
Dendrimer–tesaglitazar conjugate induces a phenotype shift of microglia and enhances β-amyloid phagocytosis†

Abstract
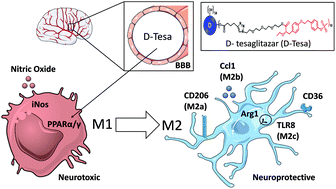
Switching microglia from a disease exacerbating, ‘pro-inflammatory’ state into a neuroprotective, ‘anti-inflammatory’ phenotype is a promising strategy for addressing multiple neurodegenerative diseases. Pro-inflammatory microglia contribute to disease progression by releasing neurotoxic substances and accelerating pathogenic protein accumulation. PPARα and PPARγ agonists have both been shown to shift microglia from a pro-inflammatory (‘M1-like’) to an alternatively activated (‘M2-like’) phenotype. Such strategies have been explored in clinical trials for neurological diseases, such as Alzheimer's and Parkinson's disease, but have likely failed due to their poor blood–brain barrier (BBB) penetration. Hydroxyl-terminated polyamidoamine dendrimers (without the attachment of any targeting ligands) have been shown to cross the impaired BBB at the site of neuroinflammation and accumulate in activated microglia. Therefore, dendrimer conjugation of a PPARα/γ dual agonist may enable targeted phenotype switching of activated microglia. Here we present the synthesis and characterization of a novel dendrimer-PPARα/γ dual agonist conjugate (D-tesaglitazar). In vitro, D-tesaglitazar induces an ‘M1 to M2’ phenotype shift, decreases secretion of reactive oxygen species, increases expression of genes for phagocytosis and enzymatic degradation of pathogenic proteins (e.g. β-amyloid, α-synuclein), and increases β-amyloid phagocytosis. These results support further development of D-tesaglitazar towards translation for multiple neurodegenerative diseases, especially Alzheimer's and Parkinson's Disease.
- This article is part of the themed collection: Editor’s Choice: Recent breakthroughs in nanobiotechnology
 Please wait while we load your content...
Please wait while we load your content...

