
Deciphering the excited-state dynamics and multicarrier interactions in perovskite core–shell type hetero-nanocrystals†
Yinjuan
Ren‡
ab,
Zhonghui
Nie‡
a,
Fei
Deng
a,
Ziming
Wang
a,
Siyang
Xia
a and
Yue
Wang
 *a
*a
aMIIT Key Laboratory of Advanced Display Materials and Devices, Institute of Optoelectronics & Nanomaterials, College of Materials Science and Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: ywang@njust.edu.cn
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore
First published on 7th December 2020
Abstract
Deciphering and modulating the carrier dynamics of perovskite nanocrystals (Pe-NCs) is crucial for their optoelectronic applications, which remains elusive to date. Herein, we, for the first time, explore the ultrafast dynamics of perovskite core–shell type NCs using CsPbBr3@ZnS as a model system. According to the transient spectroscopic characterization, a physical picture of the ultrafast dynamics in core–shell Pe-NCs is built. Specifically, we directly observed the “hot” hole transfer from CsPbBr3 to ZnS and confirmed the formation of charge-transfer state in CsPbBr3@ZnS NCs. Such ultrafast (<100 fs) hole rearrangement speeds up the carrier cooling and breaks the hot phonon bottleneck effect in Pe-NCs. Moreover, thanks to the charge separation in CsPbBr3@ZnS NCs, the Auger recombination is largely suppressed and the Auger lifetime is increased nearly 5-fold compared to that of “pure” CsPbBr3 NCs, which endows CsPbBr3@ZnS NCs with unique optical gain properties. These results are informative for halide perovskite-based applications, such as photocatalysis, hot-carrier photovoltaics and lasers.
Introduction
Deciphering the carrier dynamics of semiconductor nanocrystals (NCs) is crucial for photonic and optoelectronic applications, ranging from photocatalysis to light emission and amplification.1,2 Since the recognition of the bright prospects of NCs by virtue of the scalable solution-processability and quantum confinement effect, numerous efforts have been made to explore and tune the excited-state dynamics toward desirable photophysical properties and specific utilization fields.3 Of particular interest is the adoption of the core–shell heterostructure with different configurations for dynamics modulation.4 According to the conduction and valence band alignment, the heterostructure can be mainly categorized into two types, i.e., type-I and type-II.5 The type-I hetero-nanocrystals typically feature high photoluminescence quantum yield (PLQY) and fast carrier recombination because the electrons and holes are confined together within the core.6 In contrast, for the type-II alignment, the electrons and holes are localized in either the core or the shell. The spatial isolation of the electron and hole prolongs the excited-state carrier lifetime, and a new photophysical phenomenon may occur, exemplified by the formation of long-lived interfacial excitons.5,7Metal–halide perovskite NCs (Pe-NCs) are emerging as the attractive new-generation semiconductor NCs thanks to the large absorption cross-sections, broadband tunable emission and high PLQY.8–10 These Pe-NCs exhibit intriguing optical properties that differ from those of the traditional NCs including the bright triplet exciton emission and defect-tolerant nature.8,11 Regarding the perovskite nano-heterostructures, a plethora of core–shell and quasi-core–shell structures have been demonstrated, such as CsPbX3/ZrO2,12 CsPbBr3/SiO2,13 CsPbBr3/CdS14 and CsPbX3/PbS.15 These works mainly aimed to improve the chemical and physical stabilities of Pe-NCs due to the ionic crystalline nature and labile bonding of the surface ligands,8 and impressive progress has been made in this respect.14 However, until now, the ultrafast carrier dynamics in perovskite hetero-nanocrystal systems has rarely been investigated. Moreover, the quest for modulating the excited-state dynamics of Pe-NCs by the core–shell effect, especially for the carrier-carrier interactions, remains elusive. Such kind of study, otherwise, would be highly informative for both fundamental research and technological applications based on Pe-NCs.
In this work, we, for the first time, probe the ultrafast dynamics of perovskite core–shell NCs using CsPbBr3@ZnS with type-II-like structures as a model system. According to the comprehensive transient spectroscopic characterization (temporal resolution: ∼100 fs), the physical picture of the ultrafast dynamics in core–shell type Pe-NCs is built. We have unambiguously observed the hole transfer from the CsPbBr3 core to the ZnS shell and confirmed the formation of the charge transfer (CT) state in the CsPbBr3@ZnS NCs. It is identified that the “hot” hole transfer channel dictates the CT process where the holes directly relocate before cooling. Such an ultrafast (<100 fs) hole rearrangement speeds up the carrier cooling and breaks the hot phonon bottleneck effect in Pe-NCs. Moreover, by virtue of charge separation in CsPbBr3@ZnS NCs, the Auger recombination is largely suppressed, and the Auger lifetime (∼203 ps) is increased nearly 5-fold compared to that of “pure” CsPbBr3 NCs as a result of the reduced carrier-carrier coupling and electron–hole overlap. Such a mitigated multicarrier interaction endows the CsPbBr3@ZnS NCs with unique optical gain properties exhibiting low threshold and shift-free stimulated emissions. Our findings are insightful for metal–halide perovskite-based applications, such as photocatalysis, hot-carrier photovoltaics and lasers.
Results and discussion
The “pure” CsPbBr3 and core–shell type CsPbBr3@ZnS NCs were prepared by the reported recipe.9,16,17 In the previous literature,17 the basic properties of CsPbBr3@ZnS NCs, such as size, composition and core–shell structure, have been well studied by transmission electron microscopy (TEM), energy-dispersive X-ray spectroscopy (EDS) and steady-state absorption/emission spectroscopy. The TEM images of the present sample are shown in Fig. S1.† The zoom-in TEM (Fig. S1b†) clearly shows the core–shell-like structure. The EDS mapping manifests the uniform distribution of Zn and S along with Cs, Pb, and Br (Fig. S2†). The combined results of TEM and EDS confirm the formation of CsPbBr3@ZnS core–shell type NCs. Moreover, it is also revealed that the CsPbBr3@ZnS NCs show type-II or quasi-type-II band alignment at the CsPbBr3/ZnS interface because of the much higher conduction band minimum (CBM) of ZnS than that of CsPbBr3 and the similar valence band maximum (VBM) between them,17,18 which offers an ideal platform to study the effect of the core–shell structure on the excited-state dynamics of Pe-NCs. Herein, we move forward to investigate the ultrafast dynamics of the CsPbBr3@ZnS NCs by transient absorption (TA) spectroscopy.Fig. 1a shows the absorption and photoluminescence (PL) spectra of CsPbBr3 and CsPbBr3@ZnS NCs. After the growth of the ZnS shell, the PL spectrum redshifts from 509 to 530 nm and absorption from 500 to 512 nm with respect to that of CsPbBr3. The photographs of CsPbBr3 and CsPbBr3@ZnS NC solutions under UV irradiation are shown in the inset of Fig. 1b, manifesting the bright cyan and green emission, respectively. The temperature-dependent PL measurements have unravelled the different emission behaviours of the NCs (Fig. 1b). For the CsPbBr3 NCs (Fig. S3†), the PL intensities as a function of temperature can be well fitted by the Arrhenius function, whereas those for the CsPbBr3@ZnS NCs could only be reproduced by the Arrhenius plot with a dual activation energy model (see details in the ESI†),19 indicating the impacts of the ZnS shell on steady-state PL properties. The time-resolved PL of the CsPbBr3 and CsPbBr3@ZnS NCs are shown in Fig. 1c. The much larger (more than one order of magnitude) PL lifetime of the CsPbBr3@ZnS NCs as compared to that of the CsPbBr3 NCs is consistent with previous reports and agrees with the (quasi)-type-II band alignment in the CsPbBr3@ZnS NCs.17,18
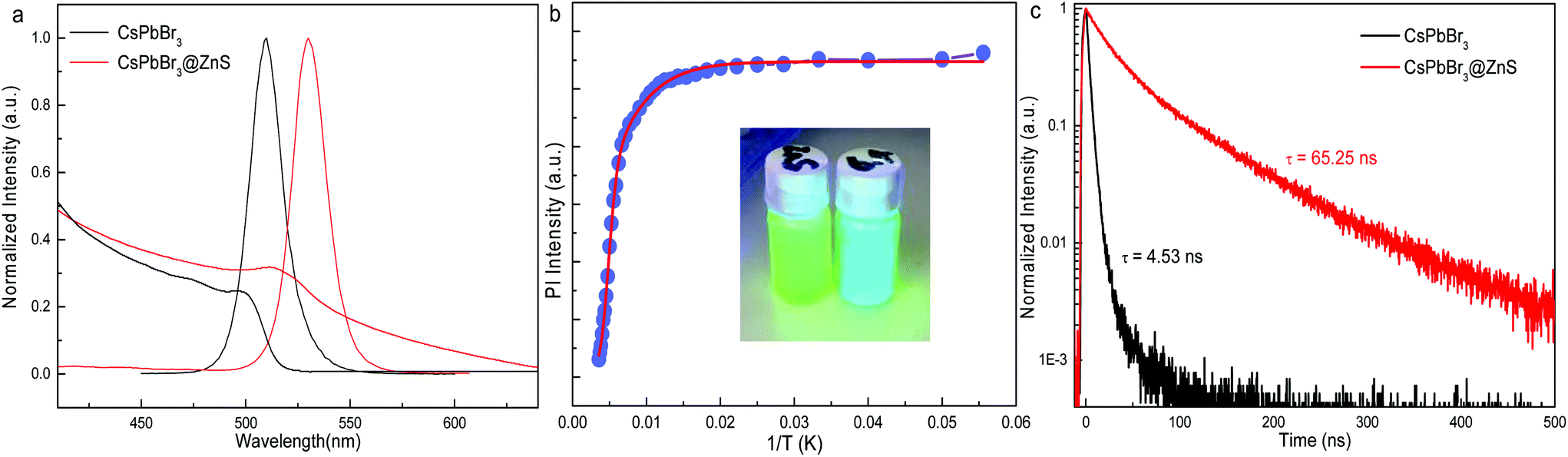 | ||
| Fig. 1 (a) Steady-state absorption and emission spectra of CsPbBr3@ZnS and CsPbBr3 NCs. (b) The integrated PL intensity as a function of temperature. The red line is the fitted curve by the Arrhenius function with dual activation energy model. The inset shows the photographs of CsPbBr3@ZnS (left) and CsPbBr3 (right) solution under UV irradiation. (c) Time-resolved PL curves of CsPbBr3@ZnS and CsPbBr3 NCs. | ||
To investigate the ultrafast carrier dynamics, TA spectroscopy was performed on the CsPbBr3@ZnS and CsPbBr3 NCs in parallel. The 400 nm femtosecond laser works as excitation, and the supercontinuum generated through nonlinear crystal is adopted to probe the ultrafast and broadband response of photocarriers (see details of the TA setup in Note 1 in the ESI). The carrier density in NCs, denoted by the average electron–hole pairs per NC 〈N0〉, is deduced from the long-term decay values in the fluence-dependent curves (Fig. S4† and Note 2 in the ESI). Fig. 2a and b show the pseudocolor TA spectra of CsPbBr3@ZnS and CsPbBr3 NCs at a low carrier density, 〈N0〉 ∼0.4. The corresponding transient spectral distributions at various delay times are plotted in Fig. 2c and d. In both cases, the dominant signals are the band-edge photobleaching (ΔOD < 0) located at ∼500 and ∼515 nm, due to the state-filling effect by the photocarriers.20,21 However, different from CsPbBr3 NCs, an obvious photobleaching signal is observed at the short-wavelength spectral regime (330–370 nm) for CsPbBr3@ZnS NCs (Fig. 2a–d). The maximum photobleaching signal is located at ∼350 nm and matches well with the bandgap of ZnS, indicating its origin from the carrier-filling of ZnS. Due to the large bandgap of ZnS (3.5 eV), the pump beam of 400 nm (∼3 eV) can merely excite the CsPbBr3 core but not the ZnS shell, and the low input intensity avoids any nonlinear optical effects (e.g., two photon absorption). Thus, the short-wavelength bleaching can only be ascribed to the charge transfer from CsPbBr3 into ZnS, that is, the hole injection driven by the band alignment.22,23 Furthermore, the decay curves at 515 nm and 350 nm, corresponding to the bleaching signals from CsPbBr3 and ZnS, respectively, are plotted in Fig. 2e. It is found that the relaxation processes of the two signals are nearly the same, which indicates that the decay of the holes in ZnS is related to the interfacial recombination with electrons in CsPbBr3, suggesting the formation of the charge transfer (CT) state.22 Consistently, the photobleaching signal in the long-wavelength region of the CsPbBr3@ZnS NCs marked in Fig. 2c is observed, which can be tentatively ascribed to the carrier-filling of the CT state.22,24 As for the “pure” CsPbBr3 NCs, only the photoinduced absorption (PIA) is detected at the similar long-wavelength side (Fig. 2d). Such a PIA signal in CsPbBr3 NCs has been previously assigned to the hot carrier-induced biexciton absorption or bandgap renormalization, and it decays rapidly with a picosecond timescale,20,25 while the long-wavelength bleaching signal in CsPbBr3@ZnS NCs is sustained over several nanoseconds. To get more insights, the decay curves at 350, 530 and 540 nm are plotted together in Fig. 2f. The complete overlap of these normalized curves demonstrates the same origins of the long-wavelength bleaching signal as that of 350 nm (bleaching of ZnS), which further confirms that the long-wavelength bleaching comes from the CT state.
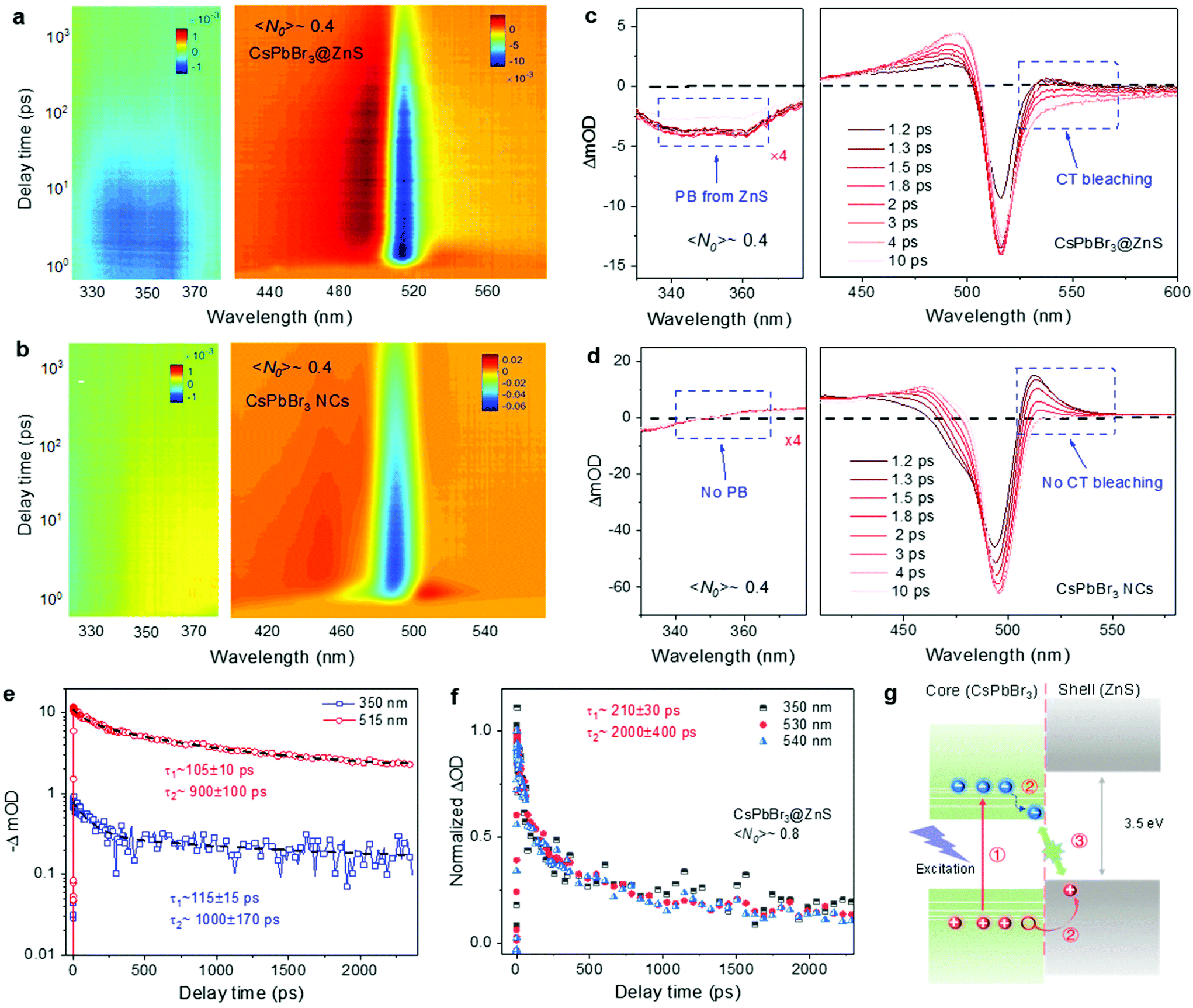 | ||
| Fig. 2 (a and b) Pseudocolor transient absorption (TA) spectroscopies of CsPbBr3@ZnS and CsPbBr3 NCs at different probe ranges, when the excitation wavelength is 400 nm. The average carrier pair number per NC 〈N0〉 is 0.4. (c and d) Broadband TA spectra of CsPbBr3@ZnS and CsPbBr3 NCs at different delay times. (e) The relaxation curves of CsPbBr3@ZnS NCs when probe wavelengths are 515 nm and 350 nm selected from (a). (f) The normalized decay curves of CsPbBr3@ZnS when probe wavelengths are 350, 530 and 540 nm. (g) The schematic illustration of photocarrier dynamics in CsPbBr3@ZnS NCs, including three main steps: ① generation of photocarriers in CsPbBr3; ② hot carrier cooling and interfacial charge transfer; and ③ interfacial recombination. | ||
Regarding the charge transfer process, there are generally two possible ways: band-edge hole transfer and hot hole transfer.26 In the first case, the photoexcited holes in the CsPbBr3 core will first relax to the valence band edge, and the “cold” holes subsequently flow into ZnS. In the second situation however, the excited hot holes will directly transfer into the ZnS valence band.26 The two different pathways will result in disparate photophysical properties (e.g., hot carrier cooling process) and application potentials.27 To explore the dominant route, we trace the rising process of the injected hole signal in ZnS. As shown in Fig. 3a, the ultrafast charge transfer (<100 fs) manifested by the bleaching build up at 350 nm uncovers that the hole transfer occurs before hot carriers cool back to the band edge, establishing the “hot” hole transfer channel. Such ultrafast hot hole transfer will benefit the carrier extraction in hetero-nanocrystals, which is desirable for applications such as photocatalysis and hot-carrier photovoltaics.1 In addition, the “hot” hole transfer suggests that the holes with excess energy will dissipate into cooling phonons in ZnS, which can reduce the hot phonon population in CsPbBr3 and speed up hot carrier relaxation. Fig. 3b directly compares the rising edges of the two samples corresponding to the cooling process of hot carriers under the same conditions. The rising time was fitted as 0.22 and 0.49 ps for CsPbBr3@ZnS and CsPbBr3 NCs, respectively. Moreover, Fig. 3c shows the rising dynamics of normalized band-edge bleaching at 515 nm of CsPbBr3@ZnS NCs with different carrier densities 〈N0〉, from 0.2 to 3.3. As previously reported, due to the effect of a hot phonon bottleneck, the rising time of the band-edge bleaching signal in the “pure” CsPbBr3 NCs was found to be extended with the increase of carrier density,21 as shown in Fig. S5.† However, the pump intensity–dependent variation of the rising edges is negligible for CsPbBr3@ZnS NCs (Fig. 3c). We further adopt the Fermi–Dirac formula to fit the high-energy tail of the band-edge bleaching signal to extract the typical electronic temperature Te (Supporting Note 3).21,28 The decay processes of Te are plotted in Fig. 3d. It is found that the high Te in CsPbBr3 NCs can last over 30 ps, which is consistent with previous literatures and results from the hot phonon bottleneck effect.28 As a comparison, the small initial value and short relaxation time of Te in CsPbBr3@ZnS NCs with single-exponential process indicates the absence of a hot phonon bottleneck during hot carrier cooling. The fast hot carrier cooling and the absence of a hot phonon bottleneck in CsPbBr3@ZnS NCs further support the “hot” hole transfer channel and suggest the importance of shell engineering to modulate the photophysical properties for desired applications.
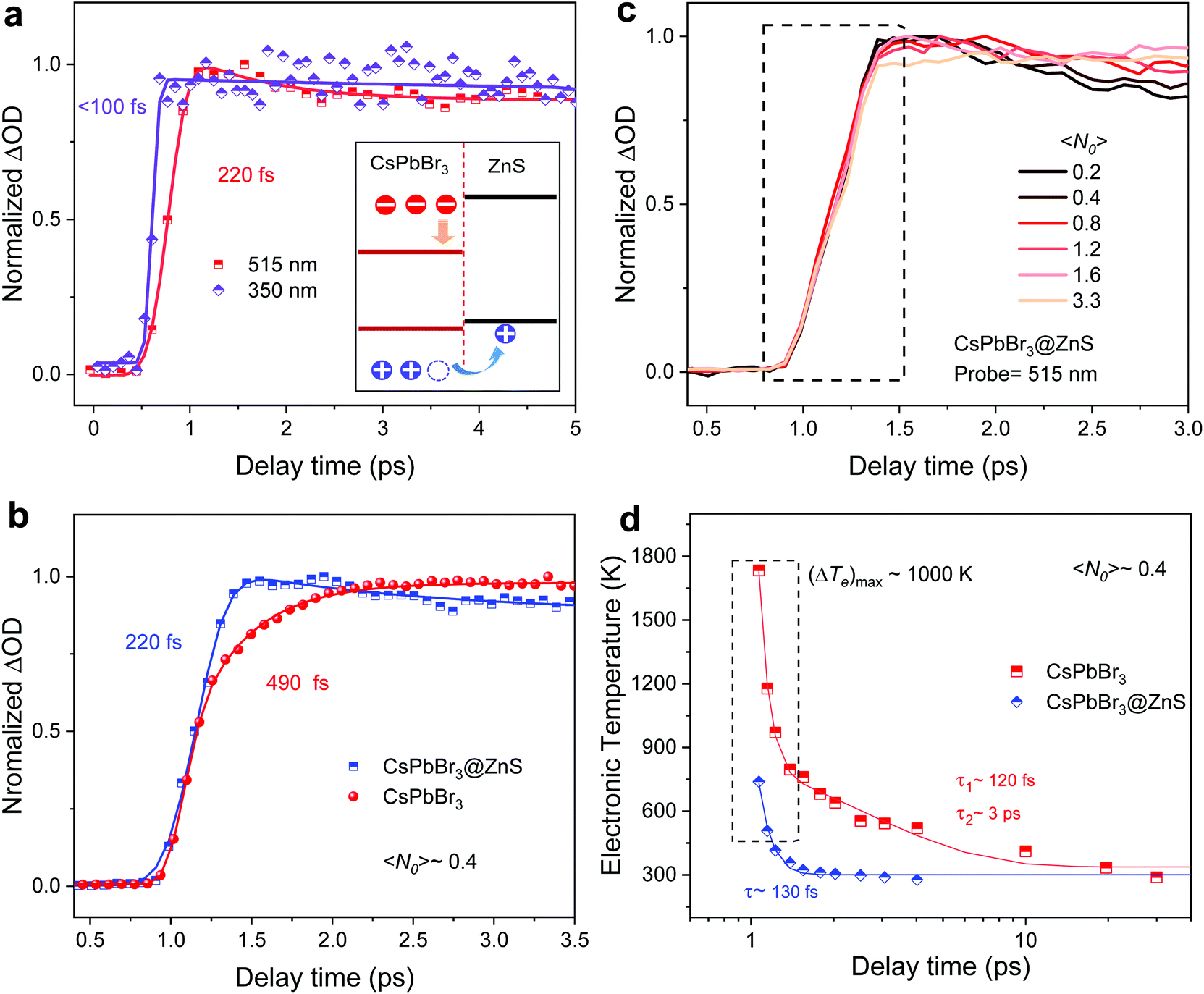 | ||
| Fig. 3 (a) The normalized decay curves of CsPbBr3@ZnS NCs within the first 5 ps when the probes are at 350 and 515 nm. The solid lines are from global fitting, where the corresponding rising times are <100 and 220 fs. The inset shows the interfacial transfer process of hot holes from CsPbBr3 to ZnS. (b) Normalized dynamics of CsPbBr3@ZnS NCs probed at the band edge, when 〈N0〉 is tuned from 0.2 to 3.3. (c) The comparison of the rising time in CsPbBr3@ZnS and CsPbBr3 NCs, where 〈N0〉 is 0.4. The rising times fitted by global fitting are 220 and 490 fs for CsPbBr3@ZnS and CsPbBr3 NCs, respectively. (d) Electronic temperatures of hot carriers in CsPbBr3@ZnS and CsPbBr3 NCs as a function of delay time when 〈N0〉 is 0.4. The solid lines represent the single- and bi-exponential functions. | ||
According to the TA spectroscopic characterization, we can build the physical picture of the ultrafast carrier dynamics in the CsPbBr3@ZnS hetero-nanocrystals (see Fig. 2g). First, the hot electrons and holes are generated by photoexcitation. Then, the hot electrons cool back to the CBM of CsPbBr3, while the hot holes directly undergo the ultrafast charge transfer from the CsPbBr3 core to the ZnS shell and form the CT state; finally, the interfacial recombination of electrons and holes in the core–shell type NCs occurs. It is noted that similar to the previous report,17 the shell growth is not homogeneous on top of all core NCs for the present sample, and a small part of the NCs are even core only. Nevertheless, the CsPbBr3@ZnS core–shell type NCs show apparent distinctions in ultrafast dynamics from those of the CsPbBr3 NCs, which may indicate the non-significant influence of inhomogeneity on the TA signals. The detailed effect of the inhomogeneity on the ultrafast dynamics may be studied in the future when homogeneous shell coating is available.
By virtue of the ultrafast charge transfer and charge separation in CsPbBr3@ZnS NCs, it can be expected that carrier–carrier interactions would be altered.3 As one of the many-body effects, the Auger process always involves multicarrier interactions, and the long Auger lifetime could benefit the high-power light-emitting and lasing applications.2 According to the pump fluence-dependent TA measurements, the biexciton Auger recombination can be derived.29,30Fig. 4a and c show the band-edge decay processes of CsPbBr3@ZnS and CsPbBr3 NCs under varied photoexcited carrier densities. We rescale the band-edge decay curves to overlap at the long timescale since the long-lived relaxation represents the single-exciton recombination. With the increase of carrier density or 〈N0〉, a new and fast relaxation component emerges at the initial stages (Fig. 4a and c), which is a typical feature of the multicarrier interaction. According to the previously reported approach,30 the Auger recombination lifetime can be obtained by subtracting the dynamics at low carrier density, which is fitted well by the single-exponential function. As such, the Auger lifetime τxx is determined to be 203 ± 15 ps and 47 ± 5 ps for CsPbBr3@ZnS and CsPbBr3 NCs, respectively. The large elongation (nearly five times) of the Auger lifetime in CsPbBr3@ZnS NCs demonstrates the significant effect of shell-coating on the multicarrier interactions in Pe-NCs. Previous studies of the Auger recombination in NCs revealed a general scaling of the Auger lifetime with NC volume (known as V-scaling),3 but recent measurements on CsPbBr3 NCs showed a strong deviation from the V-scaling.29 Such difference likely stems from the large size of NCs in the weak confinement regime. The typical length of CsPbBr3 in this work is much larger than the Bohr size (∼5.8 nm); therefore, the increase of Auger lifetime may not result from the change of volume. On the other hand, the Auger recombination rate of NCs can be described by Fermi's Golden Rule, showing that the Auger lifetime (τA) is inversely proportional to the wavefunction overlap integral between the initial and final states: τA∝1/|〈ψe|ψh〉|2.2,3 Considering the spatial separation of electrons and holes in CsPbBr3@ZnS NCs, the wavefunction overlap should be greatly reduced, which slows down the Auger recombination rate.
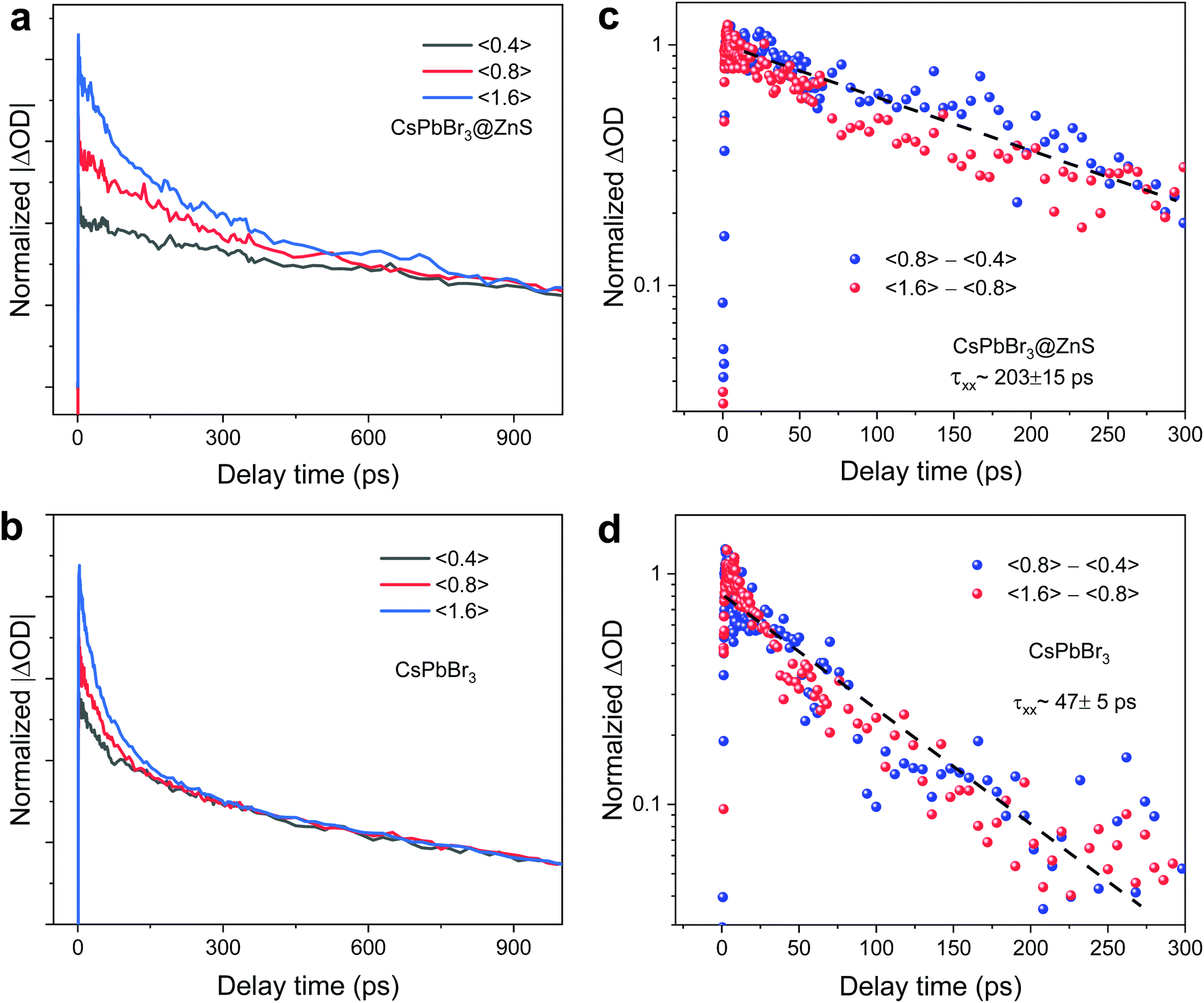 | ||
| Fig. 4 Auger recombination processes (a and b) and prominent PB dynamics of CsPbBr3@ZnS and CsPbBr3 NCs with varying average pair number per NC from 0.4 to 1.6. These curves have been normalized to overlap their longer-term values. (c and d) Biexciton recombination kinetics for CsPbBr3@ZnS and CsPbBr3 NCs, subtracted from (a and b), and the solid lines represent single-exponential decay functions, where the biexciton lifetimes are 203 and 47 ps, respectively. | ||
It is known that the Auger recombination represents the main optical loss in NCs for light amplification.3 Thanks to the suppressed Auger process, the CsPbBr3@ZnS NCs may serve as favorable optical gain media. Fig. 5a and b show the pump fluence dependent PL spectra from the CsPbBr3@ZnS and CsPbBr3 NCs, respectively, by means of the stripe pumping configuration.2 Under low pump fluence, the PL is dominated by the relatively broad spontaneous emission (SPE). With the increasing of the pump fluence, a new PL peak with a narrow linewidth (∼5 nm) emerges (Fig. 5a), indicating the occurrence of stimulated emission (STE) from CsPbBr3@ZnS NCs. The plot of the integrated PL intensity as a function of pump fluence shows a nonlinear behavior (Fig. S6†), further confirming the achievement of STE, and the threshold is extracted to be ∼35 μJ cm−2, which is among the lowest values for NC-gain media.2 Notably, the relative peak positions between STE and SPE are obviously distinct for the two NCs. In contrast to CsPbBr3 whose STE is largely red-shifted from SPE (∼11 nm), the STE of CsPbBr3@ZnS lies close to its SPE. This phenomenon originates from the intrinsically distinct carrier–carrier interactions in the samples. Specifically, in CsPbBr3, the large overlap among the multicarrier wave functions enables the strong attractive many-body interactions; thus, the multicarrier-induced STE greatly red-shifts with respect to that of SPE.2 However, for CsPbBr3@ZnS, by virtue of the spatially isolated electron and hole wave functions, the attractive interaction is weakened while the repulsive carrier interaction strengthens, which jointly leads to the similar peak positions of STE and SPE.3,31 It is noted that the ASE threshold of CsPbBr3@ZnS NCs is higher than that of CsPbBr3, which can be attributed to the reduced oscillator strength in CsPbBr3@ZnS due to the spatial separation of the electron and hole wavefunction.
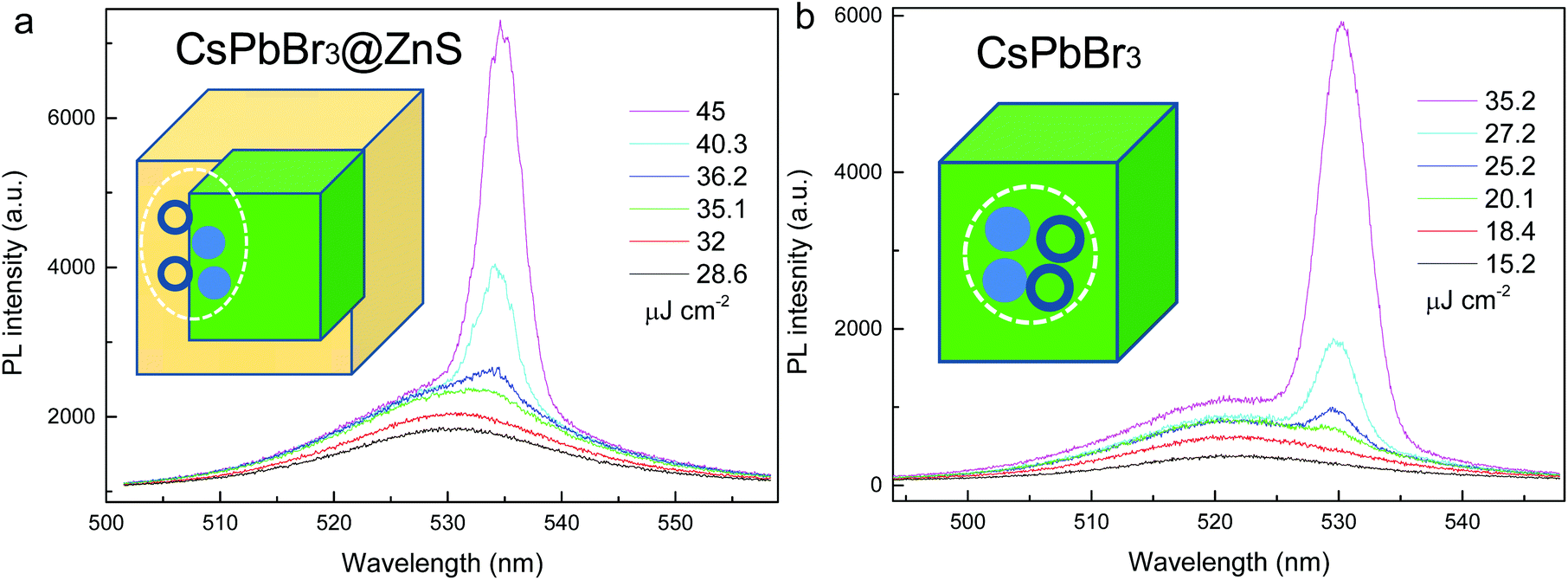 | ||
| Fig. 5 Pump fluence dependent PL spectra from CsPbBr3@ZnS (a) and CsPbBr3 NCs (b) showing the development of stimulated emission. The inset shows the schematic carrier–carrier interaction in the respective NCs. | ||
Experimental
Fabrication of CsPbBr3@ZnS and CsPbBr3 NCs
Zn(DDTC)2 was used as the zinc and sulphur source to treat CsPbBr3 NCs. Briefly, 30 mg CsPbBr3 NC powder was dispersed in 6 mL of ODE, and 32 mg of Zn(DDTC)2 powder was added along with 40 μL of OAmBr. Then, the mixture was heated at 130 °C for 1 h using an oil bath. To purify the NCs, the precipitate obtained was dispersed in toluene, MeOAc was added in equal volume, and the solution was centrifuged at 7000 rpm to remove the supernatant.Optical characterization
Time-resolved photoluminescence was measured by 375 nm pulsed laser and detected by a single photon counting photomultiplier module. For ASE characterization, the sample was pumped at 400 nm by frequency-doubling the fundamental wavelength (800 nm) from the Ti:sapphire regenerative amplifier (100 fs, 1 kHz). The laser beam was focused on the sample with a beam size of ∼3 mm × 200 μm by the standard strip pumping configuration. The emission signal was collected from the edge and detected by a monochromator coupled with a charge-coupled device (CCD).Conclusions
In summary, we have deciphered the ultrafast dynamics of perovskite hetero-nanocrystals using CsPbBr3@ZnS as a model system. We unambiguously clarified the important role of the core–shell effect on the excited-state dynamics (e.g., the ultrafast charge transfer, the formation of the CT state and hot carrier cooling processes) and multicarrier interactions in Pe-NCs. These results not only provide insightful physical information but also benefit the applications based on Pe-NCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20190446) and National Natural Science Foundation of China (11904172). Y. W thanks the support of the start-up funding from Nanjing University of Science and Technology.References
- J. B. Baxter, C. Richter and C. A. Schmuttenmaer, Annu. Rev. Phys. Chem., 2014, 65, 423–447 CrossRef CAS PubMed.
- Y. Wang and H. Sun, Prog. Quantum Electron., 2018, 60, 1–29 CrossRef.
- J. M. Pietryga, Y.-S. Park, J. Lim, A. F. Fidler, W. K. Bae, S. Brovelli and V. I. Klimov, Chem. Rev., 2016, 116, 10513–10622 CrossRef CAS PubMed.
- P. Reiss, M. Protière and L. Li, Small, 2009, 5, 154–168 CrossRef CAS PubMed.
- S. S. Lo, T. Mirkovic, C.-H. Chuang, C. Burda and G. D. Scholes, Adv. Mater., 2011, 23, 180–197 CrossRef CAS.
- J. Zhou, M. Zhu, R. Meng, H. Qin and X. Peng, J. Am. Chem. Soc., 2017, 139, 16556–16567 CrossRef CAS.
- K. Wu, W. E. Rodríguez-Córdoba, Z. Liu, H. Zhu and T. Lian, ACS Nano, 2013, 7, 7173–7185 CrossRef CAS PubMed.
- Y. Wang and H. Sun, Small Methods, 2018, 2, 1700252 CrossRef.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- F. Zhang, H. Zhong, C. Chen, X.-g. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- M. A. Becker, R. Vaxenburg, G. Nedelcu, P. C. Sercel, A. Shabaev, M. J. Mehl, J. G. Michopoulos, S. G. Lambrakos, N. Bernstein, J. L. Lyons, T. Stöferle, R. F. Mahrt, M. V. Kovalenko, D. J. Norris, G. Rainò and A. L. Efros, Nature, 2018, 553, 189–193 CrossRef CAS PubMed.
- H. Liu, Y. Tan, M. Cao, H. Hu, L. Wu, X. Yu, L. Wang, B. Sun and Q. Zhang, ACS Nano, 2019, 13, 5366–5374 CrossRef CAS PubMed.
- H. Hu, L. Wu, Y. Tan, Q. Zhong, M. Chen, Y. Qiu, D. Yang, B. Sun, Q. Zhang and Y. Yin, J. Am. Chem. Soc., 2018, 140, 406–412 CrossRef CAS PubMed.
- X. Tang, J. Yang, S. Li, Z. Liu, Z. Hu, J. Hao, J. Du, Y. Leng, H. Qin, X. Lin, Y. Lin, Y. Tian, M. Zhou and Q. Xiong, Adv. Sci., 2019, 6, 1900412 CrossRef CAS PubMed.
- X. Zhang, X. Wu, X. Liu, G. Chen, Y. Wang, J. Bao, X. Xu, X. Liu, Q. Zhang, K. Yu, W. Wei, J. Liu, J. Xu, H. Jiang, P. Wang and X. Wang, J. Am. Chem. Soc., 2020, 142, 4464–4471 CrossRef CAS PubMed.
- Y. Wang, X. Li, J. Song, L. Xiao, H. Zeng and H. Sun, Adv. Mater., 2015, 27, 7101–7108 CrossRef CAS PubMed.
- V. K. Ravi, S. Saikia, S. Yadav, V. V. Nawale and A. Nag, ACS Energy Lett., 2020, 5, 1794–1796 CrossRef CAS.
- W. Chen, J. Hao, W. Hu, Z. Zang, X. Tang, L. Fang, T. Niu and M. Zhou, Small, 2017, 13, 1604085 CrossRef PubMed.
- H. D. Sun, S. Calvez, M. D. Dawson, J. A. Gupta, G. C. Aers and G. I. Sproule, Appl. Phys. Lett., 2006, 89, 101909 CrossRef.
- N. Mondal, A. De, S. Das, S. Paul and A. Samanta, Nanoscale, 2019, 11, 9796–9818 RSC.
- Z. Nie, X. Gao, Y. Ren, S. Xia, Y. Wang, Y. Shi, J. Zhao and Y. Wang, Nano Lett., 2020, 20, 4610–4617 CrossRef CAS PubMed.
- Q. Li, Z. Xu, J. R. McBride and T. Lian, ACS Nano, 2017, 11, 2545–2553 CrossRef CAS PubMed.
- C.-H. Chuang, T. L. Doane, S. S. Lo, G. D. Scholes and C. Burda, ACS Nano, 2011, 5, 6016–6024 CrossRef CAS PubMed.
- D. B. Sulas-Kern, E. M. Miller and J. L. Blackburn, Energy Environ. Sci., 2020, 13, 2684–2740 RSC.
- A. Mondal, J. Aneesh, V. K. Ravi, R. Sharma, W. J. Mir, M. C. Beard, A. Nag and K. V. Adarsh, Phys. Rev. B, 2018, 98, 115418 CrossRef CAS.
- H. Bässler and A. Köhler, Phys. Chem. Chem. Phys., 2015, 17, 28451–28462 RSC.
- H. Chung, S. I. Jung, H. J. Kim, W. Cha, E. Sim, D. Kim, W.-K. Koh and J. Kim, Angew. Chem., Int. Ed., 2017, 56, 4160–4164 CrossRef CAS PubMed.
- M. Li, S. Bhaumik, T. W. Goh, M. S. Kumar, N. Yantara, M. Grätzel, S. Mhaisalkar, N. Mathews and T. C. Sum, Nat. Commun., 2017, 8, 14350 CrossRef CAS PubMed.
- Y. Li, T. Ding, X. Luo, Z. Chen, X. Liu, X. Lu and K. Wu, Nano Res., 2019, 12, 619–623 CrossRef CAS.
- V. I. Klimov, A. A. Mikhailovsky, D. W. McBranch, C. A. Leatherdale and M. G. Bawendi, Science, 2000, 287, 1011–1013 CrossRef CAS PubMed.
- Y. Gao, M. Li, S. Delikanli, H. Zheng, B. Liu, C. Dang, T. C. Sum and H. V. Demir, Nanoscale, 2018, 10, 9466–9475 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures. See DOI: 10.1039/d0nr06884e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |