
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoscale flow cytometry for immunophenotyping and quantitating extracellular vesicles in blood plasma†
Nikki
Salmond
 a,
Karan
Khanna
a,
Gethin R.
Owen
b and
Karla C.
Williams
*a
a,
Karan
Khanna
a,
Gethin R.
Owen
b and
Karla C.
Williams
*a
aFaculty of Pharmaceutical Sciences, The University of British Columbia, Vancouver, V6T1Z3, Canada. E-mail: karla.williams@ubc.ca
bCentre for High Throughput Phenogenomics, Faculty of Dentistry, Oral, Biological & Medical Sciences, The University of British Columbia, 2199 Wesbrook Mall, Vancouver, British Columbia V6T1Z3, Canada
First published on 15th January 2021
Abstract
Extracellular vesicles (EVs) are lipid membrane enclosed nano-sized structures released into the extracellular environment by all cell types. EV constituents include proteins, lipids and nucleic acids that reflect the cell from which they originated. The molecular profile of cancer cells is distinct as compared to healthy cells of the same tissue type, and this distinct profile should be reflected by the EVs they release. This makes EVs desirable candidates for blood-based biopsy diagnosis of cancer. EVs can be time consuming to isolate therefore, a technology that can analyze EVs in complex biological samples in a high throughput manner is in demand. Here nanoscale flow cytometry is used to analyze EVs in whole, unpurified, plasma samples from healthy individuals and breast cancer patients. A known breast cancer marker, mammaglobin-a, was evaluated as a potential candidate for expression on EVs and increased levels in breast cancer. Mammaglobin-a particles were abundantly detected in plasma by nanoscale flow cytometry but only a portion of these particles were validated as bona fide EVs. EVs could be distinguish and characterized from small protein clusters and platelets based on size, marker composition, and detergent treatment. Mammaglobin-a positive EVs were characterized and found to be CD42a/CD41-positive platelet EVs, and the number of these EVs present was dependent upon plasma preparation protocol. Different plasma preparation protocols influenced the total number of platelet EVs and mammaglobin-a was found to associate with lipid membranes in plasma. When comparing plasma samples prepared by the same protocol, mammaglobin-a positive EVs were more abundant in estrogen receptor (ER) positive as compared to ER negative breast cancer patient plasma samples. This study demonstrates the capabilities of nanoscale flow cytometry for EV and small particle analysis in whole, unpurified, plasma samples, and highlights important technical challenges that need to be addressed when developing this technology as a liquid biopsy platform.
Introduction
Extracellular vesicles (EVs) are lipid bilayer enclosed structures released by all cell types into the extracellular environment and biological fluids.1,2 EVs are released either by budding from the plasma membrane to form microvesicles (∼50 nm–1 μm) or are formed within the endosomal system and released as exosomes (∼30–100 nm).1 EV constituents include lipid, glycan, protein and nucleic acid signatures that reflect the cell from which they are derived and EV contents can be delivered to recipient cells in a functional capacity.2,33 As EV contents are proposed to reflect their cell of origin, the potential use of EVs as a biomarker platform in cancer diagnosis and prognosis is an intensive area of research. EVs offer certain advantages over circulating tumor cells and cell free DNA biomarker platforms such as abundance (approximately ∼1010 EVs per ml of plasma)3 and stability.4 However, isolation and characterization of EVs from complex biological samples such as plasma can be time consuming and requires specialized equipment which imposes limitations on their utility in a clinical setting. Thus, development of a platform that is able to analyze and quantitate EV biomarker composition without purification would support the implementation of EV-based liquid biopsies. Here, we aimed to develop the use of an emerging technology – nanoscale flow cytometry – for the analysis of EVs in human plasma samples and assessed its utility in the detection and quantification of EVs.5–10Mammaglobin-a is a secretoglobin protein with enriched expression in breast tissues.11 Mammaglobin-a expression has also been observed in normal and malignant uterine, ovarian and cervical tissues.12 Whilst the function of mammaglobin-a is unknown, mammaglobin-a expression is elevated in primary and metastatic breast tumors but is not conclusively predictive of cancer grade or subtype.13–15 Additionally, mammaglobin-a mRNA is elevated in the blood of breast cancer patients as detected by qPCR of circulating tumor cells. A meta-analysis of several studies revealed that higher levels of circulating mammaglobin-a in plasma correlates with lymph node metastasis and advanced tumor stage.16–19 Furthermore, enzyme linked immunosorbent diagnostic assay (ELISA) has been used to show that secreted mammaglobin-a protein is elevated in breast cancer patient blood.20 Predictive analysis of mammaglobin-a protein structure reveals a putative transmembrane domain suggesting that mammaglobin-a may be anchored at the plasma membrane on breast cells. Indeed histological analysis of breast cancer and healthy tissues show plasma membrane localization of mammaglobin-a.21 Due to the cell surface association of mammaglobin-a and its increased expression in breast cancer cells, interest has arisen in targeting mammaglobin-a as an imaging agent,22 drug delivery target,21 and as a vaccine.23 Membrane associated mammaglobin-a could also be present on EV membranes and used in a diagnostic capacity. Mammaglobin-a, is an interesting candidate to investigate for the development of an EV liquid blood biopsy in breast cancer.
In this study we quantify the mammaglobin-a content in plasma from healthy individuals and breast cancer patients using nanoscale flow cytometry.5–10 We find mammaglobin-a particles readily abundant in both healthy individuals and breast cancer patients but only a fraction of these particles are EVs as determined using tetraspanin CD9 and Triton-X detergent treatment. In plasma, the majority of CD9-positive EVs were found to be platelet derived and mammaglobin-a was associated with these EVs. Overall, our work details a nanoscale flow cytometry platform for the analysis of EVs in complex biological fluids such as plasma, and highlights technical considerations for the use of this technology in identifying and quantitating specific EV populations in plasma.
Methods
Human specimens and processing
Biobanked blood plasma from breast cancer patients and individuals with benign breast disease was prepared and obtained from London Tumor Biobank (London, Ontario), Alberta Biobank (Alberta, BC) and BC Cancer (Victoria, BC). Healthy plasma samples were obtained from Innovative Research Inc. Plasma samples were collected as follows: London Tumor Biobank-10 ml dipotassium EDTA collection tube centrifuged at 2500g for 15 minutes; Alberta Biobank-6 ml dipotassium EDTA collection tube centrifuged at 3500 rpm 15 minutes. BC Cancer – 3–10 ml plasma in dipotassium EDTA collection tube centrifuged at 2500g for 10 minutes. Healthy blood samples were handled in larger volumes of 450 ml, from which plasma was isolated by centrifugation at 5000g for 15 minutes. Dipotassium EDTA was used as the anticoagulant agent. All patients had pathology diagnosed stage 1–3 breast cancer or benign disease. All individuals provided informed consent in accordance with institution protocol and study approval was obtained by the institutional review board of UBC (IRB#H17-01442). Study is compliant with all relevant ethical regulations on the use of human plasma. Plasma was stored in liquid nitrogen at biobank facilities and shipped on dry ice.Nanoscale flow cytometry
Plasma was received from biobank, thawed, vortexed and aliquoted into 100 μl aliquots and stored at −80 °C. For antibody labelling of plasma EVs, plasma was thawed on ice, vortexed on low for 5 seconds, and 10 μl plasma was removed, placed in a new tube and incubated with up to four antibodies for 30 minutes in the dark at room temperature. Details of antibodies and the concentrations used can be found in ESI Table 1.† Plasma was diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 in 300 μl 0.02 μm filtered PBS (MultiCell) and transferred into a flat bottomed 96 well plate. Samples were ran on CytoFLEX S (Beckman) using MilliQ water (ultrapure filtered and deionized water obtained from the MilliQ water purification system) as the diluent instead of sheath fluid. The following settings were used: trigger on 405 nm violet side scatter, violet side scatter detection set = 1027, slow speed setting for 30 seconds (10 μl sample analyzed per minute). Manual gating was performed on populations of interest with reference to isotype control. All data was exported into an excel file and analysis was performed on the total number of events that occurred within the gated areas during the 30 seconds run. Gain settings: VSSC: 100. FITC: 500. PE: 50. APC: 50. BV421: 100. For particle size estimation silica beads between 180 nm–1.3 μm in size (Apogee) and 122 nm GFP labelled murine virus (ViroFlow Technologies) were ran on the CytoFLEX using the settings detailed above.
:30 in 300 μl 0.02 μm filtered PBS (MultiCell) and transferred into a flat bottomed 96 well plate. Samples were ran on CytoFLEX S (Beckman) using MilliQ water (ultrapure filtered and deionized water obtained from the MilliQ water purification system) as the diluent instead of sheath fluid. The following settings were used: trigger on 405 nm violet side scatter, violet side scatter detection set = 1027, slow speed setting for 30 seconds (10 μl sample analyzed per minute). Manual gating was performed on populations of interest with reference to isotype control. All data was exported into an excel file and analysis was performed on the total number of events that occurred within the gated areas during the 30 seconds run. Gain settings: VSSC: 100. FITC: 500. PE: 50. APC: 50. BV421: 100. For particle size estimation silica beads between 180 nm–1.3 μm in size (Apogee) and 122 nm GFP labelled murine virus (ViroFlow Technologies) were ran on the CytoFLEX using the settings detailed above.
Purification and analysis of extracellular vesicles
Approximately 1 ml plasma was thawed on ice, once thawed plasma was filtered using 0.8 μm filter (MilliPore). Size exclusion column (IZON qEV original/70 nm) was brought to room temperature and equilibrated using 20 ml 0.2 μm filtered PBS. 500 μl of the filtered plasma was applied to the column and 3 ml flow through was collected immediately. PBS (0.2 μm filtered) was used to elute sample from column. Once 3 ml flow through had been collected, a 1.5 ml and a 1 ml fraction was collected as the EV fraction alongside three subsequent 1 ml fractions (fractions 2, 3 and 4). EV fractions 1.5 ml and 1 ml fraction were pooled and all collected fractions were concentrated to 100 μl using a 10 kDa MWCO Amicon concentrator (Millipore). The resultant EVs were then used for downstream processes.For flow cytometry analysis, 10 μl purified EVs and subsequent fractions were incubated with antibodies, as specified in ESI Table 1,† and then ran on the CytoFLEX S using the protocol described for plasma. Protein quantification of EV fractions was performed using BCA gold assay (Pierce) according to manufacturer's instructions.
Triton-X 100 lysis of extracellular vesicles
After incubation of purified EVs with selected antibodies for 30 minutes at room temperature in the dark, EVs were lysed by diluting the EVs 1:30 in 300 μl 1% Triton-X 100 (Fisher Bioreagents) in PBS. Samples were analyzed by nanoscale flow cytometry as described above.
Platelet isolation
Platelets were pelleted from plasma samples by centrifugation at 2800g for 10 minutes. The plasma supernatant was removed and the pellet washed with PBS and pelleted again by a final centrifugation step. Pellet was used in downstream applications (electron microscopy and nanoscale flow cytometry according to the protocol stated above).Electron microscopy
EVs and platelets pre-fixed in 2% paraformaldehyde were adsorbed onto Formvar/carbon coated nickel or copper 200 mesh grids respectively (Ted Pella/electron microscopy sciences) for approximately 2 minutes. Grids were then negatively stained with pre-filtered 1% Uranyl acetate pH 4.6 (Fischer Sci) for 30 s then blotted with filter paper and allowed to dry. Grids were imaged in scanning transmission mode with a Helios NanoLab 650, fitted with an STEM detector, (Thermofisher, Systems for Research, Kanata, ON, Canada) at 30 kV using the bright field imaging mode.Nanoparticle tracking analysis
Isolated EV fractions from three healthy plasma samples were diluted 1:1000 in 0.2 μm filtered PBS. The number and size of diluted EVs was analyzed using the NanoSight LM10 with a blue 488 nm laser (Malvern Panalytical). Three 30 seconds measurements were acquired using the syringe pump to maintain EV movement through the chamber at speed 40. Camera level was adjusted according to sample. Detection threshold for analysis was always kept at 5. NTA software version 3.2.16 was used to analyze data.
Western blot
10 or 20 μg of each fraction was mixed with 10% reducing buffer (10× Novex Bolt™) and 25% loading buffer (Novex Life Technologies). For samples that were run under non-reducing conditions reducing buffer was not added (for CD63). Samples were boiled at 95 °C for 10 minutes, loaded onto Bolt 4–12% Bis Tris Plus gradient gels (Thermo Fisher Scientific), and ran at 200 V for 30 minutes in MOPS (50 mM MOPS (Sigma), 50 mM Tris Base, 0.1% SDS, 1 mM EDTA, pH 7.7) running buffer. All reagents from Fisher Bioreagents unless otherwise stated. The proteins on the gel were subject to a wet transfer (190 mM glycine, 25 mM Tris Base) onto nitrocellulose 0.45 μm membrane (BioRad). After transfer, membranes were blocked in 5% milk in TBS-T (20 mM Tris base, 160 mM NaCl, 0.1% Tween) for 1 hour. Primary antibodies were applied overnight at 4 °C in 1% milk at the dilutions given in ESI Table 1.† Membranes were washed with three (10 minutes) washes in TBS-T. Secondary Licor IRDye® 680RD /IRDye® 800CW was applied in 1% milk for 30 minutes in the dark before repeating the wash steps. Membranes were imaged on the Licor Odyssey® CLx according to manufacturer's instructions using Image Studio Lite software (5.2). For all antibody information please see ESI Table 1.† 1.2 μg recombinant mammaglobin-a protein (Origene) was used as a positive control on mammaglobin-a blots.Software and data analysis
Nanoscale flow cytometry data and images were acquired using the CytoFLEX CytExpert 2.3 software. Figures were prepared with Inkscape™ 0.92 or Adobe Illustrator. Data was handled in Excel and analyzed using GraphPad Prism 8.0.1. Distribution of data was analyzed for normality. Data proven to normally distribute was analyzed by parametric Students t-test or ordinary one way ANOVA Turkey's multiple comparison. If normality failed, data was analyzed using non-parametric Mann–Whitney t-test or Kruskal–Wallis ANOVA. *p ≤ 0.05. **p ≤ 0.01. ***p ≤ 0.001. ****p ≤ 0.0001.Results
Nanoscale flow cytometry for detection of nano-sized particles
GFP labelled murine virus particles (122 nm) and silica Apogee beads with particle size range of 180 nm–1.3 μm were ran on the CytoFLEX S flow cytometer using violet side scatter (VSSC) trigger. VSSC triggering was capable of detecting all size ranges of Apogee beads down to 180 nm and GFP labelled 122 nm virus particles (Fig. 1A). To detect EVs using nanoscale flow cytometry a simple workflow was developed whereby 10 μl plasma was incubated with desired fluorescently labelled antibodies (see ESI Table 1†) for 30 minutes at room temperature in the dark before being diluted 1:30 using 0.02 μm filtered PBS (Fig. 1B). The samples were then loaded into a 96 well plate for high throughput analysis using the CytoFLEX S (Beckman Coulter). The loading of the 96 well plate was an important consideration in order to avoid cross-contamination between samples and maintain accurate results. A PBS wash between each sample and a 1% Contrad 70™ wash followed by a water wash at the end of each row was implemented to prevent buildup of plasma EVs and proteins during sample acquisition to minimize the risk of tubing blockages from arising (Fig. 1C).
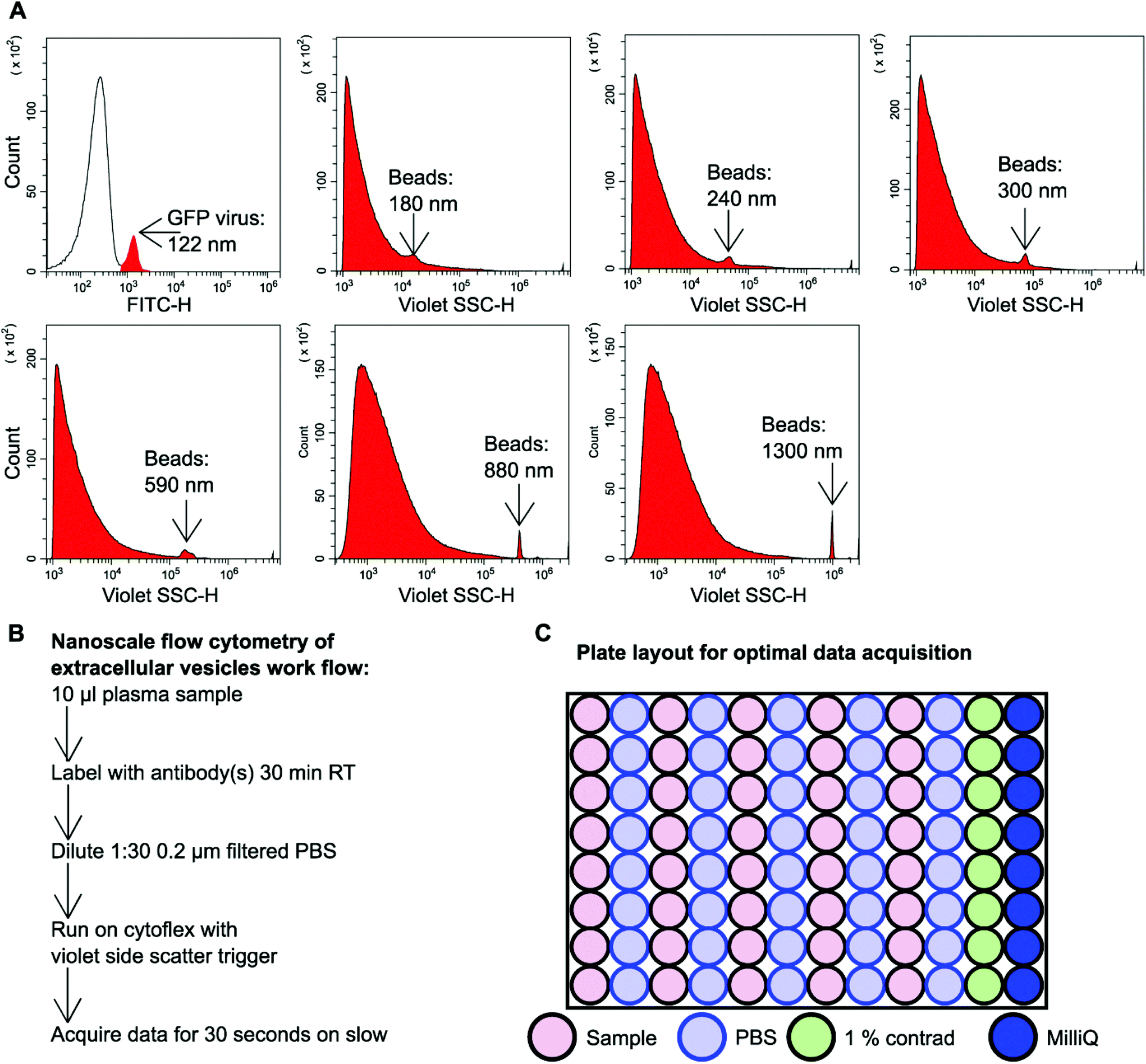 | ||
| Fig. 1 Nanoscale flow cytometry detects and analyzes particles in the nanoscale size range. (A) Representative images of violet side scatter (SSC) nanoscale flow cytometry of a GFP 122 nm virus or silica beads of different sizes 180 nm–1300 nm (Apogee). n = 3. GFP virus was detected and gated using FITC-H whereas populations of non-fluorescent beads were detected using Violet SSC-H. (B) Workflow describing the process of preparing plasma EVs for nanoscale flow cytometry. (C) Diagram of a 96 well plate set-up for high-throughput nanoscale flow cytometry analysis of plasma EVs. | ||
To ensure that signal detected by nanoscale flow cytometry was specific to antibody EV-binding and not from aggregated antibody or non-specific binding, each antibody used was subject to extensive control-based experiments. The background signal and electrical noise detected by the CytoFLEX S was confirmed to be low when PBS and plasma were ran on their own (ESI Fig. 1†). To ensure antibodies were binding to EVs and not aggregating into nano-sized structures, the antibodies were ran on their own in the absence of plasma (ESI Fig. 2†). To ensure the primary antibody was binding to the specific target and not binding non-specific targets, the isotype control for each antibody was incubated with plasma and this resulted in minimal to no signal (ESI Fig. 2†). Finally, the antibodies of choice were incubated with plasma together (ESI Fig. 2†) or separately (ESI Fig. 1†) to identify signal crossover into other fluorescent channels. Only the PE signal was found to crossover into the FITC channel and a compensation value of 3 was applied to the PE channel to eliminate false-positive FITC signal.
Detection of mammaglobin-a positive nanosized particles in plasma using nanoscale flow cytometry
Given that mammaglobin-a can associate with the plasma membrane cell surface and its expression is increased in breast cancer we sought to determine if mammaglobin-a positive EVs could be detected in breast cancer patient plasma samples. To detect breast derived EVs in plasma, plasma samples were incubated with an antibody against the mammaglobin-a protein. A distinct population of nanosized mammaglobin-a positive particles were observed in plasma samples (Fig. 2A). Antibody concentration was optimized through titration studies aimed at reducing non-specific signal and ensuring antibody saturation of available epitopes (Fig. 2B). The concentration of antibody showing a reduction in the number of mammaglobin-a events relative to the antibody titration was used in all experiments (Fig. 2B, highlighted red bar). To assess for coincidence events, plasma volume was decreased whilst keeping the concentration of antibody stable (Fig. 2C). The number of recorded events reduced by approximately 50% with each dilution demonstrating a lack of coincident detection. These controls were done for each antibody used in subsequent experiments (ESI Fig. 2D and E†).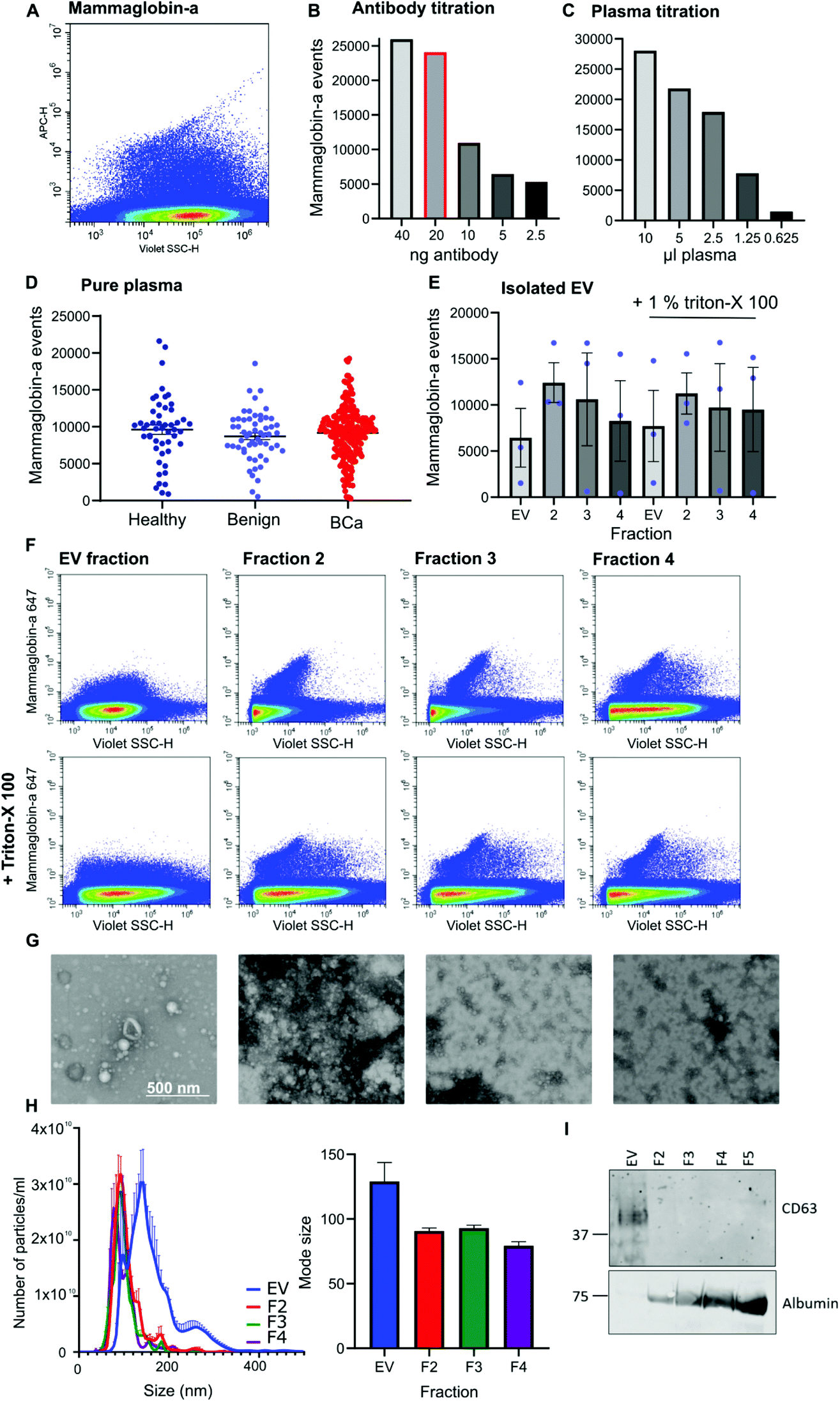 | ||
| Fig. 2 Mammaglobin-a EVs and protein particles are detected by nanoscale flow cytometry. (A) Representative image of the mammaglobin-a population as observed on the nanoscale flow cytometer. (B) 10 μl plasma was incubated with decreasing concentrations of mammaglobin-a antibody and mammaglobin-a events were quantified using nanoscale flow cytometry. Graph represents a single sample. (C) Decreasing volumes of plasma were incubated with 20 ng mammaglobin-a antibody followed by nanoscale flow cytometry analysis. Graph represents a single sample. (D) The amount of mammaglobin-a present in 50 healthy, 56 benign, and 219 breast cancer plasma samples was quantified using nanoscale flow cytometry. n = 2. ±SEM. (E) EVs were purified from healthy plasma samples by size exclusion chromatography, the EV fraction and subsequent fractions 2, 3 and 4 were analyzed for mammaglobin-a abundance using nanoscale flow cytometry in the absence and presence of Triton-X 100. n = 3 plasma samples. ±SEM. (F) Representative images of mammaglobin-a populations in size exclusion chromatography fractions: EV, F2, F3 and F4, ±Triton-X 100. (G) Isolated fractions were fixed in 2% paraformaldehyde and adsorbed onto Formvar/carbon coated nickel 200 mesh grids. Samples were negatively stained using 1% uranyl acetate before being imaged in scanning transmission mode at 30 kV using a Helios NanoLab 650. n = 2. (H) The size and concentration of isolated EVs was analyzed using nanoparticle tracking analysis. n = 3. ±SEM. (I) Western blot of EV marker CD63 and contaminant protein albumin expression in 20 μg EVs and fractions 2, 3 and 4 (2.5 μg). n = 3. | ||
Once the antibody concentration was optimized, 10 μl plasma from 50 healthy, 56 benign, and 219 breast cancer plasma samples was incubated with 20 ng mammaglobin-a antibody for 30 minutes. Sample was then diluted 30 times in 300 μl PBS and ran on the CytoFLEX flow cytometer triggering on 405 nm violet side scatter. Each sample was analyzed in two independent experiments to ensure reproducibility, and a third experiment used an isotype control antibody to ensure antibody specificity (ESI Fig. 3†). The variance between independent experiments was measured using a linear regression analysis. The R2 value was found to lie between 0.671–0.8909, giving evidence of a strong correlation and therefore reproducibility between runs (ESI Fig. 3†). Analysis of mammaglobin-a populations from each patient group found mammaglobin-a particles detected in the plasma of all patient groups with no significant differences between any of the groups (Fig. 2D).
To determine whether mammaglobin-a positive particles detected by nanoscale flow cytometry were bona fide EVs, EVs were purified from 500 μl plasma using size exclusion chromatography. The EV fraction was collected along with subsequent proteinacious fractions 2, 3 and 4. Nanoscale flow cytometry analysis revealed that all fractions contained mammaglobin-a nanoparticles, and most particles were resistant to detergent treatment indicating that the majority of mammaglobin-a signal is attributed to protein clusters or aggregates rather than EVs (Fig. 2E). Representative images of the mammaglobin-a nanoparticles detected within each fraction are shown in Fig. 2F. EV isolation was validated using transmission scanning electron microscopy, nanoparticle tracking analysis and western blot. Electron microscopy images show the presence of EV structures in the EV fraction, and smaller lipid structures and protein aggregates present in the later fractions (Fig. 2G). Nanoparticle tracking analysis shows similar EV/nanoparticle concentrations in each fraction, however the size of the detected particles decreased as the fraction elution progressed (Fig. 2H). Finally western blot analysis of fractions showed the EV marker CD63 present in the EV fraction but not in later fractions, whereas EV contaminant protein albumin was not present in the EV fraction and was abundant in later fractions (Fig. 2I). Whilst CD63 positive EVs were detected in the EV fraction mammaglobin-a was not. Mammaglobin-a however, could be detected in the later SEC fraction (ESI Fig. 4†). The inability to detect mammaglobin-a on EVs by western blot could be explained by the low number of mammaglobin-a EVs in the EV fraction which is possible below the detection limit of western blotting. Alternatively the association of mammaglobin-a with EVs could be weak or transient causing mammaglobin-a to be separated from EVs during the SEC EV purification process.
Mammaglobin-a positive CD9 EVs can be detected in plasma by nanoscale flow cytometry
To distinguish mammaglobin-a positive EVs from protein aggregates, plasma samples were immunolabeled for mammaglobin-a and EV marker CD9. This revealed a population of dual CD9-mammaglobin-a positive events, thus identifying a mammaglobin-a EV population. Surprisingly, plasma from healthy subjects contained significantly more CD9 positive EVs and CD9-mammaglobin-a dual positive EVs than plasma from subjects with benign breast disease or breast cancer (Fig. 3A and C). To ensure that the detected mammaglobin-a and CD9 positive events identified in whole plasma were bona fide EVs, EVs were isolated from plasma by size exclusion chromatography and analyzed by nanoscale flow cytometry. All fractions isolated by size exclusion chromatography were immunolabeled for CD9 and mammaglobin-a and analyzed by nanoscale flow cytometry. Nanoscale flow cytometry revealed that CD9 positive and CD9-mammaglobin-a dual positive EVs were found predominantly in the EV fraction with a smaller population in fraction 2 and negligible amounts in fractions 3 and 4 (Fig. 3B, D and E). Importantly, all CD9 positive and CD9-mammaglobin-a dual positive EVs were sensitive to detergent treatment further validating these structures as EVs (Fig. 3B and D). Representative images of the CD9 and dual positive CD9-mammaglobin-a EV populations are shown in Fig. 3E. | ||
| Fig. 3 Mammaglobin-a and CD9 positive EVs can be detected in plasma samples from healthy, benign and breast cancer subjects. (A and C) Plasma samples from healthy (50), benign (56), and breast cancer (219) subjects were incubated with CD9 and mammaglobin-a antibodies and the total number of CD9 positive events (A) and dual positive CD9-mammaglobin-a events (C) was quantified. All samples were analyzed twice and an average of the two runs taken. (B and D) EVs and subsequent fractions 2, 3 and 4 were isolated from plasma by size exclusion chromatography and analyzed for CD9 and mammaglobin-a positive EVs by nanoscale flow cytometry in the absence and presence of 1% Triton-X 100. n = 3 plasma samples. ±SEM. (E) Representative images of identified CD9 EVs and dual positive CD9-mammaglobin-A EVs. CD9 and dual populations of interest were gated for image generation. | ||
Nanoscale flow cytometry detects two CD9 populations: platelets and EVs
Nanoscale flow cytometry analysis of CD9-positive events in plasma gave rise to two distinct populations (Fig. 4A). As standard biobanking protocols generate platelet-low plasma rather than platelet free, it was hypothesized that the populations represented EVs and platelets. To confirm this, an antibody for platelet marker CD41 was used in combination with CD9. CD41 was found to co-localize with the CD9 population supporting the notion that these are indeed platelets (Fig. 4A). In addition to the larger CD9-positive structures, CD41 was also found on the majority of the smaller CD9-positive structures (Fig. 4A). In the healthy and breast cancer plasma cohort greater than 75% of CD9 EVs immunostained dual positive for platelet protein marker CD41 (83% ± 20 and 76.5% ± 15.3, respectively). This demonstrates that the majority of CD9-positive EVs in plasma were likely to be platelet derived (ESI Fig. 5†). To further validate the larger CD9-positive population as a platelet population, plasma was centrifuged at 2800g for 10 minutes to pellet platelets. The pre-spin plasma, post-spin plasma, and pellet were immunolabelled with CD9 and analyzed by nanoscale flow cytometry. The number of CD9-positive platelet events was significantly reduced in the post-spin plasma sample whereas the smaller CD9-positive EV population did not significantly change post-spin (Fig. 4B and ESI Fig. 6†). Next, the platelet pellets were subject to electron microscopy. The 2–3 μm size range and rounded morphology of the imaged structures along with their CD9 and CD41 positivity indicated that the structures had been correctly identified as platelets (Fig. 4C).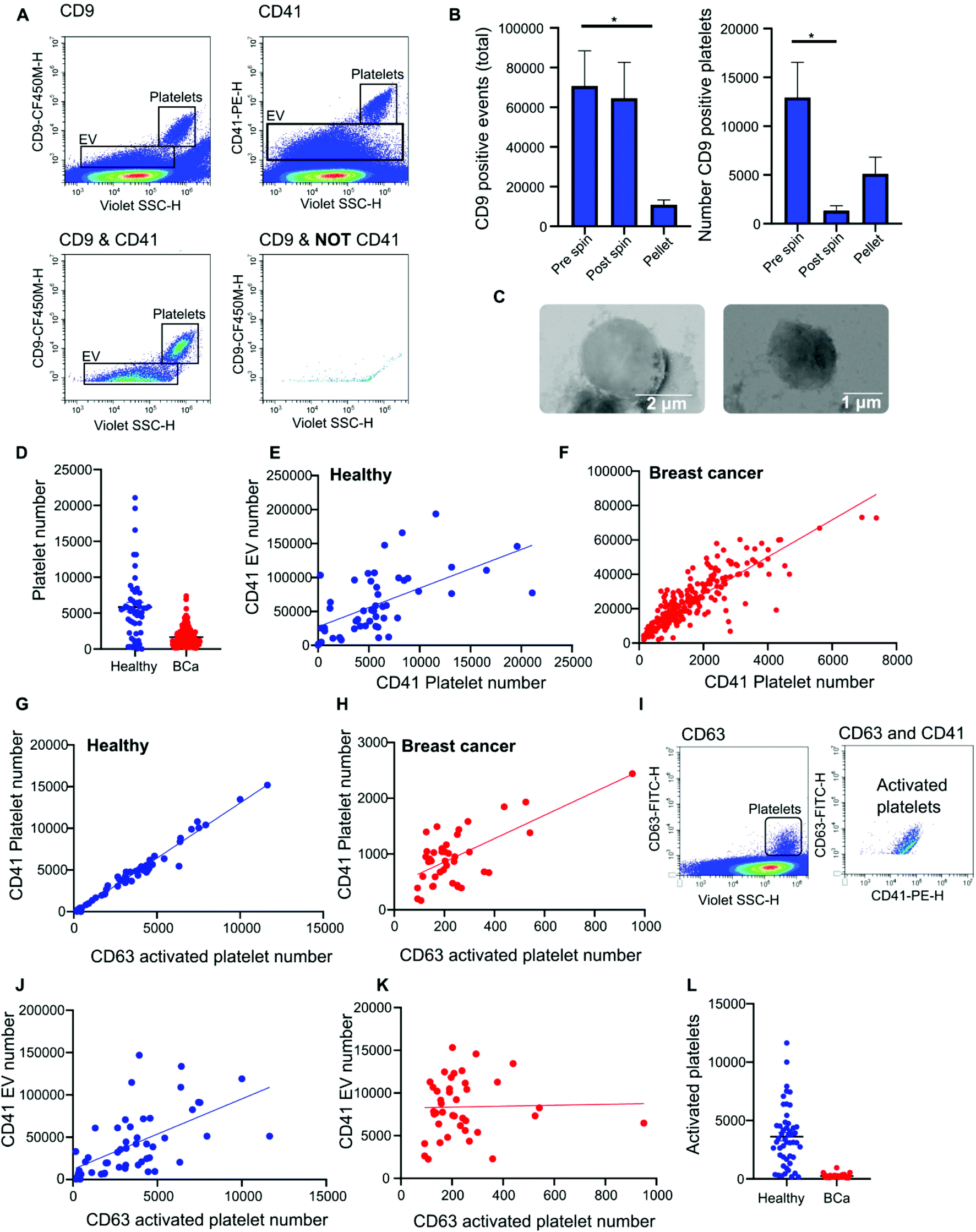 | ||
| Fig. 4 Plasma contains platelets, activated platelets and platelet derived EVs that can be detected by nanoscale flow cytometry. (A) Representative flow cytometry images of CD41 and CD9 positive platelets and platelet derived EVs. (B) Platelets were spun out of plasma samples at 2800g and the plasma (pre and post spin) as well as the platelet pellet were analyzed for CD9 positive events and CD9 positive platelets. 5 samples. (C) Representative transmission scanning electron microscopy images of platelets pelleted from plasma at 2800g. 3 samples were imaged. (D) The number of CD41 positive platelets present in healthy (50) versus breast cancer (219) patient plasma samples. All samples were analyzed in duplicate and an average value taken. ±SEM. (E and F) The correlation between the presence of CD41 positive platelets and the number of platelet derived EVs was analyzed in both healthy and breast cancer plasma samples by linear regression. n = 2. (G and H) Activated platelets in healthy and breast cancer plasma samples were identified by quantifying CD41 and CD63 dual positive events. Linear regression was used to identify a correlation between the number of CD41 positive platelets the number of activated CD63 platelets. Healthy samples: 50. Breast cancer samples: 40. (I) Representative images of CD41 and CD63 positive activated platelets as observed using nanoscale flow cytometry. (J and K) The correlation between CD63 positive activated platelets and the number of CD41 positive platelet EVs was assessed in healthy and breast cancer plasma samples by linear regression. Healthy samples: 50. Breast cancer samples: 40. (L) CD63 positive platelets present in breast cancer versus healthy patient plasma samples. Healthy samples: 50. Breast cancer samples: 40. | ||
Analysis of platelet levels in breast cancer patient plasma compared to that of healthy individual plasma found significantly more platelets in healthy individuals; breast cancer plasmas had, on average, 1500 platelets (1648 ± 80 total events) whereas healthy individuals had, on average, 6000 platelets (5847 ± 660 total events) (Fig. 4D). To further understand why the number of platelets in plasma samples was varied, we investigated the impact of blood draw volume (4, 6 and 10 ml) upon platelet number. Larger draw volumes (10 ml) correlated with higher platelet numbers remaining post-centrifugation in the plasma as compared to the smaller draw volumes (3 ml) (ESI Fig. 7A†). To confirm the effects of blood volume draw on platelet numbers, human whole blood was centrifuged at 2500g for 15 minutes using decreasing blood volumes. Plasma was isolated and immunolabeled with CD9 to analyze the platelet population. Indeed, the larger the starting volume of blood the higher the number of platelets in the plasma after centrifugation (ESI Fig. 7B†). Centrifugation time may also alter platelet number. To evaluate this, 10ml of blood was spun at 2500g for 10 or 15 minutes. Both time points gave similar levels of platelet removal (ESI Fig. 7C†) and was the condition used in the collection of benign and breast cancer patient plasma. The volume of healthy plasma, however, was 450ml and spun at 5000g for 15min. The larger starting volume of plasma therefore, accounts for the higher number of platelets in the healthy plasma samples.
While it is well recognized that blood collection protocols affect platelet number, whether or not this translates to an increase in EV number is less well described. As platelet-derived EVs make up a large proportion of EVs blood,24 differences in levels between healthy and cancer plasmas could affect downstream analysis and results. To determine if the number of platelets influences the number of platelet derived EVs present in plasma, the platelet number was assessed for correlation to platelet EV number. A significant positive correlation between platelet number and platelet EV number was found for both healthy and breast cancer plasma (Fig. 4E and F); the correlation was moderately strong for breast plasma samples (r2 = 0.715; p < 0.0001) and modest for healthy plasma samples (r2 = 0.3422; p < 0.0001). As platelets can shed EVs upon activation,25 to determine the contribution of activated platelets to total platelet EV number, the relative number of activated platelets in healthy and breast cancer patient plasma was assessed and compared to the number of platelet EVs. The number of activated, CD63 positive, platelets within a sample positively correlated with the number of CD41 positive platelets present in the sample (Fig. 4G and H). For healthy plasma, the correlation was strong (r2 = 0.97) and for breast cancer plasma the correlation was moderate (r2 = 0.44). While platelet EVs were readily detected in plasma samples, CD63 was not detected on platelet EVs by nanoscale flow cytometry (Fig. 4I). As such, the number of CD63 platelets in relation to the number of CD41 EVs was assessed. A positive correlation between increased activated platelet number and increased platelet EV number was identified in healthy plasma samples (r2 = 0.33) (Fig. 4J). There was no correlation between the number of activated, CD63 positive, platelets and the number of platelet EVs in breast cancer plasma samples (r2 = 0.0006) (Fig. 4K). While this is potentially a consequence of low levels of activated platelets in breast cancer plasmas as compared to healthy (Fig. 4L), it is also possible that platelet number rather than activation status is a better representation of platelet-derived EV number. Overall, these results demonstrate that platelet number significantly correlates with platelet-derived EV number and analysis of CD41 platelets represents a method to identify plasma samples with high or low levels of platelet-derived EV.
Mammaglobin-a is associated with platelet derived EVs
The majority of CD9-positive EVs were found to be of platelet origin, as identified by CD41. This suggests that CD9-mammaglobin-a dual positive EVs may in-fact be platelet EVs. To evaluate the origin of the CD9-mammaglobin-a dual positive population, EVs were analyzed using platelet markers CD41 and CD42a (Fig. 5A and B). The majority of CD9-mammaglobin-a dual positive events were also positive for platelet markers CD41 and CD42a (Fig. 5A and B). Thus, the majority of mammaglobin-a EVs detected by nanoscale flow cytometry in plasma are in-fact of platelet origin. Mammaglobin-a was also detected on platelets suggesting that secreted and membrane associated proteins to bind other membranous structures in plasma (ESI Fig. 8†). Mammaglobin-a association with platelet membranes in whole plasma is potentially a weak or transient interaction. To assess this, platelets were pelleted, resuspended, and analyzed for mammaglobin-a by nanoscale flow cytometry. Mammaglobin-a was not detected on isolated platelets by either flow cytometry or western blot (ESI Fig. 9†). This suggests that mammaglobin-a is weakly or transiently associated with lipid structures in whole plasma.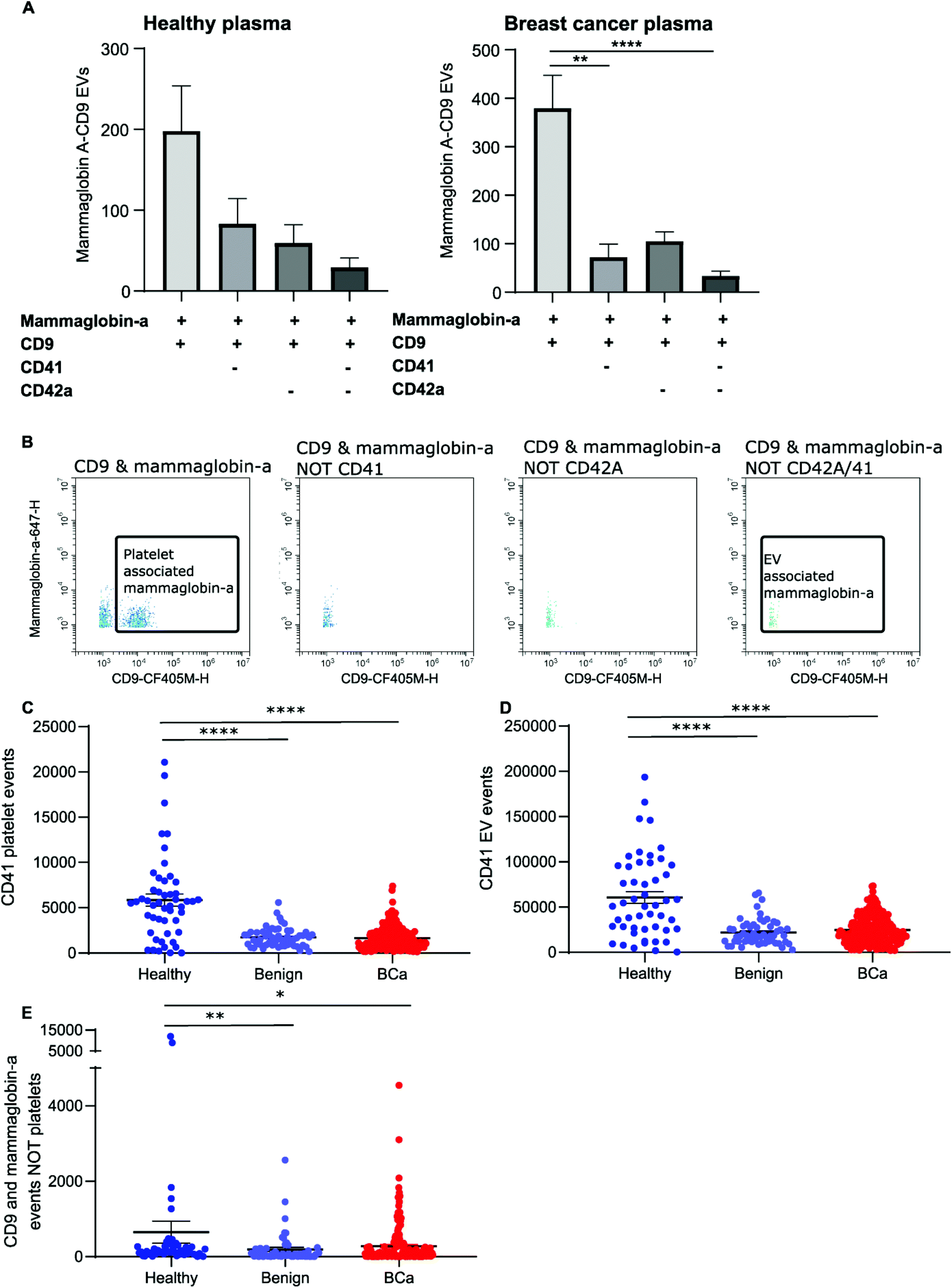 | ||
| Fig. 5 Mammaglobin-a and CD9 positive EVs detected by nanoscale flow cytometry are CD41 and CD42a positive indicating that they are platelet derived. (A) The proportion of mammaglobin-a CD9 EVs that were also positive for EV markers CD41 and CD42a in healthy and breast cancer samples was analyzed using nanoscale flow cytometry. Healthy: 10 samples. Breast cancer: 10 samples. (B) Representative images of mammaglobin-a and CD9 positive EVs before and after removal of CD41 and CD42a positive events. (C and D) The number of CD41 positive platelets and CD41 positive EVs as detected by nanoscale flow cytometry was quantified in plasmas derived from healthy (50), benign (56) and breast cancer (219) patients. n = 2. (E) Quantification of the number of CD9 and mammaglobin-a dual positive EV events (minus any CD9 and mammaglobin-A dual positive platelets) detected by nanoscale flow cytometry. n = 2. | ||
As mammaglobin-a was found to be associated with platelets and platelet EV membranes, the number of platelets and platelet EVs in each sample would strongly influence mammaglobin-a EV levels. Healthy plasma samples were found to have significantly higher levels of CD41 positive platelet and platelet-derived EVs as compared to benign and breast cancer plasmas (Fig. 5C and D). As such, it is not surprising that healthy plasma samples were found to have significantly higher levels of CD9-mammaglobin-a dual positive EVs compared to benign and breast cancer plasma samples (Fig. 5E). These results suggest that elevated CD9-mammaglobin-a dual positive EV levels in healthy plasma may solely be a consequence of high platelet and platelet EV numbers.
Mammaglobin-a positive EVs are elevated in estrogen receptor positive breast cancer
Mammaglobin-a was found to associate with platelet derived EVs. Healthy plasma samples had elevated levels of platelets and platelet EVs as a consequence of the plasma isolation protocol. As mammaglobin-a associates with platelet EV membranes, comparing mammaglobin-a EV levels between plasma cohorts that were isolated using different protocols introduces a confounding variable that could mask results or lead to inaccurate reporting. However, the cohort of breast cancer plasma samples were all prepared and stored by the same biobank and therefore the data collected within this cohort was comparable and quantifiable. As increased expression of mammaglobin-a has been demonstrated in breast tumors, particularly in estrogen receptor (ER) positive tumors, we analyzed mammaglobin-a EV levels in plasma from ER-positive and ER-negative breast cancer patients.26 The number of CD9-mammaglobin-a dual positive EVs was found to be significantly higher in ER-positive breast cancers as compared to ER-negative breast cancers (Fig. 6A). These results indicate that there is an increased amount of secreted and circulating mammaglobin-a in ER-positive breast cancers which associates with platelet derived EVs and this can be analyzed and quantified using nanoscale flow cytometry (Fig. 6B).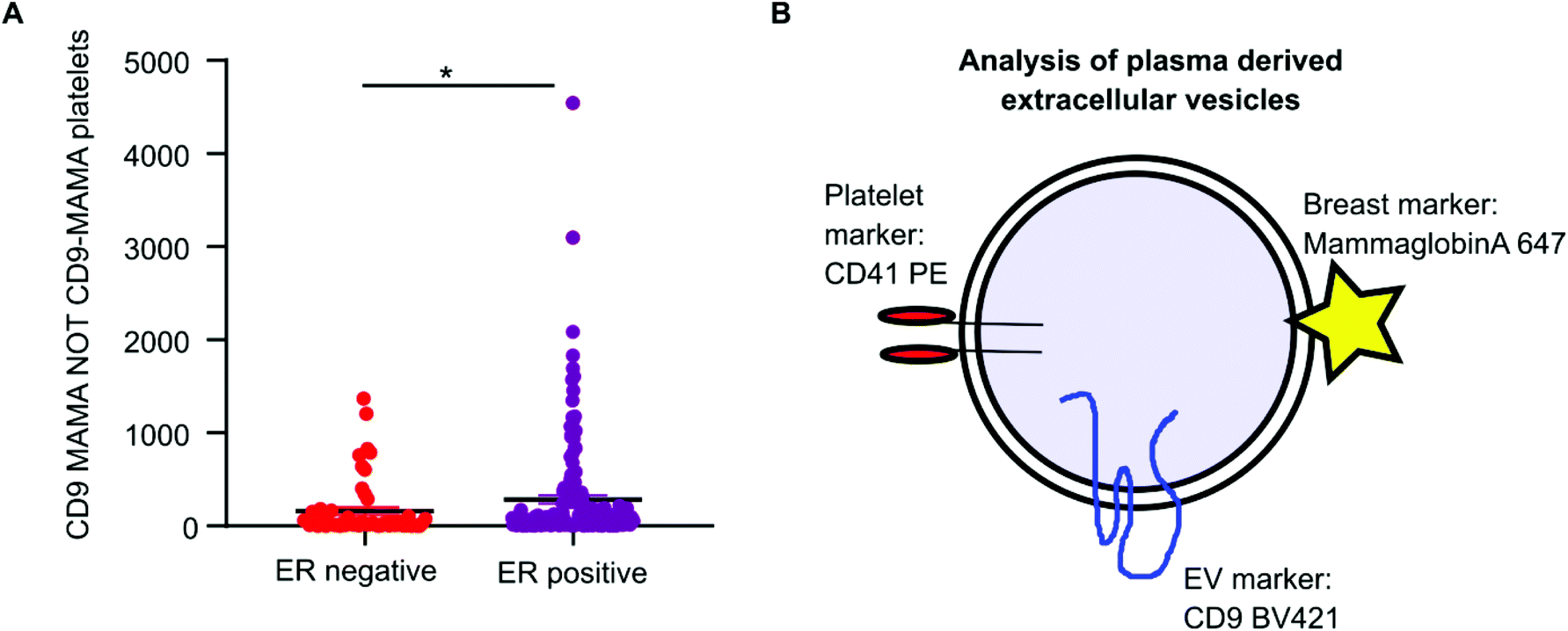 | ||
| Fig. 6 Mammaglobin-a and CD9 dual positive EVs are elevated in plasma samples from ER-positive breast cancer patients. (A) Nanoscale flow cytometry data from the breast cancer plasma cohort, n = 219, (all plasma samples were handled and treated the same from collection to storage) was analyzed for mammaglobin-a and CD9 positive EVs in ER-negative versus ER-positive breast cancers. Mammaglobin-a positive CD9 platelets were excluded from the data set. n = 2. ±SEM. (B) Schematic images representing an EV with a labelling strategy for biomarker discovery and cell of origin identification using nanoscale flow cytometry. | ||
Discussion
Nanoscale flow cytometry is an emerging technology that can identify and quantitate protein expression on nano-sized particles such as EVs. As EVs are time consuming to isolate by ultracentrifugation or size exclusion chromatography,27,28 nanoscale flow cytometry offers a significant advantage by being able to measure EVs in plasma without isolation or purification. In addition, a 96 well plate format supports its feasibility as a technology for use in high-throughput clinical diagnostic tests. Another powerful aspect of nanoscale flow cytometry is the capability to immunophenotype EVs and identify unique EV populations. Indeed it has been demonstrated by others that prostate cancer derived EVs in patient plasma and in cell line purified EVs can be detected using nanoscale flow cytometry.7,8 Detection and quantification of tumor cell derived EVs in plasma holds great promise for the development of liquid blood-based biopsy test to diagnose, prognosticate, predict, and monitor disease.Our study aimed to optimize nanoscale flow cytometry for the analysis of tissue specific EVs in unpurified plasma. We successfully identified CD9-positive EVs that also expressed the breast enriched protein mammaglobin-a. Further characterization of CD9-mammaglobin-a EVs identified two critical factors important for the analysis of EVs in plasma: (1) The majority of CD9-mammaglobin-a positive EVs were in-fact platelet EVs suggesting that plasma proteins can associate with EV membranes, and (2) Platelet number directly correlated with platelet-derived EV number, thus plasma preparation method affects not only the quantity of platelets but also platelet EVs. Therefore a critical assessment of the quantity of platelet and platelet derived EVs are of upmost importance when comparing between different patient cohorts which have undergone different isolation protocols. This can be expected when carrying out retrospective biomarker discovery studies using plasmas stored by different biobanks or institutions.
Our work has identified important factors to consider when designing experiments for nanoscale flow cytometry analysis of EVs in plasma samples; such as, plasma preparation method, presence of platelets and platelet EVs, the membrane association, or binding, capabilities of biomarker of interest and detection of protein aggregates or particles. The number of platelets in plasma samples was influenced by blood draw volume. Another important consideration is the number of freeze thaw cycles the plasma undergoes. Whilst studies report that serum/plasma can undergo up to four freeze thaw cycles without negative impact upon EVs,29,30 others have found that platelet derived EV levels increase upon freeze/thaw cycles.31 In our study, samples were freeze/thawed three times to achieve an n = 2 for each antibody and one antibody control run. Assessment of the correlation between the two runs was found to be strong suggesting that any changes in EV levels due to freeze/thaw cycles was not significantly affecting our EV levels. However, additional freeze/thaw cycles could potentially result in changes to EV levels. To avoid these challenges, the use of platelet poor plasma would be advisable. However, biobanks have different protocols that they follow during plasma isolation, most of which result in platelet low plasma. The use of platelet poor plasma would reduce high levels of platelets and platelet-derived EVs in plasma; however, it is not always possible to obtain platelet poor plasma especially in retrospective studies using historical biobanks of cancer patient plasmas. Therefore, consideration should be given when assessing plasma samples from different biobanks. This would be of crucial importance if the biomarker of interest is not an integral membrane protein but a secreted protein with membrane binding capacity such as mammaglobin-a which may be able to integrate into, or bind, other membranous structures in plasma.
Through our analysis of mammaglobin-a events we found that nanoscale flow cytometry not only detects EVs and platelets but also protein aggregates. Size exclusion chromatography isolation of EVs and analysis by nanoscale flow cytometry showed, as expected, CD9-positive EVs abundant in EV fraction 1, a minor amount in fraction 2, and negligible amounts detected in the protein rich fractions (fraction 3 and 4). However, mammaglobin-a was detected by nanoscale flow cytometry in each fraction and found in the protein rich fraction by western blot. Furthermore, only a fraction of mammaglobin-a events detected by nanoscale flow cytometry were able to be lysed by Triton-X 100 treatment establishing that mammaglobin-a protein aggregates were detected in plasma by nanoscale flow cytometry. Only a small proportion of mammaglobin-a events were associated with CD9 and identified as bona fide EVs. Additional EV validation strategies such as western blotting, nanoparticle tracking analysis, and electron microscopy were employed to validate that the isolated EVs being analyzed were indeed EVs. Thus, while nanoscale flow cytometry is capable of accurately detecting and quantifying EVs, this technology also detects small particle structures, such as protein aggregates or clusters, and requires additional validation studies to authenticate detected particles as EVs.
Interestingly, while we could detect CD41 and CD63 on platelets we could only detect CD41-positive EVs using nanoscale flow cytometry. CD63 was found on Western blots of EVs purified from plasma suggesting that CD63 is present on EVs but not detectable by nanoscale flow cytometry. There are several reasons as to why CD63 is not detected on EVs by nanoscale flow cytometry. Residual antibody may create high fluorescent background making smaller vesicles difficult to detect above threshold. Alternatively, CD63 EV signal may be hidden by the presence of large numbers of EVs or other particles in the plasma generating a high level of background noise requiring a strong fluorescence signal to break above the detection threshold. Surface expression of CD63 would also an important consideration for detection in complex biofluids. Consideration for the number of CD41 versus CD63 molecules found on an average platelet may explain our results. An average platelet (surface area ∼8 μm2) displays ∼40000 copies of CD41, so a ∼100 nm diameter EV (∼0.04 μm2) would display ∼200 molecules of CD41.6 However, the average number of CD63 molecules per platelet has been estimated to be 2200, meaning that an EV could contain as few as 11 CD63 molecules, far below the detection limits of most flow cytometers.6,32 This means that CD41 EVs will be detected more frequently and will be brighter than CD63 EVs. Indeed, we have found that CD41 EVs are detected by nanoscale flow cytometry, but we are unable to detect CD63 EVs. Thus, nanoscale flow cytometry likely has serious limitations in its sensitivity to low-abundance biomarkers. There may be only a few copies of a given biomarker per EV, and this challenge is exacerbated by the modest brightness and rapid photobleaching of many fluorescent dyes. Overcoming the limitations imposed by low antigen levels and dim fluorescence will likely require the development of new materials or methods that can be implemented into a nanoscale flow cytometry platform.
Conclusion
Nanoscale flow cytometry provides a rapid platform for immunophenotyping and quantitating EVs in complex biofluids. We developed this platform to quantitate the levels of CD9-mammaglobin-a dual positive EVs in breast cancer patient plasma and found elevated levels significantly associated with ER-positive breast cancer. However, nanoscale flow cytometry detected not only mammaglobin-a EVs but also non-EV mammaglobin-a particles highlighting the need for careful validation when using this technique to quantitate EVs. Additionally, by immunophenotyping mammaglobin-a positive EVs, mammaglobin-A positive EVs were found to be CD41 and CD42a positive indicating that they were in-fact platelet EVs. Plasma preparation techniques were found to significantly impact not only the number of platelets but also the number of platelet derived EVs present in the plasma. Therefore, analysis of platelet EVs required plasma sample preparation to be carried out in the same manor for samples to be comparable. Nanoscale flow cytometry analysis of low-abundance EV markers, such as CD63, in whole plasma may be poor but the development of new brighter materials could overcome this challenge. Continued improvements to this technology and our understanding of plasma EV composition could develop this technology for use as a high-through platform in liquid biopsy for cancer.Conflicts of interest
K.C.W has a collaborative research agreement with Beckman Coulter, Inc to provide guidance on optimizing nanoparticle detection; no financial conflict of interest.Acknowledgements
The authors would like to acknowledge the Centre for High-Throughput Phenogenomics group in the Faculty of Dentistry for assistance in acquiring electron microscopy images during this study. The authors also thank Dr. Russ Algar for use of their NanoSight instrument for nanoparticle tracking analysis of extracellular vesicles. This work was supported by the Department of Defense Congressionally Directed Medical Research Programs, Breast Cancer Research Program (W81XWH-16-1-0525), and NSERC Discovery Grants Program. K. C. W holds a Tier 2 Canada Research Chair and a Michael Smith Foundation for Health Research Scholar award. K. K. is a recipient of a UBC Doctoral Recruitment award.References
- G. van Niel, G. D'Angelo and G. Raposo, Shedding light on the cell biology of extracellular vesicles, Nat. Rev. Mol. Cell Biol., 2018, 19(4), 213–228 CrossRef CAS
. PubMed PMID: 29339798. Epub 2018/01/18. eng.
- S. L. N. Maas, X. O. Breakefield and A. M. Weaver, Extracellular Vesicles: Unique Intercellular Delivery Vehicles, Trends Cell Biol., 2017, 27(3), 172–188 CrossRef CAS
- K. B. Johnsen, J. M. Gudbergsson, T. L. Andresen and J. B. Simonsen, What is the blood concentration of extracellular vesicles? Implications for the use of extracellular vesicles as blood-borne biomarkers of cancer, Biochim. Biophys. Acta, Rev. Cancer, 2019, 1871(1), 109–116 CrossRef CAS
- A. Jeyaram and S. M. Jay, Preservation and Storage Stability of Extracellular Vesicles for Therapeutic Applications, AAPS J., 2017, 20(1), 1 Search PubMed
- A. Morales-Kastresana, B. Telford, T. A. Musich, K. McKinnon, C. Clayborne and Z. Braig,
et al., Labeling Extracellular Vesicles for Nanoscale Flow Cytometry, Sci. Rep., 2017, 7(1), 1878 CrossRef
- J. P. Nolan and J. C. Jones, Detection of platelet vesicles by flow cytometry, Platelets, 2017, 28(3), 256–262 CrossRef CAS
- R. S. Padda, F. K. Deng, S. I. Brett, C. N. Biggs, P. N. Durfee and C. J. Brinker,
et al., Nanoscale flow cytometry to distinguish subpopulations of prostate extracellular vesicles in patient plasma, Prostate, 2019, 79(6), 592–603 CrossRef CAS
- A. Morales-Kastresana, T. A. Musich, J. A. Welsh, W. Telford, T. Demberg and J. C. S. Wood,
et al., High-fidelity detection and sorting of nanoscale vesicles in viral disease and cancer, J. Extracell. Vesicles, 2019, 8(1), 1597603 CrossRef CAS
- G. C. T. Brittain, Y. Q. Chen, E. Martinez, V. A. Tang, T. M. Renner and M. A. Langlois,
et al., A Novel Semiconductor-Based Flow Cytometer with Enhanced Light-Scatter Sensitivity for the Analysis of Biological Nanoparticles, Sci. Rep., 2019, 9(1), 16039 CrossRef
- J. A. Welsh, E. Van Der Pol, G. J. A. Arkesteijn, M. Bremer, A. Brisson and F. Coumans,
et al., MIFlowCyt-EV: a framework for standardized reporting of extracellular vesicle flow cytometry experiments, J. Extracell. Vesicles, 2020, 9(1), 1713526 CrossRef CAS
- M. A. Watson and T. P. Fleming, Mammaglobin, a mammary-specific member of the uteroglobin gene family, is overexpressed in human breast cancer, Cancer Res., 1996, 56(4), 860–865 CAS
- M. Zafrakas, B. Petschke, A. Donner, F. Fritzsche, G. Kristiansen and R. Knuchel,
et al., Expression analysis of mammaglobin A (SCGB2A2) and lipophilin B (SCGB1D2) in more than 300 human tumors and matching normal tissues reveals their co-expression in gynecologic malignancies, BMC Cancer, 2006, 6, 88 CrossRef
- Z. Wang, B. Spaulding, A. Sienko, Y. Liang, H. Li and G. Nielsen,
et al., Mammaglobin, a valuable diagnostic marker for metastatic breast carcinoma, Int. J. Clin. Exp. Pathol., 2009, 2(4), 384–389 CAS
- M. A. Watson, S. Dintzis, C. M. Darrow, L. E. Voss, J. DiPersio and R. Jensen,
et al., Mammaglobin expression in primary, metastatic, and occult breast cancer, Cancer Res., 1999, 59(13), 3028–3031 CAS
- J. H. Han, Y. Kang, H. C. Shin, H. S. Kim, Y. M. Kang and Y. B. Kim,
et al., Mammaglobin expression in lymph nodes is an important marker of metastatic breast carcinoma, Arch. Pathol. Lab. Med., 2003, 127(10), 1330–1334 CAS
- Y. Hu, P. Liu, D. Wu and Y. Jiang, Prognostic role of plasma mammaglobin A expression in breast carcinoma patients: a meta-analysis, OncoTargets Ther., 2018, 11, 3245–3255 CrossRef
- G. W. Lee, J. Y. Kim, E. H. Koh, D. Kang, D. S. Choi and K. Y. Maeng,
et al., Plasma human mammaglobin mRNA associated with poor outcome in patients with breast cancer, Genet. Mol. Res., 2012, 11(4), 4034–4042 CrossRef CAS
- P. Ferro, M. C. Franceschini, B. Bacigalupo, P. Dessanti, E. Falco and V. Fontana,
et al., Detection of circulating tumour cells in breast cancer patients using human mammaglobin RT-PCR: association with clinical prognostic factors, Anticancer Res., 2010, 30(6), 2377–2382 CAS
- S. Zhao, H. Yang, M. Zhang, D. Zhang, Y. Liu and Y. Liu,
et al., Circulating tumor cells (CTCs) detected by triple-marker EpCAM, CK19, and hMAM RT-PCR and their relation to clinical outcome in metastatic breast cancer patients, Cell Biochem. Biophys., 2013, 65(2), 263–273 CrossRef CAS
- J. L. Bernstein, J. H. Godbold, G. Raptis, M. A. Watson, B. Levinson and S. A. Aaronson,
et al., Identification of mammaglobin as a novel serum marker for breast cancer, Clin. Cancer Res., 2005, 11(18), 6528–6535 CrossRef CAS
- L. Zuo, L. Li, Q. Wang, T. P. Fleming and S. You, Mammaglobin as a potential molecular target for breast cancer drug delivery, Cancer Cell Int., 2009, 9, 8 CrossRef
- N. K. Tafreshi, S. A. Enkemann, M. M. Bui, M. C. Lloyd, D. Abrahams and A. S. Huynh,
et al., A mammaglobin-A targeting agent for noninvasive detection of breast cancer metastasis in lymph nodes, Cancer Res., 2011, 71(3), 1050–1059 CrossRef CAS
- S. W. Kim, P. Goedegebuure and W. E. Gillanders, Mammaglobin-A is a target for breast cancer vaccination, OncoImmunology, 2016, 5(2), e1069940 CrossRef
- N. Arraud, R. Linares, S. Tan, C. Gounou, J. M. Pasquet and S. Mornet,
et al., Extracellular vesicles from blood plasma: determination of their morphology, size, phenotype and concentration, J. Thromb. Haemostasis, 2014, 12(5), 614–627 CrossRef CAS
- H. F. Heijnen, A. E. Schiel, R. Fijnheer, H. J. Geuze and J. J. Sixma, Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules, Blood, 1999, 94(11), 3791–3799 CrossRef CAS
- X. F. Guan, M. K. Hamedani, A. Adeyinka, C. Walker, A. Kemp and L. C. Murphy,
et al., Relationship between mammaglobin expression and estrogen receptor status in breast tumors, Endocrine, 2003, 21(3), 245–250 CrossRef CAS
- A. N. Boing, E. van der Pol, A. E. Grootemaat, F. A. Coumans, A. Sturk and R. Nieuwland, Single-step isolation of extracellular vesicles by size-exclusion chromatography, J. Extracell. Vesicles, 2014, 3(1), 23430 CrossRef
- C. Thery, S. Amigorena, G. Raposo and A. Clayton, Isolation and characterization of exosomes from cell culture supernatants and biological fluids, Curr. Protoc. Cell Biol., 2006, 22 Search PubMed
- R. Baek, E. K. Sondergaard, K. Varming and M. M. Jorgensen, The impact of various preanalytical treatments on the phenotype of small extracellular vesicles in blood analyzed by protein microarray, J. Immunol. Methods, 2016, 438, 11–20 CrossRef CAS
- M. Jayachandran, V. M. Miller, J. A. Heit and W. G. Owen, Methodology for isolation, identification and characterization of microvesicles in peripheral blood, J. Immunol. Methods, 2012, 375(1–2), 207–214 CrossRef CAS
- L. Ayers, M. Kohler, P. Harrison, I. Sargent, R. Dragovic and M. Schaap,
et al., Measurement of circulating cell-derived microparticles by flow cytometry: sources of variability within the assay, Thromb. Res., 2011, 127(4), 370–377 CrossRef CAS
- M. B. Protty, N. A. Watkins, D. Colombo, S. G. Thomas, V. L. Heath and J. M. Herbert,
et al., Identification of Tspan9 as a novel platelet tetraspanin and the collagen receptor GPVI as a component of tetraspanin microdomains, Biochem. J., 2009, 417(1), 391–400 CrossRef CAS
- B. S. Batista, W. S. Eng, K. T. Pilobello, K. D. Hendricks-Muñoz and L. K. Mahal, Identification of a conserved glycan signature for microvesicles, J. Proteome Res., 2011, 10(10), 4624–4633, DOI:10.1021/pr200434y
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr05525e |
| This journal is © The Royal Society of Chemistry 2021 |