
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA roadmap for metagenomic enzyme discovery
Serina L.
Robinson
 *,
Jörn
Piel
and
Shinichi
Sunagawa
*,
Jörn
Piel
and
Shinichi
Sunagawa
Eidgenössische Technische Hochschule (ETH), Zürich, Switzerland. E-mail: srobinson@ethz.ch
First published on 12th April 2021
Abstract
Covering: up to 2021
Metagenomics has yielded massive amounts of sequencing data offering a glimpse into the biosynthetic potential of the uncultivated microbial majority. While genome-resolved information about microbial communities from nearly every environment on earth is now available, the ability to accurately predict biocatalytic functions directly from sequencing data remains challenging. Compared to primary metabolic pathways, enzymes involved in secondary metabolism often catalyze specialized reactions with diverse substrates, making these pathways rich resources for the discovery of new enzymology. To date, functional insights gained from studies on environmental DNA (eDNA) have largely relied on PCR- or activity-based screening of eDNA fragments cloned in fosmid or cosmid libraries. As an alternative, shotgun metagenomics holds underexplored potential for the discovery of new enzymes directly from eDNA by avoiding common biases introduced through PCR- or activity-guided functional metagenomics workflows. However, inferring new enzyme functions directly from eDNA is similar to searching for a ‘needle in a haystack’ without direct links between genotype and phenotype. The goal of this review is to provide a roadmap to navigate shotgun metagenomic sequencing data and identify new candidate biosynthetic enzymes. We cover both computational and experimental strategies to mine metagenomes and explore protein sequence space with a spotlight on natural product biosynthesis. Specifically, we compare in silico methods for enzyme discovery including phylogenetics, sequence similarity networks, genomic context, 3D structure-based approaches, and machine learning techniques. We also discuss various experimental strategies to test computational predictions including heterologous expression and screening. Finally, we provide an outlook for future directions in the field with an emphasis on meta-omics, single-cell genomics, cell-free expression systems, and sequence-independent methods.
 Serina L. Robinson | Serina Robinson is an ETH Zürich postdoctoral fellow with Dr Jörn Piel and will start her independent career as a tenure-track group leader at the Swiss Federal Institute of Aquatic Science and Technology (Eawag) in autumn 2021. She obtained her PhD in Microbiology and MSc in Bioinformatics and Computational Biology from the University of Minnesota, Minneapolis, USA (advisor: Larry Wackett), where she applied machine learning and genome mining techniques to investigate β-lactone synthetases, a newly-discovered family of enzymes involved in natural product biosynthesis. Her current research focuses on the discovery of new biosynthetic enzymes from marine, freshwater, and wastewater metagenomes. |
 Jörn Piel | Jörn Piel studied Chemistry at the University of Bonn, Germany, and obtained a PhD in 1998 (advisor: Wilhelm Boland). After a postdoc with Bradley S. Moore and Heinz G. Floss he became group leader at the Max Planck Institute for Chemical Ecology in Jena, Germany, in 2000. From 2004–2013 he was associate professor at the University of Bonn and subsequently full professor at the Institute of Microbiology, ETH Zürich. His lab works at the interface of Chemistry and Biology, studying bacterial metabolism with an emphasis on microbial and biosynthetic ‘dark matter’, symbiosis, marine natural products, biosynthetic engineering, and chemical ecology. |
 Shinichi Sunagawa | Shinichi Sunagawa studied Biochemistry and Marine Ecology in Germany, and obtained his PhD in 2010 at the University of California, Merced, USA. After returning to Germany, he joined the European Molecular Biology Laboratory in Heidelberg as a postdoctoral fellow, and continued to work on ocean and human gut microbial communities as a research- and staff scientist. In 2016, he established the Microbiome Research Laboratory at the Institute of Microbiology at ETH Zürich, which combines bioinformatic and experimental approaches to integrate quantitative ‘meta-omics’ readouts with contextual information to study and the role of environmental microorganisms and mechanisms of host-microbial homeostasis. |
1. Introduction
1.1. The sequence–structure–function paradigm
The 1972 Nobel laureate in Chemistry, Christian Anfinsen, ended his Nobel lecture with the line, “It is certain that major advances in the understanding of cellular organization…will occur when we can predict, in advance, the three-dimensional, phenotypic consequences of a genetic message”. Nearly 5 decades later, predicting the phenotypic consequences of protein sequences remains a complex task. Significant progress has been made on the three-dimensional prediction front, however. In 2020, the deep learning algorithm AlphaFold2 achieved landmark results for the prediction of 3D protein structure from primary sequence. In a rigorous blinded global competition, AlphaFold2 averaged within 1.6 Å of the truth, achieving an error less than the width of one atom.1 To this news, Frances Arnold, 2018 Nobel laureate in Chemistry, reacted with, “Pretty impressive! Perhaps we can now move to the protein function problem?”.While accurate predictions for the 3D structures of many proteins from primary sequence are now within our grasp, understanding function from protein structure or sequence is far from solved. Even for Escherichia coli, one of the most well-characterized organisms on earth, >35% of genes lack experimental evidence of function.2 Moreover, the pan-genome, that is, the complete set of genes found among all strains of E. coli is estimated to contain >16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 different families of homologous genes.3 By these estimates, E. coli is still considered to have an open pan-genome since the species is undergoing constant gene acquisition and diversification.4 Our limited understanding of one of the world's most intensively-studied model organisms5 emphasizes the challenge in determining the functions of coding sequences not from organisms grown in monoculture in the laboratory but from metagenomic DNA from complex environments.
000 different families of homologous genes.3 By these estimates, E. coli is still considered to have an open pan-genome since the species is undergoing constant gene acquisition and diversification.4 Our limited understanding of one of the world's most intensively-studied model organisms5 emphasizes the challenge in determining the functions of coding sequences not from organisms grown in monoculture in the laboratory but from metagenomic DNA from complex environments.
1.2. Metagenomics: promises and perils
Metagenomics, a term first coined in 1998,6 refers to the study of environmental DNA (eDNA). This is not only limited to natural environments in the classical sense, but to essentially every sampling location conceivable, including the hindguts of termites,7 cheese rinds,8 and the International Space Station.9 Enabled by next-generation sequencing technologies, metagenomics quickly became a new scientific field in its own right, contributing to exponential growth in the size of sequencing repositories. In 2007, still relatively early years for metagenomics, a single study – the Global Ocean Sampling Expedition – nearly doubled the total number of protein sequences in public databases.10 The rate of increase in next-generation sequencing has far surpassed Moore's law and number of nucleotide base pairs (bp) in public repositories is estimated to reach exabase-scale (1018 bp) well within the next five years.11One of the major advantages of metagenomics is gaining access to genetic information about the uncultivated majority of microbes which still largely lack functional characterization.12 Metagenomics studies have reshaped our view of the tree of life13,14 and led to the identification of deeply rooted and metabolically-diverse lineages such as the DPANN archaea15 and candidate phyla radiation.16 Many uncultivated microbial phyla, including ‘Candidatus Tectomicrobia’,17 ‘Eelbacter’18 and ‘Angelobacter’18 have had remarkable biosynthetic potential revealed by metagenomics. In the case of ‘Ca. Tectomicrobia,’ heterologous expression enabled the experimental characterization of new biosynthetic pathways and products.17,19–21 However, the tantalizing promises of discovering new enzymology from metagenomes goes hand-in-hand with the challenges discussed in Section 4.2 of working with DNA from organisms that have eluded laboratory cultivation.
In this review, we aim to provide a bird's-eye view of tools and strategies for metagenomic enzyme discovery. We emphasize enzymes involved in natural product biosynthesis, but many proteins outside of biosynthetic contexts will also be discussed as examples for relevant discovery strategies. We will also cover a number of examples from microbial isolates and highlight techniques which may be useful in future metagenome mining efforts.
1.3. Definitions for enzyme discovery
Before diving into methods, we will first attempt to define metagenomic enzyme discovery. The simplest definition – characterization of new enzymes from eDNA – lacks sufficient resolution. What exactly is a ‘new’ enzyme? In this review, we conceptualize metagenomic enzyme discovery as a pyramid with three tiers (Fig. 1). The tip of the pyramid, which we refer to as de novo enzyme discovery, refers to the identification of entirely new types of biocatalysts. In other words, de novo enzymes must belong to protein folds or families without any functionally characterized members. To date, most examples of de novo enzyme discovery have come from culturable bacteria and fungi rather than eDNA and uncultivated microbes. Yet it is clear that there is significant unexplored diversity in protein families identified from metagenomes. Wyman et al. recently reported >118000 different protein domain families currently lacking functional characterization.22 About 6688 of these families were conserved in at least two separate taxonomic classes of organisms and ubiquitous in the environment including Tara Oceans23 and Human Microbiome Project24 metagenomes. This analysis was used to compile a ‘most wanted’ list of unknown protein families for experimental investigation.22 With regards to this most wanted list, it is interesting to note that biosynthetic enzymes often have a more discontinuous taxonomic distribution than primarily metabolic enzymes.25,26 Therefore the remaining 111312 protein domains not on the list with a sparser taxonomic distribution may actually be of greater interest for the natural products community. Regarding de novo discovery of enzymes with new structural folds, the Baker lab recently used metagenomic sequences to model more than 614 protein families with unknown structures, 137 of which have completely new protein folds.27 This study and others predicting 3D structures from metagenomic protein sequences28 demonstrates that our experimental survey of natural protein space is far from complete.
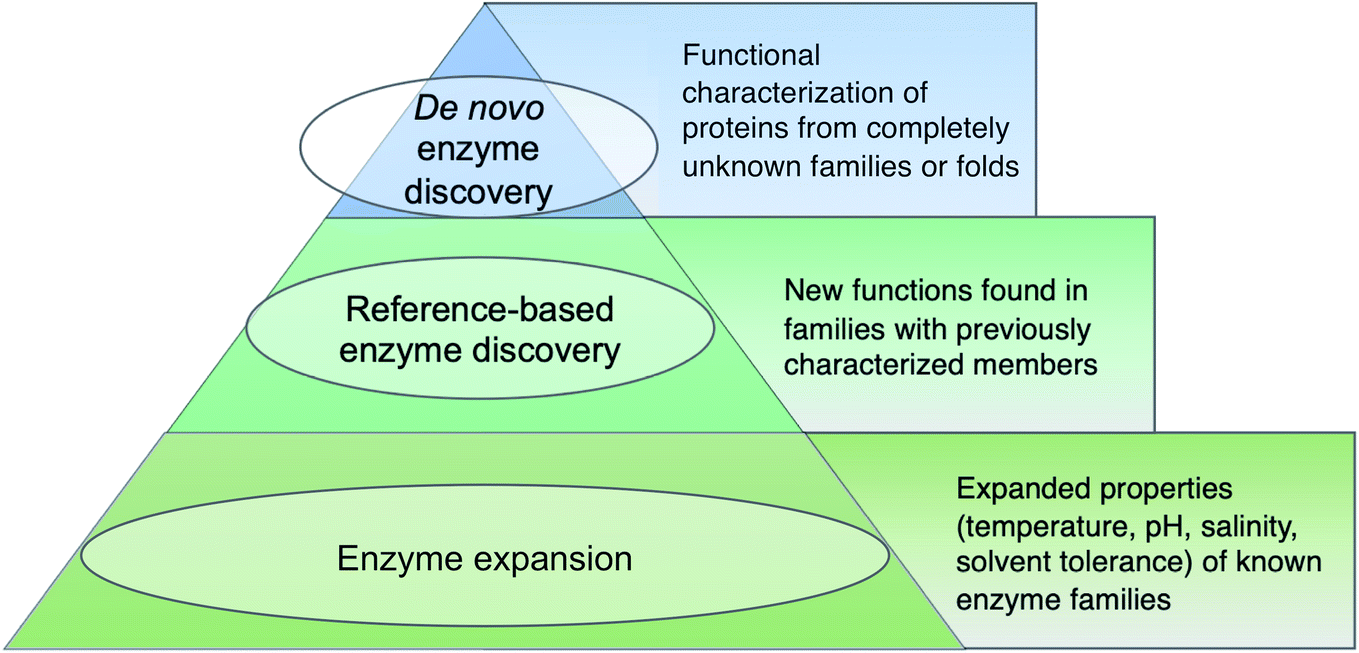 | ||
| Fig. 1 Tiered definitions of enzyme discovery. The hierarchical structure is not meant to reflect superiority of higher tiers rather it is a reference to the relative number of metagenomic enzyme studies falling within each category. | ||
The second tier in the pyramid, which we call ‘reference-based enzyme discovery’, is the characterization of new reaction types within the context of already discovered protein families (Fig. 1). One recent example of reference-based enzyme discovery is CreM, an ATP-dependent enzyme that installs diazo moieties in cremeomycin.29 CreM homologs are annotated in databases as acyl-CoA ligases but CreM from Streptomyces cremeus was experimentally found to use nitrite to catalyze N–N bond formation. Although functional discovery in this specific case was not aided by metagenomics, this is one of many reports of mis-annotated enzymes capable of catalyzing unprecedented reactions within well-established enzyme families.30,31 The distinction between reference-based and de novo discovery, although seemingly subtle, comes with unique challenges in each case. One major difficulty of de novo discovery is to determine functions for ‘hypothetical proteins’ or ‘domains of unknown function’ without any reference points for substrates, cofactors, or enzyme reaction classes. In reference-based discovery, however, one or more characterized enzymes within the protein fold or family is already known, but the newly discovered enzymes are actually functionally divergent. The comparison between these tiers is somewhat analogous to bugs in computer programming. In the de novo tier, an error is thrown with the cryptic error message: ‘hypothetical protein’. In reference-based enzyme discovery, the analogous situation is more like a ‘hidden bug’ in that the software functions normally and transfers functional predictions to proteins based on homology, but the functional annotation is incorrect.
The base of the pyramid in Fig. 1, representing the largest fraction of metagenomic studies so far, refers to the discovery of enzymes with different substrate specificities or preferred reaction conditions including temperature, pH, salinity, or solvent preferences. Although often described as ‘enzyme discovery’ in the literature, we will refer to cases where the properties of a known enzyme class are extended as ‘enzyme expansion’ for clarity. Perhaps the most famous example of enzyme expansion is the highly thermostable Taq polymerase from Thermus aquaticus.32 Substitution of the E. coli DNA polymerase with T. aquaticus polymerase for improved polymerase chain reaction (PCR) efficiency is viewed by many as one of the key breakthroughs that advanced the modern field of molecular biology. Although Taq polymerase was discovered before the advent of metagenomics, mining eDNA from extreme environments such as hot springs or hydrothermal vents to identify ‘extremozymes’ remains a useful strategy, particularly for industrial applications. Enzyme expansion studies are extensively reviewed elsewhere,33,34 and will largely not be covered here so as to focus on biosynthetic novelty.
1.4. Caveats and assumptions
Some important caveats must be mentioned for the scope of this review. We will focus on mining metagenomes for naturally occurring enzymes and will not cover non-natural enzymes accessed through engineering or directed evolution strategies. We will also focus mostly on bacterial enzymes encoded in biosynthetic gene clusters (BGCs) since these have been the most extensively studied by the natural products community, but we must emphasize the vast underexplored diversity of enzymes from archaea, fungi, plants, and other eukaryotes. Characterized biosynthetic enzymes from plants and other non-fungal eukaryotes are especially lacking. For example, the curated Minimum Information about a BGC (MIBiG) database (version 2.0)35 contains >1500 experimentally characterized BGCs from prokaryotes but less than 30 from plants and other eukaryotes, excluding fungi. This knowledge gap may be attributed to additional challenges of dealing with sequences from eukaryotes including lower genomic coverage, fewer reference genomes, exon–intron architecture, splice variants, unusual enzymology, unclustered genes, RNA editing, and the lack of methods for heterologous expression and gene inactivation. Moreover, eukaryotes also have a significantly higher percentage of intrinsically disordered proteins with long (>30 amino acid) disordered segments further complicating our understanding of the relationship between protein structure and function.36 Intrinsically disordered proteins, small proteins and peptides, and protein isoforms all lie in the gray area outside the classical field of enzymology and thus represent exciting areas for future investigation and potential enzyme discovery.Another important albeit obvious caveat for this review is that metagenomic DNA sequences are not fundamentally different from genomic DNA obtained from microbial isolates. Both are strings of nucleotides which come from biological systems. Architecturally, BGCs from metagenomic samples are largely indistinguishable from BGCs from the reference genomes of isolates apart from sometimes being more fragmented due to contig boundaries and errors introduced during assembly. Some metagenomic BGCs even have homologous clusters in the genomes of culturable organisms thereby offering promising routes to characterization as we discuss further in Section 4.2. Numerous studies have shown, however, that specialized metabolism is often limited to specific taxonomic groups.37,38 Thus, many new classes of biosynthetic enzymes and their corresponding natural products from deeply-branching, uncultivated lineages are likely only accessible through metagenomics or other cultivation-independent approaches.
2. Setting course: experimental design for metagenomics studies
In this section, we aim to provide a roadmap of in silico and experimental methods to access new enzymology from metagenomes with a focus on natural product biosynthesis. Although the main emphasis will be on enzyme discovery from shotgun metagenomic data, we will first provide a brief overview of activity-guided and PCR-based methods which are collectively referred to as functional metagenomics methods. Comprehensive reviews focusing on functional metagenomics approaches for natural products discovery are available,39,40 therefore only a brief overview of common methods is provided to allow comparisons with shotgun metagenomic sequencing.2.1. Activity-guided functional metagenomics
Activity-guided functional metagenomic library screening was one of the earliest methods developed in the field of metagenomics.6 This approach centers on the identification of clones, e.g., from fosmid, cosmid, or artificial chromosome libraries, that exhibit desired phenotypes. Common methods for detection of enzymatic activity includes using antibiotic resistance, zones of inhibition, or colorimetric or fluorimetric readouts, as will be discussed further in Section 4.3. Since this activity-forward workflow does not rely on sequence homology, it is particularly effective for de novo enzyme discovery. Activity-guided screening has also been widely used in enzyme expansion studies, particularly for industrially relevant families including lipases/esterases, cellulases/hemicellulases, chitinases, and amylases.33 There are a number of disadvantages associated with activity-based screening for natural product biosynthetic enzymes however (Table 1). Since many biosynthetic enzymes require specialized substrates or cofactors, general assays developed for primary metabolic enzymes are unlikely to detect activity. Moreover, the number of hits can be limited due to incompatibility in codon usage bias, metabolic requirements, or low expression levels in library hosts. Despite these limitations, activity-guided screening remains one of the most effective and popular methods for sequence-independent enzyme discovery.41| Methods of enzyme discovery | Shotgun metagenomic sequencing | Activity-guided screening | PCR-based screening |
|---|---|---|---|
| Pros | • Complete functional profile of an environment | • Can lead to detection of new enzymes or folds catalyzing known reactions | • Sensitive for low-abundance sequences |
| • Genomic context and taxonomy obtained through binning/assembly | • Well-developed methods to screen for industrially-relevant enzymes, e.g., lipases, cellulases | • Detect variation within a single gene family at the level of single nucleotide changes | |
| • Higher accuracy achievable with proximity-guided assembly and long-read sequencing methods | • Inexpensive | • Relatively inexpensive | |
| • Can be combined with other meta-omics analyses | • Activity-forward method guarantees enzymes are active and express well in E. coli | ||
| • Generally less biased than activity- and PCR-based methods | |||
| Cons | • High sequencing depth required to detect genes in low abundance | • Limited to genes and small to medium-sized gene clusters that are expressed in the screening host | • Requires conserved DNA motifs in target sequences |
| • Computationally-intensive assembly and binning | • Typically limited to types of reactions that can be screened rapidly | • Not effective for detecting novel enzyme seqences or folds | |
| • Challenging to infer function from sequence alone | • Can requires specific high-throughput screening equipment | • Little to no taxonomic information | |
| • No taxonomic information | • PCR-bias against GC-rich sequences | ||
| • Can only screen for one type of reaction/function at a time | Short reads make gene cluster context difficult to recover |
2.2. PCR-based functional metagenomics
As the name suggests, PCR-based functional screening relies on the use of degenerate primers for the amplification of genes from eDNA coding for protein domains of interest. PCR-based screening methods are highly-sensitive and throughput can be enhanced through the use of pooling and deconvolution strategies.42,43 Amplicon-based analysis of common biosynthetic markers including adenylation and ketosynthase domains have been used widely with success to detect new BGCs and natural products.44,45 In a notable example, a completely new class of calcium-dependent antibiotics, the malacidins, were detected by PCR-based screening of adenylation domains from soil metagenomes.45 The major drawback of this approach, however, can be summed up with the line, “you get what you screen for”. PCR-based screening relies on sequence homology to known biosynthetic domains thereby limiting the detection of entirely new enzyme classes. Moreover, PCR-based methods have inherent amplification biases against GC-rich sequences46 and for low-abundance taxa. Short functional amplicons are also typically not able to provide reliable information about the taxonomy of the source organism or co-occurrence with other neighboring genes (Table 1). To combat the latter, Libis et al. reported an innovative method termed CONKAT-Seq which relies on co-occurrence network analysis of targeted amplicon sequences.44 The core of the CONKAT-Seq workflow is position-barcoded domain amplification followed by statistical analysis of co-occurring biosynthetic domains to identify rare BGCs. Amplicon sequencing is also a relatively low-cost technique (Table 1). As sequencing costs continue to drop however, we anticipate shotgun metagenomics will further advance as a complementary alternative to functional metagenomics methods for enzyme discovery.2.3. Shotgun metagenomic sequencing
In contrast to the aforementioned methods, shotgun metagenomics refers to the direct, untargeted sequencing of eDNA. Methods for shotgun metagenomic sample preparation, sequencing, assembly, and analysis are covered in several comprehensive reviews.47–50 Compared to functional metagenomics (Table 1), less bias is typically introduced during shotgun sequencing since PCR amplification and library hosts like E. coli are not required. Shotgun sequencing is also generally less labor-intensive and yields sequencing data much faster than constructing metagenomic fosmid or cosmid libraries. However, shotgun sequencing alone will not provide phenotypic information, thus downstream cloning and heterologous expression steps are still required for biochemical characterization of enzymes from both shotgun and functional metagenomics methods. Some of the greatest challenges of shotgun metagenomics includes the requirements for sufficient quantity and quality of eDNA from complex environmental samples and adequate sequencing depth to detect and correct errors in individual reads. For the detection of BGCs from rare organisms, new workflows such as Samplix technologies,51 offer solutions for dealing with lower quantities of genetic material. Samplix techniques rely on indirect capture and sequence enrichment through microdroplet multiple displacement amplification of unknown sequences that flank short, desired detection sequences. Targeted enrichment methods for sequencing can be especially useful where longer reads from specific taxa or BGCs are sought from low amounts of eDNA.Key disadvantages of shotgun metagenomics using Illumina short-read sequencing, which is currently the most widely used technology, includes the computational cost, limitations, and inaccuracy of metagenomic assembly and binning. Complementary techniques for short-read assemblies such as Hi–C chromosome capture for proximity-guided assembly of short reads, have been used to obtain improved genome-resolved resolution of cow rumen52 and human gut microbial communities.53 Oxford Nanopore54 and PacBio HiFi55 methods for long-read sequencing56 can also be combined with short-read sequencing to dramatically improves the quality of (meta)genomic assemblies,57 particularly when dealing with large or repetitive BGCs. Regardless of the sequencing method, one key advantage of direct shotgun sequencing over large-insert libraries is that complete sequencing datasets are typically deposited in public databases. This process effectively crowdsources the analysis of metagenomes to different research groups around the world. As an example, Tara Oceans, one of the largest metagenomic sequencing initiatives to date, has prioritized making all sequencing datasets with detailed environmental metadata available for public analysis. Indeed, since the research schooner, Tara, first set sail in 2009, over 100 papers have been published by the project members alone. Different groups around the world have further analyzed the released datasets to probe countless aspects of global ocean ecosystems biology.23 This output demonstrates how a single meta-omics campaign has contributed to research findings spanning the fields of ecology, evolution, enzymology, oceanography, virology, biogeochemistry, and more.
Compared to activity- and PCR-based functional metagenomics screens, the number of studies in which enzymes were discovered from direct shotgun metagenome sequencing data are still relatively rare. In a recent review of metagenomic enzyme discovery in 2017, only seven studies identified new enzymes through direct metagenomic sequencing compared to >300 that used functional screening methods.33 With the increasing accessibility of metagenomic sequencing data, however, we predict the tide will continue to shift towards in silico enzyme prospecting of shotgun metagenomes.
2.4. Parallels with natural product research
The balance between functional metagenomics and shotgun metagenomics-driven enzyme discovery is somewhat analogous to the changing field of natural products research. Historically, microbial natural products were identified through activity-guided bioassays from cultured organisms. After the initial boom of discovery, re-isolation of the same natural product types became commonplace, particularly for better-studied taxa. In the post-genomic era, genome mining methods coupled with heterologous expression and MS-based molecular networking have emerged as powerful, complementary approaches to bioactivity screening. These techniques are useful for rapid de-replication of candidate compounds to limit rediscovery.58 Nonetheless, new natural products continue to be discovered regularly through classical bioactivity-guided screening methods. Similarly, we anticipate activity-based and PCR-based functional metagenomics techniques will remain important pillars for enzyme discovery and expansion. However, advances in bioinformatic algorithms and technologies applicable to shotgun sequencing data offers the promise of new routes for enzyme discovery.Specifically, we seek to highlight how enzymes involved in natural product biosynthesis can provide useful handles for combing through large-scale metagenomic datasets to gain functional insights into the secondary metabolism of uncultivated microbes. Our reasoning for the utility of biosynthetic gene products as handles is based on following criteria: (1) biosynthetic genes tend to cluster together. This enables taking a ‘guilt-by-association’ approach (Section 3.4) to predict enzyme function from genomic information. (2) The ability to predict chemical building blocks and moieties for many BGC types provides critical clues into the potential functions and substrates of biosynthetic enzymes. (3) Since secondary metabolism evolved from primary metabolism, secondary metabolic enzymes are particularly liable to be misannotated based on homology transfer from their primary metabolic functions. They are more likely therefore to be ‘hidden in plain sight’ by catalyzing different chemical reactions than their annotation suggests. Lastly, (4) natural products are some of the most complex non-polymeric chemical compounds known on earth. They also often contain a high number of stereocenters. Therefore, scaffolds require an exceptional diversity of biocatalysts to install regio- and stereoselective modifications. Amidst all this diversity, where do we begin?
2.5. Hotbeds for enzyme discovery
As a starting point, we will first ask the question, “are there hotbeds for enzyme discovery?” More specifically, we will investigate strategies to identify protein families with enriched biocatalytic diversity to increase chances of success for new functional discoveries. One strategy is to focus on structural folds that are easily repurposed, such as the ubiquitous TIM-barrel scaffold used by at least 15 distinct enzyme families.59 Another route is to investigate protein families that tend to be more promiscuous, that is, they are able to catalyze one or more side-reactions in addition to their main reaction. Extensive work by Tawfik, Copley, Thornton, and others have suggested alternative functions arise from a combination of changes in the protein sequence that alter both substrate binding and the overall chemical reaction.60–62 In the case of phosphatases and sulfatases, particularly promiscuous enzyme families, Pabis et al. found that increased structural and/or electrostatic flexibility in their binding pockets to allow more unspecific accommodation of substrates.63 Ding et al. and others have proposed that enzymes with radical mechanisms may be more promiscuous than other enzyme classes.64 Clearly, the reasons underlying promiscuity are often enzyme family-specific,65 making it difficult to draw broad generalizations about relationships between enzyme evolution and biocatalysis. Regarding the promiscuity of enzymes in natural product biosynthesis, we refer readers to excellent recent reviews on secondary metabolic enzyme evolution.25,66For this review, we sought to systematically explore the diversity of different reactions catalyzed by common natural product biosynthetic enzymes building on the work of Veprinskiy et al. and others.67 We first extracted all protein family (PFAM) domains from the MIBiG database35 and quantified PFAM reaction diversity based on the number of unique Enzyme Commission (EC) codes to the level of two digits that were associated with each PFAM domain. EC digits correspond to varying levels of resolution for enzyme classification. The first EC digits categorize enzymes into seven large reaction classes: (1) oxidoreductases, (2) transferases, (3) hydrolases, (4) lyases, (5) isomerases, (6) ligases and (7) translocases. The second digit covers broad reaction type, e.g., EC 2.7, the most common reaction in our dataset, indicates enzymes that transfer phosphorus-containing groups. Associations between 1931 PFAM domains extracted from MIBiG and 8256 high-confidence ECDomainMiner predictions68 were cross-referenced and visualized as a heatmap (Fig. 2). To constrain heatmap size, we only display PFAM domains associated with 10 or more different EC classes (to the level of two EC digits) and occurring in at least 30 different BGCs in MIBiG. Fig. 2 highlights that oxidoreductases (EC class 1) tend to have the highest number of distinct within-EC-class reactions. Indeed, many redox enzymes including cytochrome p450 monooxygenases, aldo-keto reductases, short chain dehydrogenases, and Rieske oxygenases are known to introduce a wide variety of modifications in natural product scaffolds.69–71 In one notable example, the NAD(P)H-dependent oxidoreductase, IkaB, works in tandem with alcohol dehydrogenase-family enzyme, IkaC, for polycyclization of the complex macrolactam structure of ikarugamycin (Fig. 4A).72,73
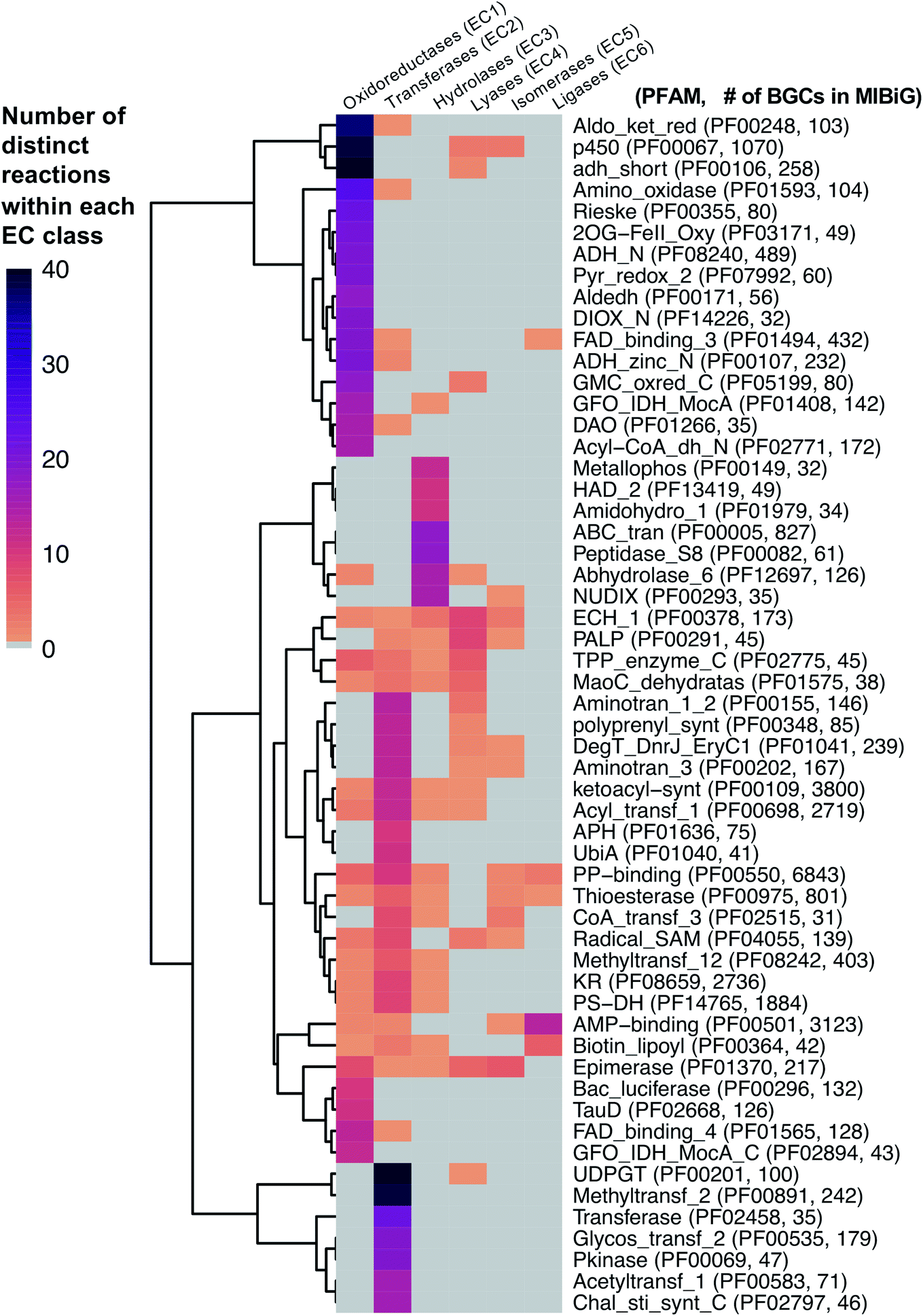 | ||
| Fig. 2 Heatmap of PFAM domains extracted from the MIBiG database35 cross-referenced with predicted EC reactions for each PFAM domain using ECDomainMiner.68 Color intensity corresponds to the number of distinct predicted reactions (at the level of two EC class digits) associated with each PFAM domain. Y-Axis heatmap labels include standard PFAM domain abbreviations and PFAM family ID and number of occurrences of each PFAM domain in MIBiG BGCs in parentheses. X-Axis heatmap labels refer to the standard top-level EC number codes (excluding EC7 translocases which were not included in this analysis). | ||
Cytochrome p450 monooxygenases stand in Fig. 2 as one of the most promiscuous and the most prevalent PFAM domains in MIBiG with over >1000 examples found in experimentally characterized BGCs. Cytochrome p450s have been shown to modify compounds from nearly every major natural product class74 and also play a central role in xenobiotic metabolism and biodegradation. Cytochrome p450s catalyze a dizzying array of transformations including epoxidation, N- and S-oxidation, C–C bond cleavage, desaturation, and N-, O-, and S-dealkylations.75 Additionally, some naturally occurring cytochrome p450s catalyze Baeyer–Villiger type oxidations or phenolic couplings.75 A new class of cytochrome p450 enzymes was recently reported to catalyze biaryl linkages of tripeptides in a BGC containing the smallest synthesized and post-translationally modified peptide (RiPP) precursor-encoding gene (18 bp) reported to date.76 Engineered p450s have dramatically expanded beyond the limits of naturally occurring biocatalysts to catalyze olefin cyclopropanation,77 carbon–silicon,78 and carbon–boron bond formation.79 Structural analysis of cytochrome p450 monooxygenases has provided insights into the reasons underlying their remarkably wide reaction range including the highly-reactive activated oxygen species generated during the catalytic cycle and unusually dynamic elements of the core protein scaffold.69
Transferases (EC class 2) also stand out in Fig. 2 as catalyzing the highest number of across-EC-class reactions as well as remarkable within-EC-class diversity. Among many possible examples, we highlight radical S-adenosyl-L-methionine (SAM) enzymes (PF04055) for their across-EC-class promiscuity. Radical SAM enzymes are notorious for catalyzing C–C bond formation and breakage to install diverse modifications across a wide range of natural product scaffolds.80 In particular, radical SAM enzymes post-translationally modify many RiPPs through epimerization of L- to D-amino acids,19,81 excision of tyramine to form α-keto moieties,82 and formation of intramolecular crosslinks including strained cyclophane macrocycles.83 Radical SAMs also play a role in the biosynthesis of hypermodified tRNA bases84 and nucleoside-based natural products through C–C bond extension at C5′ of ribose rings to connect nucleosides to structurally diverse functional groups.85
A number of other enzyme classes not covered in detail here also were predicted to have remarkable across-EC-class reaction diversity. Thioesterases, phosphopantetheine-binding domains, epimerases, and crotonases are predicted to catalyze reactions spanning 5 different EC classes. Overall, our analysis suggests that targeted characterization of hotbed PFAM domains such as cytochrome p450s and radical SAM enzymes from candidate metagenomic BGCs can be a strategy to hedge bets for the identification of new biochemistry. Moreover, it is clear we have only uncovered the tip of the iceberg even for reference-based discovery of new enzymology from BGCs.86 To further facilitate de novo enzyme discovery, applying ECDomainMiner or similar tools to predict EC classes for PFAMs of unknown functions may yield initial insights into relative within-EC-class or across-EC-class reaction diversity of underexplored areas of sequence space.
3. On the road: computational methods for enzyme function prediction
3.1. Querying metagenomic databases
In the next sections, we will cover computational methods to predict new enzyme functions within protein families, such as the hotbeds identified in the previous section. Most shotgun metagenomics studies start with sampling the environment, extracting eDNA, and sequencing. Downstream bioinformatic processing steps must then be carried out make metagenomes publicly available in public repositories such as the Joint Genome Institute Integrated Microbial Genomes and Microbiomes resource (JGI IMG/M),87 iMicrobe,88 or MGnify.89 We specifically highlight MGnify as a consolidated resource which the authors highlight as being developed for ‘searching the microbial dark matter’. One benefit of MGnify is the ability to query metagenomes with Hidden Markov Models (HMMs) rather than using basic sequence alignment-based search methods such as BLAST90 or DIAMOND.91 While both of these are effective and quick methods for a first pass analysis, HMMs are particularly useful for the identification of more remote homologs. Profile HMMs can detect distant sequences more sensitively based on their underlying probabilistic models, enabling detection of enzymes at the boundaries of protein families. Rather than being based on just one single query sequence, HMMs are built from sets of aligned sequences and custom HMMs can easily be built for smaller clades of evolutionarily related proteins to more accurately mine metagenomes for specific subfamilies. For example, Neubauer et al. built a custom HMM based on known tryptophan halogenase sequences.92 The authors then queried metagenomes from public metagenomic databases and identified 254 HMM hits. One of these flavin-dependent halogenases was found to convert indole to 3-bromoindole. Notably, the enzyme preferred bromination even in the presence of excess chloride. The authors note, however, that the relatively low specific activity (2.5 mU mg−1) suggests indole may not be the natural substrate, which further highlights the challenges of determining substrate and function solely based on sequence homology.A complementary approach to gain genome-resolved information about shotgun metagenomic datasets is the reconstruction of metagenome-assembled genomes (MAGs). Nayfach et al. recently published >52000 medium- to high-quality MAGs from >10000 metagenomes from various environments on earth.93 This study was estimated to have expanded the known phylogenetic diversity of bacteria and archaea by 44% and provided insights into their predicted biosynthetic potential. Analysis by antiSMASH94 led to identification of >100000 BGCs including the single largest candidate BGC known with 62 different modules containing polyketide synthase (PKS) or nonribosomal peptide synthetase (NRPS) domains in the soil-derived MAG for an Acidobacterium. This large BGC still awaits functional characterization. Studies of this scale underpin both the challenges and opportunity of metagenomics from the sheer quantity of data that are generated. Scientists face a Sisyphean task of novel functional enzyme discovery from such large metagenomic resources. There is a distinct need for improved platforms to facilitate and accelerate novel enzyme discovery, building on the foundation of existing targeted tools like MGnify89 and ANASTASIA.95 In the next sections, we will provide an overview of additional in silico and experimental methods which can be used to systematically probe large metagenomic datasets (Fig. 3).
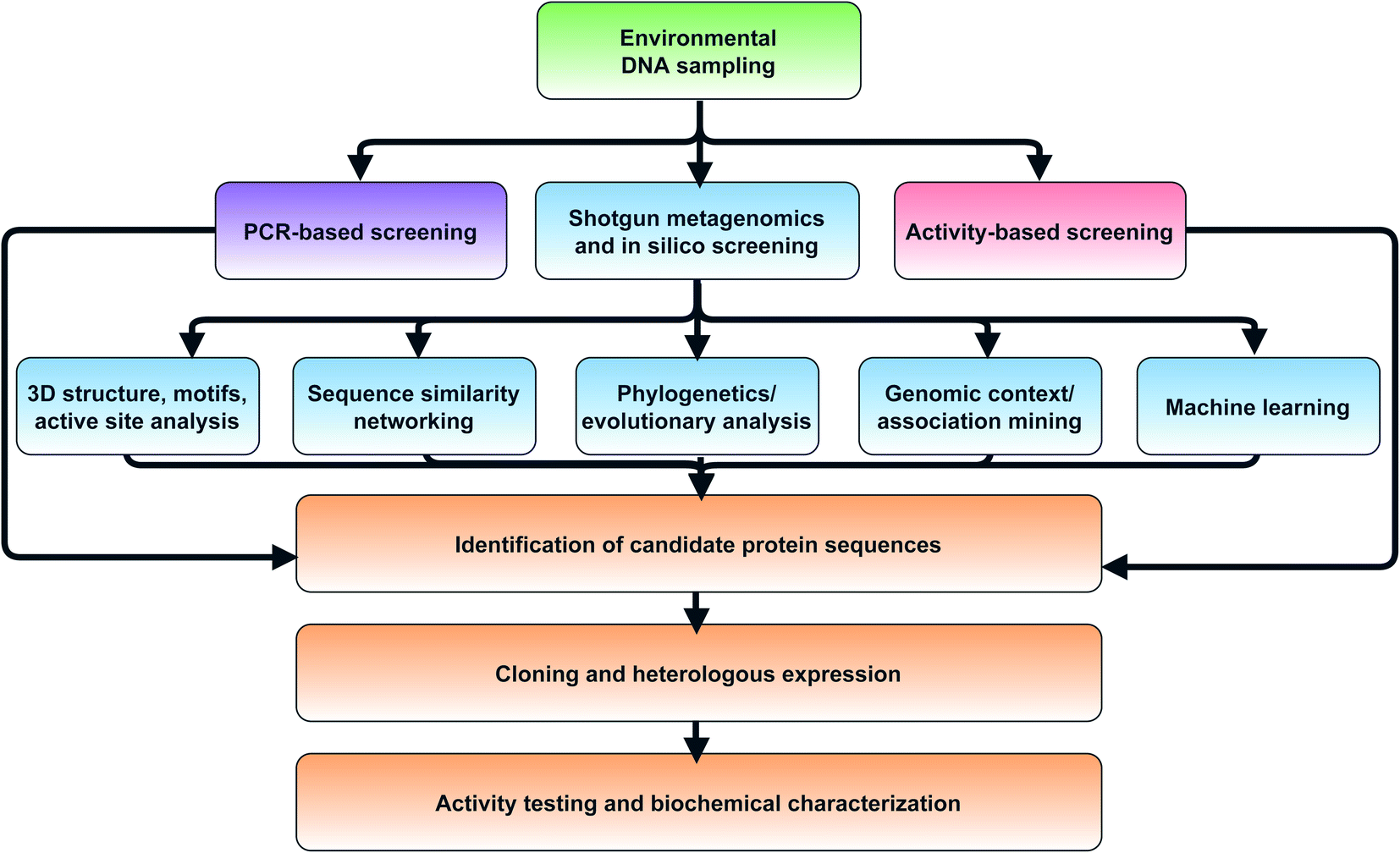 | ||
| Fig. 3 Flowchart of strategies for in silico selection and experimental characterization of candidate metagenomic enzymes. | ||
3.2. Phylogenetics
Dating back to Darwin's first sketches of phylogenetic trees,96 the study of evolutionary relationships has long been a central tenet of biology. After the genetic code was cracked, phylogenetic analysis could be conducted at DNA and protein level instead of only morphological traits. Across these different scales, the overarching goal of phylogenetics has remained constant: to understand relationships between shared functional traits, which includes functionally related proteins. Unlike standard taxonomic markers like 16s rRNA genes, many classes of biosynthetic enzymes tend to group by preferred substrates and/or functions rather than source organism.97,98 This makes phylogenetics a useful approach for reference-based biosynthetic enzyme discovery, particularly when seed sequences of characterized enzymes are aligned with uncharacterized (meta)genomic sequences. Curated databases such as Swiss-Prot,99 the Protein Data Bank100 and literature searches are useful to acquire characterized seed sequences for protein families. Sequences that form distinct phylogenetic clades without seed sequences are often interesting places to start for experimental characterization as they may prefer different substrates or perform new functions.Detailed methods for phylogenetic and phylogenomic analysis of metagenomic sequencing data are reviewed elsewhere.101,102 Here we will briefly touch on commonly used tools and their limitations in the context of metagenomic enzyme discovery. One key disadvantage is that phylogenetic trees are only as accurate as the underlying multiple sequence alignments. Countless tools for generating sequence alignments including MUSCLE,103 MAFFT,104 and Clustal Omega105 are available. Independent of alignment method, an often overlooked but important intermediate step is manually inspection of sequence alignments and trimming large gap regions with tools such as trimAl106 or Gblocks before treeing.107 Another limitation of phylogenetic analysis is the computational cost of estimating trees from large sequence alignments. FastTree108 overcomes this disadvantage by using heuristic methods to constrain the tree search space and make approximate maximum-likelihood estimations thereby dramatically cutting treeing time. Surprisingly, for many applications, FastTree is often nearly as accurate as more rigorous maximum-likelihood methods109,110 such as PhyML or RaxML that make fewer assumptions but require orders of magnitude more time to run.111 The recently released RaxML-NG also combines the improved accuracy of RaxML with computational scalability for the analysis of large (meta)genome-scale datasets.112 Another popular phylogenetic tool is IQ-Tree, which includes the added features of automated model selection and ultra-fast bootstrap approximation.113 For visualization and advanced annotation options of these phylogenetic trees, we recommend the widely-used Python ETE 3 toolkit114 or ggtree in R.115
Ancestral sequence reconstruction adds another dimension to phylogenetic analysis by using contemporary protein sequences to infer their evolutionary history116 such as how biosynthetic enzymes might have arisen from primary metabolic enzymes. Ancestral reconstruction of adenylate-forming enzymes suggested that secondary metabolic enzymes such as β-lactone synthetases and nonribosomal peptide synthases arose from protein scaffolds similar to contemporary primary metabolic enzymes such as CoA ligases.97 Hendrikse et al. reconstructed the evolutionary history of diterpene cyclases and experimentally characterized the predicted ancestral sequences. They reported the ancestral enzymes had increased thermostability and broader substrate specificity, both of which are common features of ancestral sequences that may promote the evolution of new functions.117 Probabilistic web-based tools like FastML make ancestral sequence reconstruction accessible to non-experts.118 Bayesian phylogenetic methods are also powerful for understanding evolutionary relationships, as exemplified by a phylogenomic study of lanthipeptide synthetases, a family of RiPP maturases that introduce sulfur bridges into peptides.119 Through Bayesian phylogenomic analysis of lanthipeptide BGCs, Zhang et al. unexpectedly found that the sequences of lanthipeptide precursors as well as maturases played a decisive role in determining the structure of the final natural products. Overall, phylogenetics remains one of the first and most fundamental stops on the roadmap for enzyme bioprospecting from metagenomes (Fig. 3 and Table 2).
| In silico methods for enzyme discovery | Phylogenetics | Sequence similarity networking | Genome neighborhoods and protein interaction networks | 3D-structural methods, motifs, and active site residues | Machine learning |
|---|---|---|---|---|---|
| Pros | • Longstanding, well-established methods to investigate functional relationships between proteins | • Intuitive graphical representation of thousands of protein sequences simultaneously | • Guilt-by-association methods can reveal new functional relationships for proteins independent of primary sequence | • Variations in active site architecture can have large consequences for biocatalysis → handles for discovery | • Deep learning, transfer learning, and autoencoding methods useful to learn complex or hidden relationships for functional inference |
| • Insights into evolution of protein families, e.g., through ancestral sequence reconstruction | • Allows users to quickly identify clusters without known representatives in sequence space | • Unusual co-occurring domains or interacting proteins are new targets for enzyme discovery | • Structural motifs are useful for searches independent of full-length primary sequence | • Capable of recognizing patterns in big metagenomic datasets | |
| Cons | • Heavily influenced by the quality of the underlying sequence alignment | • Pruning of SSNs by BLAST e-value can be subjective | • Analysis of gene neighborhoods from metagenomes requires assembly → introduces errors and not always possible to recover flanking genes for lowly-abundant organisms | • Similar structural folds catalyze a wide range of different reactions | • Requires a large quantity of ‘labeled’ e.g., experimentally-verified training data |
| • Not all biosynthetic domains have a consistent or strong phylogenetic signal | • Unclear how to handle or gain functional insights from ‘singletons’ | • Relatively few structures solved from metagenomic sources | • Classification systems limited in their ability to predict entirely new enzyme functions |
In the context of natural product biosynthesis, many tools have been developed to predict biosynthetic enzyme substrate or function using phylogenetic methods, as recently reviewed by Adamek et al.120 The Natural Product Domain Seeker (NaPDoS) makes structural inferences about natural products based on phylogenetic analysis of ketosynthase and condensation domains.121 Other phylogeny-based methods such as PrediCAT122 for NRPS adenylation domains and TransATor for trans-acyltransferase PKS prediction123 both enable natural product structural predictions for these respective classes. Other classes of biosynthetic domains, however, are less amenable to making phylogeny-based structural or functional inferences. For example, type I thioesterase domains do not have a strong phylogenetic signal for the substrate class or offloading chemistry.124,125 Plant sesquiterpene synthases are similar and tend to group based on taxonomy of the source organism rather than chemical similarity of carbocation product type.126 Even for biosynthetic domains with a strong signal, there are always phylogenetic outliers which present challenges for substrate or final natural product structure classification.
For phylogeny-based genome mining to detect new enzyme classes, we highlight two complementary software tools, EvoMining and CORASON.127,128 EvoMining is based on the premise that primary metabolic enzymes often undergo duplication or horizontal gene transfer events, both of which may lead to the emergence of new enzyme functions in secondary metabolic pathways. EvoMining has been used for example to find enzymes that catalyze similar chemical reactions but perform different cellular functions,129 or to discover new enzymes involved in the biosynthesis of arseno-organic metabolites.130 A related tool, CORe Analysis of Syntenic Orthologs to prioritize Natural product BGCs (CORASON),128 generates cluster variation databases for intuitive phylogenetic visualization of core and ancillary genes in BGC families. Overall, while phylogenetic analysis is a key first step, it is often more informative when used in combination with other approaches as will be discussed herein (Table 2).
3.3. Sequence similarity networking
Compared to phylogenetics, sequence similarity networks (SSNs) are relatively new methods for the visualization of protein families and superfamilies. First published for the purpose of protein superfamily analysis in 2009,131 SSNs are graphs that display relationships between protein families. SSNs are usually generated with an all-by-all BLAST search of a custom sequence set and visualized as a graph where nodes are protein sequences, and each edge represents pairwise sequence similarity. Typically, SSNs are pruned by setting different protein similarity score thresholds to reveal smaller clusters of protein subfamilies. As with phylogenetics, it is useful to include seed sequences of characterized enzymes in SSNs to serve as anchor points when seeking to identify relationships between enzyme families or subfamilies. In a massive enzyme screening study from soil and vanilla pod metagenomes, SSNs were used to identify the location of new functional triesterase hits in multiple unexplored protein family subclusters spread across three different protein superfamilies.132 SSNs have also been used in combination with phylogenetics to propose the nitroreductase protein superfamily arose from the radial divergence of functional diversity from a minimal cofactor-binding scaffold.133 These examples demonstrate the utility of SSNs to identify both known and unknown protein subfamily clusters as candidates for experimental characterization.A major advantage of SSNs is the ability to quickly visualize the relationships between thousands of protein sequences simultaneously. Compared to a bootstrapped maximum-likelihood phylogenetic tree, SSNs are typically faster to compute and can be interactively visualized using the open-source software, Cytoscape, which provides a friendly Graphical User Interface.134 A downside of the point-and-click Cytoscape software is that workflows are often tedious to reproduce, particularly for large networks with thousands of nodes. With the release of the CyREST API, popular high-level languages such as Python and R can now be used to program reproducible SSN workflows.135,136 Alternative network analysis packages such as igraph are also popular and available for Python, R, and C/C++.137 For users without programming experience, the Enzyme Function Initiative Enzyme Similarity Tool (EFI-EST) was the first web-based application enabling automated construction of sequence similarity networks.138
A key downside of SSNs is the bias that can be introduced during the selection of similarity thresholds to prune networks, most commonly based on BLAST e-value. BLAST e-values are dependent on the size of the sequence database and comparisons of e-value thresholds between SSNs generated using databases of different sizes is misleading. Moreover, various types of graph layouts for SSNs can lead to different interpretations. Therefore, we recommend users make the sequences, code, and networks over the full range of possible layouts and BLAST e-values available on a publicly available scientific image repository such as Zenodo. This promotes data transparency and limits the cherry-picking of specific e-values or network topologies.
3.4. Gene context and interactions
Gene context is an often underemphasized but highly effective method for enzyme discovery especially for natural product biosynthesis. Flanking genes can often provide insights into substrates, cofactors, and natural product bioactivity. For example, a new family of cobamide-remodeling enzymes widespread in the human gut microbiome was identified based on genome context analysis of a coding sequence of unknown function flanked by cobamide biosynthesis and salvaging genes.139 To automate genome neighborhood analysis, a widely used addition to the EFI-EST is the Genome Neighborhood Tool (GNT).140 EFI-GNT generates genome neighborhood networks and allows for rapid visual assessment of genome context. It also conducts statistical analysis of gene co-occurrence to identify possible functional linkages. For natural product BGCs we also recommend specific tools such as BiG-SLICE141 and BiG-SCAPE128 designed to identify and group BGCs into gene cluster families. BiG-SCAPE is integrated with CORASON (Section 3.2), thus combining the power of phylogenetics with neighborhood clustering methods. BiG-SLICE is specifically designed to handle massive numbers of BGCs by representing them in Euclidean space rather than by pairwise comparison.141 This dramatically cut runtime to enable clustering of over one million BGCs from metagenome-assembled genomes. Based on its ‘BiG’ savings in computational cost, BiG-SLICE is therefore particularly well-suited for analysis of metagenomes for genome-context guided enzyme discovery. There are also numerous genome context tools available for specific natural product classes. For example, RODEO142 and RiPPeR98 are useful to identify new RiPPs and maturases based on genomic context. Although RODEO is targeted towards RiPPs, it is not restricted to them and can be used generally to rapidly pull genome neighborhoods for any set of query sequences from public databases. Flanking genes are provided in tabular format for downstream PFAM co-occurrence analysis, phylogenetics, and SSN generation.Genome neighborhood context can also provide insights into natural product bioactivity and guide the identification of new targets and self-resistance genes. The Antibiotic Resistance Target Seeker (ARTS) is one automated approach to identify known and potentially new self-resistance targets through analysis of gene proximity, duplication, and diversification events.143 Culp et al. recently used genome context-guided detection of known resistance genes combined with phylogenetic analysis to identify a divergent clade of glycopeptide antibiotic BGCs lacking well-characterized self-resistance genes.144 This led to the discovery of a completely new mode of action for a divergent clade of glycopeptides represented by complestatin and a novel antibiotic, carbomycin. This multi-pronged approach of genome context mining and phylogenetic analysis often yields a more holistic picture of BGC divergence and evolution, thereby guiding selection of candidate enzymes and cellular targets for experimental characterization.
More generally, the identification of gene functions based on genomic context has been termed a ‘guilt-by-association’ approach.145 One broad use platform that relies on guilt-by-association methods is the STRING web resource.146 STRING provides an intuitive interface for functional analysis of proteins including the prediction of protein–protein interactions through text mining of scientific literature and associations inferred from genomic context, co-expression data, or gene orthology to model organisms. Although STRING is not specifically targeted towards metagenomics or natural product biosynthesis, it can be used to predict–protein interactions such as for MbtH-like proteins in NRPS systems.147 A more specific tool, CO-ED, is useful for network analysis and identification of unusual co-occurring domains in multi-domain proteins including megasynthases commonly involved in natural product biosynthesis.148 CO-ED relies on PFAM information as inputs which can be extracted from (meta)genomes using PfamScan.149 CO-ED highlights which co-occurring enzyme domains are already found in public databases (e.g. MIBiG,35 UniPROT,150 or BRENDA151), and which combinations have not yet been characterized. As a proof-of-principle, CO-ED analysis of the Pseudoalteromonas rubra genome identified an unusual nitroreductase-ThiF PFAM domain pair in a protein termed OxzB. Heterologous expression of oxzB and its upstream gene oxzA in 5 different organisms resulted in production of pigmented yellow natural products with unusual oxazolone moieties. In vitro characterization of OxzB revealed the nitroreductase and ThiF-like domains catalyze the oxidation and cyclization of N-acyl amino acid substrates, respectively, to form oxazolone heterocycles (Fig. 4B). Oxazolone-forming enzymes were previously unknown in nature, thus CO-ED analysis of protein domains facilitated biochemical discovery of the first oxazolone synthase.148
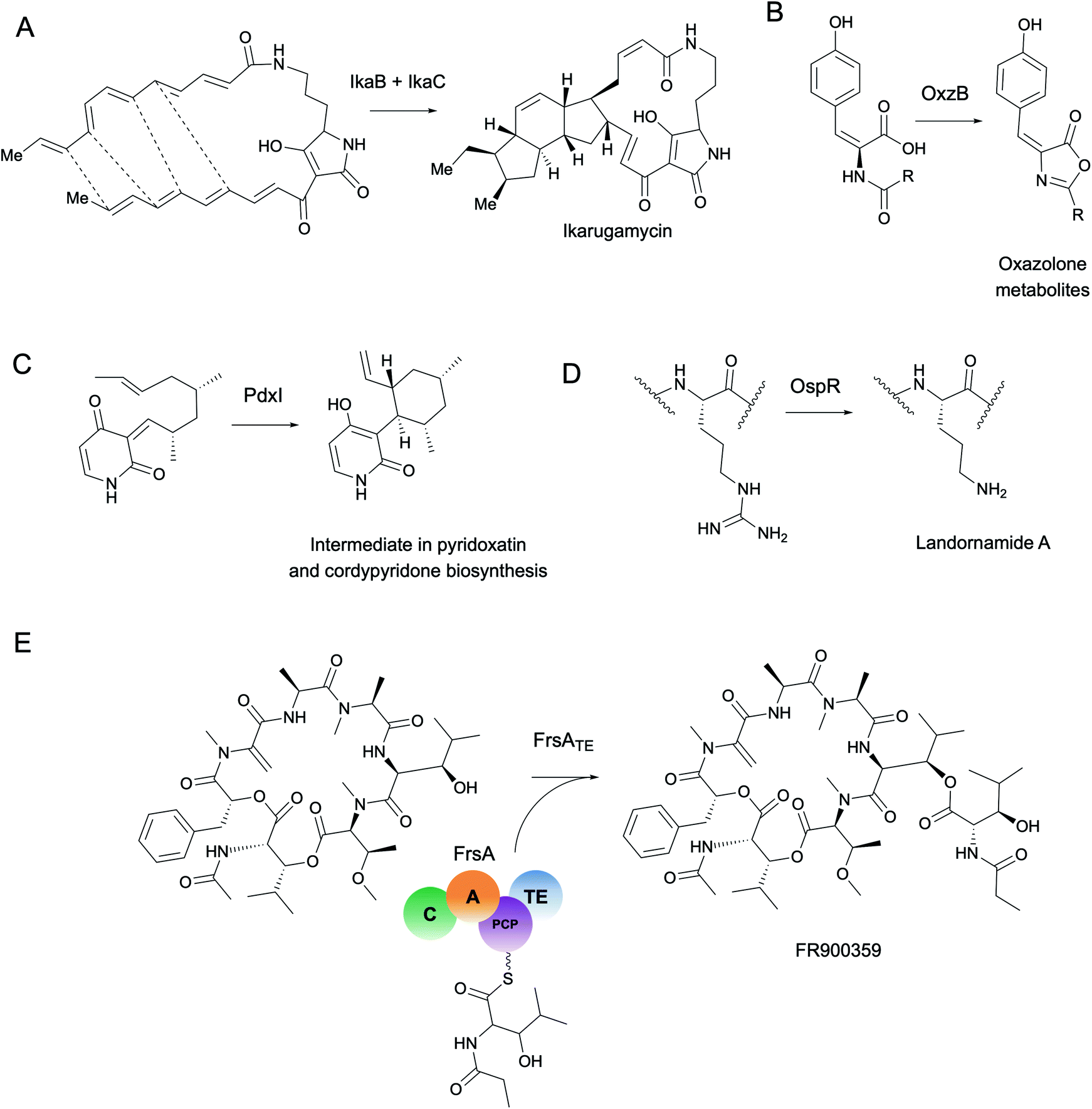 | ||
| Fig. 4 Selected enzymes highlighted in this review. (A) IkaB oxidoreductase involved in ikarugamycin polycyclization. (B) ThiF-nitroreductase di-domain enzyme, OxzB, catalyzes cyclization of oxazolone-containing metabolites with homologs detected in metagenomes from various environments (mainly marine). (C) PdxI catalyzes an alder–ene reaction to form a vinyl cyclohexane intermediate in biosynthetic pathways for fungal alkaloids including pyridoxatin and cordypyridones. (D) Arginase-family enzyme, OspR, promiscuously installs ornithines in the backbones of peptide natural products. OspR homologs were characterized from various microbial isolates and from the uncultivated phylum ‘Candidatus Wallbacteria’ from groundwater metagenomes. (E) FrsA thioesterase domain originally detected in an uncultivated leaf symbiont catalyzes intramolecular thioesterification of the Gq protein inhibitor FR900359. | ||
3.5. 3D-structure based methods
Previously, the inclusion of structural information to infer metagenomic enzyme function was hampered by the lack of solved protein structures. Rooted in the assumption that novel protein folds are more likely to perform novel functions, high-throughput protein structural characterization campaigns were initiated around the globe to catalogue protein structural space.152 Still, these efforts focused disproportionately on culturable organisms. As of January 2021, less than 0.3% of entries in the PDB were tagged as belonging to metagenomes or uncultured organisms. Moreover, while these high-throughput structural genomics initiatives have solved thousands of structures, they surprisingly yielded far fewer completely new protein folds than expected.153 Out of the vastness of protein conformations given all possible amino acid combinations, only a small fraction of this is represented in biological macromolecules, at least in organisms interrogated to date. It remains to be seen if and how much of structural and functional protein space still awaits discovery within the uncultivated majority of microbial life. The fact that even the most conserved protein folds identified to date are able to catalyze a variety of different reactions further underpins that we are only at the beginning of understanding how the multi-dimensional space of enzymes affects catalytic diversity. It is clear that even powerful structure prediction tools like AlphaFold2 will not solve the ‘function’ aspect of the sequence–structure–function problem alone.Nonetheless, secondary, tertiary, and quaternary structures of proteins can yield critical insights into function beyond primary sequence. Many protein families involved in natural product biosynthesis including RiPP recognition elements,154 adenylate-forming enzymes,155 and thioesterase domains124 share the same highly conserved structural fold but relatively low amino acid sequence similarity with other members of the family. Not surprisingly, for many enzyme families, structural alignment tools such as MAMMOTH,156 MATRAS,157 and Caretta158 yield significantly more accurate alignments than purely sequence-based alignment methods.159 Although AlphaFold2 is currently not publicly available, existing web-based homology modeling tools including Phyre2,160 I-TASSER,161 and SWISS-MODEL162 can be used to provide insights into predicted structural fold of metagenomic sequences. Recently, a deep-learning structure prediction pipeline was used to model Tara Oceans metagenomic sequences across different ocean depths and implicated the involvement of a ubiquitous protein family (PF15461) in photosynthesis.28 Structural modeling is often the first step towards detecting active site residues and structural motifs which can play a disproportionately large role in determining protein function as will be discussed in the next section.
3.6. Motifs and active site residues
Enzyme active sites only occupy a small fraction of the volume of a full-length protein folded in 3D space. Compared to the rest of the protein, catalytic residues are typically limited in their identity and arranged in conserved architectures.163 Perhaps the most famous example of active site conservation is the Ser-His-Asp catalytic triad used by alpha/beta hydrolases as well as several other protein folds including the subtilisin and chymotrypsin folds.164 This same triad hydrolyzes over 17 different reaction mechanisms spanning nearly every type of EC class. The multifunctionality of the Ser-His-Asp triad in particular is attributed to its ability to accommodate a wide range of substrates which can have different chemical interactions with the same key catalytic residues.165 Only about half of the enzymes with Ser-His-Asp triads had architectural differences in the active site such as changes in hydrogen bond partners or acids/bases for new mechanisms; the rest were driven by substrate chemistry alone.165 This study is just one example of how the same active site architecture can catalyze remarkable chemical diversity, making total prediction of function from protein active site alone challenging, if not impossible.Often altering even one residue can be sufficient to change the substrate specificity or enantioselectivity of an enzyme.166,167 Protein engineers are well aware of the fact, however, that making changes in the active site can have dire consequences for enzyme activity. The high-risk, high-reward task of active site modification often leads to countless evolutionary dead-ends. As a complement to engineering studies, characterization of naturally occurring active site variants that are conserved across different (meta)genomes provides an alternative route for enzyme discovery. As a striking example of the importance of active site variants, Ohashi et al. discovered several new enzymes originally annotated as O-methyltransferases, e.g., LepI168 and PdxI,31 which catalyze various types of pericyclic reactions in the biosynthesis of fungal alkaloids (Fig. 4C). Alteration of a single residue (V413M) in PdxI was able to shift the selectivity away from the Alder-ene reaction towards a more energetically favorable hetero-Diels–Alder reaction.31 Mutations of other residues in the PdxI active site could further tune periselectivity and regioselectivity and highlighted how even subtle changes can dramatically affect the final structures of natural products.
Studies targeting active site variants have not yet been widely applied to the task of enzyme discovery from shotgun metagenomes. Aberrant active site architectures are typically only remarked on during enzyme characterization following activity-based screening. For example, a divergent catalytic triad in an acid-stable endoglucanase was reported from activity-based screening of an soil metagenomic library.169 For detection of active site residues without knowledge of the enzyme class or function, tools such as CASTp for automated detection of active site pockets are useful.170 Comprehensive databases such as the Mechanism and Catalytic Site Atlas (M-CSA) catalogue known active site architectures and mechanisms.171 As of December 2020, the M-CSA contains nearly 1000 hand-curated entries representative of >73k Swiss-Prot entries and >15k PDB structures. However, with >176k structures in the PDB and the number growing daily, M-CSA still represents less than 10% of known structural space. UniProt also provides predicted active site information which can be useful for structural alignments to identify divergent active site architectures in metagenome sequences.
In addition to the active site, other conserved motifs or cofactor binding sites are also important for protein function and can be detected with tools such as ScanProsite.172 For example, structural alignment of promiscuous RiPP maturases that install ornithine residues into peptide backbones revealed a conserved ‘DXHXD’ motif which was then used to detect and characterize new RiPP-modifying arginases from groundwater metagenomes and culturable isolates (Fig. 4D).173 In this study and many others, motif searching is used combination with full-length sequence homology searches for improved accuracy. For a different approach, however, motif searches can be used to identify conserved cofactor binding sites or structural features independent of protein family or fold from metagenomic sequences.
3.7. Machine learning
Machine learning offers the promise of moving beyond simple homology transfer methods to learn hidden relationships between protein sequences, structures, and functions. Advances in computing power and algorithms, have led to a renaissance of machine learning in many fields including biology and chemistry. Just in the past decade, >35 different machine learning-based methods have been published for protein function prediction.174 Rather than compare individual algorithms, we will focus on key steps and common pitfalls in a generalized machine learning workflow (Fig. 5).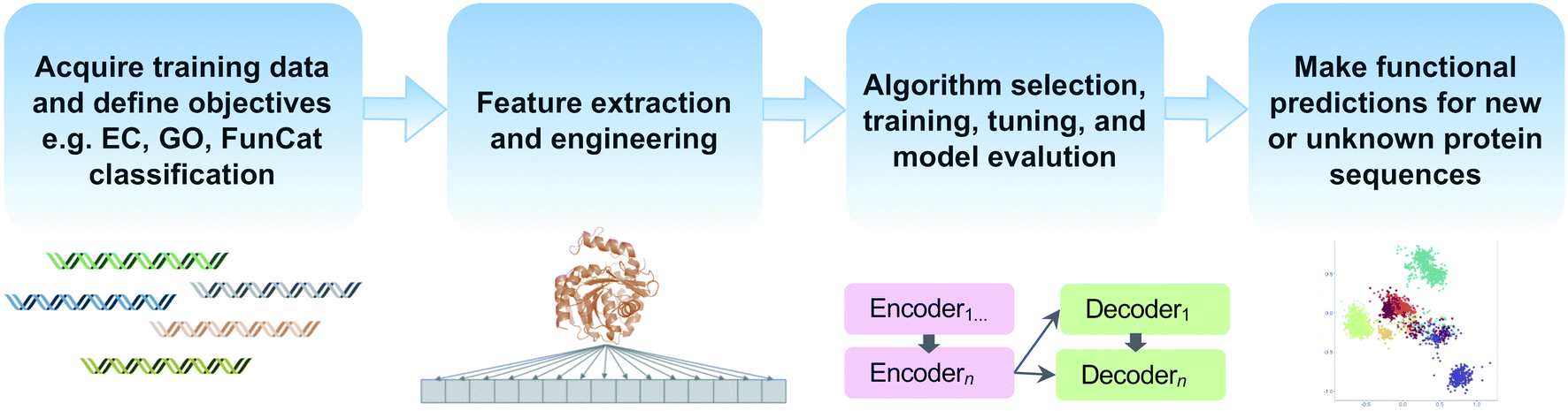 | ||
| Fig. 5 Common steps in a machine learning workflow for protein function prediction covered in this review. | ||
Although machine learning has received a significant amount of hype in recent years, it is not a panacea. One key disadvantage is that machine learning techniques are extremely data hungry. Even the most sophisticated of machine learning models are only as good as the underlying quantity and quality training data. In fact, increasing model complexity requires larger amounts of data. Deep neural networks, in particular, commonly suffer from overfitting, that is, they cannot be generalized to other studies or data sets. Therefore, continued support and curation of public databases which provide high-quality training data, such as MIBiG35 and Swiss-Prot,175 are essential for machine learning to enable future enzyme discoveries. One active area of research that seeks to handle the paucity of ‘labeled’ or experimentally verified data points biology is known as transfer learning. During transfer learning, models are pre-trained on large quantities of unlabeled data, e.g., unknown metagenomic sequences, to learn features that are general to these sequences and thereby improve performance on separate, related tasks such as enzyme function prediction through transfer of knowledge. A generalized transfer learning model pre-trained on short metagenomic reads was recently used for the identification of new candidate oxidoreductases from Tara Oceans metagenomes.176 Further advances in transfer learning and other semi-supervised learning techniques will help us leverage big metagenomic datasets with few labeled examples in the future.
Feature extraction and engineering is another critical step in machine learning workflows (Fig. 5). In addition to using protein sequences and structural information as features, some methods incorporate physicochemical amino acid properties or protein–protein interaction information to improve functional prediction accuracy.177,178 Natural language processing methods for text mining have also been used to extract features since biochemical knowledge continues largely to be stored in text format in journal articles.179,180 Recently, the use of autoencoders for unsupervised encoding of protein features has emerged. Autoencoders are artificial neural networks to automate the manual process of feature extraction and engineering, thereby removing human biases during the feature engineering process. One downside, however, is that autoencoders require even larger datasets and increased compute time.181,182
Machine learning algorithms used to predict protein function also vary from simple logistic regression and random forest models to multi-layer neural networks.174 However, benchmarking performance across studies can be complicated by inconsistent classification systems for the objective, e.g., protein function prediction. Most machine learning models use hierarchical tree-based structures such as Gene Ontology (GO),183 Functional Catalogue (FunCat),184 or EC classification systems185 as objectives but comparisons between models remains a challenge. Initiatives such as the Critical Assessment of Functional Annotation (CAFA) challenge, now in its fourth year, are making inroads to standardize the field.186 In recent comparisons of protein function prediction models, however, even simple homology transfer and logistic regression models were still able to outperform deep neural networks for some protein function prediction tasks.174 Unlike AlphaFold2's performance for protein structure prediction, the function prediction problem is far from solved and the field is still developing.
In terms of machine learning applications for natural product biosynthesis, BGCs have a unique advantage since biosynthetic logic and linkage to natural product structures can help narrow the range of potential substrates and functions. There are a growing number of BGC-specific machine learning tools available to predict natural product structure and bioactivity from metagenomes. These include BGC detection and classification software reviewed elsewhere187 such as antiSMASH,94 PRISM,188 DeepBGC189 and most recently, GECCO (https://gecco.embl.de/). Unfortunately, researchers tend to work either on the more general protein function prediction problem or on natural products biosynthesis, but they do not often communicate with each other. Increasing integration between these distinct research communities such as through joint conferences and workshops would advance progress for the prediction of new secondary metabolic enzyme functions.
A major limitation of machine learning methods available to date is the task of predicting truly novel enzyme functions. In many of the methods described, models are trained on a range of possible objectives, e.g., EC, FunCat, or GO terms, or BGCs for known natural product classes, but they are not capable of predicting entirely new classes. One alternative to multi-label classification is the use of negative selection algorithms which can label a protein as performing a particular function or not.190 The benefit of this approach is that negative selection does not force a protein to fit into a previously defined class. Rather a sequence can be predicted to not fit into any known functional classes or to fit into multiple classes, thus suggesting new function(s), potential promiscuity or even moonlighting.191 However, the best-case scenario still only provides negative or positive predictions. Will machine learning algorithms ever reach the stage of making completely de novo protein function predictions? Only time will tell, but if AlphaFold2 is any indication, then the future is bright.
4. Reaching the destination: characterizing new enzymes
4.1. Cloning and heterologous expression
Up until this point, our roadmap has explored various in silico methods for enzyme function prediction. However, while these methods can be helpful for identification and prioritization of new areas of protein sequence space they are, at best, only computational predictions. They do not provide functional validation, which requires experimental characterization. When selecting proteins to characterize in the lab, an important first step is quality control to remove chimeras and truncated sequences that may have sequencing errors or do not encode fully functional proteins. Outliers in sequence length visible from multiple-sequence alignments can also indicate a misprediction of start or stop codons. Particularly for Actinobacteria which are prolific natural product producers, gene products with non-canonical start and stop codons192 can be mis-predicted by gene annotation tools leading to frustrations in the laboratory.When dealing with hundreds to thousands of metagenomic sequences as candidates to choose from, many tools exist to cluster proteins by similarity and automatically select representative sequences. Early pioneers in the field that are still widely used include CD-HIT193 and UCLUST.194 Newer clustering algorithms such as Linclust available through the MMSeqs2 software suite also can perform clustering for metagenomic protein sequences in a fraction of the CD-HIT runtime.195 SSN analysis and selection of cluster representatives using Cytoscape or igraph can also be used to select protein subfamily representatives. Independent of clustering strategy, the underlying assumption is that highly similar proteins will also perform the same functions, which is often, but not always, true.196,197 One striking example where homology did not determine function was exemplified by the discovery of a completely new enzyme within the well-known NifH protein family. Based on previous observations of unexpected ethylene gas production from freshwater and soil bacteria, North et al. used a combination of differential gene expression and knockouts to determine the new function of a Rhodospirillum rubrum NifH homolog as a methylthio-alkane reductase.198 This enzyme is part of a biosynthetic pathway for methionine and a new route for anaerobic ethylene and methane production completely independent from nitrogenase activity. Thus, homology determining function does not always hold, even for famous and well-characterized families like NifH proteins. Nonetheless, clustering remains a useful method to choose protein representatives.
Depending on dataset size, further filtering steps may be required. For many enzyme activity assays without high-throughput capabilities, screening may be the bottleneck allowing for the selection of only a handful of metagenomic sequences to express and test. In this case, the decision of which few sequences to select becomes more tailored to the research question. In general, one of the most obvious strategies is to choose metagenomic sequences also found in culturable organisms, since this can permit functional characterization in the native host. Another popular strategy is the selection of proteins from thermophilic organisms which tend to encode enzymes with increased thermostability. We note this is also a generalization, however, since analysis of the ‘meltomes’ of complete proteomes of diverse organisms across the tree of life showed high variability in protein melting temperature even for organisms adapted to temperature extremes.199
Alternative approaches include selecting candidates that are more likely to be stable and express well including filtering for proteins that do not have high GC-content, transmembrane regions, or long disordered regions. Automated approaches to predict protein stability have been developed mainly for predicting crystallization propensity including tools like XtalPred,200 XANNPred,201 OB-Score,202 CrystalP2,203 ParCrys,204 and Crysf.205 A recent comparison of these tools was published by Wang et al.205 Given the abundance of different approaches to predict protein stability, we recommend using multiple criteria to rank and prioritize protein sequences for experimental characterization. In this way, individual biases in prediction tools may partially be offset by an ensemble-based approach to identify the most promising candidates.
Another useful step to increase the likelihood of obtaining soluble protein is the removal of signal peptides, that is, regions 16–30 amino acids in length at the N-terminus of many prokaryotic and eukaryotic proteins.206 These short regions of DNA typically direct the export of proteins from the cytosol. Signal peptides can influence protein solubility and export in heterologous expression experiments, particularly with N-terminal His tags. The gold standard for signal peptide detection and removal has been the software tool, SignalP,207 but more advanced machine learning methods for signal peptide detection and design are emerging.208 Recently, attention-based neural networks were successfully used to detect and generate diverse, functional signal peptides for a variety of protein families.208 As our understanding of the relationships between signal peptides and protein functions deepens, these short stretches of amino acids could also as serve features for enzyme discovery efforts in the future.
4.2. Heterologous expression
Once enzymes or BGCs of interest have been identified, constructs for heterologous expression must be designed. Unfortunately, most vectors that work for metagenomic library preparation for functional metagenomics methods are not typically suitable for heterologous expression. Many complete BGCs are also not fully captured in metagenomic libraries since fosmid/cosmid vectors have a maximum insert size of 45 kb. In addition to classical restriction cloning and Gibson assembly methods, new methods have been developed to improve the efficiency and ease of cloning large BGCs into heterologous hosts.209 One popular method, transformation-associated recombination (TAR) cloning, relies on natural homologous recombination in yeast to piece together overlapping eDNA cosmid/fosmid clones from soil and sponge metagenomes.210 Genetic recombineering,209,211 uses a variety of bacteriophage proteins to mediate homologous recombination in E. coli, including a new rapid method for efficient cloning of large BGCs using RecET direct cloning coupled to Redαβ recombination.212 For a comprehensive review of cloning methods for BGCs, we refer readers to Zhang et al.209To obtain sufficient genetic material for cloning, PCR amplification is still often the most cost-effective method if original eDNA is still available or if source organisms are culturable. In the event genetic material from the source is not available, the costs of DNA synthesis have dropped significantly in recent years. Moreover, gene synthesis enables complete codon optimization to match codon usage preferences of the heterologous host, which is particularly useful for expression of metagenomic sequences from taxonomically distant, uncultivated organisms.213
Even with constructs that are properly designed, many heterologous expression experiments still fail. From our personal experience, the activity of some biosynthetic enzymes is only detectable through the expression of complete BGCs rather than expressing genes individually from a cluster. This further underscores the importance of protein–protein interactions for enzyme activity (Section 3.4). In other cases where expression fails, enzymes may require cofactors or other metabolic machinery not found in model organisms such as E. coli. For example, methylmalonyl-CoA is needed for complex polyketide biosynthesis in actinomycetes, but it is not produced by E. coli.214 Modular PKS and NRPS clusters also typically require co-expression of secondary metabolism-type phosphopantetheine transferases (PPTases) from source organisms since the proper PPTases for post translational modification of these complex natural products often differ from the PPTases present in E. coli.215 Many cobalamin-dependent radical SAM enzymes such as C-methyltransferases involved in maturation of proteusin-family RiPPs are also inactive in E. coli.216 In all of these cases, engineered E. coli strains have been developed,217 including recently published plasmids to improve cobalamin uptake in E. coli.218 Even using engineered strains, many natural products still are not detectable in their final modified form from expression in E. coli.
As an alternative to using model organisms as heterologous hosts, non-model hosts can often be identified through genome mining. When heterologous expression in E. coli yielded low amounts of FR900359, a potent Gq protein inhibitor first uncovered from metagenomic eDNA of an uncultivated leaf symbiont,219 Hermes et al. identified a homologous BGC in the genome of the culturable bacterium Chromobacterium vaccinii. Knockout studies of the native cluster in C. vaccinii and successful heterologous expression of the C. vaccinii enzymes in E. coli enabled characterization of the unusual thioesterase domain catalyzing intermolecular thioesterification of FR900359 (Fig. 4E).220 In another example, a homologous BGC to the RiPP-family polytheonamide cluster from the uncultivated sponge symbiont ‘Candidatus Entotheonella’ was found in the culturable betaproteobacterium, Microvirgula aerodenitrificans. In particular, the cobalamin-dependent radical SAM C-methyltransferases that were largely inactive in E. coli were found to be highly active in M. aerodenitrificans, enabling production of fully modified final products.221 The polygeonoides, polytheonamide-like compounds from a metagenomic bin of a deep-rock subsurface environment, were also produced and characterized from M. aerodenitrificans. The generalized strategy of searching for metagenomic BGC hits in the genomes of culturable organisms can be especially fruitful in the case that heterologous expression in model organisms is unsuccessful.
Selecting the closest culturable taxonomic relative, particularly if genetic tools are available for this strain, can also be another promising method to select heterologous hosts. This strategy enabled the discovery of Fe–S flavoenzymes involved in bile acid dehydroxylation produced by the gut microbiome commensal, Clostridium scidens.222 Funabashi et al. characterized these enzymes by introducing them into a closely related Clostridium strain amenable to genetic manipulation. This approach has long been used to express diverse BGCs from Streptomyces spp. in the model Streptomyces coelicolor A3(2).223 In other cases, heterologous expression of genes from taxonomically distant organisms can still work in E. coli, such as in the case of expressing a BGC from a diatom for domoic acid production.224 As with many experimental systems, the selection of a heterologous host is still largely a process of trial-and-error. In the future, we anticipate design-build-test-learn workflows used in synthetic biology and already being applied for the optimization of hosts will reduce this tedious trial-and-error process.225
4.3. Screening for enzyme activity
Once enzymes of interest have been expressed, the next challenge comes in assaying them for in vivo or in vitro for activity. There is often a trade-off between throughput and generalizability for enzyme screening methods (Fig. 6). Activity-based screening of metagenomic libraries typically involves searching for zones of inhibition around bacterial colonies or using cleavable substrates that produce a color or fluorescence. However, for many biosynthetic enzymes, appropriate substrate analogs may not be available, or the presence of a large fluorophore or chromophore may interfere with enzyme activity. Mass spectrometry (MS) is a sensitive and highly generalizable technique to monitor activity for many different enzymes since it does not require the use of substrate analogs. Due to the need for chromatographic separation steps, however, MS-based methods have historically suffered from low throughput. A number of alternative workflows to bypass the chromatographic separation step allow for scaling up MS-based screening of enzyme activity. For example, nanostructure-initiator mass spectrometry (NIMS) substitutes column-based separation with an in situ washing step over a perfluoroalkylated surface. The wash step promotes non-covalent fluorous interactions enabling compound separation.226 Previously, NIMS has only been successfully applied to enzymes with active sites able to accommodate bulky perfluoroalkylated tails such as acetyltransferases and carbohydrate-degrading enzymes.227 A more generalized solution, termed PECAN (Probing Enzymes with Click-Assisted NIMS), relies on click chemistry to expand NIMS technology to other enzyme classes.228 Although still requiring substrate analogs with ‘clickable’ alkynes or azides, these small handles are far less bulky than perfluoroalkylated tails. Another label-free alternative to NIMS is known as SAMDI-MS (Self-Assembled Monolayers for matrix-assisted Desorption/Ionization Mass Spectrometry). SAMDI-MS relies on the immobilization of proteins or metabolites on self-assembled monolayers of alkanethiolates on gold.229 Importantly, the immobilization process for SAMDI-MS combines the sensitivity and generalizability of MS-based methods with significantly higher throughput. Since SAMDI-MS does not require labeling of the substrates, it is especially useful for enzyme classes which are challenging to screen such as glycosyltransferases.230 SAMDI-MS has mainly been applied for directed evolution or metabolic engineering studies to date,231,232 but it is also a well-suited method for rapid and sensitive MS-based screening of metagenomic enzymes. The specialized expertise and equipment required for adoption of NIMS and SAMDI-MS methods are current barriers that must be overcome for the techniques to be used more widely. | ||
| Fig. 6 Trade-off between generalizability and throughput for common enzyme screening approaches. | ||
Biosensors are another area of active research for the detection of new biocatalysts.233 One recent study developed a genetic screening system for the detection of capro-lactam ring-forming enzymes. A biosensor based on the highly specific activation of the NitR regulatory protein from Alcaligenes faecalis in response to the presence of ε-caprolactam compounds was optimized and used to screen a marine sediment metagenomic library. In combination with FACS sorting, this biosensor enabled the identification of a new enzyme capable of cyclizing ω-amino fatty acids.234 mRNA display is another screening method to efficiently link peptides to their own encoding RNA through in vitro ribosomal translation. Most commonly used for directed evolution studies, mRNA display was also recently employed to screen activity of a naturally occurring highly promiscuous RiPP maturase, PaaA.235 PaaA modifies glutamic acid residues to form fused bicyclic cores in a wide range of peptide substrate analogs. We note, however, that both the biosensor and mRNA display methods described here are tailored to specific enzyme functions. Therefore, these approaches are extremely high-throughput but less generalizable than MS-based approaches. Overall, the methods described in this section present an overwhelming array of new tools which can be adapted to screen metagenomic enzymes from diverse environments. In the next section, we will focus on marine systems as a case study for exploring new enzymology.
5. Scenic drives: a case study on marine metagenomics
5.1. Global ocean microbiomics
Marine systems are treasure troves for new biosynthetic enzymes for a number of reasons: (1) due to the harsh and fluctuating environmental conditions, enzymes from marine organisms can often tolerate extremes in salinity, temperature, pH, and atmospheric pressure.236 (2) The chemical composition of oceans includes relatively high abundances of elements including boron, bromine, and chlorine compared to terrestrial systems. This naturally results in an enrichment of biosynthetic enzymes for the tailoring of boron-containing and halogenated natural products.237 Finally, (3) less than 10% of microbial marine metagenomes can currently be matched to cultivated reference genomes at the species level,238 highlighting metagenomics as a critical strategy to probe the uncultivated majority of the world's oceans.To enable marine bioprospecting, several studies have published meta-omics data resources239–242 including the Tara Oceans sampling expeditions which sequenced over 600 metatranscriptomes and 1000 metagenomes from >200 globally-distributed sampling stations.23 The combination of Tara Oceans data with Global Ocean Sampling,10 Malaspina,241 and bioGEOTRACES expeditions240 enabled the assembly of the largest genome-resolved ocean resource to date.239 From these data, >25000 metagenome-assembled genomes were constructed and, together with ∼10000 marine single and isolate genomes, processed with antiSMASH,94 leading to the identification and functional characterization of new BGCs and biosynthetic enzymes.242 The scale of this metagenomics repository for one specific environment type is perhaps only paralleled by the Human Microbiome Project.24 Compared to the human gut microbiome, however, the variable ocean conditions promote a greater diversity of enzymes, particularly along the water column where a gradient of different temperature and pressure conditions exist. Indeed, analysis of microbial metagenomes from the Tara Oceans sampling expedition revealed the ocean harbors more than four times the number of unique genes found in the human gut.243 Accordingly, we anticipate marine systems harbor significant biosynthetic potential and new enzymology.
In a recent example of enzyme expansion, a new member of the amine dehydrogenase family was detected by mining marine metagenomes.244 Caparco et al. first constructed HMMs from eight characterized amine dehydrogenase sequences. They identified hundreds of metagenomic hits and systematically narrowed the search space by focusing on distant homologs which the authors reasoned were more likely to have altered substrate specificity. Eighteen homologs were cloned and heterologously expressed in E. coli. One eukaryotic enzyme from the Marine Atlas of Tara Oceans Unigenes245 was found to have an unusually high specific activity for isobutyraldehyde and represented the first eukaryotic amine dehydrogenase to be experimentally characterized.244 This enzyme was discovered from metagenomes from the open ocean, which has generally been an understudied ecosystem by natural products researchers compared to the microbiomes of marine sponges and other invertebrates.
5.2. Microbiomes of marine invertebrates
Marine sponges, tunicates, bryozoans, and molluscs are sessile or slow-moving animals that commonly benefit from chemical defenses provided by a wide range of natural products. A growing body of evidence has implicated invertebrate microbiota as an important source of bioactive substances found in these animals.246 Producers and their biosynthetic enzymes were initially identified by PCR screening metagenomic libraries and more recently by metagenomic sequencing as first steps towards functional studies. Intriguingly, the bacterial producers identified to date mostly belong to unusual taxa distinct from common natural product sources, such as actinomycetes. An example is ‘Candidatus Entotheonella’, belonging of the uncultivated phylum ‘Tectomicrobia’, a group of filamentous sponge symbionts with a rich specialized metabolism.17 Notable examples of enzymatic transformations in these bacteria are the polytheonamide peptide maturation system that installs up to 50 posttranslational modifications,19,247 RiPP S-methylation,248 diverse trans-AT PKS systems that assemble complex polyketides,17,20cis-AT PKS modules catalyzing single-carbon extensions (e.g., calyculins,21 orbiculamides,17 keramamides,249 konbamides17), a promiscuous halogenation,249 and a to-date unique and biosynthetically unassigned peptide cross-link involving a histidine imidazole moiety.250In addition to ‘Entotheonella’, other producers in sponges have been identified, all of which remain uncultured. These include a multiproducer community providing the cytotoxic pelorusides, pateamines, mycalamides, and other compounds in the sponge Mycale hentscheli, an intracellular producer of renieramycin with a highly reduced genome.251,252Hormoscilla (formerly Oscillatoria) spongeliae, is a cyanobacterial sponge symbiont and a source of halogenated compounds, including polybrominated diphenyl ethers (PDBE). The combination of de novo metagenomic sequencing of the sponge metagenome and heterologous expression of the candidate biosynthetic locus for PDBE in a non-standard host, Synechococcus elongatus PCC 7942, ultimately led to successful characterization of this BGC.253 Cyanobacterial symbionts from other marine animals, such as tunicates, are also rich sources of biosynthetic diversity. Biosynthetic pathways for the cyanobactins, a class of cytotoxic RiPPs including the patellamides and trunkamides, were originally discovered from metagenomic eDNA.254–256 The promiscuity of RiPP maturases encoded in cyanobactin pathways, e.g., heterocyclases for azoline installation257 or macrocyclases for cyclic peptide formation,258 have been exploited to generate libraries of synthetic peptides.
Another biosynthetic treasure trove are shipworms, the bivalve molluscs famous for boring holes in wooden boat hulls and piers. Recent shotgun metagenomic analysis coupled with cultivation strategies revealed more than 150–200 distinct BGCs from shipworm gill endosymbionts.259 In addition to being biosynthetically talented, shipworm symbionts are of biotechnological interest for biomass degradation due to their wood-based diet. A new enzyme involved in lignocellulose degradation was recently isolated and characterized from the shipworm symbiont Teredinibacter turnerae.260 This represents a case of enzyme expansion within the family of oxidative enzymes known as lytic polysaccharide monooxygenases (LPMOs) that degrade chitin and cellulose-like polymers.261 Since the relatively recent discovery of the LPMOs in 2010, this enzyme family has been of great interest for biotechnological applications including the oxidative degradation of recalcitrant polymers.261 Perhaps the greatest finding from shipworm metagenomic studies to date, however, is that most members of the shipworm gill endosymbiont microbial communities are culturable.259 The ability to cultivate and genetically manipulate nearly-complete microbial consortia from shipworm gills provides an exciting experimentally-tractable system within which to study host-endosymbiont co-evolution of secondary metabolism.
6. Gearing up for the future: new frontiers in enzyme discovery
In this section, we will provide an outlook on the future of the field and highlight emerging techniques which can be paired with metagenomics workflows to accelerate enzyme discovery.6.1. Meta-omics
The integration of various meta-omics techniques, including metatranscriptomics, metaproteomics, and metabolomics, into enzyme discovery workflows can be a powerful framework connecting genotype to phenotype for hypothesis generation. RNA-Seq, for example, provides a global snapshot of differentially expressed genes under conditions of interest to implicate coding sequences of unknown function in specific cellular processes. Maini-Rekdal et al. used RNA-Seq to characterize the involvement of an unknown molybdenum-dependent enzyme, DadH, in the catabolism of dopamine in the human gut.262 Differential expression analysis of the gut bacterium Eggerthella lenta revealed dadH was upregulated >2500-fold in the presence of dopamine. Although DadH from E. lenta only exhibited narrow substrate specificity for L-dopa and close analogs, metagenome mining using DadH as a query sequence expanded the protein family to other molybdenum-dependent enzymes capable of degrading other classes of neurotransmitters and diet-derived catechols.263 In another example, RNA-Seq analysis of the marine diatom Pseudo-nitzschia under phosphate limitation and high CO2 guided the identification of the biosynthetic pathway for domoic acid, a harmful neurotoxin.224 Heterologous expression of the domoic acid BGC and structural analysis yielded insights into how the unusual biosynthetic enzyme, DabA, that catalyzes N-prenylation of a primary amine arose within the ubiquitous terpene cyclase protein fold.264Although both of these examples used RNA-Seq to discover new enzymes from organisms in monoculture, similar strategies can be applied to metatranscriptomes. Surprisingly, the number of studies using metatranscriptomics for enzyme discovery are still relatively rare but increasing. Recently, metatranscriptomic analysis of a compost microbial community resulted in expansion of the glycoside hydrolase family to include an unusual enzyme with exo-1,4-β-xylanase activity.265 A new tool, BiG-MAP, was released to facilitate differential expression analysis of BGCs from (meta)transcriptomic datasets.266 BiG-MAP results can also further be integrated with metabolomics data. As a proof-of-principle, BiG-MAP was used to link differentially expressed BGCs from healthy and caries-associated oral microbiome samples with mass fragments associated with reuterin, a natural product inhibiting growth of the opportunistic pathogen Streptococcus mutans involved in tooth decay. With increasing availability of paired metatranscriptomic and metabolomic datasets and analysis pipelines like BiG-MAP, we anticipate meta-omic mining will accelerate the discovery of new biosynthetic enzymes.
Relative to the other -omics techniques, metaproteomics remains particularly underexploited. Sukul et al. proposed a workflow for functional metaproteomics relying on the direct isolation of proteins from soil samples followed by separation using 2D-polyacrylamide gel electrophoresis. Refolded proteins were assayed in-gel using a fluorogenic lipase substrate to detect new lipolytic enzymes. Hits were then excised from the gel, digested and subjected to MS analysis. Extracting eDNA from the same soil samples for shotgun metagenomic sequencing allowed mass spectra from in-gel lipolytically-active enzymes to be compared to a custom environmental database to identify full-length sequences and permit their taxonomic assignment.267 While in-gel metaproteomics workflows are viable strategies for well-characterized enzymes such as lipases, they are more challenging for enzyme functions lacking established colorimetric or fluorimetric substrates. The limited availability of functional assays is one downside of metaproteomics in addition to difficulty with directly isolating proteins from environmental samples and a low likelihood of proper in-gel re-folding of enzymes. Technical challenges notwithstanding, it is clear that the integration of different multi-omics datasets offers promising new routes for enzyme discovery.
6.2. Single-cell genomics
Single-cell genomic sequencing is an alternative and complementary approach to shotgun metagenomics. Single-cell genomics relies on the sorting of microbial cells, usually with microfluidics (Section 6.3) or FACS methods, followed by lysis and whole genome multiple displacement amplification with high-fidelity polymerases.268 Despite enabling over a billion-fold amplification of genetic material from a single cell, the quantity of DNA is often still low, resulting in poor genome quality and a risk of contamination from extracellular DNA. Optimized protocols using thermostable polymerases have been developed to improve these issues and also correct for biases against amplification of GC-rich templates which is especially relevant for BGCs from Actinobacteria and other organisms with high GC-content.269 The benefits of directly linking taxonomic classification to genomic functional content without requiring binning provides a clear advantage of single-cell genomics over shotgun metagenomic sequencing. Optimally, single-cell genomics and metagenomics methods are best applied in combination since they have different sampling biases which minimizes their overlap. Previous analysis in our lab also found that reference genomes from cultivated marine isolates rarely overlapped with marine SAGs or MAGs, indicating that multi-pronged approaches of cultivation and multiple types of sequencing contributes to a greater genome-resolved understanding of ecological community composition.242There are still relatively few cases of biosynthetic enzyme discovery from SAGs. As the earliest example applied to natural product studies, Grindberg et al. detected the cyanobacterial apratoxin biosynthetic pathway in a mixed bacterial assemblage through a combination of single-cell sequencing, in silico mining, and a metagenomic fosmid library screening.270 Based on biosynthetic logic and a priori knowledge of the apratoxin chemical structure, the authors used known conserved motifs from hydroxymethylglutaryl-CoA synthase-like enzymes to detect homologs in their SAG contig library. The motifs were then used to design degenerate primers and PCR screen their metagenomic library to identify overlapping contigs and assemble the complete apratoxin biosynthetic cluster. This study illustrated how the combination of single-cell genomics and functional metagenomics ultimately revealed the complete BGC for apratoxin. Skiba et al. later built on these findings to characterize an unusual mononuclear iron-dependent di-methylating methyltransferase that initiates apratoxin biosynthesis through production of branched polyketide starter units.271 More recently, Mori et al. used single-cell sequencing to characterize the remarkable biosynthetic potential of ‘Candidatus Entotheonella’ symbionts from marine sponges (Section 5.2).20
Importantly, single-cell genomics does not rely on the assumption that populations of similar cells are clonal. As a result, single-cell genomics studies have revealed remarkable within-population genome variability and evolution in systems ranging from marine phytoplankton272 to cancer cells.273,274 BGCs are also notoriously strain-specific and single-cell sequencing is useful to detect needle BGCs in the haystack. For example, Sugimoto et al. developed a new HMM-based computational strategy to mine BGCs from human microbiomes and found that some BGCs are only found in one out strain out >1000–5500 bacterial isolates from the same taxon.275 Advances in single-cell RNA sequencing in prokaryotes has further demonstrated how even genetically-identical populations of bacteria exhibit spatial transcriptional heterogeneity in communities.276 Spatial heterogeneity and division of labor has been documented for secondary metabolite production in a variety of systems both in microbial communities and monocultures.277 This nascent area of research calls for further applications of single-cell and spatial transcriptomics methods to better understand how microbial community structure and microenvironment affects biosynthetic gene expression.
6.3. Microfluidics
Microfluidic technologies have revolutionized biomedicine, particularly for devices where ‘lab on a chip’ compactness is desired. Microfluidic-based sorting methods have been widely applied for directed evolution and protein engineering studies,278 but have only rarely been used for metagenomic enzyme discovery. Colin et al. screened >1250000 water-in-oil droplets using a microfluidic system to identify metagenomic enzymes that hydrolyze sulfate monoesters and phosphotriesters.132 This study highlighted microfluidics as a useful technique to probe the promiscuity of metagenomic enzymes in a sequence-independent manner. Another recent study used optical tweezers and microfluidics to sort complex microbial communities based on the Raman spectra of individual cells.279 This has numerous applications for downstream single-cell sequencing or cultivation efforts including sorting microbes based on stable isotope labels or natural Raman signals from storage compounds or carotenoids. By processing sorted cells for downstream single-cell sequencing, chemical phenotypes of live individual cells can be directly linked to their genotypes. Like most other new techniques discussed in this section, however, the application of microfluidics to discover new biosynthetic enzymes from metagenomes has not yet been widely applied.
6.4. Cell-free platforms
An elegant alternative to heterologous expression and protein purification is the use of cell-free systems such as filtered lysate from E. coli or another host. Since cellular machinery remains in the lysate, exogenous addition of components including cofactors, amino acids, and DNA is all that is required to express enzymes or pathways of interest.280 Cell-free systems create conditions for rapid transcription and translation of desired DNA sequences without the constraints of maintaining cellular growth. Unlike in vivo expression systems, cell-free platforms also allow for the production of toxic metabolites that normally kill heterologous hosts. To further increase throughput, screening methods including mRNA display, matrix-assisted desorption/ionization-MS and in-droplet reaction microfluidics have already been integrated with cell-free platforms.280 For some biosynthetic pathways, high yields are produced in just a few hours from DNA templates.281,282 In practice, however, low yields are a common challenge especially when working with DNA from organisms that are taxonomically-distant from E. coli.280 Fast degradation of mRNA templates and other necessary reactions components is another challenge commonly faced when working with cell extract-based systems. Nonetheless, these systems have seen explosive popularity in recent years, and we anticipate future exploitation of cell-free platforms for metagenomic enzyme discovery.6.5. Sequence-independent methods
On the whole, the vast majority of techniques described in this review rely on either sequence-based or structure-based homology to infer protein function. However, these approaches often fall short when making predictions for the ‘unknown unknowns’, that is, for the de novo discovery of enzymes that do not share sequence or structural similarity with one or more characterized protein families. Sequence- or structure-independent approaches are also rarely used in natural products research, since most computational methods to identify BGCs rely on homology to common biosynthetic domains.283 In a departure from sequence-based methods, decRiPPter (Data-driven Exploratory Class-independent RiPP TrackER) was developed for the explicit purpose of detecting new RiPP classes without relying on homology to known RiPP classes or enzymatic machinery.284 The core filtering step of the decRiPPter algorithm uses pan-genomic comparisons to detect operons that are sparsely distributed within taxonomic groups and thus are likely involved in secondary rather than primary metabolic functions. Kloosterman et al. analyzed 1295 Streptomyces genomes with decRiPPter to identify a new family of RiPP maturases catalyzing dehydration and cyclization reactions for a new lanthipeptide class of natural products.284 While this singular example is a proof-of-principle that sequence-independent methods can be used successfully for enzyme and natural product discovery, the authors emphasize a key limitation of this approach is the large number of false positives when searching for novelty rather than homology. Beyond RiPPs, the field remains open and poised for the emergence of new sequence- and structure-independent methods for enzyme discovery.7. Conclusions
A major takeaway from this review is the surprising paucity of studies of de novo and reference-based enzyme discovery studies that have used shotgun metagenomics rather than functional metagenomics. Even as we amass petabytes of meta-omics data in public databases, there is a disconnect between the relative ease of next-generation sequencing and the difficulty of gaining insights into new protein families and their functions. Based on a meta-analysis of this review, we will attempt to offer some general recommendations to advance future efforts in the field:7.1. Discoveries often occur at the boundaries of protein families
Although not a universal rule, proteins with low sequence identities to reference proteins of known functions are more likely than enzymes with high sequence identities to accommodate different substrates and catalyze new reaction types. To detect distantly related sequences, we recommend moving beyond a basic BLAST search and using tools that are more sensitive for remote homology detection such as PSI-BLAST285 or HMMs. MetaHMM286 or HMMSearch tools in the EBI MGnify portal89 allow non-experts to query metagenomes with protein queries or custom HMMs through a web interface. Expert users may be interested in applying an iterative HMM search strategy. In this approach, an initial HMM model is used and combined with BLAST searches to identify more distant homologs of the same gene family. The newly identified sequences are then used to update the initial model and the procedure repeated until no additional homologs are identified. This strategy has been applied to identify new β-defensin members in humans and mice279 and to discover a cysteine-rich gene family in corals.2877.2. Think outside the colorimetric assay box to move into unexplored protein space
A previous meta-analysis of metagenomic enzymes discovered between Jan 2014 and March 2017 found that >84% belong to either the lipase/esterase or cellulase/hemicellulase classes.33 Similarly, >82% were discovered by activity-based screening. Clearly, there is a bias in current metagenomic screening methods towards industrially relevant enzyme classes that can readily be detected with standard colorimetric assays. Although less well-understood and more challenging to screen, protein superfamilies for which remarkable diversity is already documented, including, but not limited to, radical SAMs enzymes, methyltransferases, thioesterases, and cytochrome p450 monooxygenases, represent hotbeds for new enzymology.7.3. Move beyond E. coli into new hosts
E. coli has been the workhorse for the vast majority of functional metagenomics and heterologous expression efforts to date. The benefits of using a model organism are undeniable for the relative ease of cloning, expression, and screening. However, a controlled study found that only 30–40% of genes from environmental bacteria in general and only 7% of high GC-content DNA could be expressed in E. coli.288 Unfortunately, many secondary metabolites are produced by organisms classified as high GC-content thus classical functional metagenomics methods likely fail to capture large swaths of diversity in eDNA. As discussed in Section 4.2, a suite of other problems such as the lack of proper coenzymes, substrates, cofactors, posttranslational modification systems, self-resistance genes, protein folding factors, and additional proteins required for megasynthases (e.g., MbtH-like proteins, PPTases) all can cause heterologous expression in E. coli to fail. One strategy to troubleshoot these issues is using alternative hosts. For functional metagenomics, Pseudomonas, Streptomyces, Rhodococcus, Bacillus, and even archaea have been used as library hosts as well as multi-host expression systems with shuttle vectors.289,290 Similarly, non-traditional heterologous expression hosts such as M. aerodenitrificans221 have been developed to access new enzymology from metagenomic BGCs.7.4. (Genome) context is everything
There has been an explosion of new tools devoted to examination of genes in the context of their genome neighborhoods rather than in isolation. This has been a particularly fruitful strategy for the discovery of new multi-domain enzyme functions148 or RiPP maturases.173 Taking this one step further, deep learning methods for embedding genes as vectors in their genomic context (e.g., pfam2vec) have led to improvements in BGC prediction.189 Although the current reliance on short-read sequencing methods requires binning and assembly to extract genomic context from shotgun metagenomes, we anticipate that advances in long-read metagenome sequencing will pave the way for genome neighborhoods to be analyzed more directly and accurately from eDNA. Just as bacteria act differently in communities than in isolation, we propose that enzymes are best studied and understood in the complete milieu of their flanking genes. Identification of gene coexpression modules from (meta)transcriptomic datasets can additionally be used reconstruct transcriptional units and predict the function of unknown genes through coexpression of genes with known function. Coexpression networks can also guide hypotheses regarding protein–protein interactions and complex formation. Overall, the protein interactome is an underexplored avenue to uncover new enzyme functions.Perhaps one day we will see the emergence of AlphaFunction2 as a successor to AlphaFold2. But in order to train artificial intelligence models to tackle the sequence–structure–function problem, continued exploration of new areas of protein space is an important task for experimental enzymologists and computational researchers alike. As a final remark, we emphasize that AlphaFold2, and many other computational tools described in this review, were only made possible through the biochemical characterization of thousands of proteins by experimentalists at the bench. Experimental work is more critical than ever to enable new data-driven discoveries.
8. Conflicts of interest
The authors declare no conflicts of interest.9. Acknowledgements
S. L. R. is supported by an ETH Zurich Postdoctoral Fellowship (20-1 FEL-07). Work in J. P.'s lab on uncultivated bacteria was supported by grants from the Swiss National Science Foundation (205321_165695), the Gordon and Betty Moore Foundation (#9204, DOI: 10.37807/GBMF9204), the Helmut Horten Foundation, and the Promedica Foundation. Work on the ocean microbiome and bioinformatics tool development in S. S.'s lab has been supported by grants of the Swiss National Science Foundation (205321_184955), the NCCR Microbiomes (51NF40_180575) and the ETH domain (PHRT #521).10. References
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, K. Tunyasuvunakool, O. Ronneberger, R. Bates, A. Zidek, A. Bridgland and C. Meyer, Fourteenth Critical Assessment of Techniques for Protein Structure Prediction, 2020, pp. 22–24 Search PubMed.
- S. Ghatak, Z. A. King, A. Sastry and B. O. Palsson, Nucleic Acids Res., 2019, 47, 2446–2454 Search PubMed.
- R. S. Kaas, C. Friis, D. W. Ussery and F. M. Aarestrup, BMC Genomics, 2012, 13, 577 Search PubMed.
- D. A. Rasko, M. J. Rosovitz, G. S. A. Myers, E. F. Mongodin, W. F. Fricke, P. Gajer, J. Crabtree, M. Sebaihia, N. R. Thomson, R. Chaudhuri, I. R. Henderson, V. Sperandio and J. Ravel, J. Bacteriol., 2008, 190, 6881–6893 Search PubMed.
- Z. D. Blount, eLife, 2015, 4, e05826 Search PubMed.
- J. Handelsman, M. R. Rondon, S. F. Brady, J. Clardy and R. M. Goodman, Chem. Biol., 1998, 5, R245–R249 Search PubMed.
- F. Warnecke, P. Luginbühl, N. Ivanova, M. Ghassemian, T. H. Richardson, J. T. Stege, M. Cayouette, A. C. McHardy, G. Djordjevic, N. Aboushadi, R. Sorek, S. G. Tringe, M. Podar, H. G. Martin, V. Kunin, D. Dalevi, J. Madejska, E. Kirton, D. Platt, E. Szeto, A. Salamov, K. Barry, N. Mikhailova, N. C. Kyrpides, E. G. Matson, E. A. Ottesen, X. Zhang, M. Hernández, C. Murillo, L. G. Acosta, I. Rigoutsos, G. Tamayo, B. D. Green, C. Chang, E. M. Rubin, E. J. Mathur, D. E. Robertson, P. Hugenholtz and J. R. Leadbetter, Nature, 2007, 450, 560–565 Search PubMed.
- B. E. Wolfe, J. E. Button, M. Santarelli and R. J. Dutton, Cell, 2014, 158, 422–433 Search PubMed.
- N. A. Be, A. Avila-Herrera, J. E. Allen, N. Singh, A. Checinska Sielaff, C. Jaing and K. Venkateswaran, Microbiome, 2017, 5, 81 Search PubMed.
- S. Yooseph, G. Sutton, D. B. Rusch, A. L. Halpern, S. J. Williamson, K. Remington, J. A. Eisen, K. B. Heidelberg, G. Manning, W. Li, L. Jaroszewski, P. Cieplak, C. S. Miller, H. Li, S. T. Mashiyama, M. P. Joachimiak, C. van Belle, J.-M. Chandonia, D. A. Soergel, Y. Zhai, K. Natarajan, S. Lee, B. J. Raphael, V. Bafna, R. Friedman, S. E. Brenner, A. Godzik, D. Eisenberg, J. E. Dixon, S. S. Taylor, R. L. Strausberg, M. Frazier and J. C. Venter, PLoS Biol., 2007, 5, e16 Search PubMed.
- Z. D. Stephens, S. Y. Lee, F. Faghri, R. H. Campbell, C. Zhai, M. J. Efron, R. Iyer, M. C. Schatz, S. Sinha and G. E. Robinson, PLoS Biol., 2015, 13, e1002195 Search PubMed.
- L. Solden, K. Lloyd and K. Wrighton, Curr. Opin. Microbiol., 2016, 31, 217–226 Search PubMed.
- L. A. Hug, B. J. Baker, K. Anantharaman, C. T. Brown, A. J. Probst, C. J. Castelle, C. N. Butterfield, A. W. Hernsdorf, Y. Amano, K. Ise, Y. Suzuki, N. Dudek, D. A. Relman, K. M. Finstad, R. Amundson, B. C. Thomas and J. F. Banfield, Nat. Microbiol., 2016, 1, 16048 Search PubMed.
- D. H. Parks, M. Chuvochina, D. W. Waite, C. Rinke, A. Skarshewski, P.-A. Chaumeil and P. Hugenholtz, Nat. Biotechnol., 2018, 36, 996–1004 Search PubMed.
- C. J. Castelle, K. C. Wrighton, B. C. Thomas, L. A. Hug, C. T. Brown, M. J. Wilkins, K. R. Frischkorn, S. G. Tringe, A. Singh, L. M. Markillie, R. C. Taylor, K. H. Williams and J. F. Banfield, Curr. Biol., 2015, 25, 690–701 Search PubMed.
- A. L. Jaffe, C. J. Castelle, P. B. Matheus Carnevali, S. Gribaldo and J. F. Banfield, BMC Biol., 2020, 18, 69 Search PubMed.
- M. C. Wilson, T. Mori, C. Rückert, A. R. Uria, M. J. Helf, K. Takada, C. Gernert, U. A. E. Steffens, N. Heycke, S. Schmitt, C. Rinke, E. J. N. Helfrich, A. O. Brachmann, C. Gurgui, T. Wakimoto, M. Kracht, M. Crüsemann, U. Hentschel, I. Abe, S. Matsunaga, J. Kalinowski, H. Takeyama and J. Piel, Nature, 2014, 506, 58–62 Search PubMed.
- A. Crits-Christoph, S. Diamond, C. N. Butterfield, B. C. Thomas and J. F. Banfield, Nature, 2018, 558, 440–444 Search PubMed.
- M. F. Freeman, M. J. Helf, A. Bhushan, B. I. Morinaka and J. Piel, Nat. Chem., 2017, 9, 387–395 Search PubMed.
- T. Mori, J. K. B. Cahn, M. C. Wilson, R. A. Meoded, V. Wiebach, A. F. C. Martinez, E. J. N. Helfrich, A. Albersmeier, D. Wibberg, S. Dätwyler, R. Keren, A. Lavy, C. Rückert, M. Ilan, J. Kalinowski, S. Matsunaga, H. Takeyama and J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1718–1723 Search PubMed.
- T. Wakimoto, Y. Egami, Y. Nakashima, Y. Wakimoto, T. Mori, T. Awakawa, T. Ito, H. Kenmoku, Y. Asakawa, J. Piel and I. Abe, Nat. Chem. Biol., 2014, 10, 648–655 Search PubMed.
- S. K. Wyman, A. Avila-Herrera, S. Nayfach and K. S. Pollard, PLoS One, 2018, 13, e0205749 Search PubMed.
- S. Sunagawa, S. G. Acinas, P. Bork, C. Bowler, Tara Oceans Coordinators, D. Eveillard, G. Gorsky, L. Guidi, D. Iudicone, E. Karsenti, F. Lombard, H. Ogata, S. Pesant, M. B. Sullivan, P. Wincker and C. de Vargas, Nat. Rev. Microbiol., 2020, 18, 428–445 Search PubMed.
- J. Lloyd-Price, A. Mahurkar, G. Rahnavard, J. Crabtree, J. Orvis, A. Brantley Hall, A. Brady, H. H. Creasy, C. McCracken, M. G. Giglio, D. McDonald, E. A. Franzosa, R. Knight, O. White and C. Huttenhower, Nature, 2017, 550, 61–66 Search PubMed.
- M. G. Chevrette, K. Gutiérrez-García, N. Selem-Mojica, C. Aguilar-Martínez, A. Yañez-Olvera, H. E. Ramos-Aboites, P. A. Hoskisson and F. Barona-Gómez, Nat. Prod. Rep., 2020, 37, 566–599 Search PubMed.
- A. M. Kloosterman, P. Cimermancic, S. S. Elsayed, C. Du, M. Hadjithomas, M. S. Donia, M. A. Fischbach, G. P. van Wezel and M. H. Medema, PLoS Biol., 2020, 18, e3001026 Search PubMed.
- S. Ovchinnikov, H. Park, N. Varghese, P.-S. Huang, G. A. Pavlopoulos, D. E. Kim, H. Kamisetty, N. C. Kyrpides and D. Baker, Science, 2017, 355, 294–298 Search PubMed.
- Y. Wang, Q. Shi, P. Yang, C. Zhang, S. M. Mortuza, Z. Xue, K. Ning and Y. Zhang, Genome Biol., 2019, 20, 229 Search PubMed.
- A. J. Waldman and E. P. Balskus, J. Org. Chem., 2018, 83, 7539–7546 Search PubMed.
- J. K. Christenson, S. L. Robinson, T. A. Engel, J. E. Richman, A. N. Kim and L. P. Wackett, Biochemistry, 2017, 56, 5278–5287 Search PubMed.
- M. Ohashi, C. S. Jamieson, Y. Cai, D. Tan, D. Kanayama, M.-C. Tang, S. M. Anthony, J. V. Chari, J. S. Barber, E. Picazo, T. B. Kakule, S. Cao, N. K. Garg, J. Zhou, K. N. Houk and Y. Tang, Nature, 2020, 586, 64–69 Search PubMed.
- A. Chien, D. B. Edgar and J. M. Trela, J. Bacteriol., 1976, 127, 1550–1557 Search PubMed.
- F. Berini, C. Casciello, G. L. Marcone and F. Marinelli, FEMS Microbiol. Lett., 2017, 364(21), fnx211 Search PubMed.
- L. Ufarté, G. Potocki-Veronese and É. Laville, Front. Microbiol., 2015, 6, 563 Search PubMed.
- S. A. Kautsar, K. Blin, S. Shaw, J. C. Navarro-Muñoz, B. R. Terlouw, J. J. J. van der Hooft, J. A. van Santen, V. Tracanna, H. G. Suarez Duran, V. Pascal Andreu, N. Selem-Mojica, M. Alanjary, S. L. Robinson, G. Lund, S. C. Epstein, A. C. Sisto, L. K. Charkoudian, J. Collemare, R. G. Linington, T. Weber and M. H. Medema, Nucleic Acids Res., 2020, 48, D454–D458 Search PubMed.
- C. J. Oldfield, V. N. Uversky, A. Keith Dunker and L. Kurgan, Intrinsically Disord. Proteins, 2019, 1–34 Search PubMed.
- M. G. Chevrette, C. M. Carlson, H. E. Ortega, C. Thomas, G. E. Ananiev, K. J. Barns, A. J. Book, J. Cagnazzo, C. Carlos, W. Flanigan, K. J. Grubbs, H. A. Horn, F. M. Hoffmann, J. L. Klassen, J. J. Knack, G. R. Lewin, B. R. McDonald, L. Muller, W. G. P. Melo, A. A. Pinto-Tomás, A. Schmitz, E. Wendt-Pienkowski, S. Wildman, M. Zhao, F. Zhang, T. S. Bugni, D. R. Andes, M. T. Pupo and C. R. Currie, Nat. Commun., 2019, 10, 516 Search PubMed.
- A. B. Chase, D. Sweeney, M. N. Muskat and D. Guillén-Matus, bioRxiv, 2021 DOI:10.1101/2020.12.19.423547.
- J. Piel and M. Rust, Compr. Nat. Prod. Chem., 2020, 50–89 Search PubMed.
- M. Katz, B. M. Hover and S. F. Brady, J. Ind. Microbiol. Biotechnol., 2016, 43, 129–141 Search PubMed.
- M. Ferrer, M. Martínez-Martínez, R. Bargiela, W. R. Streit, O. V. Golyshina and P. N. Golyshin, Microb. Biotechnol., 2016, 9, 22–34 Search PubMed.
- J. G. Owen, B. V. B. Reddy, M. A. Ternei, Z. Charlop-Powers, P. Y. Calle, J. H. Kim and S. F. Brady, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11797–11802 Search PubMed.
- S. Hrvatin and J. Piel, J. Microbiol. Methods, 2007, 68, 434–436 Search PubMed.
- V. Libis, N. Antonovsky, M. Zhang, Z. Shang, D. Montiel, J. Maniko, M. A. Ternei, P. Y. Calle, C. Lemetre, J. G. Owen and S. F. Brady, Nat. Commun., 2019, 10, 3848 Search PubMed.
- B. M. Hover, S.-H. Kim, M. Katz, Z. Charlop-Powers, J. G. Owen, M. A. Ternei, J. Maniko, A. B. Estrela, H. Molina, S. Park, D. S. Perlin and S. F. Brady, Nat. Microbiol., 2018, 3, 415–422 Search PubMed.
- M. F. Laursen, M. D. Dalgaard and M. I. Bahl, Front. Microbiol., 2017, 8, 1934 Search PubMed.
- M. Ayling, M. D. Clark and R. M. Leggett, Briefings Bioinf., 2020, 21, 584–594 Search PubMed.
- Z. Wang, Y. Wang, J. A. Fuhrman, F. Sun and S. Zhu, Briefings Bioinf., 2020, 21, 777–790 Search PubMed.
- O. G. G. Almeida and E. C. P. De Martinis, Appl. Microbiol. Biotechnol., 2019, 103, 69–82 Search PubMed.
- R. R. Pal, R. P. More and H. J. Purohit, Soft Computing for Biological Systems, 2018, pp. 91–110 Search PubMed.
- T. Kvist, L. Sondt-Marcussen and M. J. Mikkelsen, PLoS One, 2014, 9, e106817 Search PubMed.
- R. D. Stewart, M. D. Auffret, A. Warr, A. H. Wiser, M. O. Press, K. W. Langford, I. Liachko, T. J. Snelling, R. J. Dewhurst, A. W. Walker, R. Roehe and M. Watson, Nat. Commun., 2018, 9, 870 Search PubMed.
- E. Yaffe and D. A. Relman, Nat. Microbiol., 2020, 5, 343–353 Search PubMed.
- M. Jain, H. E. Olsen, B. Paten and M. Akeson, Genome Biol., 2016, 17, 239 Search PubMed.
- T. Hon, K. Mars, G. Young, Y.-C. Tsai, J. W. Karalius, J. M. Landolin, N. Maurer, D. Kudrna, M. A. Hardigan, C. C. Steiner, S. J. Knapp, D. Ware, B. Shapiro, P. Peluso and D. R. Rank, Sci. Data, 2020, 7, 399 Search PubMed.
- D. Lang, S. Zhang, P. Ren, F. Liang, Z. Sun, G. Meng, Y. Tan, X. Li, Q. Lai, L. Han, D. Wang, F. Hu, W. Wang and S. Liu, GigaScience, 2020, 9, giaa123 Search PubMed.
- J. Risse, M. Thomson, S. Patrick, G. Blakely, G. Koutsovoulos, M. Blaxter and M. Watson, GigaScience, 2015, 4, 60 Search PubMed.
- D. B. B. Trivella and R. de Felicio, mSystems, 2016, 3, 18 Search PubMed.
- R. K. Wierenga, FEBS Lett., 2001, 492, 193–198 Search PubMed.
- S. Martínez Cuesta, S. A. Rahman, N. Furnham and J. M. Thornton, Biophys. J., 2015, 109, 1082–1086 Search PubMed.
- S. D. Copley, Curr. Opin. Struct. Biol., 2017, 47, 167–175 Search PubMed.
- O. Khersonsky and D. S. Tawfik, Annu. Rev. Biochem., 2010, 79, 471–505 Search PubMed.
- A. Pabis, F. Duarte and S. C. L. Kamerlin, Biochemistry, 2016, 55, 3061–3081 Search PubMed.
- W. Ding, X. Ji, Y. Li and Q. Zhang, Front. Chem., 2016, 4, 27 Search PubMed.
- S. Martinez Cuesta, N. Furnham, S. A. Rahman, I. Sillitoe and J. M. Thornton, Curr. Opin. Struct. Biol., 2014, 26, 121–130 Search PubMed.
- L. Noda-Garcia and D. S. Tawfik, Curr. Opin. Chem. Biol., 2020, 59, 147–154 Search PubMed.
- V. Veprinskiy, L. Heizinger, M. G. Plach and R. Merkl, BMC Evol. Biol., 2017, 17, 36 Search PubMed.
- S. Z. Alborzi, M.-D. Devignes and D. W. Ritchie, BMC Bioinf., 2017, 18, 107 Search PubMed.
- L. M. Podust and D. H. Sherman, Nat. Prod. Rep., 2012, 29, 1251 Search PubMed.
- P. Wang, X. Gao and Y. Tang, Curr. Opin. Chem. Biol., 2012, 16, 362–369 Search PubMed.
- C. Perry, E. L. C. de Los Santos, L. M. Alkhalaf and G. L. Challis, Nat. Prod. Rep., 2018, 35, 622–632 Search PubMed.
- G. Zhang, W. Zhang, Q. Zhang, T. Shi, L. Ma, Y. Zhu, S. Li, H. Zhang, Y.-L. Zhao, R. Shi and C. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4840–4844 Search PubMed.
- J. Antosch, F. Schaefers and T. A. M. Gulder, Angew. Chem., Int. Ed., 2014, 53, 3011–3014 Search PubMed.
- A. Greule, J. E. Stok, J. J. De Voss and M. J. Cryle, Nat. Prod. Rep., 2018, 35, 757–791 Search PubMed.
- V. B. Urlacher and M. Girhard, Trends Biotechnol., 2019, 37, 882–897 Search PubMed.
- M. M. Zdouc, M. M. Alanjary, G. S. Zarazúa, S. I. Maffioli, M. Crüsemann, M. H. Medema, S. Donadio and M. Sosio, Cell Chem. Biol., 2020 DOI:10.1016/j.chembiol.2020.11.009.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 Search PubMed.
- S. B. J. Kan, S. B. Jennifer Kan, R. D. Lewis, K. Chen and F. H. Arnold, Science, 2016, 354, 1048–1051 Search PubMed.
- S. B. J. Kan, X. Huang, Y. Gumulya, K. Chen and F. H. Arnold, Nature, 2017, 552, 132–136 Search PubMed.
- B. Fu and E. P. Balskus, Curr. Opin. Biotechnol., 2020, 65, 94–101 Search PubMed.
- A. L. Vagstad, T. Kuranaga, S. Püntener, V. R. Pattabiraman, J. W. Bode and J. Piel, Angew. Chem., Int. Ed., 2019, 58, 2246–2250 Search PubMed.
- B. I. Morinaka, E. Lakis, M. Verest, M. J. Helf, T. Scalvenzi, A. L. Vagstad, J. Sims, S. Sunagawa, M. Gugger and J. Piel, Science, 2018, 359, 779–782 Search PubMed.
- T. Q. N. Nguyen, Y. W. Tooh, R. Sugiyama, T. P. D. Nguyen, M. Purushothaman, L. C. Leow, K. Hanif, R. H. S. Yong, I. Agatha, F. R. Winnerdy, M. Gugger, A. T. Phan and B. I. Morinaka, Nat. Chem., 2020, 12, 1042–1053 Search PubMed.
- V. Bandarian, Biochim. Biophys. Acta, 2012, 1824, 1245–1253 Search PubMed.
- E. A. Lilla and K. Yokoyama, Nat. Chem. Biol., 2016, 12, 905–907 Search PubMed.
- T. A. Scott and J. Piel, Nat. Rev. Chem., 2019, 3, 404–425 Search PubMed.
- I.-M. A. Chen, K. Chu, K. Palaniappan, A. Ratner, J. Huang, M. Huntemann, P. Hajek, S. Ritter, N. Varghese, R. Seshadri, S. Roux, T. Woyke, E. A. Eloe-Fadrosh, N. N. Ivanova and N. C. Kyrpides, Nucleic Acids Res., 2021, 49(D1), D751–D763 Search PubMed.
- K. Youens-Clark, M. Bomhoff, A. J. Ponsero, E. M. Wood-Charlson, J. Lynch, I. Choi, J. H. Hartman and B. L. Hurwitz, GigaScience, 2019, 8, giz083 Search PubMed.
- A. L. Mitchell, A. Almeida, M. Beracochea, M. Boland, J. Burgin, G. Cochrane, M. R. Crusoe, V. Kale, S. C. Potter, L. J. Richardson, E. Sakharova, M. Scheremetjew, A. Korobeynikov, A. Shlemov, O. Kunyavskaya, A. Lapidus and R. D. Finn, Nucleic Acids Res., 2020, 48(D1), D570–D578 Search PubMed.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 Search PubMed.
- B. Buchfink, C. Xie and D. H. Huson, Nat. Methods, 2015, 12, 59–60 Search PubMed.
- P. R. Neubauer, C. Widmann, D. Wibberg, L. Schröder, M. Frese, T. Kottke, J. Kalinowski, H. H. Niemann and N. Sewald, PLoS One, 2018, 13, e0196797 Search PubMed.
- S. Nayfach, S. Roux, R. Seshadri, D. Udwary, N. Varghese, F. Schulz, D. Wu, D. Paez-Espino, I.-M. Chen, M. Huntemann, K. Palaniappan, J. Ladau, S. Mukherjee, T. B. K. Reddy, T. Nielsen, E. Kirton, J. P. Faria, J. N. Edirisinghe, C. S. Henry, S. P. Jungbluth, D. Chivian, P. Dehal, E. M. Wood-Charlson, A. P. Arkin, S. G. Tringe, A. Visel, IMG/M Data Consortium, T. Woyke, N. J. Mouncey, N. N. Ivanova, N. C. Kyrpides and E. A. Eloe-Fadrosh, Nat. Biotechnol., 2020, 1–11 Search PubMed.
- K. Blin, S. Shaw, K. Steinke, R. Villebro, N. Ziemert, S. Y. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2019, 47, W81–W87 Search PubMed.
- T. Koutsandreas, E. Ladoukakis, E. Pilalis, D. Zarafeta, F. N. Kolisis, G. Skretas and A. A. Chatziioannou, Front. Genet., 2019, 10, 469 Search PubMed.
- C. Darwin, On the Origin of Species, 1871 Search PubMed.
- S. L. Robinson, B. R. Terlouw, M. D. Smith, S. J. Pidot, T. P. Stinear, M. H. Medema and L. P. Wackett, J. Biol. Chem., 2020, 295, 14826–14839 Search PubMed.
- J. Santos-Aberturas, G. Chandra, L. Frattaruolo, R. Lacret, T. H. Pham, N. M. Vior, T. H. Eyles and A. W. Truman, Nucleic Acids Res., 2019, 47, 4624–4637 Search PubMed.
- B. Boeckmann, A. Bairoch, R. Apweiler, M.-C. Blatter, A. Estreicher, E. Gasteiger, M. J. Martin, K. Michoud, C. O'Donovan, I. Phan, S. Pilbout and M. Schneider, Nucleic Acids Res., 2003, 31, 365–370 Search PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 Search PubMed.
- A. Sánchez-Reyes and J. L. Folch-Mallol, in Metagenomics, ed. W. N. Hozzein, IntechOpen, Rijeka, 2020 Search PubMed.
- A. D. Young and J. P. Gillung, Syst. Entomol., 2020, 45, 225–247 Search PubMed.
- R. C. Edgar, Nucleic Acids Res., 2004, 32, 1792–1797 Search PubMed.
- K. Katoh, K. Misawa, K.-I. Kuma and T. Miyata, Nucleic Acids Res., 2002, 30, 3059–3066 Search PubMed.
- F. Sievers and D. G. Higgins, Protein Sci., 2018, 27, 135–145 Search PubMed.
- S. Capella-Gutiérrez, J. M. Silla-Martínez and T. Gabaldón, Bioinformatics, 2009, 25, 1972–1973 Search PubMed.
- D. M. Portik and J. J. Wiens, Syst. Biol., 2020, syaa064, DOI:10.1093/sysbio/syaa064.
- M. N. Price, P. S. Dehal and A. P. Arkin, PLoS One, 2010, 5, e9490 Search PubMed.
- X. Zhou, X.-X. Shen, C. T. Hittinger and A. Rokas, Mol. Biol. Evol., 2018, 35, 486–503 Search PubMed.
- K. Liu, C. R. Linder and T. Warnow, PLoS One, 2011, 6, e27731 Search PubMed.
- A. Stamatakis, Bioinformatics, 2014, 30, 1312–1313 Search PubMed.
- A. M. Kozlov, D. Darriba, T. Flouri, B. Morel and A. Stamatakis, Bioinformatics, 2019, 35, 4453–4455 Search PubMed.
- B. Q. Minh, H. A. Schmidt, O. Chernomor, D. Schrempf, M. D. Woodhams, A. von Haeseler and R. Lanfear, Mol. Biol. Evol., 2020, 37, 1530–1534 Search PubMed.
- J. Huerta-Cepas, F. Serra and P. Bork, Mol. Biol. Evol., 2016, 33, 1635–1638 Search PubMed.
- G. Yu, Curr. Protoc. Bioinf., 2020, 69, e96 Search PubMed.
- G. K. A. Hochberg and J. W. Thornton, Annu. Rev. Biophys., 2017, 46, 247–269 Search PubMed.
- N. M. Hendrikse, G. Charpentier, E. Nordling and P.-O. Syrén, FEBS J., 2018, 285, 4660–4673 Search PubMed.
- H. Ashkenazy, O. Penn, A. Doron-Faigenboim, O. Cohen, G. Cannarozzi, O. Zomer and T. Pupko, Nucleic Acids Res., 2012, 40, W580–W584 Search PubMed.
- Q. Zhang, Y. Yu, J. E. Vélasquez and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18361–18366 Search PubMed.
- M. Adamek, M. Alanjary and N. Ziemert, Nat. Prod. Rep., 2019, 36, 1295–1312 Search PubMed.
- N. Ziemert, S. Podell, K. Penn, J. H. Badger, E. Allen and P. R. Jensen, PLoS One, 2012, 7, e34064 Search PubMed.
- M. G. Chevrette, F. Aicheler, O. Kohlbacher, C. R. Currie and M. H. Medema, Bioinformatics, 2017, 33, 3202–3210 Search PubMed.
- E. J. N. Helfrich, R. Ueoka, A. Dolev, M. Rust, R. A. Meoded, A. Bhushan, G. Califano, R. Costa, M. Gugger, C. Steinbeck, P. Moreno and J. Piel, Nat. Chem. Biol., 2019, 15, 813–821 Search PubMed.
- M. E. Horsman, T. P. A. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 Search PubMed.
- T. P. A. Hari, P. Labana, M. Boileau and C. N. Boddy, ChemBioChem, 2014, 15, 2656–2661 Search PubMed.
- J. Durairaj, A. Di Girolamo, H. J. Bouwmeester, D. de Ridder, J. Beekwilder and A. D. van Dijk, Phytochemistry, 2019, 158, 157–165 Search PubMed.
- N. Sélem-Mojica, C. Aguilar, K. Gutiérrez-García, C. E. Martínez-Guerrero and F. Barona-Gómez, Microb. Genomics, 2019, 5, 445270 Search PubMed.
- J. C. Navarro-Muñoz, N. Selem-Mojica, M. W. Mullowney, S. A. Kautsar, J. H. Tryon, E. I. Parkinson, E. L. C. De Los Santos, M. Yeong, P. Cruz-Morales, S. Abubucker, A. Roeters, W. Lokhorst, A. Fernandez-Guerra, L. T. D. Cappelini, A. W. Goering, R. J. Thomson, W. W. Metcalf, N. L. Kelleher, F. Barona-Gomez and M. H. Medema, Nat. Chem. Biol., 2020, 16, 60–68 Search PubMed.
- J. K. Schniete, P. Cruz-Morales, N. Selem-Mojica, L. T. Fernández-Martínez, I. S. Hunter, F. Barona-Gómez and P. A. Hoskisson, mBio, 2018, 9, e02283-17 Search PubMed.
- P. Cruz-Morales, J. F. Kopp, C. Martínez-Guerrero, L. A. Yáñez-Guerra, N. Selem-Mojica, H. Ramos-Aboites, J. Feldmann and F. Barona-Gómez, Genome Biol. Evol., 2016, 8, 1906–1916 Search PubMed.
- H. J. Atkinson, J. H. Morris, T. E. Ferrin and P. C. Babbitt, PLoS One, 2009, 4, e4345 Search PubMed.
- P.-Y. Colin, B. Kintses, F. Gielen, C. M. Miton, G. Fischer, M. F. Mohamed, M. Hyvönen, D. P. Morgavi, D. B. Janssen and F. Hollfelder, Nat. Commun., 2015, 6, 10008 Search PubMed.
- E. Akiva, J. N. Copp, N. Tokuriki and P. C. Babbitt, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9549–E9558 Search PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 Search PubMed.
- J. A. Gustavsen, S. Pai, R. Isserlin, B. Demchak and A. R. Pico, F1000Research, 2019, 8, 1774 Search PubMed.
- K. Ono, T. Muetze, G. Kolishovski, P. Shannon and B. Demchak, F1000Research, 2015, 4, 478 Search PubMed.
- G. Csardi and T. Nepusz, InterJournal, 2006, 1695, 1–9 Search PubMed.
- J. A. Gerlt, J. T. Bouvier, D. B. Davidson, H. J. Imker, B. Sadkhin, D. R. Slater and K. L. Whalen, Biochim. Biophys. Acta, 2015, 1854, 1019–1037 Search PubMed.
- K. C. Mok, O. M. Sokolovskaya, A. M. Nicolas, Z. F. Hallberg, A. Deutschbauer, H. K. Carlson and M. E. Taga, mBio, 2020, 11, e02507–e02520 Search PubMed.
- R. Zallot, N. Oberg and J. A. Gerlt, Biochemistry, 2019, 58, 4169–4182 Search PubMed.
- S. A. Kautsar, J. J. J. van der Hooft, D. de Ridder and M. H. Medema, GigaScience, 2021, 10, giaa154 Search PubMed.
- J. I. Tietz, C. J. Schwalen, P. S. Patel, T. Maxson, P. M. Blair, H.-C. Tai, U. I. Zakai and D. A. Mitchell, Nat. Chem. Biol., 2017, 13, 470–478 Search PubMed.
- M. D. Mungan, M. Alanjary, K. Blin, T. Weber, M. H. Medema and N. Ziemert, Nucleic Acids Res., 2020, 48, W546–W552 Search PubMed.
- E. J. Culp, N. Waglechner, W. Wang, A. A. Fiebig-Comyn, Y.-P. Hsu, K. Koteva, D. Sychantha, B. K. Coombes, M. S. Van Nieuwenhze, Y. V. Brun and G. D. Wright, Nature, 2020, 578, 582–587 Search PubMed.
- L. Aravind, Genome Res., 2000, 10, 1074–1077 Search PubMed.
- D. Szklarczyk, A. L. Gable, D. Lyon, A. Junge, S. Wyder, J. Huerta-Cepas, M. Simonovic, N. T. Doncheva, J. H. Morris, P. Bork, L. J. Jensen and C. von Mering, Nucleic Acids Res., 2019, 47, D607–D613 Search PubMed.
- R. D. Zwahlen, C. Pohl, R. A. L. Bovenberg and A. J. M. Driessen, ACS Synth. Biol., 2019, 8, 1776–1787 Search PubMed.
- T. de Rond, J. E. Asay and B. S. Moore, bioRxiv, 2020 DOI:10.1101/2020.06.11.147165.
- F. Madeira, Y. M. Park, J. Lee, N. Buso, T. Gur, N. Madhusoodanan, P. Basutkar, A. R. N. Tivey, S. C. Potter, R. D. Finn and R. Lopez, Nucleic Acids Res., 2019, 47, W636–W641 Search PubMed.
- UniProt Consortium, Nucleic Acids Res., 2019, 47, D506–D515 Search PubMed.
- A. Chang, L. Jeske, S. Ulbrich, J. Hofmann, J. Koblitz, I. Schomburg, M. Neumann-Schaal, D. Jahn and D. Schomburg, Nucleic Acids Res., 2021, 49, D498–D508 Search PubMed.
- A. Joachimiak, Curr. Opin. Struct. Biol., 2009, 19, 573–584 Search PubMed.
- L. Jaroszewski, Z. Li, S. S. Krishna, C. Bakolitsa, J. Wooley, A. M. Deacon, I. A. Wilson and A. Godzik, PLoS Biol., 2009, 7, e1000205 Search PubMed.
- B. J. Burkhart, G. A. Hudson, K. L. Dunbar and D. A. Mitchell, Nat. Chem. Biol., 2015, 11, 564–570 Search PubMed.
- A. M. Gulick, ACS Chem. Biol., 2009, 4, 811–827 Search PubMed.
- A. R. Ortiz, C. E. M. Strauss and O. Olmea, Protein Sci., 2002, 11, 2606–2621 Search PubMed.
- T. Kawabata, Nucleic Acids Res., 2003, 31, 3367–3369 Search PubMed.
- M. Akdel, J. Durairaj, D. de Ridder and A. D. J. van Dijk, Comput. Struct. Biotechnol. J., 2020, 18, 981–992 Search PubMed.
- M. Carpentier and J. Chomilier, Bioinformatics, 2019, 35, 3970–3980 Search PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 10, 845–858 Search PubMed.
- J. Yang, R. Yan, A. Roy, D. Xu, J. Poisson and Y. Zhang, Nat. Methods, 2015, 12, 7–8 Search PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 Search PubMed.
- A. J. M. Ribeiro, J. D. Tyzack, N. Borkakoti, G. L. Holliday and J. M. Thornton, J. Biol. Chem., 2020, 295, 314–324 Search PubMed.
- G. Dodson and A. Wlodawer, Trends Biochem. Sci., 1998, 23, 347–352 Search PubMed.
- A. Rauwerdink and R. J. Kazlauskas, ACS Catal., 2015, 5, 6153–6176 Search PubMed.
- O. May, P. T. Nguyen and F. H. Arnold, Nat. Biotechnol., 2000, 18, 317–320 Search PubMed.
- E. T. Johnson, S. Ryu, H. Yi, B. Shin, H. Cheong and G. Choi, Plant J., 2001, 25, 325–333 Search PubMed.
- M. Ohashi, F. Liu, Y. Hai, M. Chen, M.-C. Tang, Z. Yang, M. Sato, K. Watanabe, K. N. Houk and Y. Tang, Nature, 2017, 549, 502–506 Search PubMed.
- L. Xiang, A. Li, C. Tian, Y. Zhou, G. Zhang and Y. Ma, Protein Expression Purif., 2014, 102, 20–26 Search PubMed.
- W. Tian, C. Chen, X. Lei, J. Zhao and J. Liang, Nucleic Acids Res., 2018, 46, W363–W367 Search PubMed.
- A. J. M. Ribeiro, G. L. Holliday, N. Furnham, J. D. Tyzack, K. Ferris and J. M. Thornton, Nucleic Acids Res., 2018, 46, D618–D623 Search PubMed.
- E. de Castro, C. J. A. Sigrist, A. Gattiker, V. Bulliard, P. S. Langendijk-Genevaux, E. Gasteiger, A. Bairoch and N. Hulo, Nucleic Acids Res., 2006, 34, W362–W365 Search PubMed.
- S. Mordhorst, B. I. Morinaka, A. L. Vagstad and J. Piel, Angew. Chem., Int. Ed., 2020, 59, 21442–21447 Search PubMed.
- R. Bonetta and G. Valentino, Proteins, 2020, 88, 397–413 Search PubMed.
- S. Poux, C. N. Arighi, M. Magrane, A. Bateman, C.-H. Wei, Z. Lu, E. Boutet, H. Bye-A-Jee, M. L. Famiglietti, B. Roechert and UniProt Consortium, Bioinformatics, 2017, 33, 3454–3460 Search PubMed.
- A. Hoarfrost, A. Aptekmann, G. Farfañuk and Y. Bromberg, bioRxiv, 2021 DOI:10.1101/2020.12.23.424215.
- Q. Ni, Z.-Z. Wang, Q. Han, G. Li, X. Wang and G. Wang, 3rd International Conference on Bioinformatics and Biomedical Engineering, 2009 Search PubMed.
- M. Kulmanov, M. A. Khan, R. Hoehndorf and J. Wren, Bioinformatics, 2018, 34, 660–668 Search PubMed.
- R. You, X. Huang and S. Zhu, Methods, 2018, 145, 82–90 Search PubMed.
- K. M. Verspoor, Methods Mol. Biol., 2014, 95–108 Search PubMed.
- V. Gligorijevic, M. Barot and R. Bonneau, Bioinformatics, 2018, 34, 3873–3881 Search PubMed.
- M. Nauman, H. U. Rehman, G. Politano and A. Benso, J. Grid Comput., 2019, 17, 225–237 Search PubMed.
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler, J. M. Cherry, A. P. Davis, K. Dolinski, S. S. Dwight, J. T. Eppig, M. A. Harris, D. P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J. C. Matese, J. E. Richardson, M. Ringwald, G. M. Rubin and G. Sherlock, Nat. Genet., 2000, 25, 25–29 Search PubMed.
- A. Ruepp, A. Zollner, D. Maier, K. Albermann, J. Hani, M. Mokrejs, I. Tetko, U. Güldener, G. Mannhaupt, M. Münsterkötter and H. W. Mewes, Nucleic Acids Res., 2004, 32, 5539–5545 Search PubMed.
- E. C. Webb, Enzyme Nomenclature 1992, Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology and the Nomenclature and Classification of Enzymes, Academic Press, 1992 Search PubMed.
- N. Zhou, Y. Jiang, T. R. Bergquist, A. J. Lee, B. Z. Kacsoh, A. W. Crocker, K. A. Lewis, G. Georghiou, H. N. Nguyen, M. N. Hamid, L. Davis, T. Dogan, V. Atalay, A. S. Rifaioglu, A. Dalkıran, R. Cetin Atalay, C. Zhang, R. L. Hurto, P. L. Freddolino, Y. Zhang, P. Bhat, F. Supek, J. M. Fernández, B. Gemovic, V. R. Perovic, R. S. Davidović, N. Sumonja, N. Veljkovic, E. Asgari, M. R. K. Mofrad, G. Profiti, C. Savojardo, P. L. Martelli, R. Casadio, F. Boecker, H. Schoof, I. Kahanda, N. Thurlby, A. C. McHardy, A. Renaux, R. Saidi, J. Gough, A. A. Freitas, M. Antczak, F. Fabris, M. N. Wass, J. Hou, J. Cheng, Z. Wang, A. E. Romero, A. Paccanaro, H. Yang, T. Goldberg, C. Zhao, L. Holm, P. Törönen, A. J. Medlar, E. Zosa, I. Borukhov, I. Novikov, A. Wilkins, O. Lichtarge, P.-H. Chi, W.-C. Tseng, M. Linial, P. W. Rose, C. Dessimoz, V. Vidulin, S. Dzeroski, I. Sillitoe, S. Das, J. G. Lees, D. T. Jones, C. Wan, D. Cozzetto, R. Fa, M. Torres, A. Warwick Vesztrocy, J. M. Rodriguez, M. L. Tress, M. Frasca, M. Notaro, G. Grossi, A. Petrini, M. Re, G. Valentini, M. Mesiti, D. B. Roche, J. Reeb, D. W. Ritchie, S. Aridhi, S. Z. Alborzi, M.-D. Devignes, D. C. E. Koo, R. Bonneau, V. Gligorijević, M. Barot, H. Fang, S. Toppo, E. Lavezzo, M. Falda, M. Berselli, S. C. E. Tosatto, M. Carraro, D. Piovesan, H. Ur Rehman, Q. Mao, S. Zhang, S. Vucetic, G. S. Black, D. Jo, E. Suh, J. B. Dayton, D. J. Larsen, A. R. Omdahl, L. J. McGuffin, D. A. Brackenridge, P. C. Babbitt, J. M. Yunes, P. Fontana, F. Zhang, S. Zhu, R. You, Z. Zhang, S. Dai, S. Yao, W. Tian, R. Cao, C. Chandler, M. Amezola, D. Johnson, J.-M. Chang, W.-H. Liao, Y.-W. Liu, S. Pascarelli, Y. Frank, R. Hoehndorf, M. Kulmanov, I. Boudellioua, G. Politano, S. Di Carlo, A. Benso, K. Hakala, F. Ginter, F. Mehryary, S. Kaewphan, J. Björne, H. Moen, M. E. E. Tolvanen, T. Salakoski, D. Kihara, A. Jain, T. Šmuc, A. Altenhoff, A. Ben-Hur, B. Rost, S. E. Brenner, C. A. Orengo, C. J. Jeffery, G. Bosco, D. A. Hogan, M. J. Martin, C. O'Donovan, S. D. Mooney, C. S. Greene, P. Radivojac and I. Friedberg, Genome Biol., 2019, 20, 244 Search PubMed.
- D. Prihoda, J. M. Maritz, O. Klempir, D. Dzamba, C. H. Woelk, D. J. Hazuda, D. A. Bitton and G. D. Hannigan, Nat. Prod. Rep., 2021 10.1039/d0np00055h.
- M. A. Skinnider, C. W. Johnston, M. Gunabalasingam, N. J. Merwin, A. M. Kieliszek, R. J. MacLellan, H. Li, M. R. M. Ranieri, A. L. H. Webster, M. P. T. Cao, A. Pfeifle, N. Spencer, Q. H. To, D. P. Wallace, C. A. Dejong and N. A. Magarvey, Nat. Commun., 2020, 11, 6058 Search PubMed.
- G. D. Hannigan, D. Prihoda, A. Palicka, J. Soukup, O. Klempir, L. Rampula, J. Durcak, M. Wurst, J. Kotowski, D. Chang, R. Wang, G. Piizzi, G. Temesi, D. J. Hazuda, C. H. Woelk and D. A. Bitton, Nucleic Acids Res., 2019, 47, e110 Search PubMed.
- N. Youngs, D. Penfold-Brown, R. Bonneau and D. Shasha, PLoS Comput. Biol., 2014, 10, e1003644 Search PubMed.
- S. D. Copley, Curr. Opin. Chem. Biol., 2003, 7, 265–272 Search PubMed.
- F. Belinky, I. B. Rogozin and E. V. Koonin, Sci. Rep., 2017, 7, 12422 Search PubMed.
- L. Fu, B. Niu, Z. Zhu, S. Wu and W. Li, Bioinformatics, 2012, 28, 3150–3152 Search PubMed.
- R. C. Edgar, Bioinformatics, 2010, 26, 2460–2461 Search PubMed.
- M. Steinegger and J. Söding, Nat. Commun., 2018, 9, 2542 Search PubMed.
- K. Verdel-Aranda, S. T. López-Cortina, D. A. Hodgson and F. Barona-Gómez, Microb. Biotechnol., 2015, 8, 239–252 Search PubMed.
- A. Khanal, S. Yu McLoughlin, J. P. Kershner and S. D. Copley, Mol. Biol. Evol., 2015, 32, 100–108 Search PubMed.
- J. A. North, A. B. Narrowe, W. Xiong, K. M. Byerly, G. Zhao, S. J. Young, S. Murali, J. A. Wildenthal, W. R. Cannon, K. C. Wrighton, R. L. Hettich and F. R. Tabita, Science, 2020, 369, 1094–1098 Search PubMed.
- A. Jarzab, N. Kurzawa, T. Hopf, M. Moerch, J. Zecha, N. Leijten, Y. Bian, E. Musiol, M. Maschberger, G. Stoehr, I. Becher, C. Daly, P. Samaras, J. Mergner, B. Spanier, A. Angelov, T. Werner, M. Bantscheff, M. Wilhelm, M. Klingenspor, S. Lemeer, W. Liebl, H. Hahne, M. M. Savitski and B. Kuster, Nat. Methods, 2020, 17, 495–503 Search PubMed.
- L. Slabinski, L. Jaroszewski, L. Rychlewski, I. A. Wilson, S. A. Lesley and A. Godzik, Bioinformatics, 2007, 23, 3403–3405 Search PubMed.
- I. M. Overton, C. A. J. van Niekerk and G. J. Barton, Proteins, 2011, 79, 1027–1033 Search PubMed.
- I. M. Overton and G. J. Barton, FEBS Lett., 2006, 580, 4005–4009 Search PubMed.
- L. Kurgan, A. A. Razib, S. Aghakhani, S. Dick, M. Mizianty and S. Jahandideh, BMC Struct. Biol., 2009, 9, 50 Search PubMed.
- I. M. Overton, G. Padovani, M. A. Girolami and G. J. Barton, Bioinformatics, 2008, 24, 901–907 Search PubMed.
- H. Wang, L. Feng, G. I. Webb, L. Kurgan, J. Song and D. Lin, Briefings Bioinf., 2017, 18, 1092 Search PubMed.
- G. von Heijne, J. Mol. Biol., 1985, 184, 99–105 Search PubMed.
- J. J. Almagro Armenteros, K. D. Tsirigos, C. K. Sønderby, T. N. Petersen, O. Winther, S. Brunak, G. von Heijne and H. Nielsen, Nat. Biotechnol., 2019, 37, 420–423 Search PubMed.
- Z. Wu, K. K. Yang, M. J. Liszka, A. Lee, A. Batzilla, D. Wernick, D. P. Weiner and F. H. Arnold, ACS Synth. Biol., 2020, 9, 2154–2161 Search PubMed.
- J. J. Zhang, X. Tang and B. S. Moore, Nat. Prod. Rep., 2019, 36, 1313–1332 Search PubMed.
- J. H. Kim, Z. Feng, J. D. Bauer, D. Kallifidas, P. Y. Calle and S. F. Brady, Biopolymers, 2010, 93, 833–844 Search PubMed.
- Y. Zhang, F. Buchholz, J. P. Muyrers and A. F. Stewart, Nat. Genet., 1998, 20, 123–128 Search PubMed.
- H. Wang, Z. Li, R. Jia, Y. Hou, J. Yin, X. Bian, A. Li, R. Müller, A. F. Stewart, J. Fu and Y. Zhang, Nat. Protoc., 2016, 11, 1175–1190 Search PubMed.
- A. M. Kunjapur, P. Pfingstag and N. C. Thompson, Nat. Commun., 2018, 9, 4425 Search PubMed.
- H. Zhang, B. A. Boghigian and B. A. Pfeifer, Biotechnol. Bioeng., 2010, 105, 567–573 Search PubMed.
- J. Beld, E. C. Sonnenschein, C. R. Vickery, J. P. Noel and M. D. Burkart, Nat. Prod. Rep., 2014, 31, 61–108 Search PubMed.
- M. F. Freeman, Methods Enzymol., 2018, 604, 259–286 Search PubMed.
- B. A. Pfeifer and C. Khosla, Microbiol. Mol. Biol. Rev., 2001, 65, 106–118 Search PubMed.
- N. D. Lanz, A. J. Blaszczyk, E. L. McCarthy, B. Wang, R. X. Wang, B. S. Jones and S. J. Booker, Biochemistry, 2018, 57, 1475–1490 Search PubMed.
- M. Crüsemann, R. Reher, I. Schamari, A. O. Brachmann, T. Ohbayashi, M. Kuschak, D. Malfacini, A. Seidinger, M. Pinto-Carbó, R. Richarz, T. Reuter, S. Kehraus, A. Hallab, M. Attwood, H. B. Schiöth, P. Mergaert, Y. Kikuchi, T. F. Schäberle, E. Kostenis, D. Wenzel, C. E. Müller, J. Piel, A. Carlier, L. Eberl and G. M. König, Angew. Chem., Int. Ed., 2018, 57, 836–840 Search PubMed.
- C. Hermes, R. Richarz, D. A. Wirtz, J. Patt, W. Hanke, S. Kehraus, J. H. Voß, J. Küppers, T. Ohbayashi, V. Namasivayam, J. Alenfelder, A. Inoue, P. Mergaert, M. Gütschow, C. E. Müller, E. Kostenis, G. M. König and M. Crüsemann, Nat. Commun., 2021, 12, 144 Search PubMed.
- A. Bhushan, P. J. Egli, E. E. Peters, M. F. Freeman and J. Piel, Nat. Chem., 2019, 11, 931–939 Search PubMed.
- M. Funabashi, T. L. Grove, M. Wang, Y. Varma, M. E. McFadden, L. C. Brown, C. Guo, S. Higginbottom, S. C. Almo and M. A. Fischbach, Nature, 2020, 582, 566–570 Search PubMed.
- M. M. Zhang, Y. Wang, E. L. Ang and H. Zhao, Nat. Prod. Rep., 2016, 33, 963–987 Search PubMed.
- J. K. Brunson, S. M. K. McKinnie, J. R. Chekan, J. P. McCrow, Z. D. Miles, E. M. Bertrand, V. A. Bielinski, H. Luhavaya, M. Oborník, G. Jason Smith, D. A. Hutchins, A. E. Allen and B. S. Moore, Science, 2018, 361, 1356–1358 Search PubMed.
- P. Carbonell, A. J. Jervis, C. J. Robinson, C. Yan, M. Dunstan, N. Swainston, M. Vinaixa, K. A. Hollywood, A. Currin, N. J. W. Rattray, S. Taylor, R. Spiess, R. Sung, A. R. Williams, D. Fellows, N. J. Stanford, P. Mulherin, R. Le Feuvre, P. Barran, R. Goodacre, N. J. Turner, C. Goble, G. G. Chen, D. B. Kell, J. Micklefield, R. Breitling, E. Takano, J.-L. Faulon and N. S. Scrutton, Commun. Biol., 2018, 1, 66 Search PubMed.
- T. R. Northen, J.-C. Lee, L. Hoang, J. Raymond, D.-R. Hwang, S. M. Yannone, C.-H. Wong and G. Siuzdak, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3678–3683 Search PubMed.
- T. de Rond, M. Danielewicz and T. Northen, Curr. Opin. Biotechnol., 2015, 31, 1–9 Search PubMed.
- T. de Rond, J. Gao, A. Zargar, M. de Raad, J. Cunha, T. R. Northen and J. D. Keasling, Angew. Chem., Int. Ed., 2019, 58, 10114–10119 Search PubMed.
- M. Mrksich, ACS Nano, 2008, 2, 7–18 Search PubMed.
- L. Ban, N. Pettit, L. Li, A. D. Stuparu, L. Cai, W. Chen, W. Guan, W. Han, P. G. Wang and M. Mrksich, Nat. Chem. Biol., 2012, 8, 769–773 Search PubMed.
- P. T. O'Kane, Q. M. Dudley, A. K. McMillan, M. C. Jewett and M. Mrksich, Sci. Adv., 2019, 5, eaaw9180 Search PubMed.
- A. J. Pluchinsky, D. J. Wackelin, X. Huang, F. H. Arnold and M. Mrksich, J. Am. Chem. Soc., 2020, 142, 19804–19808 Search PubMed.
- E. van der Helm, H. J. Genee and M. O. A. Sommer, Nat. Chem. Biol., 2018, 14, 752–759 Search PubMed.
- S.-J. Yeom, M. Kim, K. K. Kwon, Y. Fu, E. Rha, S.-H. Park, H. Lee, H. Kim, D.-H. Lee, D.-M. Kim and S.-G. Lee, Nat. Commun., 2018, 9, 5053 Search PubMed.
- S. R. Fleming, P. M. Himes, S. V. Ghodge, Y. Goto, H. Suga and A. A. Bowers, J. Am. Chem. Soc., 2020, 142, 5024–5028 Search PubMed.
- A. Trincone, Mar. Drugs, 2011, 9, 478–499 Search PubMed.
- C. J. Carrano, S. Schellenberg, S. A. Amin, D. H. Green and F. C. Küpper, Mar. Biotechnol., 2009, 11, 431–440 Search PubMed.
- S. Nayfach, B. Rodriguez-Mueller, N. Garud and K. S. Pollard, Genome Res., 2016, 26, 1612–1625 Search PubMed.
- G. Salazar, L. Paoli, A. Alberti, J. Huerta-Cepas, H.-J. Ruscheweyh, M. Cuenca, C. M. Field, L. P. Coelho, C. Cruaud, S. Engelen, A. C. Gregory, K. Labadie, C. Marec, E. Pelletier, M. Royo-Llonch, S. Roux, P. Sánchez, H. Uehara, A. A. Zayed, G. Zeller, M. Carmichael, C. Dimier, J. Ferland, S. Kandels, M. Picheral, S. Pisarev, J. Poulain, Tara Oceans Coordinators, S. G. Acinas, M. Babin, P. Bork, C. Bowler, C. de Vargas, L. Guidi, P. Hingamp, D. Iudicone, L. Karp-Boss, E. Karsenti, H. Ogata, S. Pesant, S. Speich, M. B. Sullivan, P. Wincker and S. Sunagawa, Cell, 2019, 179, 1068–1083 Search PubMed.
- S. J. Biller, P. M. Berube, K. Dooley, M. Williams, B. M. Satinsky, T. Hackl, S. L. Hogle, A. Coe, K. Bergauer, H. A. Bouman, T. J. Browning, D. De Corte, C. Hassler, D. Hulston, J. E. Jacquot, E. W. Maas, T. Reinthaler, E. Sintes, T. Yokokawa and S. W. Chisholm, Sci. Data, 2018, 5, 180176 Search PubMed.
- S. G. Acinas, P. Sánchez, G. Salazar, F. M. Cornejo-Castillo, M. Sebastián, R. Logares, S. Sunagawa, P. Hingamp, H. Ogata, G. Lima-Mendez, S. Roux, J. M. González, J. M. Arrieta, I. S. Alam, A. Kamau, C. Bowler, J. Raes, S. Pesant, P. Bork, S. Agustí, T. Gojobori, V. Bajic, D. Vaqué, M. B. Sullivan, C. Pedrós-Alió, R. Massana, C. M. Duarte and J. M. Gasol, bioRxiv, 2019 DOI:10.1101/635680.
- L. Paoli, H. J. Ruscheweyh, C. C. Forneris, S. Kautsar, Q. Clayssen, G. Salazar, A. Milanese, D. Gehrig, M. Larralde, L. Carroll, P. Sánchez, A. Zayed, D. R. Cronin, S. G. Acinas, P. Bork, C. Bowler, T. O. Delmont, M. B. Sullivan, P. Wincker, G. Zeller, S. L. Robinson, J. Piel and S. Sunagawa, bioRxiv, 2021 DOI:10.1101/2021.03.24.436479.
- S. Sunagawa, L. P. Coelho, S. Chaffron, J. R. Kultima, K. Labadie, G. Salazar, B. Djahanschiri, G. Zeller, D. R. Mende, A. Alberti, F. M. Cornejo-Castillo, P. I. Costea, C. Cruaud, F. d'Ovidio, S. Engelen, I. Ferrera, J. M. Gasol, L. Guidi, F. Hildebrand, F. Kokoszka, C. Lepoivre, G. Lima-Mendez, J. Poulain, B. T. Poulos, M. Royo-Llonch, H. Sarmento, S. Vieira-Silva, C. Dimier, M. Picheral, S. Searson, S. Kandels-Lewis, Tara Oceans Coordinators, C. Bowler, C. de Vargas, G. Gorsky, N. Grimsley, P. Hingamp, D. Iudicone, O. Jaillon, F. Not, H. Ogata, S. Pesant, S. Speich, L. Stemmann, M. B. Sullivan, J. Weissenbach, P. Wincker, E. Karsenti, J. Raes, S. G. Acinas and P. Bork, Science, 2015, 348, 1261359 Search PubMed.
- A. A. Caparco, E. Pelletier, J. L. Petit, A. Jouenne, B. R. Bommarius, V. Berardinis, A. Zaparucha, J. A. Champion, A. S. Bommarius and C. Vergne-Vaxelaire, Adv. Synth. Catal., 2020, 362, 2427–2436 Search PubMed.
- Q. Carradec, E. Pelletier, C. Da Silva, A. Alberti, Y. Seeleuthner, R. Blanc-Mathieu, G. Lima-Mendez, F. Rocha, L. Tirichine, K. Labadie, A. Kirilovsky, A. Bertrand, S. Engelen, M.-A. Madoui, R. Méheust, J. Poulain, S. Romac, D. J. Richter, G. Yoshikawa, C. Dimier, S. Kandels-Lewis, M. Picheral, S. Searson, Tara Oceans Coordinators, O. Jaillon, J.-M. Aury, E. Karsenti, M. B. Sullivan, S. Sunagawa, P. Bork, F. Not, P. Hingamp, J. Raes, L. Guidi, H. Ogata, C. de Vargas, D. Iudicone, C. Bowler and P. Wincker, Nat. Commun., 2018, 9, 373 Search PubMed.
- U. Hentschel, J. Piel, S. M. Degnan and M. W. Taylor, Nat. Rev. Microbiol., 2012, 10, 641–654 Search PubMed.
- M. F. Freeman, C. Gurgui, M. J. Helf, B. I. Morinaka, A. R. Uria, N. J. Oldham, H.-G. Sahl, S. Matsunaga and J. Piel, Science, 2012, 338, 387–390 Search PubMed.
- M. J. Helf, A. Jud and J. Piel, ChemBioChem, 2017, 18, 444–450 Search PubMed.
- D. R. M. Smith, A. R. Uria, E. J. N. Helfrich, D. Milbredt, K.-H. van Pée, J. Piel and R. J. M. Goss, ACS Chem. Biol., 2017, 12, 1281–1287 Search PubMed.
- E. W. Schmidt, A. Y. Obraztsova, S. K. Davidson, D. J. Faulkner and M. G. Haygood, Mar. Biol., 2000, 136, 969–977 Search PubMed.
- M. Rust, E. J. N. Helfrich, M. F. Freeman, P. Nanudorn, C. M. Field, C. Rückert, T. Kündig, M. J. Page, V. L. Webb, J. Kalinowski, S. Sunagawa and J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9508–9518 Search PubMed.
- M. A. Storey, S. K. Andreassend, J. Bracegirdle, A. Brown, R. A. Keyzers, D. F. Ackerley, P. T. Northcote and J. G. Owen, mBio, 2020, 11, e02997–e03019 Search PubMed.
- V. Agarwal, J. M. Blanton, S. Podell, A. Taton, M. A. Schorn, J. Busch, Z. Lin, E. W. Schmidt, P. R. Jensen, V. J. Paul, J. S. Biggs, J. W. Golden, E. E. Allen and B. S. Moore, Nat. Chem. Biol., 2017, 13, 537–543 Search PubMed.
- E. W. Schmidt, J. T. Nelson, D. A. Rasko, S. Sudek, J. A. Eisen, M. G. Haygood and J. Ravel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7315–7320 Search PubMed.
- P. F. Long, W. C. Dunlap, C. N. Battershill and M. Jaspars, ChemBioChem, 2005, 6, 1760–1765 Search PubMed.
- J. Koehnke, A. F. Bent, W. E. Houssen, G. Mann, M. Jaspars and J. H. Naismith, Curr. Opin. Struct. Biol., 2014, 29, 112–121 Search PubMed.
- J. A. McIntosh and E. W. Schmidt, ChemBioChem, 2010, 11, 1413–1421 Search PubMed.
- S. Sarkar, W. Gu and E. W. Schmidt, ACS Catal., 2020, 10, 7146–7153 Search PubMed.
- M. A. Altamia, Z. Lin, A. E. Trindade-Silva, I. D. Uy, J. Reuben Shipway, D. V. Wilke, G. P. Concepcion, D. L. Distel, E. W. Schmidt and M. G. Haygood, mSystems, 2020, 5(3), e00261-20 Search PubMed.
- C. A. Fowler, F. Sabbadin, L. Ciano, G. R. Hemsworth, L. Elias, N. Bruce, S. McQueen-Mason, G. J. Davies and P. H. Walton, Biotechnol. Biofuels, 2019, 12, 1–11 Search PubMed.
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sørlie and V. G. H. Eijsink, Science, 2010, 330, 219–222 Search PubMed.
- V. Maini-Rekdal, E. N. Bess, J. E. Bisanz, P. J. Turnbaugh and E. P. Balskus, Science, 2019, 364, 1055 Search PubMed.
- V. M. Rekdal, P. N. Bernadino, M. U. Luescher, S. Kiamehr, C. Le, J. E. Bisanz, P. J. Turnbaugh, E. N. Bess and E. P. Balskus, eLife, 2020, 9, e50845 Search PubMed.
- J. R. Chekan, S. M. K. McKinnie, J. P. Noel and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12799–12805 Search PubMed.
- B. L. Mello, A. M. Alessi, D. M. Riaño-Pachón, E. R. deAzevedo, F. E. G. Guimarães, M. C. Espirito Santo, S. McQueen-Mason, N. C. Bruce and I. Polikarpov, Biotechnol. Biofuels, 2017, 10, 254 Search PubMed.
- V. P. Andreu, H. E. Augustijn, K. van den Berg, J. J. J. van der Hooft, M. A. Fischbach and M. H. Medema, bioRxiv, 2020 DOI:10.1101/2020.12.14.422671.
- P. Sukul, S. Schäkermann, J. E. Bandow, A. Kusnezowa, M. Nowrousian and L. I. Leichert, Microbiome, 2017, 5(1), 1–5 Search PubMed.
- T. Woyke, D. F. R. Doud and F. Schulz, Nat. Methods, 2017, 14, 1045–1054 Search PubMed.
- R. Stepanauskas, E. A. Fergusson, J. Brown, N. J. Poulton, B. Tupper, J. M. Labonté, E. D. Becraft, J. M. Brown, M. G. Pachiadaki, T. Povilaitis, B. P. Thompson, C. J. Mascena, W. K. Bellows and A. Lubys, Nat. Commun., 2017, 8, 84 Search PubMed.
- R. V. Grindberg, T. Ishoey, D. Brinza, E. Esquenazi, R. C. Coates, W.-T. Liu, L. Gerwick, P. C. Dorrestein, P. Pevzner, R. Lasken and W. H. Gerwick, PLoS One, 2011, 6, e18565 Search PubMed.
- M. A. Skiba, A. P. Sikkema, N. A. Moss, C. L. Tran, R. M. Sturgis, L. Gerwick, W. H. Gerwick, D. H. Sherman and J. L. Smith, ACS Chem. Biol., 2017, 12, 3039–3048 Search PubMed.
- M. G. Pachiadaki, J. M. Brown, J. Brown, O. Bezuidt, P. M. Berube, S. J. Biller, N. J. Poulton, M. D. Burkart, J. J. La Clair, S. W. Chisholm and R. Stepanauskas, Cell, 2019, 179, 1623–1635 Search PubMed.
- U. Ben-David, B. Siranosian, G. Ha, H. Tang, Y. Oren, K. Hinohara, C. A. Strathdee, J. Dempster, N. J. Lyons, R. Burns, A. Nag, G. Kugener, B. Cimini, P. Tsvetkov, Y. E. Maruvka, R. O'Rourke, A. Garrity, A. A. Tubelli, P. Bandopadhayay, A. Tsherniak, F. Vazquez, B. Wong, C. Birger, M. Ghandi, A. R. Thorner, J. A. Bittker, M. Meyerson, G. Getz, R. Beroukhim and T. R. Golub, Nature, 2018, 560, 325–330 Search PubMed.
- K. Morita, F. Wang, K. Jahn, T. Hu, T. Tanaka, Y. Sasaki, J. Kuipers, S. Loghavi, S. A. Wang, Y. Yan, K. Furudate, J. Matthews, L. Little, C. Gumbs, J. Zhang, X. Song, E. Thompson, K. P. Patel, C. E. Bueso-Ramos, C. D. DiNardo, F. Ravandi, E. Jabbour, M. Andreeff, J. Cortes, K. Bhalla, G. Garcia-Manero, H. Kantarjian, M. Konopleva, D. Nakada, N. Navin, N. Beerenwinkel, P. A. Futreal and K. Takahashi, Nat. Commun., 2020, 11, 5327 Search PubMed.
- Y. Sugimoto, F. R. Camacho, S. Wang, P. Chankhamjon, A. Odabas, A. Biswas, P. D. Jeffrey and M. S. Donia, Science, 2019, 366, eaax9176 Search PubMed.
- F. Imdahl, E. Vafadarnejad, C. Homberger, A.-E. Saliba and J. Vogel, Nat. Microbiol., 2020, 5, 1202–1206 Search PubMed.
- N. J. Tobias and H. B. Bode, J. Mol. Biol., 2019, 431, 4589–4598 Search PubMed.
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J.-C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4004–4009 Search PubMed.
- K. S. Lee, F. C. Pereira, M. Palatinszky, L. Behrendt, U. Alcolombri, D. Berry, M. Wagner and R. Stocker, Nat. Protoc., 2020, 1–43, DOI:10.1038/s41596-020-00427-8.
- J. W. Bogart, M. D. Cabezas, B. Vögeli, D. A. Wong, A. S. Karim and M. C. Jewett, ChemBioChem, 2020, 22, 84–91 Search PubMed.
- L. Zhuang, S. Huang, W.-Q. Liu, A. S. Karim, M. C. Jewett and J. Li, Metab. Eng., 2020, 60, 37–44 Search PubMed.
- Q. M. Dudley, C. J. Nash and M. C. Jewett, Synth. Biol., 2019, 4, ysz003 Search PubMed.
- N. Ziemert, M. Alanjary and T. Weber, Nat. Prod. Rep., 2016, 33, 988–1005 Search PubMed.
- A. M. Kloosterman, P. Cimermancic, S. S. Elsayed, C. Du, M. Hadjithomas, M. S. Donia, M. A. Fischbach, G. P. van Wezel and M. H. Medema, PLoS Biol., 2020, 18, e3001026 Search PubMed.
- S. F. Altschul, T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller and D. J. Lipman, Nucleic Acids Res., 1997, 25, 3389–3402 Search PubMed.
- B. Szalkai and V. Grolmusz, Genomics, 2019, 111, 883–885 Search PubMed.
- S. Sunagawa, M. K. DeSalvo, C. R. Voolstra, A. Reyes-Bermudez and M. Medina, PLoS One, 2009, 4, e4865 Search PubMed.
- E. M. Gabor, W. B. L. Alkema and D. B. Janssen, Environ. Microbiol., 2004, 6, 879–886 Search PubMed.
- W. Liebl, A. Angelov, J. Juergensen, J. Chow, A. Loeschcke, T. Drepper, T. Classen, J. Pietruszka, A. Ehrenreich, W. R. Streit and K.-E. Jaeger, Appl. Microbiol. Biotechnol., 2014, 98, 8099–8109 Search PubMed.
- N. Katzke, A. Knapp, A. Loeschcke, T. Drepper and K.-E. Jaeger, Methods Mol. Biol., 2017, 1539, 159–196 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |