
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeparin length in the coating of extremely small iron oxide nanoparticles regulates in vivo theranostic applications†
Hugo
Groult
 *a,
Susana
Carregal-Romero
bc,
David
Castejón
d,
Mikel
Azkargorta
e,
Ana-Beatriz
Miguel-Coello
b,
Krishna Reddy
Pulagam
b,
Vanessa
Gómez-Vallejo
b,
Rémi
Cousin
a,
María
Muñoz-Caffarel
fg,
Charles H.
Lawrie
fg,
Jordi
Llop
bc,
Jean-Marie
Piot
a,
Felix
Elortza
e,
Thierry
Maugard
*a,
Jesús
Ruiz-Cabello
*bcgh and
Ingrid
Fruitier-Arnaudin
a
*a,
Susana
Carregal-Romero
bc,
David
Castejón
d,
Mikel
Azkargorta
e,
Ana-Beatriz
Miguel-Coello
b,
Krishna Reddy
Pulagam
b,
Vanessa
Gómez-Vallejo
b,
Rémi
Cousin
a,
María
Muñoz-Caffarel
fg,
Charles H.
Lawrie
fg,
Jordi
Llop
bc,
Jean-Marie
Piot
a,
Felix
Elortza
e,
Thierry
Maugard
*a,
Jesús
Ruiz-Cabello
*bcgh and
Ingrid
Fruitier-Arnaudin
a
aBCBS team (Biotechnologies et Chimie des Bioressources pour la Santé), LIENSs Laboratory (Littoral environment et Sociétés), UMR CNRS 7266, University of La Rochelle, La Rochelle, France. E-mail: hugo.groult@univ-lr.fr; thierry.maugard@univ-lr.fr
bCIC biomaGUNE and Basque Research and Technology Alliance (BRTA), Donostia-San Sebastián, Gipuzkoa, Spain. E-mail: jruizcabello@cicbiomagune.es
cCIBER de Enfermedades Respiratorias (CIBERES), Madrid, Spain
dUnidad de RMN – CAI Bioimagen Complutense, Universidad Complutense de Madrid, Spain
eProteomics Platform CIC bioGUNE, Bizkaia Science and Technology, Derio, Spain
fMolecular Oncology Group, Biodonostia Health Research Institute, San Sebastian, Spain
gIkerbasque, Basque Foundation for Science, 48013 Bilbao, Spain
hDepartamento de Química en Ciencias Farmacéuticas, Universidad Complutense de Madrid, Madrid, Spain
First published on 22nd December 2020
Abstract
The positive contrast of extremely small iron oxide nanoparticles (ESIONP) in magnetic resonance imaging (MRI) rejuvenates this class of metal nanoparticles (NP).Yet, the current synthesis often lacks the possibility of adjusting the core size (while it is a key element for ESIONP-based MRI contrast behaviour), and also involved multiple complex steps before obtaining a ready-to-use probe for medical applications. In this study, we faced these challenges by applying heparin oligosaccharides (HO) of different lengths as coatings for the preparation of HEP-ESIONP with a one-pot microwave method. We demonstrated that the HO length could control the core size during the synthesis to achieve optimal positive MRI contrast, and that HEP-ESIONP were endowed directly with anticoagulant properties and/or a specific antitumor activity, according to the HO used. Relevantly, positron emission tomography (PET)-based in vivo biodistribution study conducted with 68Ga core-doped HEP-ESIONP analogues revealed significant changes in the probe behaviours, the shortening of HO promoting a shift from hepatic to renal clearance. The different conformations of HO coatings and a thorough in vitro characterisation of the probes’ protein coronas provided insight into this crucial impact of HO length on opsonization-mediated immune response and elimination. Overall, we were able to identify a precise HO length to get an ESIONP probe showing prolonged vascular lifetime and moderate accumulation in a tumor xenograft, balanced with a low uptake by non-specific organs and favourable urinary clearance. This probe met all prerequisites for advanced theranostic medical applications with a dual MRI/PET hot spot capability and potential antitumor activity.
1. Introduction
Far from decline, the long-standing iron oxide nanoparticles (NP) have recently made a new breakthrough in the medical field.1 Compared to their previous use as negative contrast agents for magnetic resonance imaging (MRI) applications, a new generation based on extremely small iron oxide nanoparticles (ESIONP, with a magnetic core < 5 nm) has been developed to provide a positive contrast.2 This contrast makes these NPs closer to the current clinical practices for MRI-based diagnosis, providing a higher resolution for an easier detection.3 ESIONP might be a promising biocompatible alternative to gadolinium chelates that are currently the reference positive MRI contrast agents but are known to have safety problems.4 In comparison, ESIONP can also warrant prolonged blood circulation time5–7 and be easily engineered for multifunctional applications.8–10 In this context, research has been conducted to implement synthesis conditions with an increased control of ESIONP core size, which is a key parameter for their magnetic properties.11–13 However, most of these approaches have used “neutral” coatings and further steps were needed to provide ESIONP enhanced colloidal stability and vascular lifetime, targeting features, or a specific bioactivity. Based on our previous results, we considered herein the use of heparin oligosaccharides (HO) as bioactive coating agents to produce multifunctional ESIONP with a one-pot microwave synthesis that allowed control of the core size.14Heparin is a linear polysaccharide, which has long been used in clinical practice as an anticoagulant.15 It is also currently investigated in wound healing, lung disorders, chronic inflammation, or as an anti-angiogenic and anti-metastatic agent to control tumour growth.16,17 Heparin has been extensively used in the surface chemistry of NP appreciated for its colloidal stability, bioacceptability and easy chemical modification.18 It is mostly applied as a scaffold to be functionalized with drug conjugates or as a suitable template for self-assembly due to its high negative charge.19 In these formulations, it is rather included as a backbone for multifunctional NP carrying an exogenous drug or intended for diagnostic purposes that are used in various medical areas such as antiviral, anti-inflammatory or cancer management.20,21 The inclusion of a solitary heparin coating as an active ingredient has been more sporadic due to the possible risk of internal bleeding, except when the anticoagulant properties or hemocompatibility have been intentionally sought.22–24 Several studies have described the use of heparin-coated NP in cell labelling, transduction or for their anti-angiogenic properties, but thorough in vivo proofs are lacking.25–27 One of the current challenges in this regard is to develop nanomedicines with newly generated heparin oligosaccharide derivatives showing limited antithrombotic properties but with other selected bioactivities.16,22 For example, it has been suggested that low molecular weight heparins could provide a targeting capability to NP through interactions with extracellular enzymes or cell membrane biomarkers, as well as provide an antitumour activity.28–31
These HO derivatives are produced by controlled depolymerisation and chemical modifications, including desulfation, N-acetylation or glycol-splitting.17 In addition to modulating HO-related bioactivities of NP, the reduction in molecular weight (Mw) caused by depolymerisation may have spectacular effects on other physicochemical features and in vivo fate of these NP, starting with their blood pharmacokinetics and clearance.32–34 Indeed, after intravenous administration most of polymer-coated metallic NP are rapidly captured by the different organs of the mononuclear phagocyte system (especially the liver, spleen and bone marrow), where they are very slowly processed before excretion.35 A prolonged circulation time in the blood is then decisive to promote NP accumulation in the target organ and it is well described that the coating charge, nature and also dimension modulate the vascular lifetime.36,37 Hence, with the aim of extending NP vascular duration, several studies have been conducted to understand the role of the polymer length and configuration in the interactions with the cells of the phagocytic system or on the adsorption of plasma proteins (i.e. the protein corona)38,39 responsible for opsonisation.40,41 Secondly, the polymer length also strongly influence NP crossing of endogenous biological barriers, penetration in tumor stroma or cell internalization.33,42 Interestingly, the use of shorter HO may also reduce enough the final NP hydrodynamic size (HS) until the glomerular filtration threshold for renal elimination, estimated at about 8–10 nm. This route of urinary clearance is highly appreciated because it reduces toxicity issues and translational impairments, whereas the rapid removal of the NP from circulation by the filtration system may limit the time to accumulate in the targeted tissue or to exert their effect.43 As general acceptance, creating metallic NP with a prolonged vascular lifetime to enhance targeting properties but mostly undergoing optimal renal clearance will represent a breakthrough in the field.44
To our knowledge, a detailed comparative study about the impact of heparin length on all these aspects has not yet been addressed, in contrast with other well-known NP coatings such as polyethylene glycol.45 Given the clinical potential of the new HO derivatives,46 this evaluation may be crucial to support the decision to apply the most appropriate HO Mw as coating in the NP design, depending on the intended application. In this work, the physicochemical and biological features of ESIONP functionalised with five HO of decreasing length (HEP-ESIONP) were analysed. We showed that HO length could modulate the core diameter obtained during the synthesis, and how the in vivo positive MRI contrast performance of HEP-ESIONP were consequently affected. Next, using HEP-ESIONP analogues core-doped with 68Ga as positron emission tomography (PET) tracers, we investigated the HO length related changes in the vascular lifetime and biodistribution of the probes in a mice model of low-vascularized BT-474 breast cancer xenograft.47 We aimed to determine whether these changes were due to the protein corona composition and/or HO conformation at the NP surface. Finally, to complete the picture of the most suitable length, we also observed the influence of the HO length on HEP-ESIONP anti-thrombotic properties as well as antitumor activity via the inhibition of heparanase (HPSE), a key enzyme involved in angiogenesis and invasion. All these results were useful to demonstrate that heparin should not be considered as a simple NP stabilizer suitable for further adaptations, but on the contrary that by adjusting its length, it could play a central role in NP specific functions and in vivo behaviour.
2. Experimental
2.1 Synthesis of HO derivatives
A 40 mL solution of native heparin (Heparin sodium salt, Molekula) at 25 mg mL−1 was prepared with Milli-Q water. To start depolymerisation, 460 μL of hydrogen peroxide (H2O2 30%, Merck) were then added, corresponding to a final H2O2/native heparin (w/w) ratio of 0.15. Immediately after the addition, the reaction mixture was sealed and placed in a heating incubator at 60 °C under stirring at 500 rpm. Finally, 10 mL aliquots were collected at four different timepoints (4 h, 8 h, 12 h and 24 h) and were freeze-dried before analysis and preparation of HEP-ESIONP.2.2 Structural analysis of HO by size exclusion-high-performance liquid chromatography
The structural analysis of HO was performed using a high-performance liquid chromatography system from Agilent (1100 LC) with one TSK-GEL G4000SW (30 cm × 7.5 mm) column and two TSK-GEL G3000SW (30 cm × 7.5 mm) columns mounted in series and maintained at 25 °C. HO were eluted in a 0.1 M sodium nitrate (NaNO3) buffer at a flow rate of 0.8 mL min−1 and detected by differential refractometry. The refractometry signal was then processed and quantified using a HP Chemstation software off-line. A calibration curve of heparin oligosaccharide standards (Iduron, Manchester, UK) ranging from 8 to 32 DP (degree of polymerisation) of the monomer unit (300 Da) was used. It allowed calculating the number average molecular weight (Mn), Mw, polydispersity index (I) and DP according to a previously published method48 and using the following equations: | (1) |
 | (2) |
 | (3) |
 | (4) |
2.3 Quantification of the degree of sulfation of HO
The degree of sulfation was calculated using a colorimetric assay based on absorbance reading at 640 nm of the (7-aminophenothiazin-3-ylidene)-dimethylazanium chloride (Azure A) that disappears after binding to the sulfated groups on the polysaccharide backbones.49 In a 96-well plate, 200 μL of an aqueous Azure A solution (0.01 mg mL−1) were added to 20 μL of HO samples diluted at 6 different concentrations ranging from 6.25 to 25 mg L−1, and absorbance was read after 15 min of incubation. A dextran sulfate standard with a known sulfur content of 17% was used to build a calibration curve using the absorbance values obtained from a serial dilution (0–30 mg L−1).2.4 Synthesis of HEP-ESIONP
In a microwave-adapted flask, HO (100 mg for HEP0-ESIONP/HEP4-ESIONP/HEP8-ESIONP or 75 mg for HEP12-ESIONP/HEP24-ESIONP) were dissolved in 1.25 mL of distilled water and mixed with 1 mL of iron(III) chloride hexahydrate (17.5 mg). Then, 0.25 mL of hydrazine monohydrate (64–65%) were added and the flask was immediately sealed before being placed in the microwave unit (CEM Discover CP). The mixture was heated under magnetic stirring up to 100 °C within 60 seconds with power set at 300 W, and the “flash” reaction took place at this temperature for 225 seconds. After cooling at room temperature, the HEP-ESIONP were purified twice by gel filtration onto a PD10 desalting column (GE Healthcare) with Milli-Q water used as an eluent, to obtain a final solution of 4 mL. All samples were stored at +4 °C before physicochemical characterisation, in vitro biological and cellular assays or in vivo experiments.2.5 Physicochemical characterization of HEP-ESIONP
High contrast transmission electronic microscopy (TEM) images were provided by a LaB6-TEM of type JEOL JEM-1400PLUS (40 kV–120 kV) equipped with a GATAN US1000 CCD camera (2k × 2k). For ultra high resolution studies a FEG-TEM of type JEOL JEM-2100F UHR (80 kV–200 kV) equipped with a TVIPS F216 CMOS camera (2k × 2k) was used.The HS and zeta potential of HEP-ESIONP were measured using a NanoZS90 device (Malvern Instrument, UK) using respectively a 12 mm2 polystyrene cuvette and folded capillary cells (DTS 1070, Malvern Instrument, UK).
Iron concentrations of HEP-ESIONP were calculated using two methods. HEP-ESIONP solutions (100 μL) were first digested overnight at 30 °C in 1 mL of a H2O2/HNO3 mixture (1/1 in volume ratio). In the first method, the T1 relaxivity values of the Fe3+ ions in the resulting dilution were measured using a Bruker MQ60 device (Bruker Biospin, Germany) at 1.5 T and 37 °C, using an Inversion Recovery sequence and the mono-exponential curve-fitting option. The final iron concentration was calculated using a T1 calibration curve of Fe3+ (0–2 mM) obtained from FeCl3·6H2O solutions that underwent the same acidic treatment as the samples. In the second method, iron concentrations were determined after digestion overnight in a 60% solution of HNO3 and further dilution (1/10) in nanopure H2O with a iCAP-Q inductively coupled plasma-mass spectrometry from Thermo equipped with collision/reaction cell and kinetic energy discrimination.
The heparin content in the HEP-ESIONP solution was calculated as previously published,14 by measuring the amount of sulfate groups in solution with the “Azure A” method and by comparing it to the degree of sulfation of the respective HO.
The density σ of HO on each HEP-ESIONP was estimated (Appendix A†) by calculating the ESIONP core volume from the mean diameters determined on the TEM images and by taking into account the spherical shape. Then, the total weight of each core was deducted from the density of bulk Fe3O4 magnetite (5.17 g cm−3), which also allowed obtaining the weight of irons in each core. The number of ESIONP cores in solution was determined by comparison with the iron concentration measured in solution. In parallel, the number of HO species in solution was calculated from the heparin content measured in solution and the Mw. The σ was finally estimated as the HO species/ESIONP cores ratio.
In the discussion about the HO configuration at the ESIONP surface, the transition distance hc between the concentrated polymer brush (CPB) and semi-diluted polymer brush (SDPB) regions was calculated from the Daoud-Cotton model using the following equation:
 | (5) |
2.6 Magnetic characterisation of HEP-ESIONP
The longitudinal (r1) and transversal (r2) relaxation values of HEP-ESIONP were calculated using a Bruker MQ60 device (Bruker Biospin, Germany) at 1.5 T and 37 °C. The T1 and T2 relaxation times of serial dilutions of HEP-ESIONP (iron concentration from 0.5 to 20 mM) were measured using the standard T2 Carr–Purcell Meiboom Gill and T1 inversion-recovery spin echo sequences. The longitudinal and transversal relaxation rates, respectively 1/T1 and 1/T2 in s−1 obtained from the relaxation times were corrected by subtracting the water relaxation rate. The linear fit of this data according to the iron concentration in mM provided straight lines the slope of which corresponded to the relaxation values r1 and r2 in s−1 mM−1.DC magnetometric measurements as a function of temperature and field have been performed in a Commercial SQUID magnetometer (MPMS3, Quantum Design Inc.) with a typical sensitivity better than 1e−12 A m2 and a maximum field of 7 T.
2.7 In vivo positive contrast performance of HEP-ESIONP on 1 T MRI
MRI was performed at the NMR Centre of the Universidad Complutense (CAI de RMN) using a 1 Tesla benchtop MRI scanner (ICON 1T-MRI; Bruker BioSpin GmbH, Ettlingen, Germany). The system consisted of a 1 T permanent magnet with a gradient system capable of supplying a 450 mT m−1 gradient strength. A solenoid mouse body RF-coil was used. Animals (6-week old female Balb/c mice, n = 3) were anesthetised using 2% isofluorane (IsoFlo, Zoetis, NJ, USA). The main MRI experiment consisted of three-dimensional T1 weighted images used to monitor the evolution of the contrast enhancements before and after HEP-ESIONP intravenous injection. The isotropic T1WI experiments (0.5 × 0.5 × 0.5 mm) were acquired using a gradient echo sequence with a repetition time of 21 ms, an echo time of 3 ms and a flip angle of 20°. The total acquisition time was 6 minutes.2.8 PET imaging of the in vivo biodistribution of 68Ga-HEP-ESIONP in mice
2.9 HEP-ESIONP protein coronas
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g and 4 °C.
:protein ratio of 1:50, and the mixture was incubated overnight at 37 °C, dried in a RVC2 25 speedvac concentrator (Christ), and resuspended in 0.1% formic acid. Peptides were desalted and resuspended in 0.1% FA using C18 stage tips (Millipore).
000g and 4 °C and washed with 400 μL of nanopure H2O each time. Bradford analysis was performed into microplate wells (triplicates) were 250 μL of Bradford reagent (Sigma) was mixed with 5 μL of each of the measured samples. Absorbance at 595 nm was measured with a plate reader (TECAN, Genios Pro). We used as blank, solutions of HEP-ESIONP with similar concentrations.
000g and 4 °C.
:protein ratio of 1:50, and the mixture was incubated overnight at 37 °C, dried in a RVC2 25 speedvac concentrator (Christ), and resuspended in 0.1% formic acid. Peptides were desalted and resuspended in 0.1% FA using C18 stage tips (Millipore).
000g and 4 °C and washed with 400 μL of nanopure H2O each time. Bradford analysis was performed into microplate wells (triplicates) were 250 μL of Bradford reagent (Sigma) was mixed with 5 μL of each of the measured samples. Absorbance at 595 nm was measured with a plate reader (TECAN, Genios Pro). We used as blank, solutions of HEP-ESIONP with similar concentrations.
2.10 Cell toxicity and uptake of HEP-ESIONP
MTT cell viability assays. The effect of HEP-ESIONP on the proliferation of HEK 293 cells was assessed using a MTT assay. Cells (2 × 103 per well) were seeded into a 96-well microplate in 50 μL of OptiMEM® medium (supplemented with 10% FBS, 1% penicillin/streptomycin) and let grown for 24 h at 37 °C in a 5% CO2 humidified atmosphere (Heracell, Thermofischer). Then, 50 μL of the same culture medium containing HEP-ESIONP at 100 μg mL−1 of HO content or water (positive proliferation control) were added. After 24 h or 72 h of incubation under the same conditions, 20 μL of a MTT solution (5 g L−1) were added and the microplate was further incubated for 4 h. Thereafter, the medium was removed and the formazan crystals formed were dissolved in 100 μL of DMSO. Absorbance of the DMSO solution was measured at 550 nm using a BMG Labtech FLUOstar Omega spectrometer and related to the value of the positive proliferation control to express a % of cell viability.Iron uptake quantification. MDA-MB-231 and HSkMEC cells were cultured in a 24-well plate in 2 mL of OptiMEM® medium (supplemented with 10% FBS, 1% penicillin/streptomycin) at 37 °C in a 5% CO2 humidified atmosphere (Heracell, Thermofischer). Once approximatively 80% of confluence was reached, the cells were incubated for 6 h with 50 μL of the same culture medium containing HEP-ESIONP at ∼0.1 mg mL−1 of iron content. Thereafter, the cells were trypsinized and after cell centrifugation, the cell pellet was washed two times with PBS. Cells were then lysed with 300 μL of lysis buffer (50 mM NaCl, 50 mM TrisCl at pH 8, 0.2% SDS) for 3 h at 55 °C and mixed with the same volume of 1.5 M HCl. Then, 300 μL of a 1.5 M HCl/4.5% KMnO4 mixture (1/1 in volume ratio) were added. After 2 h at 60 °C, 90 μL of a detection solution containing 6.5 mM ferrozine, 6.5 mM neocuproine, 2.5 M ammonium acetate and 1 M ascorbic acid were added. The absorbance was read at 550 nm after an additional 30 min of incubation. The concentration of internalized Fe was calculated from a standard curve of FeCl3 (0–300 μM) under the same incubation conditions. The cell lysates were digested in 0.8 mL of a H2O2/HNO3 mixture (1/1 in volume ratio) overnight at 30 °C before direct measurement of the iron content by inductively coupled plasma-mass spectrometry.
2.11 Bioactivities of HEP-ESIONP
The anti-factor Xa and anti-factor IIa activities of HO and HEP-ESIONP were measured using a colorimetric assay based on the conversion of a chromogenic substrate in the presence of these coagulation factors. For the anti-factor Xa assay, antithrombin AT-III (25 μL, 0.625 μg μL−1) was incubated at 37 °C in 96-well plates for 2 minutes with 25 μL of HO or HEP-ESIONP in Milli-Q water at 0.5 mg L−1. Then, 25 μl of factor Xa (11.25 nkat mL−1) were added and the plate was incubated for an additional 2 min. Finally, the factor Xa chromogenic substrate (CH2SO2-D205 Leu-Gly-Arg-pNA-AcOH, 3.25 nM, 25 μL) was added and the absorbance of the reaction mixture was read for 3 min at 405 nm every 8 seconds. The initial velocity was determined as the slope of the linear segment of the kinetics curve and the % of inhibition was calculated based on the initial velocity of the same reaction performed without HO or HEP-ESIONP inhibitor. The experimental procedure for the anti-factor IIa assay was the same except that HO or HEP-ESIONP were tested at 1 mg L−1 and a different chromogenic substrate (EtM-SPro-Arg-pNA-AcOH, 1.4 nM, 25 μL) was used. At least three independent experiments of triplicates were performed.The anti-HPSE activities of HO and HEP-ESIONP were measured (one experiment of four replicates) using the HPSE assay toolbox (Cisbio Assay, France) which is based on a heparan sulfate substrate labelled with both biotin and Eu3+ cryptate (biotin-heparan sulfate-Eu(K)). Briefly, upon excitation at 337 nm, the Eu3+ cryptate produces a fluorescent emission at 620 nm that can be converted into an emission at 665 nm by fluorescence resonance energy transfer thanks to a streptavidin-XL665 (SA-XL665) bound to the biotin. Hydrolysis and cleavage of the heparan sulfate substrate by HPSE result in a loss of the fluorescence resonance energy transfer, and thus in the emission of SA-XL665 at 665 nm. Briefly, the enzymatic reaction was performed in white 96-well half-area plates (Corning® #3693) and monitored using a spectrofluorometer (BMG Labtech FLUOstar Omega) with a high time resolved fluorescence module.
First, 15 μL of HO or HEP-ESIONP at 2.5 mg L−1 in Milli-Q water were added into the wells followed by 15 μL of HPSE solution (recombinant human HPSE-1, R&D systems, 400 ng mL−1 in Tris-HCl at pH 7.5, 0.15 M NaCl and 0.1% CHAPS). After a 10 min preincubation at 37 °C, the reaction was initiated by adding 30 μL of a biotin–heparan sulfate-Eu(K) solution (1.0 ng μL−1 in 0.2 M sodium acetate buffer, pH 4.5) and the plate was incubated at 37 °C for 15 min. At the end of the reaction, 30 μL of streptavidin-XL665 solution (SA-XL665, 10 ng μL−1 in 0.1 M NaPO4 buffer at pH 7.4, 0.8 M KF, 0.1% BSA) were added. After 5 min of incubation for biotin–streptavidin conjugation, the fluorescence was recorded at λem1 = 620 nm and λem2 = 665 nm after a 60 μs excitation at λex = 337 nm. According to the manufacturer's instructions, the percentage of heparan sulfate degradation (%D) was calculated using the following equations:
 | (6) |
 | (7) |
 | (8) |
3. Results and discussion
3.1 Preparation of HO derivatives
Five different HO with decreasing average molecular weight (Mw) were prepared from a native 12 kDa heparin (degree of polymerisation [DP] ∼ 44), using a H2O2-based radical hydrolysis depolymerisation method over progressively longer incubation times.48 To designate the different HO derivatives, each HO was named “HEP plus the depolymerisation time (in hours)”, HEP0 being the native heparin. The number average molecular weight (Mn) and Mw were calculated by a size exclusion chromatography analysis using a calibration curve of HO standards ranging from 8 to 32 DP of the monomer unit. As expected, the kinetics of the chain length reduction was faster in the first hours with a high drop in Mw, followed by a gradual decrease in depolymerisation rate (Fig. S1†). HO Mw were distributed up to 4.8 kDa corresponding to a DP of ∼13, guaranteeing a wide range of Mw for studying the polymer length impact on the physicochemical and biological properties of the HEP-ESIONP (Table 1). The polydispersity index (I) of the polymers, a parameter that may affect the HS of the ESIONP once the HO is functionalized, was estimated from the weights obtained and was not greater than 1.35, to ensure a limited influence in the following study about the chain conformation.55 Some of the depolymerisation techniques, in particular the non-enzymatic ones, could induce changes in chemical structure of heparin, especially the desulfatation.48 This could complicate the analysis since the comparisons would not only be limited to the individual heparin length parameter but would also depend on the polymeric charge or chemical nature. The degree of sulfation was stable with the depolymerisation method chosen as shown in Table 1, except for the smallest HO that underwent a significant desulfation of about 20% to be taken into account in the discussion.| Time (h) | M n (KDa) | M w (Da) | I | DP | Degree of sulfation |
|---|---|---|---|---|---|
| 0 | 12.7 ± 0.2 | 14.2 ± 1.2 | 1.12 | 44.1 ± 0.7 | 43.9 ± 1.9 |
| 4 | 7.0 ± 0.2 | 8.8 ± 0.3 | 1.25 | 24.3 ± 0.7 | 43.7 ± 1.6 |
| 8 | 5.5 ± 0.3 | 7.5 ± 0.5 | 1.35 | 19.2 ± 1.0 | 41.3 ± 2.0 |
| 12 | 4.5 ± 0.2 | 5.9 ± 0.4 | 1.32 | 15.5 ± 1.0 | 38.4 ± 2.0 |
| 24 | 3.8 ± 0.3 | 4.8 ± 0.4 | 1.29 | 13.0 ± 1.1 | 35.2 ± 2.3 |
3.2 Physicochemical characterisation and core size control of HEP-ESIONP
Each of the five heparin species was then used for the synthesis of HEP-ESIONP by a microwave-assisted bottom-up method previously described.14 Briefly, it consisted in the hydrazine reduction of iron salt, followed by an ultrafast thermic treatment for crystal growth with the HO incorporated in situ for ESIONP stabilization. Transmission electron microscopy (TEM) images (Fig. 1A) showed well-dispersed sphere-shaped NP obtained with all heparin species, although for HEP0- and HEP4-ESIONP, the cores were occasionally assembled into small groupings (Fig. S2†). High-resolution TEM images showed the lattice fringes on each of the HEP-ESIONP core, demonstrating excellent crystallinity (Fig. S3†). Analysis of the d-spacing corroborated magnetite phase, as compared with the standard atomic spacing for Fe3O4 and the respective hkl indexes from JCPS card (Table S1†), without excluding possible presence of maghemite phase.56 It is important to note that the core size distributions, computed from the images (Fig. S4A†), revealed that the use of heparins of different Mw allowed controlling the ESIONP core diameter (Table 2). As shown in Fig. 1B, the core diameters unexpectedly decreased linearly with the reduction of HO sizes, from 6.6 nm for HEP0-ESIONP synthetized with the native heparin of the highest Mw, to 2.2 nm for HEP24-ESIONP produced with heparin of the smallest Mw. In addition, the mean deviations followed the same trend, narrowing when HO Mw was reduced. Therefore, sample quality measured as NP size uniformity can be improved using low Mw HO. Compared to previous studies in which the polymer length barely interfered with the final size of NP cores,57 the observed size control in our study could be due to the increasing number of heparin chains introduced for the HO of lower Mw, and in particular, to the number of reducing ends that could arbitrate the nucleation and/or growth phase.58 The HS of the HEP-ESIONP measured by dynamic light scattering showed as well a correlation with the length of the heparin, as expected (Fig. S4B† and Fig. 1C). The HEP-ESIONP composition was characterized through FTIR spectroscopy and compared with the free HO (Fig. S4C†). FTIR spectra of the different free HO species were sensibly similar and FTIR spectra of HEP-ESIONP showed the characteristic bands of heparin in all cases.59,60 | ||
| Fig. 1 Physicochemical characterization of the HEP-ESIONP. (A) TEM images of the different HEP-ESIONP, scale bar is 100 nm. (B) Core size of HEP-ESIONP according to the length in DP of their HO coating. (C) Hydrodynamic size of the HEP-ESIONP according to the length in DP of their HO. Triangles represent the extrapolated hydrodynamic size of the HEP-ESIONP, if the HO coating had a free configuration without steric constraints. Dashed line represents the extrapolated hydrodynamic size of the HEP-ESIONP, if the HO coating were fully stretch under steric constraint. | ||
| HEP-ESIONP | Core size (nm) | HS (nm) | Z-potential (mV) | [Fe] (mg ml−1) | [HEP] (mg ml−1) | σ (chains per nm2) | h (nm) | Theoretical hc (nm) |
|---|---|---|---|---|---|---|---|---|
| a h = (HS − core size)/2. | ||||||||
| HEP0-ESIONP | 6.6 ± 2.6 | 43.4 | −37.2 | 0.9 ± 0.2 | 12.6 ± 0.8 | 2.3 | 18.4 | 9.3 |
| HEP4-ESIONP | 4.3 ± 1.7 | 22.4 | −36.3 | 1.0 ± 0.2 | 10.3 ± 0.5 | 2.0 | 9.1 | 5.5 |
| HEP8-ESIONP | 3.9 ± 1.2 | 20.7 | −34.4 | 1.1 ± 0.2 | 9.6 ± 0.5 | 1.9 | 8.4 | 5.0 |
| HEP12-ESIONP | 3.4 ± 0.9 | 17.7 | −30.3 | 1.1 ± 0.2 | 7.2 ± 0.2 | 1.6 | 7.1 | 3.6 |
| HEP24-ESIONP | 2.2 ± 0.6 | 16.1 | −32.1 | 1.1 ± 0.2 | 6.2 ± 0.4 | 1.0 | 7.0 | 1.7 |
Usual large band in the 3200–3600 cm−1 region assigned to the deformation of the –OH hydroxyls were retrieved. Carbonyl symmetric stretch and adsorbed water's asymmetric stretch are typical of carboxyl groups and were found at 1400 cm−1 and 1600 cm−1, respectively. The bands that appeared at 1235 cm−1 and 1060 cm−1 are due to the S–O bonds, while the ones at 980 cm−1 and 800 cm−1 are representative of the C–O–S strechts. Shoulder at 980 cm−1 and small bands at 890 cm−1 may be attributed to coupling components of the C–O–C ring and glycosidic bonds. Relative signal intensity of the carboxyl bands paralleled with sulfate group bands decreased for the HEP-ESIONP in comparison with the free HO. This may be indicative of the complexation of the heparin coating to iron oxide core through –COOH groups rather than –SO3– groups. Interestingly, a spliting of the peak at 2900 cm−1 found in the spectra of the free HO was observed in case of HEP-ESIONP analysis. This band is usually assigned to C–H vibration. However, although it is very difficult to detect, we believe that this splitting may be illustrative of the presence of aldehyde groups of oxidised polysaccharides. This assumption is supported by the appearance of a peak at 1740 cm−1 in the FTIR spectra (ν C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and support a conjugation to the iron oxide core at the terminal hemi-acetal sugar, as discussed later.58,61
O) and support a conjugation to the iron oxide core at the terminal hemi-acetal sugar, as discussed later.58,61
We then investigated the configuration of the oligosaccharides present on the surface of ESIONP. The length, together with the adsorbed density of material, configures the polymeric structures and dynamics on the surface of the metallic core, which directly determines the coating height of the NP (h) (Fig. S6†). It may vary from a mushroom regime, where the polymeric chains are not affected among themselves and adopt a conformation similar to the free structure in solution, up to a semi-diluted polymer brush (SDPB) regime where h begins to be sensitive to σ.62 In the SDPB regime, one of the most popular models used for predicting polymer height, that has been supported experimentally, is defined by the MWC-WZ equation where h ∼ (DP3rNP2/5σ)1/5.63 Under high grafting conditions, the structure becomes more complex and consists of an inner region of highly stretched chains (CPB) near the nanoparticle surface, followed by a SDPB region in the outer layer. In this case, the Daoud–Cotton model has been shown to correctly provide the theoretical transition distance hc between the CPB and SDPB regions.64 Thus, we first estimated a theoretical h of the different HO attached on the ESIONP in the absence of steric constraints, derived from the sizes of several HO free in solution obtained from a study by S. Khan and coworkers (Fig. S7† and Appendix A†).65 Due to their relatively small sizes, in high solubility condition the free HO already adopt a linear extended semi-rigid conformation with n = 0.65 in a DPn-dependent relationship, typical of the SDPB regime. In parallel, we also estimated the theoretical maximum height if the HO was fully stretched, resting on a 0.5 nm heparin monomeric unit according to previous works (Fig. S7†).65,66 The HS of HEP-ESIONP were compared according to these two limits, and it appeared that, except for HEP0-ESIONP, HO followed an apparent SDPB regime in relation to the free solution sizes of heparin segments (Fig. 1C). For HEP0-ESIONP, their greatest chain length and σ lead to a transition to the presence of a CPB region and the stretching of the polymer under steric constraints. Since σ and the HO size varied simultaneously, it was difficult to further characterise and build a complete picture of the HO configuration adopted on ESIONP surface. In addition, in our study, the HO Mw and NP radii were relatively smaller than those traditionally used in works assessing the polymeric structure on NP surface, so that our values were at the limits of the models proposed in the literature.67 On the one hand, the measured coating sizes followed fairly well the MWC-WZ equation for polymers grafted onto a surface in a SDPB regime (Appendix A†), which was here related to the relaxing configuration of the HO because they were in a small-size range. However, we assumed that, in fact, all the HEP-ESIONP were under high-grafted conditions but with a limited CPB region, except for HEP0-ESIONP. In our case, due to the sharp surface curvature of very small ESIONP cores, the steric freedom of HO could be rapidly restored in the outer sphere and return to its relaxing state configuration typical of a SDPB region. To support this assumption, we calculated and found coherent small theoretical transition distance hc between a CPB and SDPB regions of the Daoud–Cotton model (listed in Table 2).
Finally, zeta potential measurements showed for all HEP-ESIONP a similar high negative surface potential of about −35 mV, due to the multiple negative charges carried by the HO sulfate and carboxylate groups. The slightly higher values measured for HEP12- and HEP24-ESIONP could therefore be due to the small desulfatation observed during the preparation of these two HO (Table 2).
3.3 Magnetic characterisation and determination of HEP-ESIONP optimal for positive MRI contrast
Direct current magnetometry studies showed that magnetite cores were superparamagnetic at room temperature. For example, magnetization versus temperature curves obtained by the well-known zero-field-cooling–field-cooling protocol exhibited a characteristic pattern undoubtedly related to the thermal relaxation of the particle magnetic moment (Fig. 2A). The thermal energy becomes higher than the anisotropy energy above certain temperature threshold, usually referred as the blocking temperature. The so-called blocking temperature, which is located at the maximum of the zero-field-cooling curve, was clearly correlated with the magnetite core size of the different HEP-ESIONP, falling from 60 K for HEP0-ESIONP to 6 K for HEP24-ESIONP. | ||
| Fig. 2 Magnetic behaviour and relaxometric parameters. (A) Direct current magnetometry study, zero-field-cooling-field-cooling protocol of the HEP-ESIONP. (B) Longitudinal relaxivity r1 (black dot) and relaxivity ratio (r2/r1) of the HEP-ESIONP according to their core size. | ||
In addition, the superparamagnetic behavior of the NP was also very visible in the magnetization versus field measurements (Fig. S8A†), which were totally un-hysteretic. These M(H) curves fall into the characteristic Langevin-type behavior, when the approach to saturation is strongly size-dependent. It is known that MRI contrast agents based on iron oxide NP reduce the longitudinal relaxation processes T1 (spin–lattice) and transverse T2 (spin–spin) responsible, respectively, for the brightness and darkening of surrounding tissues.1 To identify which of the two relaxation times had the prevailing effect, the following parameters were investigated: longitudinal relaxation r1, transverse relaxation r2 and r2/r1 relaxation ratio. For a good positive contrast behaviour based on T1, a contrast agent shall have the highest possible r1, with a low r2/r1 ratio, generally less than 5.68 To find the most suitable HEP-ESIONP for T1 positive MRI contrast, these three parameters were therefore calculated. The relaxation rates 1/T1 and 1/T2 (s−1) according to the iron concentrations (mM) were measured at 37 °C in a 1.5 T field and the slopes of the linear fittings provided the r1 and r2 relaxations (Fig. S8B and S8C†). As shown in Fig. 2B, r1 decreased progressively along with the core diameter of HEP-ESIONP, while the r2/r1 ratio dropped between HEP0-ESIONP (Dcore = 6.6 nm) and HEP4-ESIONP (Dcore = 4.3 nm) before stabilizing at about 3–3.4. HEP12-ESIONP and HEP24-ESIONP showed a low T1 and T2 contrast capability, while HEP0-ESIONP showed a slightly too high r2/r1 ratio for a positive contrast. In accordance with previous works, HEP4-ESIONP and HEP8-ESIONP appeared to have the most suitable adjusted balance between r1 and the r2/r1 ratio to be the most optimal T1 positive contrast agents. Their r1 is twofold higher than that of the standard gadolinium-based positive MRI contrast agents such as Magnevist® or Multihance®, but their r2/r1 ratio is slightly higher (∼3.3 versus ∼1.2 for commercial products).69 Their core sizes were closed to the 3.6 nm optimal core size proposed for T1-weighted imaging by Shen et al. after evaluation of seven poly(acrylic acid)-coated ESIONP with cores of less than 5 nm.11 For the iron oxide NP, the ordered spins of the magnetic core create a strong magnetization closely related to the T2 performance, with the induced dipole moment shortening the transverse spin–spin relaxation time of the neighbouring water protons (outer-sphere model). The main feature that provides ESIONP with the T1 contrast capacity is derived from a layer of disordered/canted spins at the NP surface due to incomplete coordination and altered symmetry.70 When the core size decreases, the magnetization is lower because the volume occupied by well-organized spins is reduced and the proportion of the magnetically-dead layer is increased.12 This leads to a decrease in transverse relaxivity r2 as illustrated in Fig. S8D,† and therefore in the r2/r1 ratio, so that ESIONP are converted into more suitable T1 contrast agents. The longitudinal relaxation r1 is also affected by the effect related to the dipole interaction with water protons of the outer-sphere model, but to a lesser extent. In addition, r1 is influenced at the same time by direct interactions between free paramagnetic iron ions present on the surface and the nearest water molecules (inner-sphere model). Therefore, a decrease in core size can also improve T1 performance due to a greater surface-to-volume ratio for small NP, increasing the possible links with water molecules compared to large NP.68,70 However, in our case, the outer-sphere model was predominant as shown in Fig. 2B by the r1 decrease with the reduction of the HEP-ESIONP core size. Probably, this is due to the high heparin σ that limits the accessibility of water molecules very close to the ESIONP surface for direct interactions (inner-sphere model). The organic stabilizing coating is crucial to provide colloidal stability and also to modulate the exposure and mobility of water molecules according to its physicochemical features (binding affinity, thickness, hydrophobicity, configuration, etc.); and therefore directly affects both r1 and r2/r1.68,71,72 For instance, in the outer sphere model high-Mw HO could promote T1 and T2 performance compared to the smallest HO (HEP12 and HEP24) because it has been shown that long polymers slow down the diffusion/mobility of water molecules inside the coating, so that they undergo the effect related to the dipole interaction for longer.73 Interestingly, in accordance with this, when plotting r2versus DP, a straight linear slope was observed, whereas this was not the case when plotting r2versus the core size (Fig. S8D†).
To validate the results obtained in the relaxometry study and deepen the identification of the best HEP-ESIONP to be used as a positive contrast agent, we conducted in vivo MRI experiments. The different capacities of HEP0, HEP8 and HEP24-ESIONP to produce T1-weighted imaging were assessed after intravenous injection in 6-week old female Balb/c mice and compared to a commercial gadolinium-based product (MultiHance®). The experiments were performed using a low-field 1 T device, that is closer to human clinical practice and at which the relaxometric properties were obtained to facilitate the comparison. As shown in Fig. 3, the main vascular architecture (in particular the abdominal aorta and hepatic vessels) was revealed immediately after injection in all cases, confirming the T1-weighted imaging capability of the three HEP-ESIONP. The contrast enhancements were consistent with their different r1 values, the brightest signal being obtained with HEP0-ESIONP (r1 = 12.8 mM−1 s−1) and then, the intensity slightly decreased with HEP8-ESIONP (r1 = 4.3 mM−1 s−1) and almost disappeared with HEP24-ESIONP (r1 = 0.6 mM−1 s−1). The good result of HEP0-ESIONP as a positive contrast agent could appear surprising with respect to its high r2 value and apparently unfavourable r2/r1 ratio. However, excellent results have been obtained in T1-weighted MRI imaging with other iron-based agents with similar relaxometric parameters such as Ferumoxytol® (r1 = 15 mM−1 s−1, r2 = 89 mM−1 s−1, r2/r1 = 5.9 at 1.5 T).74 Nevertheless, when examining the images 15 min after injection, the signal rapidly disappeared with HEP0-ESIONP compared to the other two HEP-ESIONP for which it persisted in the vascular system for up to 60 min. This result suggests a rapid elimination by the mononuclear phagocyte system, which was confirmed by a strong negative signal observed in the liver compared to the basal image (Fig. S9†), showing HEP0-ESIONP accumulation and aggregation in hepatic macrophages. In this case, the signal was negative due to the aggregation of the ESIONP core that will largely increase the T2* effect. On the contrary, for HEP8-ESIONP and HEP24-ESIONP a slight increase of signal intensity in bladder was noticed for the longest acquisitions, suggesting renal clearance as one of the elimination routes (data not shown). The commercial gadolinium-chelate MultiHance® displayed a contrast enhancement similar to that of HEP0-ESIONP. Noteworthy, it was so rapidly eliminated that after 15 min most of the signal was found in the bladder and a much lesser proportion in the liver. The potential long vascular lifetime of ESIONP-based positive contrast agents is an advantage over the gadolinium complexes that may have a greater contrast performance but are rapidly eliminated by the kidneys. Specifically, it could be used in T1 blood pool MRI, using long acquisition times (steady-state imaging) to obtain informative high-resolution images useful in various clinical applications, including the diagnosis of cardiovascular diseases, the characterisation of tumour angiogenesis or the detection of renal failure.2,68 Therefore, despite its slightly lower T1 contrast capability, HEP8-ESIONP appeared to be the best agent rather than HEP0-ESIONP, due to its prolonged vascular lifetime (HEP24-ESIONP being set aside because of a too low signal enhancement).
 | ||
| Fig. 3 In vivo positive contrast MRI of the HEP0, HEP8 and HEP24-ESIONP. Coronal and sagittal images of mice vascular system performed on a 1 T MRI (ICON 1 T-MRI; Bruker BioSpin GmbH) with a T1-weighted gradient echo sequence after i.v.a of HEP-ESIONP (50 μl, [Fe] = 1 mg ml−1) or Gadobenate dimeglumine (Multihance®; 529 mg mL−1). | ||
3.4 In vivo biodistribution study
We investigated the influence of the HO length on the in vivo behaviour of HEP-ESIONP in mice bearing BT-474 breast cancer xenograft in order to explore their pharmacokinetics and possible accumulation in tumours. Nuclear imaging is by far the most sensitive technique for studying the qualitative and quantitative biodistribution. We therefore took advantage of the easy-tailoring ESIONP platform, one of their advantage over molecular gadolinium-contrast agents, to develop 68Ga core-doped HEP-ESIONP, using a method developed by Bhavesh et al.75 The radioisotope 68Ga is advantageous because it can be produced using a home generator and has a short radioactive half-life, which limits radiation exposure.76 However, this short half-life can be a double-edged sword, requiring the rapid preparation of the final probe, and we benefited here from our approach based on a rapid and simple one-step microwave synthesis of HEP-ESIONP.77 The reaction was similar to those used for HEP-ESIONP except that a fraction of 68GaCl3 (in 0.05 M HCl) eluted from the 68Ge/68Ga generator was added to the iron precursor solution. After purification, the probes were immediately administered intravenously to the mice before performing a PET-scan for a 3 h monitoring. The same three HO (HEP0, HEP8 and HEP24) used in the in vivo MRI experiments were assessed. We observed significant differences between the different 68Ga-HEP-ESIONP biodistributions, as it could be suspected from MRI results (Fig. 4A). After injection, 68Ga-HEP0-ESIONP was almost immediately eliminated by the liver and more than half of the injected dose (ID) was found in this organ after 60 min, while less than 5% of the ID per cm3 was found in the heart, indicating that only low amounts of these NP were present in the bloodstream. Conversely, a fast excretion through urine occurred for 68Ga-HEP24-ESIONP and more than 60% of the ID per cm3 was found in the bladder after 60 min, while less than 8% was found in the heart (Fig. S10†). Yet, the most promising results were found for 68Ga-HEP8-ESIONP, functionalized with the intermediate coating length. The hepatic elimination was limited to ∼15% of the ID per cm3 and more signal was found in the kidneys that translocated quickly into the bladder (Fig. 4B). This fast transfer into the urine may ensure a low nephrotoxicity. Even more interesting, around 12% of the ID per cm3 was still present in the bloodstream after 60 minutes and more than 5% persisted even after 3 hours (estimated from the signal in the heart, Fig. 4B and Fig. S10†). We therefore had a closer look to the tumoral accumulation for this probe and found a small retention around 3–4% that is maintained overtime (Fig. 4B and C). Although limited, this accumulation was in the range of the renal clearable inorganic NP44 and unexpected towards the low vascularized BT-474 xenograft model used.47 Further studies will be required to know whether the accumulation observed for 68Ga-HEP8-ESIONP was due to the EPR effect or also in part to an “active inherent targeting” of the heparin coating, which is known to be able to interact with various components of the tumour microenvironment (VEGFs, HPSE, etc.). The observed clearance can be first interpreted according to the hydrodynamic sizes of the nanoparticles. The full HS distribution of HEP0-ESIONP was higher than the 8–10 nm limit for kidney excretion (Table 2 and Fig. S4B†), explaining the final and rapid predominant biodistribution in the liver in the PET study. Regarding HEP8-ESIONP and HEP24-ESIONP biodistribution, since part of these NP HS distributions have a size below the renal clearance threshold (Fig. S4B†), it is expected to observe excretion by the kidneys, as confirmed by PET imaging. Nevertheless, the differences observed in pharmacokinetics were more inquiring. For 68Ga-HEP8-ESIONP, the part of HS distribution of which was higher than the kidney threshold, a circulating fraction persisted in the bloodstream with a delayed clearance and was not immediately eliminated in a few minutes as for 68Ga-HEP0-ESIONP. A few studies have described the in vivo biodistribution of NP coated with only native heparin and they all observed a rapid uptake by the liver.78,79 This could explain why in most of the research works heparin is usually used in NP formulation with other polymers or conjugated with biomolecules. For instance, several studies based on imaging have reported an increased vascular lifetime and tumour accumulation using native heparin in combination with polyethylene glycol, poly(lactic-co-glycolic acid), chitosan, deoxycholate, retinoid acid or doxorubicin.18,20,21 Others have reported heparin-coated NP with stealth properties and delayed uptake by liver, preparing a dense brush coating conformation that prevent blood opsonin adsorption.80–82 So far, selecting the in vivo behaviour of NP coated with bare heparin by only modification of the polysaccharide length have not been reported yet. Our findings are very promising in the application of HO derivatives in nanomedicine, since in the case of HEP8-ESIONP the rapid kidney clearance of excess probes could help to prevent toxicity while the fraction maintained in the bloodstream for longer could promote the EPR effect. In order to get more insights into the effect of the HO coating size and conformations on the pharmacokinetics of the probes, we next characterised the protein coronas. | ||
| Fig. 4 In vivo biodistribution study. (A) PET images of the 68Ga-HEP-ESIONP. (B) In vivo quantification of the biodistribution of the 68Ga-HEP8-ESIONP in the major organs. (C) Image of the moderate tumour accumulation of 68Ga-HEP8-ESIONP at 3 hours (L: liver). | ||
3.5 Influence of the coating length on HEP-ESIONP protein coronas
Blood protein corona refers to the dynamic set of plasma proteins that is adsorbed at a NP surface during its vascular life. When NP enter the bloodstream, they are considered external agents, and the protein corona has been shown to strongly contribute to the immune response, by mediating opsonisation, and controlling the duration and type of NP elimination by the mononuclear phagocyte system.38,40 It is also involved in the passage of NP across several biological barriers under physiological or pathological conditions, including the blood brain barrier, atheroma plaques or tumour stroma. The protein corona composition is a competitive balance over time between the almost 3000 blood proteins, governed by strong NP-protein and weak protein–protein binding affinities. It therefore represents a real fingerprint that is unique to each NP, and depends on multiple parameters such as the NP composition, morphological properties, surface charge and coating properties (e.g. size, density, etc.).83,84To better understand the difference in HEP-ESIONP biodistributions and half-life in the bloodstream obtained in the nuclear imaging study, we studied in vitro the protein corona composition after a 60 min incubation in mouse serum. A typical citrate-coated ESIONP (Cit-ESIONP) was used for comparison because its core size, HS and surface charge are within the ranges of those of HEP-ESIONP (the physicochemical properties of Cit-ESIONP are summarized in Fig. S11†). Cit-ESIONP are also known for its anticoagulant properties as a calcium-chelating agent, but it lacks of antithrombotic activity like heparin. Proteomics experiments were performed using a last generation TIMS-powered quadrupole time-of-flight mass spectrometer having the feature for parallel accumulation/serial fragmentation (PASEF) scan modes, and identification was performed using Mascot (Matrixscience) search engine against the Uniprot/Swissprot database. The analysis identified more than 150 proteins for each ESIONP, expressed as a percentage of the total protein corona using the Normalized Spectral Abundance Factor relative quantification method. At first glance, the results for Cit-ESIONP and HEP0-ESIONP, HEP8-ESIONP and HEP24-ESIONP showed many similarities as illustrated in Fig. 5A showing the protein classification according to their biological activity in the bloodstream. This was somewhat expected given the many close features shared by these NP. In all cases, the immunoglobulins (Ig) used by the adaptive immune system, mainly the IgM and IgG, accounted for more than half of the protein corona composition. We also observed that almost only kappa light chains composed the fab region of these Ig. IgM is the first antibody present at the initiation of the immune response and, together with IgG, is very effective for binding the C1-complex in order to activate the complement system. This part of the innate immune system involves opsonins that enhance in particular phagocytosis by liver's Kupffer cells or other macrophage types, and it is closely related to the hepatic elimination that we have observed in some cases in the biodistribution study.85 Accordingly, opsonins of the complement system represented the second major family identified in all ESIONP coronas. Compared to Cit-ESIONP protein corona, protein coronas for HEP-ESIONP contained a higher proportion of lectins and a lower proportion of C1q subunit and IgM/IgG constant domains, suggesting the activation of the alternative lectin pathway in addition to the classical complement activation pathway. These findings are in accordance with previous works that have reported interactions between heparin and mannose-binding proteins triggering the lectin pathway.86,87
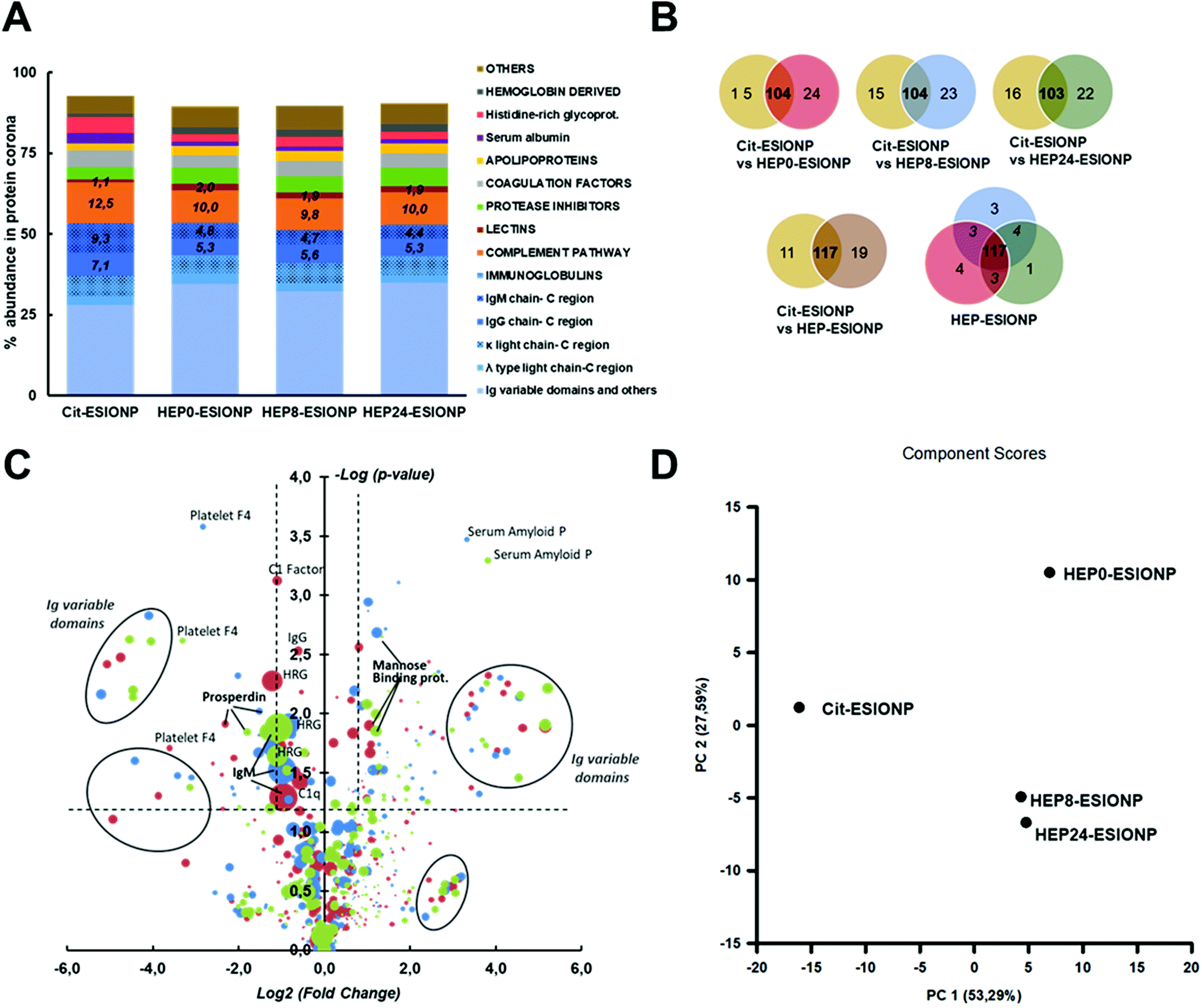 | ||
Fig. 5 Protein corona characterization of Cit-ESIONP and Hep-ESIONP. (A) Relative abundance of proteins in the coronas classified in family according to their biological activity in blood (proteins with % abundance below 0.15% in all the four ESIONP coronas were excluded). (B) Venn diagrams of the unique proteins (proteins with % abundance below 0.15% in all the four ESIONP coronas were excluded). (C) Plot of the fold-changes in abundance of each protein alongside their p-value significance, assessing the coronas of the Cit-ESIONP vs. the three different HEP-ESIONP. Positive changes means a higher abundance of the protein in HEP-ESIONP coronas than in the Cit-ESIONP corona, while negative changes means a higher abundance of the protein in the Cit-ESIONP corona than in the HEP-ESIONP. Y-Axis indicates significance of the change according to the p-value obtained with a Student's t-test statistic. Surface of the dot represented are proportional to the abundance of the protein in the HEP-ESIONP or Cit-ESIONP coronas depending whether the fold-change is, respectively, positive or negative.  Cit-ESIONP vs. HEP0-ESIONP, Cit-ESIONP vs. HEP0-ESIONP,  Cit-ESIONP vs. HEP8-ESIONP and Cit-ESIONP vs. HEP8-ESIONP and  Cit-ESIONP vs. HEP24-ESIONP. (D) Principal component analysis of the multivariate protein systems of the HEP-ESIONP and Cit-ESIONP. Cit-ESIONP vs. HEP24-ESIONP. (D) Principal component analysis of the multivariate protein systems of the HEP-ESIONP and Cit-ESIONP. | ||
More differences were found between Cit-ESIONP and HEP-ESIONP considering the Venn diagram (Fig. 5B and Table S2†). Most of the unique proteins were variable domains of kappa light chains and, to a lesser extent, of mu heavy chains. The heat map also showed qualitative and quantitative changes in several others Ig subpopulations (full protein classification in Appendix B†). To go further in the analysis, the fold-change in each protein content according to their p-value was plotted (Fig. 5C), to compare the coronas of Cit-ESIONP versus the three different HEP-ESIONP. The dot surface was proportional to the protein content in HEP-ESIONP and Cit-ESIONP coronas depending on whether the fold change was positive or negative, respectively. Above the confidence threshold (−logp-value > 1.3, corresponding to p < 0.05), as expected various Ig subunit populations located in the upper fold changes appeared (log2 fold change >2.3, corresponding to a fold change >5). They corresponded to variable domains of heavy and light Ig chains. Also, a significant decrease in IgM constant domains was found in HEP-ESIONP coronas compared to Cit-ESIONP corona with a fold change within the 0.5 range. This finding was associated for all HEP-ESIONP with a decrease in histidine rich-glycoprotein, that has been described to be associated with IgM and IgG.88,89 This could suggest a lesser initiation of the immune response or the activation of other pathways. Among the proteins with significant positive fold changes that were common to the three HEP-ESIONP coronas (Table S3†), an enrichment of mannose-binding protein A was observed. This result could support the involvement of the lectin pathway for which no preliminary recognition by Ig is needed to activate the complement system. On the other hand, higher proportions of C1 subcomponents and properdin (a stabilizing protein of the C3bBb convertase obtained from the hydrolysis of C3) were observed in Cit-ESIONP corona, suggesting respectively an involvement of the alternative and classical pathways. To summarize, we concluded that the immune response was initiated by IgM/IgG mobilisation with involvement of the kappa light chain that triggered the complement response. Variable domains were adapted according to the nature of the coating to recognise either citrate or heparin oligosaccharides. Regarding HEP-ESIONP, the complement response involved a part of the lectin pathway at the detriment of the alternative pathway, which was more activated with Cit-ESIONP, due to the direct interaction between mannose-binding proteins and heparin. A recent study by V. P. Vu et al. has emphasised this relationship between the Ig deposition in the protein corona and the efficiency of complement opsonisation.90
Regarding the comparison between the three HEP-ESIONP protein coronas Venn diagrams showed 87% of homology (Fig. 5B and Table S4†). After statistical analysis, only a few proteins showed significant changes (p < 0.05) (Table S5†). Because the shift between hepatic to renal clearance was observed for NP obtained with heparin lengths below 7.5 kDa, we compared HEP0-ESIONP to HEP8 and HEP24-ESIONP. The results shown that only two Ig subunits and two complement factors decrease with a low variation in HEP8-ESIONP or HEP24-ESIONP corona as compared to HEP0-ESIONP corona. We therefore considered the proteins with a strong positive or negative change in their mean abundance (fold change >2 or <0.5, respectively), without taking into account the p-value confidence index. A set of proteins that were strongly decreased both in HEP8-ESIONP and HEP24-ESIONP coronas compared to HEP0-ESIONP corona was identified, including 5 variable domains of Ig, the C 1s subunit and ferritin (Fig. S12† and Appendix B†). The slightly higher content of these proteins in HEP0-ESIONP corona could provide a small specific biological identity to HEP0-ESIONP compared to the other two HEP-ESIONP. It could be assumed, with caution due to the low statistical significance, that the presence of some Ig domains could be related to the size of HO coating and that the activation of the complement pathway could be facilitated when the HO coating used had a high Mw.
A principal component analysis of the multivariate protein system could be used to provide another general view of these results. The first two principal components captured more than 80% of the variance, represented on the two-dimension graph in Fig. 5D. It revealed that Cit-ESIONP corona was different from HEP-ESIONP coronas according to the first principal component (PC1) that gathered a specific population of proteins that accounted for 53.3% of the variance in the dataset. On the other hand, HEP0-ESIONP corona was slightly different from HEP8-ESIONP and HEP24-ESIONP coronas based on the second principal component (PC2) that gathered a smaller cluster of proteins, accounting for 27.6% of the variance in the dataset. Overall, the hepatic accumulation of Cit-ESIONP and HEP0-ESIONP could be explained by their protein coronas, densely charged with potent opsonins. However, a high degree of similarity was found between the three different HEP-ESIONP protein coronas with almost no qualitative change observed according to the length of their HO coating. Thus, our proteomic characterization was useful to conclude that the longer vascular life of HEP8-ESIONP and HEP24-ESIONP as compared to HEP0-ESIONP was not due to the protein corona composition. It has been shown that HS of the NP, or even just its polymer height and σ, can modulate the total amount of proteins adsorbed without changing the qualitative composition of the corona, resulting in opsonized NP more furtive against macrophages lasting longer in the bloodstream.91,92 Several studies have reported that dense concentrated brush conformations of polysaccharide coatings at the NP surface can prevent the adsorption of blood proteins93 (in particular those belonging to the complement system) whereas SDPB conformation can favour it.94,95 For HEP-ESIONP, based on the proposed CPB-SDPB conformation hypothesis, the SDPB region was much larger in HEP0-ESIONP than in HEP8-ESIONP and HEP24-ESIONP (respectively 9.1 nm versus 3.4 and 5.3 nm) and could explain the differences observed. Two studies using native heparin have reported the stealthiness of heparin-coated NP on the account of a dense brush coating conformation rather than the heparin nature itself.80,81 Also, for renal excretion that relies on glomerular filtration with an estimated filter size threshold of about 8 nm, the final size to calculate should include the additional thickness provided by the blood proteins adsorbed in vivo at the NP surface. To explore these aspects, we measured the total amount of protein found in the HEP-ESIONP coronas after a 60 min in vitro incubation in mouse serum. Results indeed showed a decrease of the amount of protein adsorbed per nanoparticle surface area for the HEP-ESIONP with coatings of shorter length (Fig. S13†). These data could be additional information to understand the longer circulation time and renal clearance observed for HEP8-ESIONP and HEP24-ESIONP.
3.6 Cell toxicity and uptake of HEP-ESIONP
Compared to gadolinium-chelates, the use of ESIONP-based contrast agents could prevent several toxicity issues. It has been also reported that low molecular weight heparins bind with less affinity to the macrophages and the endothelial cells than the native heparin, explaining difference in the clearance routes.96 So we performed a preliminary cellular study of the different HEP-ESIONP to explore this aspects. First, an MTT cell viability assay was performed to assess the proliferation of human embryonic kidney cells HEK293 (a standard cell line to evaluate cytotoxicity) in the presence of HEP-ESIONP. The growth of these cells incubated for 24 h with HO at a concentration of 100 μg ml−1 was similar to that of control cells (treated with a H2O vehicle), as shown in Fig. 6A. A slight induction of proliferation was observed after 72 hours. The HO concentration of 100 μg ml−1 was chosen because it is often used in the literature as the upper limit to assess in vitro polysaccharide effects on cellular models.97 As we fixed the HO concentration, the iron doses ranged between 5 to 20 μg ml−1, according to the HEP-ESIONP. These doses were representative to the ones that have been administrated in vivo. The preliminary data obtained showed that HEP-ESIONP had negligible cytotoxicity effect on HEK293 non-tumour cell line. We next looked at the uptake and interactions of HEP-ESIONP by two different cell lines: the MDA-MB-231 breast cancer cells and the HSkMEC micro-endothelial cells. These cell lines were selected because they could also provide information on HEP-ESIONP behaviour in the surroundings of a tumour stroma, where neoangiogenesis takes place. After 6 h incubation of subconfluent cells with HEP-ESIONP, the iron concentration in the cell lysates (directly indicative of NP uptake) was measured using both a colorimetric method and inductively coupled plasma-mass spectrometry. The amounts of internalized HEP-ESIONP were different for the different cell lines (Fig. 6B). MDA-MB-231 cells, internalized similar amounts of iron regardless their HO coating length. On the contrary, HSkMEC cells internalized higher amounts of HEP-ESIONP compared to MDA-MB-231 cells. Moreover, the uptake decreased along with the length of the HO coating. This experiment is in accordance with the two different pharmacokinetic mechanisms described for free native heparin and low molecular weight heparins, respectively the saturable and the non-saturable mechanism. The saturable mechanism represents a clearance by the mononuclear phagocyte system and endothelial cells, to which heparin binds with a high affinity. On the contrary, low molecular weight heparins are known to undergo a non-saturable mechanism represented by renal excretion.96 Gathered with the HO coating conformation considerations, this represents a second important explanation about the variation observed in the biodistribution study. | ||
| Fig. 6 Cell viability and cell uptake of the HEP-ESIONP. (A) Viability of HEK 293 measured by MTT assay after 24 h and 72 h incubation with the HEP-ESIONP (HO = 100 μg ml−1). (B) % of iron internalized by MDA-MB-231 and HSkMEC cells after 6 h incubation with the HEP-ESIONP ([Fe] = 10 μg ml−1). | ||
3.7 Modulation of HEP-ESIONP biological activities
Native polysaccharides are widely used as coating agents for metallic NP because they provide a colloidal stability and an easy further functionalisation. They can also confer pharmacological properties to the NP although it is hard-to-control because their high Mw, complex and heterogeneous structures lead to divergent range of bioactivities.19 During the last years, novel low molecular weight polysaccharide derivatives with better-defined structures and bioactivities have been proposed.98 In this context, one of the crucial aspects of our approach was to demonstrate that the HO could be used as ESIONP stabilizers with an efficacy similar or even greater to that of the widely used native heparin, and, at the same time, to learn how to take advantage of its best-defined pharmacological activities.Among the various bioactivities of heparin, we selected two representative ones: its anticoagulant property used in clinical practice for years and its more recently identified anti-tumour activity which is based in part on its inhibitory effect on heparanase (HPSE), an enzyme involved in tumour progression.16 Interestingly for this study, these two activities may be contradictory, especially in the treatment of cancer where the anticoagulant property may be risky for patients with fragile vascular homeostasis. The anticoagulant activity is mainly due to a specific pentasaccharide sequence present in the heparin chain that binds and activates the antithrombin protein AT-III by a conformational change. AT-III is an endogenous coagulation cascade controller that inhibits factor Xa and thrombin IIa, two key serine proteases involved in the coagulation cascade, that intervene a step before the conversion of fibrinogen into fibrin thrombus. The allosteric activation of AT-III by the pentasaccharide sequence of heparin is sufficient for an effective inhibition of factor Xa, while for thrombin IIa, a supplementary bridging assembly is needed, consisting of a steric interaction between the protease and another part of the long enough heparin chain (minimum of 17 saccharides). Therefore, Mw reduction/depolymerisation techniques are commonly applied to modulate these anticoagulant activities.17
As shown in Fig. 7A, the radical-mediated hydrolytic depolymerisation technique used progressively decreased the anti-Xa activities of the HO produced, using a kinetic enzymatic assay with a chromogenic substrate. The same was observed for anti-IIa activities but with a much more pronounced effect (Fig. 7B). This loss of anti-factor Xa and IIa activities was anticipated since depolymerisation randomly cuts the polymer chain and can thus cleave the pentasaccharide sequence, limiting the likelihood of AT-III activation by low molecular weight derivatives. Desulfation, which occurred for the two smallest HO, also induced structural changes in the sequence that inactivates the binding to AT-III. In the case of thrombin IIa, depolymerisation had a stronger influence on the loss of the HO activity because, in order to achieve the additional bridging assembly of heparin on IIa, a long polymer chain is needed for the inhibition. Incidentally, the smallest HEP 24 derivative had a mean DP of about 13, below the minimum required length established at 17 saccharides and, accordingly, did not have any inhibitory effect on factor IIa. When the HO played the role as ESIONP stabilizers, two distinct consequences were observed on these inhibitory activities. On the one hand, the anti-Xa activity remained unchanged, reflecting the accessibility of the AT-III binding sequence to the pentasaccharide motif present on HEP-ESIONP surface and the consecutive inhibitory interaction with Xa protein (Fig. 7A). This conservation could be the result of different effects, as pointed out in the literature.99 The NP core and σ may produce a strong steric hindrance and prevent accessibility to molecular interaction sites, even as found here, the very small size of ESIONP cores limit this effect.24 Also, the surface effect and packaging of the polymer in a restricted fixed conformation can enhance the adsorption and affinity of the protein of interest.100 In that regard, Fig. 7B indicates that the anti-IIa activity of HEP-ESIONP was drastically reduced compared to that of the free HO. This could be due precisely to the loss of HO steric freedom, arranged in a restricted conformation in the coating, that impede a bridged disposition where the HO chains have to bend to interact at the same time with AT-III and IIa.
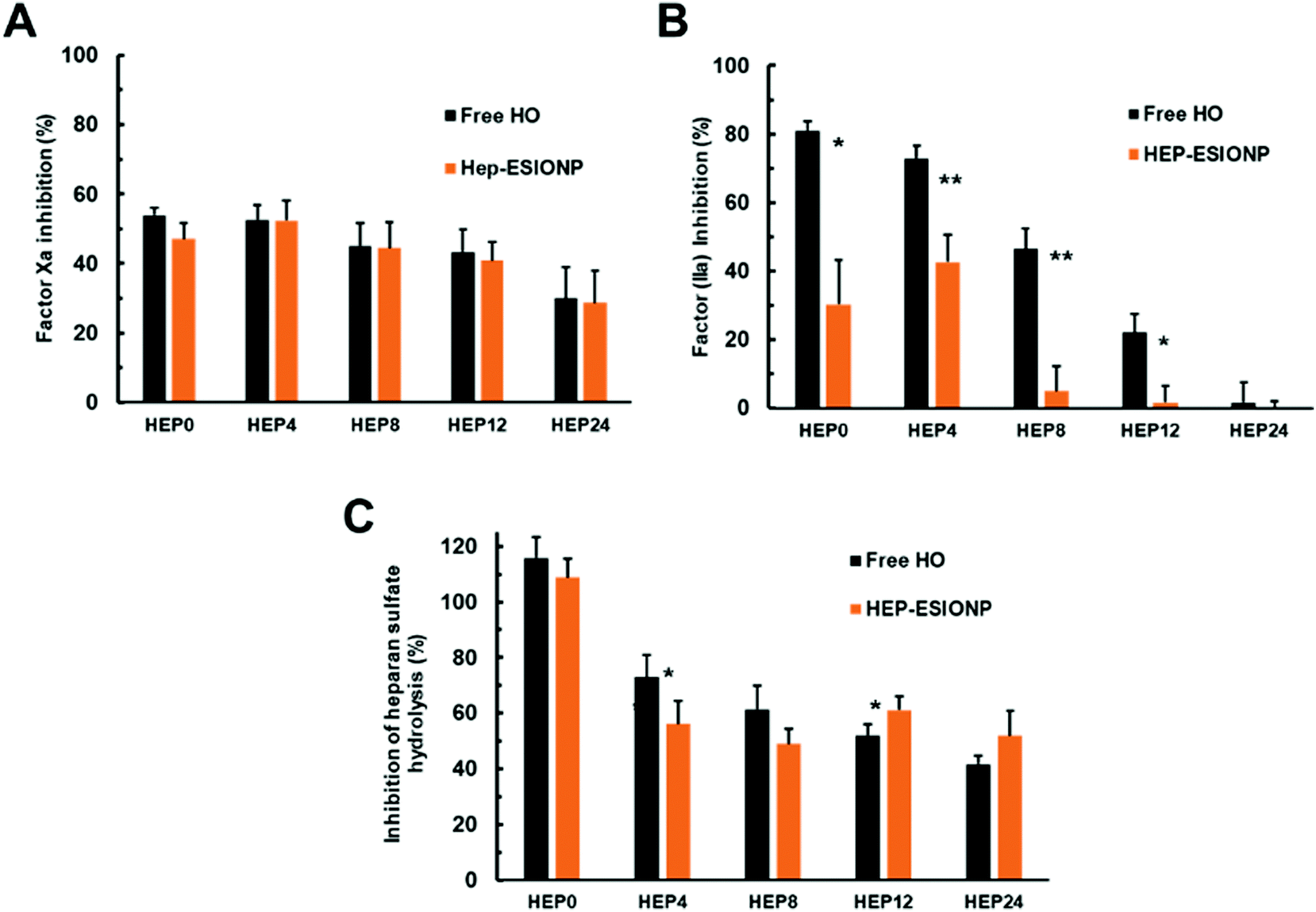 | ||
| Fig. 7 In vitro biological properties of the HEP-ESIONP. (A) Anti-factor Xa activity of the free HO and corresponding HEP-ESIONP measured at 0.125 mg L−1. (B) Anti-factor IIa activity of the free HO and corresponding HEP-ESIONP measured at 0.25 mg L−1.(±SEM). (C) Anti-HPSE activity of the free HO and corresponding HEP-ESIONP measured at 0.625 mg L−1 (±SD, n = 4). | ||
As indicated above, the second bioactivity of heparin we studied was the inhibition capability on HPSE of the different ESIONP. HPSE is a pro-tumour enzyme excreted in many tumour microenvironments. It is the only known enzyme capable to cleave the heparan sulfate chains of proteoglycans present in the extracellular matrix. It plays a central role in the biology of tumour progression, contributing especially in cancer cell migration and invasion, as well as in the release of heparan sulfate-sequestered signalling factors that promote angiogenesis. HPSE has become a promising new drug target and heparin, as a very close structural mimic of heparan sulfate, is the reference inhibitor.101 However, the anti-tumoral use of heparin is limited by its anticoagulant activity that can lead to possible adverse effect, so none-anticoagulant low molecular weight or chemically-modified HO have been proposed, and some of them are currently assessed in clinical trials.102 Herein, the anti-HPSE activities of the various HO and respective HEP-ESIONP were evaluated using a Förster Resonance Energy-based assay with a labelled-heparan sulfate (Fig. 7C). As expected, the HPSE inhibition induced by the HO decreased with its Mw because the depolymerisation method affected randomly the bonds and some of these are part of the inhibitory sequences. Also, for the smallest HEP12 and HEP24 counterparts, depolymerisation changed the sulfation pattern, which plays a key role in coordinating the interaction between heparin and HPSE. When HO was used as ESIONP stabilizer, except for HEP0-ESIONP, the anti-HPSE activity was about similar to the respective free heparin species, probably for the same reasons as the anti-factor Xa activity. Altogether, these results revealed how depolymerisation allowed modulating the balance between HO-related anticoagulant and anti-HPSE activities, but even more attractive that HO functionalisation as a coating on ESIONP could also control this balance. The rapid suppression of HEP-ESIONP anti-IIa activity as HO became smaller could be interesting for cardiovascular medical applications. In fact, in an effort to prevent the numerous side effects of the first generation of heparin-based anticoagulants, low molecular weight derivatives that only target factor Xa have been marketed such as Lovenox® or Fondaparinux®.17 Thus, HEP8-ESIONP, which was almost devoid of anti-IIa activity but maintained a satisfactory anti-Xa activity could be, for example, an interesting potential anti-thrombotic agent with limited side effects and MRI-based diagnostic capabilities.
4. Conclusions
In this work, we showed that the ESIONP core size could be controlled with HO stabilizers of various lengths obtained by a reaction of depolymerisation. Based on the relaxometric parameters, the 3.9–4.3 nm size range obtained for HO with DP between 19.2 and 44.1 respectively appeared optimal for T1-weighted MRI. In vivo MRI experiments were used to confirm which HEP-ESIONP could be used as efficient positive contrast agents. Similarly to the relaxometric measurements, the optimal T1 contrast agents where those with a degree of polymerisation above 19. However, a more thorough biodistribution study was performed and appeared as game-changer. Applying a synthesis method to get 68Ga core-doped ESIONP, PET imaging revealed a rapid internalisation of HEP0-ESIONP in the liver while a rapid excretion through urine was observed for HEP24-ESIONP, the probes coated respectively with the longest (DP 44.1, Mn ∼ 12 kDa) and the shortest (DP 13, Mn ∼ 3.8 kDa) heparin. These fast clearances prevented the probes from playing their role and accumulating in the area of interest. Total hepatic elimination is also disadvantageous in terms of pharmacokinetics because the product will undergo a slow endogenous metabolization with toxicity issues. Interestingly, in the case of HEP8-ESIONP (DP 19, Mn ∼ 5.5 kDa) we observed a balance between a limited accumulation in liver, a predominant excretion through urine, as well as a persistent fraction of circulating ESIONP without rapid uptake by the organs of the mononuclear phagocyte system or by the size-related filtration system of the kidneys. This prolonged blood half-life enabled a moderate accumulation in tumour, so that the heparin coating of intermediate length finally appeared as the good compromise between T1-MRI contrast ability and pharmacokinetics properties. To our knowledge, a such comparative study was missing about the impact of the heparin length on metallic NP biodistributions and pharmacokinetics. Deeper analysis showed that the variation observed (that should be bear in mind in the future design of heparin based NP) originated from coating density and conformation, as well as cellular interactions, rather than qualitative changes in the protein coronas.The advantage of our approach was also to produce multifunctional HEP-ESIONP in a one-step microwave synthesis. Indeed, in addition to the colloidal stability, the heparin species had specific bioactivities. We showed in vitro that for the HO below DP 19 (Mn ∼ 5 kDa), their inhibitory effect on heparanase, a pro-tumour enzyme, was preserved when they were used as ESIONP coating. At the same time, their anticoagulant properties that are not acceptable in cancer management were better regulated, in particular by limiting the anti-factor IIa activity. Therefore, from an oncological point of view, they could be good candidates for theranostic applications, by combining an antitumor activity and in vivo imaging performance. This is especially true for HEP8-ESIONP because it had optimized pharmacokinetics properties, as stressed above. Such renal clearable inorganic NP with synergetic integration of tumour accumulation capacity and bioactivity could afford unique strength like better-defined contrast enhancement imaging, low toxicity properties and simple formulation that meet the clinical translational requirements. Thus, HEP8-ESIONP could represent a promising ready-to-use theranostic platform, endowed with easy tailoring possibilities that allow the grafting of an additional targeting ligand if an increase in the accumulation in the tumor a is sought.
Overall, this study demonstrated that heparin coating should not be restrained to the native polymer limited by uncontrolled diverging bioactivities. By fine tailoring of the heparin length, a real functional coating could be obtained, fulfilling various advanced functions. This study also contributed to identify suitable coatings for the new generation of ESIONP-based positive MRI contrast agents, in order to improve their applications in guided drug delivery and/or targeted diagnostic imaging.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the Ligue contre le Cancer of Charente-Maritime CD17, Ligue contre le Cancer CSIRGO in particular CD16, CD17. Région Nouvelle Aquitaine (Projet “Nanovect”).This work was supported by grants from the Ministerio de Economía, Industria y Competitividad (MEIC) (SAF2017-84494-C2-R), and from the Gobierno Vasco, Dpto. Industria, Innovación, Comercio y Turismo under the ELKARTEK Program (Grant No. KK-2019/bmG19). JR-C received funding from the BBVA Foundation (Ayudas a Equipos de investigación científica Biomedicina 2018). JR-C received a funding from the Fundación contra la Hipertensión Pulmonar (2018). JL received funding from the Ministerio de Economía, Industria y Competitividad (MEIC) (CTQ2017-87637-R). CIC biomaGUNE is supported by the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency – Grant No. MDM-2017-0720.
We thank Xabier Ríos, Zuriñe Baz, Kepa Zamacola from CICBiomagune for their support to the in vivo experiments. We thank M. Moller from CICBiomagune for his advices and support for the HRTEM measurements. We thank C. Chot-Plassot for her support to the in vitro enzymatic assays and G. Prunier from the BCBS team (LIENSs laboratory) for their support to the in vitro cell assays. We warmly thank Iñaki Orue for its very kind availability and the useful discussion he made about the magnetic measurements.
Notes and references
- N. Lee, D. Yoo, D. Ling, M. H. Cho, T. Hyeon and J. Cheon, Chem. Rev., 2015, 115, 10637–10689 CrossRef CAS.
- Z. Shen, A. Wu and X. Chen, Mol. Pharm., 2017, 14, 1352–1364 CrossRef CAS.
- Y. Lu, Y.-J. Xu, G. Zhang, D. Ling, M. Wang, Y. Zhou, Y.-D. Wu, T. Wu, M. J. Hackett, B. Hyo Kim, H. Chang, J. Kim, X.-T. Hu, L. Dong, N. Lee, F. Li, J.-C. He, L. Zhang, H.-Q. Wen, B. Yang, S. Hong Choi, T. Hyeon and D.-H. Zou, Nat. Biomed. Eng., 2017, 1, 637–643 CrossRef CAS.
- H. Wei, O. T. Bruns, M. G. Kaul, E. C. Hansen, M. Barch, A. Wiśniowska, O. Chen, Y. Chen, N. Li, S. Okada, J. M. Cordero, M. Heine, C. T. Farrar, D. M. Montana, G. Adam, H. Ittrich, A. Jasanoff, P. Nielsen and M. G. Bawendi, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2325–2330 CrossRef CAS.
- E.-A. Park, W. Lee, Y. H. So, Y.-S. Lee, B. Jeon, K. S. Choi, E. Kim and W.-J. Myeong, Invest. Radiol., 2017, 52, 128–133 CrossRef CAS.
- Y. Cai, Y. Wang, H. Xu, C. Cao, R. Zhu, X. Tang, T. Zhang and Y. Pan, Nanoscale, 2019, 11(6), 2644–2654 RSC.
- J. Sherwood, M. Rich, K. Lovas, J. Warram, M. S. Bolding and Y. Bao, Nanoscale, 2017, 9, 11785–11792 RSC.
- J. Pellico, J. Llop, I. Fernández-Barahona, R. Bhavesh, J. Ruiz-Cabello and F. Herranz, Contrast Media Mol. Imaging, 2017, 2017, 1–24 CrossRef.
- L. Zhu, Z. Zhou, H. Mao and L. Yang, Nanomedicine, 2017, 12, 73–87 CrossRef CAS.
- Z. Jia, L. Song, F. Zang, J. Song, W. Zhang, C. Yan, J. Xie, Z. Ma, M. Ma, G. Teng, N. Gu and Y. Zhang, Theranostics, 2016, 6, 1780–1791 CrossRef CAS.
- Z. Shen, T. Chen, X. Ma, W. Ren, Z. Zhou, G. Zhu, A. Zhang, Y. Liu, J. Song, Z. Li, H. Ruan, W. Fan, L. Lin, J. Munasinghe, X. Chen and A. Wu, ACS Nano, 2017, 11, 10992–11004 CrossRef CAS.
- B. H. Kim, N. Lee, H. Kim, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. B. Na, J.-G. Park, T.-Y. Ahn, Y.-W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624–12631 CrossRef CAS.
- J. Huang, L. Wang, X. Zhong, Y. Li, L. Yang and H. Mao, J. Mater. Chem. B, 2014, 2, 5344–5351 RSC.
- H. Groult, N. Poupard, F. Herranz, E. Conforto, N. Bridiau, F. Sannier, S. Bordenave, J.-M. Piot, J. Ruiz-Cabello, I. Fruitier-Arnaudin and T. Maugard, Biomacromolecules, 2017, 18, 3156–3167 CrossRef CAS.
- E. Gray, B. Mulloy and T. W. Barrowcliffe, Thromb. Haemostasis, 2008, 99, 807–818 CrossRef CAS.
- E. I. Oduah, R. J. Linhardt and S. T. Sharfstein, Pharmaceuticals, 2016, 9(3), 38 CrossRef.
- L. Fu, M. Suflita and R. J. Linhardt, Adv. Drug Delivery Rev., 2016, 97, 237–249 CrossRef CAS.
- X. Yang, H. Du, J. Liu and G. Zhai, Biomacromolecules, 2015, 16, 423–436 CrossRef CAS.
- M. Swierczewska, H. S. Han, K. Kim, J. H. Park and S. Lee, Adv. Drug Delivery Rev., 2016, 99, 70–84 CrossRef CAS.
- A. Garg, R. Sharma, V. Pandey, V. Patel and A. K. Yadav, Crit. Rev. Ther. Drug Carrier Syst., 2017, 34, 1–33 CrossRef.
- M. del P. Rodriguez-Torres, L. S. Acosta-Torres and L. A. Diaz-Torres, J. Nanomater., 2018, 2018, 1–8 CrossRef.
- K.-O. Doh and Y. Yeo, Ther. Delivery, 2012, 3, 1447–1456 CrossRef CAS.
- H. Khurshid, S. H. Kim, M. J. Bonder, L. Colak, B. Ali, S. I. Shah, K. L. Kiick and G. C. Hadjipanayis, J. Appl. Phys., 2009, 105, 07B308 CrossRef.
- T.-Y. Liu, L.-Y. Huang, S.-H. Hu, M.-C. Yang and S.-Y. Chen, J. Biomed. Nanotechnol., 2007, 3, 353–359 CrossRef CAS.
- J. Lee, M. J. Jung, Y. H. Hwang, Y. J. Lee, S. Lee, D. Y. Lee and H. Shin, Biomaterials, 2012, 33, 4861–4871 CrossRef CAS.
- M. M. Kemp, A. Kumar, S. Mousa, E. Dyskin, M. Yalcin, P. Ajayan, R. J. Linhardt and S. A. Mousa, Nanotechnology, 2009, 20, 455104 CrossRef.
- M. M. Kemp, A. Kumar, S. Mousa, T.-J. Park, P. Ajayan, N. Kubotera, S. A. Mousa and R. J. Linhardt, Biomacromolecules, 2009, 10, 589–595 CrossRef CAS.
- L. Hou, Y. Fan, J. Yao, J. Zhou, C. Li, Z. Fang and Q. Zhang, Carbohydr. Polym., 2011, 86, 1157–1166 CrossRef CAS.
- T. Ye, X. Jiang, J. Li, R. Yang, Y. Mao, K. Li, L. Li, F. Chen, J. Yao, Y. Wu, X. Yang, S. Wang and W. Pan, Carbohydr. Polym., 2015, 122, 26–38 CrossRef CAS.
- T. Ye, H. Zhang, G. Chen, L. Shang and S. Wang, Contrast Media Mol. Imaging, 2016, 11, 482–491 CrossRef CAS.
- K. Lee, H. Lee, K. H. Bae and T. G. Park, Biomaterials, 2010, 31, 6530–6536 CrossRef CAS.
- C. Tao, Y. Chen, D. Wang, Y. Cai, Q. Zheng, L. An, J. Lin, Q. Tian and S. Yang, Nanomaterials, 2019, 9, 699 CrossRef CAS.
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673–692 CrossRef CAS.
- X. Duan and Y. Li, Small, 2013, 9, 1521–1532 CrossRef CAS.
- J. Bourquin, A. Milosevic, D. Hauser, R. Lehner, F. Blank, A. Petri-Fink and B. Rothen-Rutishauser, Adv. Mater., 2018, 30, 1704307 CrossRef.
- Q. Feng, Y. Liu, J. Huang, K. Chen, J. Huang and K. Xiao, Sci. Rep., 2018, 8, 2082 CrossRef.
- Y. Wei, L. Quan, C. Zhou and Q. Zhan, Nanomedicine, 2018, 13, 1495–1512 CrossRef CAS.
- P. C. Ke, S. Lin, W. J. Parak, T. P. Davis and F. Caruso, ACS Nano, 2017, 11, 11773–11776 CrossRef CAS.
- J. Lazarovits, Y. Y. Chen, E. A. Sykes and W. C. W. Chan, Chem. Commun., 2015, 51, 2756–2767 RSC.
- V. H. Nguyen and B.-J. Lee, Int. J. Nanomed., 2017, 12, 3137–3151 CrossRef CAS.
- K. Saha, M. Rahimi, M. Yazdani, S. T. Kim, D. F. Moyano, S. Hou, R. Das, R. Mout, F. Rezaee, M. Mahmoudi and V. M. Rotello, ACS Nano, 2016, 10, 4421–4430 CrossRef CAS.
- M. J. Ernsting, M. Murakami, A. Roy and S.-D. Li, J. Controlled Release, 2013, 172, 782–794 CrossRef CAS.
- B. Du, M. Yu and J. Zheng, Nat. Rev. Mater., 2018, 3, 358–374 CrossRef.
- M. Yu and J. Zheng, ACS Nano, 2015, 9, 6655–6674 CrossRef CAS.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti, S. Palchetti, S. De Grossi, A. Riccioli, H. Amenitsch and A. Laganà, Nanoscale, 2014, 6, 2782 RSC.
- B. Casu, I. Vlodavsky and R. D. Sanderson, Pathophysiol. Haemostasis Thromb., 2009, 36, 195–203 CrossRef.
- Y. Liang, I. Benakanakere, C. Besch-Williford, R. S. Hyder, M. R. Ellersieck and S. M. Hyder, Menopause, 2010, 17, 1040–1047 CrossRef.
- O. Achour, N. Poupard, N. Bridiau, S. Bordenave Juchereau, F. Sannier, J.-M. Piot, I. Fruitier Arnaudin and T. Maugard, Carbohydr. Polym., 2016, 135, 316–323 CrossRef CAS.
- H. Groult, R. Cousin, C. Chot-Plassot, M. Maura, N. Bridiau, J.-M. Piot, T. Maugard and I. Fruitier-Arnaudin, Mar. Drugs, 2019, 17, 140 CrossRef CAS.
- J. R. Wiśniewski, A. Zougman, N. Nagaraj and M. Mann, Nat. Methods, 2009, 6, 359–362 CrossRef.
- F. Meier, S. Beck, N. Grassl, M. Lubeck, M. A. Park, O. Raether and M. Mann, J. Proteome Res., 2015, 14, 5378–5387 CrossRef CAS.
- F. Meier, A.-D. Brunner, S. Koch, H. Koch, M. Lubeck, M. Krause, N. Goedecke, J. Decker, T. Kosinski, M. A. Park, N. Bache, O. Hoerning, J. Cox, O. Räther and M. Mann, Mol. Cell. Proteomics, 2018, 17, 2534–2545 CrossRef CAS.
- B. L. Zybailov, L. Florens and M. P. Washburn, Mol. BioSyst., 2007, 3, 354 RSC.
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein, T. Geiger, M. Mann and J. Cox, Nat. Methods, 2016, 13, 731–740 CrossRef CAS.
- P. M. Dodd and A. Jayaraman, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 694–705 CrossRef CAS.
- S. F. Chin, K. S. Iyer and C. L. Raston, Lab Chip, 2008, 8, 439 RSC.
- J. Manson, D. Kumar, B. J. Meenan and D. Dixon, Gold Bull., 2011, 44, 99–105 CrossRef CAS.
- K. G. Paul, T. B. Frigo, J. Y. Groman and E. V. Groman, Bioconjugate Chem., 2004, 15, 394–401 CrossRef CAS.
- D. Grant, W. F. Long and F. B. Williamson, Biochem. J., 1987, 244, 143–149 CrossRef CAS.
- D. Grant, W. F. Long, C. F. Moffat and F. B. Williamson, Biochem. J., 1991, 275, 193–197 CrossRef CAS.
- R. Chokkareddy and G. G. Redhi, in Green Metal Nanoparticles, ed. S. Kanchi and S. Ahmed, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2018, pp. 113–139 Search PubMed.
- M. J. A. Hore, Soft Matter, 2019, 15, 1120–1134 RSC.
- C. M. Wijmans and E. B. Zhulina, Macromolecules, 1993, 26, 7214–7224 CrossRef CAS.
- K. Ohno, T. Morinaga, S. Takeno, Y. Tsujii and T. Fukuda, Macromolecules, 2007, 40, 9143–9150 CrossRef CAS.
- S. Khan, J. Gor, B. Mulloy and S. J. Perkins, J. Mol. Biol., 2010, 395, 504–521 CrossRef CAS.
- G. Pavlov, S. Finet, K. Tatarenko, E. Korneeva and C. Ebel, Eur. Biophys. J., 2003, 32, 437–449 CrossRef CAS.
- D. Dukes, Y. Li, S. Lewis, B. Benicewicz, L. Schadler and S. K. Kumar, Macromolecules, 2010, 43, 1564–1570 CrossRef CAS.
- Y. Bao, J. A. Sherwood and Z. Sun, J. Mater. Chem. C, 2018, 6, 1280–1290 RSC.
- M. Rohrer, H. Bauer, J. Mintorovitch, M. Requardt and H.-J. Weinmann, Invest. Radiol., 2005, 40, 715–724 CrossRef.
- Y.-K. Peng, S. C. E. Tsang and P.-T. Chou, Mater. Today, 2016, 19, 336–348 CrossRef CAS.
- J. Zeng, L. Jing, Y. Hou, M. Jiao, R. Qiao, Q. Jia, C. Liu, F. Fang, H. Lei and M. Gao, Adv. Mater., 2014, 26, 2694–2698 CrossRef CAS.
- J. Pellico, J. Ruiz-Cabello, I. Fernández-Barahona, L. Gutiérrez, A. V. Lechuga-Vieco, J. A. Enríquez, M. P. Morales and F. Herranz, Langmuir, 2017, 33, 10239–10247 CrossRef CAS.
- U. I. Tromsdorf, O. T. Bruns, S. C. Salmen, U. Beisiegel and H. Weller, Nano Lett., 2009, 9, 4434–4440 CrossRef CAS.
- G. B. Toth, C. G. Varallyay, A. Horvath, M. R. Bashir, P. L. Choyke, H. E. Daldrup-Link, E. Dosa, J. P. Finn, S. Gahramanov, M. Harisinghani, I. Macdougall, A. Neuwelt, S. S. Vasanawala, P. Ambady, R. Barajas, J. S. Cetas, J. Ciporen, T. J. DeLoughery, N. D. Doolittle, R. Fu, J. Grinstead, A. R. Guimaraes, B. E. Hamilton, X. Li, H. L. McConnell, L. L. Muldoon, G. Nesbit, J. P. Netto, D. Petterson, W. D. Rooney, D. Schwartz, L. Szidonya and E. A. Neuwelt, Kidney Int., 2017, 92, 47–66 CrossRef CAS.
- R. Bhavesh, A. Lechuga-Vieco, J. Ruiz-Cabello and F. Herranz, Nanomaterials, 2015, 5, 1880–1890 CrossRef CAS.
- F. Rösch, in Theranostics, Gallium-68, and Other Radionuclides, ed. R. P. Baum and F. Rösch, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, vol. 194, pp. 3–16 Search PubMed.
- I. Velikyan, Molecules, 2015, 20, 12913–12943 CrossRef CAS.
- Y.-I. Chung, J. C. Kim, Y. H. Kim, G. Tae, S.-Y. Lee, K. Kim and I. C. Kwon, J. Controlled Release, 2010, 143, 374–382 CrossRef CAS.
- I.-C. Sun, D.-K. Eun, J. H. Na, S. Lee, I.-J. Kim, I.-C. Youn, C.-Y. Ko, H.-S. Kim, D. Lim, K. Choi, P. B. Messersmith, T. G. Park, S. Y. Kim, I. C. Kwon, K. Kim and C.-H. Ahn, Chem. – Eur. J., 2009, 15, 13341–13347 CrossRef CAS.
- C. Passirani, G. Barratt, J. Devissaguet and D. Labarre, Pharm. Res., 1998, 15, 1046–1050 CrossRef CAS.
- C. Passirani, G. Barratt, J.-P. Devissaguet and D. Labarre, Life Sci., 1998, 62, 775–785 CrossRef CAS.
- M. Socha, P. Bartecki, C. Passirani, A. Sapin, C. Damgé, T. Lecompte, J. Barré, F. E. Ghazouani and P. Maincent, J. Drug Targeting, 2009, 17, 575–585 CrossRef CAS.
- G. Caracciolo, O. C. Farokhzad and M. Mahmoudi, Trends Biotechnol., 2017, 35, 257–264 CrossRef CAS.
- U. Sakulkhu, M. Mahmoudi, L. Maurizi, J. Salaklang and H. Hofmann, Sci. Rep., 2015, 4, 5020 CrossRef.
- L. Landgraf, C. Christner, W. Storck, I. Schick, I. Krumbein, H. Dähring, K. Haedicke, K. Heinz-Herrmann, U. Teichgräber, J. R. Reichenbach, W. Tremel, S. Tenzer and I. Hilger, Biomaterials, 2015, 68, 77–88 CrossRef CAS.
- H. Yu, E. Munoz, R. Edens and R. Linhardt, Biochim. Biophys. Acta, Gen. Subj., 2005, 1726, 168–176 CrossRef CAS.
- A. Zaferani, D. Talsma, M. K. S. Richter, M. R. Daha, G. J. Navis, M. A. Seelen and J. van den Born, Nephrol., Dial., Transplant., 2014, 29, 515–522 CrossRef CAS.
- C. Fedeli, D. Segat, R. Tavano, L. Bubacco, G. De Franceschi, P. P. de Laureto, E. Lubian, F. Selvestrel, F. Mancin and E. Papini, Nanoscale, 2015, 7, 17710–17728 RSC.
- G. A. Manderson, M. Martin, P. Önnerfjord, T. Saxne, A. Schmidtchen, T. E. Mollnes, D. Heinegård and A. M. Blom, Mol. Immunol., 2009, 46, 3388–3398 CrossRef CAS.
- V. P. Vu, G. B. Gifford, F. Chen, H. Benasutti, G. Wang, E. V. Groman, R. Scheinman, L. Saba, S. M. Moghimi and D. Simberg, Nat. Nanotechnol., 2019, 14, 260–268 CrossRef CAS.
- N. Bertrand, P. Grenier, M. Mahmoudi, E. M. Lima, E. A. Appel, F. Dormont, J.-M. Lim, R. Karnik, R. Langer and O. C. Farokhzad, Nat. Commun., 2017, 8, 777 CrossRef.
- A. Vonarbourg, C. Passirani, P. Saulnier and J.-P. Benoit, Biomaterials, 2006, 27, 4356–4373 CrossRef CAS.
- D. Labarre, C. Vauthier, C. Chauvierre, B. Petri, R. Muller and M. Chehimi, Biomaterials, 2005, 26, 5075–5084 CrossRef CAS.
- K. Alhareth, C. Vauthier, F. Bourasset, C. Gueutin, G. Ponchel and F. Moussa, Eur. J. Pharm. Biopharm., 2012, 81, 453–457 CrossRef CAS.
- I. Bertholon, C. Vauthier and D. Labarre, Pharm. Res., 2006, 23, 1313–1323 CrossRef CAS.
- B. Boneu, C. Caranobe and P. Sie, Baillière's Clin. Haematol., 1990, 3, 531–544 CrossRef CAS.
- N. Poupard, P. Badarou, F. Fasani, H. Groult, N. Bridiau, F. Sannier, S. Bordenave-Juchereau, C. Kieda, J.-M. Piot, C. Grillon, I. Fruitier-Arnaudin and T. Maugard, Mar. Drugs, 2017, 15(5), 134, DOI:10.3390/md15050134.
- S. Li, Q. Xiong, X. Lai, X. Li, M. Wan, J. Zhang, Y. Yan, M. Cao, L. Lu, J. Guan, D. Zhang and Y. Lin, Compr. Rev. Food Sci. Food Saf., 2016, 15, 237–250 CrossRef CAS.
- C. Argyo, V. Cauda, H. Engelke, J. Rädler, G. Bein and T. Bein, Chem. – Eur. J., 2012, 18, 428–432 CrossRef CAS.
- A. K. A. Silva, D. Letourneur and C. Chauvierre, Theranostics, 2014, 4, 579–591 CrossRef CAS.
- V. Masola, G. Zaza, G. Gambaro, M. Franchi and M. Onisto, Semin. Cancer Biol., 2019, 62, 86–98 CrossRef.
- C. D. Mohan, S. Hari, H. D. Preetham, S. Rangappa, U. Barash, N. Ilan, S. C. Nayak, V. K. Gupta, Basappa, I. Vlodavsky and K. S. Rangappa, iScience, 2019, 15, 360–390 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr06378a |
| This journal is © The Royal Society of Chemistry 2021 |