
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchical approach for the rational construction of helix-containing nanofibrils using α,β-peptides†
Monika
Szefczyk
 *a,
Natalia
Szulc
b,
Marlena
Gąsior-Głogowska
b,
Anna
Modrak-Wójcik
c,
Agnieszka
Bzowska
c,
Wojciech
Majstrzyk
d,
Michał
Taube
e,
Maciej
Kozak
ef,
Teodor
Gotszalk
d,
Ewa
Rudzińska-Szostak
a and
Łukasz
Berlicki
*a
*a,
Natalia
Szulc
b,
Marlena
Gąsior-Głogowska
b,
Anna
Modrak-Wójcik
c,
Agnieszka
Bzowska
c,
Wojciech
Majstrzyk
d,
Michał
Taube
e,
Maciej
Kozak
ef,
Teodor
Gotszalk
d,
Ewa
Rudzińska-Szostak
a and
Łukasz
Berlicki
*a
aDepartment of Bioorganic Chemistry, Faculty of Chemistry, Wroclaw University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: monika.szefczyk@pwr.edu.pl; lukasz.berlicki@pwr.edu.pl
bDepartment of Biomedical Engineering, Faculty of Fundamental Problems of Technology, Wroclaw University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
cDivision of Biophysics, Faculty of Physics, Institute of Experimental Physics, University of Warsaw, Ludwika Pasteura 5, 02-093 Warsaw, Poland
dFaculty of Microsystem Electronics and Photonics, Wroclaw University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
eDepartment of Macromolecular Physics, Adam Mickiewicz University, Uniwersytetu Poznańskiego 2, 61-614 Poznań, Poland
fNational Synchrotron Radiation Centre SOLARIS, Jagiellonian University, Czerwone Maki 98, 30-392 Kraków, Poland
First published on 13th January 2021
Abstract
The rational design of novel self-assembled nanomaterials based on peptides remains a great challenge in modern chemistry. A hierarchical approach for the construction of nanofibrils based on α,β-peptide foldamers is proposed. The incorporation of a helix-promoting trans-(1S,2S)-2-aminocyclopentanecarboxylic acid residue in the outer positions of the model coiled-coil peptide led to its increased conformational stability, which was established consistently by the results of CD, NMR and FT-IR spectroscopy. The designed oligomerization state in the solution of the studied peptides was confirmed using analytical ultracentrifugation. Moreover, the cyclopentane side chain allowed additional interactions between coiled-coil-like structures to direct the self-assembly process towards the formation of well-defined nanofibrils, as observed using AFM and TEM techniques.
Introduction
The rational design of self-assembled nanostructures based on peptides has recently attracted much attention.1,2 In this context, helical structures possess numerous advantages, including rational design, high conformational stability and the possibility of a wide range of modifications that could lead to the formation of higher-order structures with different complexities and functions.3,4 A coiled-coil is one of the most widely studied helix-containing peptide motifs because of the well-understood relationship between its sequence and structure,5–7 which allows the construction of several nanostructures, i.e., bundles,8 nanofibers,9 nanotubes10 or cages.11 Fibrous nanostructures based on coiled-coil peptides have been most often obtained by the formation of salt bridges, disulfide bonds or complementary acidic/basic components.12 However, the construction of coiled-coil-based nanofibers exploiting hydrophobic interactions between constituent elements remains virtually unexplored.Rational and effective control over the three-dimensional structures of peptides can be achieved by incorporating conformationally constrained amino acid residues. Among numerous approaches, the application of cycloalkane-based β-amino acids has already been proven to be very successful, and several highly stable helical conformations have been found for peptides containing cyclopropane, cyclobutane, cyclopentane and cyclohexane β-amino acids.13–15 Furthermore, peptides containing β-amino acid residues have been used for the construction of supramolecular self-assemblies, including bundles and coiled-coils.15–20 Such peptides have also been proven to self-associate to form various structures including: hydrogels stable for proteolysis,21,22 liquid crystals23 or the vitamin B derivatives releasing nanoparticles.24
The methodology for the rational construction of secondary structures based on α,β-peptides, in particular helices, has already been elaborated25,26 and is based on the tendency of individual residues to form ψ and ϕ dihedral angles. Stereochemical patterning indicates that a helix will be obtained if the ψ and ϕ dihedral angles flanking the amide bond are of the same sign.27 However, rational methodologies for the construction of higher-order structures or assemblies using α,β-peptides are scarce. The most commonly used approach is based on α → β replacement, assuming that the incorporation of a β-amino acid is well tolerated within a α-helix with local distortions.28,29 Several α,β-peptides have been investigated,30 including modifications of the coiled-coil motif.31
In this paper, we investigate the possibility of rationally constructing nanostructures using α,β-peptides. We are expanding the methodology of α → β replacement in such a way that the incorporation of a constrained β-amino acid residue not only favors the formation of a structure analogous to the original one but also induces the development of larger, nanometer-size structures. In particular, we investigated helical oligomers based on coiled-coil motifs and modified them with cyclic β-amino acids, namely, trans-(1S,2S)-2-aminocyclopentanecarboxylic acid (trans-ACPC). We assume that the incorporation of a helix-favoring trans-ACPC residue will increase the propensity of the obtained peptides for coiled-coil-like structure formation. Moreover, appropriate positioning of trans-ACPC residues on the coiled-coil surface provides the possibility of additional hydrophobic interactions between coiled-coil units to form nanostructures. The designed peptides were obtained by automated solid-phase peptide synthesis and evaluated both in solution: circular dichroism (CD), analytical ultracentrifugation (AUC) and nuclear magnetic resonance spectroscopy (NMR) and in solid state: Fourier-transform infrared spectroscopy (FT-IR), Raman spectroscopy, atomic force microscopy (AFM), transmission electron microscopy (TEM) and wide-angle X-ray scattering (WAXS).
Results and discussion
Design
Hydrophobically driven self-assembly is promoted by the precise arrangements of lipophilic elements on the peptide surface and their specific associations at intermolecular contacts, as in the coiled-coil motif.32 These twisted left-handed supercoiled structures are composed of helices built up with a (HPPHPPP)n repeat pattern, usually denoted as abcdefg.33 Hydrophobic residues (H) at positions a and d are important to establish the tight knobs into hole packing and control the oligomer state of the coiled-coil, while polar residues (P) at positions e and g are responsible for the formation of salt bridges between the helices.6,7 Positions b, c and f also play a role in determining the stability of coiled-coils.34 Following these general rules governing the design of coiled-coil motifs, we endeavored to construct a trimeric coiled-coil with three heptad repeats to be a model peptide 1 (Fig. 1A). Thus, isoleucine was introduced at positions a and d, as it is a hydrophobic residue that ensures trimer formation.5 Lysine and glutamic acid were incorporated at positions e and g, respectively, since they can assure additional structure stabilization by the formation of salt bridges. Alanine was chosen to occupy the b and c positions, as this is a small, helix-favoring amino acid that prevents unexpected influences of side-chain interactions. Glutamine, a polar but charge-neutral amino acid, or tryptophan, a UV chromophore, was introduced in the most external f position. Additionally, both ends of the peptide were capped with a glycine residue, and the sequence was appended with a two-residue sequence outside the helical region, in reference to the results of a similar but longer peptide synthesized by Fletcher and coworkers.35 Additionally, the N-terminus was acetylated to reduce the net charge. | ||
| Fig. 1 (A) Helical wheel representing the model coiled-coil peptide 1. (B) The sequences of designed peptides, where the red pentagon denotes the trans-2-aminocyclopentanecarboxylic acid (trans-ACPC) residue. The letter in the peptide name indicates the position in the model coiled-coil peptide where the β-amino acid was introduced. | ||
Subsequently, model peptide 1 was subjected to further modifications. First, we performed single α → cyclic β residue replacement of the chosen position in every heptad repeat of model peptide 1 with trans-2-aminocyclopentanecarboxylic acid (trans-ACPC), producing seven α,β-peptides (Fig. 1B). The influence of this structurally constrained residue on the conformational behavior of helical structures and coiled-coil arrangements in solution as well as possible nanostructures in the solid state will be evaluated. We presumed that trans-ACPC residues present in the outer positions (b, c and f) of the coiled-coil will provide the additional possibility of interactions between coiled-coil units to favor the formation of larger, nanometer-size structures.
Synthesis and structure elucidation
All designed peptides were successfully synthesized using an automated peptide synthesizer with microwaves by applying Fmoc chemistry and purified using a preparative HPLC system with a C18 column. Initially, circular dichroism (CD) spectroscopy was used to elucidate the secondary structure and conformational stability of the obtained peptides. The CD spectrum of model peptide 1 shows two minima, at 208 and 222 nm, typical for the α-helix (Fig. 2A). The ratio molar ellipticities at the abovementioned wavelengths of θ222/θ208 equals 1, and θ222 equals ca. −40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deg cm2 dmol−1, indicating a near-complete α-helix structure.36 The obtained α,β-peptides show two different types of conformational behavior, which depend on the placement of cyclic β residues in the sequence. The CD spectra of peptides 1_b, 1_c and 1_f (dissolved in water, pH 7) have two minima, at 208 and 222 nm, but θ222/θ208 < 1, equal to 0.4, 0.7 and 0.7, respectively, indicating the presence of structures similar to the α-helix but disturbed by the presence of the β residue (Fig. 2A). In the case of peptides 1_c and 1_f, the CD spectra show a significant Cotton effect, which suggests the formation of well-defined secondary structures. Very similar CD spectra were also observed for peptides dissolved in methanol (Fig. S3_1†). The CD spectra of peptides 1_a, 1_d, 1_e and 1_g show a minimum in the 204–205 nm range (Fig. 2A), which indicates a random coil.
000 deg cm2 dmol−1, indicating a near-complete α-helix structure.36 The obtained α,β-peptides show two different types of conformational behavior, which depend on the placement of cyclic β residues in the sequence. The CD spectra of peptides 1_b, 1_c and 1_f (dissolved in water, pH 7) have two minima, at 208 and 222 nm, but θ222/θ208 < 1, equal to 0.4, 0.7 and 0.7, respectively, indicating the presence of structures similar to the α-helix but disturbed by the presence of the β residue (Fig. 2A). In the case of peptides 1_c and 1_f, the CD spectra show a significant Cotton effect, which suggests the formation of well-defined secondary structures. Very similar CD spectra were also observed for peptides dissolved in methanol (Fig. S3_1†). The CD spectra of peptides 1_a, 1_d, 1_e and 1_g show a minimum in the 204–205 nm range (Fig. 2A), which indicates a random coil.
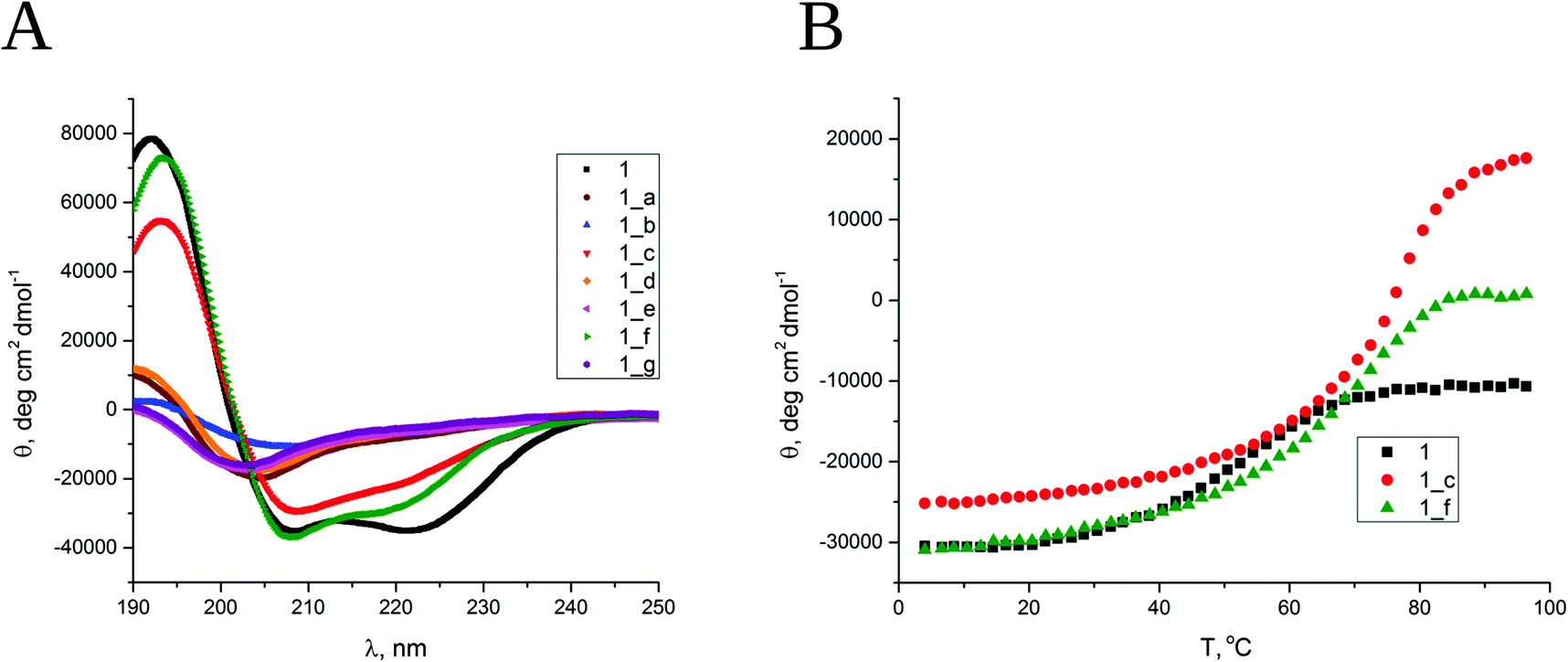 | ||
| Fig. 2 (A) CD spectra of all obtained peptides dissolved in water, pH 7. (B) Thermal unfolding curves followed by CD signal at 208 nm. Cpep = 80 μM. | ||
The conformational stability was examined for helical peptides, namely, 1, 1_b, 1_c and 1_f. The changes in CD spectra were followed in the range of 190–240 nm by a gradual increase in temperature from 4 to 98 °C (Fig. S3_1†). Thermal unfolding curves followed at 222 nm (Fig. 2B) were used to determine the melting temperatures (Table S3_2†). As expected, the short coiled-coil peptide 1 has moderate thermal stability in water with a cooperative melting transition at 56 °C. The obtained α,β-peptides also demonstrate cooperative melting transitions but at significantly higher temperatures, 76 °C and 71 °C for 1_c and 1_f, respectively. Peptide 1_b strongly aggregates under the measurement conditions (increase in the temperature), so its melting temperature was not possible to determine. The conformational stability of the peptides was additionally confirmed in the presence of 3 M guanidine hydrochloride, which was used as a denaturing agent, and it was observed that the obtained α,β-peptides exhibit higher conformational stability than peptide 1 under these conditions (Fig. S3_3†). In summary, the results of CD spectroscopy indicate the formation of well-ordered structures for peptides 1_c and 1_f that exceed the stability of model short coiled-coil peptide 1. Moreover, peptide 1_b shows a partially folded helical structure. Importantly, these results correlate well with the trans-ACPC substitution pattern. Only peptides with trans-ACPC placed in position designed to be external in the coiled-coil show the formation of a helix. The substitution of positions a, d, e and g that form hydrophobic cores or surface hydrogen bond networks leads to complete destruction of the helix.
To elucidate the three-dimensional structure in solution of peptides 1_b, 1_c and 1_f, detailed NMR analysis applying 2D TOCSY and 2D ROESY spectra was performed. Due to the insufficient solubility in water, the measurements for α,β-peptides were performed in methanol-d3 solution. Spectral analysis allowed the assignment of all resonances in all measured peptides and the detection of numerous nonsequential contacts between residues (Table S4†), namely, i → (i − 2), i → (i − 3) and i → (i − 4), which indicate the presence of stable helical structures. In the 2D ROESY spectra of all studied peptides, the most abundant are medium range i → (i − 3) contacts (HA-HN, HA-HA and HA-HB). Moreover, peptide 1_f exhibited a significantly higher number of i → (i − 3) nonsequential interproton contacts than two other peptides (36 versus 22 in 1_c and 19 in 1_b). Using NMR restraints (interproton distances and J coupling constants), the three-dimensional structures of peptides 1_b, 1_c and 1_f were modeled (Fig. 3 and S4_1†). As expected, the peptide 1_f is fully helical, with a structure highly similar to α-helix stabilized by the network of i → (i − 4) hydrogen bonds. Although the overall structure is slightly bent due to the disturbances observed at the N-terminal side of each trans-ACPC residue, where some hydrogen bonds are elongated or broken, the whole structure is very well defined. Structures of peptide 1_b and 1_c are also helical, although their C-terminal part is less defined then in case of peptide 1_f (Fig. S4_1†). Moreover, the structure of peptide 1_f trimer was modelled (Fig. S4_2†) and it was found that overall structure is highly similar to native coiled coil with small changes in conformation close to β-amino acid residues.
 | ||
| Fig. 3 (A) Average NMR structure and (B) superimposition of the ten lowest energy structures obtained for peptide 1_f. | ||
Aggregate formation
Analytical ultracentrifugation (AUC) was used to verify the oligomerization state of the obtained peptides in solution. For all peptides, sedimentation velocity experiments were carried out in water and in AUC buffer (see Experimental section), except for 1_f, which strongly aggregates in the buffer. For peptide 1, sedimentation equilibrium experiments in AUC buffer were also conducted. The c(s) distributions, resulting from the analysis of radial absorption profiles for peptide 1 (Fig. S5_1†), contain a single peak at 0.85–0.97 S slightly depending on the concentration and solvent (s-values are lower in water; Fig. 4 and Table 1). The value of the sedimentation coefficient corresponds to an intermediate mass between the mass of the dimer and trimer in water, and it increases slightly as the concentration of the peptide increases, which indicates that peptide 1 exists as a trimer in rapid equilibrium with smaller oligomeric forms. The value of the sedimentation coefficient obtained for 100 μM solution in AUC buffer corresponds clearly to that calculated for a trimer. The best fit to the radial concentration gradients is best described by dimer-tetramer and monomer–dimer–tetramer self-association models, but self-association models with the trimeric form, namely, monomer–trimer and monomer–dimer–trimer, show only slightly higher χ2 and similar residual plots (Fig. S5_2 and Table S5_3†). The model assuming a single species indicates that the mass between the dimer and trimer and the fit of this model has a higher χ2 value than the self-association models listed above (Table S5_3†). Taken together, the results from sedimentation velocity and equilibrium methods indicate that peptide 1 contains a mixture of different oligomeric forms in rapid equilibrium, but at a concentration of at least 100 μM, the trimeric form prevails.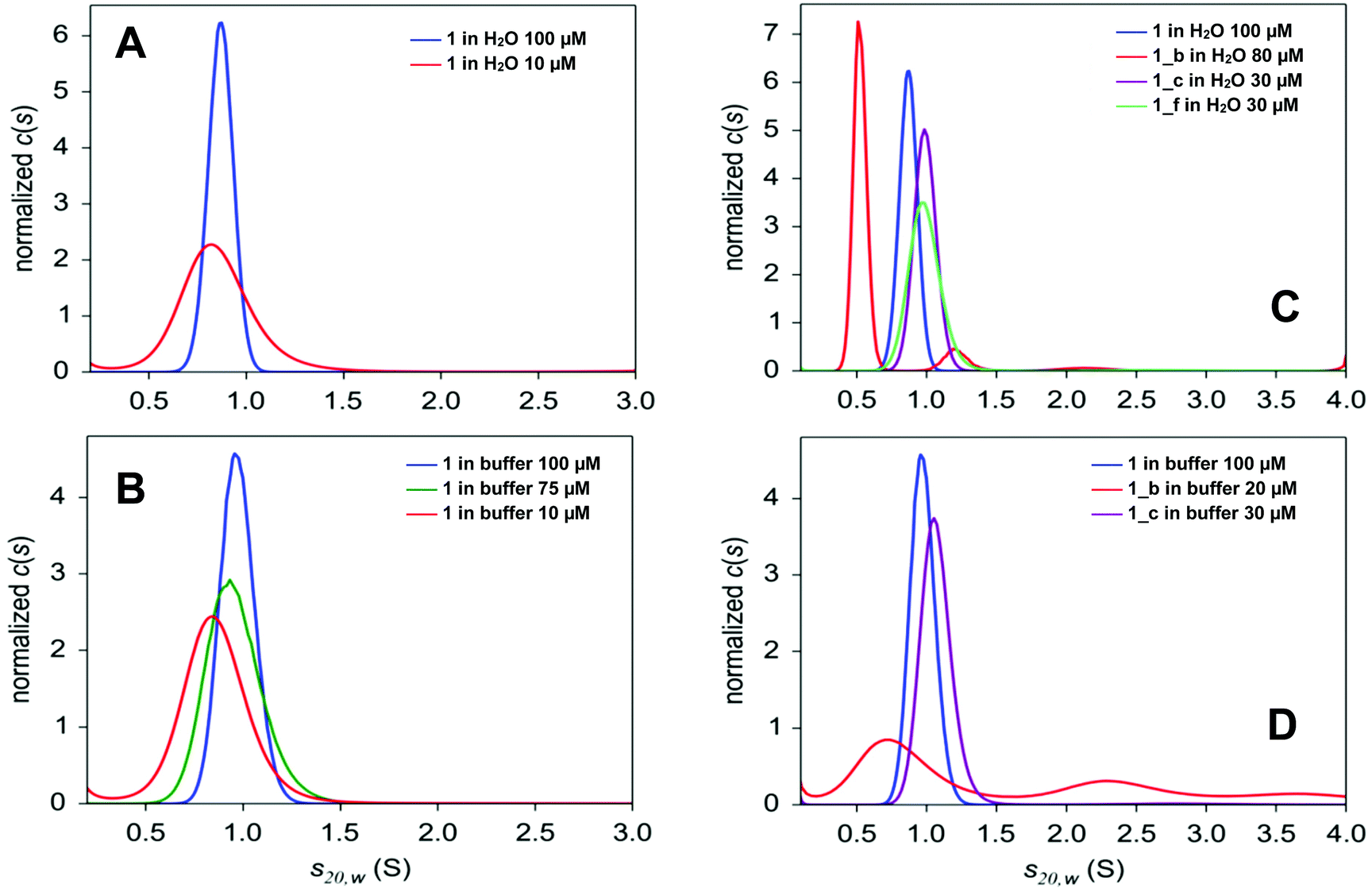 | ||
| Fig. 4 Normalized sedimentation coefficient distributions c(s20,w) for different concentrations of peptide 1 obtained in water (A), in 50 mM phosphate pH 7.0 and 50 mM NaCl (B), as well as for peptides 1, 1_b, 1_c and 1_f in water (C), in 50 mM phosphate pH 7.0 and 50 mM NaCl (D). Centrifugation was performed at 50000 rpm and 20 °C. Radial profiles were measured at 5- or 10 min intervals at 280 nm. | ||
| Peptide | [c] (μM) | Solvent b | s 20,w (S) (content %)c | f/f0d | Dominant oligomerization state (theoretical s20,w value)e |
|---|---|---|---|---|---|
|
a 50000 rpm, scans every 5 or 10 min, 20 °C, A (280 nm), duration of experiment ∼24 h.
b Buffer: 50 mM potassium phosphate pH 7.0 + 50 mM NaCl.
c When more than one species is present in the c(s) distribution, their s20,w and percentage are given.
d Values of f/f0 determined for the peptides dissolved in H2O may be biased by electrostatic effects caused by the fact that charges of peptides are not shielded.
e Theoretical values of sedimentation coefficients in the standard conditions (s20,w) for various oligomeric forms of studied peptides are presented in Table S5_E.†
f In brackets concentration of peptide in the AUC centerpiece observed after centrifugation of large aggregates.
g In the c(s) distribution third peak is observed, s = 2.1 S (3%).
h In the c(s) distribution, two more peaks are observed ∼1.1 S (7%) i ∼2.1 S (1%) (not well separated from each other and from the main peak).
i Peaks are not well separated; in the c(s) distribution, two more, not well separated peaks are present, spread up to ∼10 S (max ∼3.5 and 6.5 S, 13% and 7%, respectively); when the range 0–15 S is analyzed, large oligomers are visible, exhibiting a continuous distribution of s.
|
|||||
| 1 | 100 | H2O | 0.868 ± 0.065 | 1.078 | Dimer/trimer (0.768/1.006) |
| 10 | 0.853 ± 0.194 | 1.492 | Dimer/trimer (0.768/1.006) | ||
| 100 | Buffer | 0.972 ± 0.088 | 1.213 | Trimer (1.006) | |
| 75 | 0.957± 0.147 | 1.260 | Trimer (1.006) | ||
| 10 | 0.860 ± 0.176 | 1.289 | Dimer/trimer (0.768/1.006) | ||
| 1_b | 80 (80)f | H2O | 0.524 ± 0.049 (87) | 1.521 | Dimer (0.562) |
| 1.214 ± 0.100 (10)g | Hexamer (1.170) | ||||
| 30 (25) | 0.514 ± 0.098 (92)h | 1.695 | Dimer (0.562) | ||
| 80 (20) | Buffer | 0.812 ± 0.295 (50) | 1.831 | Trimer/tetramer (0.737/0.893) | |
| 2.346 ± 0.371 (26)i | Bundles | ||||
| 1_c | 38 (32) f | H2O | 0.989 ± 0.080 | 1.161 | Trimer (0.972) |
| 38 (27) | Buffer | 1.069 ± 0.111 | 1.271 | Trimer (0.972) | |
| 1_f | 31 | H2O | 0.988 ± 0.115 | 1.138 | Trimer (0.945) |
The α,β-peptides are not well soluble in water and are even less soluble in AUC buffer. They are also much less stable in solution compared to peptide 1, so only sedimentation velocity experiments were performed in their case. The c(s) distributions obtained for water solutions of peptides 1_c and 1_f (Fig. 4C) contain a single peak, similar to peptide 1. Both peptides, however, sediment faster (s ∼1 S) than the model peptide (s ∼0.9 S). These values of the sedimentation coefficient correspond to the trimeric forms of 1_c and 1_f (Table 1). Unlike the peptides described above, peptide 1_b dissolved in water and in AUC buffer sediments, giving a c(s) distribution with several peaks (Fig. 4C and D). Moreover, its hydrodynamic parameters are different in water and in the AUC buffer. The species with a sedimentation coefficient of 0.5 S (attributed to dimer) is dominant in water (Fig. 4C and Table 1). Small amounts of larger oligomers are detected at 1.2 S (hexamer, 7–10%) and 2.1 S (bundles, 1–3%). The content of the oligomeric forms depends on peptide concentration in the sample, which indicates that major small species (dimer) are in equilibrium with higher oligomers.37 The AUC buffer promotes the formation of large aggregates of 1_b. Only 20% of the peptide is present as small oligomers, which can be detected at a rotor speed of 50000 rpm (Table 1). Among them, half (50%) are in arrangements with a sedimentation coefficient of 0.85 S (trimer). Larger oligomers at 2.4 S (26%), 3.5 S (13%) and 6.5 S (7%) are also visible. The peaks observed in the c(s) distribution are broad and not well separated (Fig. 4D), indicating that several types of large oligomers are in rapid equilibrium. The high value of the friction ratio, f/f0, is another difference between 1_b and other peptides (f/f0 ∼1.7, compared with f/f0 ∼1.25 for 1, 1_c and 1_f; Table 1), which indicates deviation from the globular shape for 1_b.
Sedimentation velocity experiments proved the presence of higher oligomers in the case of all studied peptides. The analysis of radial profiles served to determine sedimentation coefficients, which match for the trimer model 1 and for the designed peptides 1_c and 1_f, while peptide 1_b showed a complicated mixture of oligomers. Therefore, α → cyclic β residue replacement of outer positions c and f of the coiled-coil does not interfere significantly with its oligomerization state in solution, while the substitution of position b increases the aggregation propensity, and larger aggregates are observed in solution.
In addition to AUC experiments, the thioflavin T (ThT) assay was applied for the analysis of aggregate formation in solution.38–40 The ThT assay performed for model (1) and α,β-peptides (1_b, 1_c and 1_f) showed increased fluorescence intensity compared to the ThT dye alone immediately after peptide dissolution (Table S6†), suggesting the formation of aggregates in each case. Slight enhancement of the fluorescence was observed for peptide 1 (relative fluorescence of 20.5), while α,β-peptides showed a significant increase in relative fluorescence (values of 5831, 269 and 119 for 1_b, 1_c and 1_f, respectively). Subsequently, the kinetic studies were performed as the ThT fluorescence assay was often used for kinetic studies of amyloid or collagen fibrils formation.41–44 In case of peptide 1_b and 1_c sigmoidal shape of fluorescence signal was observed (Table S6†) with lag time of 4 h and 2 h, respectively. The lag phase indicates the unfavorable energetic state of peptide which corresponds to not sufficient amount of nuclei for the next phase. Peptides 1_b and 1_c showed similar kinetics of fibril formation with τ equal to 75 min and 90 min. In case of peptide 1_f slow increase of fluorescence signal was observed, indicating that fibril formation in solution is very slow. The difference between peptides 1_b/1_c and 1_f can be attributed to the fact that β-residues in peptides 1_b and 1_c are located at the same turn of the helix, while in case of peptide 1_f, beta-residue is on the subsequent turn. Therefore, inter-helical interactions involving β-residues are similar for 1_b and 1_c and different for 1_f. Even though the peptide 1 showed a slight enhancement in the ThT emission, the fibrils were not observed under AFM and TEM. Binding to peptide 1 may be caused by unspecificity of the THT dye, which can bind to many structures, such as: nucleic acid, keratin, collagen.
The secondary structures of peptides 1, 1_b, 1_c and 1_f were studied by μIR, ATR-FTIR and FT-Raman techniques both in powder form and in solution (Table S7_1†). The amide I band (1700–1600 cm−1) characteristic of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the peptide backbone is sensitive to conformational changes.45,46 All the studied peptides possess the maximum amide I band in the range 1660–1650 cm−1 (Tables S7_1 and S7_2†) measured for powder form, which is typical for α-helical peptides. However, ATR-FTIR (Fig. S7_3†) and FT-Raman (Fig. S7_3†) spectra indicate slightly different amide I band positions for particular peptides, most likely due to the differences in the helical packing of amino acid chains.40,47,48 Therefore, α,β-peptides show maxima of the amide I band at lower frequencies than model peptide 1, and the presence of trans-ACPC in sequence caused significant broadening of amide I bands and an increase in full width at half maximum (FWHM). More accurate information about the structure is usually provided by the second derivative and decomposition of the amide I band into sub-bands. The deconvolved spectra were fitted with Voigtian or Lorentzian bands for ATR-FTIR and FT-Raman, respectively.49 In the ATR-FTIR spectra of peptides 1_b, 1_c and 1_f, instead of one dominant helical band in the amide I range, two components are clearly visible (Fig. 5), signifying a more complex structure. The lower band with a maximum near 1630 cm−1 is usually associated with aggregates.50 We propose to assign the amide I band component at 1630 cm−1 to hydrogen-bonded carbonyl groups and the higher band (at 1675 cm−1) to free CO groups of exposed amino acids into the external environment. Trans-ACPC alone possesses an absorption band at 1691 cm−1 (Fig. S7_4†) and affects the area of the band with a maximum at 1675 cm−1 present in α,β-peptides. Deconvolution of the amide I band in the FT-Raman spectra also reveals the presence of few components in the range of 1700–1600 cm−1 (Fig. 6) with origins similar to those in the FTIR spectra. The broad peak at 1615 cm−1 arises from the modes of the aromatic amino acids.51
O stretching vibrations of the peptide backbone is sensitive to conformational changes.45,46 All the studied peptides possess the maximum amide I band in the range 1660–1650 cm−1 (Tables S7_1 and S7_2†) measured for powder form, which is typical for α-helical peptides. However, ATR-FTIR (Fig. S7_3†) and FT-Raman (Fig. S7_3†) spectra indicate slightly different amide I band positions for particular peptides, most likely due to the differences in the helical packing of amino acid chains.40,47,48 Therefore, α,β-peptides show maxima of the amide I band at lower frequencies than model peptide 1, and the presence of trans-ACPC in sequence caused significant broadening of amide I bands and an increase in full width at half maximum (FWHM). More accurate information about the structure is usually provided by the second derivative and decomposition of the amide I band into sub-bands. The deconvolved spectra were fitted with Voigtian or Lorentzian bands for ATR-FTIR and FT-Raman, respectively.49 In the ATR-FTIR spectra of peptides 1_b, 1_c and 1_f, instead of one dominant helical band in the amide I range, two components are clearly visible (Fig. 5), signifying a more complex structure. The lower band with a maximum near 1630 cm−1 is usually associated with aggregates.50 We propose to assign the amide I band component at 1630 cm−1 to hydrogen-bonded carbonyl groups and the higher band (at 1675 cm−1) to free CO groups of exposed amino acids into the external environment. Trans-ACPC alone possesses an absorption band at 1691 cm−1 (Fig. S7_4†) and affects the area of the band with a maximum at 1675 cm−1 present in α,β-peptides. Deconvolution of the amide I band in the FT-Raman spectra also reveals the presence of few components in the range of 1700–1600 cm−1 (Fig. 6) with origins similar to those in the FTIR spectra. The broad peak at 1615 cm−1 arises from the modes of the aromatic amino acids.51
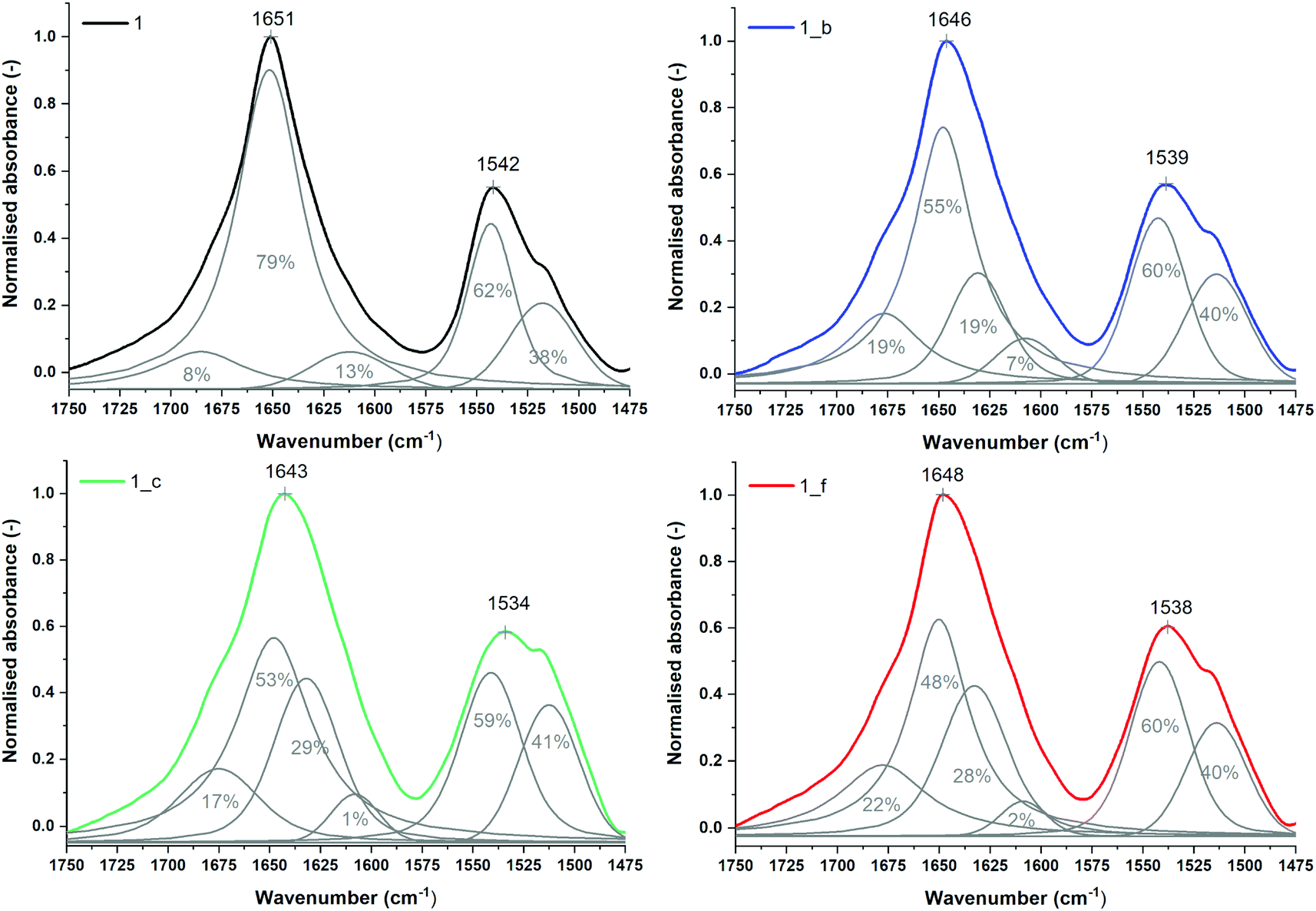 | ||
| Fig. 5 Normalized ATR-FTIR spectra of powder peptides with sub-bands and percentage area values obtained from curve fitting based on their second derivatives (amide I and II regions). | ||
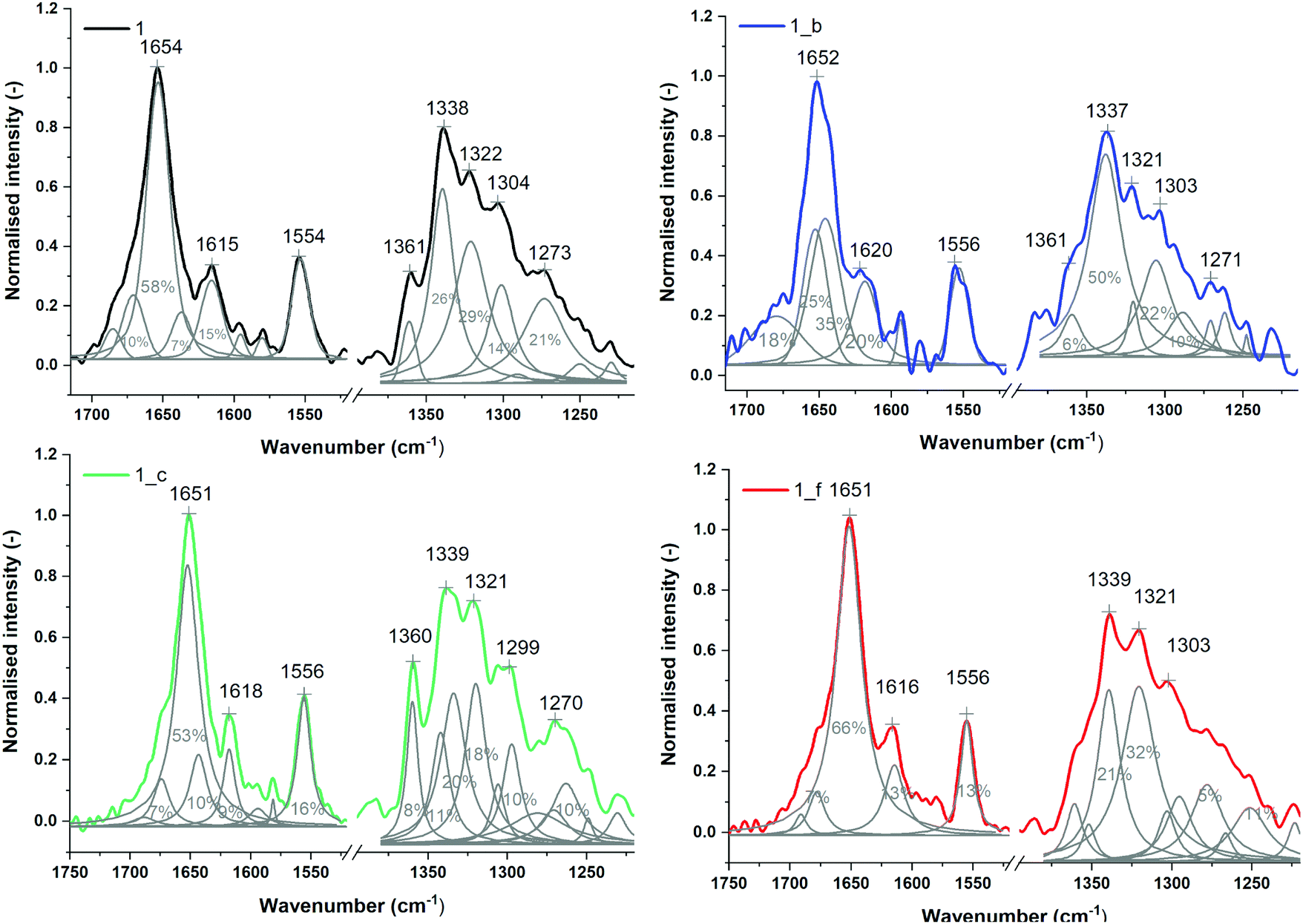 | ||
| Fig. 6 Normalized FT-Raman spectra of powder peptides with sub-bands and percentage area values obtained from curve fitting based on their second derivatives (amide I and III regions). | ||
The amide II band observed in the range of 1600–1500 cm−1, derived mainly from in-plane N–H bending, also provides valuable structural information regarding secondary structures.52 The location of its maximum near 1542 cm−1 for all studied peptides is attributed to the helix structure.40 There was no significant difference in the distribution of amide II components for the studied peptides; however, second derivatives of amide II revealed the presence of spectral features of trans-ACPC in the ATR-FTIR spectra of α,β-peptides (Fig. S7_3†). The amide II band is absent in the Raman spectra. In this wavenumber range, only an intense band at 1556 cm−1 derived from tryptophan residues is observed (Fig. 6).53
No significant changes were observed in the positions of the amide bands in the infrared spectra (Fig. S7_7 and Tables S7_1, S7_8†) obtained directly after the dissolution of the studied peptides in water, indicating preservation of the helical structure in solution. However, the shift of the maximum amide I band in the ATR-FTIR spectrum, especially evident in the case of peptide 1_b, may suggest self-association (Table S7_8†). The ATR mode can accelerate the aggregation process due to the contact of the peptide molecules with diamond, which is why in μIR spectra, the maximum amide I mode is observed at slightly different positions. Temperature dependence spectra (Fig. S7_9†) indicate that from all studied sequences, peptide 1_b formed the most stable structures. In the case of peptide 1_f, the shift of the amide I maximum to higher wavenumbers suggests that it creates less unpacked structures.
No visible change in the maxima of amide I peak positions in ATR-FTIR spectra can be observed after 7 days of incubation of the peptide solutions at 37 °C, which demonstrates no alteration in the peptide secondary structures. On the other hand, in the infrared spectra of peptides 1_c and 1_f, the peak area of the sub-band was strongly increased at approximately 1630 cm−1, which may indicate aggregate formation (Fig. S7_10 and Table S7_11†).50 Moreover, the blueshift in the amide A maximum position (∼3290 cm−1) of approximately 10 cm−1 suggests an increase in the strength of hydrogen interactions.54,55 The aggregate formation was confirmed with infrared techniques since the analysis performed after 30 days of incubation showed that for α,β-peptides, the maximum amide I was moved from 1635 cm−1 to 1627 cm−1, which is characteristic of long, rigid fibrils.50,56,57 The percentage of this sub-band was increased for samples 1, 1_b and 1_c (Table S7_12†). Only peptide 1_f did not show a significant decline in percentage (2.43%).
All peptides demonstrated spectral features characteristic of helical secondary structures, both in powder and in water solutions. Peptides 1_b, 1_c and 1_f containing cyclic β-amino acids tend to adopt more compact structures than model peptide 1. Additionally, changes in the band positions of α,β-peptides after several days of incubation may indicate self-association and aggregate formation. Similar shifting behaviors of amide I position and self-aggregation were observed for phenol-soluble modulin α3 (PSMα3) from Staphylococcus aureus58 and de novo designed peptides59 forming a cross-α architecture.
Nanostructure characterization
We used atomic force microscopy (AFM) and transmission electron microscopy (TEM) to observe the results of peptide self-aggregation. Basing on AFM measurements, we established that peptide 1 was not able to self-assemble molecularly on a mica surface (Fig. S8_1†). The aggregation process was not observed even after two weeks of sample incubation. On the other hand, α,β-peptides 1_b, 1_c and 1_f self-assemble immediately, and over a period of time, the formation of long fibrils can be observed (Fig. 7). After one hour of adsorption on the mica surface, we detected the formation of globular structures, which created fibrils after a few hours. After several days of incubation, the fibrils became crosslinked, and the average diameter of the structures changed from 3 to 6.4 nm. We presume that such a phenomenon may be caused by secondary nucleation of the aggregation process, similar to the one observed in amyloid proteins.60 The self-association process occurs faster for 1_b than for 1_c and 1_f; however, in all cases, fibrils are formed even at low concentrations (<80 μM). Morphological differences between the formed fibers, such as the smoothness of the fiber edges, can also be observed (Fig. 7). The average heights of the helical structures for all peptides ranged from 1.35 to 4.34 nm (Fig. S8_2†). The tallest structures were formed by peptide 1_c, directly after absorption on the mica surface (Fig. 7 and S8_2†). As the incubation time increased, the height of the peptides decreased, with one exception: the height of peptide 1_f increased from 1.35 to 1.53 nm. The observed fluctuation in the height of fibrils can be a sign of different stages of peptide aggregation, as mentioned above.61 | ||
| Fig. 7 AFM images of peptides 1_b, 1_c and 1_f on the mica surface after various incubation times (cpeptide = 1.3 mM). | ||
TEM analysis confirmed the pattern of nanostructure formation in water, starting from globular aggregates to fibrils, after a short period of time (Fig. 8). TEM allows us to determine more accurate values of average diameter, which are 10.42 ± 2.72 nm for 1_b, 10.18 ± 1.93 nm for 1_c and 12.08 ± 2.65 nm for 1_f (Fig. S9†). The average contour length of fibrils amounts to 412 ± 61.53 nm for 1_c and 49.8 ± 27.9 nm for 1_f (Fig. S9†). Fibers formed by peptide 1_b were too long and tangled to determine their length. In terms of fibril architecture, nanostructures formed by peptide 1_f differ from structures assembled by peptides 1_b and 1_c (Fig. 8); in the case of peptide 1_f, there are visible small, globular aggregates that interact with each other to form fibril-like structures. However, after 7 days of incubation, long fibrils can be also visible for peptide 1_f (Fig. S10†). The fibers created by peptides 1_b and 1_c are more regular, showing a twisted rope-like hierarchical structure. It appears that the fibers form predepositions as several of the fibers lay across the top of each other.
 | ||
| Fig. 8 TEM images of peptides 1_b, 1_c and 1_f on carbon grid surface after various incubation times (cpeptide = 1.3 mM). Images registered at the magnification of 50 nm. | ||
AFM and TEM analyses of the studied α,β-peptides give consistent results in terms of the type, rate and amount of nanostructure formation. It can be concluded that even at a low concentration, globular aggregates form, and after a short period of time form long fibrils. The dissimilarities in nanostructure morphology, especially for peptide 1_f, may arise from the differences in sample adsorption (mica surface in AFM versus carbon grid in TEM).
The X-ray diffraction experiments revealed presence of ordered microstructure in the dried sample of peptide 1_f deposited on SiN window. Diffraction pattern (Fig. 9A) shows multiple co-centric diffraction rings characteristic for crystallites or fibers without preferential orientation. The diffraction profile was obtained by integration of full diffraction image (Fig. 9B). It shows multiple diffraction peaks of various intensity in the Q vector range from 0.25 Å−1 to 1 Å−1. In general, such a diffraction pattern is characteristic for the α-helical assemblies. The peak corresponding to d-spacing equal to 10.4 Å is characteristic also for coiled-coil peptides assemblies heptad repeat pattern.62 The same value was observed by the use of the electron cryo-transmission microscopy for de novo designed synthetic coiled-coil peptides.63 In this case, those correspond to the packing parameter of the coiled-coil dimer into the larger fiber bundle. Observed diffraction pattern of peptide 1_f is different from the typical pattern for cross-β structure of amyloid fibrils.64 Those fibers has characteristic diffraction rings at 10–11 Å and 4.7 Å d-spacings, corresponding to the spacing between β-strands and spacing between β-sheets, respectively.
 | ||
| Fig. 9 X-ray diffraction pattern of the peptide 1_f sample at 80 μM dried on SiN window. X-ray diffraction pattern of non-aligned sample (A). Peak profile of X-ray diffraction pattern with calculated d-spacing of corresponding peaks. Q = 4πsin(θ)/λ, where θ is diffraction angle and λ is the X-ray radiation wavelength (B). | ||
Recent studies also showed that some peptides can form cross-α structure fibrils.58,65 In those structures α-helices aligns in the similar fashion to the β-strands of amyloid fibers. X-ray diffraction studies of the wild-type phenol-soluble modulin α3 (PSMα3) peptides shows characteristic diffraction patterns with d-spacing calculated for main reflections equal to: 10.5 Å, 12 Å and 25 Å. In the diffraction pattern of peptide 1_f fibrils a strong peaks at 25 Å is absent. There is only very weak diffraction peak at 25.5 Å. This phenomena suggests that peptide 1_f could form some coiled-coil structures than cross-α or cross-β structures, however further detailed studies are needed to resolve three-dimensional structure of the peptide 1_f assemblies.
Conclusions
A methodology for the rational construction of helix-containing nanofibers using the modification of coiled-coil structure outer positions with helix-promoting trans-ACPC residue was elaborated. Incorporation of one trans-ACPC residue per heptad induced the formation of helical structures only if this substitution did not interfere with further coiled-coil formation; i.e., only the outer b, c and f positions were available for this modification. The formation of helices was confirmed by various techniques, including CD, NMR and FT-IR/Raman. Importantly, significantly higher conformational stability was shown for trans-ACPC-modified peptides in comparison to model peptide 1. Subsequently, the formation of aggregates was confirmed in solution using AUC and ThT assays. In the cases of two modified peptides, namely, 1_c and 1_f, the formation of trimers was evidenced in solution, which is analogous to starting peptide 1. Again, the propensity of 1_c and 1_f for trimer formation was higher than that of unmodified peptide 1. The behavior of peptide 1_b in solution was significantly different, as a large number of larger structures were detected. Subsequently, peptides were studied in solid state on mica and carbon surfaces. Well-formed nanofibrils were observed using both AFM and TEM techniques for peptides 1_b, 1_c and 1_f. At that level of molecular organization, the difference between trans-ACPC-substituted and original coiled-coil peptides was the most substantial, as model peptide 1 did not form any nanostructures. The velocity of nanostructure formation in the studied peptides was different, which allowed us to observe the initial globular aggregates preceding the creation of nanofibers in some cases. Therefore, it could be assumed that the pathway of fibril formation includes the following stages: helices → oligomers → bundles → globular aggregates → nanofibrils. Importantly, individual features of the studied peptide sequences could be attributed to the achievement of a particular level of organization, which makes the design process rational. Structural constraints introduced by rigid trans-ACPC residues contribute to a higher propensity of the peptides to form helix structures. The residues in positions a/d and e/g in the heptad are responsible for coiled-coil formation (trimers). The formation of larger aggregates is driven by the introduced hydrophobic surface delivered by the cyclopentane side chains of trans-ACPC residues. We propose the formation of a zipper-like interaction between helices by analogy to the leucine zippers observed in transcriptional regulators.66 We consider that the presented methodology will constitute a significant step towards a better understanding of beta-amino acid-containing peptide properties and will provide a basis for future discoveries of higher-order structures of peptides from this class.Experimental section
Peptide synthesis
All commercially available reagents and solvents were purchased from Lipopharm.pl, Sigma-Aldrich or Merck and used without further purification. Fmoc-(1S,2S)-2-aminocyclopentanecarboxylic acid (trans-ACPC) was purchased from Synnovator, Inc. Described peptides were obtained with an automated solid-phase peptide synthesizer (Biotage® Initiator + Alastra™) using rink amide AM resin (loading: 0.59 mmol g−1). Fmoc deprotection was performed using 20% piperidine in DMF for 3 + 10 min. A double-coupling procedure was achieved with 0.5 M solution of DIC and 0.5 M solution of OXYMA (1:1) in DMF, for α-amino acids 2 × 15 min and for β-amino acids 30 min, at 75 °C. Acetylation reaction was carried out using NMP/DIPEA/acetic anhydride (80:15:5) mixture. Cleavage of the peptides from the resin was accomplished with the mixture of TFA/TIS/H2O (95:2.5:2.5) after 3 h of shaking. The crude peptide was precipitated with ice-cold Et2O and centrifuged (10000 rpm, 15 min, 4 °C). Peptides were purified using preparative HPLC (Knauer Prep) with a C18 column (Thermo Scientific, Hypersil Gold 12μ, 250 mm × 20 mm) with water/acetonitrile (0.05% TFA) eluent system.
Analytical high-performance liquid chromatography (HPLC)
Analytical HPLC was performed using Kinetex 5μ EVO C18 100A 150 × 4.6 mm column. Program (eluent A: 0.05% TFA in H2O, eluent B: 0.05% TFA in acetonitrile, flow 0.5 mL min−1): A: t = 0 min, 90% A; t = 30 min, 10% A.Mass spectrometry (MS)
Peptides were studied by WATERS LCT Premier XE System consisting of high resolution mass spectrometer with a time of flight (TOF).Analytical ultracentrifugation (AUC)
Analytical ultracentrifugation was run at 20 °C on Beckman Optima XL-I with 4- or 8-position An-Ti rotor and UV detection at 280 nm, in a double-sector 1.2 cm cells with charcoal-filled epon centrepieces and sapphire windows. Lyophilized peptides were dissolved in water or in AUC buffer (50 mM phosphate buffer pH 7.0, 100 mM NaCl) directly before measurements. Peptides concentration (10–140 μM) was determined spectrophotometrically (Jasco V-650 spectrophotometer) from absorption values at 280 nm using the extinction coefficients ε = 6990 M−1 cm−1 calculated from the amino acid composition by ProtParam.67 Sedimentation velocity experiments were performed at 50000 rpm. Radial absorption scans of peptide concentration profiles were measured at 5 or 10 min intervals. 390 μl of a peptide sample and 400 μl of a reference solution (water or AUC buffer) were loaded into the right and the left sector of the cell. The sedimentation velocity data were analysed using program SEDFIT with a continuous sedimentation coefficient distribution model, c(s), based on the Lamm equation.68,69 Integration of the c(s) peaks provided the signal weighted average sedimentation coefficients (s) and corresponding standard sedimentation coefficients s20,w (referring to the water and 20 °C). The density and viscosity of the AUC buffer were calculated using Sednterp program.70 The partial specific volumes of peptides were calculated from the partial specific volumes obtained in the Sednterp program (based on amino acid composition excluding ACPC residues, acetyl group at the N-terminus and NH2 group at the C-terminus) and from partial specific volumes of the ACPC residues (![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 0.929207 cm3 g−1) and of the end groups (Ac and NH2, = 0.813588 cm3 g−1) obtained according to the method of Durchschlag and Zipper71 (Table S5_D†). Multispeed sedimentation equilibrium experiments were performed at 26000, 32000, and 50000 rpm. 150 μl of a peptide samples and 160 μl of the AUC buffer (as a reference) were placed in the cells. The radial concentration gradients were collected every 3 h at the radial intervals of 0.001 cm, until the sedimentation equilibrium was attained (25–35 h per each speed). The data were analysed by means of nonlinear regression using SEDPHAT program, according to single species model, monomer–n-mer association or monomer–n-mer–m-mer association, with a global fitting to all equilibrium profiles obtained at all three rotor speeds.72 Theoretical values of sedimentation coefficients in the standard conditions (s20,w) for various oligomeric forms of studied peptides were calculated using the formula sn = s1 × n2/3, where s1 is sedimentation coefficient for a monomer.73 First, s20,w values for trimeric form (n = 3) were obtained in the SEDFIT program using its’ M(s) calculator, and introducing constraints that molecular mass calculated for the particular s20,w value must be equal to the mass of the peptide calculated form its amino acid sequence. Values f/f0 that are necessary for these calculations were f/f0 = 1.237, 1.676 and 1.271 for peptides 1, 1_b, and 1_c and 1_f, respectively, hence taken from the Table 1. In the next step s20,w for other oligomeric forms were calculated using the formula sn = s1 × n2/3.
= 0.929207 cm3 g−1) and of the end groups (Ac and NH2, = 0.813588 cm3 g−1) obtained according to the method of Durchschlag and Zipper71 (Table S5_D†). Multispeed sedimentation equilibrium experiments were performed at 26000, 32000, and 50000 rpm. 150 μl of a peptide samples and 160 μl of the AUC buffer (as a reference) were placed in the cells. The radial concentration gradients were collected every 3 h at the radial intervals of 0.001 cm, until the sedimentation equilibrium was attained (25–35 h per each speed). The data were analysed by means of nonlinear regression using SEDPHAT program, according to single species model, monomer–n-mer association or monomer–n-mer–m-mer association, with a global fitting to all equilibrium profiles obtained at all three rotor speeds.72 Theoretical values of sedimentation coefficients in the standard conditions (s20,w) for various oligomeric forms of studied peptides were calculated using the formula sn = s1 × n2/3, where s1 is sedimentation coefficient for a monomer.73 First, s20,w values for trimeric form (n = 3) were obtained in the SEDFIT program using its’ M(s) calculator, and introducing constraints that molecular mass calculated for the particular s20,w value must be equal to the mass of the peptide calculated form its amino acid sequence. Values f/f0 that are necessary for these calculations were f/f0 = 1.237, 1.676 and 1.271 for peptides 1, 1_b, and 1_c and 1_f, respectively, hence taken from the Table 1. In the next step s20,w for other oligomeric forms were calculated using the formula sn = s1 × n2/3.
Circular dichroism (CD)
CD spectra were recorded on JASCO J-815 at 20 °C between 260 and 190 nm in water with following parameters: 0.2 nm resolution, 1.0 nm bandwidth, 20 mdeg sensitivity, 0.25 s response, 50 nm min−1 scanning speed, 5 scans, 0.02 cm cuvette path length. The CD spectra of the solvents alone were recorded and subtracted from the raw data. For peptides dissolved in water pH was adjusted to 7. Typically, the samples were prepared by dilution of peptides stock solution to obtain peptide concentration of 80 μM. The CD intensity is given as mean residue molar ellipticity (θ[deg cm2 dmol−1]). The melting temperatures of studied peptides were determined by method described previously.74Temperature denaturation measurements using CD
To examine the thermal unfolding of the peptides, stock solutions were diluted to 80 μM and three types of measurements were performed: (1) in the scope 190–240 nm; (2) at 208 nm in water and (3) at 208 nm with 3 M GuHCl. The temperature was increased from 4 to 98 °C in increments of 2 °C. Ellipticity measurements were recorded with 1 mm path length cuvette, others parameters remained unchanged.Nuclear magnetic resonance (NMR)
The NMR experiments were performed on Bruker Avance™ spectrometer operating at 600.58 MHz for 1H, equipped with a 5.0 mm PA BBO probe. The NMR spectra at temperature 298 K were recorded in CD3OH. The temperature was controlled to ±0.1 K. TOCSY and NOESY experiments were performed for chemical shift and structure assignment. All NMR spectra were acquired with suppression of solvent OH signal using 3-9-19 pulse sequence with gradients. Typical TOCSY – homonuclear Hartman (Hahn transfer using mlev17 sequence for mixing using two power levels for excitation and spinlock) and 2D NOESY were recorded in phase sensitive mode with the spectral width of 6127 Hz in both dimensions using 2048 data points and relaxation delay of 2 s. These spectra were acquired with 1024 increments of 16 scans for TOCSY and 80 scans for 2D NOESY. Mixing times were set at 80 ms and 200 ms for TOCSY and 2D NOESY experiments, respectively. Processing and analysis of the NMR data were performed on Topspin 3.2 (Bruker BioSpin) software.NMR structure determination
NMR structure determination was performed using Xplor-NIH v. 2.41 program.75 NMR derived inter-proton contacts were classified by standard method with upper distance limits: strong 2.5 Å (s), medium 3.5 Å (m) and weak 5 Å (w), and the lower distance limit was set to 1.8 Å. Initially, 50 random conformations of a peptide were generated. Restraints derived from NOE contacts and 3J coupling constants of amide protons were applied. Standard protocol for NMR structure calculations implemented in Xplor-NIH was used with the following steps: (a) high temperature dynamics (3500 K, 800 ps or 8000 steps), (b) simulated annealing performed from 3500 K to 25 K with 12.5 K step, at each temperature short dynamics was done (100 steps or 0.2 ps); (c) gradient minimization of final structure. Finally, top lower energy structures were superimposed.Vibrational spectroscopy
Attenuated total reflectance – Fourier-transform infrared (ATR-FTIR) studies were performed on FT-IR spectrometer (Thermo Scientific, USA) with Golden Gate Mk II ATR Accessory with Heated Diamond Top-plate (PIKE Technologies). ATR-FTIR spectra were obtained in the range of 3600–150 cm−1. For each spectrum 128 interferograms was coadded with 4 cm−1 resolution. Directly before sampling, the background spectrum of diamond/air was recorded as a reference (512 scans, 4 cm−1). The spectra of 1.3 mM peptides solutions were registered in the temperature range 25–95 °C (77–203 °F) with a 2 °C step. Additionally, air-dried films were obtained. Initially, 10 μl of peptide aqueous solution was dropped directly on the diamond surface and was allowed to dry out. At the time, spectra were recorded at 5 minutes intervals. Infrared (IR) spectra were collected using integrated infrared microscope FTIR iN10 (Thermo Scientific, USA) equipped with a liquid nitrogen cooled mercury cadmium telluride (MCT-A) detector. All spectra were acquired in the transmission mode (10 × 10 image size, 50 micron aperture, 50 micron step size, 64 scans per point, resolution of 4 cm−1). A drop of 10 μl of each sample was deposited onto BaF2 windows and air dried prior to imaging. Measurements were conducted at room temperature, approximately 22 °C (72 °F). Raman spectra were measured using NXR FT-Raman spectrometer with MicroStage extension equipped with Nd:YVO4 laser (1064 nm, 500 mW) as an excitation source and InGaAs detector. FT-Raman were recorded over a range of 3700–150 cm−1 at 4 cm−1 resolution. In each case, an average of 1024 scans were collected. Only samples in powder form were tested. All spectra were analyzed using the OriginPro 9.0 (OriginLab Corporation, USA). The analysis included spectra baseline correction, smoothing using the Savitzky–Golay filter (parameters dependent on the type of spectra), normalization of spectra relative to selected bands and deconvolution into subcomponents using the Lorentz function based on second derivative spectra. ATR-FTIR spectra were initially preprocessed using OMNIC 8.3.103.Atomic force microscopy (AFM)
AFM images were collected using a MultiMode atomic force microscope with a NanoScope IV controller (Digital Instruments, Santa Barbara, CA). The topography was measured in tapping™ mode (TM). The tip, a force modulation etched silicon probe (Veeco Metrology, Sunnyvale, CA), had a constant of 23 N m−1, a resonance frequency of 296–351 kHz, a nominal tip radius of curvature 5 nm and a cantilever length of 110–140 μm. NCLR and TESP-SS cantilevers were used. 10 μl of the 1.3 mM peptide solution in water was applied to freshly cleaved mica sheet and after 1 minute of adsorption the sheet was rinsed with deionized water. Time of measurements: directly after one minute of adsorption on the mica surface (about 5 minutes), 1 day and 7 days. Samples were incubated at 25 °C. The resolution of the AFM scans was 512 × 512 pixels, which produces topographic images of the studied peptide on the mica surface (2 μm × 2 μm). Images were collected several times across the mica surface. The images were analyzed and processed using Gwyddion software (Czech Metrology Institute). We used first-order flattening tool – Gaussian and sharpening filters on the height images. The surface images (3D AFM) were composed of small parts of each image. The average height of nanostructures was established by performing statistical analysis of the AFM images. To determine the distribution of acquired data two normality tests Shapiro–Wilk and Kolmogorov–Smirnov were performed. All data describing vertical distance of peptides come from normal distribution (p-value <0.05). Artifacts, like curvature distortion (blowing effects), were considered during morphological analysis.Transmission electron microscopy (TEM)
The samples were suspended in water to the concentration of 1.3 mM and incubated at 37 °C. After 30 minutes, 2 and 48 hours from the incubated samples a small amount of suspension was taken and spotted on a carbon on copper grid. After 1 minute, an excess of the material was removed and 2% uranyl acetate was applied to the grid to stain negative peptide structures. After another minute, an excess of the staing was collected and the grid was allowed to dry under atmospheric conditions. Imaging was performed using transmission electron microscope Tecnai TF 20 X-TWIN (200 kV).Thioflavin T (ThT) fluorescence assay
ThT powder was dissolved in MilliQ® to final concentration 10 μM. ThT solution was dissolved in 50 mM phosphate buffer (pH = 7) to final concentration 50 mM. The 90 μl of ThT buffer was mixed with 10 μl of peptide solution (concentration 1.3 mM) in the 96 wells plate. Samples were measured on the SpectraMax® Gemini™ XPS Microplate (Molecular Devices LLC). The plate reader was heated to 37 °C. The excitation wavelength was set at 440 nm and the emission was recorded in the range from 450 to 600 nm (slit widths 3 nm). The examined peptides were measured directly after dissolving. The measurements were repeated 3 times.Kinetic measurements were done for the samples containing 10 μL of 1 mg mL−1 peptide and 90 μL of 50 μM ThT solution mixed in a 96-well plate. The fluorescence of each well was read by microplate reader CLARIOstar Plus, BMG LABTECH at 25 °C with 30 s shaking, every 30 min, during 24 hours measurements. The excitation wavelength was set at 440 nm and emission at 480 nm. Each group of experiment contained six parallel samples and the data were averaged after measurements.
The aggregation kinetic curves obtained for studied peptides represent classic example of a ThT-monitored kinetic curves and so were fitted by the sigmoidal curves using the following eqn (1):
 | (1) |
Wide-angle X-ray scattering (WAXS)
The fiber diffraction experiments were performed on laboratory SAXS/WAXS Xeuss 2.0 system (Xenocs, Sassenage, France) equipped with MetalJet D2 microfocus X-ray source (λ = 0.134 nm) with a liquid gallium alloy target (Excillum AB, Kista, Sweden). 1 mL of fibrillated peptide 1_f sample at concentration of 80 μM in H2O were centrifuged for 10 minutes at 16000g. The supernatant was discarded and the pellet was resuspended in 30 μL of H2O. About 20 μL of obtained sample was placed dried in 2 μL portions on SiN windows (Xenocs, Sassenage, France). The deposited on SiN sample was then placed on sample holder and measured in vacuum for 10 minutes. Sample-to-detector distance, as well as diffraction range, were calibrated using diffraction pattern of silver behenate as a reference sample.76 Obtained X-ray diffraction images were processed using Foxtrot package77 and background from empty SiN window was also subtracted. The real space d-spacing values were calculated from the formula: d = 2π/s, where s is a position of the peak maximum in reciprocal (Q) space.
Abbreviations
| trans-ACPC | trans (1S,2S)-2-Aminocyclopentanecarboxylic acid |
| ThT | Thioflavin T |
Funding sources
The work was financially supported by the National Science Centre, Poland, Grant No. 2017/26/D/ST5/00341 (to M. S.)Conflicts of interest
Authors declare no potential conflict of interest.Acknowledgements
The authors would like to gratefully acknowledge National Science Centre, Poland for their financial support through Grant No. 2017/26/D/ST5/00341. M. S. would like to thank to Prof. Małgorzata Kotulska for fruitful discussions. This work was partially performed in the NanoFun laboratories co-financed by the European Regional Development Fund within the Innovation Economy Operational Program, the Project No. POIG.02.02.00-00-025/09.References
- E. De Santis and M. G. Ryadnov, Peptide Self-Assembly for Nanomaterials: The Old New Kid on the Block, Chem. Soc. Rev., 2015, 44, 8288–8300 RSC.
- E. H. C. Bromley and K. J. Channon, Alpha-Helical Peptide Assemblies: Giving New Function to Designed Structures, Prog. Mol. Biol. Transl. Sci., 2011, 103, 231–275 CAS.
- L. E. R. O'Leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Multi-Hierarchical Self-Assembly of a Collagen Mimetic Peptide from Triple Helix to Nanofibre and Hydrogel, Nat. Chem., 2011, 3, 821–828 CrossRef.
- D. M. Raymond and B. L. Nilsson, Multicomponent Peptide Assemblies, Chem. Soc. Rev., 2018, 47, 3659–3720 RSC.
- P. B. Harbury, T. Zhang, P. S. Kim and T. Alber, A Switch between 2-Stranded, 3-Stranded and 4-Stranded Coiled Coils in Gcn4 Leucine-Zipper Mutants, Science, 1993, 262, 1401–1407 CrossRef CAS.
- J. M. Mason and K. M. Arndt, Coiled Coil Domains: Stability, Specificity, and Biological Implications, ChemBioChem, 2004, 5, 170–176 CrossRef CAS.
- Y. B. Yu, Coiled-Coils: Stability, Specificity, and Drug Delivery Potential, Adv. Drug Delivery Rev., 2002, 54, 1113–1129 CrossRef CAS.
- P. S. P. Wang and A. Schepartz, β-Peptide Bundles: Design. Build. Analyze. Biosynthesize, Chem. Commun., 2016, 52, 7420–7432 RSC.
- D. N. Woolfson, Coiled-Coil Design: Updated and Upgraded, Subcell. Biochem., 2017, 82, 35–61 CAS.
- C. Xu, R. Liu, A. K. Mehta, R. C. Guerrero-Ferreira, E. R. Wright, S. Dunin-Horkawicz, K. Morris, L. C. Serpell, X. Zuo, J. S. Wall and V. P. Conticello, Rational Design of Helical Nanotubes from Self-Assembly of Coiled-Coil Lock Washers, J. Am. Chem. Soc., 2013, 135, 15565–15578 CrossRef CAS.
- J. M. Fletcher, R. L. Harniman, F. R. Barnes, A. L. Boyle, A. Collins, J. Mantell, T. H. Sharp, M. Antognozzi, P. J. Booth, N. Linden, M. J. Miles, R. B. Sessions, P. Verkade and D. N. Woolfson, Self-Assembling Cages from Coiled-Coil Peptide Modules, Science, 2013, 340, 595–599 CrossRef CAS.
- Y. Wu and J. H. Collier, α-Helical Coiled-Coil Peptide Materials for Biomedical Applications, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, 1424 Search PubMed.
- D. Seebach, A. K. Beck and D. J. Bierbaum, The World of β- and γ-Peptides Comprised of Homologated Proteinogenic Amino Acids and Other Components, Chem. Biodiversity, 2004, 1, 1111–1239 CrossRef CAS.
- D. E. Mortenson, D. F. Kreitler, N. C. Thomas, I. A. Guzei, S. H. Gellman and K. T. Forest, Evaluation of β-Amino Acid Replacements in Protein Loops: Effects on Conformational Stability and Structure, ChemBioChem, 2018, 19, 604–612 CrossRef CAS.
- J. L. Price, W. S. Horne and S. H. Gellman, Structural Consequences of β-Amino Acid Preorganization in a Self-Assembling α/β-Peptide: Fundamental Studies of Foldameric Helix Bundles, J. Am. Chem. Soc., 2010, 132, 12378–12387 CrossRef CAS.
- R. D. Gopalan, M. P. Del Borgo, A. I. Mechler, P. Perlmutter and M.-I. Aguilar, Geometrically Precise Building Blocks: the Self-Assembly of β-Peptides, Chem. Biol., 2015, 22, 1417–1423 CrossRef CAS.
- K. Kulkarni, N. Habila, M. P. Del Borgo and M.-I. Aguilar, Novel Materials From the Supramolecular Self-Assembly of Short Helical β3-Peptide Foldamers, Front. Chem., 2019, 7, 70 CrossRef CAS.
- W. S. Horne, J. L. Price, J. L. Keck and S. H. Gellman, Helix Bundle Quaternary Structure from α/β-Peptide Foldamers, J. Am. Chem. Soc., 2007, 129, 4178–4180 CrossRef CAS.
- W. S. Horne, M. D. Boersma, M. A. Windsor and S. H. Gellman, Sequence-Based Design of α/β-Peptide Foldamers That Mimic BH3 Domains, Angew. Chem., Int. Ed., 2008, 47, 2853–2856 CrossRef CAS.
- S. H. Yoo and H.-S. Lee, Foldectures: 3D Molecular Architectures from Self-Assembly of Peptide Foldamers, Chem. Res., 2017, 50, 832–841 CrossRef CAS.
- Z. Yang, G. Liang, M. Ma, Y. Gao and B. Xu, In Vitro and In Vivo Enzymatic Formation of Supramolecular Hydrogels Based on Self-Assembled Nanofibers of a β-Amino Acid Derivative, Small, 2007, 3, 558–562 CrossRef CAS.
- J. Mangelschots, M. Bibian, J. Gardiner, L. Waddington, Y. Van Wanseele, A. Van Eeckhaut, M. M. Acevedo, B. Van Mele, A. Madder, R. Hoogenboom and S. Ballet, Mixed α/β-Peptides as a Class of Short Amphipathic Peptide Hydrogelators with Enhanced Proteolytic Stability, Biomacromolecules, 2016, 17, 437–445 CrossRef CAS.
- W. C. Pomerantz, V. M. Yuwono, R. Drake, J. D. Hartgerink, N. L. Abbott and S. H. Gellman, Lyotropic Liquid Crystals Formed from ACHC-Rich β-Peptides, J. Am. Chem. Soc., 2011, 133, 13604–13613 CrossRef CAS.
- J. Nanda and A. Banerjee, A Gel-Based Trihybrid System Containing Nanofibers, Nanosheets, and Nanoparticles: Modulation of the Rheological Property and Catalysis, Soft Matter, 2012, 8, 3380–3386 RSC.
- L. K. A. Pilsl and O. Reiser, Alpha/Beta-Peptide Foldamers: State of the Art, Amino Acids, 2011, 41, 709–718 CrossRef CAS.
- M. Szefczyk, E. Weglarz-Tomczak, P. Fortuna, A. Krzyszton, E. Rudzinska-Szostak and L. Berlicki, Controlling the Helix Handedness of ααβ–Peptide Foldamers through Sequence Shifting, Angew. Chem., Int. Ed., 2017, 56, 2087–2091 CrossRef CAS.
- I. M. Mandity, E. Weber, T. A. Martinek, G. Olajos, G. K. Toth, E. Vass and F. Fulop, Design of Peptidic Foldamer Helices: A Stereochemical Patterning Approach, Angew. Chem., Int. Ed., 2009, 48, 2171–2175 CrossRef CAS.
- K. Ananda, P. G. Vasudev, A. Sengupta, K. M. P. Raja, N. Shamala and P. Balaram, Polypeptide Helices in Hybrid Peptide Sequences, J. Am. Chem. Soc., 2005, 127, 16668–16674 CrossRef CAS.
- M.-R. Lee, N. Raman, S. H. Gellman, D. M. Lynn and S. P. Palecek, Incorporation of β–Amino Acids Enhances the Antifungal Activity and Selectivity of the Helical Antimicrobial Peptide Aurein 1.2, ACS Chem. Biol., 2017, 12, 2975–2980 CrossRef CAS.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Foldamers as Versatile Frameworks for the Design and Evolution of Function, Nat. Chem. Biol., 2007, 3, 252–262 CrossRef CAS.
- R. R. Araghi, C. Baldauf, U. I. M. Gerling, C. D. Cadicamo and B. Koksch, A Systematic Study of Fundamentals in α-Helical Coiled Coil Mimicry by Alternating Sequences of β- and γ-Amino Acids, Amino Acids, 2011, 41, 733–742 CrossRef.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Molecular Self-Assembly and Nanochemistry: a Chemical Strategy for the Synthesis of Nanostructures, Science, 1991, 254, 1312–1319 CrossRef CAS.
- F. Thomas, W. M. Dawson, E. J. M. Lang, A. J. Burton, G. J. Bartlett, G. G. Rhys, A. J. Mulholland and D. N. Woolfson, De Novo-Designed α-Helical Barrels as Receptors for Small Molecules, ACS Synth. Biol., 2018, 7, 1808–1816 CrossRef CAS.
- F. Lapenta, J. Aupic, Z. Strmsek and R. Jerala, Coiled Coil Protein Origami: from Modular Design Principles towards Biotechnological Applications, Chem. Soc. Rev., 2018, 47, 3530–3542 RSC.
- J. M. Fletcher, A. L. Boyle, M. Bruning, G. J. Bartlett, T. L. Vincent, N. R. Zaccai, C. T. Armstrong, E. H. Bromley, P. J. Booth, R. L. Brady, A. R. Thomson and D. N. Woolfson, A Basis Set of de Novo Coiled-Coil Peptide Oligomers for Rational Protein Design and Synthetic Biology, ACS Synth. Biol., 2012, 1, 240–250 CrossRef CAS.
- C. Toniolo, A. Polese, F. Formaggio, M. Crisma and J. Kamphuis, Circular Dichroism Spectrum of a Peptide 310-Helix, J. Am. Chem. Soc., 1996, 118, 2744–2745 CrossRef CAS.
- P. Schuck, On the Analysis of Protein Self-Association by Sedimentation Velocity Analytical Ultracentrifugation, Anal. Biochem., 2003, 320, 104–124 CrossRef CAS.
- K. Gade Malmos, L. Blancas-Mejia, B. Weber, J. Buchner, M. Ramirez-Alvarado, H. Naiki and D. Otzen, ThT 101: a Primer on the Use of Thioflavin T to Investigate Amyloid Formation, Amyloid, 2017, 24, 1–16 CrossRef CAS.
- M. Biancalana and S. Koide, Molecular Mechanism of Thioflavin-T Binding to Amyloid Fibrils, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1405–1412 CrossRef CAS.
- M. Zou, H. Yang, H. Wang, J. Zhang, B. Wei, H. Zhang and D. Xie, Detection of Type I Collagen Fibrils Formation and Dissociation by a Fluorescence Method Based on Thioflavin T, Int. J. Biol. Macromol., 2016, 92, 1175–1182 CrossRef CAS.
- E. K. Kumar, N. Hague and N. P. Prabhu, Kinetics of Protein Fibril Formation: Methods and Mechanisms, Int. J. Biol. Macromol., 2017, 100, 3–10 CrossRef CAS.
- R. Khurana, C. Coleman, V. Krishna, R. Roy and S. Singh, Mechanism of Thioflavin T Binding to Amyloid Fibrils, J. Struct. Biol., 2005, 151, 229–238 CrossRef CAS.
- K. Morimoto, K. Kawabata, S. Kunii, K. Hamano, T. Saito and B. Tonomura, Characterization of Type I Collagen Fibril Formation Using Thioflavin T Fluorescent Dye, J. Biochem., 2009, 145, 677–684 CrossRef CAS.
- M. Yan and X. Wang, Study on the Kinetic Self-Assembly of Type I Collagen from Tilapia (Oreochromis Niloticus) Skin Using the Fluorescence Probe Thioflavin T, Spectrochim. Acta, Part A, 2018, 203, 342–347 CrossRef CAS.
- A. Adochitei and G. Drochioiu, Rapid Characterization of Peptide Secondary Structure by FT-IR Spectroscopy, Rev. Roum. Chim., 2011, 56, 783–791 CAS.
- S. Spassov, M. Beekes and D. Naumann, Structural Differences between TSEs Strains Investigated by FT-IR Spectroscopy, Biochim. Biophys. Acta, 2006, 1760, 1138–1149 CrossRef CAS.
- V. Serrano, W. Liu and S. Franzen, An Infrared Spectroscopic Study of the Conformational Transition of Elastin-Like Polypeptides, Biophys. J., 2007, 93, 2429–2435 CrossRef CAS.
- R. I. Litvinov, D. A. Faizullin, Y. F. Zuev and J. W. Weisel, The α-Helix to β-Sheet Transition in Stretched and Compressed Hydrated Fibrin Clots, Biophys. J., 2012, 103, 1020–1027 CrossRef CAS.
- V. A. Lorenz-Fonfria and E. Padros, Curve-Fitting of Fourier Manipulated Spectra Comprising Apodization, Smoothing, Derivation and Deconvolution, Spectrochim. Acta, Part A, 2004, 60, 2703–2710 CrossRef.
- R. Sarroukh, E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, ATR-FTIR: A ‘Rejuvenated’ Tool to Investigate Amyloid Proteins, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2328–2338 CrossRef CAS.
- N. Kuhar, S. Sil, T. Verma and S. Umapathy, Challenges in Application of Raman Spectroscopy to Biology and Materials, RSC Adv., 2018, 8, 25888–25908 RSC.
- A. Barth, Infrared Spectroscopy of Proteins, Biochim. Biophys. Acta, Biomembr., 2007, 1767, 1073–1101 CrossRef CAS.
- T. Miura, H. Takeuchi and I. Harada, Characterization of Individual Tryptophan Side Chains in Proteins Using Raman Spectroscopy and Hydrogen-Deuterium Exchange Kinetics, Biochemistry, 1988, 27, 88–94 CrossRef CAS.
- R. W. Newberry, S. J. Orke and R. T. Raines, n → π* Interactions Are Competitive with Hydrogen Bonds, Org. Lett., 2016, 18, 3614–3617 CrossRef CAS.
- C. Mensch, P. Bultinck and C. Johannessen, The Effect of Protein Backbone Hydration on the Amide Vibrations in Raman and Raman Optical Activity Spectra, Phys. Chem. Chem. Phys., 2019, 21, 1988–2005 RSC.
- J. Seo, W. Hoffmann, S. Warnke, X. Huang, S. Gewinner, W. Schöllkopf, M. T. Bowers, G. von Helden and K. Pagel, An Infrared Spectroscopy Approach to Follow β-Sheet Formation in Peptide Amyloid Assemblies, Nat. Chem., 2017, 9, 39–44 CrossRef CAS.
- S. D. Moran and M. T. Zanni, How to Get Insight into Amyloid Structure and Formation from Infrared Spectroscopy, J. Phys. Chem. Lett., 2014, 5, 1984–1993 CrossRef CAS.
- E. Tayeb-Fligelman, O. Tabachnikov, A. Moshe, O. Goldshmidt-Tran, M. R. Sawaya, N. Coquelle, J. P. Colletier and M. Landau, The Cytotoxic Staphylococcus Aureus PSMα3 Reveals a Cross-α Amyloid-Like Fibril, Science, 2017, 355, 831–833 CrossRef.
- S. Q. Zhang, H. Huang, J. Yang, H. T. Kratochvil, M. Lolicato, Y. Liu, X. Shu, L. Liu and W. F. DeGrado, Designed Peptides that Assemble into Cross-α Amyloid-Like Structures, Nat. Chem. Biol., 2018, 14, 870–875 CrossRef CAS.
- L. Lutter, C. J. Serpell, M. F. Tuite and W.-F. Xue, The Molecular Lifecycle of Amyloid – Mechanism of Assembly, Mesoscopic Organisation, Polymorphism, Suprastructures, and Biological Consequences, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 140257 CrossRef CAS.
- W. Lee, H. Lee, G. Lee and D. S. Yoon, Advances in AFM Imaging Applications for Characterizing the Biophysical Properties of Amyloid Fibrils, in Exploring New Findings on Amyloidosis, InTech, 2016 Search PubMed.
- D. Papapostolou, A. M. Smith, E. D. Atkins, S. J. Oliver, M. G. Ryadnov, L. C. Serpell and D. N. Woolfson, Engineering nanoscale order into a designed protein fiber, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10853–10858 CrossRef CAS.
- T. H. Sharp, M. Bruning, J. Mantell, R. B. Sessions, A. R. Thomson, N. R. Zaccai, R. L. Brady, P. Verkade and D. N. Woolfson, (2012). Cryo-transmission electron microscopy structure of a gigadalton peptide fiber of de novo design, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13266–13271 CrossRef.
- O. S. Makin and L. C. Serpell, Structures for amyloid fibrils, FEBS J., 2005, 272, 5950–5961 CrossRef CAS.
- E. Tayeb-Fligelman, N. Salinas, O. Tabachnikov and M. Landau, Staphylococcus aureus PSMα3 Cross-α Fibril Polymorphism and Determinants of Cytotoxicity, Structure, 2020, 28, 301–313 CrossRef CAS.
- A. Lupas, M. Van Dyke and J. Stock, Predicting Coiled Coils from Protein Sequences, Science, 1991, 252, 1162–1164 CrossRef CAS.
- E. Gasteiger, C. Hoogland, A. Gattiker, S. Duvaud, M. R. Wilkins, R. D. Appel and A. Bairoch, Protein Identification and Analysis Tools on the ExPASy Server, in The Proteomics Protocols Handbook, Human Press, 2005, pp. 571–607 Search PubMed.
- J. Dam and P. Schuck, Calculating Sedimentation Coefficient Distributions by Direct Modeling of Sedimentation Velocity Concentration Profiles, Methods Enzymol., 2004, 384, 185–212 CAS.
- P. Schuck, Size-Distribution Analysis of Macromolecules by Sedimentation Velocity Ultracentrifugation and Lamm Equation Modeling, Biophys. J., 2000, 78, 1606–1619 CrossRef CAS.
- D. Hayes, T. Laue and J. Philo, Program Sednterp: Sedimentation Interpretation Program, Alliance Protein Laboratories, Thousand Oaks, CA, 1995 Search PubMed.
- H. Durchschlag and P. Zipper, Calculation of the Partial Volume of Organic Compounds and Polymers, Prog. Colloid Polym. Sci., 1994, 94, 20–39 CAS.
- J. Vistica, J. Dam, A. Balbo, E. Yikilmaz, R. A. Mariuzza, T. A. Rouault and P. Schuck, Sedimentation Equilibrium Analysis of Protein Interactions with Global Implicit Mass Conservation Constraints and Systematic Noise Decomposition, Anal. Biochem., 2004, 326, 234–256 CrossRef CAS.
- P. Schuck, H. Zhao, C. A. Brautigam and R. Ghirlando, Basic Principles of Analytical Ultracentrifugation, CRC Press, 2016 Search PubMed.
- D. F. Kreitler, D. E. Mortenson, K. T. Forest and S. H. Gellman, Effects of Single α-to-β Residue Replacements on Structure and Stability in a Small Protein: Insights from Quasiracemic Crystallization, J. Am. Chem. Soc., 2016, 138, 6498–6505 CrossRef CAS.
- C. D. Schwieters, J. J. Kuszewski and G. M. Clore, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 47–62 CrossRef CAS.
- T. C. Huang, H. Toraya, T. N. Blanton and Y. Wu, X-ray powder diffraction analysis of silver behenate, a possible low–angle diffraction standard, J. Appl. Crystallogr., 1993, 26(2), 180–184 CrossRef CAS.
- R. Girardot, G. Viguier, M. Ounsy and J. Pérez, Foxtrot, 2010–2017, https://www.synchrotron-soleil.fr/en/beamlines/swing.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and tables. See DOI: 10.1039/d0nr04313c |
| This journal is © The Royal Society of Chemistry 2021 |