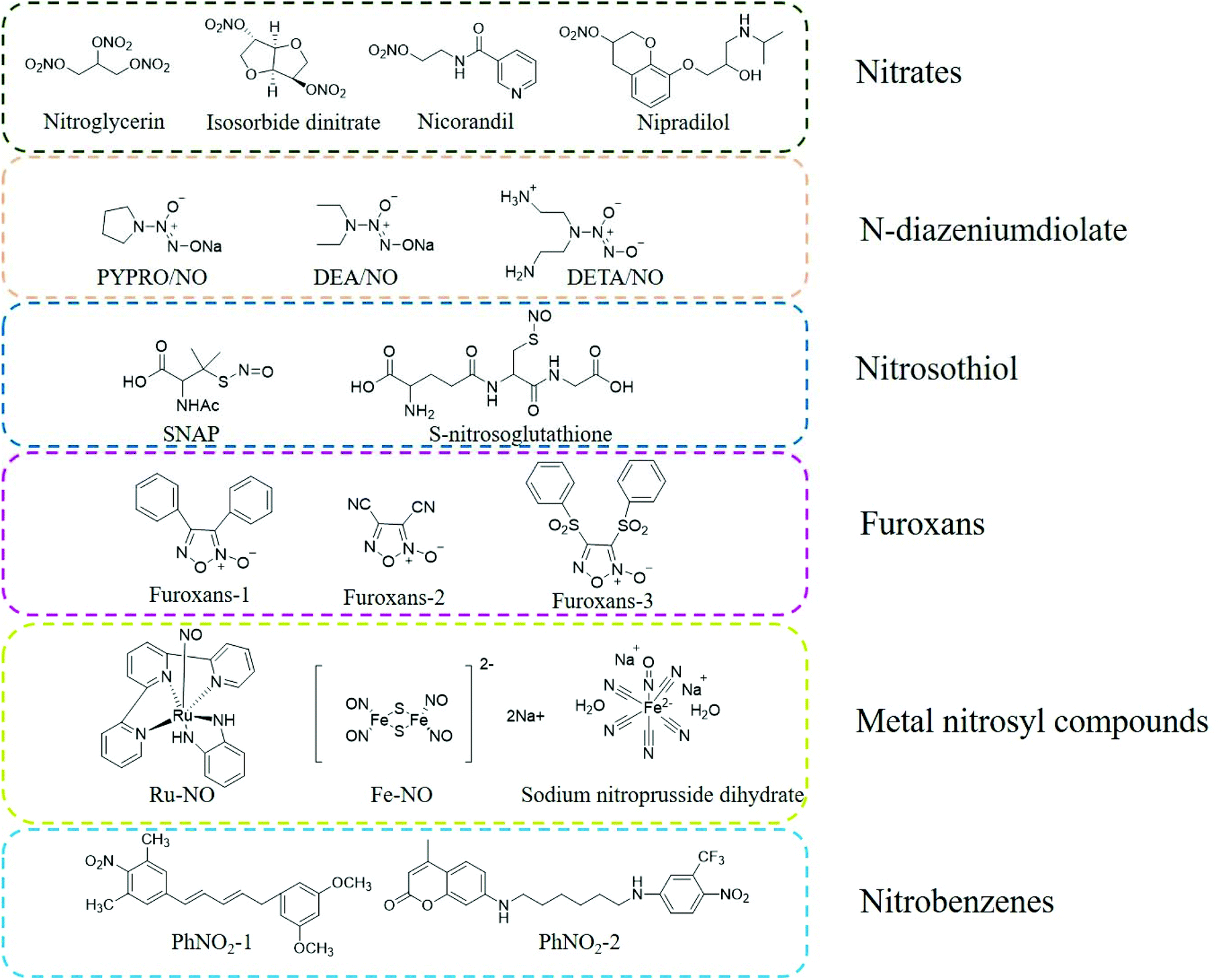 | ||
| Fig. 1 Structure of traditional NO donors. | ||
N-Diazeniumdiolates (NONOates) are formed by secondary amines and electrophilic adducts under high pressure in the presence of methanol and sodium methoxide. The half-life of NO released by NONOates ranges from a few seconds to several weeks. The parent structure of NONOates can determine the specific NO release kinetics. Besides, it is also possible to conjugate enzyme or metabolite response groups on the NONOate molecule to regulate and control the release rate of NO by a specific enzyme or metabolite. Under physiological conditions, one molecule of NONOate can be hydrolyzed to produce two molecules of NO, and the release of NO does not require unique metabolism or redox reactions, thus this type of NO donor can be conveniently used in biomedical research (Fig. 1).13,14
Compared with NONOates, nitrosothiols (RSNO) cannot release NO spontaneously but can decompose to produce NO under certain conditions. Nitrosothiols can be stimulated to release NO by transition metals (such as Cu+), ascorbic acid, ultraviolet light, heat, and enzymes (superoxide dismutase and protein disulfide isomerase). After nitrosothiol is affected by abovementioned stimuli, its S-NO bond is cleaved to generate NO and the corresponding disulfide bond, which promotes the release of NO (Fig. 1).14–16
Nitrobenzenes are the most representative organic donors for the light-controlled release of NO. These nitrobenzene derivatives are usually sterically hindered substituent groups (such as CF3, methyl, and aromatic hydrocarbons) at the ortho position. The steric hindrance of the large ortho-substituent groups results in the distortion of the spatial structure of the benzene ring. The nitro group of the aromatic ring is perpendicular to the aromatic ring plane. Under light irradiation, the nitro groups are converted into nitroso groups through light rearrangement, which induces the cleavage of oxygen and nitrogen bonds (O-NO2) in the nitroso groups to generate NO. Under physiological conditions, nitrobenzene derivatives are thermodynamically stable and undergo the light-controlled release of NO (Fig. 1).14,17
NO is a ligand with strong coordination ability with metal ions, which is significantly stronger than that with other gas molecules, such as O2 and CO. Metal nitrosyl compounds are a type of photosensitive NO donor. Under light irradiation, a photoelectron is promoted from the π orbital of the metal ion to the π anti-bond orbital of NO, resulting in the rapid release of NO from M-NO. The wavelength range of the light required to induce the release of NO can be adjusted by changing the structure of the M-NO ligand or the metal center such as iron (Fe), manganese (Mn), ruthenium (Ru), and chromium (Cr). At present, the most commonly used metallic NO donors are nitrosyl compounds with Ru and Fe as the metal center. Metal Ru nitrosyl compounds are more stable than other metal nitrosyl compounds and are more suitable for use under physiological conditions (Fig. 1).18–20
Furoxans are relatively stable to acids and bases. The 3 and 4 positions of their can be connected to the same or different groups and can also be combined with other rings (such as benzofurazan) to form different derivatives. After they enter the body, they can release NO under the action of sulfhydryl compounds (RSH, such as cysteine) (Fig. 1).21
2.2 NO donor-conjugated chemical drug
Dr Huang et al. designed and synthesized NO-donor OA derivatives (NO-OA) to study their protective effects on the liver and possible anti-liver tumor effects. The test results showed that due to the different NO donors and linking groups used, NO-OA differs in the release position and amount of NO, thus exhibiting different biological activities. Some NO-OA released a small amount of NO in liver cells, inhibiting cell apoptosis and protecting the liver. Other NO-OA selectively induced tumor cell apoptosis without harming normal liver cells, showing specific liver tumor cytotoxicity (Fig. 2A).22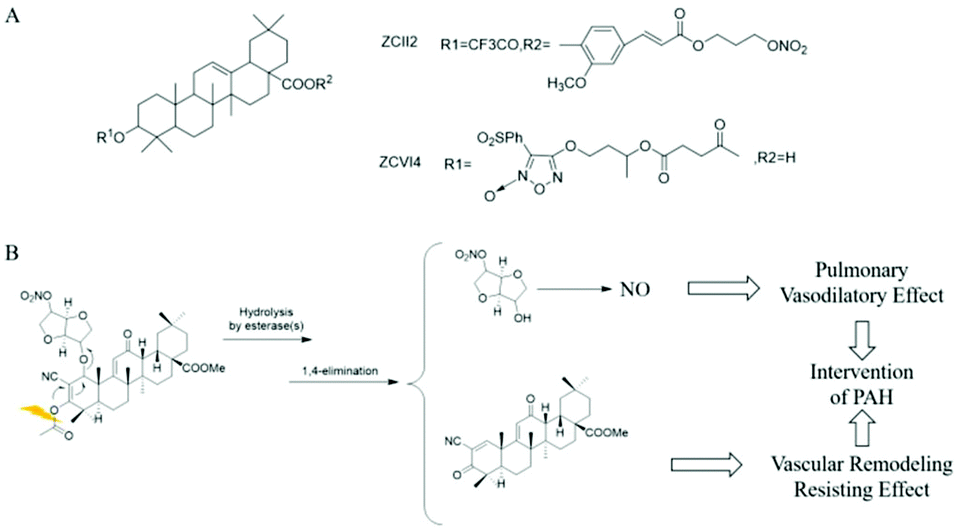 | ||
| Fig. 2 NO donor-conjugated chemical drug. A. NO donor type OA biology (NO-OA). B. Mechanism of action of CDDO-NO. | ||
Pulmonary hypertension (PAH) is a type of malignant progressive disease that increases pulmonary vascular resistance and pulmonary artery pressure, leading to heart failure and even death. Dr Huang et al. proposed that it may be better to treat PAH while inhibiting pulmonary vascular remodeling and dilating pulmonary arteries. The self-developed α-cyano-α,β-unsaturated ketone prodrug strategy was used to treat PAH in phase III trials. The drug CDDO-Me and the NO donor drug isosorbide mononitrate (ISMN) were coupled to obtain the compound CDDO-NO. Studies have found that after administering aerosol inhalation, CDDO-NO is mainly hydrolyzed in the lungs into CDDO-Me and ISMN. The former produces anti-inflammatory, anti-oxidant, and inhibiting pulmonary artery remodeling effects, while the latter slowly releases a small amount of NO to produce the activity of relaxing the pulmonary artery. The two synergistically exert a significant anti-PAH effect (Fig. 2B).23
2.3 Targeted NO prodrugs
Prodrug strategies have been very effective in solving many drug delivery problems. A key issue in the development of prodrugs is the need to balance stability while being able to transform into the original drug under physiological conditions instantly. Researchers have a long-term interest in developing prodrugs. One of the current challenges is to develop prodrugs for gas drugs including NO, hydrogen sulfide, and carbon monoxide (CO) because these gas messengers are key signaling molecules and have great prospects.24The use of specific enzymes to activate NO prodrugs can release NO at specific sites, greatly reducing its side effects. In terms of enzyme-induced NO prodrugs, Dr Keefer et al. have made great contributions in this field. Prodrugs of NO released after activation by specific enzymes have been reported to be produced by cytochrome enzymes (compound 1),25 esterases (compound 2),26 oxidoreductases (compound 3),27 reductases (compound 4)28 and glycosidases (compound 5)29,30 trigger-related work. In addition, Dr Kelso et al. designed and synthesized a cephalosporin 3′-azonium NO donor prodrug (compound 6) for the β-lactamase produced by bacterial resistance. The unique β-lactamase was activated and released NO after an elimination reaction, causing the bacterial biofilm to be dispersed and further enabling bacteria that have lost the biofilm protection to be effectively killed by antibiotics.31 Other specific activation conditions include light,32 pH,33 ultrasound.34
Dr Huang et al. also designed and synthesized a series of glutathione S-transferase π (GSTπ) activatable O2-sulfonylethyl protected diazeniumdiolates. Since these compounds are all GSH analogs, they can be recognized by GSTπ and remove the corresponding H atom to initiate a β-elimination reaction, releasing active vinylsulfone GSH derivatives and NONOates, where the latter in tumor cells released NO. The most active compound 8 released higher levels of NO and inhibited the proliferation of melanoma B16 cells, which was significantly better than the diazeniumdiolate-based anticancer molecule JS-K (compound 7) at the cellular level. At the animal level, 8 significantly inhibited the growth of melanoma B16 cells, which indicated that 8 may be used to treat melanoma (Fig. 3).35
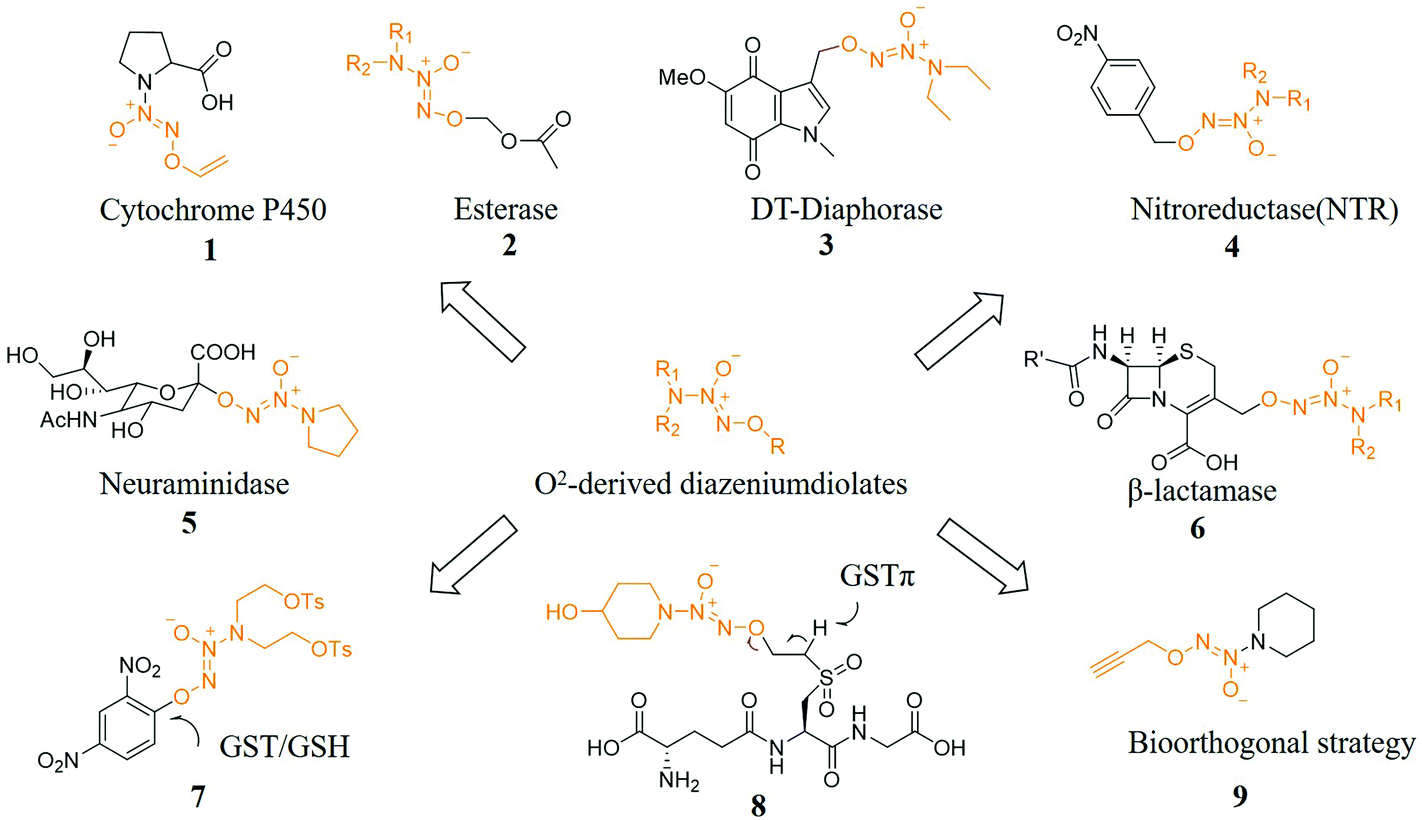 | ||
| Fig. 3 NO prodrugs activated under specific conditions. | ||
2.4 Antibody/peptides-drug conjugate NO donors
In recent years, monoclonal antibody drugs with high target recognition and affinity have attracted widespread attention in cancer treatment. However, antibody drugs face the problems of insufficient therapeutic efficacy and a narrow therapeutic window. Therefore, the strategy of antibody/peptide-drug conjugates (ADC/PDC) was devised. As the “bullet” of ADC, the antibody specifically recognizes tumor cells, and then accurately delivers the “bullet” drugs carried to the surface of tumor cells, thereby exerting a better anticancer effect and enhanced therapeutic window.36,37Encouraged by the ADC concept, Dr Huang et al. conjugated a humanized CD24 monoclonal antibody with an NO donor and designed and synthesized a new antibody-NO conjugate class (ANC). One donor molecule of NONOate can release 2 molecules of NO after cleavage. There was a disulfide bond structure between the antibody and the NO donor. The disulfide linker was stable in the blood circulatory system with a low concentration of GSH and was cleaved in reducing tumor cells with a high concentration of GSH, and then 1,6-eliminated and released NO. Studies have shown that ANC is endocytosed by tumor cells, and the disulfide bond is broken under the action of intracellular glutathione to release the active effector molecule NO. NO itself can inhibit the mitochondrial respiratory chain and make tumor cells release cytochrome c to induce apoptosis. Also, NO reacts with superoxide anions (O2˙−) and transition metal ions in tumor cells to form reactive nitrogen oxides (RNOS) such as peroxynitrite (ONOO−). Peroxynitrite interferes with the post-translational modifications of key proteins inside tumor cells, such as inducing nitration of tyrosine residues to produce 3-NT (3-nitrotyrosine). Experiments on transplanted tumors in nude mice proved that ANC has good anti-tumor activity and fewer side effects on normal tissues.38
Dr Huang et al. conjugated the N-terminus of the AMP antimicrobial peptide with the furoxan moiety through the tyrosine bond and click reaction to form the NO donor polypeptide conjugate FOTyr-AMP. Since the dispersal of biofilms is beneficial for antibacterial activity, FOTyr-AMP with a furoxan head and AMP tail can produce a better antibacterial effect on P. aeruginosa with a biofilm (Fig. 4).21
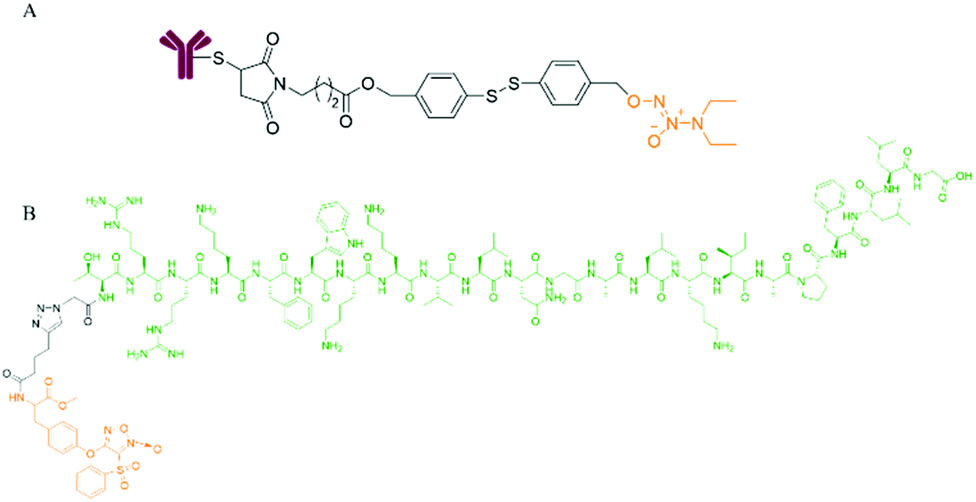 | ||
| Fig. 4 NO donor-antibody/peptide conjugates. A. Structure of antibody-NO conjugate (ANC). B. Structure of FOTyr-AMP. | ||
3. Strategies of nanomaterials prepared by NO donors
3.1 Encapsulating the NO donor in nanomaterials
Encapsulating a suitable NO donor in nanomaterials is a simple strategy for achieving NO delivery by nanomaterials. The nanomaterials are usually degraded to release the donor or some more penetrating technologies are employed to release NO from the encapsulated NO donor. As a small molecule gas messenger, NO can easily escape from the wrapping material to exert physiological effects (Fig. 5).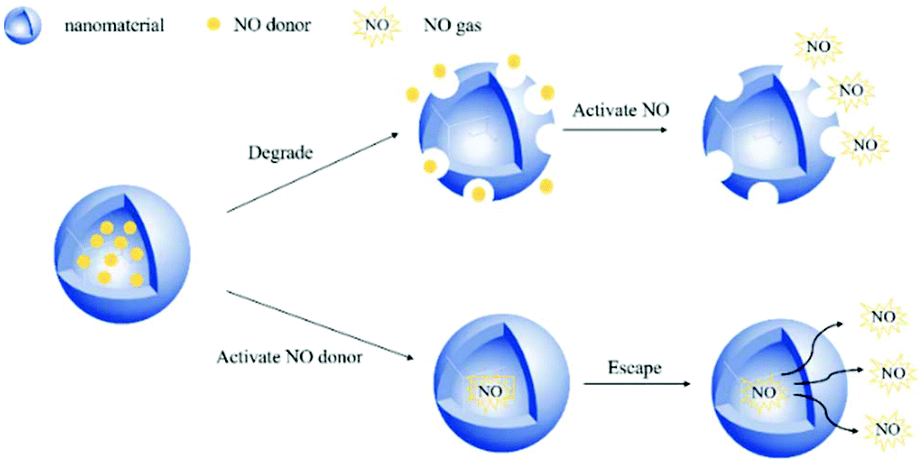 | ||
| Fig. 5 Schematic of wrapping the NO donor in nanomaterials to release NO in two ways. | ||
Previous studies have shown that the pH value in malignant tumors is 6.5–6.9, while normal tissues have a physiological pH value of 7.2–7.4. Thus, researchers hope to develop nanomedicines that release NO by pH control. Dr Yang et al. reported that calcium carbonate (CaCO3) and calcium phosphate possess unique properties, which maintain their crystal structure at physiological pH but dissolve into non-toxic ionic substances in an acidic cell environment. Therefore, based on this mineralization method, a new and highly efficient GSNO intracellular delivery system may be produced for tumor therapy. In their work, CaCO3 mineralized nanoparticles (GSNO-MNP) that could support GSNO were developed. During the mineralization process, ionized GSNO could be loaded in situ in the CaCO3 core. Experiments showed that this mineralized nanoparticle indeed enhanced the stability of GSNO in the aqueous phase and preferentially generated NO in response to the endosomal pH and intracellular reduction environment.39 Researchers have also developed some NO nanomedicines that can respond to overexpressed chemicals in tumor tissues. This satisfies the purpose of precisely regulating the release of NO. Dr Li et al. achieved the biodegradation of GSH-reactive silicone in tumors and the subsequent controlled release of NO in tumors by encapsulating the GSTπ-reactive NO release prodrug NPQ into redox-reactive silicone nanocarriers. The PEGylated nanocarrier (PDHN) allowed the silicone shell of the nanomedicine (QM-NPQ@PDHN) to degrade in the presence of high concentrations of GSH in cancer cells, causing the QM-NPQ@PDHN nanomedicine to release the NPQ prodrug. Subsequently, under the catalysis of overexpressed GSTπ in the tumor, the NPQ prodrug released in the tumor cells decomposed into NO (Fig. 6A).40
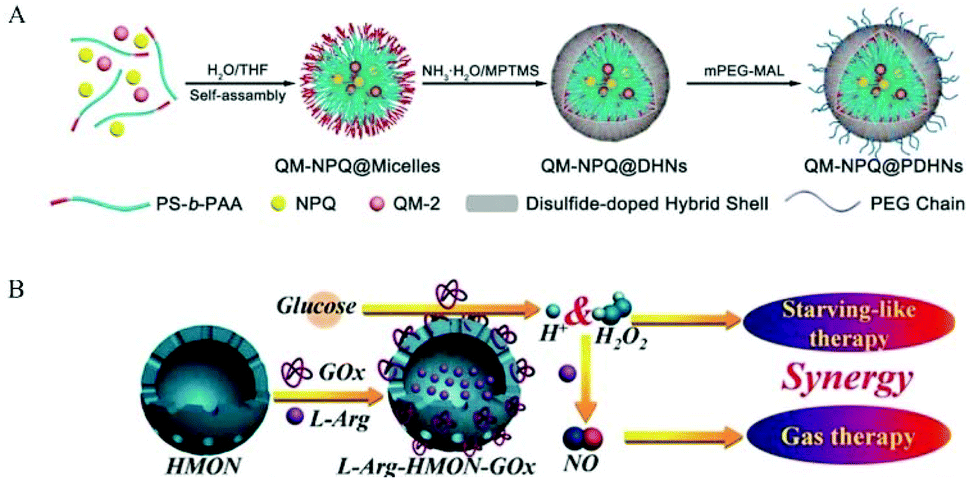 | ||
| Fig. 6 Encapsulation of NO donor in nanomaterials. A. Schematic illustration of the fabrication of QM-NPQ@PDHNs.40 Reprinted with permission from ref. 40. Copyright 2018, John Wiley & Sons. B. Schematic illustration of the construction of L-Arg-HMONGOx for synergistic cancer starving-like/gas therapy.43 Reprinted with the permission from ref. 43. Copyright 2017, John Wiley & Sons. | ||
At the same time, researchers also used some highly penetrating technologies or small molecules that can freely enter and exit nanomaterials so that the NO donor can release NO when it is wrapped, and then NO acts as a small molecule gas messenger freely diffusing out of nanomaterials. Dr Yang et al. dissolved the NO precursor diazeniumdiolate in an alkaline buffer solution (pH = 9.0) and encapsulated it in heat-sensitive liposomes. The liposome internal environment greatly delayed the spontaneous release of NO. When the liposomes were placed in physiological buffer solution and the temperature was increased to the sensitive temperature of the phospholipid membrane, protons outside began to flood in large numbers to disrupt the pH gradient, subsequently causing significant NO release.41 Dr Schoenfisch et al. also used pH to trigger the release of NO from NONOate. They prepared NO-releasing liposomes containing N-diazeniumdiolate NO donors, which released only a small amount of NO at normal physiological pH. The N-diazeniumdiolate NO donor was encapsulated in the aqueous core, and the higher internal pH limited the decomposition of the NO donor. When liposomes entered the acidic microenvironment of the tumor, the lower pH promoted the release of NO from the N-diazeniumdiolate.42
In addition, researchers used some external local triggers such as light, heat, and X-rays. However, these external triggers also encountered some inevitable shortcomings, such as poor light penetration and strong ionizing X-rays. Thus, these shortcomings severely limit their potential applicability in the clinic. Ultrasound has recently aroused increasing interest because it is non-invasive, non-ionizing, easy to control, has a high penetration depth, etc. Dr He et al. used superparamagnetic iron oxide nanoparticles (SPION) as an MRI contrast agent and synthesized a small rattle-type SPION@hMSN as an advanced multi-functional nanocarrier. In previous research work, it was found that BNN-type donors were sensitive to the release of NO by ultrasound. Thus, BNN6 was used to construct a new BNN6-SPION@hMSN nanomedicine. The BNN6-SPION@hMSN nanomedicine had the ability to target tumors passively and had high MRI performance and unique US-triggered NO release characteristics.34 Also, Dr Chen et al. used hollow mesoporous organic silica nanoparticles (HMON) for the efficient co-delivery of glucose oxidase (GOx) and L-Arg efficiency. GOx could oxidize the glucose in the tumor to H2O2, and then H2O2 oxidized L-Arg to generate NO. Simultaneously, GOx consumed a large amount of glucose in the tumor tissue, resulting in starvation therapy (Fig. 6B).43
The S-NO bond of nitrosothiols decomposes then releases NO. In addition to heat, UV/visible light triggers, Cu+, Fe2+, Hg2+, and Ag+ can also react with nitrosothiols, causing the S-NO bond to break and generate NO. Dr Yeh et al. proposed the use of djurleite (Cu2−xS) nanoparticles (NPs) to participate in the GSNO reaction to release NO. Cu1.6S NPs generated heat by laser irradiation and released NO by reacting with GSNO. The preparation included a PLGA polymer vesicle with GSNO encapsulated in the core and oil phase djurleite (Cu2−xS) nanoparticles (NPs) encapsulated in the polymer vesicle membrane. After being irradiated with photons of energy matching the absorption bands of the Cu2−xS NPs, the embedded NPs were heated, which increased the membrane permeability of PLGA, and then GSNO molecules escaped from the core of the polymer vesicle. When GSNO molecules infiltrated through the PLGA membrane, they reacted with the embedded Cu2−xS NPs to generate NO molecules, thereby relaxing cerebral blood vessels.44
Some researchers used nanomaterials to simultaneously encapsulate NO donors and anticancer drugs in materials to achieve co-delivery and exert a synergistic effect when they reach the target site. Dr Sung et al. work developed an injectable hollow microsphere (HM) system with the anticancer drug irinotecan (CPT-11) and NO-releasing donor diazeniumdiolate (DETA NONOate) in its hydrophilic core made of poly(D,L-lactic-co-glycolic acid) (PLGA), which is widely used as a drug carrier material. After being injected into the acidic tumor environment, NONOate wrapped in HMs reacted with acid to form NO bubbles, which created a permeable defect in the PLGA shell, resulting in the release of CPT-11 to the local targets adjacent to tumor cells. In addition to playing a key role in triggering drug release, the formed NO can also act as a Pgp-mediated MDR reversal agent in tumor cells.33
3.2 Surface modification of nanomaterials with NO donor
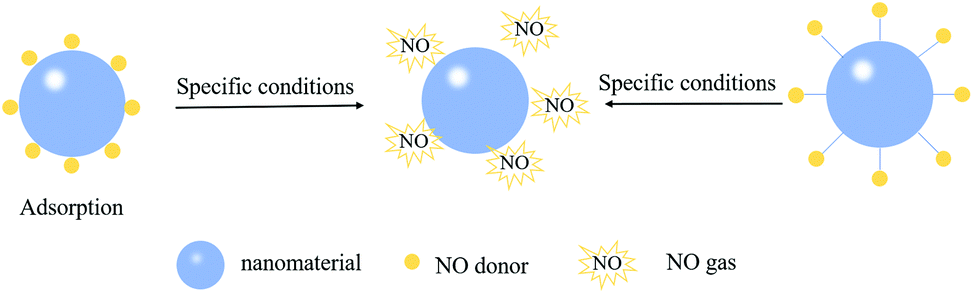 | ||
| Fig. 7 Scheme of using adsorption or covalent interactions to attach NO donors on the surface of nanomaterials. | ||
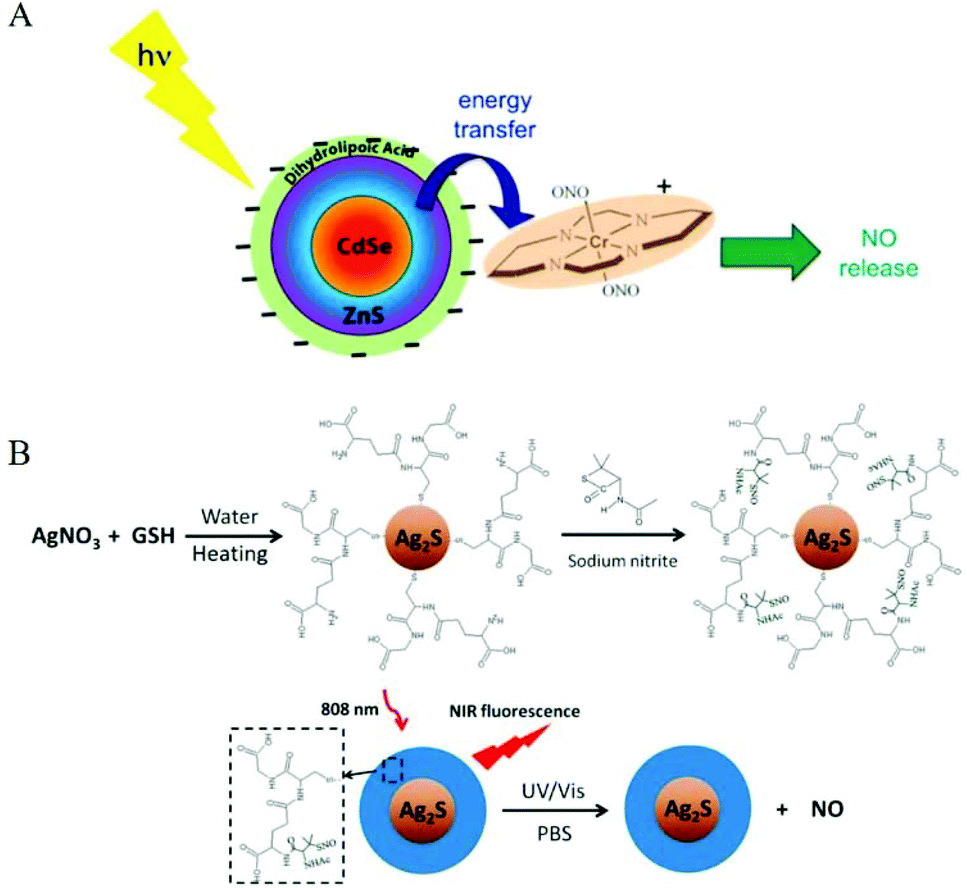 | ||
| Fig. 8 Surface modification of nanomaterials as NO donors. A. Representation of NO release from trans-Cr(cyclam)(ONO)2+ using a CdSe/ZnS core/shell QD (surface modified with dihydrolipoate) as a photosensitizer.32 Reprinted with the permission from ref. 32. Copyright 2012, the American Chemical Society. B. Schematic presentation of light-triggered NO release and NIR fluorescence imaging by Ag2S-GSH-SNO nanoparticles.54 Reprinted with permission from ref. 54. Copyright 2013, the American Chemical Society. | ||
Since most light-responsive NO donors are activated by ultraviolet and visible light, the penetration depth of ultraviolet and visible light limits their tumor application. Therefore, the main challenge for using light-releasing NO therapy for cancer treatment is to adjust the excitation wavelength so that it enters the appropriate deep tissue therapeutic window (650–1100 nm). Thus, researchers used some characteristics of nanomaterials to overcome the shortcoming of ultraviolet and visible light-activated NO donors. Dr Wan et al. used a sulfur-hydrazine hydrate complex (S-N2H4·H2O) as the sulfur source to synthesize water-dispersible Mn2+-doped ZnS QDs. Mn2+-ZnS QDs@CS was conjugated to Roussin's black salt anion Fe4S3(NO)7− (RBS) to obtain Mn2+-ZnS QDs@CS-RBS, and NO was released under NIR-II radiation.45
Due to their small size and bright blue fluorescence, N-doped graphene quantum dots (N-GQDs) are considered suitable drug carriers that can track the intracellular transport of drug delivery systems. Dr Liu et al. reported ruthenium nitrosyl and TPP-functionalized N-GQD as a new NO delivery nanoplatform {N-GQDs@Ru-NO @ TPP}. The nanoplatform was specifically located in the mitochondria and released NO simultaneously with the photothermal effect when irradiated with 808 nm light.46
Recent research showed that up-converting nanoparticles (UCNPs) doped with lanthanides can effectively collect incident near-infrared (NIR) photons and upconvert them to higher energy UV/visible photons. Specifically, Dr Zhao et al. chemically synthesized and developed mesoporous silica-coated white light-emitting UCNPs (NaYbF4:Tm@NaYF4:Yb/Er) as a near-infrared photosensitive platform. The NO donor [NH4] [Fe4S3(NO)7] (Roussin's black salt, RBS), which was adsorbed on UCNPs, performed dose-controlled NO release under 980 nm laser irradiation. By controlling the output power of the 980 nm NIR light, the NO concentration used in cancer treatment could be fine-tuned. High concentrations of NO can directly kill cancer cells, while low concentrations of NO can be used as effective P-glycoprotein (P-gp) regulators.47 Dr Ford's research group also conducted much research in this field. They prepared silica-coated hexagonal NaYF4:Yb/Er@NaYF4 UCNP with a core/shell structure and a positive outer surface and a polymer disc (PD) activated by NIR light and incorporated lanthanide-based up-conversion nanoparticles (UCNP). The PD consisted of a poly(dimethylsiloxane) (PDMS) matrix. Furthermore, they used photochemistry to release NO from an iron nitroso complex.48
The controlled release of NO at the target lesion requires some endogenous initiators, such as enzymes and hypervariable chemicals. However, some endogenous triggers are not specific to tumors but are ubiquitous in many organs, leading to the failure of an on-demand release failure. Therefore, researchers have used some controllable external response factors to stimulate the NO donor to release NO at a specific location. Dr Chen et al. reported building an ultrasound-responsive NO release system, in which the surface of hollow mesoporous silica nanoparticles (HMSN) was modified with poly(ethylene glycol) (PEG) molecules, targeting peptides, and NO donor L-arginine (LA) was loaded in the mesopore and lumen of the modified HMSN. In most tumor models, the H2O2 concentration is much higher than that in other normal organs. Local ultrasound at a frequency of 1 MHz could accelerate the reaction between the LA molecules and H2O2 in the Panc-1 tumor and release more NO.49
The co-delivery of NO and active drugs can also be achieved via adsorption. Dr Balkus et al. proposed a method of combining S-nitrosothiols with PbS quantum dots (PbS QDs) doped with TiO2 nanotubes. S-Nitrosothiol was used as a carrier to release NO. The stable photocatalytic NO donor-functionalized TiO2 nanotubes promoted the generation of singlet oxygen (1O2) by light excitation. Experiments showed that this hybrid nanomedicine could release NO and produce singlet oxygen through near-infrared light.50 In the work by Dr Gu et al., a diagnostic scintillating nanoparticle (SCNP) system containing Ce-doped LiLuF4 and Roussin's black salt (RBS) was used to generate ONOO− on demand under X-ray radiation. On the one hand, Ce-doped LiLuF4 can act as a radiosensitizer to increase the ROS yield, including O2−˙ under X-ray irradiation. On the other hand, Ce-doped LiLuF4 can convert X-rays to ultraviolet rays, thereby activating the photosensitive RBS to release NO. This simultaneous release of NO and O2−˙ ensures the efficient generation of ONOO−. ONOO− can improve the radiotherapy rate by directly destroying DNA and inhibiting the expression of DNA repair enzymes, resulting in increased DNA damage and inhibition of tumor growth.51 In the work by Dr Mosinger et al., active oxygen donor photosensitizers 5,10,15,20-tetrakis(N-methylpyridinium-4-yl)porphyrin tetra(p-toluenesulfonate) (TMPyP) or zinc(II) 2,9,16,23-tetrakis(N-methyl-pyridiumoxy) phthalocyanine tetraiodide (ZnPc) and NO photodonor 4-(N-(aminopropyl)-3-(trifluoromethyl)-4-nitrobenzenamine)-7-nitrobenzofurazan (NOP) were used to achieve antibacterial activity upon visible light irradiation.52
Dr Qu et al. reported that the system was built with UCNP loaded with Roussin's black salt (RBS). RBS is an NO donor molecule that can release NO under ultraviolet/visible light irradiation. UCNP was used as an antenna-type system that collected NIR light and transferred the collected energy to light with a specific wavelength, thereby triggering the release of NO. Quaternized chitosan (qC) can destroy the cytoplasmic membrane of pathogens, and thus is considered an indispensable weapon against drug-resistant bacteria.53
The use of X-rays can also break the covalent bond between the NO donor and nanomaterials. Many of the studies mentioned above reported a controlled release strategy for NO triggered by ultraviolet, visible, and even near-infrared light. In comparison, X-rays are expected to exhibit superiority in controlling drug release. Firstly, with precise positioning, highly penetrating X-rays can force the release of drugs only on fixed deep lesions. Secondly, on-demand drug release can be achieved only by changing the X-ray radiation dose and duration. Finally, the combination of chemotherapy and radiation therapy can be achieved to improve the therapeutic effect. Thus, researchers have developed new strategies for using highly penetrating X-rays. Dr Shi et al. successfully constructed PEG-USMSs-SNO using X-rays to control NO release using upconversion nanoparticles (UCNPs)/silica structure and then modified them with biocompatible PEG and an NO donor (SNO). High-energy X-ray radiation can decompose some low-energy bonds, such as disulfide bonds (S–S, 240 kJ mol−1) and diselenide bonds (Se–Se, 172 kJ mol−1). Considering that the S–N bond energy (about 150 kJ mol−1) in the –SNO group was relatively lower than the Se–Se bond energy, it was speculated that X-ray radiation could break the S–N bond and release NO.56
Carbon quantum dots can also be covalently connected with NO donors more conveniently. Moreover, in recent years carbon quantum dots have attracted much attention because of their excellent biocompatibility, good luminous properties, simple synthetic process, and low cost. Carbon quantum dots possess high photostability, photothermal effect, tunable emission, and two-photon excitation (TPE) cross-sections. Furthermore, the high TPE cross-section of CQDs allows the indirect excitation of the NO photodonor by NIR light at 800 nm. Thus, some NO photoactivated donors have been used in combination with carbon quantum dots. Then used the photothermal effect to continuously release NO, while generating hyperthermia to produce a synergistic anticancer effect. Dr Callan et al. reported a new type of nanostructure, in which carbon quantum dots were covalently attached to a nitroaniline derivative NO-photoactivated donor to obtain nanomedicines. They demonstrated that light-induced energy transfer occurred from the core of the carbon quantum dots to the NO photoactivated donor shell and stimulated NO release.57 In recent research, there were also many combinations of ruthenium nitrosyls and carbon quantum dots to form nanomedicines, then employing NIR light to produce a photothermal effect.58,59
Researchers have used the covalent bonding of NO donors and nanomaterials to successfully load donors and release NO at specific locations to play important physiological roles. Dr Xue et al. integrated photothermal and NO release characteristics into a single nanocomposite to achieve synergistic photothermal and NO antibacterial effects. They used Fe3O4@PDA as a light conversion agent and modified PAMAM and NONOates on its surface in sequence, thereby obtaining a multi-functional Fe3O4@PDA@PAMAM@NONOate nanocomposite. This material was irradiated with 808 nm laser light to produce synergistic photothermal and NO antibacterial effects on Gram-negative E. coli and Gram-positive S. aureus bacteria.60 Dr Herves et al. used gold nanoparticles stabilized with citrate. Both thiol-containing amino acids could be easily nitrosated, and the dissociation energy of the gold-thiol (RS-Au) bond was greater than that of the RS-NO bond (40 versus 83.7 kJ mol−1). The S-NO bond was broken in the gold nanoparticles, after which an S–Au bond was formed, and NO was released. The system allowed surface-controlled NO release, where the amount of NO released was proportional to the number of thiols bound to the surface of the gold nanoparticles. This work focused on studying the controlling effect of gold nanoparticles on the release of NO from low-molecular weight RSNOs.61 Dr Zhao et al. used a click reaction to combine the short peptide of Nap-FFGGG with galactose-modified NO donor diazeniumdiolates to form a hydrogel to deliver NO locally. After β-galactosidase hydrolyzed the galactose in the hydrogel structure, the diazeniumdiolates released NO in a controlled manner, improving wound healing.62
Researchers use covalent bonding to achieve the multi-functional effects of NO nanomedicine. Covalently connecting some substances can produce a synergistic effect with NO, such as reactive oxygen species, or connect some common antibacterial and anticancer drugs. Dr Schoenfisc et al. conducted a series of studies on the antibacterial nanomedicines of diazeniumdiolates. They used N-(6-aminohexyl)aminopropyltrimethoxysilane and tetraethoxysilane to form hybrid nanoparticles and then formed a diazeniumdiolate NO donor on the secondary amine.63 In their following work, they integrated quaternary ammonium (QA) into nanomedicines with diazeniumdiolate as NO donors. The long-chain QA had a simple structure and a broad-spectrum bactericidal effect. Therefore, it was a popular non-consumable antibacterial ingredient.64 In 2013, they reported that mesoporous silica nanoparticles were functionalized with O2-substituted N-diazeniumdiolate-modified organosilanes to produce a new class of antibacterial repair materials.65 Dr Liu et al. reported that the nanomedicines consisted of a ruthenium nitrosyl donor, [Ru(tpyCOOH)(DAMBO)NO](PF6)3, folic acid (FA) molecule, and biocompatible titanium dioxide nanoparticles (TiO2 NPs). FR is a potential cancer marker and is overexpressed in many human cancer cells. Because FA has a high affinity for FR, the {Ru-NO@TiO2 NP} nanomedicine could deliver NO under visible light to cancer cells with overexpressed folate receptors (FR). Meanwhile, under visible light, the {Ru-NO@TiO2NP} nanomedicine simultaneously produced singlet oxygen (1O2) to produce a synergistic anticancer effect.66 Dr Ji et al. conjugated S-nitrosothiol with α-cyclodextrin (α-CD) to synthesize glutathione (GSH)-sensitive NO nanomedicines and prepared supramolecular nanocarrier α-CD-Ce6-NO nanoparticles (NP). α-CD-Ce6-NO depleted GSH in tumor cells to produce NO, and the consumption of GSH increased the amount of light-activated Ce6 to produce ROS. NO and ROS continuously reacted to generate RNS (ONOO−), which, compared with ROS, greatly improved the anticancer activity (Fig. 9A).67
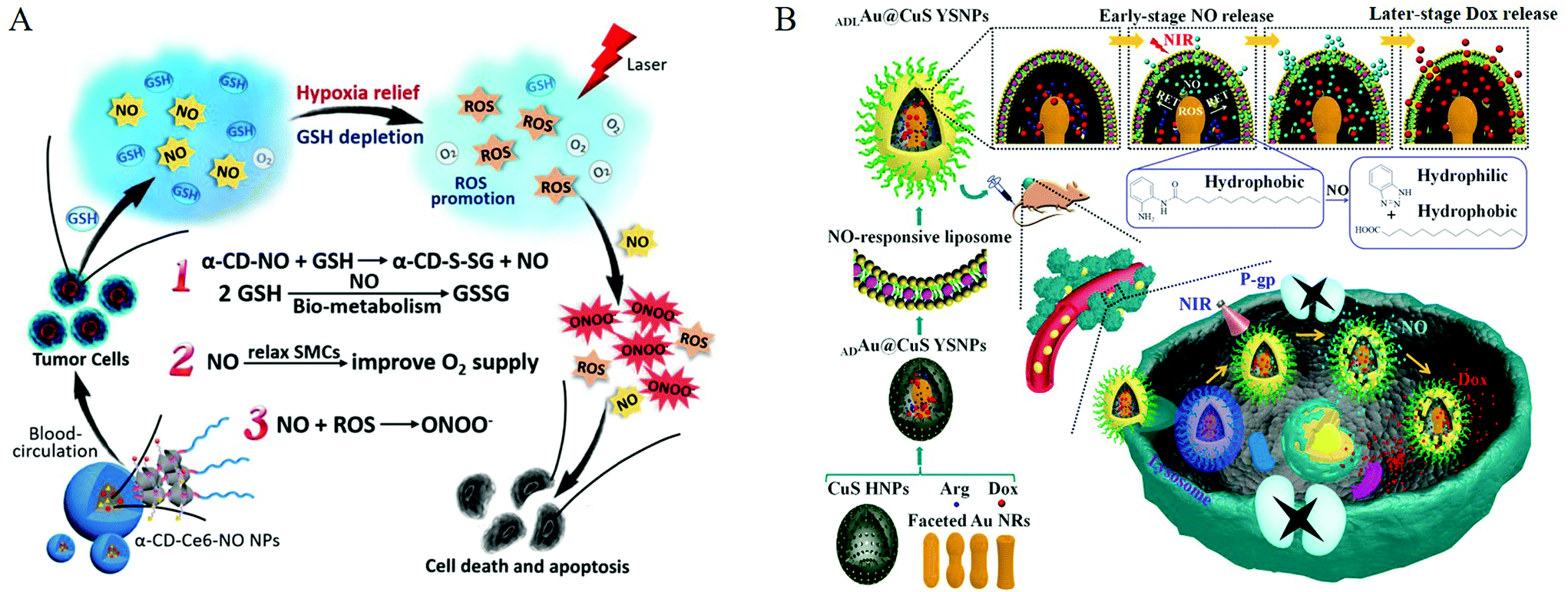 | ||
| Fig. 9 Co-delivery of nitric oxide and other active substances. A. Multiple synergistic effects between NO and PDT generated from the supramolecular α-CD-Ce6-NO NPs to improve the therapeutic efficacy.67 Reprinted with the permission from ref. 67. Copyright 2018, Elsevier. B. Schematic showing the NO and Dox programmable release and MDR cancer therapy using ADLAu2@CuS YSNPs.69 Reprinted with permission from ref. 69. Copyright 2019, the American Chemical Society. | ||
Using the advantages of covalent bonding, researchers integrated NO donors into materials and then employed encapsulation or adsorption to load some commonly used drugs for synergistic treatment. Dr Chen et al. constructed biodegradable nanomedicines based on BNN6(NO donor)/DOX-loaded monomethoxy (polyethylene glycol) poly(lactic-co-glycolic acid) (mPEG-PLGA) nanoparticles for UV-responsive NO and DOX release. The size (nano) and high surface density of mPEG endowed the nanoparticles with a relatively long blood circulation time and enhanced NO accumulation in the tumor. The generated NO gas destroyed the outer shell of the nanoparticles and promoted the release of the wrapped DOX molecules, thereby generating significant synergy between the gas and the drug.68 Then, Dr Zhang et al. designed a nanosystem that could release NO and Dox at intervals. O-Phenylenediamine-containing lipids hydrolyzed by NO were embedded in the phospholipid bilayer structure of liposomes to form NO-responsive liposomes, and L-arginine (L-Arg)/gold-loaded copper sulfide egg yolk shell nanoparticles (ADAu@CuS YSNPs) were further encapsulated to form ADLAu@CuS YSNPs. Under the irradiation from an 808 nm laser, the unique resonant energy transfer (RET) process and ROS generation in the confined space of ADLAu@CuS YSNPs effectively converted L-Arg to NO. Due to the limitation of the molecular scaffold, Dox cannot be released from YSNP at this early stage. As NO release proceeds, the reactivity of NO made the liposome layer structure more unstable, finally allowing Dox to escape (Fig. 9B).69 Also, Dr Kong et al. used high levels of GSH in tumor cells to achieve the controlled release of NO. By combining sequential galactose monomer, hydrophobic acid-sensitive DPA monomer, and the NO prodrug alkynyl-JSK, they synthesized NO-functionalized polymer nanoparticles for NO delivery. The obtained triblock copolymer could self-assemble into nanoparticles in an aqueous solution, and the anti-tumor drug doxorubicin (DOX) was simultaneously encapsulated in the nanoparticles through a co-assembly process to exert synergistic anticancer effects. High levels of GSH/GST in human liver cancer cells triggered the continuous local release of NO. Under an acidic environment, DOX was also rapidly released from the nanoparticles.70 Dr Boyer et al. reported a new type of NONOate-conjugated star polymer, which could slowly release NO. The core-crosslinked star polymer was prepared via reversible addition−fragmentation chain transfer polymerization (RAFT), and the secondary amine group reacted with NO gas to obtain an NO core crosslinked star polymer. The star polymer could completely inhibit Pseudomonas aeruginosa attachment and biofilm formation over time in a non-growth inhibiting manner. They directly obtained the NO donor by reacting the aminoglycoside antibiotic gentamicin with NO gas, thereby generating a gentamicin-NONOate complex. The nanoparticles achieved the simultaneous and sustainable release of gentamicin and NO, and these two release agents worked synergistically on the membrane of P. aeruginosa.71
3.3 NO donor Co-assembly into nanomaterials
Some researchers used NO donor co-assembly into nanomaterials to produce nanomedicine. The main forms of assembly include assembling, stacking, and aggregation (Fig. 10). Some researchers used regular self-assembly to assemble NO donors and nanomaterials. Dr Hen et al. reported a NO-driven nanomotor, in which the medical fluorescent hyperbranched polyamide (HPAM) and biocompatible zwitterionic L-arginine were selected to synthesize a hyperbranched polyamide/L-arginine (HLA) nanomotor. The nanomotor could generate NO under the catalysis of NOS, ROS, or other catalysts in the body to promote angiogenesis for cancer treatment (Fig. 11A).72 Dr Hu et al. also incorporated other antibacterial molecules into the nanosystem to achieve synergistic antibacterial effects with NO. They reported a NO-donor CouNO based on N-nitrosoamine, which contained the coumarin chromophore and could release NO upon exposure to visible light. The amphiphiles self-assembled into micelles and the light-induced NO release effectively dispersed the bacterial biofilm of P. aeruginosa. In addition, antibiotics (such as ciprofloxacin, Cip) can be loaded into NO-releasing micelles to achieve the co-delivery of NO and Cip, simultaneously disperse biofilms, and kill bacteria.73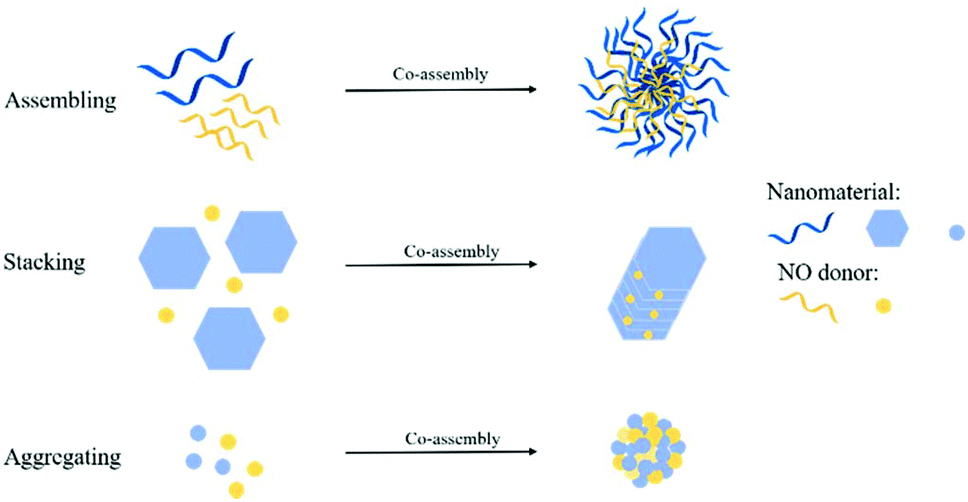 | ||
| Fig. 10 Different strategies for the co-assembly of NO donors and nanomaterials. | ||
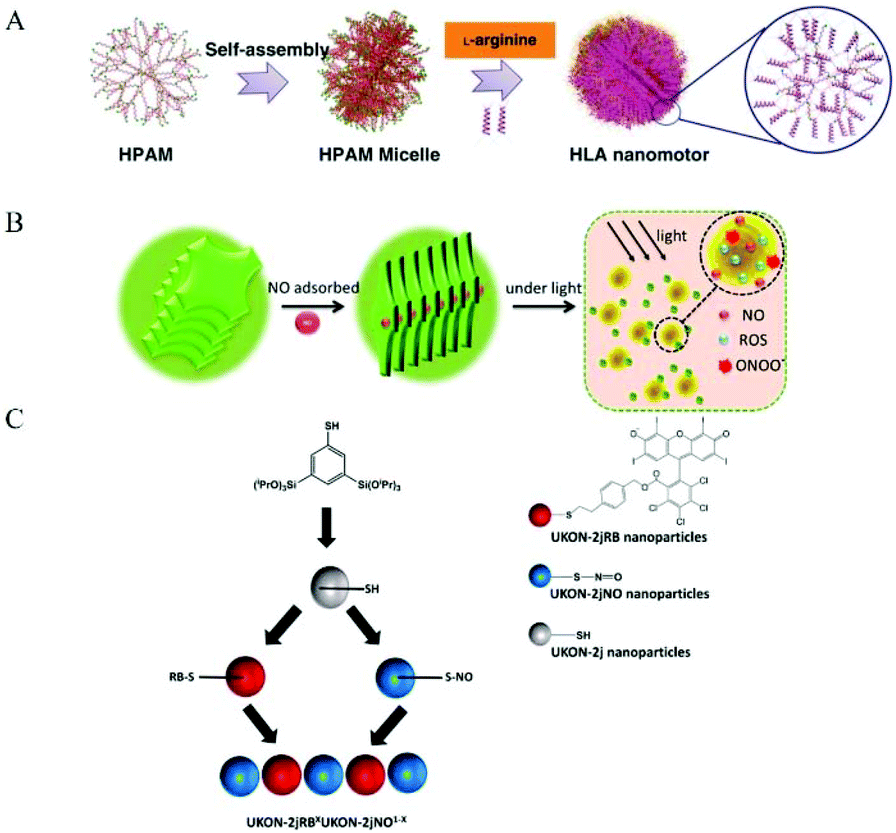 | ||
| Fig. 11 Co-assembly of NO donor and nanomaterial. A. Schematic illustration of the formation of zwitterion-based nanomotor and the NO generation principle.72 Reprinted with permission from ref. 72. Copyright 2019, Springer Nature. B. Self-assembly and NIR-responsive mechanism of the GO-BNN6 sandwich structure, which was assembled by GO nanosheets and BNN6 molecules through π–π stacking.75 Reprinted with the permission from ref. 75. Copyright 2015, The Royal Society of Chemistry. C. Schematic Representation for the preparation of NO and ROS releasing materials.79 Reprinted with permission from ref. 79. Copyright 2016, the American Chemical Society. | ||
Some researchers have used the π–π conjugation between nanomaterials to stack to form NO nanomedicine. Sortino et al. reported a modified graphene oxide (GO) nanoplatform in which GO sheets were covalently functionalized with the NO light-release agent NOP1, and the resulting hybrid nanomedicine (GO-NOP1) released a large amount of NO under visible light.74 Dr Chen et al. constructed a new type of near-infrared-responsive sandwich nano-drug (GO BNN6). Through π–π stacking, graphene oxide (GO) can absorb NIR light and convert photons into active electrons. Then the electrons enter the cage-like BNN6 and stimulate BNN6 to decompose and effectively release NO to exert anticancer activity (Fig. 11B).75 Dr Mascharak et al. loaded the photoactive manganese nitrosyl, [Mn(PaPy3)(NO)](ClO4)({Mn-NO}) into the cylindrical hole of the main body of a minosilicate-based material (MCM-41). The strong interaction between the polar nitrosyl group and the OH group on the wall led to the good attachment of the NO donor in the porous host. When the material was exposed to visible light, the rapid release of NO was observed. The NO release curve indicated that these biocompatible materials successfully eliminated the drug susceptibility and drug resistance of Acinetobacter baumannii in a soft tissue infection model.76 Dr Liu et al. used the clinical drug sodium nitroprusside and zinc ions to form a biocompatible monolayer nanosheet, which was radiolabeled with the 32P isotope to form an NO release system, ZnNO (32P). The radioactive isotope-induced Cherenkov luminescence could continuously stimulate the nanosheets to release NO, thereby regulating the hypoxic and immunosuppressive tumor microenvironment and improving the efficacy of radioisotope treatment to eliminate local tumors.77 Also, Fan et al. used the reaction of NO and active oxygen to produce peroxynitrite. They designed and synthesized ZnTPyP@NO nanoparticles with NO coordinated to the central zinc ion. The nanomedicine exhibited strong antibacterial activity against E. coli and S. aureus under visible light irradiation.78
Some researchers used a modified NO donor and nanomaterial aggregation to achieve co-assembly. Dr Polarz et al. introduced a visible light-mediated silicone nanoparticle (NP) system. Rose Bengal (RB) is an excellent photo-activated active oxygen donor with an absorption band in the visible range of 480 to 550 nm, which was covalently bonded to the mercapto group through click chemistry. Nitrosothiol nanoparticles were obtained through the reaction, which could also release NO under visible light excitation. Nitric oxide (NO) was released simultaneously with singlet oxygen and reactive oxygen species (ROS). After the reaction, peroxynitrite molecules with significantly high bactericidal activity appeared. The activity experiment proved that the NO nanomedicine had high antibacterial efficiency against Pseudomonas aeruginosa (Fig. 11C).79 Dr Huang et al. designed and synthesized a new type of carrier Fe(II)-NO nano-coordination polymer [Fe(II)-BNCP]. Firstly, they designed and synthesized a type of NO donor, BPDB (1,5-bis[(L-proline-1-yl)diazen-1-ium-1,2-diol O2-yl]-2,4-dinitrobenzene), which was chelated with ferrous ions through simple mixing, co-precipitation, and ion-exchange methods to produce a nano-sized coordination polymer, Fe(II)-BNCP. The coordination polymer only contained NO and ferrous ions, and thus can be regarded as a “zero carrier” nano-drug, which remained stable in the systemic circulation and accumulated at the tumor site through enhanced tumor penetration and retention effects. Fe(II)-BNCP released NO at the tumor site, and ferrous ions generated hydroxyl radicals and peroxynitrite through Fenton reaction and Haber–Weiss reaction, respectively. The peroxynitrite anions exerted a chemical treatment effect.80
Dr Koshiyama et al. designed a new lipophilic Ru nitrosyl complex, [Ru(L)Cl(NO)] (1, L = N,N′-ethylene-bis(4-cholesteryl-hemisuccinate-salicylideneamine)), through the specific hydrophobic interaction between cholesterol and phospholipid bilayer, where the Ru nitrosyl complex was fixed on the phospholipid bilayer of a liposome, which released NO under visible light irradiation.81
4. Biomedical applications of NO nanomedicine
4.1 Involvement in cancer progression of the metastatic cascade
It is well known that NO can affect the growth of tumors. A continued low concentration of NO can promote tumor cell growth, participating in tumor angiogenesis and inhibiting tumor cell apoptosis. However, a high concentration of NO has an anti-tumor effect. High concentrations of NO mainly achieve anti-tumor therapeutic effects through the following ways: (1) stimulation of apoptosis by up-regulating p53, (2) degradation of proteasome molecules against apoptosis, (3) cytochrome c release and mitochondrial permeability increase, (4) Smac/DIABLO release, (5) peroxynitrite or ONOO− formation, leading to an increase in p53, cell cycle arrest, cell necrosis, angiogenesis inhibition, and cytotoxicity. Therefore, the use of nanomaterials is reasonable to load NO molecules to achieve the targeted and controlled release of NO during cancer treatment.1Dr Li et al. prepared keratin hyaluronic acid nanogels (KHA-NGs) as targeted drug carriers with a high DOX loading efficiency to achieve anticancer therapeutic effects in vivo and in vitro. Changes in pH, trypsin, and GSH concentration in the intracellular environment triggered the release of DOX. Moreover, keratin can promote the production of NO in cells and make tumor cells sensitive, thereby enhancing the anticancer effect of chemotherapy drugs.82 Yang et al. used the photothermal effect of nanogenerators (PTNGs) to convert absorbed NIR light into heat to promote the ability to break S-NO bonds. Subsequently, transferrin (Tf) was conjugated to the surface of the particles to make them target cancer cells (Fig. 12).83
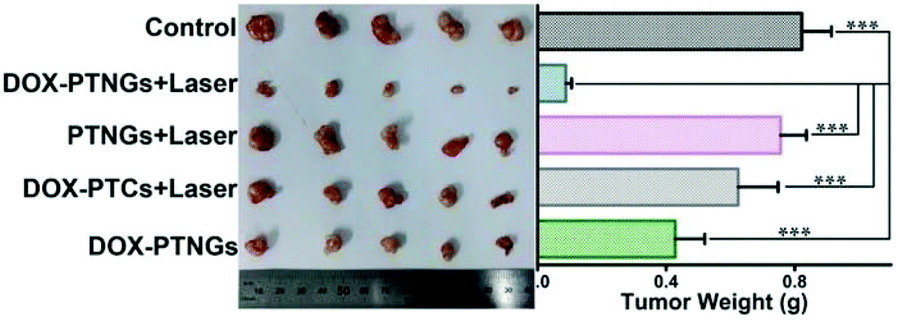 | ||
| Fig. 12 NO involvement in cancer therapy. Photograph (left) and weights (right) of tumors after excision from each group.83 Reprinted with the permission from ref. 83. Copyright 2017, John Wiley & Sons. | ||
4.2 Multifactorial damage to bacteria
In the past few years, the antibacterial properties of NO have attracted attention from many researchers, and the antibacterial mechanism of NO has been clarified. The antibacterial properties of NO are dose dependent. At high concentrations, NO has the effect of killing bacteria. As a free radical, NO is unstable in cells and can easily react with oxygen or reactive oxygen intermediates (such as superoxide (O2−) and hydrogen peroxide (H2O2)) to form products with high oxidation activity, including peroxynitrite (ONOO−), nitrogen dioxide (NO2), and nitrous oxide (N2O3). These reactive substances can interact with proteins through reactive thiols, amines, heme groups, iron–sulfur clusters, and other groups (such as phenol or aromatic amino acid residues). For example, N2O3 and ONOO− can oxidize membrane proteins at different sites through nitrosation of reactive thiols and amines or nitration of tyrosine. NO can react with metal enzymes, leading to iron depletion, and NO can also react with free sulfhydryl groups. Both of these reactions lead to the inactivation of metabolic enzymes. Reactive species can also target DNA, causing deamination and oxidative damage, leading to DNA cleavage. Besides, they can induce lipid peroxidation, thereby damaging cell membranes.84 Recent studies have shown that NO can induce the spread of bacterial biofilms at low concentrations. With an increase in the intracellular c-di-GMP concentration, the formation of biofilms will increase, and with a decrease in the intracellular c-di-GMP concentration, more bacteria will enter the planktonic mode. It has been found that NO is an important regulator signaling molecule for these processes. Specifically, later in the development of the biofilm life cycle, NO synthesizes and activates bacterial phosphodiesterase to degrade c-di-GMP through an endogenous pathway, thereby promoting biofilm diffusion.85Dr Che et al. synthesized ruthenium nanoparticles that release NO through the reaction of alkanethiolate-protected ruthenium nanoparticles with tert-butyl nitrite (tBuONO) and their water-soluble derivatives released NO in an aqueous medium under visible light irradiation.86 Kim et al. developed a combination of calcium phosphate (CaP) coating, pH-jump agent 2-nitrobenzaldehyde (o-NBA), and NO donor diazeniumdiolate. The characteristic that CaP dissolves into non-toxic ions in an acidic environment attracted researchers to develop various pH-responsive drug delivery systems. o-NBA undergoes intramolecular excited-state hydrogen transfer (ESHT) under light irradiation, which can lower the pH of its environment. The CaP coating acts as a shield for the NO donor and is degraded by acid, which would cause the NO donor to be exposed to the physiological environment to release NO (Fig. 13).87
 | ||
| Fig. 13 NO involvement in antibacterial therapy. In vivo corneal wound-healing effects of pH@MSN-CaP-NO.87 Reprinted with permission from ref. 87. Copyright 2016, the American Chemical Society. | ||
4.3 Regulating the insulin secretion
NO plays a diametrically opposite role in hyperglycemia due to its amount. Endothelial nitric oxide synthase (eNOS) dysfunction leads to a reduction in NO, which may reduce microcirculation blood flow, thereby reducing insulin delivery in hormone-sensitive organs. However, excessive NO will cause abnormalities in important molecules in the insulin signal transduction pathway and affect the normal biological function of insulin, thus mediating insulin resistance in peripheral tissues.88 Excess NO can also become an important mediator of type II diabetes by mediating pancreatic β cell dysfunction caused by high glucose, lipids, and cytokines. According to the literature, in the late stage of diabetes, L-arginine or NO donors (such as organic nitrates, nitroprusside, etc.) can be given to correct an excessive reduction in NO and enhance vascular function. Some reports also indicate that to suppress the cytotoxicity of excessive NO production in the early stage of diabetes as much as possible, appropriate iNOS inhibitors can be given simultaneously with insulin treatment, which can effectively inhibit the production of NO and reduce myocardial damage.89In the report by Dr Gu et al., each GOx-MMV structure was composed of L-arginine (NO prodrug) in the inner core, magnetic nanoparticles in the shell, and glucose oxidase (GOx) assembled on the surface. Firstly, GOx-MMV carriers can be used as an intelligent glucose stimulation system to reduce high blood sugar levels. Then, through an alternating magnetic field (AMF), the reaction of H2O2 and L-arginine was initiated to achieve the spatiotemporal control of the generation and delivery of NO (Fig. 14).90
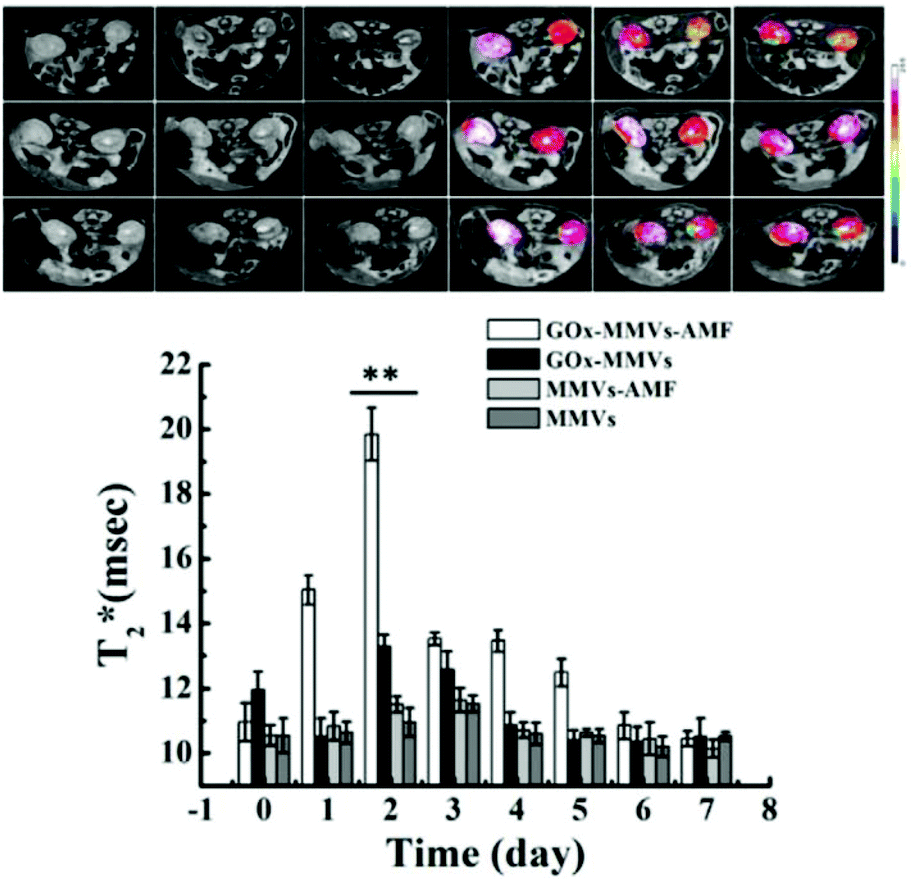 | ||
| Fig. 14 NO involvement in hyperglycemia therapy. In vivo MRI images of diabetic kidney. Before and after GOx-MMV injection triggered by AMF over time.90 Reprinted with permission from ref. 90. Copyright 2016, Elsevier. | ||
4.4 Maintain a basal vascular tone
NO participates in the regulation of intraocular pressure (IOP). Studies have found that human eye ciliary process non-pigmented epithelial cells, ciliary muscle (CM) fibers, trabecular meshwork (TM), Schlemm tube, and fluid tube endothelial cells contain eNOS, especially in the anterior segment of the TM sieve layer and CM longitudinal fibers. There is evidence that both CM and TM can continuously release NO, activate guanylate cyclase (GC), and increase the intracellular cyclic guanylate (cGMP) concentration, and K+ passes through the channel outside the cell, resulting in hyperpolarization. Consequently, this hyperpolarization leads to the inactivation of voltage-dependent Ca2+ channels, and eventually, the intracellular Ca2+ concentration decreases, resulting in CM and TM relaxation, which has a stronger relaxation effect on TM than CM, and thus the resistance of aqueous humor outflow increases. Studies have also shown that nitrate vasodilators or NO donors can reduce IOP. NO can also participate in eye blood flow regulation. Studies have shown that NO is involved in maintaining a constant choroidal vascular tone. In the retina, optic disc, and optic nerve, NO is also involved in regulating normal blood flow. When endothelial cell function is abnormal, NO production decreases, and blood vessels contract. It is worth noting that the free radical superoxide anion and a large amount of NO will combine to form the toxic compound OONO−, which triggers apoptosis of retinal ganglion cells.91In Dr Stevens's method, the enzyme can be inserted into the desired site to convert the externally administered NO donor into an active therapeutic agent. To deepen the enzyme into the TM, β-galactosidase was encapsulated in a polymer carrier. In the study, hydrogen-bonded polymer pairs based on thiol-functionalized poly(methacrylic acid) (PMASH) and poly(N-vinylpyrrolidone) (PVP) were selected as the system for encapsulating the enzyme β-galactosidase. The NO donor β-gal-NONOate was then encapsulated in liposomes because these lipid nanocarriers can continuously release biomolecules. The liposomes were delivered to the outflow pathway, and the NO donor was slowly released into the TM after the lipid vesicles were degraded (Fig. 15).92
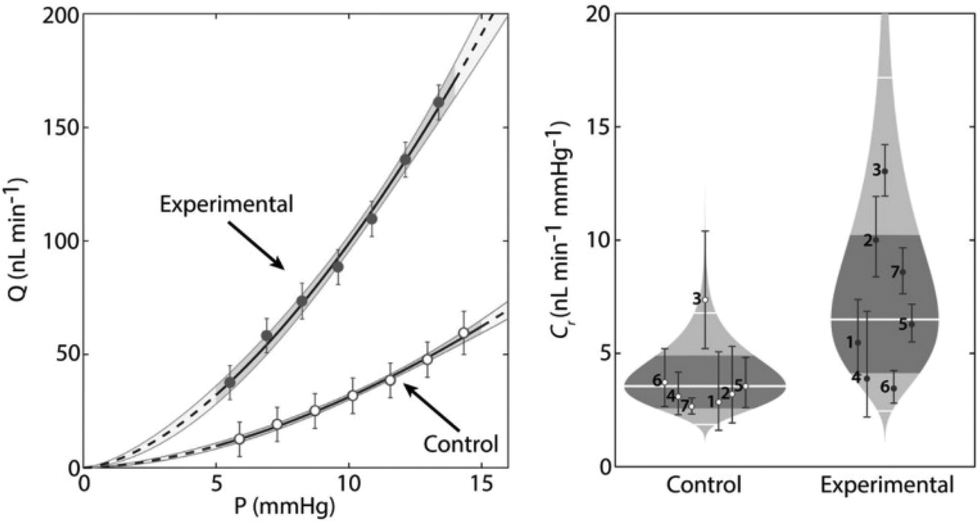 | ||
| Fig. 15 NO involvement in glaucoma therapy. Effects of localized delivery of nitric oxide (NO) on conventional outflow facility in eyes obtained from C57BL/6 mice. Paired eyes were injected with either capsules containing β-galactosidase (experimental) or empty capsules (control) 48 h prior to ex vivo mouse eye perfusions. Enucleated eyes were perfused with β-gal-NONOate-loaded liposomes (50 × 10−6 M) to determine the flow-pressure relationships for paired eyes.92 Reprinted with the permission from ref. 92. Copyright 2017, John Wiley & Sons. | ||
4.5 Major endogenous vasodilator
NO is the main endogenous vasodilator. The pathophysiological environment of the human body is closely related to changes in vascular health. Therefore, NO plays a key role in regulating many pathological diseases. When dilating agents such as acetylcholine and bradykinin act on receptors in endothelial cells, calcium influx binds calmodulin to activate eNOS. The newly synthesized NO diffuses into the adjacent smooth muscle cells and activates guanylate cyclase (GC). GC increases the intracellular cGMP content, and elevated cGMP-dependent protein kinases cause phosphorylation of various proteins and muscle relaxation. It has been reported in the literature that NO is related to various vascular reflexes. Thus, many therapies using NO have been developed to treat diseases related to pathological vasoconstriction. For example, nitroglycerin (NO donor) is used in patients with coronary artery disease. Inhaled NO is used to treat pulmonary hypertension and reverse hypoxic pulmonary vasoconstriction.93Dr Chen et al. reported that NanoNO, a nanoscale carrier, can continuously release NO. The NO released from NanoNO produces a NO gradient around blood vessels, which may normalize tumor blood vessels and promote changes in the tumor microenvironment. Firstly, the polyethylene glycol (PEG) polymer on the surface of NanoNO prevents monocytes and macrophages from recognizing and evading interaction with serum proteins, thereby extending the circulation time of NanoNO. Secondly, the biodegradable poly(lactic-co-glycolic acid) (PLGA) polymer, the main component of NanoNO, can control the continuous release of NO by the NO donor dinitrosyl iron complex (DNIC). Thirdly, NanoNO with a size of approximately 120 nm showed increased accumulation in tumors through enhanced permeability (Fig. 16).94
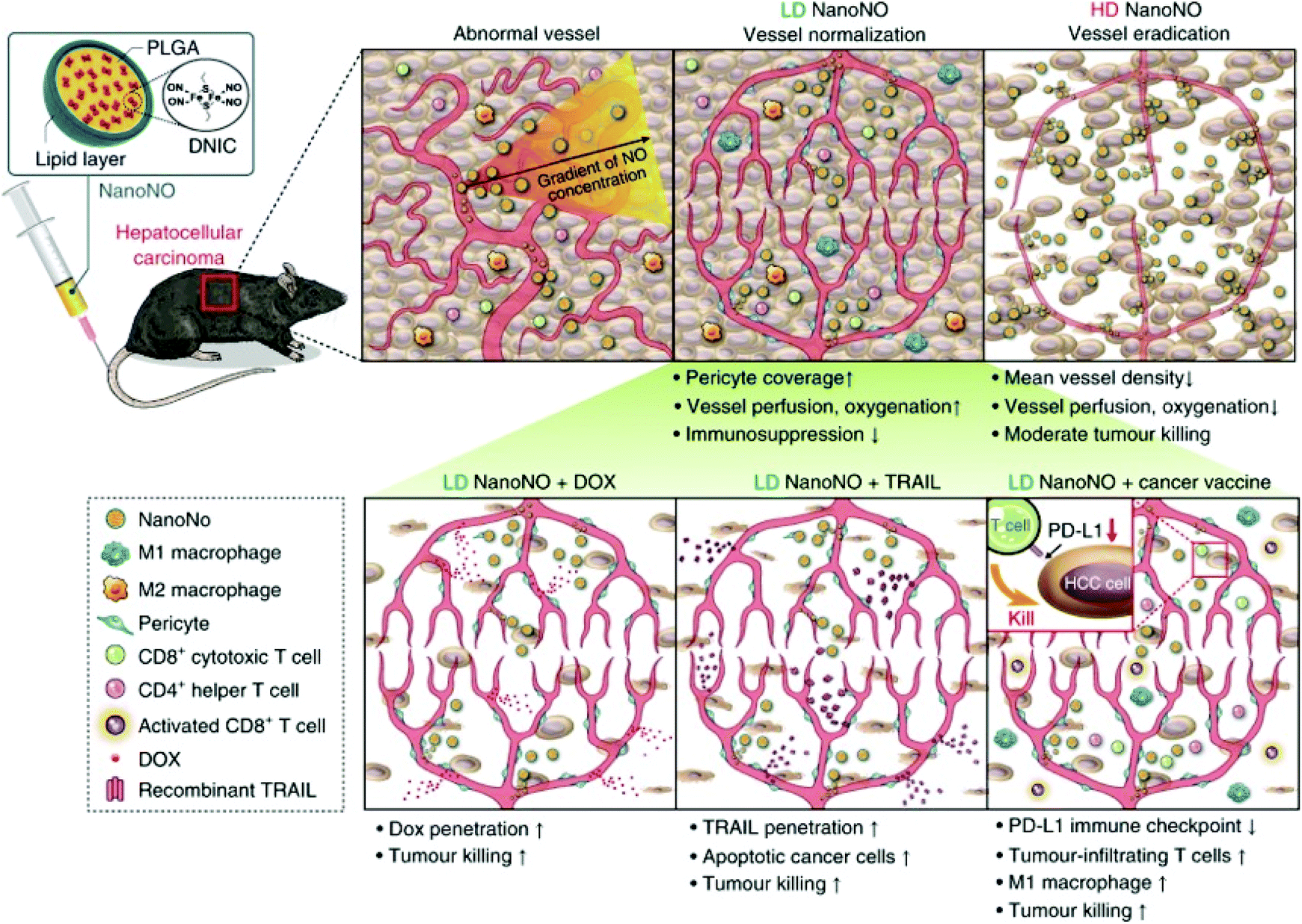 | ||
| Fig. 16 NO involvement in vasodilation therapy. Schematic showing the mechanism by which NanoNO suppresses HCC progression in mice.94 Reprinted with permission from ref. 94. Copyright 2019, Springer Nature. | ||
5. Conclusion and outlook
Since NO has been found to participate in numerous physiological and pathological processes in the human body, it has stimulated great interest from researchers. However, as a gas messenger, NO has a relatively short half-life of only 3–5 s and has no targeting ability. It can chemically react with many atoms and free radicals in the physiological environment. These chemical reactions can be used by researchers to regulate the pathological process of the human body, but simultaneously, chemical reactions in non-pathological parts are likely to cause adverse reactions. Also, different concentrations of NO have different physiological effects. For example, a low concentration of NO can be used to regulate vasodilation, nerve transmission, anti-inflammatory response and antioxidant response, but a low concentration of NO will promote tumor growth. In contrast, a high concentration of NO can inhibit the proliferation of tumor cells and accelerate the apoptosis of tumor cells. Because of the double-edged sword the role of NO plays in the physiological regulation process, it has attracted many researchers to develop various molecular structures and chemical materials to achieve controllable NO release spatially and temporally.This review mainly summarized NO nanomedicines. Because of their small size, surface, and interface effects, nanomedicines can penetrate freely in some tissue gaps, and thus many researchers wrapped appropriate NO donors in nanomedicines to overcome the above-mentioned shortcomings of NO to achieve its controlled release at a specific location. However, NO nanomedicine still faces many challenges, especially in the process of clinical trials. In clinical applications, NO, as a gas messenger that can act on the whole body, requires careful consideration of its physical and chemical properties and its accumulation in non-target organs and tissues to consider its potential toxicity. Also, some NO nanomedicines may cause oxidative stress, immune response, protein misfolding, and DNA damage. The existing NO nanomedicine uses the targeting properties of materials to release NO at a specific site, and the characteristics of nanomaterials can be used for slow release over time, but they still cannot precisely control the release concentration of NO and cannot be based on the characteristic of the lesion site releasing the appropriate concentration of NO. Furthermore, NO is a double-edged sword. If an appropriate concentration of NO cannot be used to treat the diseased area, it is very likely to promote further aggravation of the disease. Therefore, it is necessary to design and develop smarter NO nanomedicines that can release the appropriate concentration of NO according to the pathophysiological requirements, and can be turned on and off. When they are turned on, they should continuously release the appropriate concentration of NO at the pathological site. When they are turned off, the release of NO should stop. Also, they should remain in the diseased area until the next time they are turned on. In reality, the characteristics of nanomaterials can be used to achieve the coordinated release of NO.
In summary, although the development of nanomedicines can solve many of the shortcomings of the gas messenger NO as an emerging material, because of insufficient research, it also brings insecurity and deficiency due to its own characteristics. Therefore, to broaden the biological application prospects of NO nanomedicines, the characteristics of nanomedicines need to be studied more deeply.
Author contributions
The three authors contributed the same to this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key R&D Program of China (2018YFE0205400), National Natural Science Foundation of China (31671028, 81822041, 21977116, 81673305, 81773573 and 51873045), National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program” (Number: 2018ZX09711002-006-013), the Funding of Double First-Rate Discipline Construction (CPU2018GY06 and CPU2018GY18), open project of State Key Laboratory of Natural Medicines, No. SKLNMZZCX201824 and SKLNMZZ202029, and State Key Laboratory of Pathogenesis, Prevention and Treatment of High Incidence Diseases in Central Asia Fund (SKL-HIDCA-2018-1). Youth Innovation Promotion CAS (2017053).Notes and references
- Z. Huang, J. Fu and Y. Zhang, J. Med. Chem., 2017, 60, 7617–7635 CrossRef CAS.
- T. A. Heinrich, R. S. da Silva, K. M. Miranda, C. H. Switzer, D. A. Wink and J. M. Fukuto, Br. J. Pharmacol., 2013, 169, 1417–1429 CrossRef CAS.
- D. Basudhar, L. A. Ridnour, R. Cheng, A. H. Kesarwala, J. Heinecke and D. A. Wink, Coord. Chem. Rev., 2016, 306, 708–723 CrossRef CAS.
- D. A. Wink and J. B. Mitchell, Free Radical Biol. Med., 1998, 25, 434–456 CrossRef CAS.
- L. J. Ignarro, C. Napoli and J. Loscalzo, Circ. Res., 2002, 90, 21–28 CrossRef CAS.
- Y. Wang, T. Yang and Q. He, Natl. Sci. Rev., 2020, 7, 1485–1512 CrossRef CAS.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS.
- V. Wagner, A. Dullaart, A.-K. Bock and A. Zweck, Nat. Biotechnol., 2006, 24, 1211–1217 CrossRef CAS.
- L. M. Russell, C. H. Liu and P. Grodzinski, Biomaterials, 2020, 242, 119926 CrossRef CAS.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS.
- C. V. Rao, B. S. Reddy, V. E. Steele, C.-X. Wang, X. Liu, N. Ouyang, J. M. R. Patlolla, B. Simi, L. Kopelovich and B. Rigas, Mol. Cancer Ther., 2006, 5, 1530–1538 CrossRef CAS.
- M. C. Jen, M. C. Serrano, R. van Lith and G. A. Ameer, Adv. Funct. Mater., 2012, 22, 239–260 CrossRef.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS.
- S. P. Nichols, W. L. Storm, A. Koh and M. H. Schoenfisch, Adv. Drug Delivery Rev., 2012, 64, 1177–1188 CrossRef CAS.
- J. Kim, G. Saravanakumar, H. W. Choi, D. Park and W. J. Kim, J. Mater. Chem. B, 2014, 2, 341–356 RSC.
- D. A. Riccio and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3731–3741 RSC.
- M. J. Rose and P. K. Mascharak, Coord. Chem. Rev., 2008, 252, 2093–2114 CrossRef CAS.
- M. J. Rose and P. K. Mascharak, Curr. Opin. Chem. Biol., 2008, 12, 238–244 CrossRef CAS.
- N. L. Fry and P. K. Mascharak, Acc. Chem. Res., 2011, 44, 289–298 CrossRef CAS.
- Y. Fei, J. Wu, H.-W. An, K. Zhu, B. Peng, J. Cai, Y. Zhang, L.-L. Li, H. Wang and Z. Huang, J. Med. Chem., 2020, 63, 9127–9135 CrossRef CAS.
- L. Chen, Y. Zhang, X. Kong, E. Lan, Z. Huang, S. Peng, D. L. Kaufman and J. Tian, J. Med. Chem., 2008, 51, 4834–4838 CrossRef CAS.
- Y. Cheng, Y. Gong, S. Qian, Y. Mou, H. Li, X. Chen, H. Kong, W. Xie, H. Wang, Y. Zhang and Z. Huang, J. Med. Chem., 2018, 61, 1474–1482 CrossRef CAS.
- X. Ji, C. Zhou, K. Ji, R. E. Aghoghovbia, Z. Pan, V. Chittavong, B. Ke and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 15846–15851 CrossRef CAS.
- J. E. Saavedra, T. R. Billiar, D. L. Williams, Y.-M. Kim, S. C. Watkins and L. K. Keefer, J. Med. Chem., 1997, 40, 1947–1954 CrossRef CAS.
- J. E. Saavedra, P. J. Shami, L. Y. Wang, K. M. Davies, M. N. Booth, M. L. Citro and L. K. Keefer, J. Med. Chem., 2000, 43, 261–269 CrossRef CAS.
- K. Sharma, A. Iyer, K. Sengupta and H. Chakrapani, Org. Lett., 2013, 15, 2636–2639 CrossRef CAS.
- K. Sharma, K. Sengupta and H. Chakrapani, Bioorg. Med. Chem. Lett., 2013, 23, 5964–5967 CrossRef CAS.
- T. B. Cai, D. Lu, M. Landerholm and P. G. Wang, Org. Lett., 2004, 6, 4203–4205 CrossRef CAS.
- X. Wu, X. Tang, M. Xian and P. G. Wang, Tetrahedron Lett., 2001, 42, 3779–3782 CrossRef CAS.
- N. Barraud, B. G. Kardak, N. R. Yepuri, R. P. Howlin, J. S. Webb, S. N. Faust, S. Kjelleberg, S. A. Rice and M. J. Kelso, Angew. Chem., Int. Ed., 2012, 51, 9057–9060 CrossRef CAS.
- P. T. Burks, A. D. Ostrowski, A. A. Mikhailovsky, E. M. Chan, P. S. Wagenknecht and P. C. Ford, J. Am. Chem. Soc., 2012, 134, 13266–13275 CrossRef CAS.
- M.-F. Chung, H.-Y. Liu, K.-J. Lin, W.-T. Chia and H.-W. Sung, Angew. Chem., Int. Ed., 2015, 54, 9890–9893 CrossRef CAS.
- Z. Jin, Y. Wen, Y. Hu, W. Chen, X. Zheng, W. Guo, T. Wang, Z. Qian, B.-L. Su and Q. He, Nanoscale, 2017, 9, 3637–3645 RSC.
- Z. Huang, J. Wu, Y. Zou, H. Yuan, Y. Zhang, Y. Fei, A. Bhardwaj, J. Kaur, E. E. Knaus and Y. Zhang, J. Med. Chem., 2018, 61, 1833–1844 CrossRef CAS.
- R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS.
- E. L. Sievers and P. D. Senter, Annu. Rev. Med., 2013, 64, 15–29 CrossRef CAS.
- F. Sun, Y. Wang, X. Luo, Z. Ma, Y. Xu, X. Zhang, T. Lv, Y. Zhang, M. Wang, Z. Huang and J. Zhang, Cancer Res., 2019, 79, 3395–3405 CAS.
- H. J. Lee, D. E. Kim, D. J. Park, G. H. Choi, D.-N. Yang, J. S. Heo and S. C. Lee, Colloids Surf., B, 2016, 146, 1–8 CrossRef CAS.
- X. Jia, Y. Zhang, Y. Zou, Y. Wang, D. Niu, Q. He, Z. Huang, W. Zhu, H. Tian, J. Shi and Y. Li, Adv. Mater., 2018, 30, 1704490 CrossRef.
- L.-A. Tai, Y.-C. Wang and C.-S. Yang, Nitric Oxide, 2010, 23, 60–64 CrossRef CAS.
- D. J. Suchyta and M. H. Schoenfisch, Mol. Pharmaceutics, 2015, 12, 3569–3574 CrossRef CAS.
- W. Fan, N. Lu, P. Huang, Y. Liu, Z. Yang, S. Wang, G. Yu, Y. Liu, J. Hu, Q. He, J. Qu, T. Wang and X. Chen, Angew. Chem., Int. Ed., 2017, 56, 1229–1233 CrossRef CAS.
- P.-T. Kao, I. J. Lee, I. Liau and C.-S. Yeh, Chem. Sci., 2017, 8, 291–297 RSC.
- L. Tan, A. Wan, X. Zhu and H. Li, Chem. Commun., 2014, 50, 5725–5728 RSC.
- M. Guo, H.-J. Xiang, Y. Wang, Q.-L. Zhang, L. An, S.-P. Yang, Y. Ma, Y. Wang and J.-G. Liu, Chem. Commun., 2017, 53, 3253–3256 RSC.
- X. Zhang, G. Tian, W. Yin, L. Wang, X. Zheng, L. Yan, J. Li, H. Su, C. Chen, Z. Gu and Y. Zhao, Adv. Funct. Mater., 2015, 25, 3049–3056 CrossRef CAS.
- J. V. Garcia, J. Yang, D. Shen, C. Yao, X. Li, R. Wang, G. D. Stucky, D. Zhao, P. C. Ford and F. Zhang, Small, 2012, 8, 3800–3805 CrossRef CAS.
- K. Zhang, H. Xu, X. Jia, Y. Chen, M. Ma, L. Sun and H. Chen, ACS Nano, 2016, 10, 10816–10828 CrossRef CAS.
- C. Ratanatawanate, A. Chyao and K. J. Balkus, J. Am. Chem. Soc., 2011, 133, 3492–3497 CrossRef CAS.
- Z. Du, X. Zhang, Z. Guo, J. Xie, X. Dong, S. Zhu, J. Du, Z. Gu and Y. Zhao, Adv. Mater., 2018, 30, 1804046 CrossRef.
- J. Dolanský, P. Henke, Z. Malá, L. Žárská, P. Kubát and J. Mosinger, Nanoscale, 2018, 10, 2639–2648 RSC.
- K. Dong, E. Ju, N. Gao, Z. Wang, J. Ren and X. Qu, Chem. Commun., 2016, 52, 5312–5315 RSC.
- L. Tan, A. Wan and H. Li, ACS Appl. Mater. Interfaces, 2013, 5, 11163–11171 CrossRef CAS.
- J. Xu, F. Zeng, H. Wu, C. Hu, C. Yu and S. Wu, Small, 2014, 10, 3750–3760 CrossRef CAS.
- W. Fan, W. Bu, Z. Zhang, B. Shen, H. Zhang, Q. He, D. Ni, Z. Cui, K. Zhao, J. Bu, J. Du, J. Liu and J. Shi, Angew. Chem., Int. Ed., 2015, 54, 14026–14030 CrossRef CAS.
- C. Fowley, A. P. McHale, B. McCaughan, A. Fraix, S. Sortino and J. F. Callan, Chem. Commun., 2015, 51, 81–84 RSC.
- H.-J. Xiang, Q. Deng, L. An, M. Guo, S.-P. Yang and J.-G. Liu, Chem. Commun., 2016, 52, 148–151 RSC.
- H.-J. Xiang, M. Guo, L. An, S.-P. Yang, Q.-L. Zhang and J.-G. Liu, J. Mater. Chem. B, 2016, 4, 4667–4674 RSC.
- S. Yu, G. Li, R. Liu, D. Ma and W. Xue, Adv. Funct. Mater., 2018, 28, 1707440 CrossRef.
- P. Taladriz-Blanco, V. Pastoriza-Santos, J. Pérez-Juste and P. Hervés, Langmuir, 2013, 29, 8061–8069 CrossRef CAS.
- J. Gao, W. Zheng, J. Zhang, D. Guan, Z. Yang, D. Kong and Q. Zhao, Chem. Commun., 2013, 49, 9173–9175 RSC.
- A. W. Carpenter, D. L. Slomberg, K. S. Rao and M. H. Schoenfisch, ACS Nano, 2011, 5, 7235–7244 CrossRef CAS.
- A. W. Carpenter, B. V. Worley, D. L. Slomberg and M. H. Schoenfisch, Biomacromolecules, 2012, 13, 3334–3342 CrossRef CAS.
- A. W. Carpenter, K. P. Reighard, J. E. Saavedra and M. H. Schoenfisch, Biomater. Sci., 2013, 1, 456–459 RSC.
- H.-J. Xiang, L. An, W.-W. Tang, S.-P. Yang and J.-G. Liu, Chem. Commun., 2015, 51, 2555–2558 RSC.
- Y. Deng, F. Jia, S. Chen, Z. Shen, Q. Jin, G. Fu and J. Ji, Biomaterials, 2018, 187, 55–65 CrossRef CAS.
- J. Fan, Q. He, Y. Liu, F. Zhang, X. Yang, Z. Wang, N. Lu, W. Fan, L. Lin, G. Niu, N. He, J. Song and X. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 13804–13811 CrossRef CAS.
- L. Wang, Y. Chang, Y. Feng, X. Li, Y. Cheng, H. Jian, X. Ma, R. Zheng, X. Wu, K. Xu and H. Zhang, Nano Lett., 2019, 19, 6800–6811 CrossRef CAS.
- J. Zhang, H. Song, S. Ji, X. Wang, P. Huang, C. Zhang, W. Wang and D. Kong, Nanoscale, 2018, 10, 4179–4188 RSC.
- H. T. T. Duong, K. Jung, S. K. Kutty, S. Agustina, N. N. M. Adnan, J. S. Basuki, N. Kumar, T. P. Davis, N. Barraud and C. Boyer, Biomacromolecules, 2014, 15, 2583–2589 CrossRef CAS.
- M. Wan, H. Chen, Q. Wang, Q. Niu, P. Xu, Y. Yu, T. Zhu, C. Mao and J. Shen, Nat. Commun., 2019, 10, 966 CrossRef.
- Z. Shen, K. He, Z. Ding, M. Zhang, Y. Yu and J. Hu, Macromolecules, 2019, 52, 7668–7677 CrossRef CAS.
- N. Marino, S. Petralia, M. Perez-Lloret, J. Mosinger, S. Conoci and S. Sortino, J. Mater. Chem. B, 2016, 4, 5825–5830 RSC.
- J. Fan, N. He, Q. He, Y. Liu, Y. Ma, X. Fu, Y. Liu, P. Huang and X. Chen, Nanoscale, 2015, 7, 20055–20062 RSC.
- B. J. Heilman, J. St. John, S. R. J. Oliver and P. K. Mascharak, J. Am. Chem. Soc., 2012, 134, 11573–11582 CrossRef CAS.
- L. Tian, Y. Wang, L. Sun, J. Xu, Y. Chao, K. Yang, S. Wang and Z. Liu, Matter, 2019, 1, 1061–1076 CrossRef.
- D. Wang, L. Niu, Z.-Y. Qiao, D.-B. Cheng, J. Wang, Y. Zhong, F. Bai, H. Wang and H. Fan, ACS Nano, 2018, 12, 3796–3803 CrossRef CAS.
- J. Gehring, B. Trepka, N. Klinkenberg, H. Bronner, D. Schleheck and S. Polarz, J. Am. Chem. Soc., 2016, 138, 3076–3084 CrossRef CAS.
- Y. Hu, T. Lv, Y. Ma, J. Xu, Y. Zhang, Y. Hou, Z. Huang and Y. Ding, Nano Lett., 2019, 19, 2731–2738 CrossRef CAS.
- K. Nakanishi, T. Koshiyama, S. Iba and M. Ohba, Dalton Trans., 2015, 44, 14200–14203 RSC.
- Z. Sun, Z. Yi, X. Cui, X. Chen, W. Su, X. Ren and X. Li, Nanoscale, 2018, 10, 12109–12122 RSC.
- R. Guo, Y. Tian, Y. Wang and W. Yang, Adv. Funct. Mater., 2017, 27, 1606398 CrossRef.
- D. O. Schairer, J. S. Chouake, J. D. Nosanchuk and A. J. Friedman, Virulence, 2012, 3, 271–279 CrossRef.
- F. Rong, Y. Tang, T. Wang, T. Feng, J. Song, P. Li and W. Huang, Antioxidants, 2019, 8, 556 CrossRef CAS.
- C.-M. Ho, K.-J. Liao, C.-N. Lok and C.-M. Che, Chem. Commun., 2011, 47, 10776–10778 RSC.
- H. W. Choi, J. Kim, J. Kim, Y. Kim, H. B. Song, J. H. Kim, K. Kim and W. J. Kim, ACS Nano, 2016, 10, 4199–4208 CrossRef CAS.
- G. A. Spinas, R. Laffranchi, I. Francoys, I. David, C. Richter and M. Reinecke, Diabetologia, 1998, 41, 292–299 CrossRef CAS.
- F. Paneni, S. Costantino and F. Cosentino, World J. Diabetes, 2015, 6, 326–332 CrossRef.
- F. Yang, M. Li, Y. Liu, T. Wang, Z. Feng, H. Cui and N. Gu, J. Controlled Release, 2016, 228, 87–95 CrossRef CAS.
- F. Becquet, Y. Courtois and O. Goureau, Surv. Ophthalmol., 1997, 42, 71–82 CrossRef CAS.
- R. Chandrawati, J. Y. H. Chang, E. Reina-Torres, C. Jumeaux, J. M. Sherwood, W. D. Stamer, A. N. Zelikin, D. R. Overby and M. M. Stevens, Adv. Mater., 2017, 29, 1604932 CrossRef.
- D. S. Bredt and S. H. Snyder, Annu. Rev. Biochem., 1994, 63, 175–195 CrossRef CAS.
- Y.-C. Sung, P.-R. Jin, L.-A. Chu, F.-F. Hsu, M.-R. Wang, C.-C. Chang, S.-J. Chiou, J. T. Qiu, D.-Y. Gao, C.-C. Lin, Y.-S. Chen, Y.-C. Hsu, J. Wang, F.-N. Wang, P.-L. Yu, A.-S. Chiang, A. Y.-T. Wu, J. J.-S. Ko, C. P.-K. Lai, T.-T. Lu and Y. Chen, Nat. Nanotechnol., 2019, 14, 1160–1169 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |