
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlock copolymer–nanodiamond coassembly in solution: towards multifunctional hybrid materials†
Burhannudin
Sutisna
 *,
Stoffel D.
Janssens
,
Alessandro
Giussani
,
David
Vázquez-Cortés
and
Eliot
Fried
*
*,
Stoffel D.
Janssens
,
Alessandro
Giussani
,
David
Vázquez-Cortés
and
Eliot
Fried
*
Mathematics, Mechanics, and Materials Unit (MMMU), Okinawa Institute of Science and Technology Graduate University (OIST), 1919-1 Tancha, Onna-son, Kunigami-gun, Okinawa 904-0495, Japan. E-mail: burhannudin.sutisna@oist.jp; eliot.fried@oist.jp
First published on 5th January 2021
Abstract
Polymer–nanodiamond composites are excellent candidates for the fabrication of multifunctional hybrid materials. They integrate polymer flexibility and exceptional properties of nanodiamonds (NDs), such as biocompatibility, mechanical strength, color centers, and chemically-tailored surfaces. However, their development is hindered by the challenge of ensuring that NDs are homogeneously distributed in the composites. Here, we exploit colloidal coassembly between poly(isoprene-b-styrene-b-2-vinyl pyridine) (ISV) block copolymers (BCPs) and NDs to avoid ND self-agglomeration and direct ND spatial distribution. NDs were first air oxidized at 450 °C to obtain stable dispersions in dimethylacetamide (DMAc). By adding ISV into the dispersions, patchy hybrid micelles were formed due to H-bonds between NDs and ISV. The ISV–ND coassembly in DMAc was then used to fabricate nanocomposite films with a uniform sub-50 nm ND distribution, which has never been previously reported for an ND loading (ϕND) of more than 50 wt%. The films exhibit good transparency due to their well-defined nanostructures and smoothness and also exhibit an improved UV-absorption and hydrophilicity compared to neat ISV. More intriguingly, at a ϕND of 22 wt%, ISV and NDs coassemble into a network-like superstructure with well-aligned ND strings via a dialysis method. Transmission electron microscopy and dynamic light scattering measurements suggest a complex interplay between polymer–polymer, polymer–solvent, polymer–ND, ND–solvent, and ND–ND interactions during the formation of structures. Our work may provide an important foundation for the development of hierarchically ordered nanocomposites based on BCP–ND coassembly, which is beneficial for a wide spectrum of applications from biotechnology to quantum devices.
1 Introduction
Amalgams of hard and soft materials with hierarchical architectures exhibiting advantageous mechanical properties are omnipresent in nature, with common examples being wood,1 nacre,2 and bone.3 The mechanical effects of their molecular composition and multilevel assembly have evidenced the importance of designing hierarchically ordered composites for advanced materials. Analogously, polymer–nanoparticle composites have also been a benchmark in industry to produce tougher yet light and processable materials as in Bakelite,4 Toyota's nylon-mica nanocomposites,5 and carbon fiber reinforced polymers.6 Moreover, recent advances in nanotechnology have stimulated the development of bio-inspired artificial composites and hierarchical materials for biomedical applications and beyond.7,8There has been a growing attention over the past two decades for the development of polymer–nanodiamond composites to create advanced multifunctional materials, owing to the superior mechanical, optical and thermo-electrical properties of nanodiamonds (NDs) as well as their biocompatibility and rich surface chemistry.9,10 NDs are highly relevant in the coating, tribological, and biotech industries11,12 and are frequently used as seeding materials to grow polycrystalline diamond thin films for microdevices.13,14 Morimune et al.15 reported that a small addition of NDs (1 wt%) into poly(vinyl alcohol) (PVA) provided a higher reinforcement compared to that of carbon nanotubes and graphene oxide and increased the Young's modulus by a factor of 2.5 relative to that of neat PVA. The high transparency of the ND-PVA film was also maintained and accompanied by a significant improvement of thermal conductivity. NDs can also improve the performance and stability of supercapacitors when embedded into an electrochemically active polymer of polyanyline.16 More recently, Kulvelis and coworkers17 have also shown that NDs in a prefluorinated membrane used in hydrogen fuel cells can act as a proton reservoir at low temperatures and increase the membrane conductivity at high temperatures. Moreover in biotechnology, polymer–ND composites have shown great potential applications for gene delivery,18 bone scaffold materials,19 post-treatment of root canal therapy,20 and membrane bioreactors.21
Nevertheless, there is still a great challenge in controlling the nanoscopic distribution of NDs in polymeric matrices, as NDs tend to self-aggregate due to the interparticle van der Waals and electrostatic forces.22,23 Previous studies of polymer–ND composites have mainly focused on ND reinforcement properties with lack of nanostructural control, leading to poor nanoscopic distribution and reduced performance at high ND loadings.9,10 Meanwhile, scientists have tailored spatial distribution of various nanoparticles (NPs)24–26 using block copolymers (BCPs), which comprise chains of two or more incompatible macromolecules that self-assemble into ordered mesophases.27 The self-assembly of BCPs in bulk and as colloidal particles with internal morphology can be used to guide gold and silver NPs into hierarchical assembly within the BCP domains.28–30 The extent to which NP size influences their localization within BCP domains has also been discussed.31 BCP-guided NPs can improve reinforcement due to a higher interphase region and possible formation of ordered networks, in contrast to what can be achieved with NP-homopolymer. The nanoscopic arrangement of NPs can also affect optical, magnetic, chemical, electrical, and quantum properties of NPs, which may lead to advanced applications of nanocomposites.32,33 Surprisingly, the use of BCP self-assembly to guide nanoscopic distribution of NDs has rarely been explored. Research on BCP–ND nanocomposites has been limited to ND dispersion stability,34–36 biological applications,37 and elastomeric BCP reinforcements.38,39 Furthermore, in contrast to other spherical NPs, exploring BCP–ND nanocomposites may present a greater challenge due to the self-aggregation behavior of NDs.
In this work, the possibility of controlling the spatial distribution of NDs at the nanoscale is investigated using amphiphilic triblock terpolymers of poly(1,4-isoprene-b-styrene-b-2-vinyl pyridine) (PI-b-PS-b-P2VP). The overview of the materials systems and fabrication methods investigated here is visualized in Fig. 1a. Regular arrangements of NDs on the shells of ISV micelles that are dispersed in dimethylacetamide (DMAc) were obtained by promoting H-bond interactions between NDs and P2VP chains of the terpolymer. Such interactions were established by initially functionalizing NDs with carboxyls through air oxidation. The hybrid micelles can further be used as materials for building transparent and UV-protective films that accommodate well-dispersed NDs at a high ND loading of more than 50 wt%. We also show that ISV and NDs can coassemble into an ordered superstructure with a network-like polymeric matrix and well-aligned ND strings via a step-wise method, which demonstrates the potential ability of directing NDs into arrays over widely separated length scales.
 | ||
| Fig. 1 (a) Schematic of the investigated materials systems and fabrication methods. (b) FTIR and (c) Raman spectra of pristine and oxidized NDs, confirming the increase of O–H content and ND purity upon air oxidation. (d) High resolution DF-STEM (dark field scanning transmission electron microscopy) image unveiling the atomic structures of NDs oxidized at 450 °C. Some crystalline domains are highlighted with circles as a guide for the eye. | ||
2 Experimental
2.1 Materials
Detonation nanodiamond powders (NanoAmando Hard Hydrogel, primary particle size 3.9 ± 0.7 nm) were purchased from NanoCarbon Research Institute, Ltd. Poly(1,4-isoprene-block-styrene-block-2-vinyl pyridine) (ISV) block copolymers listed in Table 1 were procured from Polymer Source, Inc. (Canada). Dimethylacetamide (DMAc) (SAJ special grade), osmium tetroxide (OsO4, 4 wt% in H2O), acetone (≥99.0%), and ethanol (95%) were supplied by Sigma-Aldrich and used as received. Corning Lotus™ NXT Glass (alkaline earth boro-aluminosilicate, thickness 200 μm) and UV-grade fused silica glasses (thickness 520 μm) were used as substrates for BCP–ND film fabrication and Si wafers were used as substrates for Raman spectroscopy measurements. All substrates were cleaned by sonication in acetone (10 min) and isopropanol (10 min), followed by plasma cleaning in air for 60 s before solution casting.2.2 Preparation of ND dispersions and BCP–ND nanocomposites
The as-received ND powders were first subjected to air oxidation40 in a furnace at varying temperatures from 400 to 525 °C. After 5 h of oxidation, the oxidized NDs were collected and stored in a glass vial. Weighted amounts of oxidized NDs (50, 100, 150, 200, or 300 mg) were dispersed in 10 mL DMAc via sonication for 60 min using an Epishear probe sonicator with 1/8′′ probe operating at 70% amplitude. To avoid overheating, the sonication was run every 5 min with 1 s/1 s ON/OFF pulsing signal and a time interval of at least 10 min between each run. Alternatively, overheating was avoided by conducting the sonication with 1 s/3 s ON/OFF signal until reaching 60 min sonication in total. Sonicated dispersions were then centrifuged with a rotating speed of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rpm at 20 °C for 60 min to remove large aggregates. The supernatant around 75–80 vol% of the total centrifuged volume was transferred into a glass vial using a syringe and stored as a stock ND dispersion for the preparation of BCP–ND solution. The concentrations of NDs in the dispersions were analyzed using a thermogravimetric technique. The analysis was performed by a complete solvent-evaporation of 300 μL ND dispersion casted on a substrate that is heated on a hotplate at ≥60 °C overnight. Precise mass measurements of samples before and after solvent-evaporation were performed using an analytical balance with 0.01 mg resolution.
500 rpm at 20 °C for 60 min to remove large aggregates. The supernatant around 75–80 vol% of the total centrifuged volume was transferred into a glass vial using a syringe and stored as a stock ND dispersion for the preparation of BCP–ND solution. The concentrations of NDs in the dispersions were analyzed using a thermogravimetric technique. The analysis was performed by a complete solvent-evaporation of 300 μL ND dispersion casted on a substrate that is heated on a hotplate at ≥60 °C overnight. Precise mass measurements of samples before and after solvent-evaporation were performed using an analytical balance with 0.01 mg resolution.
To obtain self-assembled BCP–ND nanocomposites, weighted amounts of ISV terpolymer were dissolved in the previously-prepared ND dispersion in DMAc with a polymer concentration (Cpol) of 10 g L−1. By using different ND concentrations in the dispersion, the ND loading ϕND of the nanocomposites was varied from 22 to 55 wt%. Furthermore, BCP–ND films were fabricated by dropping 30 μL (for thin films) or 90 μL (for thick films) BCP–ND dispersion onto a 1 × 1 cm2 glass substrate, followed by complete solvent-evaporation overnight at an ambient temperature in a fumehood.
2.3 Dialysis of BCP–ND dispersions
For dialysis experiments, BCP–ND dispersions in DMAc (Cpol = 10 g L−1) were first diluted to Cpol = 1 g L−1 by adding an appropriate amount of DMAc. The experiments were conducted according to a method similar to one previously reported41 using a regenerated cellulose membrane with a molecular weight cut-off of 12–14 kDa (Spectrum Laboratories, Inc., Spectra/Por). The membrane was first soaked in Milli-Q water for >10 min, and was then washed with DMAc several times.Dialysis was performed using 3 mL BCP–ND dispersion (Cpol = 1 g L−1) against 300 mL acetone or ethanol. Proper stirring was applied in the dialysate chamber to maintain a uniform bulk concentration. A solvent-resistant nylon wire was used to hold the enclosed dialysis membrane (bag) ensuring that it remained completely immersed in the dialysate solution without touching the stirrer bar. The system was tightly capped to avoid the dialysate evaporation. The dialysate was changed with a fresh one after overnight dialysis and the operation was continued for several more hours. The dialyzed dispersion was carefully recovered using a syringe and was then stored in a glass vial for further characterizations.
Experimental details on characterization methods are provided in the ESI.†
3. Results and discussion
3.1 ND functionalization and dispersion
Stable dispersions of NDs in DMAc were obtained by first functionalizing ND surfaces with carboxyls via air oxidation.40 The completion of oxidation reactions was confirmed using Fourier-transform infrared (FTIR) spectroscopy. Fig. 1b displays FTIR spectra of ND powders treated at different oxidation temperatures for 5 hours. The displayed spectra have been normalized with respect to the O–H peak that is situated at ≈3400 cm−1. The result shows that as the temperature increases, the O–H stretch band (2700–3700 cm−1) broadens, indicating an increase in the O–H content on ND surfaces. The occurrence of the oxidation is also confirmed by the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak shifting from ≈1720 cm−1 for the pristine NDs to ≈1780 cm−1 after the oxidation, indicating the conversion of ketones and aldehydes into esters, lactones, anhydrides, or carboxylic acids. The conversion intensifies as the temperature increases from 400 to 475 °C as evidenced by the increasing CO peak intensity. However, the CO peak decreases from 500 °C, presumably due to further oxidation of the already-oxidized carbonyls (esters, lactones, anhydrides) into carboxylic acids, which increases the O–H content while maintaining that of CO, thereby reducing the CO to O–H ratio. The removal of graphitic carbons cannot be clearly seen from the C–H stretch bands (2850–2960 cm−1) since they are overlapped with the O–H band as the oxidation progresses. However, a slight decrease in the intensity of the CC peak (≈1625 cm−1) might indicate that the graphitic carbons are gradually removed as the oxidation temperature increases.
O peak shifting from ≈1720 cm−1 for the pristine NDs to ≈1780 cm−1 after the oxidation, indicating the conversion of ketones and aldehydes into esters, lactones, anhydrides, or carboxylic acids. The conversion intensifies as the temperature increases from 400 to 475 °C as evidenced by the increasing CO peak intensity. However, the CO peak decreases from 500 °C, presumably due to further oxidation of the already-oxidized carbonyls (esters, lactones, anhydrides) into carboxylic acids, which increases the O–H content while maintaining that of CO, thereby reducing the CO to O–H ratio. The removal of graphitic carbons cannot be clearly seen from the C–H stretch bands (2850–2960 cm−1) since they are overlapped with the O–H band as the oxidation progresses. However, a slight decrease in the intensity of the CC peak (≈1625 cm−1) might indicate that the graphitic carbons are gradually removed as the oxidation temperature increases.
Raman spectroscopy was then performed (Fig. 1c) to confirm the removal of graphitic carbons during the oxidation process. The measurements were carried out using three different specimens prepared by drop casting and drying DMAc-based suspensions of pristine NDs, NDs annealed at 450 °C, and NDs annealed at 525 °C. The spectra obtained from these specimens were normalized with respect to the graphite G-band peak (≈1600 cm−1); hence, the removal of graphitic carbons is evidenced by the increase in the intensity of the diamond peak (≈1325–1328 cm−1) as the oxidation temperature increases from 450 to 525 °C. The spectra also show that the characteristic disorder-induced double-resonance D-band of NDs peaks at ≈1400 cm−1. Since the Raman cross-section of the sp2 phase is about 50 to 230 times larger than that of diamond,42 the higher intensity of the diamond peak compared to G-band peak after the oxidation clearly shows that the annealing treatment has significantly improved the sp3 phase purity. It is important to recognize that the pristine ND spectra shows some inhomogeneity when measured at different specimen locations in contrast to the oxidized NDs. This suggests that the air oxidation leads to more homogenous distributions of NDs. Although similar spectra as those obtained for 450 °C-oxidized NDs can occasionally be found in pristine ND samples, the pristine ND spectra exhibit a larger shift of the diamond peak (with respect to the single crystal diamond) in comparison to those obtained from oxidized NDs, as shown in Fig. S1a of the ESI.† This shift indicates the presence of tensile strain in the NDs and from the magnitude of the shift we infer that on average the strain gradually reduces as the oxidation temperature increases from 450 to 525 °C.
The XRD spectra presented in Fig. S1b of the ESI† show that all the samples exhibit a diamond (111) Bragg reflection. The peaks are slightly shifted towards smaller 2θ angles with respect to the expected position of unstrained bulk diamond, revealing that the deposited nanodiamonds are characterized by a tensile strain in the range of 0.1–0.2%. In line with the Raman data, the specimen prepared from NDs oxidized at 525 °C displays the smallest shift relative to unstrained bulk diamond and hence the smallest strain. Furthermore, the XRD peak of the sample also features the narrowest full width at half maximum (FWHM), suggesting that annealing at 525 °C also narrows the distribution of lattice constants arising from crystal imperfections and impurities.
Although NDs oxidized at 525 °C exhibit the highest ND purity and O–H content, oxidation at 525 °C results in a significant weight loss, making it less economically viable. We also found that more particles were removed after centrifugation of the sonicated 525 °C-oxidized ND dispersion in DMAc compared to that of 450 °C-oxidized NDs at the same initial concentration and centrifugation conditions. This suggests that larger aggregates were formed by 525 °C-oxidized NDs after sonication in DMAc, presumably affected by an excessive O–H content that prompts additional interparticle H-bonds. As discussed hereinafter, 450 °C was accordingly chosen as the optimum temperature for ND oxidation during the preparation of BCP–ND nanocomposites. The atomic resolution scanning transmission electron microscopy (STEM) image of the ND particles oxidized at 450 °C, depicted in Fig. 1d, shows that a high crystallinity of the annealed NDs is maintained after oxidation. The average size of the crystallites can be estimated from the peak's FWHM of the XRD spectra using the Scherrer formula, suggesting an average size of 4 ± 1 nm, which is consistent with the size of the crystallites shown in the STEM image.
3.2 BCP–ND micellar coassembly
We further demonstrate the formation of BCP–ND hybrid micelles in DMAc using ISV1 (see Table 1). Before mixing with ISV1, oxidized NDs were first dispersed in DMAc via sonication and the dispersions were subsequently centrifuged. The 450 °C-annealed NDs can be dispersed well in DMAc with an ND mass concentration (Cf) of up to 17 g L−1, as indicated by the semi-transparent brownish solutions in Fig. 2 that are stable for months in contrast to that of unannealed NDs. The image also shows that the opacity of the dispersions increases as Cf increases, which is in qualitative agreement with the Beer–Lambert law. DLS measurements of the dispersions showed that for Cf values between 3 and 13 g L−1, the average hydrodynamic radii (![[R with combining macron]](https://www.rsc.org/images/entities/i_char_0052_0304.gif) ND) vary from 14 to 20 nm (see Table S1 of the ESI† for statistical data). These radii are comparable to the commonly reported BCP domain sizes that self-assemble in bulk or in solution, which suggests the presence of an entropically desirable condition for incorporating NDs into BCP domains. Also, a hydrodynamic radius of approximately 35 nm (see Table S1 of the ESI†) was observed at Cf = 17 g L−1, indicating a higher self-agglomeration tendency due to a closer interparticle distance and particle collisions by Brownian motion.
ND) vary from 14 to 20 nm (see Table S1 of the ESI† for statistical data). These radii are comparable to the commonly reported BCP domain sizes that self-assemble in bulk or in solution, which suggests the presence of an entropically desirable condition for incorporating NDs into BCP domains. Also, a hydrodynamic radius of approximately 35 nm (see Table S1 of the ESI†) was observed at Cf = 17 g L−1, indicating a higher self-agglomeration tendency due to a closer interparticle distance and particle collisions by Brownian motion.
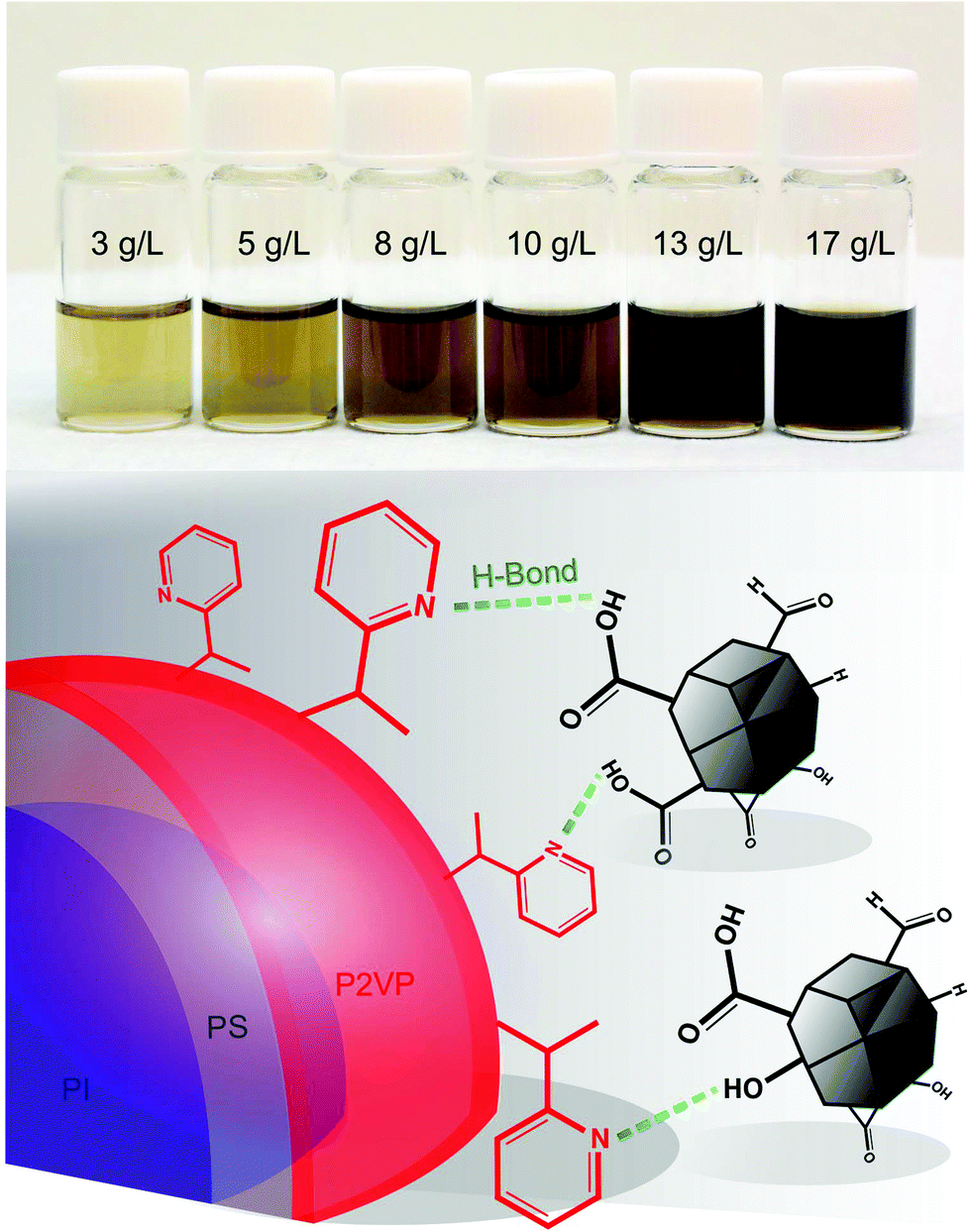 | ||
| Fig. 2 (Top) Photograph of oxidized-ND dispersions in DMAc at various mass concentrations after centrifugation and (bottom) schematic diagram of H-bond interactions between oxidized NDs and the P2VP corona (red color) of a PI-b-PS-b-P2VP micelle. The blue and gray regions correspond to PI and PS, respectively. | ||
ND dispersions with a Cf of 3–13 g L−1 were mixed with ISV1 at a polymer concentration (Cpol) of 10 g L−1, resulting in ND loading (ϕND) of 22–55 wt% in ISV1–ND nanocomposites (Table S1†). The mixture of ND dispersion at Cf = 17 g L−1 with ISV1 led to precipitation and turbidity and therefore was not used for the investigation. Calculation of the Flory–Huggins interaction parameters (χi,DMAc) presented in Table S2 of the ESI† show that χP2VP,DMAc < χPS,DMAc < χPI,DMAc; thus, while DMAc is a good solvent for P2VP it is a nonsolvent for PS and PI. Hence, ISV1 can be used to form core–shell micelles in DMAc. In these micelles, the PI chains form the micelle core, the core is encapsulated by PS domains, and P2VP corona is located on the outer shell. The pyridine groups of the P2VP corona can therefore interact with the NDs via H-bonds, as illustrated in Fig. 2.
The nanostructures of the ISV1–ND hybrid micelles in DMAc at various ϕND are displayed in Fig. 3. The TEM images show that the NDs form patches on the BCP micelle corona and that this effect becomes more pronounced as ϕND increases. It is noteworthy that the TEM images show aggregates, which presumably formed due to the relatively slow evaporation of DMAc during TEM sample preparation. As the morphology of the hybrid micelles at ϕND values of 22 and 34 wt% can be resolved from the individual micelles that are spotted next to the aggregates. Moreover, since the aggregates mostly feature strings of micelles with NDs on the interfaces between the micelles and at the end of the strings, we infer that the formation of symmetrical patches on the individual micelles is preferred, as illustrated by the schematic. Similarly, at a ϕND of 50 wt%, the micellar constituents of the aggregates are used to interpret the hybrid micelle morphology. To complement the TEM results and analyze the particle size distribution of the hybrid micelles in the dispersions, DLS measurements were performed as discussed hereinafter. The attachment of NDs onto the micelle corona is governed mainly by the H-bond interaction rather than by electrostatic forces, as illustrated in Fig. 2. This is supported by zeta potential measurements, which show that the ISV1 micelles in DMAc have a relatively neutral potential of approximately −4.7 mV, while the oxidized NDs in DMAc exhibit a negatively charged surface of −55.9 ± 3.5 mV. The localization of ND patches on ISV1 micelles at ϕND of 22 and 34 wt% is not yet understood. From the TEM images we infer that the tendency exists for NDs to form patches on two opposing sides of a micelle, as illustrated by the schematic in Fig. 3. However, cases where one or four patches are formed also exist.
 | ||
| Fig. 3 Particle size distributions (a, c and e) and micrographs (b, d and f) of ISV1–ND nanocomposites obtained from DLS and TEM measurements, respectively, of ISV1–ND dispersions in DMAc. The Gaussian distribution curves show how the particle hydrodynamic radii () vary at the polymer concentration (Cpol) of 5 g L−1 and 10 g L−1 and ND loading (ϕND) of 22, 34, and 50 wt%. The TEM samples were stained with OsO4 (darker phases represent the PI) and confirm the formation of core–shell hybrid micelles with ND patches that azimuthally distribute on the micelle corona depending on ϕND as depicted in the schematic drawings. | ||
DLS analyses of ISV1–ND dispersions are presented in Fig. 3a, c, and e as probability density functions of the particle hydrodynamic radii. At a Cpol of 10 g L−1 (blue-filled curves), the particle sizes increase as ϕND increases, as shown by the shift of average hydrodynamic radii () from approximately 28 nm for ϕND = 22 wt% to approximately 36 nm and 113 nm for ϕND = 34 and 50 wt%, respectively. Additionally, peak broadening indicates an increase in dispersity of the dispersed particles, as driven by an increasing tendency towards particle agglomeration with larger values of ϕND. At Cpol = 10 g L−1 and high ϕND, the interparticle distance appears to be sufficiently close for interparticle ND attraction to cause agglomeration. However, the agglomeration does not spoil the spherical shape of the individual micelles, as can be witnessed from the TEM images. We additionally found that diluting the 10 g L−1 dispersions to 5 g L−1 arrests interparticle agglomeration and leads to the recovery of the individual micelles. The DLS results of the 5 g L−1 dispersions reveal the size recovery of the individual micelles with = 28 ± 4 nm, consistent with the dimensions of the micellar constituents observed from the TEM images. Since ND coverage of micelles is proportional to ϕND, at Cpol = 5 g L−1 is found to increase slightly with ϕND.
Our results suggest that patchy hybrid micelles can be fabricated using BCP–ND coassembly via a simple dissolution method. The hybrid micelles may be beneficial as multicompartments for catalyis/biological applications, or can serve as building bricks for designing hierarchically ordered nanocomposites. In this work, we further demonstrate that the hybrid micelles can be utilized to fabricate nanocomposite films with BCP-guided ND distribution. Fig. 4 shows photographs of ISV1–ND films prepared by drop casting the dispersions on glass substrates followed by drying. The pristine ISV1 film (0 wt% NDs) exhibits some opacity caused by light scattering of the film nanostructure and surface roughness. The atomic force microscopy (AFM) depicted in Fig. 4a and b and SEM images depicted in Fig. 5 reveal that elongated structures with sizes of more than 100 nm are formed in the pristine ISV1 film. We hypothesize that these originate from micelle coalescence during solvent evaporation. Also, in the ND containing films, the presence of NDs stabilizes the micelles and preserves the spherical shape of the micelles during the film fabrication. This yields well-defined nanostructures with uniform domain sizes of approximate diameter 50 nm, as estimated from the SEM images, leading to a strong reduction in opacity evidenced from comparing the photographs of the ISV1 and ISV1–ND films. The SEM images in Fig. 5 also show that a uniform distribution of NDs with agglomerate sizes less than 50 nm can be obtained along the film surfaces and cross sections. The ND self-aggregation within the polymer can be significantly reduced, even at a ϕND as high as 55 wt%. However, at ϕND >55 wt%, precipitates were observed in the dispersions and severe cracks appeared in the films. The photographs in Fig. 4 show that the composite films have a brown color, indicating the presence of graphitic phases in the NDs. The reduction of transparency accompanied by these phases can be avoided in the future by using chemical vapor deposition (CVD)-based NDs43 that are highly transparent. Our nanocomposite films can however maintain acceptable levels of transparency at high values of ϕND, an outcome attributable to their nanostructures and the smoothness of their surfaces. As confirmed by the mean square roughness (Sq) and mean roughness (Sa) measurements from 5 × 5 μm2 AFM images that are depicted in Fig. 4b, d, f, and h, the film surfaces become smoother as ϕND increases.
 | ||
| Fig. 4 (Top) Photographs of ISV1–ND thin films (4 ± 1 μm thickness) at various ϕND casted on ca. 1 × 1 cm2 glass substrates, displaying the film's transparency after ND reinforcement accompanied by the increase in brownish color due to the sp2 carbon phase. (Bottom) Topography images of the corresponding ISV1–ND films at ϕND of (a and b) 0 wt%, (c and d) 22 wt%, (e and f) 50 wt%, and (g and h) 55 wt% at high and low magnifications, confirming the improvement in film's smoothness measured from the 5 × 5 μm2 AFM images. | ||
 | ||
| Fig. 5 SEM surface (top) and cross-sectional (bottom) images of ISV1–ND films at different ϕND casted on glass substrates. The images reveal the possibility of creating uniform ND distribution (white spots) in the polymeric matrix up to 55 wt% ND content both on the surface and along the film cross-sections. Scale bars on the insets are 100 nm. | ||
The fabricated films are capable of filtering harmful UV radiation in line with previous reports44,45 on nanodiamond–polymer composite films. These findings were confirmed by UV-Vis spectroscopy measurements yielding data presented in Fig. 6a and b. Although neat ISV1 can absorb UV radiation of wavelength below 280 nm, the addition of NDs can further extend the range of radiation filtering to approximately 400 nm. The spectra also show that the UV protection capacity is proportional to ϕND and the film thickness. This is evidenced by the highest absorption achieved by the thickest ISV1–ND film fabricated in this work with the largest ϕND, namely 44 wt%, as shown in Fig. 6b. The film nearly absorbs 85% of the UV radiation down to a wavelength of 320 nm. The high UV absorption is also accompanied by the film transparency, where around 60–70% of the visible light are transmitted according to the UV-Vis spectra. It is worth notifying that films with a thickness of 13 ± 1 μm, here referred to as thick films, with ϕND > 44 wt% were not characterized due to severe crack formation. The decrease in visible light (340–780 nm) transmittance as ϕND increases is consistent with the visual observation of the films discussed heretofore, which can be attributed to the sp2 inclusions of NDs.
 | ||
| Fig. 6 UV-Vis spectra of (a) 4 ± 1 μm and (b) 13 ± 1 μm thick ISV1–ND films with different ϕND, and (c) water contact angles of pristine ISV1 (0 wt% ϕND) and ISV1–ND films for various ϕND. Films with a thickness of 13 ± 1 μm and ϕND > 44 wt% were not characterized due to severe crack formation. | ||
Furthermore, the presence of NDs also improves the surface hydrophilicity, as shown in Fig. 6c. The contact angle θ between a water droplet and film surface decreases with ϕND, which is beneficial for anti-fouling properties in biological applications. To explain this trend qualitatively, we assume that the surface is homogeneously wetted and apply the Cassie equation46
| cosθ = xNDcosθND + (1 − xND)cosθISV1, | (1) |
| cosθ = rcosθ*, | (2) |
θ in eqn (1) takes the role of cosθ* in eqn (2). From the latter equation it can be inferred that for a hydrophobic surface (θ > 90°), surface roughness enlarges θ. On the contrary, for a hydrophilic surface (θ < 90°), surface roughness diminishes θ. From this, we deduce that θISV1 and θND obtained in this work might be overestimated and underestimated, respectively. We conclude that under the assumption of homogeneous wetting, the presence of polar functional groups on the surface of the NDs is responsible for the decrease in θ and that surface roughness might influence θ but not invert the observed trend.
3.3 BCP–ND self-assembled superstructures
We further studied the prospect of using our ISV1–ND nanocomposites to form hierarchically ordered superstructures. A step-wise dialysis method was applied to promote the structure formation in which the solvent (DMAc) of the ISV1–ND dispersion was gradually replaced by acetone or ethanol via a semi-permeable membrane. A similar method has been previously studied by Müller and Gröschel groups to fabricate multicompartment nanostructures (MCNs) based on triblock terpolymers.41,48,49 By tailoring the polymer–solvent interaction and BCP architectures, various MCN geometries with a wide variety of internal morphologies can be achieved. Adaptation of such a concept to hybrid systems containing BCPs and gold nanoparticles (AuNPs) has also been demonstrated, resulting in regular arrangements of AuNPs within self-assembled polymeric matrices via a step-wise nonsolvent addition50 or emulsification.51 Leveraging the step-wise coassembly to BCP–ND amphiphiles provides a greater challenge due to the self-aggregating nature of NDs, yet it can be beneficial for the fabrication of multifunctional hybrid materials with complex nanostructures.By applying the step-wise method, we find that the copolymer blocks and NDs rearrange simultaneously into a superstructure with a morphology completely different from that of neat ISV1, as evidenced by Fig. 7a–c. To more clearly visualize the suggested superstructures, overfocus TEM images are provided in Fig. S3 of the ESI,† as are lower magnification images. In the absence of NDs, ISV1 self-assembled, in acetone, into a superstructure with a lamellar order. The copolymer chain mobility in acetone is higher since acetone is a good solvent for P2VP and it has a lower Florry–Huggins interaction parameter χij with PS and PI relative to the interaction parameter χij of DMAc with PS/PI (Table S2 of the ESI†). The higher chain mobility of PS and PI in acetone compared to that in DMAc causes the micelles to merge and form a more ordered morphology, namely the lamellar superstructure. It is noteworthy that ordering is also influenced by the molecular weight composition of ISV1. In the presence of NDs, coassembly between ISV1 and NDs occured and resulted in a network-like superstructure at ϕND = 22 wt% as shown in Fig. 7b. However, as ϕND increased to 50 wt% (Fig. 7c), a less-ordered morphology with randomly distributed NDs formed, indicating an order-to-disorder transition at high values of ϕND. Similar phenomena were observed in previous reports describing techniques for using H-bond forming molecules to tailor the morphologies of bulk BCPs52–54 and BCP membranes.55,56 The molecules were found to influence BCP chain stretching and micelle rigidity, or to form subphases that are selectively miscible with one of the copolymer blocks. In our case, NDs containing H-bonding moieties (COOH and OH) are found to affect the ISV1 chain mobility and to form a distinct phase which is selectively miscible with P2VP domains, resulting in morphological transitions of the terpolymer.
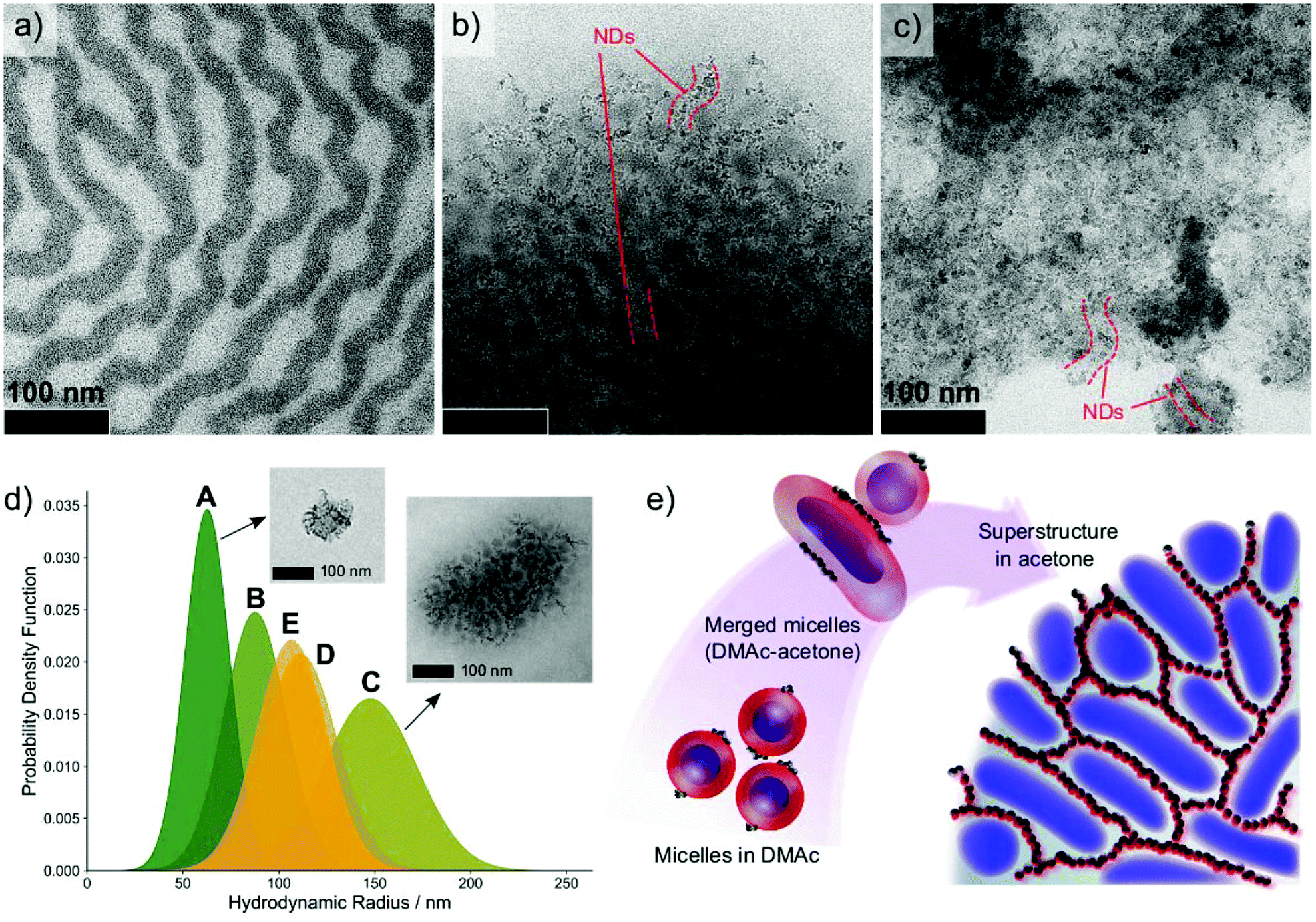 | ||
| Fig. 7 TEM images of (a) ISV1 terpolymer, (b) ISV1–ND at ϕND = 22 wt%, and (c) ISV1–ND at ϕND = 50 wt% from the dispersions in acetone after dialysis. The dark phases are PI domains stained with OsO4, while ND domains are partially highlighted by dashed red lines. For more clarity, overfocus TEM images are provided in Fig. S3 of the ESI† along with lower magnification images. (d) Particle size distribution of various acetone-dialyzed BCP–ND dispersions after passing through a 5 μm syringe filter: A. ISV1–ND at ϕND = 50 wt%, B. ISV1–ND at ϕND = 55 wt%, C. ISV1–ND at ϕND = 22 wt%, D. ISV2–ND at ϕND = 22 wt%, and E. ISV3–ND at ϕND = 22 wt%. The TEM image insets are the typical aggregates formed in the dispersions. (e) Proposed mechanism of the BCP–ND nanocomposite (22 wt% NDs) superstructure formation upon solvent exchange from DMAc to acetone. The schematic illustration elucidates how the oxidized NDs attached on the P2VP corona (red color) arrange into strings guided by the BCP self-assembly. The presence of acetone increases the mobility of PS (gray color), which leads the micelle corona to merge and the soft PI core (blue color) to coalesce. As the dispersion is richer with acetone, the BCP–ND coassembles to a 3D network with well-organized interconnected ND strings. | ||
We emphasize that the dialysis against acetone resulted in the formation of turbid supernatant accompanied by some precipitates. The TEM images depicted in Fig. 7a–c represent local observations of precipitate morphology. Concurrently, the particles that are dispersed in the supernatant, further called aggregates, exhibit a morphology similar to that of the precipitates with average particle diameters between 100–300 nm, as discussed hereinafter. The formation micron scale precipitates with uniform internal morphology indicates that the generated superstructures exhibit a long-range order. Additionally, it implies that the BCP–ND coassembly can be directed to form arrayed nanostructures over manifold length scales, which is essential for the fabrication of functional devices.
Since both the precipitates and the suspended aggregates have the same internal morphology, we assume that the precipitates were formed kinetically from the aggregates as their concentration increases during the dialysis process. To better understand the colloidal coassembly of ISV1–ND during the dialysis process, the supernatants were analyzed using DLS after filtering the dialyzed ISV1–ND dispersions through 5 μm pore-size polytetrafluoroethylene (PTFE) syringe filters. The DLS results displayed in Fig. 7d show that the ISV1–ND dispersion with ϕND = 22 wt% (curve C) comprises larger aggregates than those obtained for higher values of ϕND (curves A and B). This finding is supported by the TEM image insets of curves A and C, which display the morphology of aggregates formed in the corresponding dispersions. This phenomenon can be related to the influence of NDs on polymer chain mobility, as discussed below.
A sufficiently high ϕND (50 and 55 wt%) stabilizes the BCP micelles in the dispersion and obstructs the copolymer chain mobility to further coalesce and coassemble into a larger aggregate as in the case of ϕND = 22 wt% and, hence, to form smaller aggregates. Fig. 7d also shows that aggregates from ISV1–ND with ϕND = 55 wt% (curve B) are larger than those obtained for ϕND = 50 wt% (curve A); this is because the initial micelle size and ND–ND interactions increase with ϕND. At ϕND = 22 wt%, larger aggregates with an ordered morphology were formed, indicating that, at such ND loading, chain mobility is sufficient to allow micelles to merge and self-assemble into a superstructure with cylindrical PI cores in an interconnected PS matrix, while the P2VP domain stabilizes the NDs at the interfaces. Accordingly, NDs that are attached onto the P2VP chains by H-bonds assemble into interconnected strings, as illustrated in Fig. 7e.
At ϕND = 22 wt%, similar morphologies (shown in Fig. S3 of the ESI†) were also attained using ISV terpolymers with different molecular weight compositions, namely ISV2 and ISV3 (see Table 1). The DLS measurements of ISV2–ND and ISV3–ND dispersions in acetone (curves D and E of Fig. 7d, respectively) reveal the formation of smaller aggregates compared to that of ISV1–ND. This result can be explained by analysing the molar volume ratio of P2VP to PS (VP2VP/VPS) in the corresponding ISV terpolymers. The molar volume of block i was calculated by dividing its molecular weight by its mass density ρi, with ρPS = 1.04 kg L−1and ρP2VP = 1.14 kg L−1.57 We find that as VP2VP/VPS decreases from ISV1 to ISV3 (ISV1 = 0.21, ISV2 = 0.20, and ISV3 = 0.17), the average hydrodynamic radii of the aggregates decrease. The compositional analysis shows P2VP and PS are more compatible with acetone than is PI. This implies that P2VP and PS dominate the copolymer chain mobility during the formation of aggregates. Moreover, of the polymer block components we considered, P2VP is the most soluble in acetone, and the attachment of NDs on P2VP further increases its influence on aggregate formation due to the interparticle ND attraction. We accordingly infer from the experimental results that with higher P2VP to PS ratios the tendency to form larger aggregates is enhanced.
The strong influence of NDs in the formation of agglomerates during ISV1–ND coassembly was also corroborated by dialyzing neat ISV1 and ISV1–ND dispersions in DMAc against ethanol (EtOH). TEM and DLS measurements of neat ISV1 dialyzed against EtOH (Fig. S5a and S5c†) suggest that a stable and monodispersed ISV1 dispersion in EtOH with of 29 nm was obtained without spoiling the core–shell morphology of the micelles. Since EtOH is a good solvent for P2VP and a nonsolvent for PS and PI (Table S1†), the P2VP corona can stabilize neat ISV1 core–shell micelles and micelle agglomeration is avoided when changing the solvent from DMAc to EtOH. On the other hand, due to the gluing effect of NDs, the ISV1–ND micelles tend to agglomerate, without changes to their individual micellar shape, after exchanging the solvent from DMAc to EtOH (Fig. S5b and S5c†). The formation of agglomerates might be affected by the intermicelle-ND attraction, which is more favorable in EtOH due to the lower polarity index of EtOH (P′EtOH = 4.3) relative to that of DMAc (P′DMAc = 6.5).58
In summary, our experimental results suggest a complex interplay between BCP self-assembly in solutions, BCP–ND interactions, and ND colloidal behaviors. Whereas a proper interaction between BCPs and a selective solvent is required to generate a desired BCP self-assembled morphology, a preferential compatibility between NDs and BCPs is also crucial to achieving thermodynamically stable ordered structures with well dispersed NDs. The presence of preferential H-bond interactions between BCPs and NDs revealed in this work qualitatively demonstrates the importance of minimizing the enthalpy of mixing between BCP and NDs. Furthermore, our experimental results show the influence of ND loading ϕND on the polymer chain mobility, which suggests a similar behavior to the incorporation of NPs into BCPs in bulk, that involves polymer-chain conformational entropy and translational entropy of NPs.59,60 Our findings also suggest that ND self-agglomeration further complicates not only the thermodynamics but also the kinetics of BCP–ND coassembly. On the thermodynamic side, this might give rises to a free energy penalty by ND–ND interactions qualitatively similar to energetic contributions due to rod–rod interactions in BCP-nanorod blends investigated by Thorkelsson and co-workers.61 On the kinetic side, ND self-agglomeration that occurs for sufficiently high values of ϕND might increase the activation energy barrier for interdomain diffusion and thereby impede long-range diffusion, as observed for high loadings of AuNPs in supramolecular nanocomposites reported by Kao et al.62 Additionally, apart from the fact that BCP self-assembly can guide ND arrays, the formation of ND strings observed in our experiments, either in the patchy hybrid micelles or in the self-assembled superstructures, may point to a tendency for NDs to self-assemble in solution. This resonates with a recent report by Chang et al.63 concerning the dynamic self-assembly of NDs in water that can form strings due to electrostatic forces, chemical functionalities, and surrounding medium. Such colloidal behavior of NDs may contribute to an entropic factor related to the aggregate orientation as described previously for NP localization in BCP micelles.64
4 Conclusions
The formation of hierarchically ordered nanocomposites based on coassembly between block copolymers (BCPs) and nanodiamonds (NDs) has been investigated by initially fabricating patchy hybrid micelles in DMAc. H-Bond interaction between BCPs and NDs promoted by the chemical functionalization of the ND surface is one of the key factors in the patchy structure formation, in tandem with the sonication and centrifugation processes. Fig. 8 summarizes the advantages and intrinsic importance of the BCP–ND coassembly investigated in our work. Firstly, the hybrid micelles can be used as building materials for the manufacture of UV-protective, hydrophilic, and transparent films with a remarkably uniform ND distribution, even at high ND loadings exceeding 50 wt%. Previously reported polymer–ND composites44,45 exhibited a trade-off between ND loadings (namely UV shielding performance) and film transparency due to ND self-aggregation. These studies reported that with UV absorption (320 nm wavelength) of approximately 60–85% at an ND loading of 40 wt%, the visible light transmittance of the composites was lower than 40%. In contrast, our BCP–ND nanocomposites maintain visible light transmittance of more than 60% with UV-absorption (320 nm wavelength) of 85% at an ND loading of 44 wt%, making it comparable with UV-shielding and transparency performance of nanocomposite films incorporated with inorganic nanoparticles, such as TiO2, ZnO, and CeO2.65–67 This result clearly corroborates a key advantage of our BCP–ND nanocomposites. | ||
| Fig. 8 Advantages and necessity of the BCP–ND colloidal coassembly investigated in this work, which comprise as-cast film formation and step-wise coassembly of BCP–ND. The as-cast films prepared via drop casting exhibit high performance UV-protective and transparent coating, and the step-wise coassembly demonstrates the possibility of forming hybrid multicompartment micelles and superstructures with nanoscale control of ND arrangement over multiple length scales. | ||
Secondly, our patchy hybrid micelles also exhibit potential utility as objects for building self-assembled superstructures and hybrid multicompartment micelles (aggregates) with well-organized ND arrays via a step-wise dialysis method against acetone. The BCP–ND nanocomposites with an ND loading of 22 wt% were found to possess a fascinating network-like morphology which exhibits long-range order. The structure features interconnected ND strings within a self-assembled polymeric matrix as hybrid multicompartment micelles and self-assembled superstructures. This result also underscores the possibility of controlling nanoscale ND arrangement in polymeric nanocomposites, which has never been previously investigated. The formation mechanism of the reported structures was studied by performing TEM and DLS analyses which testify to the indispensable roles of polymer–polymer, polymer–solvent, polymer–ND, ND–solvent, and ND–ND interactions in determining the nanocomposite morphology. Our work may lead to a scalable bottom-up strategy for guiding ND assembly into arrays. Such a strategy would benefit a wide spectrum of applications in biotechnology, high performance coating, energy harvesting and storage, and quantum devices.
Despite the complexity of BCP–ND colloidal coassembly, we suppose that wet-based techniques are preferable to dry alternatives like extrusion and milling since they can improve polymer chain mobility and reduce ND self-agglomeration by solvation. Further study is, however, needed to discover pathways to different possible combinations of ND surface chemical modifications, polymer functionalities, solvent polarity, and fabrication routes. Owing to the biocompatibility of NDs, it might also be interesting to investigate methods for forming multicompartment nanostructures68,69 based on BCP and NDs. Since BCPs have been recognized as synthetic materials that can be used to construct models for biological systems,70,71 our findings may also provide a foundation for advanced bio-applications of NDs. Furthermore, a systematic theoretical study of BCP–ND coassembly in solution is also highly desired to better understand the kinetics of structure formation, predict other possible thermodynamically stable structures, and obtain optimum conditions for achieving targeted material properties.
In addition, since assembling NDs into arrays is crucial for their applications in quantum sensings72,73 and quantum information processings,74 our work may open a new avenue for directing arrays of NDs with quantum properties. Previous works have been performed to direct fluorescent ND assembly including the use of lithography techniques,75,76 or biological molecules.77,78 However, to the best of our knowledge, our approach is an unprecedented strategy to guide ND nanoscopic arrangement by using BCP self-assembly, which can be more scalable for designing quantum devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the Okinawa Institute of Science and Technology Graduate University (OIST) with subsidy funding from the Cabinet Office, Government of Japan. B. Sutisna acknowledges funding from the Japanese Society for the Promotion of Science (Grants-in-Aid for Early-Career Scientists, Grant No. 20K15347). The authors thank T. Sasaki for the technical guidance on TEM & SEM measurements, N. Ishizu for the assistance on SEM microscopy, A. Matsumoto and Micro/Bio/Nanofluidics Unitat OIST for the guidance and accesses on the DLS and zeta potential measurements, and N. Moreno for arranging the availability of ISV block copolymers.References
- D. Fengel and G. Wegener, Wood: Chemistry, Ultrastructure, Reactions, Walter de Gruyter, Berlin, 2011, pp. 6–25 Search PubMed.
- A. Jackson, J. F. Vincent and R. Turner, Proc. R. Soc. London, Ser. B, 1988, 234, 415–440 Search PubMed.
- J.-Y. Rho, L. Kuhn-Spearing and P. Zioupos, Med. Eng. Phys., 1998, 20, 92–102 CrossRef CAS.
- L. H. Baekeland, Sci. Am., 1909, 68, 322–323 CrossRef.
- A. Usuki, Y. Kojima, M. Kawasumi, A. Okada, Y. Fukushima, T. Kurauchi and O. Kamigaito, J. Mater. Res., 1993, 8, 1179–1184 CrossRef CAS.
- E. Fitzer, Carbon, 1989, 27, 621–645 CrossRef.
- K. Ariga, X. Jia, J. Song, J. P. Hill, D. T. Leong, Y. Jia and J. Li, Angew. Chem., Int. Ed., 2020, 59, 15424–15446 CrossRef CAS.
- G. Yang, X. Li, Y. He, J. Ma, G. Ni and S. Zhou, Prog. Polym. Sci., 2018, 81, 80–113 CrossRef CAS.
- V. N. Mochalin and Y. Gogotsi, Diamond Relat. Mater., 2015, 58, 161–171 CrossRef CAS.
- P. Karami, S. S. Khasraghi, M. Hashemi, S. Rabiei and A. Shojaei, Adv. Colloid Interface Sci., 2019, 269, 122–151 CrossRef CAS.
- A. Krüger, Nanodiamonds, John Wiley & Sons, Weinheim, 2010, pp. 329–387 Search PubMed.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2012, 7, 11–23 CrossRef CAS.
- S. D. Janssens, D. Vázquez-Cortés, A. Giussani, J. A. Kwiecinski and E. Fried, Diamond Relat. Mater., 2019, 98, 107511 CrossRef CAS.
- S. D. Janssens, B. Sutisna, A. Giussani, J. A. Kwiecinski, D. Vázquez-Cortés and E. Fried, Appl. Phys. Lett., 2020, 116, 193702 CrossRef CAS.
- S. Morimune, M. Kotera, T. Nishino, K. Goto and K. Hata, Macromolecules, 2011, 44, 4415–4421 CrossRef CAS.
- I. Kovalenko, D. G. Bucknall and G. Yushin, Adv. Funct. Mater., 2010, 20, 3979–3986 CrossRef CAS.
- Y. V. Kulvelis, O. Primachenko, A. Odinokov, A. Shvidchenko, V. Y. Bayramukov, I. Gofman, V. Lebedev, S. Ivanchev, A. Y. Vul and A. Kuklin, et al. , Fullerenes, Nanotubes, Carbon Nanostruct., 2020, 28, 140–146 CrossRef CAS.
- X.-Q. Zhang, M. Chen, R. Lam, X. Xu, E. Osawa and D. Ho, ACS Nano, 2009, 3, 2609–2616 CrossRef CAS.
- Q. Zhang, V. N. Mochalin, I. Neitzel, I. Y. Knoke, J. Han, C. A. Klug, J. G. Zhou, P. I. Lelkes and Y. Gogotsi, Biomaterials, 2011, 32, 87–94 CrossRef CAS.
- D.-K. Lee, T. Kee, Z. Liang, D. Hsiou, D. Miya, B. Wu, E. Osawa, E. K.-H. Chow, E. C. Sung and M. K. Kang, et al. , Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9445–E9454 CrossRef CAS.
- H. Etemadi, R. Yegani and M. Seyfollahi, Sep. Purif. Technol., 2017, 177, 350–362 CrossRef CAS.
- L.-Y. Chang, E. Ōsawa and A. S. Barnard, Nanoscale, 2011, 3, 958–962 RSC.
- A. Shvidchenko, E. Eidelman, A. Y. Vul, N. Kuznetsov, D. Y. Stolyarova, S. Belousov and S. Chvalun, Adv. Colloid Interface Sci., 2019, 268, 64–81 CrossRef CAS.
- A. Haryono and W. H. Binder, Small, 2006, 2, 600–611 CrossRef CAS.
- B. Sarkar and P. Alexandridis, Prog. Polym. Sci., 2015, 40, 33–62 CrossRef CAS.
- C. Yi, Y. Yang, B. Liu, J. He and Z. Nie, Chem. Soc. Rev., 2020, 49, 465–508 RSC.
- F. S. Bates and G. Fredrickson, Phys. Today, 2000, 52, 32–38 CrossRef.
- S. Sanwaria, A. Horechyy, D. Wolf, C.-Y. Chu, H.-L. Chen, P. Formanek, M. Stamm, R. Srivastava and B. Nandan, Angew. Chem., Int. Ed., 2014, 53, 9090–9093 CrossRef CAS.
- J. Kao, K. Thorkelsson, P. Bai, Z. Zhang, C. Sun and T. Xu, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- M. Xu, K. H. Ku, Y. J. Lee, J. J. Shin, E. J. Kim, S. G. Jang, H. Yun and B. J. Kim, Chem. Mater., 2020, 32, 7036–7043 CrossRef CAS.
- M. R. Bockstaller, Y. Lapetnikov, S. Margel and E. L. Thomas, J. Am. Chem. Soc., 2003, 125, 5276–5277 CrossRef CAS.
- S. I. Stupp and P. V. Braun, Science, 1997, 277, 1242–1248 CrossRef CAS.
- M. R. Bockstaller and E. L. Thomas, Phys. Rev. Lett., 2004, 93, 166106 CrossRef.
- J. Cheng, J. He, C. Li and Y. Yang, Chem. Mater., 2008, 20, 4224–4230 CrossRef CAS.
- R.-M. Chin, S.-J. Chang, C.-C. Li, C.-W. Chang and R.-H. Yu, J. Colloid Interface Sci., 2018, 520, 119–126 CrossRef CAS.
- T. Tiainen, T. T. Myllymäki, T. Hatanpää, H. Tenhu and S. Hietala, Diamond Relat. Mater., 2019, 95, 185–194 CrossRef CAS.
- J. W. Lee, S. Lee, S. Jang, K. Y. Han, Y. Kim, J. Hyun, S. K. Kim and Y. Lee, Mol. BioSyst., 2013, 9, 1004–1011 RSC.
- A. Ozerin, T. Kurkin, G. Alkhanishvili, A. Kechek'yan, O. Gritsenko, N. Perov, L. Ozerina, M. Beshenko and V. Y. Dolmatov, Nanotechnol. Russ., 2009, 4, 480–488 CrossRef.
- A. Shakun, J. Vuorinen, M. Hoikkanen, M. Poikelispää and A. Das, Composites Part, A, 2014, 64, 49–69 CrossRef CAS.
- S. Osswald, G. Yushin, V. Mochalin, S. O. Kucheyev and Y. Gogotsi, J. Am. Chem. Soc., 2006, 128, 11635–11642 CrossRef CAS.
- A. H. Gröschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther and A. H. Müller, Nat. Commun., 2012, 3, 710 CrossRef.
- A. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 121405 CrossRef.
- S. Heyer, W. Janssen, S. Turner, Y.-G. Lu, W. S. Yeap, J. Verbeeck, K. Haenen and A. Krueger, ACS Nano, 2014, 8, 5757–5764 CrossRef CAS.
- K. D. Behler, A. Stravato, V. Mochalin, G. Korneva, G. Yushin and Y. Gogotsi, ACS Nano, 2009, 3, 363–369 CrossRef CAS.
- N. Attia, J. Rao and K. Geckeler, J. Nanopart. Res., 2014, 16, 2361 CrossRef.
- A. Cassie, Discuss. Faraday Soc., 1948, 3, 11–16 RSC.
- R. N. Wenzel, Ind. Eng. Chem., 1936, 28, 988–994 CrossRef CAS.
- A. H. Gröschel, A. Walther, T. I. Löbling, F. H. Schacher, H. Schmalz and A. H. Müller, Nature, 2013, 503, 247–251 CrossRef.
- T. I. Löbling, O. Borisov, J. S. Haataja, O. Ikkala, A. H. Gröschel and A. H. Müller, Nat. Commun., 2016, 7, 12097 CrossRef.
- Y. Liu, Y. Li, J. He, K. J. Duelge, Z. Lu and Z. Nie, J. Am. Chem. Soc., 2014, 136, 2602–2610 CrossRef CAS.
- N. Yan, X. Liu, J. Zhu, Y. Zhu and W. Jiang, ACS Nano, 2019, 13, 6638–6646 CrossRef CAS.
- S. Valkama, H. Kosonen, J. Ruokolainen, T. Haatainen, M. Torkkeli, R. Serimaa, G. ten Brinke and O. Ikkala, Nat. Mater., 2004, 3, 872–876 CrossRef CAS.
- S. Valkama, T. Ruotsalainen, A. Nykänen, A. Laiho, H. Kosonen, G. Ten Brinke, O. Ikkala and J. Ruokolainen, Macromolecules, 2006, 39, 9327–9336 CrossRef CAS.
- A. J. Soininen, I. Tanionou, N. ten Brummelhuis, H. Schlaad, N. Hadjichristidis, O. Ikkala, J. Raula, R. Mezzenga and J. Ruokolainen, Macromolecules, 2012, 45, 7091–7097 CrossRef CAS.
- P. Madhavan, K.-V. Peinemann and S. P. Nunes, ACS Appl. Mater. Interfaces, 2013, 5, 7152–7159 CrossRef CAS.
- B. Sutisna, G. Polymeropoulos, E. Mygiakis, V. Musteata, K.-V. Peinemann, D.-M. Smilgies, N. Hadjichristidis and S. P. Nunes, Polym. Chem., 2016, 7, 6189–6201 RSC.
- H. Kim, M. M. Arras, J. P. Mahalik, W. Wang, D. M. Yu, S. Chernyy, M. Goswami, R. Kumar, B. G. Sumpter and K. Hong, et al. , Macromolecules, 2018, 51, 7491–7499 CrossRef CAS.
- L. Snyder, J. Chromatogr. Sci., 1978, 16, 223–234 CAS.
- R. B. Thompson, V. V. Ginzburg, M. W. Matsen and A. C. Balazs, Science, 2001, 292, 2469–2472 CrossRef CAS.
- A. C. Balazs, T. Emrick and T. P. Russell, Science, 2006, 314, 1107–1110 CrossRef CAS.
- K. Thorkelsson, A. J. Mastroianni, P. Ercius and T. Xu, Nano Lett., 2012, 12, 498–504 CrossRef CAS.
- J. Kao and T. Xu, J. Am. Chem. Soc., 2015, 137, 6356–6365 CrossRef CAS.
- S. L. Chang, P. Reineck, D. Williams, G. Bryant, G. Opletal, S. A. El-Demrdash, P.-L. Chiu, E. Ōsawa, A. S. Barnard and C. Dwyer, Nanoscale, 2020, 12, 5363–5367 RSC.
- J. Wang, W. Li and J. Zhu, Polymer, 2014, 55, 1079–1096 CrossRef CAS.
- R. Matsuno, H. Otsuka and A. Takahara, Soft Matter, 2006, 2, 415–421 RSC.
- Y.-Q. Li, S.-Y. Fu and Y.-W. Mai, Polymer, 2006, 47, 2127–2132 CrossRef CAS.
- O. Parlak and M. M. Demir, ACS Appl. Mater. Interfaces, 2011, 3, 4306–4314 CrossRef CAS.
- A. H. Gröschel and A. H. Müller, Nanoscale, 2015, 7, 11841–11876 RSC.
- N. Moreno, B. Sutisna and E. Fried, Nanoscale, 2020, 12, 22059–22069 RSC.
- B. Sutisna, P. Bilalis, V. Musteata, D.-M. Smilgies, K.-V. Peinemann, N. Hadjichristidis and S. P. Nunes, Macromolecules, 2018, 51, 8174–8187 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS.
- S. Steinert, F. Dolde, P. Neumann, A. Aird, B. Naydenov, G. Balasubramanian, F. Jelezko and J. Wrachtrup, Rev. Sci. Instrum., 2010, 81, 043705 CrossRef CAS.
- Y. Wu, F. Jelezko, M. B. Plenio and T. Weil, Angew. Chem., Int. Ed., 2016, 55, 6586–6598 CrossRef CAS.
- G. Fuchs, G. Burkard, P. Klimov and D. Awschalom, Nat. Phys., 2011, 7, 789–793 Search PubMed.
- M. Kianinia, O. Shimoni, A. Bendavid, A. W. Schell, S. J. Randolph, M. Toth, I. Aharonovich and C. J. Lobo, Nanoscale, 2016, 8, 18032–18037 RSC.
- S. G. Rao, A. Karim, J. Schwartz, N. Antler, T. Schenkel and I. Siddiqi, ACS Appl. Mater. Interfaces, 2014, 6, 12893–12900 CrossRef CAS.
- A. Albrecht, G. Koplovitz, A. Retzker, F. Jelezko, S. Yochelis, D. Porath, Y. Nevo, O. Shoseyov, Y. Paltiel and M. B. Plenio, New J. Phys., 2014, 16, 093002 CrossRef.
- T. Zhang, A. Neumann, J. Lindlau, Y. Wu, G. Pramanik, B. Naydenov, F. Jelezko, F. Schüder, S. Huber and M. Huber, et al. , J. Am. Chem. Soc., 2015, 137, 9776–9779 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/D0NR07441A |
| This journal is © The Royal Society of Chemistry 2021 |