
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntracellular optical probing with gold nanostars†
Cecilia
Spedalieri
a,
Gergo Peter
Szekeres‡
 ab,
Stephan
Werner
c,
Peter
Guttmann
c and
Janina
Kneipp
*ab
ab,
Stephan
Werner
c,
Peter
Guttmann
c and
Janina
Kneipp
*ab
aHumboldt-Universität zu Berlin, Department of Chemistry, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: janina.kneipp@chemie.hu-berlin.de
bHumboldt-Universität zu Berlin, School of Analytical Sciences Adlershof, Albert-Einstein-Str. 5-9, 12489 Berlin, Germany
cHelmholtz-Zentrum Berlin für Materialien und Energie GmbH, Department X-ray Microscopy, Albert-Einstein-Str. 15, 12489 Berlin, Germany
First published on 9th December 2020
Abstract
Gold nanostars are important nanoscopic tools in biophotonics and theranostics. To understand the fate of such nanostructures in the endolysosomal system of living cells as an important processing route in biotechnological approaches, un-labelled, non-targeted gold nanostars synthesized using HEPES buffer were studied in two cell lines. The uptake of the gold nanostructures leads to cell line-dependent intra-endolysosomal agglomeration, which results in a greater enhancement of the local optical fields than those around individual nanostars and near aggregates of spherical gold nanoparticles of the same size. As demonstrated by non-resonant surface-enhanced Raman scattering (SERS) spectra in the presence and absence of aggregation, the spectroscopic signals of molecules are of very similar strength over a wide range of concentrations, which is ideal for label-free vibrational characterization of cells and other complex environments. In 3T3 and HCT-116 cells, SERS data were analyzed together with the properties of the intracellular nanostar agglomerates. Vibrational spectra indicate that the processing of nanostars by cells and their interaction with the surrounding endolysosomal compartment is connected to their morphological properties through differences in the structure and interactions in their intracellular protein corona. Specifically, different intracellular processing was found to result from a different extent of hydrophobic interactions at the pristine gold surface, which varies for nanostars of different spike lengths. The sensitive optical monitoring of surroundings of nanostars and their intracellular processing makes them a very useful tool for optical bionanosensing and therapy.
Introduction
Gold nanostars, which are anisotropic nanostructures with sharp tips, are promising candidates for a wide range of biotechnological and theranostic applications. Important examples of their utilization are photothermal applications,1–4 sensing,5–7 and imaging.8,9 Their localized surface plasmon resonances can be tuned in the visible and near-infrared wavelength range by modifying their size and spike morphology.10,11 The dependence of optical properties of gold nanostars on their geometry is getting better understood.12–15 The high enhancement in optical signatures, not only particularly visible by the strong enhancement of Raman scattering and surface-enhanced Raman scattering (SERS)16,17 but also from the metal-enhanced fluorescence,18 as well as their two-photon luminescence3,19 makes them the basis of efficient optical labels for imaging and multifunctional theranostic tools.20–22 The high SERS enhancement factors of gold nanostars allow their use in SERS tags with different label molecules in animal cells, e.g., for the tracing of cells in co-cultures23 and targeting of antigens on cancer cells,24–26 or of nucleic acid sequences.27 Conjugated with molecules that serve as reporter species, SERS from immobilized gold nanostars was used to retrieve information on the pH28 or the oxygen level29 from the region of the cell membrane and inside the cells. Signals of molecules in the cell membrane were reported from nanostars that are brought into the proximity of the membrane.30,31 Recently, SERS imaging data also indicated the internalization of receptor-bound gold nanostars by the cells.9 As with other nanoparticles, their surface composition and size change the uptake, processing, and toxicity of nanostars as well.3,32,33 Detailed knowledge on their surface properties when inside different types of cells, e.g., on processing in a harsh endolysosomal environment or regarding integration into membranous structures, is needed to develop and optimize theranostic and biotech applications.34,35 Since the processing of nanomaterials in cells is determined by the biomolecular interactions at their surface, the molecular information must be combined with the ultramicroscopic information from cells.Here, we report the data on the distribution of gold nanostars in the cellular ultrastructure using 3D nanotomography together with detailed information from SERS on the structure and composition of the surface of nanostars residing in the cells of two cell lines. Both cell lines, 3T3 and HCT-116, have been used in the past to study the uptake and processing of gold36–38 or gold-composite39,40 nanostructures. SERS can give information on the interaction of the molecules at the immediate surface of the gold nanostructures and has been recently shown to indicate details about the structure and interactions of and within the hard corona of plasmonic nanoparticles in living cells.38,41 Adding the localization in the cellular ultrastructure by cryo soft X-ray nanotomography (cryo-SXT) of the vitrified cell samples, which is a powerful tool to study the interaction of gold nanoparticles with the cell and with each other,37,38,42 we link the interactions at the gold nanostar surface to their fate inside the cells. Of the known surfactant-free synthetic routes,11,43–45 the generation of the nanostars in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer for reduction, as a capping agent and a growth director, is used herein to ensure compatibility with the cell culture conditions.34,46 Unlike the previous work that aimed to circumvent the formation of a corona determined by the biological environment due to its potential interference with other targeting tasks,31 here, we study the molecular structure and interactions of such a biomolecular corona of different nanostars in the cell culture and when they are contained in the endolysosomal system of the cells.
As will be shown, direct interaction of the biomolecules with the gold surface occurs when the nanostars reside in endolysosomal structures in cells and their interactions are driven by hydrophobic interactions of proteins and lipids. Based on nanotomography imaging together with simulations of the electromagnetic enhancement of SERS and its comparison with the spectral data, the characterization of intra-endolysosomal aggregates was undertaken. Data obtained for the two different cell lines will be discussed in order to delineate the extent up to which the details of the surface interactions of the gold nanostars are determined by the cell culture, the intracellular environment, and by the nanostar morphology. The results underpin the potential role of label-free gold nanostars as probes of cellular biochemistry for a wide range of theranostic applications.
Experimental section
Gold(III) chloride trihydrate, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), adenine, and 4-mercaptobenzoic acid (pMBA) were purchased from Sigma-Aldrich (Steinheim, Germany). Dulbecco's Modified Eagle Medium (DMEM), fetal calf serum (FCS), and phosphate buffered saline (PBS) were purchased from Biochrom (Berlin, Germany).Gold nanostar synthesis and characterization
Gold nanostars were synthesized using a published protocol with slight modifications.47 Initially, a solution of 0.375 M HEPES at pH 7.4 was prepared with MilliQ water and filtered through a 0.22 μm-pore filter. According to three different final concentrations of HEPES of 25 mM, 50 mM, and 75 mM, the HEPES solution was diluted with MilliQ water up to 12 mL final volume. Vials containing the diluted solutions were placed in a thermostatic bath at 22–24 °C. Then, an aliquot of 336 μL 2 mM HAuCl4 was added to each vial, the solution was quickly mixed and left undisturbed for 80 min. After that time, 864 μL of the same gold solution was added, the solution was quickly mixed and left undisturbed in the thermostatic bath overnight. The nanostars were stored as prepared at 4 °C until further use. Before each experiment, if any precipitate was observed, the suspension was briefly sonicated (∼1 min) in an ultrasound bath at room temperature.For experiments with SERS probe molecules, aqueous solutions of pMBA and adenine of different concentrations were added to 100 μL of the nanostar suspension. The probe molecule solution volume was added sequentially to the same nanostar suspension from 1 to 10 μL to avoid excessive dilution. Before the experiments with pMBA, the nanostars were centrifuged and resuspended in water before the addition of the probe molecule. The pH in these samples was modified by adding different amounts of 1 M HCl or 1 M NaOH. For aggregation experiments, 5 μL 1 M NaCl were added to the adenine-nanostar suspension.
The absorbance spectra of all the samples were measured with a UV-vis-NIR double-beam spectrophotometer (Jasco V-670, Germany) in the wavelength range of 250–1200 nm using quartz cuvettes of 10 mm path length. Transmission electron micrographs were taken using a Tecnai G2 20 TWIN instrument operating at 200 kV acceleration voltage. Particle dimensions were measured in the TEM micrographs using the ImageJ software.48 The concentration of the nanostars (∼5 × 10−11 M) was estimated using the average size (Fig. S1†) and the gold concentration of the nanostar preparation.
Cell cultures and nanostar incubation
Swiss albino mouse fibroblast cell line 3T3 (DSMZ, Braunschweig, Germany) and human colorectal carcinoma cell line HCT-116 (LGC Standards, Wesel, Germany) were grown in DMEM supplemented with 10% FCS and maintained in a humidified environment at 37 °C with 5% CO2. Cells were incubated for 24 hours with the dilution of gold nanostars in DMEM–FCS of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (estimated nanostar concentration 5 × 10−12 M). For SERS, cells were grown on glass cover slips, washed two times with phosphate buffer before measurements to eliminate the remaining culture medium and nanostars, and kept in PBS. For 3T3 cells incubated with 25 mM HEPES nanostars, 602 spectra displaying SERS signal from 8 cells were obtained; for 50 mM HEPES nanostars, 120 spectra were obtained from 4 cells; for 75 mM HEPES nanostars, 68 spectra were obtained from 3 cells. For HCT-116 cells incubated with 25 mM HEPES gold nanostars, 245 spectra from 17 cells were collected. The SERS spectra were also measured from the cell culture medium using the nanostars at a ratio of 1:10 in DMEM–FCS and incubated for 24 h at 4 °C. For cryo soft X-ray nanotomography, cells grown on Formvar-coated tomography gold grids and incubated with the nanostars for 24 h were washed three times with PBS, excess of the buffer was removed with a filter paper, and the grid with the cells was plunge-frozen using liquid ethane.
:10 (estimated nanostar concentration 5 × 10−12 M). For SERS, cells were grown on glass cover slips, washed two times with phosphate buffer before measurements to eliminate the remaining culture medium and nanostars, and kept in PBS. For 3T3 cells incubated with 25 mM HEPES nanostars, 602 spectra displaying SERS signal from 8 cells were obtained; for 50 mM HEPES nanostars, 120 spectra were obtained from 4 cells; for 75 mM HEPES nanostars, 68 spectra were obtained from 3 cells. For HCT-116 cells incubated with 25 mM HEPES gold nanostars, 245 spectra from 17 cells were collected. The SERS spectra were also measured from the cell culture medium using the nanostars at a ratio of 1:10 in DMEM–FCS and incubated for 24 h at 4 °C. For cryo soft X-ray nanotomography, cells grown on Formvar-coated tomography gold grids and incubated with the nanostars for 24 h were washed three times with PBS, excess of the buffer was removed with a filter paper, and the grid with the cells was plunge-frozen using liquid ethane.
SERS experiments
The SERS spectra were obtained using a single-stage spectrograph equipped with a CCD detector (Horiba, Munich, Germany) and a diode laser operated at 785 nm (Toptica, Graefelfing, Germany). To focus the excitation light and collect the Raman scattered light in 180 degree backscattering geometry, a 10× objective was used for the nanostar suspension samples and a 60× water immersion objective (N.A. 1.2) was used for the experiments with the culture media and the cells. The excitation intensity was 3 × 105 W cm−2 and 8 × 105 W cm−2 in these two experiments, respectively. All the spectra were obtained with an acquisition time of 1 s. Considering the whole recorded spectral range from 400 to 1800 cm−1, the spectral resolution was 4–7 cm−1. All spectrum pre-processing and band occurrence calculations were done using Matlab R2018a (The MathWorks, Inc.) and Wolfram Mathematica 12 software, respectively.Cryo soft X-ray nanotomography
Vitrified cell monolayers with a thickness of approximately 10 μm were examined using a transmission X-ray microscope equipped with a cryostage.49 X-ray microscopy was performed at beamline U41-PGM1-XM50 at the electron storage ring BESSY II (Helmholtz-Zentrum Berlin für Materialien und Energie, Berlin, Germany). The tilt series of the individual cells were acquired at different angle ranges in increments of 1° at a pixel size of 9.8 nm (25 nm zone plate objective) and a photon energy of 510 eV. Depending on the sample thickness, the exposure time was adjusted for each tilt angle series between 1 and 15 s. The tilt series consists of up to 130 images. Tomograms were obtained by alignment of the corrected tilt series and tomographic reconstruction using the Etomo software (IMOD© 4.9.0, Colorado, USA). The reconstruction of the sample volume was carried out by back-projection or simultaneous iterative reconstruction technique (SIRT). The images of the tilt series were aligned using the intracellular gold nanostars as fiducial markers.Finite difference time domain (FDTD) simulations
Simulations were carried out with Lumerical FDTD Solutions 8.6.3 software using a mesh of 0.2 nm for all nanostructures. For excitation, a plane wave was used with a wavelength of 785 nm, propagating along the negative z-axis direction. The frequency-dependent dielectric function of gold was taken from Johnson and Christy51 and the refractive index of the surrounding medium was set to 1.33. All the output images are normalized to the intensity of the excitation source.Results and discussion
Plasmonic properties and SERS enhancement
The gold nanostars for intracellular application were synthesized via a two-step process using the HEPES buffer, which is commonly used in biochemistry. The synthesis consisted of a first step where mainly the seeds are formed, followed by the addition of gold ions in the second step, thus allowing further growth of the anisotropic structures.47 In the reaction, the piperazine ring generates nitrogen-centered free radicals that reduce the gold ions52 and has a weaker adsorption on the (111) planes of the gold nanocrystals, thereby allowing preferential growth in this direction.11,45 The nanostar suspensions presented blue or green color depending on the HEPES concentration used for their synthesis. Fig. 1A shows the absorbance spectra of the gold nanostars synthesized with HEPES at three concentrations. Two plasmon bands can be observed (Fig. 1A), which are in agreement with the previous reports, corresponding to a high-frequency plasmon mode associated with the inner core, here at 533 nm, and to a low-frequency mode with the individual tips of the nanostars, respectively,10,53 the latter showing a variation in the nanostars obtained at different HEPES conditions here (Fig. 1A). The red shift in the lower-energy plasmon band can be interpreted as an increase in the tip length44,54 with increasing HEPES concentration. The TEM micrographs (Fig. 1B) confirm this. With an increase in the HEPES concentration, the length and distribution of the tips change considerably (Fig. 1B, left to right), while the overall size of the nanostars is similar on average (Fig. S1†). For the lowest HEPES concentration, the tips are short and evenly distributed. When doubling the HEPES concentration to 50 mM, the nanostars show longer tips but an overall similar size (Fig. 1B, compare leftmost and middle column and Fig. S1B and S1C†). For 75 mM of the reducing agent, the tips become slightly longer and some nanostructures show ramifications on the tips (Fig. 1B, rightmost column). In agreement with this, comparing the absorbance spectra of the nanostars at 50 mM and 75 mM HEPES concentrations, the shift to longer wavelengths is small (Fig. 1A, blue and black spectrum). In addition, the broadening of the lower-energy plasmon band is observed with increasing HEPES concentration, which is accentuated between the bands of nanostars prepared with 50 mM and 75 mM HEPES (Fig. 1A, blue and black spectrum). As previously discussed,12,16,53 this was associated with the heterogeneity in the shape of the nanostars, while the overall size is similar for all the conditions (Fig. S1†). The washing of the nanostars by centrifugation and re-suspension in water does not change the plasmonic properties (compare Fig. 1A with Fig. S4†).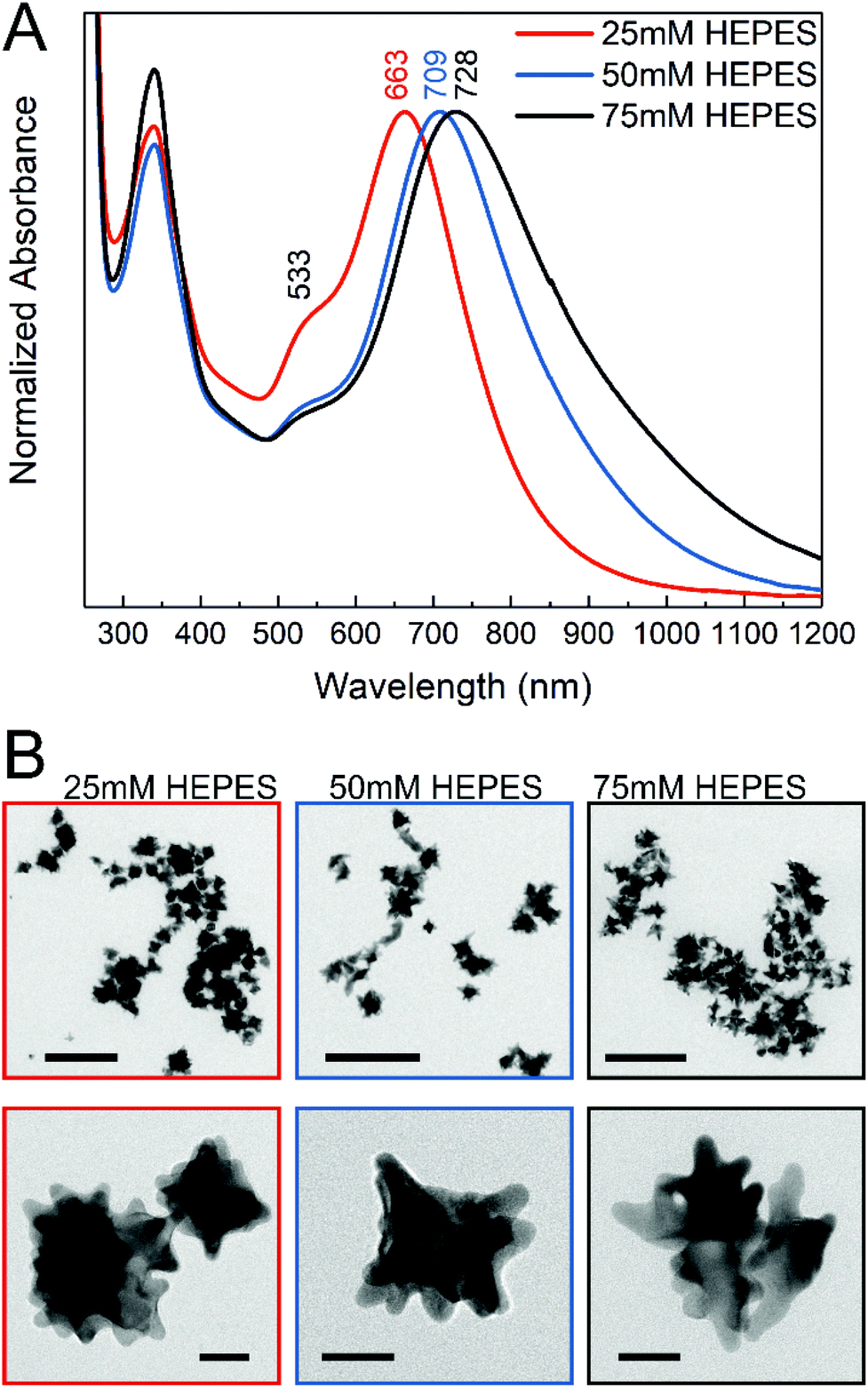 | ||
| Fig. 1 (A) UV-vis-NIR spectra of gold nanostars synthesized with different HEPES concentrations. Maxima assigned to plasmon resonances are labeled for each nanostar condition. The maximum at 533 nm is common to all the spectra. (B) TEM micrographs of the nanostars. Color code is maintained. Scale bars: upper row 200 nm, lower row 20 nm. | ||
The molecular composition at the surface of the nanostars is indicated by their SERS spectra (Fig. S2A†). The spectra have bands at 1142 cm−1, 1268 cm−1, 1348 cm−1, 1376 cm−1, 1445 cm−1, and 1504 cm−1, where the C–N stretching vibration around 1270 cm−1 and the asymmetric stretching mode of the sulfonate at 1348 cm−1 indicate the presence of unmodified HEPES molecules in the surroundings of the nanostars.55 The bands at 1142 cm−1, 1445 cm−1, and 1504 cm−1 suggest that an undetermined product of HEPES oxidation is also present in the solution, also supported by the presence of a band at about 340 nm in the UV-vis spectra of the nanostars (Fig. 1A), which can be assigned to a nitro-compound,52 and which disappears when the HEPES is removed by centrifugation and resuspension in water (Fig. S4†).
The SERS enhancement of the gold nanostars was assessed using 4-mercaptobenzoic acid (pMBA) and adenine as the probe molecules under non-resonant excitation using a 785 nm laser. The spectra of pMBA (Fig. S3A†) are in good agreement with the spectra of this molecule obtained with other nanostar structures.17,53,56 The spectra obtained at different concentrations (Fig. S3A†) indicate that the orientation of pMBA on the metal surface does not change with altered concentration.57 The intensity of the pMBA ring breathing SERS signal at 1078 cm−1 as a function of pMBA total concentration was determined for all the nanostar samples (Fig. 2). It increases with pMBA concentration (Fig. S3A†) until saturation for concentrations higher than ∼5 × 10−7 M. This is similar regardless of the HEPES concentration used in the synthesis and, therefore, of the tip length and the overall structure. Such small differences are in good agreement with a small variation in the enhancement factors achieved for pMBA with individual nanostars of different sizes.53 The amount of gold seeds can be different for each respective synthetic condition and due to the anisotropy of the structures, a straightforward estimation of the amount of pMBA on the surface is challenging. However, all samples were prepared with the same gold ion concentration and their overall size does not show significant differences (cf.Fig. 1B and Fig. S1†). As a consequence of both, a very similar nanostar concentration of ∼5 × 10−11 M is estimated. Assuming the direct chemisorption on the gold surface,57 the similarity of the pMBA concentration at which no further increase in the signal is found (Fig. 2) and hence the one at which all the gold surface is completely covered indicates that the surface that is available is similar in all the preparations. The nanostars show no sign of agglomeration for all the synthetic conditions, resulting in an un-altered electromagnetic contribution to the enhancement with pMBA present in this concentration range (Fig. S4†). The characteristic distinction of SERS signals of the protonated or deprotonated carboxylate group of pMBA56–58 were not observed in the experiment here when the pMBA concentration was altered at a constant pH of 7 (Fig. S3A†). When adding acid or base to the pMBA-nanostar suspension, there is a shift in the frequency of the ring stretching vibration,56 indicating that pH-sensitive groups may be exposed (Fig. S3B†), and the band at 1422 cm−1 assigned to the COO− stretching and the band at 1702 cm−1 attributed to the COOH stretching do appear (Fig. S3C†).56,57
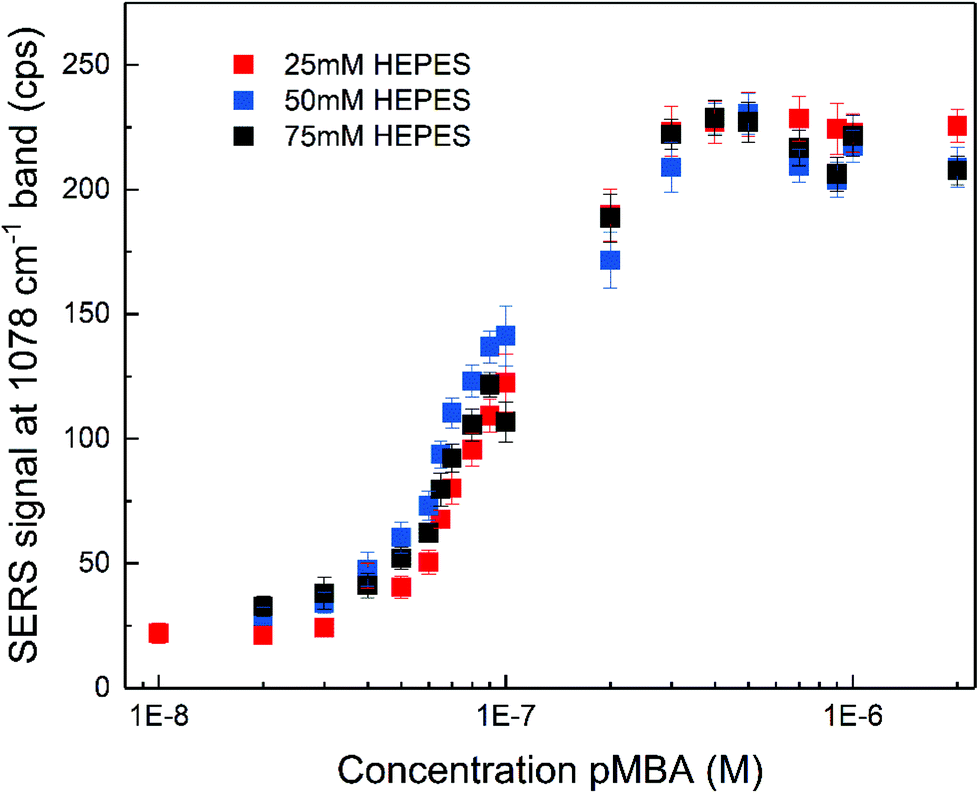 | ||
| Fig. 2 SERS signal of the 1078 cm−1 band in the spectrum of pMBA as a function of pMBA concentration for nanostars synthesized with different HEPES concentration. Each signal value was obtained by averaging 30 spectra and the error bars represent the standard deviation. | ||
The SERS spectra of adenine obtained with the nanostars (Fig. S5†) show typical bands for vibrations of the adenine molecule (see Table S1† for all the assignments)59,60 with slightly varying relative intensities for different HEPES concentrations and in the presence and absence of sodium chloride as the aggregating agent (compare scales in Fig. S5A and S5B†). The signal strength of the pronounced 738 cm−1 band assigned to the ring breathing mode changes for different concentrations of adenine with nanostars synthesized with 25 mM and 50 mM HEPES (Fig. 3). When no sodium chloride was added, the nanostars synthesized at higher HEPES concentration result in larger signals for a wide range of adenine concentrations (Fig. 3, compare solid blue and red markings). Here, both the different nanoparticle morphology, e.g., the longer tips (Fig. 1B)16 and also the changed interaction of the probe molecule with the surface that is known to depend on charge and local pH,60,61 can play a role. The different signals, especially at higher probe molecule concentration, can be the result of the different extent of aggregate formation induced by the analyte and/or of different maximum concentration of molecules that can participate in SERS, e.g., by full occupation of the available hot spots or full coverage of the surface.62,63 Also, in agreement with the latter, the signals are on the same order of magnitude over a wide concentration range of the analyte molecule, indicating that the number of molecules participating in the SERS process must be similar for different nanostar preparations and analyte concentrations. When sodium chloride is added, a signal increase is observed for both nanostar samples (Fig. 3, compare the filled and open markings within each color). For the nanostars synthesized with 25 mM HEPES (Fig. 3, red markings), which were observed to have shorter tips (cf.Fig. 1B), the signals are the highest, suggesting that the addition of sodium chloride induces the formation of nanostar aggregates very efficiently and the major role of the electromagnetic enhancement as the contributor to SERS in these samples. The different signals must be a consequence of different properties of the aggregates, e.g., due to a different spatial arrangement of the nanostars in the aggregates. They could be related to the slightly different morphologies of the individual nanostars in the different preparations as well as the HEPES concentration in the solution.
 | ||
| Fig. 3 SERS signal of the 738 cm−1 band in the spectrum of adenine as a function of adenine concentration for nanostars synthesized with different HEPES concentration in the presence and absence of NaCl. Each signal value was obtained by averaging 30 spectra; the error bars represent the standard deviation. | ||
The formation of aggregates of the nanostars in cells is a very important aspect in their application, since processing and endolysosomal fusion and maturation take place.37 To obtain an idea on the impact of aggregate formation of the nanostars on the distribution of the local optical fields that are responsible for the electromagnetic enhancement in SERS, we performed 3D-FDTD calculations on simplified geometrical models of the nanostars. In Fig. 4, the distribution of the intensity enhancement of the excitation field with respect to that of the incoming field at a wavelength of 785 nm, as used in the experiments here, is shown for nanostars with slightly different morphology. Similar to the samples in the experiment (cf.Fig. 1B and Fig. S1†), the model considers structures of a similar outer diameter but the spikes and the core have different sizes. Comparing the two geometries (Fig. 4A and B), the longer spikes give rise to high field intensity at their tips that will lead to a higher SERS intensity. With shorter spikes, the field intensity at the tips of the spikes is smaller in comparison, which is in full agreement with previous reports.10,12,13 There is also higher enhancement at the base between the longer spikes, close to the core (Fig. 4B). This supports the experimental data shown in Fig. 3, where a higher signal is observed for nanostars with longer tips. When considering two nanostars separated by a small gap (Fig. 4C and D) as a simple model for agglomeration, e.g., in the cellular context, the localization of a high field intensity in the gap between the nanostars with shorter spikes is found (Fig. 4C) for the polarization of incident light along the long axis of the nanostar dimer, exceeding the intensity around the individual nanostar by ∼3 orders of magnitude (compare Fig. 4A with Fig. 4C). This is similar to the distribution of the field around the dimers of spherical particles of the same size, showing the formation of a hot spot between the particles,63,64 as also confirmed in Fig. S6† for spherical nanoparticles of the same outer diameter. The field enhancement has a lower value and remains evenly distributed around the tips of the nanostars when the spikes are longer, with only a slightly higher field in the gap between those spikes that are aligned in the propagation direction of the excitation wave (Fig. 4D). The intensity values for the electric field in the gap between the particles with the longer spikes (Fig. 4D) show similar values to the values in the gap between the smooth gold spheres (Fig. S6†). The different possibilities of the tip-to-tip and tip-to-core plasmonic coupling of the nanostars in close proximity and the extent to which modes other than localized surface plasmon modes influence the spectrum when small changes occur in nanostar geometry13 can lead to a strong variation in the field distribution and the performance of the nanostars in an actual SERS experiment that uses more heterogeneous structures. Here, the small differences in the signal that occur when particles with longer spikes are aggregated by an external agent (Fig. 3, filled and open blue markings) are in good agreement with the smaller increase in the field intensity found for dimers of such structures compared to single particles (compare Fig. 4B and Fig. D). In contrast, the signal observed for the short-spike nanostars increases considerably not only compared to single particles (Fig. 3, open and filled red markings) but also to aggregated stars with longer spikes (Fig. 3, open red and blue markings), which is explained by the hot spots that are obtained in the gaps between the nanostars that have shorter spikes (Fig. 4C).
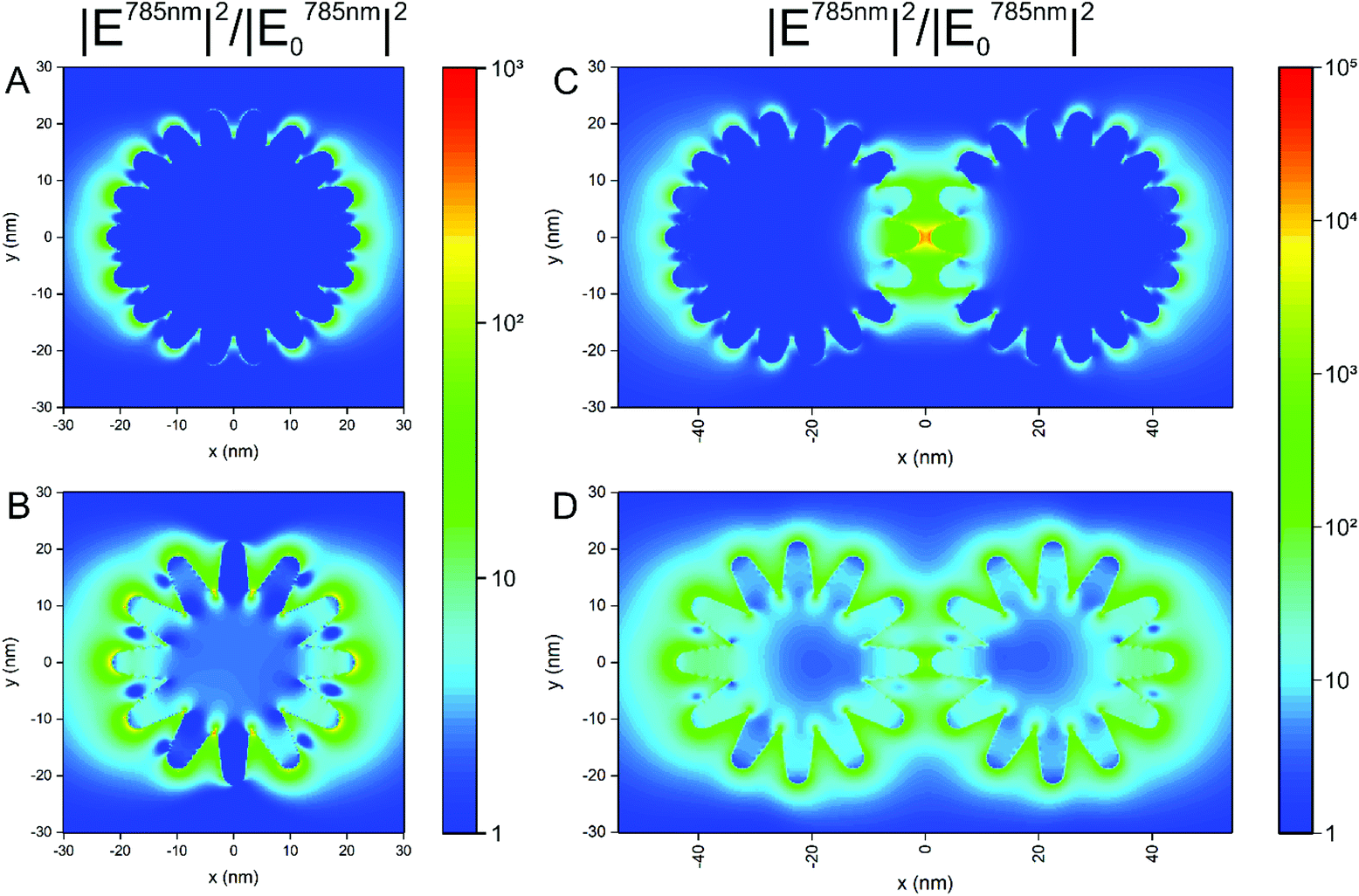 | ||
| Fig. 4 Electric field intensity distribution (normalized to the intensity of the incident field) at 785 nm in the xy plane as obtained by FDTD simulation for (A) a single nanostar with 15 nm radius of the core and 7.5 nm long spikes, (B) a single nanostar with 10 nm radius of the core and 11.5 nm long spikes, (C) two nanostars with 15 nm radius of the core and 7.5 nm long spikes separated by a 2 nm gap, and (D) two nanostars with 10 nm radius of the core and 11.5 nm long spikes separated by a 2 nm gap. The excitation wavelength was set to 785 nm. The polarization of the incident plane wave was in the x direction. The monitor was placed in the equatorial plane. Common scale bar for (A) and (B) (middle), and common scale bar for (C) and (D) (rightmost). | ||
Biomolecular interactions at the nanostar surface in living cells
Three different nanostar samples were incubated with cells of the fibroblast cell line 3T3 by adding them to the cell cultures. When immersing them in the typical cell culture medium, here DMEM–FCS, the intensity of the signals ascribed to the HEPES at the surface diminishes, band ratios observed in the pristine nanostars spectra change, and bands that can be assigned to protein components in the culture medium become visible (compare Fig. S2A and S2B†). This indicates that the capping groups of the nanostars are easily interchangeable with the biomolecular content of the cell culture medium or other environments, such as their immediate cellular surroundings. Rather than being determined by the original capping agent, the uptake of the nanostars by the cells is therefore dictated by the acquired biomolecular corona, whose structure and modifications inside the cells can be characterized by SERS.38,41To study the nanostar–biomolecule interactions in the living cells, the SERS spectra were obtained after incubation of the cell cultures with gold nanostars for 24 h. After this incubation time, endolysosomes of many different maturation stages are known to form, which are distributed across the cytoplasm.37 The average spectra with the nanostars synthesized at different HEPES concentration differ (Fig. 5A–C). To account for the known variability between the individual spectra from the same cell culture batch that is expected due to (i) the biological variation, (ii) the particular local composition in the environment of many different nanostars within each probed cell, and (iii) the variation that is inherent to each SERS experiment,42,65 the occurrence of bands in each individual spectrum was analyzed regardless of their absolute or relative intensities (Fig. 5D–F). The simultaneous analysis of the average spectra and the band occurrences is helpful to draw conclusions on the interactions between the biomolecules and particular types of nanoparticles, here, the nanostars of different preparations and their effect on cellular processing.38 Many similarities are found that are indicative of a direct interaction of the proteins at the hard corona-gold interface of the nanostars with the occurrence of bands in the SERS spectra measured in cells incubated with spherical, citrate-stabilized gold nanoparticles.38 The signals mainly attributed to proteins observed in all the cases are expected because of the known formation of a protein corona in the culture medium and its processing in the cells.41 The tentative assignments of the bands are given in Table S2.†
 | ||
| Fig. 5 SERS average spectra (left column) and relative band occurrence (right column) for 3T3 cells incubated for 24 h with gold nanostars synthesized with 25 mM HEPES (A and D), 50 mM HEPES (B and E) and 75 mM HEPES (C and F). Tentative band assignments are provided in Table S2.† Scale bars (A–C): 10 cps. | ||
The intensity ratios for some bands change in the average spectra (Fig. 5A–C), which is in agreement with the frequency of finding the bands in the individual spectra (Fig. 5D–F). The results suggest that the biomolecules or functional groups in the surroundings of the nanostars inside the cells are similar but interact differently with the nanostars of each of the samples. As a first very obvious difference, the interaction of proteins with gold nanostars by their aromatic side chains must be named. Typical bands assigned to tryptophan at 1353 cm−1 and 424 cm−1 occur only in the spectra of the nanostars made at the lowest HEPES concentration of 25 mM (Fig. 5D). Those of tyrosine, e.g., represented by a characteristic signal at about 840 cm−1 in the data sets as well as the pronounced ring breathing vibration of phenylalanine at 1003 cm−1 are also obtained with the nanostars prepared at 50 mM HEPES concentration (Fig. 5D and E). However, no signals assigned to these amino acids appear in the spectra of the cells incubated with nanostars synthesized at 75 mM HEPES concentration (compare Fig. 5F with Fig. 5D and E), indicating that they are not located in the proximity of the gold surface due to different protein structure or interaction. Secondly, bands that are assigned to S–S stretching (∼503 cm−1) and C–S stretching modes (∼650 cm−1) that could be associated both with an intact secondary structure of the adsorbed proteins66 as well as to the fragmentation of proteins in the corona of gold nanoparticles,41 depending on particular features, are found much more rarely in these samples as well (compare Fig. 5F with Fig. 5D and E). As a third element, signals from the protein backbone, represented by the vibrations in the amide II (1480–1580 cm−1) and amide III (1220–1320 cm−1) regions are very different, e.g., including a lack of bands assigned to the NH3+ vibration at 1489 cm−1 in all but the cells incubated with nanostars synthesized at the lowest HEPES concentration (Fig. 5D). The interaction with the protein backbone is also indicated by the strong representation of bands associated with C–C and C–N stretching vibrations, e.g., the band at ∼1130 cm−1, which is observed more frequently in the cells with nanostars synthesized at lower HEPES concentration (Fig. 5A and D). The concerted interactions of proteins through aromatic side chains, sulfur containing groups, and backbone bonds on the surface of the nanostars are also observed, concluding the observation that some of the above-mentioned bands occur together in many single spectra (data not shown).
The high abundance of a signal at 1123 cm−1 indicates the presence of lipids in the local environment of the nanostars obtained at an HEPES concentration of 50 mM (Fig. 5E), in accordance with the expected uptake of the nanoparticles into endolysosomal structures where they can interact with the endolysosomal membrane.42 In these cells, a few spectra also show signals at the typical frequencies of the lipid ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration at about 1740 cm−1, and most bands associated with C–H deformation vibrations have a Raman shift of 1432 cm−1 or of 1440 cm−1, which is characteristic of CH2 groups that could be a part of long lipid chains (Fig. 5E).67 The interaction of the nanostars with a lipid-rich environment is also visible in the characteristic Raman shift for the CH2 deformation of lipids that occurs at about 1365 cm−1 (Fig. 5F) or the C–H deformation at about 1273 cm−1.67
O stretching vibration at about 1740 cm−1, and most bands associated with C–H deformation vibrations have a Raman shift of 1432 cm−1 or of 1440 cm−1, which is characteristic of CH2 groups that could be a part of long lipid chains (Fig. 5E).67 The interaction of the nanostars with a lipid-rich environment is also visible in the characteristic Raman shift for the CH2 deformation of lipids that occurs at about 1365 cm−1 (Fig. 5F) or the C–H deformation at about 1273 cm−1.67
Such interfacial interactions can be related to the processing of gold nanoparticles by the cells38 since the protein corona plays an important role as mediator between the pristine nanostructure and the cellular environment. The SERS data obtained with the nanostars here also support this. Since the nanostars carry the same molecular species at their surface when they are immersed in the cell culture medium regardless of the HEPES concentration used in their synthesis (Fig. S2B†), the different functional groups interacting at the surface observed after their incubation in the cells result from their different processing, which must be the consequence of different physicochemical properties, e.g., surface properties and geometry of the pristine gold nanostructures. Rather than a straightforward conclusion on one specific type of protein–nanostar interaction, the spectra indicate that depending on the morphological properties (spike length, branched tips), proteins and lipids interact differently, either in their native state or denatured and exposing hydrophobic groups. The latter can be connected to the fragmentation of the hard corona proteins of the gold nanostructures that was shown to occur in the endolysosomal system41 and to a localization of the nanostars in hydrophobic, membrane-rich environments in the endolysosomal vesicles. Studies on nanostars with suitable molecular models, such as liposomes and protein models that explain the biomolecular interactions on other gold nanoparticles41,67,68 and suitable modelling approaches69 will be needed to find out about the influence of nanostar geometry on particular biomolecular surface interactions.
Interaction with the intracellular environment in different cell lines
The cells of the HCT-116 cell line in the same culture medium were also incubated with the nanostars obtained with the reduction of 25 mM HEPES, and both the average spectra (Fig. 6A) and the band occurrences were analyzed in all the individual spectra (Fig. 6B). In comparison to the spectra obtained with 3T3 cells with these nanostars (Fig. 5A and D), the bands assigned to aromatic rings from hydrophobic amino acid side chains at 1357 cm−1, 424 cm−1, 1003 cm−1, and 840 cm−1 are much less abundant or are not shown at all (Fig. 6). A smaller frequency of occurrence is also observed for bands associated with C–S and S–S stretching modes of ∼650 cm−1 and ∼503 cm−1, respectively, which are found less often (compare Fig. 5D and 6B). This indicates that these functional groups interact less frequently with the nanostar surface in the HCT-116 cells than in the 3T3 fibroblasts, probably as a consequence of less fragmentation or denaturation occurring and/or a smaller fraction of proteins with disulfide bonds present at the nanostar surface when in these cells. In contrast, the band that can be assigned to the amide III component at 1315 cm−1 is found more frequently in the spectra from the HCT-116 cells than in the fibroblast cells, indicating a close interaction of the backbone of proteins with the nanostar surface. This band is also very pronounced in the spectra of the corona of spherical, citrate-stabilized gold nanoparticles in HCT-116 cells38 and its observation here indicates that the specific structure and interaction of the protein corona is related to the processing by the HCT-116 cells. Interestingly, in the spectra obtained from spherical gold nanoparticles in the HCT-116 cells, the interaction of disulfide groups from their protein corona at about ∼500 cm−1 is found much more frequently38 than here in the spectra obtained after incubation with the gold nanostars. This supports a previous discussion of the link of the physicochemical properties of the gold nanostructures, determining the composition and interaction in the protein corona, with the processing of the nanostructures by the cells.38 In addition, the close interaction with C–C bonds of lipids and with methyl groups is evidenced by the co-occurrence of the bands at 1120 cm−1 with 1440 cm−1 in the HCT-116 cells (data not shown). These differences show that the interaction of the biomolecules in the cellular environment are different in the two cell lines and point towards an increased interaction with lipid components, for instance, of vesicle membranes, in these cells compared to the interactions observed for the short spiked nanostars in the fibroblast cells. Since the incubation conditions are the same for both the cell lines, this is the result of distinct processing mechanisms of the nanostars.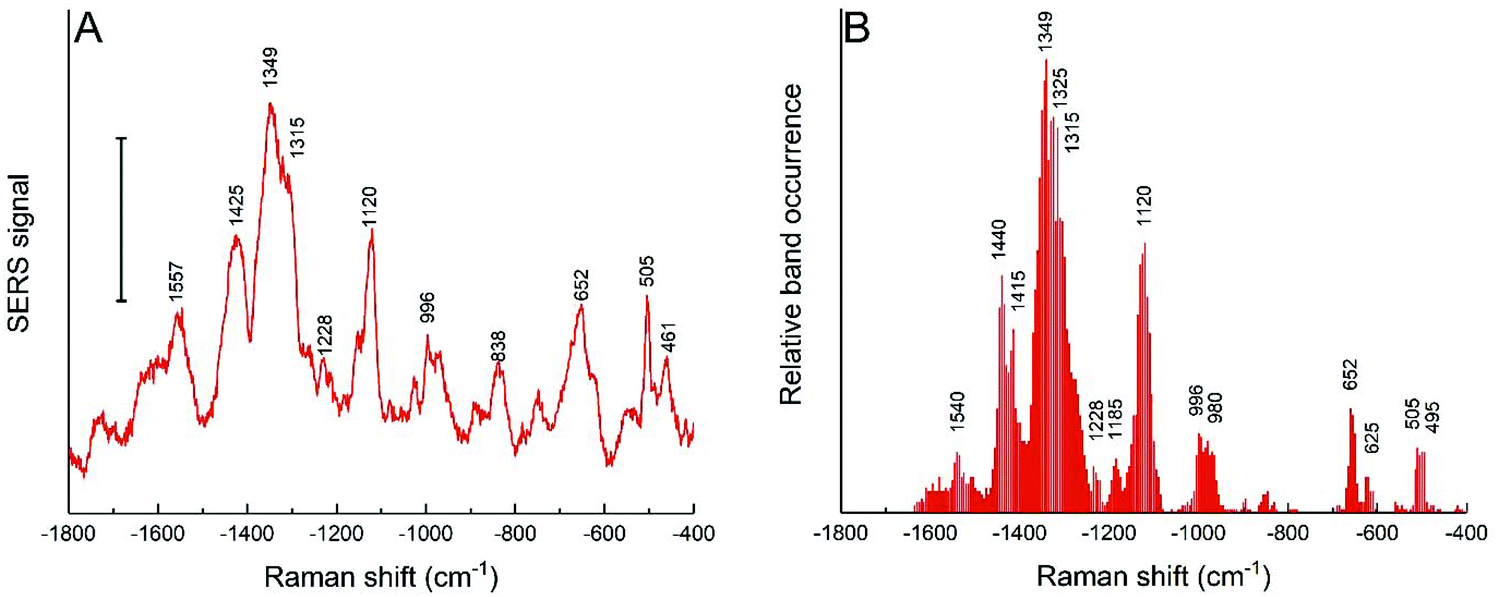 | ||
| Fig. 6 (A) SERS average spectra and (B) relative band occurrence for HCT-116 cells incubated for 24 h with gold nanostars synthesized with 25 mM HEPES. Scale bar in (A): 10 cps. | ||
To obtain an insight into the processing of the nanostars by the cells of both cell lines and to observe their interactions at the ultrastructural level, 3T3 fibroblast and HCT-116 cells were incubated with the nanostars and plunge-frozen for analysis by soft X-ray nanotomography. In the samples of the 3T3 cells (Fig. 7A and S7A†), the nanostars are contained inside well-defined vesicular structures that are both single and multi-lamellar, indicating typical endolysosomal processing, and in many cases, are in close contact with the vesicles’ membranes. The localization within and close to the membranous structures supports the observation of hydrophobic interactions of the proteins at the nanostar surface and of SERS signals that point directly towards the interaction of lipids with the gold in the 3T3 cells (Fig. 5). Since the uptake of nanostars is continuous throughout the 24 h incubation period, both individual nanostars and agglomerates are expected to be found inside the cells. Even though some single particles can be observed (Fig. 7A), a majority of the nanostars are a part of agglomerates of sizes between 100 and 450 nm (corresponding to ∼10–1000 individual nanostars) without a particular spatial arrangement, which is in agreement with the strong SERS signals that were obtained for all the cells. These arrangements are similar to those found for spherical gold nanoparticles37,38 that, unlike the linear arrangements of silver nanostructures,70 form agglomerates with their biomolecular environment. Interparticle gaps are visible, which indicate a loose configuration of the individual stars in the agglomerates in both the cell lines (Fig. 7A and B). No nanostars were found inside the nucleus or free in the cytosol, which indicates that during the incubation period, the nuclear membrane is not crossed by the particles and that the particles have not escaped the endolysosomal vesicles. In HCT-116 cells (Fig. 7B and S7B†), the distribution of particle accumulations is clearly different, with agglomerates larger in size (between 200 nm and 800 nm, corresponding to ∼90–5700 individual nanostars) than those observed for the 3T3 cells (Fig. 7A and S7A†). In the HCT-116 cells, there are also single particles distributed in the cytoplasm, indicating that the nanostructures are endocytosed as individual nanoparticles, similar to other gold nanostructures.37,71,72
 | ||
| Fig. 7 Slices of X-ray tomographic reconstruction of (A) 3T3 and (B) HCT-116 cells incubated for 24 h with gold nanostars synthesized with 25 mM HEPES (cf.Fig. 1B). The red arrows indicate single particles or aggregates of nanostars. More examples are provided in Fig. S7.† Abbreviations: Nu, nucleus; NM, nuclear membrane; V, vesicles; M, mitochondria; L, lipid droplets. Scale bars: 2 μm. | ||
The cellular compartments can be clearly visualized in the tomographic reconstructions, such as vesicular bodies, mitochondria, and lipid droplets. For both cell lines used, neither of the sampled cells show an obvious indication of cell death, such as dilated perinuclear cisternae,73 suggesting that the uptake of nanostars does not induce adverse cellular responses. This is interesting, as the sensitivity of the HCT-116 cells towards incubation with citrate-stabilized gold nanoparticles in the same culture medium was recently demonstrated.38 The absence of indicators of cell death here may be due to the different shape and specific structure and interactions of the corona of the nanostars, as revealed by the SERS spectra.
Conclusions
The cellular processing of un-labelled, non-functionalized gold nanostars synthesized by a mild, biocompatible method was studied, and their optical properties were assessed via experiments and by theory under conditions that resemble the situation in living cells. As evidenced by X-ray nanotomography, the uptake of single nanostars results in their processing through the endolysosomal pathway, where they are prone to aggregation, without the evidence of endosomal escape. The number and length of spikes were found to vary slightly for nanostars prepared at different concentrations of HEPES, at a very similar outer diameter of the nanostructures. While the enhancement of Raman scattering in non-resonant SERS experiments was very similar in the absence of aggregation of the nanostars, the optical properties of aggregates formed by the nanostars yielded very different effects, with higher enhancements found for nanostars of shorter spike length. The experimental data obtained with probe molecules outside the cells are in good agreement with the results of FDTD simulations under excitation conditions that are similar to those in the SERS experiments conducted in live cells. Non-resonant SERS spectra were obtained both in the presence and absence of aggregation, and under both conditions, the spectroscopic signals of the molecules are of similar magnitude over a wide range of concentrations, which is ideal for label-free SERS in cells and other complex environments. As indicated by the formation of nanostar aggregates in the endolysosomal system of 3T3 and HCT-116 cells, the high local fields in the aggregates of nanostars with shorter spikes found in the experimental and theoretical model must contribute significantly to the high SERS signals that were obtained from the endolysosomal system in the live cells. Understanding the endolysosomal processing and intra-endolysosomal aggregate formation has implications for the application of the nanostars in photothermal therapy and drug delivery.The processing of nanoparticles and their surface species was studied by the qualitative analysis of SERS spectra incubated with the cells, independent of the (quantitative) enhancement of the Raman signals. A comparison of the biomolecular environment of the nanostars of slightly different geometry in the same cell line (3T3) indicated differences in the structure and interaction of the proteins in the hard corona of the nanoparticles, and also a different interaction with the membranous structures in the endolysosomal compartment. The very similar SERS data obtained in the cell culture medium with three different nanostar preparations, yet the very different spectral patterns in the endolysosomal system leads us to conclude that the specific molecular environment observed inside the cells is a consequence of post-endocytotic processing. In agreement with the important function of the protein corona to mediate between the physical properties of the pristine gold surface and the processing in the cellular ultrastructure,38 the characteristic structure and interaction at the nanoparticle surface, which is evident from the spectra, are likely the result of small differences in the nanostar shape and geometry.
A comparison of the surface molecular structure and nanoparticle processing in the 3T3 and HCT-116 cell lines indicated differences in the size and density of the intra-endolysosomal nanostar agglomerates, connected to a different extent of hydrophobic interactions at the nanostar surface, which must be further elucidated with suitable lipid model systems as they will be important for the development of theranostic applications using gold nanostars. The similarities observed in the interacting functional groups and the structure of the protein corona of gold nanostars with that of the spherical gold nanoparticles in a previous study in the HCT-116 cell line38 indicates the importance of similar, cell line-specific processing pathways that could also be related to the initial structure and processing of their protein corona.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank HZB for the allocated beam time at beamline U41-PGM1-XM and experimental support at BESSY II. We also thank Jan Simke and Sören Selve (ZELMI, Technical University Berlin) for TEM measurements. C.S. gratefully acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008-390540038 – UniSysCat, G.P.S. by a fellowship of School of Analytical Sciences Adlershof (DFG GSC 1013), and J. K. by ERC grant no. 259432.References
- I. Baginskiy, T.-C. Lai, L.-C. Cheng, Y.-C. Chan, K.-Y. Yang, R.-S. Liu, M. Hsiao, C.-H. Chen, S.-F. Hu, L.-J. Her and D. P. Tsai, J. Phys. Chem. C, 2013, 117, 2396–2410 CrossRef CAS.
- P. Pallavicini, A. Dona, A. Taglietti, P. Minzioni, M. Patrini, G. Dacarro, G. Chirico, L. Sironi, N. Bloise, L. Visaie and L. Scarabelli, Chem. Commun., 2014, 50, 1969–1971 RSC.
- L. Sironi, S. Freddi, M. Caccia, P. Pozzi, L. Rossetti, P. Pallavicini, A. Donà, E. Cabrini, M. Gualtieri, I. Rivolta, A. Panariti, L. Dalfonso, M. Collini and G. Chirico, J. Phys. Chem. C, 2012, 116, 18407–18418 CrossRef CAS.
- H. Yuan, A. M. Fales and T. Vo-Dinh, J. Am. Chem. Soc., 2012, 134, 11358–11361 CrossRef CAS.
- M. Bhamidipati, H. Y. Cho, K. B. Lee and L. Fabris, Bioconjugate Chem., 2018, 29, 2970–2981 CrossRef CAS.
- D. Jana, C. Matti, J. He and L. Sagle, Anal. Chem., 2015, 87, 3964–3972 CrossRef CAS.
- A. S. Indrasekara, S. Meyers, S. Shubeita, L. C. Feldman, T. Gustafsson and L. Fabris, Nanoscale, 2014, 6, 8891–8899 RSC.
- F. Tian, J. Conde, C. Bao, Y. Chen, J. Curtin and D. Cui, Biomaterials, 2016, 106, 87–97 CrossRef CAS.
- C. D. L. De Albuquerque and Z. D. Schultz, Anal. Chem., 2020, 92, 9389–9398 CrossRef CAS.
- F. Hao, C. L. Nehl, J. H. Hafner and P. Nordlander, Nano Lett., 2007, 7, 729–732 CrossRef CAS.
- J. A. Webb, W. R. Erwin, H. F. Zarick, J. Aufrecht, H. W. Manning, M. J. Lang, C. L. Pint and R. Bardhan, J. Phys. Chem. C, 2014, 118, 3696–3707 CrossRef CAS.
- E. Hao, R. C. Bailey, G. C. Schatz, J. T. Hupp and S. Li, Nano Lett., 2004, 4, 327–330 CrossRef CAS.
- T. V. Tsoulos and L. Fabris, J. Phys. Chem. C, 2018, 122, 28949–28957 CrossRef CAS.
- C. Hrelescu, T. K. Sau, A. L. Rogach, F. Jackel, G. Laurent, L. Douillard and F. Charra, Nano Lett., 2011, 11, 402–407 CrossRef CAS.
- Z. Xu, W. Kong, L. Cheng, A. Ma, X. Lu, Y. Wang, X. Zhang and X. Y. Zhang, Plasmonics, 2015, 10, 1185–1193 CrossRef CAS.
- P. Senthil Kumar, I. Pastoriza-Santos, B. Rodriguez-Gonzalez, F. Javier Garcia de Abajo and L. M. Liz-Marzan, Nanotechnology, 2008, 19, 015606 CrossRef.
- C. Hrelescu, T. K. Sau, A. L. Rogach, F. Jäckel and J. Feldmann, Appl. Phys. Lett., 2009, 94, 153113 CrossRef.
- I. G. Theodorou, P. Ruenraroengsak, D. A. Gonzalez-Carter, Q. Jiang, E. Yagüe, E. O. Aboagye, R. C. Coombes, A. E. Porter, M. P. Ryan and F. Xie, Nanoscale, 2019, 11, 2079–2088 RSC.
- H. Yuan, J. A. Gomez, J. S. Chien, L. Zhang, C. M. Wilson, S. Li, A. M. Fales, Y. Liu, G. A. Grant, M. Mirotsou, V. J. Dzau and T. Vo-Dinh, J. Biophotonics, 2016, 9, 406–413 CrossRef CAS.
- Y. Liu, Z. Chang, H. Yuan, A. M. Fales and T. Vo-Dinh, Nanoscale, 2013, 5, 12126–12131 RSC.
- J. Chen, Z. Sheng, P. Li, M. Wu, N. Zhang, X. F. Yu, Y. Wang, D. Hu, H. Zheng and G. P. Wang, Nanoscale, 2017, 9, 11888–11901 RSC.
- C. Carrillo-Carrión, R. Martínez, M. F. Navarro Poupard, B. Pelaz, E. Polo, A. Arenas-Vivo, A. Olgiati, P. Taboada, M. G. Soliman, Ú. Catalán, S. Fernández-Castillejo, R. Solà, W. J. Parak, P. Horcajada, R. A. Alvarez-Puebla and P. del Pino, Angew. Chem., Int. Ed., 2019, 58, 7078–7082 CrossRef.
- D. Jimenez de Aberasturi, A. B. Serrano-Montes, J. Langer, M. Henriksen-Lacey, W. J. Parak and L. M. Liz-Marzán, Chem. Mater., 2016, 28, 6779–6790 CrossRef CAS.
- M. Li, S. R. Banerjee, C. Zheng, M. G. Pomper and I. Barman, Chem. Sci., 2016, 7, 6779–6785 RSC.
- X. P. Wang, B. Walkenfort, M. König, L. König, S. Kasimir-Bauer and S. Schlücker, Faraday Discuss., 2017, 205, 377–386 RSC.
- T. R. Nayak, C. Andreou, A. Oseledchyk, W. D. Marcus, H. C. Wong, J. Massagué and M. F. Kircher, Nanoscale, 2017, 9, 1110–1119 RSC.
- K. Dardir, H. Wang, B. E. Martin, M. Atzampou, C. B. Brooke and L. Fabris, J. Phys. Chem. C, 2020, 124, 3211–3217 CrossRef CAS.
- X. Zhao, S. Campbell, G. Q. Wallace, A. Claing, C. G. Bazuin and J. F. Masson, ACS Sens., 2020, 5, 2155–2167 CrossRef CAS.
- T. D. Nguyen, M. S. Song, N. H. Ly, S. Y. Lee and S. W. Joo, Angew. Chem., Int. Ed., 2019, 58, 2710–2714 CrossRef CAS.
- Q. Jin, M. Li, B. Polat, S. K. Paidi, A. Dai, A. Zhang, J. V. Pagaduan, I. Barman and D. H. Gracias, Angew. Chem., Int. Ed., 2017, 56, 3822–3826 CrossRef CAS.
- S. Sloan-Dennison and Z. D. Schultz, Chem. Sci., 2019, 10, 1807–1815 RSC.
- M. Bhamidipati and L. Fabris, Bioconjugate Chem., 2017, 28, 449–460 CrossRef CAS.
- M. Enea, E. Pereira, M. P. de Almeida, A. M. Araujo, M. L. Bastos and H. Carmo, Nanomaterials, 2020, 10 CrossRef CAS.
- G. Plascencia-Villa, D. Bahena, A. R. Rodríguez, A. Ponce and M. José-Yacamán, Metallomics, 2013, 5, 242–250 RSC.
- A. Balfourier, N. Luciani, G. Wang, G. Lelong, O. Ersen, A. Khelfa, D. Alloyeau, F. Gazeau and F. Carn, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 103–113 CrossRef CAS.
- D. Drescher, H. Traub, T. Büchner, N. Jakubowski and J. Kneipp, Nanoscale, 2017, 9, 11647–11656 RSC.
- D. Drescher, T. Büchner, P. Guttmann, S. Werner, G. Schneider and J. Kneipp, Nanoscale Adv., 2019, 1, 2937–2945 RSC.
- G. P. Szekeres, S. Werner, P. Guttmann, C. Spedalieri, D. Drescher, V. Zivanovic, M. Montes-Bayon, J. Bettmer and J. Kneipp, Nanoscale, 2020, 12, 17450–17461 RSC.
- D. Drescher, I. Zeise, H. Traub, P. Guttmann, S. Seifert, T. Büchner, N. Jakubowski, G. Schneider and J. Kneipp, Adv. Funct. Mater., 2014, 24, 3765–3775 CrossRef CAS.
- T. Buchner, D. Drescher, V. Merk, H. Traub, P. Guttmann, S. Werner, N. Jakubowski, G. Schneider and J. Kneipp, Analyst, 2016, 141, 5096–5106 RSC.
- G. P. Szekeres, M. Montes-Bayón, J. Bettmer and J. Kneipp, Anal. Chem., 2020, 92, 8553–8560 CrossRef CAS.
- V. Zivanovic, S. Seifert, D. Drescher, P. Schrade, S. Werner, P. Guttmann, G. P. Szekeres, S. Bachmann, G. Schneider, C. Arenz and J. Kneipp, ACS Nano, 2019, 13, 9363–9375 CrossRef CAS.
- K. Chandra, K. S. B. Culver, S. E. Werner, R. C. Lee and T. W. Odom, Chem. Mater., 2016, 28, 6763–6769 CrossRef CAS.
- H. Yuan, C. G. Khoury, H. Hwang, C. M. Wilson, G. A. Grant and T. Vo-Dinh, Nanotechnology, 2012, 23, 075102 CrossRef.
- J. Xie, J. Y. Lee and D. I. C. Wang, Chem. Mater., 2007, 19, 2823–2830 CrossRef CAS.
- P. M. Favi, M. Gao, L. J. Sepúlveda Arango, S. P. Ospina, M. Morales, J. J. Pavon and T. J. Webster, J. Biomed. Mater. Res., Part A, 2015, 103, 3449–3462 CrossRef CAS.
- S. Saverot, X. Geng, W. Leng, P. J. Vikesland, T. Z. Grove and L. R. Bickford, RSC Adv., 2016, 6, 29669–29673 RSC.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- G. Schneider, P. Guttmann, S. Heim, S. Rehbein, F. Mueller, K. Nagashima, J. B. Heymann, W. G. Muller and J. G. McNally, Nat. Methods, 2010, 7, 985–987 CrossRef CAS.
- P. Guttmann, S. Werner, F. Siewert, A. Sokolov, J.-S. Schmidt, M. Mast, M. Brzhezinskaya, C. Jung, R. Follath and G. Schneider, Microsc. Microanal., 2018, 24, 206–207 CrossRef.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Solid State, 1972, 6, 4370–4379 CrossRef CAS.
- A. Habib, M. Tabata and Y. G. Wu, Bull. Chem. Soc. Jpn., 2005, 78, 262–269 CrossRef CAS.
- C. G. Khoury and T. Vo-Dinh, J. Phys. Chem. C, 2008, 112, 18849–18859 CrossRef CAS.
- H. de Puig, J. O. Tam, C. W. Yen, L. Gehrke and K. Hamad-Schifferli, J. Phys. Chem. C, 2015, 119, 17408–17415 CrossRef CAS.
- W. Xi and A. J. Haes, J. Am. Chem. Soc., 2019, 141, 4034–4042 CrossRef CAS.
- Y. Liu, H. Yuan, A. M. Fales and T. Vo-Dinh, J. Raman Spectrosc., 2013, 44, 980–986 CrossRef CAS.
- A. Michota and J. Bukowska, J. Raman Spectrosc., 2003, 34, 21–25 CrossRef CAS.
- J. Kneipp, H. Kneipp, B. Wittig and K. Kneipp, Nano Lett., 2007, 7, 2819–2823 CrossRef CAS.
- B. Giese and D. McNaughton, J. Phys. Chem. B, 2002, 106, 101–112 CrossRef CAS.
- E. Papadopoulou and S. E. J. Bell, J. Phys. Chem. C, 2010, 114, 22644–22651 CrossRef CAS.
- F. Madzharova, Z. Heiner, M. Gühlke and J. Kneipp, J. Phys. Chem. C, 2016, 120, 15415–15423 CrossRef CAS.
- V. Joseph, A. Matschulat, J. Polte, S. Rolf, F. Emmerling and J. Kneipp, J. Raman Spectrosc., 2011, 42, 1736–1742 CrossRef CAS.
- F. Madzharova, Z. Heiner, J. Simke, S. Selve and J. Kneipp, J. Phys. Chem. C, 2018, 122, 2931–2940 CrossRef CAS.
- P. Nordlander, C. Oubre, E. Prodan, K. Li and M. I. Stockman, Nano Lett., 2004, 4, 899–903 CrossRef CAS.
- H. Xu, J. Aizpurua, M. Käll and P. Apell, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 4318–4324 CrossRef CAS.
- G. P. Szekeres and J. Kneipp, Analyst, 2018, 143, 6061–6068 RSC.
- V. Živanović, Z. Kochovski, C. Arenz, Y. Lu and J. Kneipp, J. Phys. Chem. Lett., 2018, 9, 6767–6772 CrossRef.
- J. Broda, J. Setzler, A. Leifert, J. Steitz, R. Benz, U. Simon and W. Wenzel, Nanomedicine, 2016, 12, 1409–1419 CrossRef CAS.
- S. Salassi, F. Simonelli, D. Bochicchio, R. Ferrando and G. Rossi, J. Phys. Chem. C, 2017, 121, 10927–10935 CrossRef CAS.
- D. Drescher, P. Guttmann, T. Buchner, S. Werner, G. Laube, A. Hornemann, B. Tarek, G. Schneider and J. Kneipp, Nanoscale, 2013, 5, 9193–9198 RSC.
- M. Liu, Q. Li, L. Liang, J. Li, K. Wang, J. Li, M. Lv, N. Chen, H. Song, J. Lee, J. Shi, L. Wang, R. Lal and C. Fan, Nat. Commun., 2017, 8, 15646 CrossRef CAS.
- X. Xie, J. Liao, X. Shao, Q. Li and Y. Lin, Sci. Rep., 2017, 7, 3827 CrossRef.
- M. K. Sakamoto, S. Mima, T. Kihara and T. Tanimura, Anat. Rec., Part A, 2004, 279, 652–663 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr07031a |
| ‡ Current address: Department of Molecular Physics, Fritz Haber Institute of the Max Planck Society, Faradayweg 4-6, 14195 Berlin, Germany. |
| This journal is © The Royal Society of Chemistry 2021 |