
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bottromycins - biosynthesis, synthesis and activity
Laura
Franz
 a,
Uli
Kazmaier
b,
Andrew W.
Truman
c and
Jesko
Koehnke
*ad
a,
Uli
Kazmaier
b,
Andrew W.
Truman
c and
Jesko
Koehnke
*ad
aHelmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research (HZI), Saarland University Campus, 66123 Saarbrücken, Germany
bSaarland University, Organic Chemistry, Campus Geb. C4.2, 66123 Saarbrücken, Germany
cDepartment of Molecular Microbiology, John Innes Centre, Norwich, UK
dSchool of Chemistry, University of Glasgow, Glasgow, UK. E-mail: Jesko.koehnke@glasgow.ac.uk
First published on 23rd February 2021
Abstract
Covering: 1950s up to the end of 2020
Bottromycins are a class of macrocyclic peptide natural products that are produced by several Streptomyces species and possess promising antibacterial activity against clinically relevant multidrug-resistant pathogens. They belong to the ribosomally synthesised and post-translationally modified peptide (RiPP) superfamily of natural products. The structure contains a unique four-amino acid macrocycle formed via a rare amidine linkage, C-methylation and a D-amino acid. This review covers all aspects of bottromycin research with a focus on recent years (2009–2020), in which major advances in total synthesis and understanding of bottromycin biosynthesis were achieved.
 Laura Franz | Laura Franz studied biochemistry and received her Bachelor's and Master's degree at Bielefeld University, Germany. She is currently a PhD student at the Helmholtz Institute for Pharmaceutical Research Saarland under supervision of Jesko Koehnke. Her research focuses on the elucidation of RiPP pathways, characterisation of biosynthetic enzymes and production of new-to-nature ribosomal peptides. |
 Uli Kazmaier | Uli Kazmaier studied chemistry at the University of Stuttgart where he obtained his PhD 1990 while working with U. Schmidt. Afterwards he joined the groups of M. T. Reetz (Marburg) and B. M. Trost (Stanford) as postdoctoral fellow. In 1992, he moved to Heidelberg, starting his own scientific work under the mentorship of G. Helmchen. In 2000, he received a Novartis Chemistry Lectureship and in 2001 an offer for a full professorship at Saarland University. His current research interests focuses on new organometallic reagents and reactions especially for amino acid and peptide synthesis, and their application to natural product synthesis. |
 Andrew W. Truman | Andrew Truman obtained his PhD at the University of Cambridge in 2008, where he remained to conduct postdoctoral research. In 2013, he established his research group at the John Innes Centre as a Royal Society University Research Fellow. His laboratory use genomics to guide the discovery of new natural products from bacteria, with a major focus on ribosomally synthesised and post-translationally modified peptides. His group aim to understand the biosynthesis and natural functions of these specialised metabolites. |
 Jesko Koehnke | Jesko Koehnke studied Biochemistry at Leibniz University Hannover. After graduating in 2005, he obtained his PhD at Columbia University under the supervision of Prof. L. Shapiro. After graduating in 2010, he joined Prof. J. H. Naismith at the University of St Andrews as a postdoctoral researcher. In 2015 he established his independent junior group leader at the Helmholtz Institute for Pharmaceutical Research Saarland, where his work focused on the structural biology and biochemistry of RiPPs biosynthesis. In 2020 he moved to the University of Glasgow as a Reader to continue his research centered around RiPPs. |
1 Introduction
Bottromycins were originally isolated from the fermentation broth of Streptomyces bottropensis in the late 1950s and described as peptidic natural products with antibacterial activity against Gram-positive pathogens.1,2 A series of mechanism of action (MoA) studies revealed that bottromycins inhibit protein synthesis by binding to the aminoacyl-tRNA binding site (A-site) of the prokaryotic 50S ribosome.3–5 The bottromycin target site is currently not addressed by any antibiotic used in the clinic, which makes cross-resistance unlikely. As a result, bottromycins are effective against the problematic human pathogens methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococci (VRE).The chemical structure of bottromycins proved very difficult to elucidate and consequently underwent several revisions that ultimately led to the assignment of 1. It showed that bottromycins are highly modified heptapeptides that are comprised of an N-terminal, four-amino acid macrocycle formed via a unique amidine linkage, several C-methylated residues, D-aspartate and a C-terminal thiazole (Fig. 1A). This structure was confirmed by total synthesis in 2009.10 Shortly after the successful total synthesis, several groups reported the discovery of the bottromycin biosynthetic gene cluster (BGC), which revealed that they are unusual ribosomally synthesized and post-translationally modified peptides (RiPPs).6,8,11,12
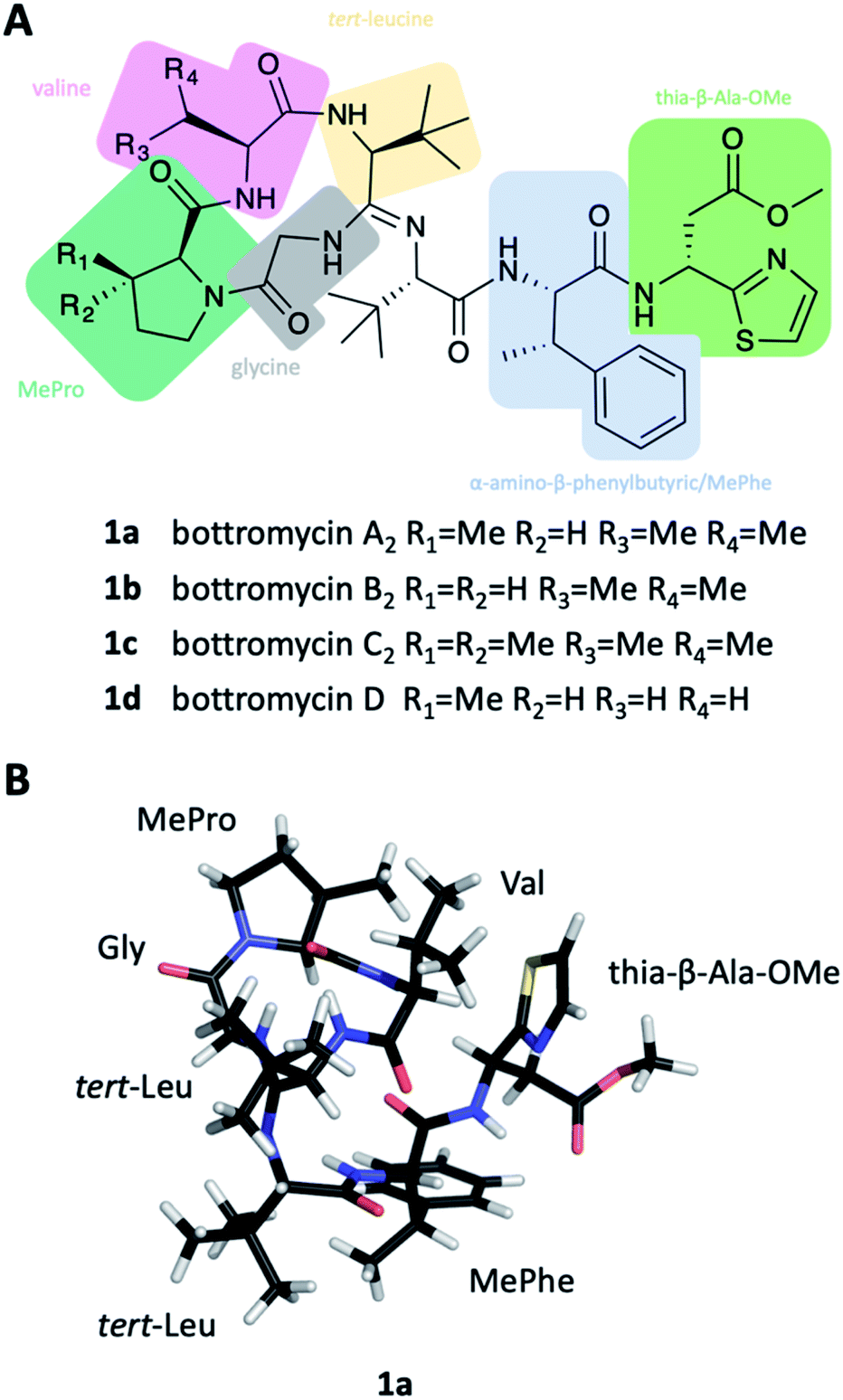 | ||
| Fig. 1 Structures of bottromycins. (A) Structural formular of bottromycin A2, B2, C2 and D. (B) NMR structure of bottromycin A2 in CDCl3. Coloured fragements in (A) represent the identified ninhydrin positive compounds after hydrolysis of bottromycin during the structure elucidation process of bottromycin. | ||
The emerging RiPP superfamily encompasses highly diverse molecules with interesting bioactivities.13 The unifying feature is their biosynthetic logic: a short structural gene is expressed and yields the precursor peptide, which consists of one or more core peptide(s) (the eventual natural product(s)) and an N-terminal leader or C-terminal follower peptide that is important for recognition of the precursor peptide by parts of the biosynthetic machinery. The post-translational modifications introduced in the core peptide have been reviewed extensively elsewhere and expand the chemical and structural features far beyond the 20 canonical amino acids.13,15 These modifications include, but are not limited to, heterocyclisation of Ser/Thr and Cys residues to oxazolines and thiazolines, oxidation of these heterocycles to the corresponding azoles, epimerisation of amino acids to give D-stereocentres, methylation, Ser/Thr/Tyr prenylation, dehydration, hydroxylation, macrocycle formation and the formation of new C–C bonds through different chemistries. In fact, in some RiPPs such as pyrroloquinoline quinone, the final product does not contain any peptide bonds.15 Bottromycins are the only RiPP of bacterial origin that utilises a follower peptide rather than the canonical leader peptide for biosynthesis.6,8,11,12
This review aims to cover all aspects of bottromycin research with a focus on the recent years (2009–2020). We will place particular emphasis on the total synthesis of bottromycins, studies conducted to investigate the biosynthesis and produce derivatives in vivo and very recent progress on the enzymology of individual steps involved in bottromycin biosynthesis.
2 Discovery, structure elucidation and activity
In 1955, a new Streptomyces species, Streptomyces bottropensis, isolated from a soil sample taken in the German town Bottrop, was reported.16 Its fermentation broth displayed antibiotic activity and led to the discovery of a new antibiotic, named bottromycin.1,16 Waisvisz and coworkers were the first group to investigate the bottromycin structure and reported that although bottromycin itself gave a negative ninhydrin test, hydrolysis of bottromycins gave 7 ninhydrin positive compounds. Four remained unspecified, but two that could be identified as glycine and valine.1,17–19 Acetylation of bottromycin yielded two degradation products, which were further analysed. This resulted in the identification of two ninhydrin-positive compounds, α-amino-β-phenylbutyric (MePhe) acid and β-(2-thiazole)-β-alanine methyl ester (thia-β-Ala-OMe)18 as well as the previously identified glycine and valine.19 Thus, only two of the unknown hydrolysis products remained obscure. As part of these studies, a methyl ester moiety was identified and further analysed: mild alkaline hydrolysis yielded a biologically inactive bottromycin,17 while re-esterification in methanolic HCl solution yielded a biologically active compound identical with bottromycin. Esterification with ethanol or n-butyl alcohol produced less active bottromycin derivatives.17In 1965, Nakamura and colleagues reported the isolation of bottromycin from Streptomyces No. 3668-L2.20 They were able to identify the two remaining unknown ninhydrin-positive substances from acid hydrolysis as L-β,β-dimethyl-alpha-aminobutyric acid (tert-leucine, t-Leu) and L-cis-3-methylproline (MePro).21 Analysis of the isolated antibiotic by thin-layer chromatography revealed it to contain a major component, which was identical to the previously studied bottromycin and designated as bottromycin A, and two minor components, designated as bottromycin B and C.21,22 Bottromycin B and C are almost identical to bottromycin A, but contain L-proline (bottromycin B) and L-3,3-dimethylproline (Me2Pro) (bottromycin C) instead of MePro. Bottromycins B and C are biologically active, but bottromycin B displayed 3–4 times less potency than bottromycin A and C (see Tables 1 and 5).22 Nakamura and colleagues also reported the recovery of pivalic acid after hydrolysis of bottromycin beside the 6 previously described compounds.21 Different tests (i.e. van Slyke test) also suggested the existence of an amidine group in bottromycin in the tetrapeptide moiety.21 It was concluded that the N-terminus of bottromycins must not be free, because they are negative in ninhydrin reactions, Edman degradation and Sanger decomposition.21 The structure (2) that was proposed based on these data harboured pivalic acid at the N-terminus of the tetrapeptide (t-Leu, Val, MePro, Gly) (Fig. 2). A subsequent revision of the structure postulated 1-Δ1-caproic acid instead of pivalic acid at the N-terminus.23 Bottromycin with pivalic acid was designated as bottromycin A1, and bottromycin containing 1-Δ1-caproic acid was designated as bottromycin A2, but synthetic attempts by Yamada et al. indicated that the proposed structure was incorrect.24
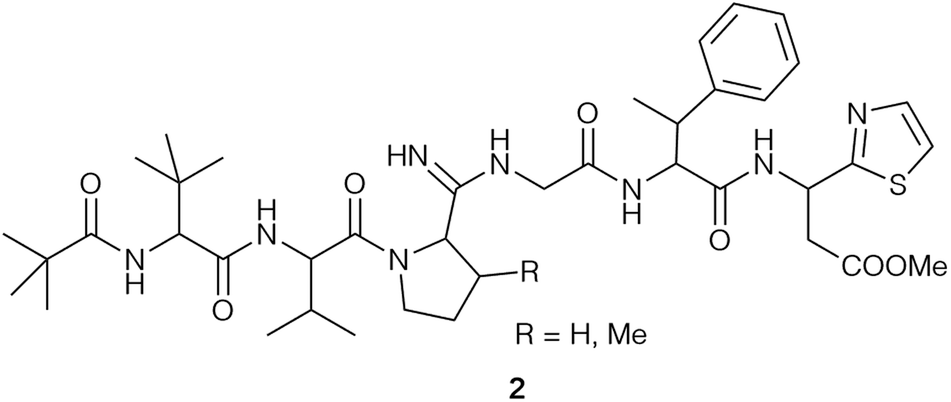 | ||
| Fig. 2 Structure of bottromycin (2) according to Nakamura et al.21 | ||
Ten years later, Takita and colleagues proposed a new structure for bottromycin, based on mass spectrometry and 1H NMR data.25 This cyclic structure of the tetrapeptide was revised by Shipper in 1983,26 who demonstrated that an unusual amidine moiety links the cyclic tetrapeptide and the linear chain and is formed by condensation of N-terminal amino group and the backbone amide carbonyl. In spite of these iterative revisions, and the fact that bottromycin A1 and A2 were actually identical, the designation bottromycin A2 (as well as B2 and C2) for bottromycin A, B and C was retained. It took until 2009 before the correct structure of bottromycin A2 (1a) (see Fig. 1A) was determined by Shimamura et al. through the total synthesis of bottromycin A2, demonstrating the D-configuration of the thia-β-Ala-OMe.10 In 2012, Gouda et al. determined the three-dimensional structure of bottromycin A2 in CDCl3 based on NMR data.27 In this structure (Fig. 1B) the C-terminal residues fold back on the macrocycle made by the four N-terminal amino acids. Hence, the MePro and the thia-β-Ala-OMe, which are essential for activity, are on one side of the three-dimensional structure, which suggests an involvement of this region in target engagement.
Biological data for bottromycins A2–C2 were reported by Nakamura et al.22 They determined the minimal growth inhibitory concentrations (MIC) towards a wide range of bacterial strains (Table 1). In addition, bottromycin A2 also showed strong inhibition against mycoplasma (0.001–0.01 μg mL−1),2,28 the multidrug-resistant human pathogens MRSA (1 μg mL−1) and VRE (0.5 μg mL−1)2 and Xanthomonas oryzae pv. oryzae KACC 10331, a pathovar that causes rice bacterial blight,29 which makes bottromycins potentially interesting for agrochemical use.
| Strain | MIC (μg ml−1) | ||
|---|---|---|---|
| A2 | B2 | C2 | |
| a S. aureus BR4, R1, R5 and R6 are clinical isolates resistant to antibiotics: BR4 is erythromycin–carbomycin resistant, R1 and R6 are penicillin-tetracycline resistant. | |||
| Staphylococcus aureus (Smith) | 0.2 | 0.8 | 0.1 |
| Staphylococcus aureus (209 P) | 0.1 | 0.8 | 0.1 |
| Staphylococcus aureus (BR4) | 0.4 | 0.8 | 0.4 |
| Staphylococcus aureus (R1) | 0.4 | 1.5 | 0.4 |
| Staphylococcus aureus (R5) | 0.4 | 1.5 | 0.4 |
| Staphylococcus aureus (R6) | 0.4 | 1.5 | 0.2 |
| Micrococcus flavus | 0.4 | 1.5 | 1.5 |
| Bacillus subtilis (PCI 219) | 0.06 | 0.2 | 0.06 |
| Bacillus cereus (IAM 1729) | 0.4 | 0.8 | 0.25 |
| Corynebacterium xerosis | 0.06 | 0.2 | 0.06 |
| Mycobacterium phlei | 0.1 | 1.5 | 0.1 |
| Mycobacterium 607 | 25 | (25) | (12) |
| Shigella dysenteria | 12 | 25 | 25 |
| Shigella sonnei | 25 | >100 | 50 |
| Salmonella typhosa | 50 | >100 | >100 |
| Salmonella paratyphi | 25 | 6 | 6 |
| Escherichia coli B | 3 | 25 | 6 |
| Escherichia coli K12 | 25 | 100 | 50 |
| Klebsiella pneumoniae 602 | 50 | >100 | >100 |
| Pseudomonas aeruginosa A3 | 50 | >100 | 100 |
| Sarcina lutea | 0.4 | 0.8 | |
| Proteus vulgaris OX-19 | 12 | 100 | 50 |
Although highly active in vitro, the bottromycins showed no convincing in vivo efficiency because of their instability in oral and parenteral administration,30 which is mainly the result of the lability of the methyl ester under physiological conditions.2 Synthetic approaches to change the methyl ester moiety could increase plasma stability without decreasing the activity (see Tables 2–5). In vivo studies using bottromycin derivatised at the methyl ester moiety displayed in vivo activity against staphylococcal and streptococcal infection in animals,30 and Mycoplasma gallispetium (pleuropneumonia-like organisms) in chicken using subcutaneous administration.28,31,32
3 Mechanism of action
In MoA studies, it was demonstrated that bottromycin A2 inhibits protein synthesis,33 which is a common target of antibiotics (e.g. aminoglycosides and macrolides). How bottromycin inhibits protein synthesis was investigated by different groups, who came to divergent conclusions.Tanaka and co-workers were the first group that studied the MoA of bottromycin and demonstrated the inhibition of protein biosynthesis in vivo and in vitro.33 The inhibition was reported to be highly dependent on the base composition.33 They showed that bottromycin inhibits neither aminoacyl-tRNA synthesis nor its binding to the ribosome.33 The puromycin reaction, which is regarded as an analogous reaction to the peptide bond formation, was not significantly affected by bottromycin A2 in the absence of guanosine triphosphate (GTP) and G factor. This reaction does not require GTP or G factor. Addition of GTP and G factor stimulate translocation of the peptidyl-tRNA from the A- to the P-site. In the presence of GTP and G factor the puromycin reaction was inhibited by bottromycin A2.34,35 They concluded from these results that bottromycin A2 interferes with the translocation of peptidyl-tRNA and movement of mRNA on the ribosomes.35,36 In a cell-free system, it was determined on which subunit of the ribosome the antibiotic acts. Examining the inhibitory effect of bottromycin A2 in a protein synthesising system containing excess of either 30S or 50S ribosomal subunit, the excess of 50S over 30S subunit decreased the inhibitory effect by bottromycin. This effect could not be observed using an excess of 30S over 50S subunit. From these results it was concluded that bottromycin interacts with the 50S subunit of the ribosome.37
Pestka and Brot also examined the effect of bottromycin on several steps of the protein synthesis.38 They also determined an effect of bottromycin on the translocation process, using an oligophenylalanine formation assay. An inhibitory effect was observed in the absence and presence of G protein and GTP. In contrast to Tanaka et al., they also observed an effect of bottromycin on peptide bond synthesis using an acetyl-phenylalanyl-puromycin formation assay.38 As the degree of inhibition on oligophenylalanine synthesis and the puromycin reaction were comparable, they suggested that the inhibition of the peptide bond formation may be the primary action of bottromycin A2.38
The latest studies examining the MoA of bottromycins were carried out in the early 1980s by Otaka and coworkers.3–5 They reported that bottromycin interferes with the interaction of aminoacyl- or peptidyl-tRNA with the A (aminoacyl) site of ribosomes3,5 and proposed the hypothesis that bottromycin binds to (or close to) the A site of the ribosome and lowers the affinity of aminoacyl-, peptidyl-tRNA or puromycin.4,5
The proposed MoA of bottromycin from Otaka and coworkers is similar to the mechanism of tetracyclines. Tetracyclines block the binding of aminoacyl-tRNAs to the A site of the ribosome,39 but only bottromycins are able to release bound tRNA from the A site. While tetracyclines bind to the 30S ribosomal subunit,39,40 bottromycins are reported to bind the 50S subunit of the ribosome.37 The different MoAs and binding sites between tetracycline and bottromycin are also supported by the observation that no cross-resistance to the tetracycline-resistant strains S. aureus R1 and R6 (Table 1) is observed. Other antibiotics that act at the A-site can also have different functions, such as negamycin, which inhibits translocation and stimulates miscoding.40–42 In the past decade, multiple structures of the 70S ribosome or its subunits in complex with antibiotics have been determined, which provided insights into their mechanism of action.42,43 Unfortunately, no ribosome–bottromycin complex structure has been published yet, so our understanding of the MoA of bottromycin remains limited.
4 Synthetic approaches and total syntheses towards bottromycins
4.1 Synthesis of the unusual amino acids44
Based on the early structure proposals, the first syntheses of the unusual amino acid building blocks were reported from the mid 1970's on. A first stereospecific synthesis of (2S,3R)-3-methylproline (MePro) was reported by Titouani et al. in 1980 using a Hofmann–Löffler–Freitag reaction.45 Herdeis et al. reported the syntheses of both, the (2S,3S)- and the (2S,3R)-isomer of MePro from a pyroglutaminol derivate,46 while Karoyan and Chassaing used a 5-exo trig cyclisation approach to generated the five-membered ring.47 Kamenecka et al. developed a protocol starting from commercially available 3-hydroxy-(S)-proline using Stille cross coupling chemistry,48 and a stereoselective cuprate addition was the key step in the synthesis by Flamant-Robin et al.493,3-Dimethyl-(2S)-proline (Me2Pro) is one of the unusual amino acids found in bottromycin C2. A first enantioselective synthesis was reported by Sharma and Lubell in 1996.50 A regioselective enolisation of a 4-oxo-proline derivative followed by alkylation with different alkyl halides allowed the synthesis of a variety of proline derivatives. Two approaches towards racemic Me2Pro were described by Medina51 and Bott et al.52
Most investigations focused on the synthesis of (2S,3S)-3-methylphenylalanine (MePhe) because it also appears in some other natural products, such as mannopeptimycin53 and the isoleucyl-tRNA-synthetase inhibitor SB-203208.54 In connection with one of the first synthetic studies towards bottromycins, Kataoka et al. described the synthesis and optical resolution of MePhe via condensation of racemic 1-bromo-1-phenyl-ethane with acetaminomalonate.55 Many attempts have been undertaken to separate the stereoisomers more easily using modern chromatographic techniques.56–64 Ogawa et al. reported an enzymatic approach to MePhe,65 while Tsuchihashi et al. used the Michael addition of malonate onto a chiral vinyl sulfoxide as a key step.66 Dharanipragada et al.67,68 and Fioravanti et al.69 described the asymmetric syntheses of MePhe using auxiliary-controlled enolate chemistry, while the groups of Pericas and Rieva developed a protocol using a Sharpless epoxidation as a stereo-controlling step.70 O'Donnell et al. reported an acyclic stereoselective boron alkylation as a key step using a chiral boron reagent in the presence of cinchona alkaloids,71 while the group of Turner developed a chemo-enzymatic route towards enantiomerically pure MePhe derivatives, based on an oxidation-reduction sequence.72 Ooi et al. described a phase transfer-catalysed alkylation of a glycinate Schiff base with 1-bromo-1-phenylethane under the influence of chiral quaternary ammonium bromide and 18-crown-6.73 And finally, Zhang et al. reported a palladium-catalysed C–H functionalisation of C(sp3)–H bonds using 8-aminoquinoline (AQ) as a directing group, giving access to fully protected MePhe derivatives.
The unusual C-terminal thiazolyl amino acid thia-β-Ala was the last one whose configuration was determined. It required the total synthesis of the bottromycins to establish it definitively. The problem arose from the structure elucidation of bottromycin. By hydrolysis of the natural product with conc. HCl, Waisvisz et al. obtained a “sulfur-containing amino acid”, which unfortunately showed no optical activity.18 Umezawa's group subsequently obtained an optically active amino acid ([α]18D: +9) by hydrolysing the antibiotic with acetic anhydride.74 To determine the structure of the C-terminal amino acid Waisvisz prepared racemic thia-β-Ala by addition of hydroxylamine towards β-2-thiazolacrylic acid, unfortunately only with moderate yield.75
Seto et al. tried to obtain optically active (S)-thia-β-Ala starting from (S)-aspartic acid.75 Thiazole formation was performed by condensation of the corresponding protected aspartic acid thioamide with bromoacetaldehyde, but unfortunately, these derivatives were also optically inactive. Obviously, complete epimerisation occurred in the thiazole formation step. The racemic amino acid, however, could be resolved into its enantiomers by treating the Phth-derivative with brucine.49 After cleavage of the Phth-protecting group, the (+)-amino acid, the constituent of bottromycin, was isolated in pure form. It should be mentioned that the thia-β-Ala derivatives prepared also lost their optical activity after heating under reflux in 6 N HCl for 8 h, while the same compounds were stable at room temperature or under slightly basic conditions, illustrating the configurational lability of these compounds. The only enantioselective synthesis of enantiomerically pure (S)-and (R)-thia-β-Ala so far was reported by the groups of Sunazuka and Ōmura,10 taking advantage of the chiral sulfinamide chemistry developed by Davis and Ellman,76,77 which allowed the synthesis of both enantiomers in a highly stereoselective fashion.
4.2 Synthetic studies towards bottromycins
The total synthesis of bottromycins was adversely affected by the long-standing erroneous structure proposals for the compounds, and so far only one total synthesis exists.10,78 It confirmed that the C-terminal thia-β-Ala is (R)-configured and not (S)- as originally reported.While the wrong structural assumptions meant that early synthetic work could only ever be unsuccessful, significant efforts were directed towards the synthesis of the partial structure of this rather unique peptide. The first investigations were already reported by Yamada et al. in 1997.79 Their synthetic route was based on the linear hexapeptide 2 proposed by Nakamura et al. (Fig. 2).22,74,80,81
Yamada et al. focused on the synthesis and properties of the central amidine unit. Several amidines were prepared by condensation of Cbz-protected amino acid imido esters with amino acid esters (Scheme 1).24 The desired amidine 3 could be obtained without problems, but it was impossible to extend the dipeptides at the C-terminus. On activation, or even on standing under basic conditions cyclisation occurred to the corresponding imidazolone 4. Therefore, the authors decided to form the amidine unit of 7 by coupling two model tripeptide fragments, the tripeptide imido ester 5 and tripeptide 6.24
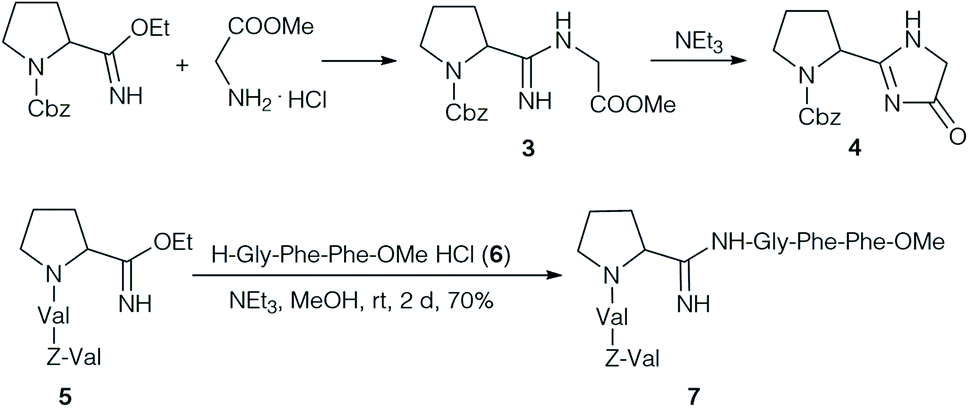 | ||
| Scheme 1 Formation of amidine 7. | ||
Interestingly, the pKa of all synthesised amidines (pKa ∼ 9.3) were around 1 pKa higher than in the natural product (∼8.2), a first indication that the structure proposal might not be correct. The antimicrobial activities of these amidines were examined, but no activity was observed.
Based on the revised structures by Schipper26 and Kaneda,82 who proposed a cyclic tetrapeptide with a tripeptide chain connected via an unusual amidine moiety, Kazmaier et al. focused on the synthesis of the corresponding peptide ring and the highly substituted amidine.83 A key step of their approach was an Ugi reaction using a protected thioamino acid and NH3 as the amine compound (Scheme 2). Although Ugi reactions with NH3 are often non-specific and yield a range of side products, good results were obtained with sterically demanding aldehydes.84,85 With thiocarboxylic acids this approach allowed the synthesis of endothiopeptides.86–88 With isocyanoacetate the linear tripeptide 8 was obtained, which could be extended to the desired tetrapeptide 9 under standard conditions. Attempts to cyclise 9 or to connect the side chain via peptide coupling failed, because the thioamide underwent cyclisation to the thiazolinone 10, comparable to the imidazolone formation reported by Yamada (Scheme 1).24
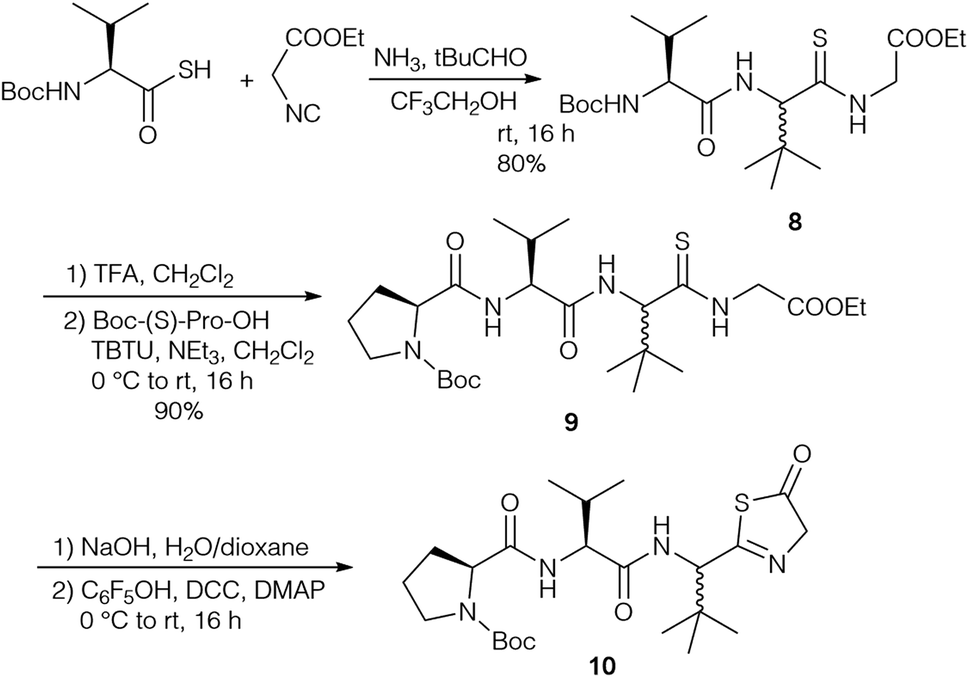 | ||
| Scheme 2 Synthesis of endothiopeptides via Ugi reaction. | ||
To figure out if amidine formation is possible between sterically demanding amino acids, thiopeptide 11 was synthesised in an analogous fashion (Scheme 3). Attempts to couple 11 directly with amines failed, so the thioamide was converted into the corresponding thioimidoester 12. In the presence of Hg(OOCCF3)212 could be coupled with valine methyl ester to obtain amidine 13 in good yield.
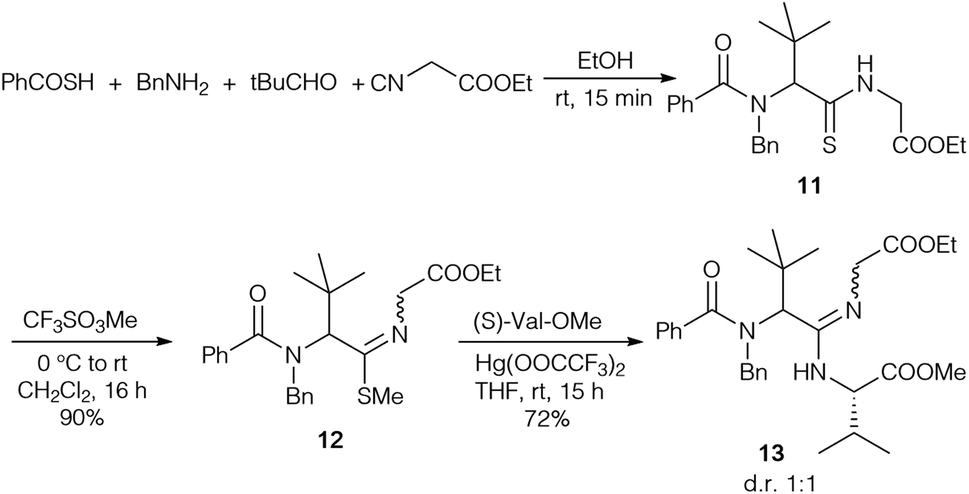 | ||
| Scheme 3 Synthesis of amidine 13via Ugi reaction. | ||
The diastereomers formed could be separated by flash chromatography, but unfortunately this protocol could not be carried out with endothiopeptide 8. This resulted in a change in the strategy, replacing the intermolecular amidine formation with an intramolecular one by using the isocyanide of tLeu-OMe (Scheme 4). The endothiopeptide 14 was obtained in high yield and could be extended on the N-terminus. S-Methylation and cyclisation in the presence of Hg(OOCCF3)2 gave access to cyclic amidine 15.
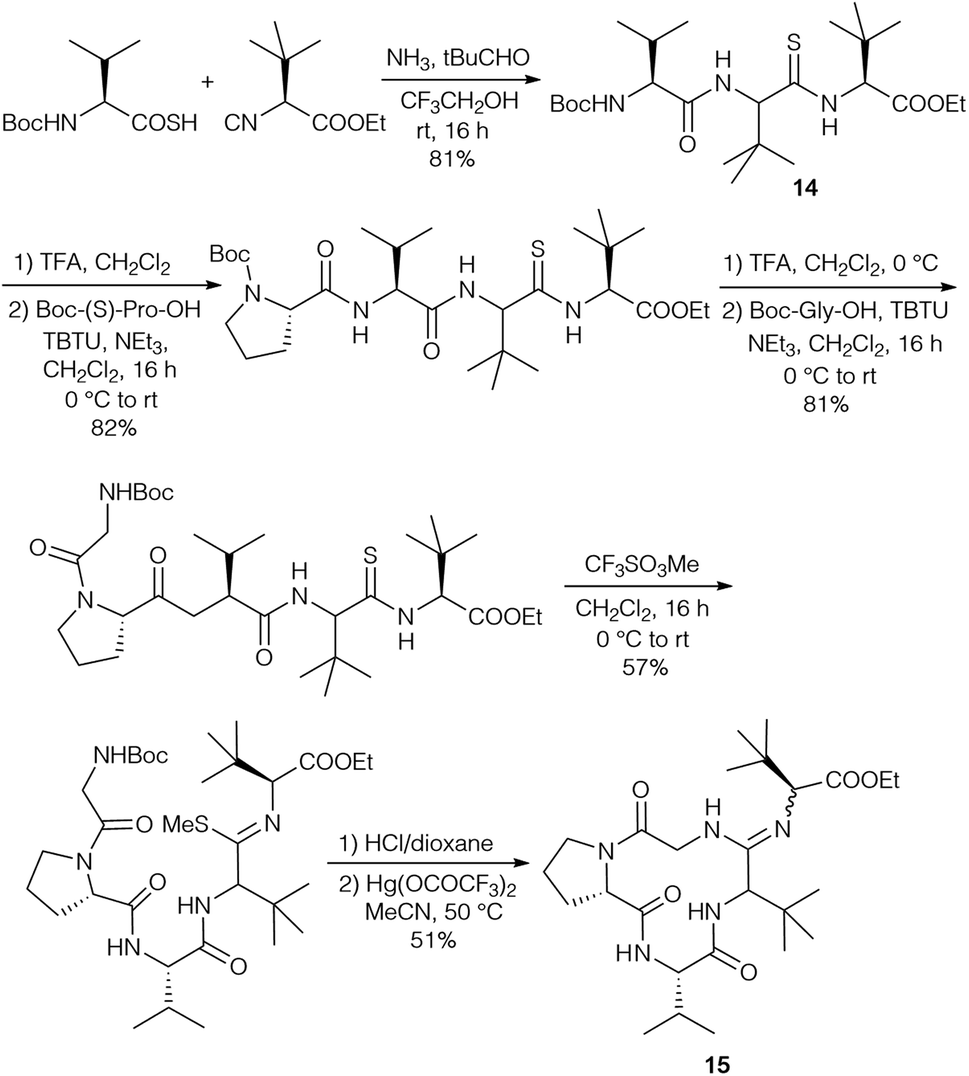 | ||
| Scheme 4 Synthesis of cyclic amidine 15via Ugi reaction. | ||
Amidine formation as the key step was also investigated in detail by Ōmura and Sunazuka et al. during their synthesis of bottromycin A2 (1a) and B2 (1b) (Scheme 5).78 They investigated the reaction of thioamide 16 with the tripeptide side chain 17. No reaction was observed in THF using NEt3 as a base, while in the presence of Hg(OAc)2 the desired amidine 18 was not obtained, and instead the amide 19 was produced. Better results were obtained using HgCl2 and Hg(OTf)2 as Lewis acids. Finally, 2,6-lutidine in acetonitrile was the method of choice to yield 19.
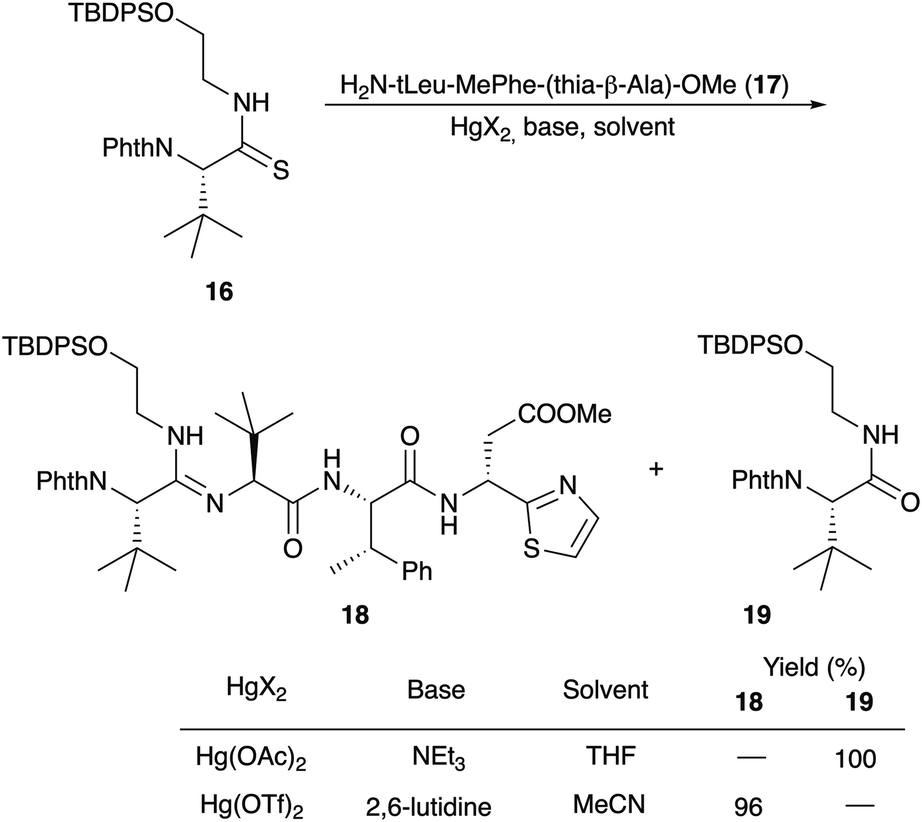 | ||
| Scheme 5 Synthesis of linear amidine 18. | ||
The same groups also performed degradation studies of bottromycin obtained by fermentation (Scheme 6).89 They subjected 1a to pyrolysis in MeOH in a sealed tube at 130 °C, resulting in cleavage of the tripeptide side chain. Besides dipeptide 20, cyclic product 21 was also obtained as a diastereomeric mixture. Obviously, the epimerisation of the tLeu in the side chain occurred via the enol-form of 21. This could explain why the tLeu obtained by total hydrolysis of the bottromycins has a lower optical rotation than the synthetic enantiopure amino acid. Reduction under mild conditions converted the natural product into alcohol 22, which could be used to investigate cyclisation conditions.
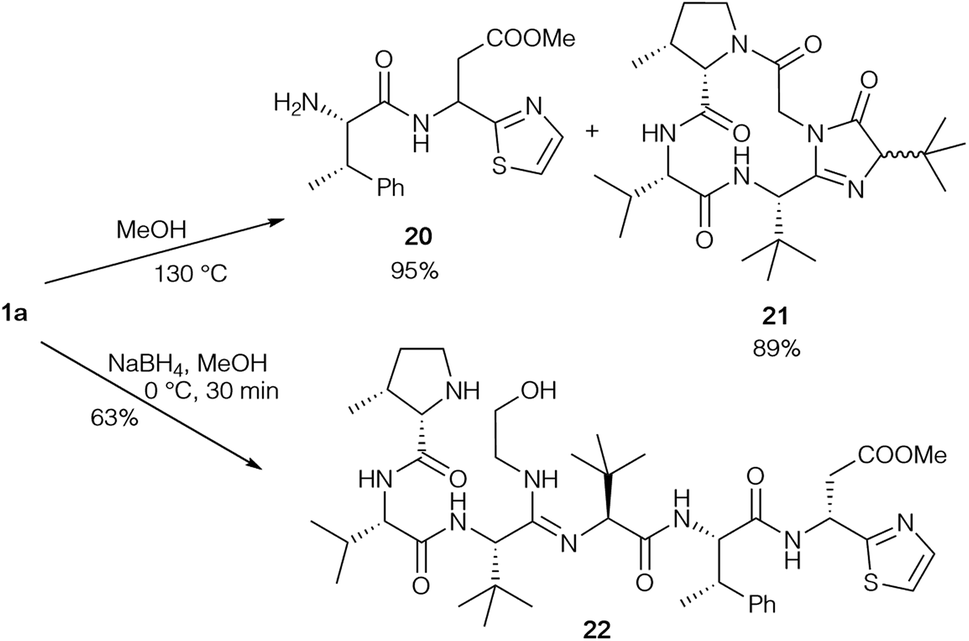 | ||
| Scheme 6 Degradation of bottromycin A2 (1a). | ||
Dipeptide 20 was also used to determine the configuration of the thia-β-Ala, an amino acid that is rather configurational labile.75 Both enantiomers of thia-β-Ala-OMe were synthesised via the sulfinamide protocol and subsequently coupled with azido-MePhe 23 (Scheme 7). Reduction of the azido functionality of 24 provided the two diastereomeric dipeptides 20. Comparison of their 1H NMR spectra with the spectrum of 20 obtained via pyrolysis clearly indicated that the (R)-isomer is incorporated into the bottromycins and that the original structure proposal (S) was incorrect. Coupling of 20 with Boc-(S)-tLeu and subsequent Boc-cleavage provided tripeptide 17, which was also used in the amidine formation experiments (Scheme 5).
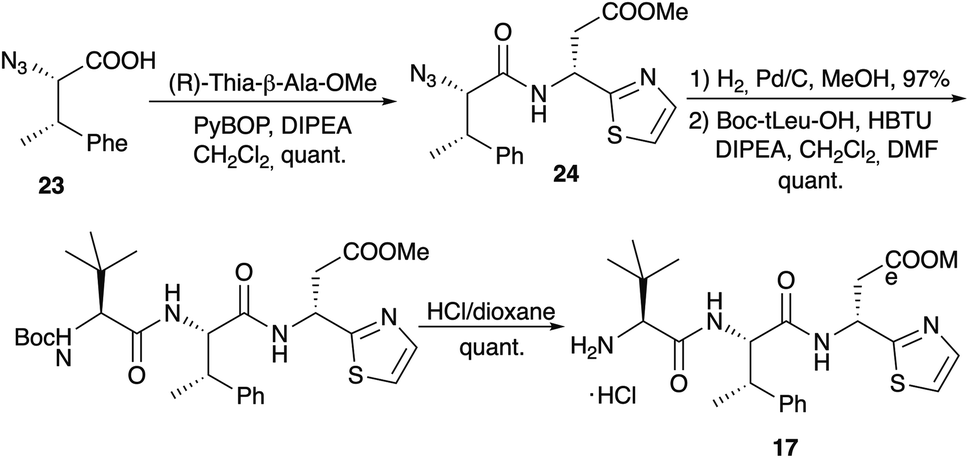 | ||
| Scheme 7 Synthesis of tripeptide side chain 17. | ||
4.3 Total synthesis of bottromycin and analogs
Based on their own synthetic studies and with all building blocks in hand, Sunazuka and Ōmura et al. developed the first and so far only complete total synthesis of bottromycin (Scheme 8).10,78 To extend the peptide chain, the Phth-group of amidine 18 was removed and the free amine was coupled with Boc-(S)-Val. Further elongation gave rise to hexapeptide 25, which was subjected to desilylation and oxidation. These last two steps had to be carried out on the stage of the hexapeptide, as attempts to oxidise tetrapeptide 18 resulted in the formation of a diketopiperazine. The oxidation was a very critical step due to the nucleophilicity of the internal amidine. Thus all oxidation methods proceeding via an aldehyde failed, because this aldehyde intermediate was trapped by the amidine forming an imidazole. Only Jones oxidation was successful, providing an acceptable yield of the desired carboxylic acid 26. The amidine was also problematic in the final macrocyclisation step, and the best results were obtained using EDCI/DIPEA in CH2Cl2, although the yield of 1a was only moderate.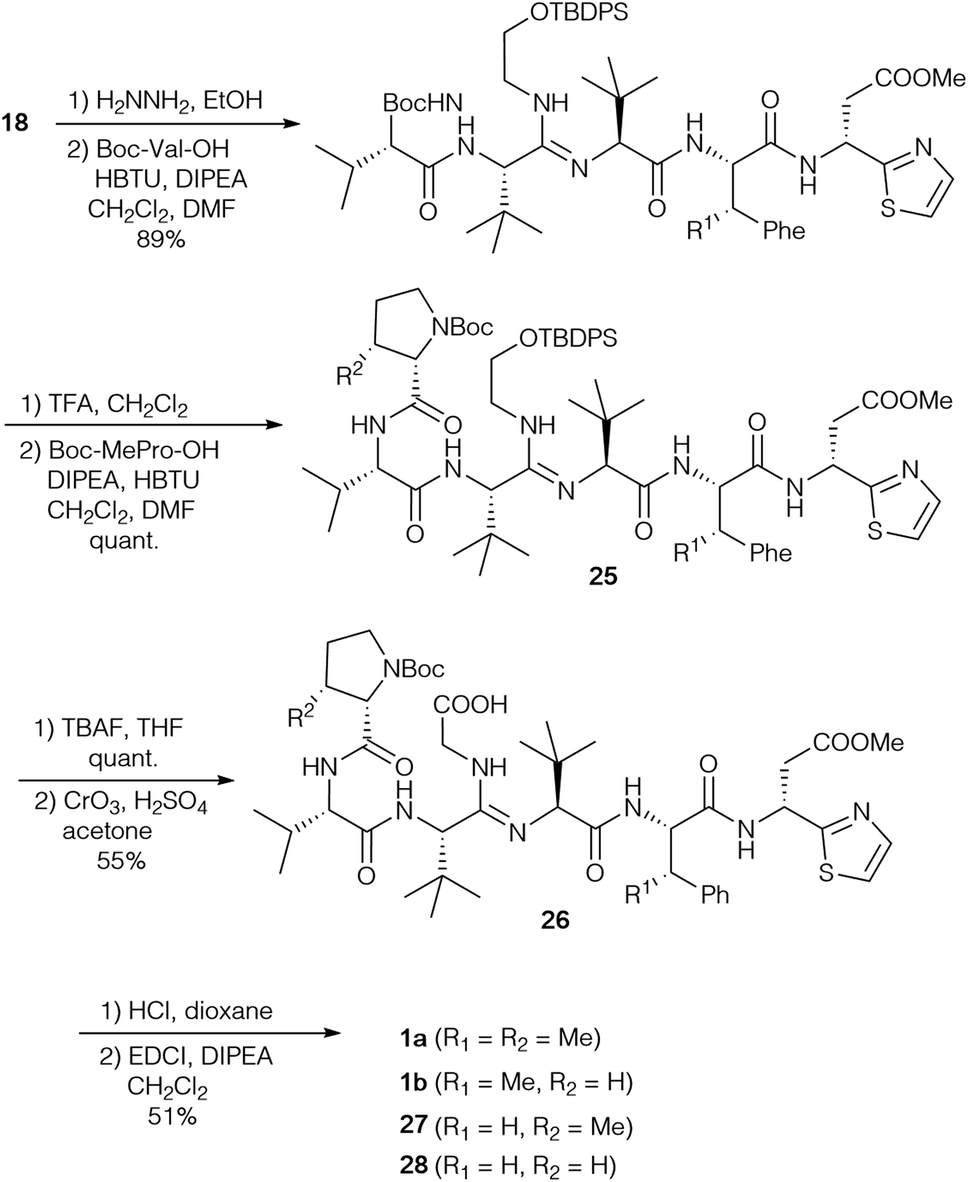 | ||
| Scheme 8 Total synthesis of bottromycin A2 (1a). | ||
This protocol was also used to generate derivatives missing some β-methyl groups, such as bottromycin B2 (1b) (Pro instead of MePro), or derivatives where β-MePhe was replaced by Phe [Phe-BotA2 (27), PheBotB2 (28)]. Their NMR spectra were rather complicated (existence of conformers), which suggests that the methyl group of the β-MePhe is important for the three-dimensional structure of the bottromycins.
Since it is known that the methyl ester of the thia-β-Ala has an effect on the biological activity of bottromycins in vitro and in vivo2 Sunazuka and Ōmura considered the synthesis of a bottromycin derivative missing the C-terminal amino acid, so that this position can be varied in the last step by coupling a wide range of amines to the “shortened” hexapeptide. Although this is a highly interesting approach, it was not as trivial as hoped. Azido-MePhe 23 was converted into the corresponding benzyl ester and, after reduction of the azide, coupled to Boc-(S)-tLeu (Scheme 9). The dipeptide 29 was incorporated into bottromycin derivative 30 according to Scheme 9. The benzyl ester could be cleaved easily to the carboxylic acid, the key intermediate for the synthesis of analogs. To validate the concept, the acid was coupled with (R)-thia-β-Ala-OMe to the original natural product 1a. The reaction proceeded smoothly, but 1a was only a side product. The main product was derivative 31 containing an imidazole on the tetrapeptide ring.
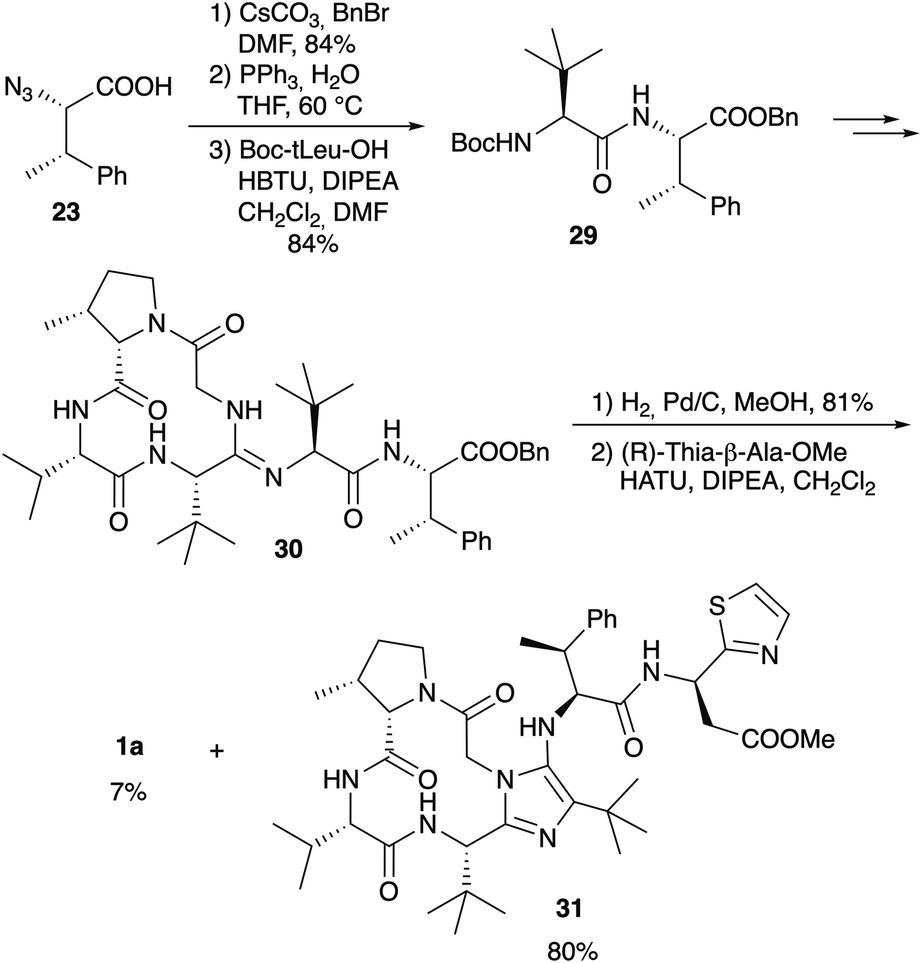 | ||
| Scheme 9 Variable synthesis of 1a. | ||
So far, HATU as the coupling reagent gave the best yields for bottromycin A2 analogs (32) and was used to generate a range of amides (Scheme 10), but the corresponding imidazole was the main product in all cases.
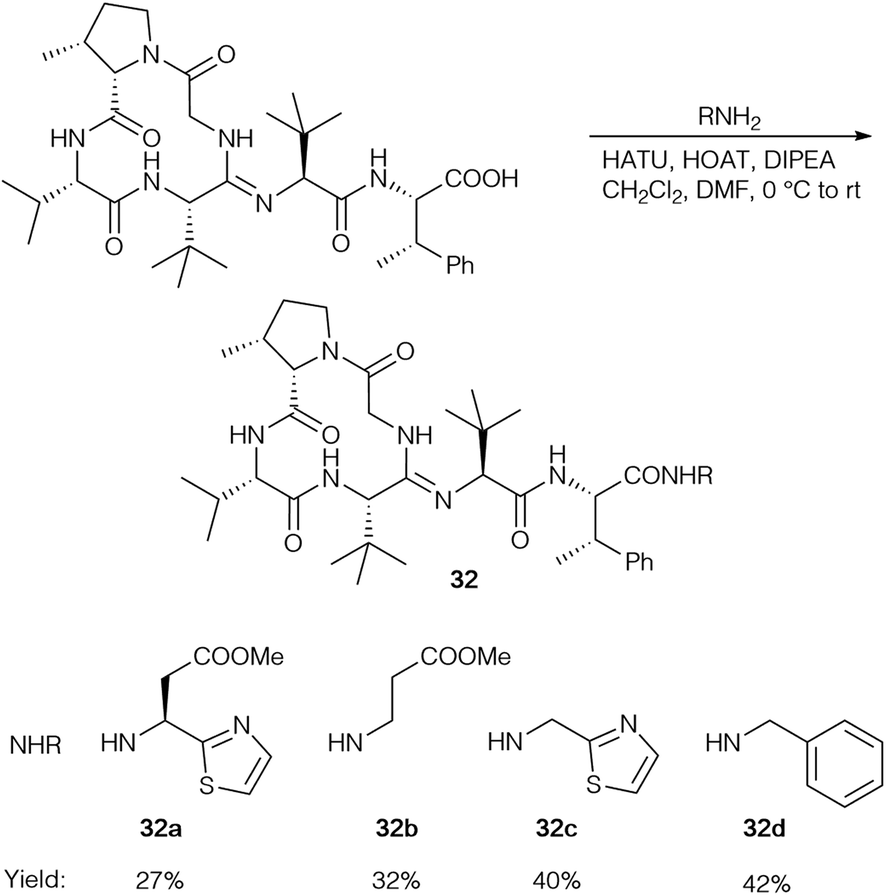 | ||
| Scheme 10 Synthesis of bottromycin A2 analogs 32. | ||
Further bottromycin derivatives were obtained by saponification of the natural product 1a at the C-terminus and coupling the free acid 33 with suitable nucleophiles (Scheme 11). Researchers at AiCuris used this approach for the synthesis of “Weinreb-amide”-type N-alkyl-N-alkoxyamides 34 by reaction with linear or cyclic N,O-dialkylhydroxylamines (Table 2).90
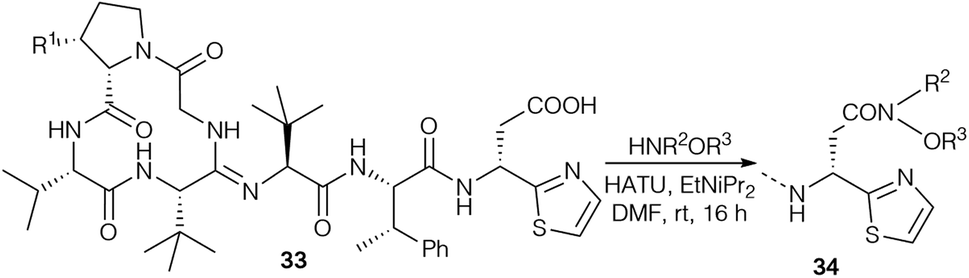 | ||
| Scheme 11 Synthesis of bottromycin A2 analogs 34. | ||
| 34 | R1 | R2 | R3 | Yield (%) |
|---|---|---|---|---|
| 34a | Me | Me | Me | 70 |
| 34b | Me | –(CH2)3– | 40 | |
| 34c | Me | –(CH2)4– | 35 | |
| 34d | H | –(CH2)4– | 14 | |
| 34e | Me | Et | Et | 50 |
| 34f | Me | Me | Et | 47 |
| 34g | Me | –CH2–CHOH–CH2– | 63 | |
| 34h | Me | –(CH2)3–CH(COOEt)– | 52 | |
| 34i | Me | –(CH2)3–CH(COOEt)– | 84 | |
| 34k | Me |
|
90 |
The groups of Ōmura and Sunazuka synthesised a range of different derivatives via the corresponding hydrazide 35 as a common intermediate (Scheme 12).2 The hydrazide was easily obtained by heating the solution of bottromycin A2 with hydrazine. Nitrosation gave rise to acyl azide 36 as an active intermediate, which could be coupled with a range of amines to the corresponding amides 37. Application of mono Boc-protected piperazine allowed further modification by replacing the Boc-protecting group (38). On the other hand, heating the acyl azide to 60 °C resulted in a Curtius rearrangement that gave rise to an isocyanate 39, which on treatment with amines provided ureas 40. Reacting 36 with thiols gave rise to thioesters such as 41 which could be subjected to palladium-catalysed cross coupling reactions with organozinc reagents generating ketones 42.
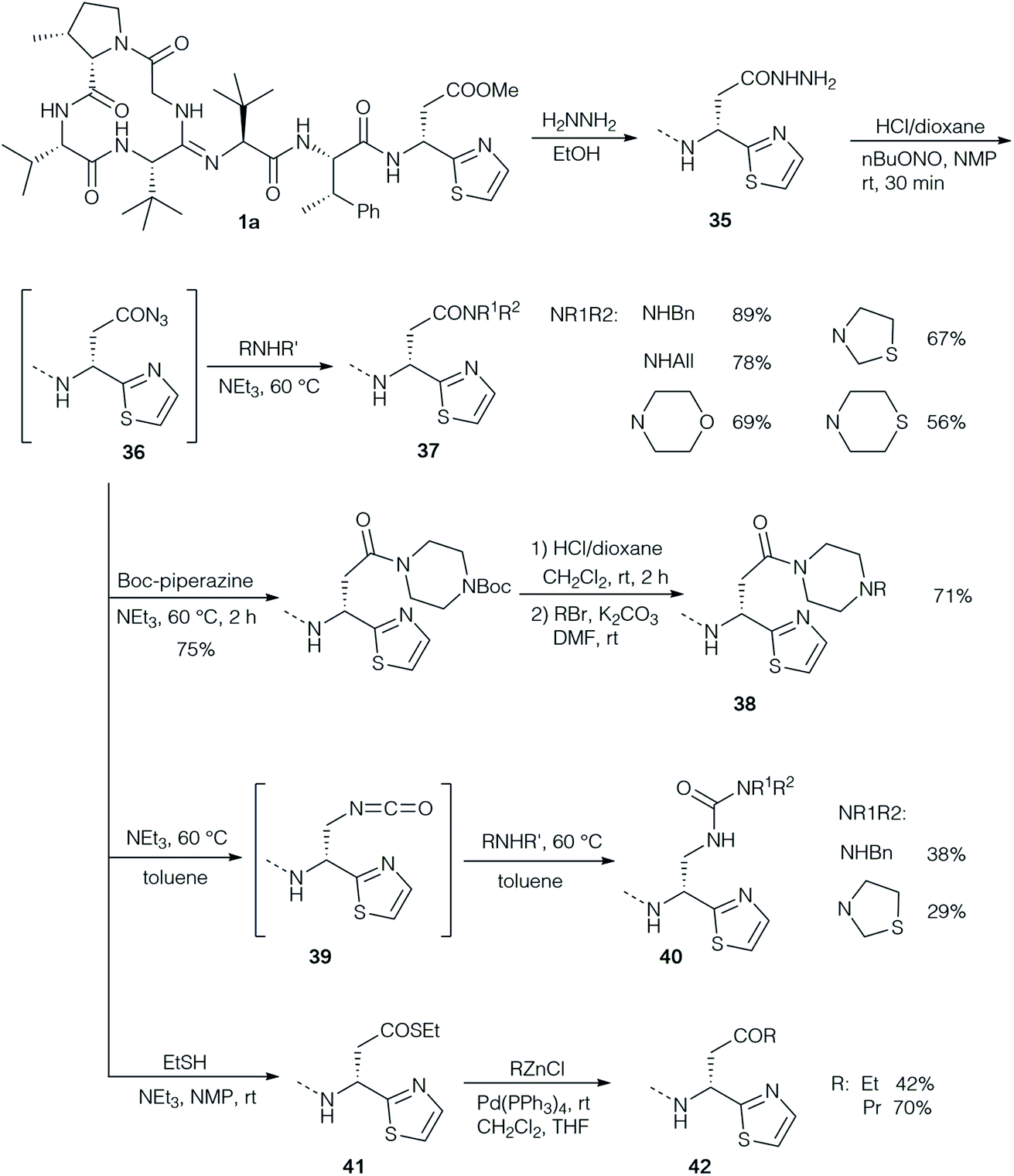 | ||
| Scheme 12 Synthesis of bottromycin A2 analogs via hydrazine 35. | ||
4.4 Biological activities and structure–activity relationship (SAR) studies of bottromycins and derivatives
Based on the importance of the methyl ester for bioactivity and its lability in vivo a range of amide derivatives 37 were prepared and their in vitro and in vivo activity towards S. aureus (Table 3) was compared.32 The tests were either carried out in vitro using a rapid tube dilution test, or in an in vivo mouse model, where compounds were administered intraperitoneally once. All compounds were active, but the primary and secondary amides were less active than the esters in the in vitro test, but more active in vivo. Aromatic amides (37k–m) and those with a basic substituent (37n) were less active to almost inactive in vivo, and tertiary amides (37o,p) were also significantly less active, while hydrazides (35, 37q) and hydroxamates (37r) were relatively active in vivo.| Compound | R1 | R2 | In vitro MIC (μg ml−1) | In vivo ED50 (μg per dose) | Ratio in vivo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :in vitro :in vitro |
|---|---|---|---|---|---|
| 1a | 0.01 | 50 | 5000 | ||
| 1b | 0.04 | 200 | 5000 | ||
| 37a | H | H | 0.10 | 25 | 250 |
| 37b | Me | H | 0.05 | 10 | 200 |
| 37c | Et | H | 0.05 | 15 | 300 |
| 37d | nPr | H | 0.5 | 10 | 20 |
| 37e | iPr | H | 1.0 | 10 | 10 |
| 37f | tBu | H | 0.25 | 10 | 40 |
| 37g | Bn | H | 0.5 | 18 | 36 |
| 37h | CH2CHOHCH2OH | H | 1.0 | 25 | 25 |
| 37i | CH2CH2OH | H | 0.5 | 18 | 36 |
| 37k | Ph | H | 60 | ||
| 37l | p-F-C6H4 | >100 | |||
| 37m | α-Naphthyl | H | >100 | ||
| 37n | NH(CH2)2NEt3 | H | >100 | ||
| 37o | iPr | iPr | 82 | ||
| 37p | –CH2CH2CH2CH2– | 95 | |||
| 37q | NMe2 | H | 35 | ||
| 37r | Me | OH | 28 | ||
| 35 | NH2 | H | 46 |
In a patent, researchers at AiCuris described the synthesis of N,O-dialkylated bottromycin hydroxamates 34a–k (Table 2) and their biological evaluation (Table 4).90
| 34 | MIC (μM) | |||
|---|---|---|---|---|
| S. aureus 133 | S. pneumoniae G9a | E. faecium BM4147 | E. faecalis ICB27159 | |
| 34a | 0.78 | <0.05 | 0.39 | 0.78 |
| 34c | 0.39 | <0.05 | 0.1 | 0.39 |
| 34d | 3.13 | <0.05 | 0.39 | 1.56 |
By far the most detailed SAR studies were reported by Ōmura and Sunazuka, who also investigated the desmethyl derivatives 27 and 28, which were obtained by total synthesis (Scheme 8). A wide range of different derivatives such as amides 32 (Scheme 12), 37 and 38, hydrazide 35, ureas 40, thioester 41 and ketones 42 were prepared from bottromycins obtained by fermentation (Scheme 12). Their activity was tested towards a panel of Gram-positive strains, using vancomycin (VCM) and linezolid (LZD) as references (Table 5).2,78 The results of the SAR studies are summarised schematically in Fig. 3.
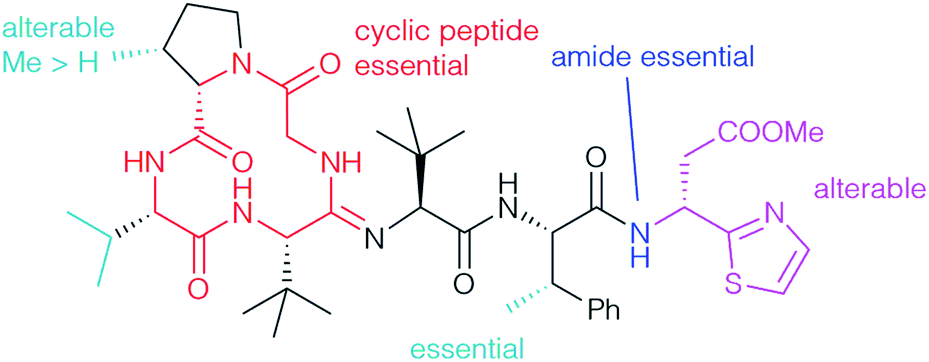 | ||
| Fig. 3 Summary of SAR for bottromycin derivatives. | ||
The unusual methylation pattern (cyan) has a significant effect on the bioactivity towards S. aureus. Bottromycin D (1d), where the valine is replaced by an alanine, and bottromycin B2 (1b), which does not have the methyl group at the proline, were less active than bottromycin A2 (1a) (Tables 2 and 5).22 Bottromycin C2 (1c), the analog dimethylated on proline, was roughly as active as bottromycin A2. The β-methyl group on the Phe seems to be essential and its removal (27, 28) causes a dramatic drop in activity (Table 5). It appears that this methyl group influences the conformation of the side chain and controls the three-dimensional structure of the whole molecule, an assumption which is supported by 1H NMR.10,78 Bicyclic derivatives such as 31 and linear peptides do not show significant activity, probably due to an undesired three dimensional conformation, which clearly indicates that the cyclic peptide ring (red) is essential.78 No activity was observed for derivatives with either a COOH-group at the C-terminus, such as 33, or if the thia-β-Ala is missing completely. This might be caused by a drop in the hydrophobicity. Interestingly, incorporating the opposite (S)-isomer of thia-β-Ala (32a) had no significant effect on activity (2 μg mL−1). The thia-β-Ala (purple) was not essential at all for activity – derivatives missing the acetate side chain (32c) or the thiazole unit (32b) were only slightly less active.
| Comp. | R1 | R2 | MIC (μg ml−1) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| S. aureus FDA209Pa | S. aureus Smitha | MRSA HH-1b | MRSA 92-1191b | VRE NCTC12201c | VRE NCTC12203c | Rates of residual anti-MRSA activity (%) | |||
| a S. aureus FDA209P and Smith: susceptible strains. b MRSA HH-1 and 92-1191: MRSA strains isolated from clinical patients. c Vancomycin resistant Enterococcus faecalis NCTC12201 and NCTC12203: encoded by vanA gene. d Vancomycin. e Linezolid. | |||||||||
| 1a | 1 | 1 | 1 | 2 | 1 | 0.5 | 0 | ||
| 1b | 4 | 4 | 4 | 4 | |||||
| 27 | 32 | >32 | >32 | 32 | |||||
| 28 | >32 | >32 | >32 | >32 | |||||
| 30 | >32 | >32 | >32 | 32 | |||||
| 31 | >32 | >32 | >32 | >32 | |||||
| 32a | 2 | 2 | 2 | 2 | |||||
| 32b | 8 | 8 | 8 | 8 | |||||
| 32c | 4 | 4 | 4 | 2 | |||||
| 32d | 2 | 4 | 2 | 2 | |||||
| 33 | Me | H | 64 | 64 | 64 | 128 | 128 | 32 | — |
| 35 | 16 | 16 | 16 | 32 | 8 | 4 | 86 | ||
| 37g | Bn | H | 8 | 8 | 8 | 8 | 8 | 2 | 71 |
| 37s | CH2CCH | H | 8 | 8 | 8 | 16 | 4 | 2 | 100 |
| 37t | –(CH2)2O(CH2)2– | 16 | 8 | 16 | 32 | 16 | 4 | 100 | |
| 37u | –(CH2)2SCH2– | 4 | 4 | 8 | 8 | 8 | 2 | 100 | |
| 37v | –(CH2)2S(CH2)2– | 8 | 4 | 8 | 8 | 4 | 4 | 100 | |
| 38a | Boc | H | 8 | 4 | 8 | 8 | 8 | 4 | 42 |
| 38b | H | 64 | 32 | 64 | 128 | 32 | 32 | — | |
| 38c | CH2CCH | 16 | 16 | 16 | 32 | 16 | 16 | 67 | |
| 38d | Bn | 4 | 4 | 4 | 4 | 4 | 4 | 84 | |
| 40a | –(CH2)2SCH2– | 4 | 4 | 4 | 4 | 4 | 2 | 100 | |
| 40b | Bn | 8 | 16 | 16 | 16 | 8 | 4 | 100 | |
| 41 | <0.25 | 0.5 | <0.25 | 0.5 | <0.25 | <0.25 | 0 | ||
| 42a | Et | 1 | 1 | 2 | 2 | 2 | 1 | 100 | |
| 42b | nPr | 1 | 1 | 1 | 2 | 2 | 0.5 | 100 | |
| VCM | 1 | 1 | 0.5 | 1 | >128 | >128 | — | ||
| LZD | 2 | 2 | 2 | 2 | 2 | 2 | — | ||
Surprisingly benzyl amide 32d is almost as active as 1a, while the corresponding benzyl ester 30 is not very effective. The data showed that the amide functionality (blue) is necessary for good activities. Benzyl amide 32d is more active than the dethiazolyl analog 32b, which indicates that an (hetero)aromatic substituent at the C-terminus has a positive effect on activity. The moderate in vivo activity of the methyl ester in the natural products probably results from its low hydrolytic stability under physiological conditions and its cleavage towards the almost inactive carboxylic acid 33. Although significantly less active in vitro, better in vivo stabilities were observed for secondary aliphatic amides (Table 3, 37a-i), while aromatic (37j–m) and tertiary amides (37o,p) as well as those with basic side chains (37n) were almost inactive.32 Piperazino derivatives 38 and ureas 40 exhibited 4- to 32-fold weaker activity in vitro, but better stability.2 Thioesters such as 41 were significantly more active than 1a, but due to their great reactivity completely unstable in mouse plasma. Ketones 42, which cannot undergo hydrolysis, are perfectly stable and showed activities comparable to 1a and vancomycin, but importantly were also active against vancomycin-resistant strains. Subsequent biological evaluation using MRSA-infected mice showed that propyl ketone 42b might be a good candidate for drug development. 100 mg kg−1 given to mice orally resulted in survival for at least five days after administration, while all non-treated animals died in the same time frame. Hydroxamates 34 (Table 4) might also be suitable for this purpose.90
5 Biosynthetic gene cluster and biosynthesis
The biosynthetic origin of the bottromycins was unknown for decades following their discovery. They could feasibly be synthesised via either ribosomal or non-ribosomal pathways, as all residues could theoretically be produced from proteinogenic amino acids. Early studies of bottromycin biosynthesis involved elegant isotope labelling experiments from Arigoni and colleagues, where it was shown that the methyl groups at the β-positions of proline, two valines and phenylalanine, along with the thia-β-Ala-OMe, were derived from methionine.91 They also showed that β-methylated valine and phenylalanine derive from L-valine and L-phenylalanine. Feeding S. bottropensis with methionine featuring an isotopically labelled chiral methyl group ([methyl-(2H,3H)]-(2S,methyl-R)-methionine) showed that the β-methylation occurs with a retention of configuration. This was consistent with double inversion of configuration via a radical SAM mechanism.92 Additional experiments using isotopically labelled amino acids were also consistent with a radical SAM mechanism.91In 2012, four independent research teams identified bottromycin biosynthetic gene clusters (BGCs) in four different Streptomyces species: the known producers S. bottropenesis11 and Streptomyces sp. BC16019,8 the plant pathogen Streptomyces scabies6 and the marine ascidian-derived Streptomyces sp. WMMB27212 (Fig. 4A). These reports corroborated the earlier feeding studies by showing that bottromycins are ribosomally synthesised and post-translationally modified peptides (RiPPs)15 and that the BGCs encode three radical SAM methyltransferases. In each study, the BGC was identified by BLAST searches for genes that could encode a putative bottromycin core peptide, GPVVVFDC (or GPAVVFDC for bottromycin D in S. sp. WMMB272) (Fig. 4B). RiPPs originate from a larger ribosomally synthesised precursor peptide that usually consists of a leader peptide and a core peptide that is post-translationally modified by tailoring enzymes. However, the discovery of the bottromycin BGC provided the first (and still only) example of a bacterial RiPP that derives from an N-terminal core peptide that has no leader peptide and is attached to a “follower” peptide (Fig. 4C).
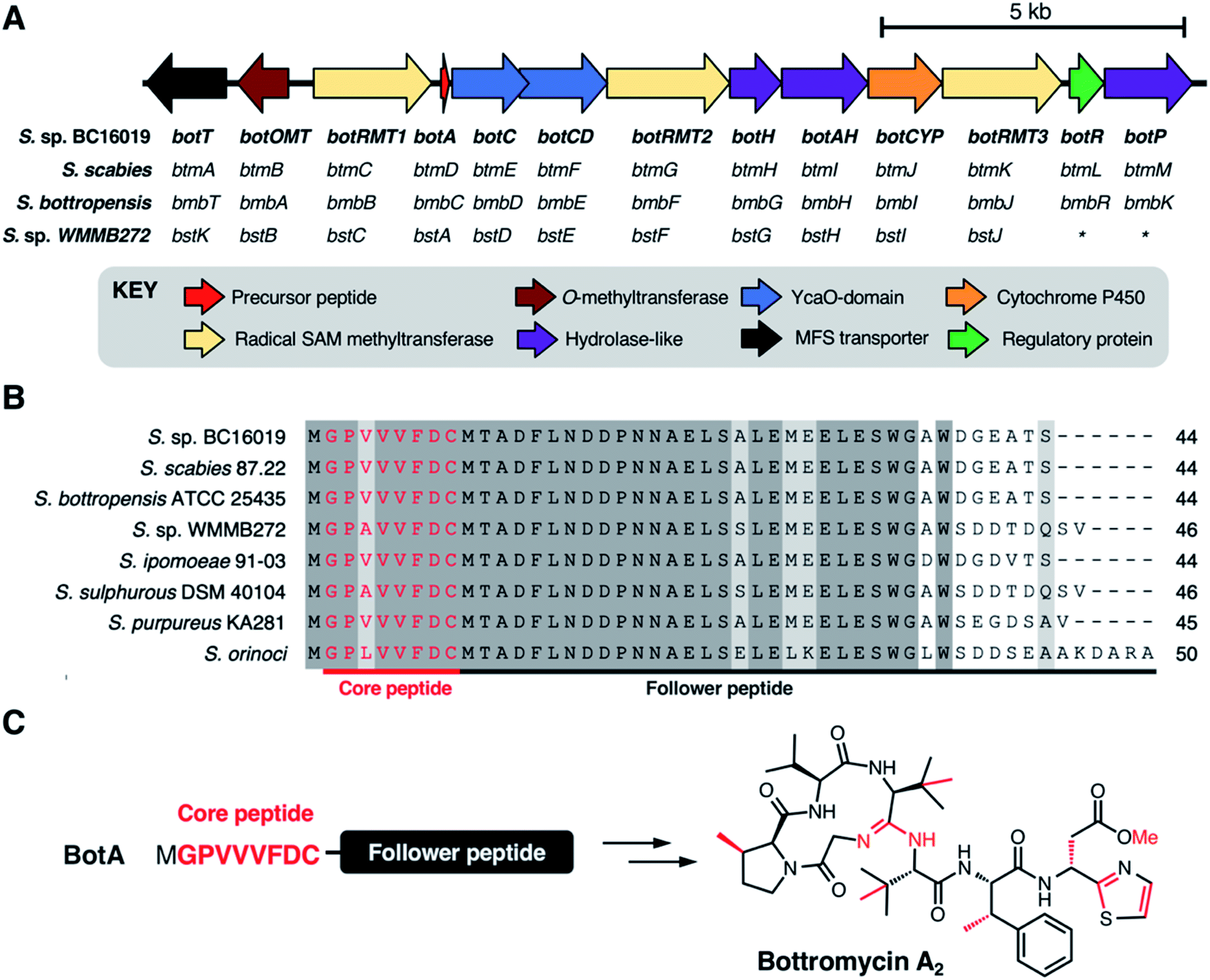 | ||
| Fig. 4 Identification of the bottromycin BGC. (A) Organisation of the BGC. Gene nomenclature from each strain is shown (* = genes not identified in the S. sp. WMMB272 study as this BGC was at the end of a contig). (B) Sequence alignment of precursor peptides from every bottromycin BGC identified bioinformatically (all are Streptomyces species). Identical residues are shown with a dark grey background, similar residues are shown with a light grey background (Risler matrix score >0.7) and the core peptide is highlighted in red text. (C) Schematic showing the conversion of the BotA precursor peptide into bottromycin A2, where all post-translational modifications are coloured red in the final product. | ||
The genetic organisation of these BGCs is effectively identical, and while there are significant differences in protein sequence identity between each BGC, S. sp. BC16019 nomenclature will be used here onwards for clarity. The bottromycin BGC encodes 13 proteins (Fig. 4A): one precursor peptide (BotA), two YcaO-domain proteins (BotC and BotCD), three radical SAM methyltransferases (BotRMT1-3), three putative hydrolases (BotH, BotAH, BotP), one cytochrome P450 (BotCYP), one O-methyltransferase (BotOMT), one putative regulatory protein (BotR) and one major facilitator superfamily transporter (BotT). These initial studies revealed a number of key details relating to bottromycin biosynthesis. Gene inactivation experiments in S. bottropenesis,11S. scabies6 and S. sp. BC160198 confirmed the identity of the BGC, which was further validated by heterologous expression of the S. sp. BC16019 BGC. The identity of the BGC in S. sp. WMMB272 was demonstrated by the production of bottromycin A2 upon expression of a mutant precursor peptide gene that encoded the bottromycin A2 core peptide instead of the natural bottromycin D core peptide.12
Notably, gene deletions in the S. scabies BGC6 and insertional inactivation of genes in the S. sp. BC16019 BGC8 demonstrated the roles of the radical SAM methyltransferases BotRMT1-3 via the production of differentially methylated bottromycin derivatives by each mutant, thereby validating the earlier isotopic labelling studies.91 BotRMT1 catalyses radical C-methylation of Phe6, BotRMT2 catalyses radical C-methylation of both Val4 and Val5, and BotRMT3 catalyses radical C-methylation of Pro2. At the time, this represented one of the first examples of radical β-methylation of amino acid residues, along with the polytheonamides, 49-amino acid RiPPs produced by ‘Candidatus Entotheonella factor’, a member of a marine sponge microbiome.93 Multiple non-ribosomal peptides contain β-methylated amino acids, but these are generated via conventional methylation of a precursor keto acid.94 Inactivation of botOMT in the S. sp. BC16019 BGC confirmed its role in O-methylation of Asp78. However, little else was known about the biosynthetic steps required to convert BotA into mature bottromycin, although plausible routes were initially proposed based on the predicted catalytic roles of bot proteins. In-frame gene deletions in the S. scabies BGC had demonstrated the essentiality of numerous putative biosynthetic genes,6 including botC and botCD (encoding the two YcaO-domain proteins), and botCYP (encoding a P450), but no bottromycin related metabolites could be initially identified from these mutants. The challenge with identifying molecules related to RiPPs following gene deletions is that a pathway may “stall” if a key step is disrupted, with the core peptide still attached to the leader/follower peptide. This therefore is likely to undergo further degradation into a very short modified peptide that may be distantly related to the final product and therefore difficult to detect.
To improve the detection of bottromycin-related metabolites from pathway mutants, Truman and co-workers used mass spectrometry-based molecular networking95 and untargeted metabolomics to study in-frame deletions of orthologues of botA, botC, botCD, botAH, botRMT1, botRMT2 and botCYP in S. scabies.96 This analysis identified a series of bottromycin-related molecules (intermediates or shunt metabolites) associated with each mutant strain, which were then used to propose a feasible pathway based on where the pathway stalled (Fig. 5). This indicated that radical methylation by BotRMT1 and BotRMT2 were early steps in the pathway in S. scabies, as was heterocyclisation of Cys8 by the standalone YcaO-domain protein BotC. Additionally, this study showed that the M17-family leucine aminopeptidase BotP removes methionine from the N-terminus of BotA.
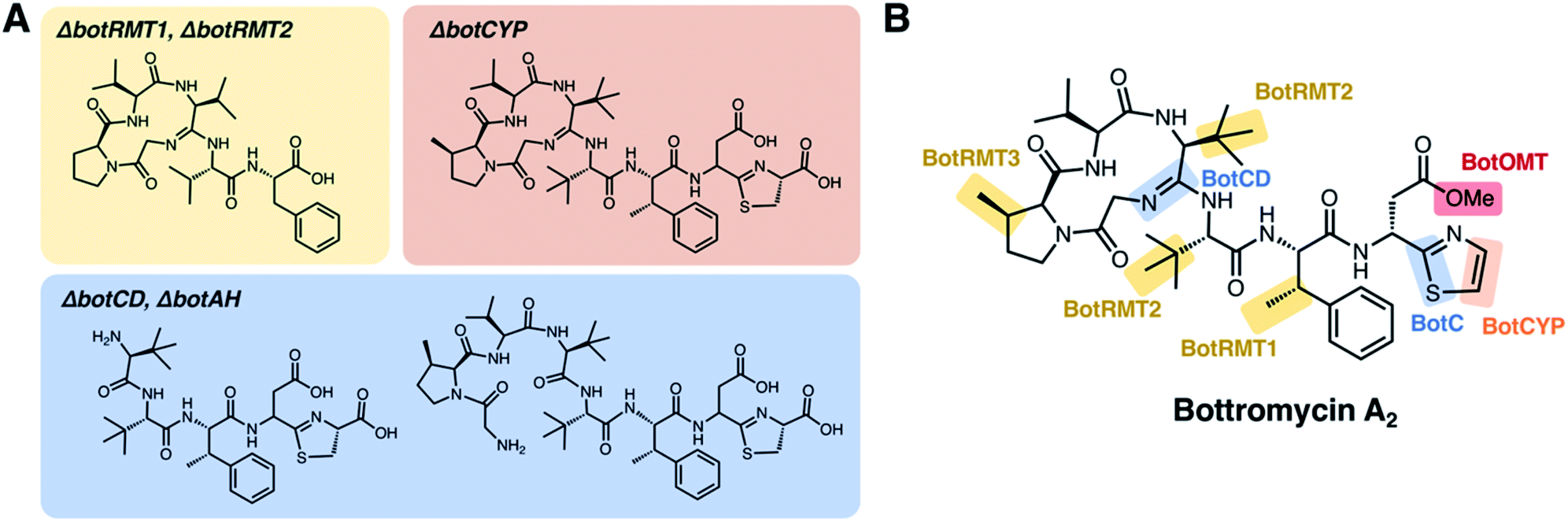 | ||
| Fig. 5 Key metabolites identified by mutational analysis of bot biosynthetic genes. (A) Structures of metabolites based on tandem MS data from Crone et al. (B) Proposed roles of biosynthetic proteins based on the studies of Crone et al.6 and Huo et al.8 | ||
Following removal of the N-terminal methionine by BotP, the unique macrocyclic amidine of bottromycin was proposed to be formed by BotCD (YcaO-domain protein) and BotAH (hydrolase), based on the production of linear bottromycin-related peptides by each mutant (Fig. 5). Deletion of botCYP led to the accumulation of O-desmethyl bottromycins A2 and B2 carboxylated at their C-termini. This was consistent with BotCYP catalysing late-stage oxidative decarboxylation of the thiazoline moiety to generate a terminal thiazole. Each compound mass appeared as twin peaks via liquid chromatography -mass spectrometry (LC-MS), suggesting a mixture of aspartate epimers, which was supported by deuterium labelling and therefore provided a potential route to D-aspartate in mature bottromycin. Epimerisation was shown to happen spontaneously, but it was not clear whether other proteins were involved in accelerating this key step. No mutant strains produced metabolites that are O-methylated on Asp7. This suggested that O-methylation is the final biosynthetic step, and it was shown that purified BotOMT could methylate O-desmethyl bottromycin A2. This was in agreement with the earlier gene inactivation work by Huo et al.8
6 Biosynthetic enzymes
The following paragraphs will be dedicated to the enzymes involved in the biosynthesis of the bottromycin core scaffold (desmethyl bottromycin): BotP, BotC, BotCD, BotAH, BotH, and BotCYP. The activity of these enzymes has been reconstituted in vitro, and structural information is available for several of them. Although the enzymes were studied using enzyme homologues from different BGC containing Streptomyces species (see Fig. 4B), the S. sp. BC16019 nomenclature will be used for clarity.6.1 Aminopeptidase BotP
The aminopeptidase BotP belongs to the family of hexameric M17 leucine aminopeptidases (LAP). LAPs are metallo-exopeptidases found in all kingdoms of life and cleave N-terminal residues from proteins and peptides. They do not only hydrolyse N-terminal leucine residues, but often are promiscuous. The function of LAPs is diverse and goes beyond the function of recycling amino acids and includes the processing of bioactive peptides and peptides for major histocompatibility complex (MHC) class I antigen presentation, gene regulation and vesicular trafficking.97,98Usually, N-terminal methionine is hydrolysed by endogenous aminopeptidases, but these do not function efficiently with the MGP sequence found at the N-terminus of BotA.99 The aminopeptidase BotP was predicted6,8 and confirmed96,100 to remove the N-terminal methionine from the precursor peptide BotA (43), which generates the free glycine amino group (44) (Fig. 7) that is necessary for the cyclisation onto an internal amide carbonyl to generate the unique amidine macrocycle found in bottromycins.
Koehnke and coworkers determined the crystal structure of BotP (Fig. 6B) and assessed the substrate promiscuity of BotP using pentapeptide mimics of BotA (Fig. 6A).100 BotP showed a hexameric structure typical for M17 LAPs and the activity of recombinant BotP, isolated from E. coli, could be reconstituted in presence of Co2+ (or Mn2+) ions. RiPP enzymes catalysing the initial biosynthetic steps often bind to the follower (or for other RiPPs leader) peptide to aid substrate recognition and enzyme activity.101 For BotP modelling suggests that only the first 3–4 amino acids contribute to substrate binding, but an in vitro assay using pentapeptides showed that these truncated substrates were processed slower than full-length BotA. The reasons for this discrepancy remain to be determined. BotP tolerates several amino acid changes in P1-P3′, but processing is reduced drastically.
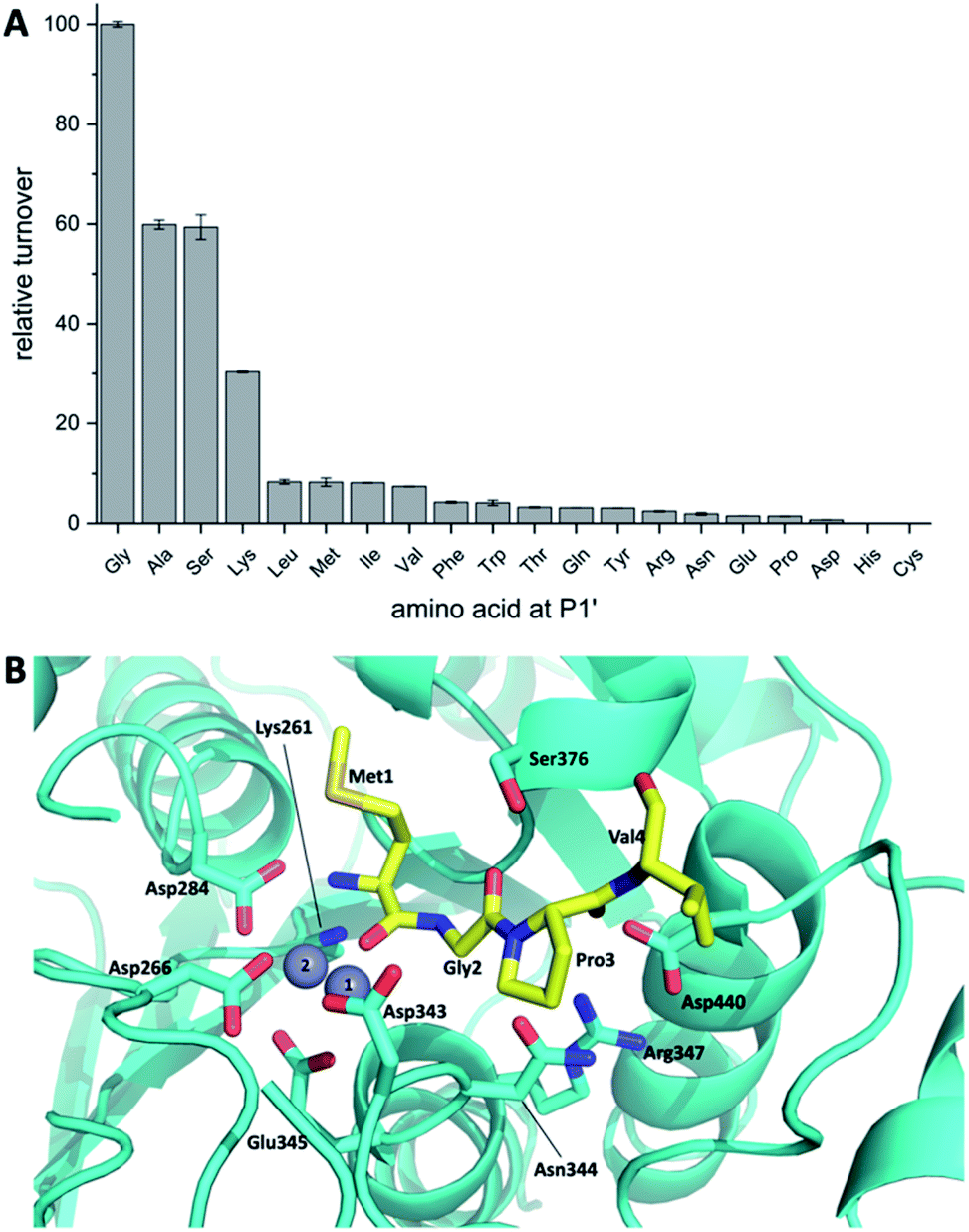 | ||
| Fig. 6 (A) BotP substrate promiscuity using pentapeptide mimics of BotA with amino acid changes in P1′position. (B) Model of BotP-Mn2+ (cyan) with the peptide MGPV (yellow). | ||
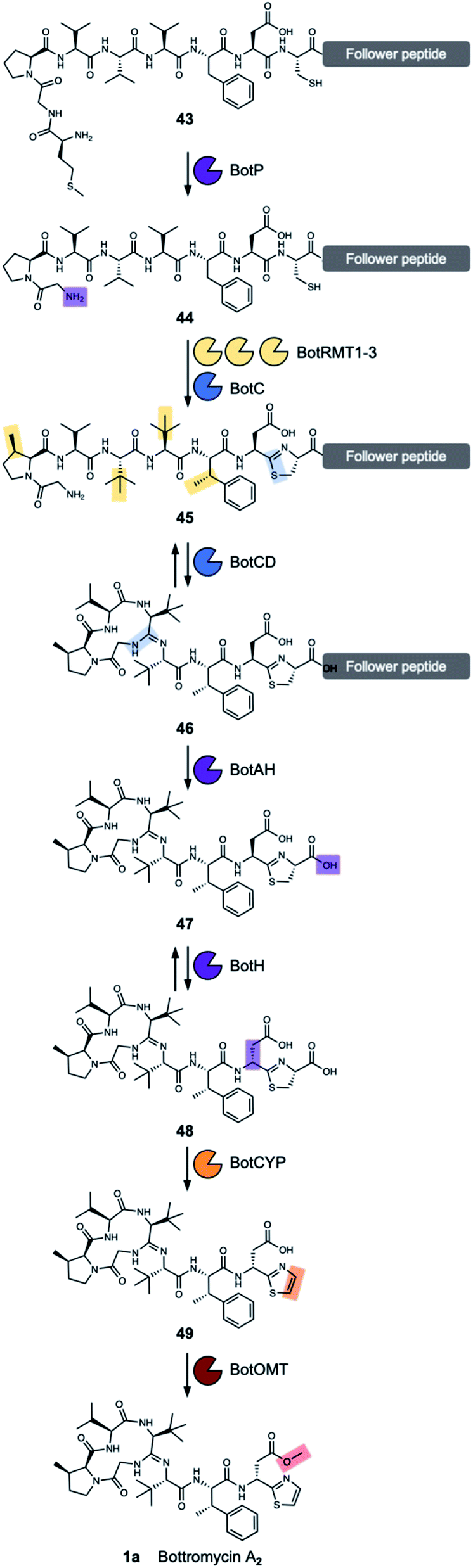 | ||
| Fig. 7 Biosynthetic pathway for bottromycin A2. | ||
6.2 YcaO domain enzymes BotC and BotCD
The bottromycin biosynthetic gene cluster contains two genes encoding for YcaO-domain enzymes, BotC and BotCD.6,8,11,12 YcaO proteins can be found in bacteria and archaea. Their function is best characterised in RiPPs such as linear azol(in)e-containing peptides (LAPs), thiopeptides and cyanobactins, where the YcaO enzymes catalyse the ATP dependent cyclodehydration reaction of Cys, Ser or Thr side chains onto the preceding backbone amide carbonyl to form azoline heterocycles.102–104 They can however also be used to generate non-natural heterocycles.105,106From an untargeted metabolomic approach it was predicted that BotC catalyses the heterocyclisation of Cys to thiazoline and BotCD, together with BotAH, catalyses the formation of the unique macroamidine linkage.96 The function of the two bottromycin YcaO enzymes were independently studied in in vitro approaches by the Mitchell and Koehnke groups.107,108 It was demonstrated that BotC catalyses the heterocyclisation reaction that converts the core peptide's Cys residue to a thiazoline. The second YcaO enzyme, BotCD, was sufficient to catalyse macroamidine formation. Both proteins bind to the follower peptide, but with low affinity.107 BotC and BotCD were quite tolerant to changes in the core peptide sequence but recalcitrant to changes of the nucleophile.107,108 BotC was unable to utilise Ser or Thr instead of Cys to generate oxazolines.107 While the turnover of all bottromycin biosynthetic enzymes has been shown to be relatively fast, heterocyclisation by BotC was shown to be slow and could be the rate limiting step of the pathway, since BotCD strongly prefers a heterocyclised substrate for macrocyclisation.108 In contrast to all other YcaO enzymes studied to date, the BotCD reaction was shown to be reversible: the enzyme catalysed amidine formation and ring opening, both in an ATP and Mg2+-dependent fashion.108 Thus BotCD expanded the catalytic scope of YcaO enzymes in RiPP pathways to amidine formation, but also raised questions as to possible partner proteins for BotCD, which may prevent ring opening.
Based on the biochemical data it was proposed that macroamidine formation proceeds analogously to heterocyclization: after nucleophilic attack of the N-terminal amino group onto the amide carbon a hemiorthoamide intermediate is formed, which is then ATP-dependent O-phosphorylated, followed by subsequent phosphate elimination to form the macroamidine (Fig. 8).
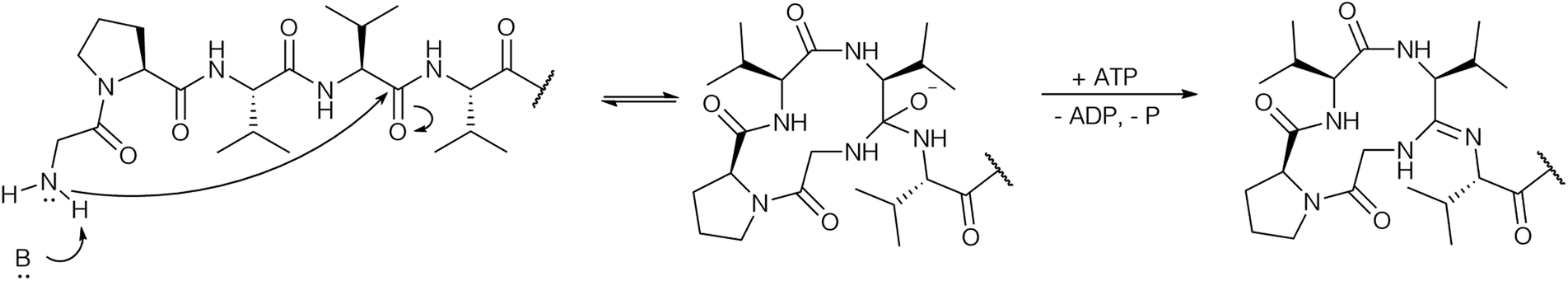 | ||
| Fig. 8 Proposed mechanism for macroamidine formation by the YcaO domain enzyme BotCD. | ||
6.3 Amidohydrolase BotAH
Analysis of the metabolomic network from S. scabies ΔbtmI (botAH orthologue) and ΔbtmF (botCD orthologue) revealed two new bottromycin related molecules, which could not be found in S. scabies WT and that were not macrocyclised. From this observation it was proposed that BotCD and BotAH are involved in macrocyclisation.96 However, in vitro data showed that BotCD is sufficient to catalyse macrocyclisation. Pfam-analysis classifies the protein encoded by the gene botAH as a putative metallo-dependent amidohydrolase. From its putative function it was assumed that either BotAH, or the putative α/β hydrolase BotH, catalyses the liberation of the modified core from the follower peptide for the final biosynthetic steps. The crystal structure of the BotAH homologue PurAH confirmed the Pfam prediction. When the role of BotAH was probed in vitro it was discovered that it cleaved the follower peptide off the hetero- and macrocyclised core peptide, and a mechanism was proposed (Fig. 9).109 As observed for BotP, the highest activity of BotAH was observed after addition of Co2+. This may explain increasing bottromycin production levels in S. scabies by supplementation of production medium with Co2+.7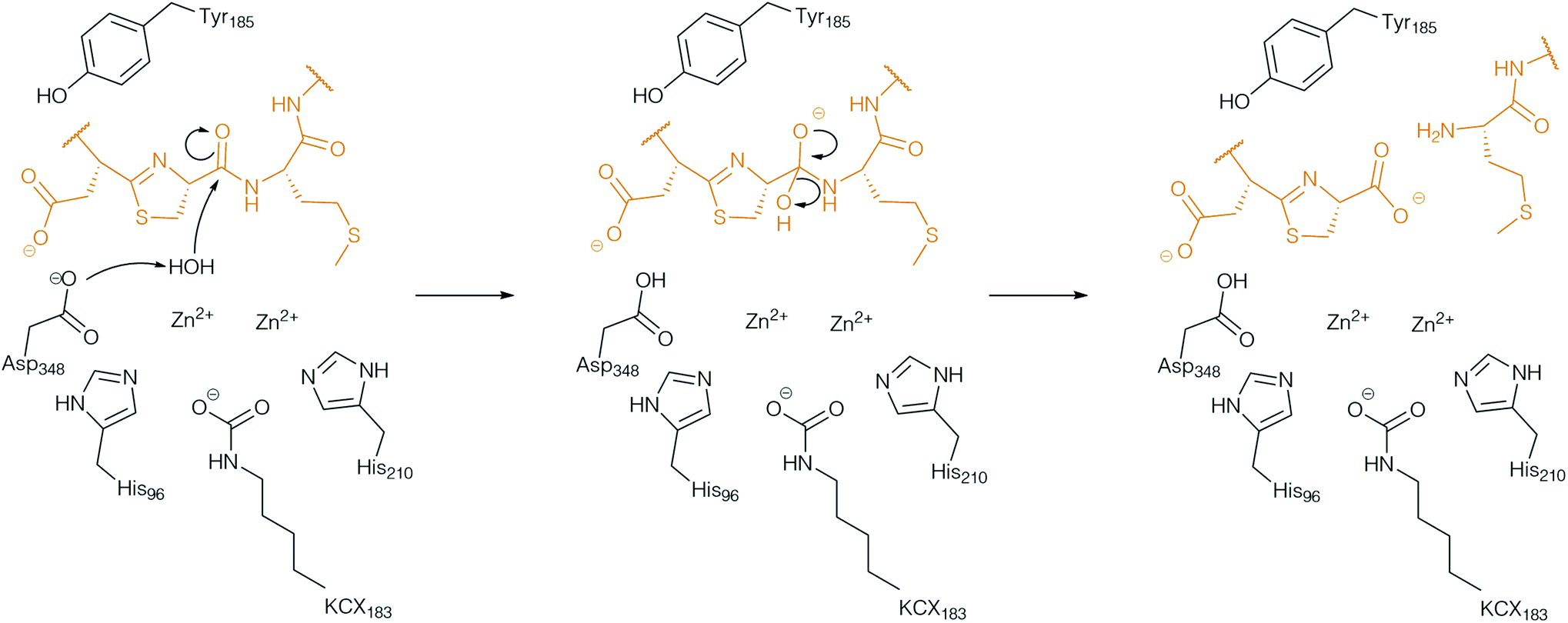 | ||
| Fig. 9 Proposed mechanism of the follower peptide cleavage by the amidohydrolase AH. BotAH active side residues are shown in back, the peptide substrate is shown in orange. | ||
The biosynthetic role of BotAH raised the question of whether it could aid macrocyclisation by influencing the equilibrium of the BotCD reaction. After all, the activity of BotCD was dependent on the follower peptide (attached to the modified core peptide). Indeed, when added to macrocyclisation reactions, BotAH's activity precluded ring opening and pulled the macroamidine formation equilibrium to the side of macrocyclised product.109 As a result, BotAH can be viewed as a YcaO accessory protein, the first hydrolase for which such a function has been reported. These observations also rationalise why the amidohydrolase is essential for macroamidine formation in vivo.
6.4 Atypical α/β hydrolase BotH
The function of the α/β hydrolase (ABH) BotH was not studied in vivo by gene deletion experiments and remained unclear until very recently. Enzymes of the α/β hydrolase family catalyse the hydrolysis of (thio)ester and peptide bonds, but some members have diverse functions, such as dehalogenases, dioxygenases and decarboxylases.110,111BotH is annotated as an ABH, and its crystal structure revealed a typical ABH fold, but the canonical Ser–His–Asp catalytic triad residues, which the majority of the ABH family members possess, is mutated (Ser to Phe, His to Ile) or missing (Asp).112 Accordingly, no hydrolytic activity was detected for BotH. Instead, the follower-cleaved intermediate 47 binds to the large cavity of the designated BotH active site and in vitro biochemical assays determined BotH to function, unexpectedly, as the epimerase of the Asp residue found in bottromycins (1).112 Deuteron labelling experiments revealed, that the enzyme catalyses the rapid epimerisation of L-Asp to D-Asp, and its back-reaction (Fig. 7). The action of BotH leads to a mixture of 47 and 48, but provides a much greater abundance of the D-Asp containing intermediate 48 than the spontaneous epimerization, which proceeds a glacial speeds and favours 47. In the complex crystal structure of BotH with 48, no potential catalytic residues of BotH were in a reasonable distance of the substrate Asp Cα proton. In addition, mutation of the core peptide Asp residue to Ala or Asn prevented epimerisation, but not binding. Substitution of Asp with Glu still allowed epimerization, which led the suggestion of substrate-assisted catalysis. BotH is the first reported ABH to catalyse peptide epimerisation.
In other RiPP pathways, epimerisation usually involves radical SAM enzymes or a two-step dehydration–hydrogenation process to generate D-alanine from L-serine.113–116 BotH is thus the founding member of a group of atypical ABH enzymes that may be able to epimerise amino acids post-translationally and but also other secondary metabolites. Further, BotH binds the pathway product bottromycin A2, but is not able to epimerise it. The complex crystal structure of BotH and bottromycin A2 was the first crystal structure of any bottromycin (Fig. 10). The resulting orthosteric inhibition of BotH by bottromycin A2 may results in a biosynthetic feedback mechanism to prevent self-poisoning.112
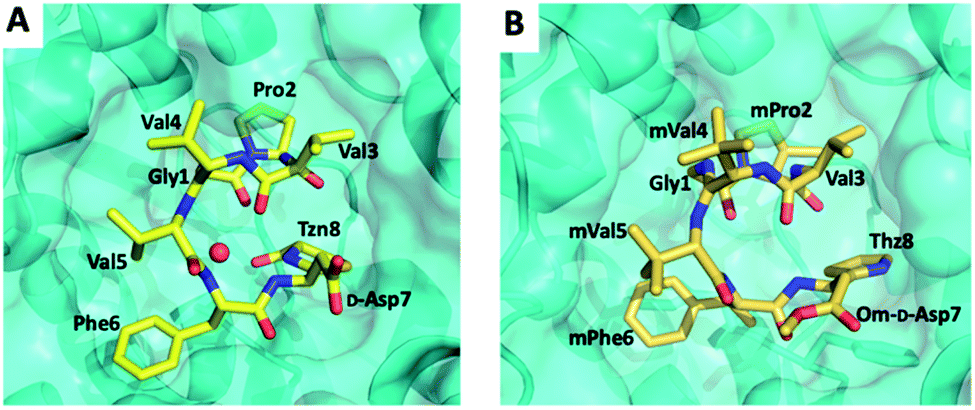 | ||
| Fig. 10 BotH-48 (A) and BotH-bottromycin A2 (B) complex structures. | ||
6.5 Cytochrome P450 enzyme BotCYP
Thiazoles (and oxazoles) as heterocycles derived from cysteine (or serine/threonine) are a common motif in RiPPs and are associated with linear azol(in)e-containing peptides (LAPs), cyanobactins, thiopeptides, and bottromycins. Thiazoline oxidation is more frequently observed than oxazoline oxidation and usually involves an FMN-dependent dehydrogenase.13,15 In bottromycin biosynthesis the oxidative carboxylation of the C-terminal thiazoline is catalysed by the P450 enzyme found in the gene cluster.96 P450 enzymes are not very common in RiPP pathways, although a recent large-scale survey of the RiPP landscape identified ∼1800 P450 genes associated with putative RiPP BGCs.117 The handful of P450 enzymes that had been characterized from RiPP biosynthetic pathways predominantly catalysed amino acids hydroxylation,118–121 but other functions had also been reported.120,122,123 The P450-catalyzed oxidative decarboxylation of a heterocycle in RiPP biosynthesis had not been investigated in vitro but recent work by the Koehnke group demonstrated that BotCYP selectively acts on D-Asp containing intermediate 48 (Fig. 11).117 Given the substrate-assisted mechanism of BotH, this observation rationalizes why all attempts to produce bottromycin analogs with residues other than Asp/Glu were unsuccessful (Fig. 13). Without a D-amino acid in core peptide position 7 the pathway stalls at the point of oxidative decarboxylation. Unfortunately the crystal structure of a close BotCYP homologue did not provide an answer as to how this enzyme may provide stereochemical resolution for the pathway. The reconstitution of this step has nevertheless enabled the production of the bottromycin core scaffold, which may aid the production of bottromycin analogs for bioactivity testing in the future.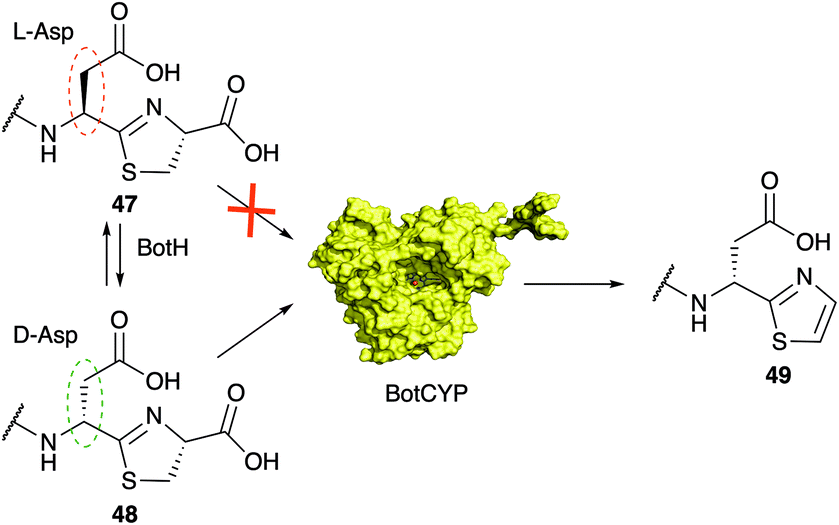 | ||
| Fig. 11 BotCYP catalyses the oxidative decarboxylation reaction of the thiazoline in 48 to the thiazole found in the bottromycin core structure. The cytochrome P450 enzyme is stereoselective for the D-Asp containing intermediate and thus provides stereochemical resolution for the pathway. | ||
7 Heterologous production of bottromycins and derivatives
Early studies on bottromycin benefitted from large-scale fermentation to aid with product isolation and characterisation, such as a 3000 litre fermentation used by Waisvisz and colleagues in 1957 to first characterise bottromycin.1 However, more recently, numerous groups have reported very low bottromycin yields with laboratory-scale fermentations,8,9,96 which has presented challenges for understanding bottromycin biosynthesis and engineering the pathway to generate derivatives.7.1 Regulation of the bottromycin biosynthetic gene cluster
To identify the transcriptional organisation of the bottromycin BGC, Vior et al.7 used 5′-tag-RNA-seq in S. scabies. This revealed transcriptional start sites preceding botRMT1 and botOMT (Fig. 12A), as well as an internal transcriptional start site within botRMT1, which precedes the precursor peptide gene, botA. This is hypothesised to increase expression of the precursor peptide in relation to the tailoring enzymes. RT-PCR (reverse transcription - polymerase chain reaction) and 5′-RACE (rapid amplification of cDNA ends) experiments were consistent with the 5′-tag-RNA-seq analysis, although inferred a possible additional transcriptional start site before botT that was not detected by 5′-tag-RNA-seq. This study showed that the bottromycin BGC does not encode a pathway-specific master regulator, and instead encodes a regulatory protein, BotR, that specifically modulates the expression of botA but not other genes in the BGC. The precise mechanism of BotR-mediated modulation remains enigmatic, especially as no strong promoter activity could be detected from the internal transcriptional start site that precedes botA.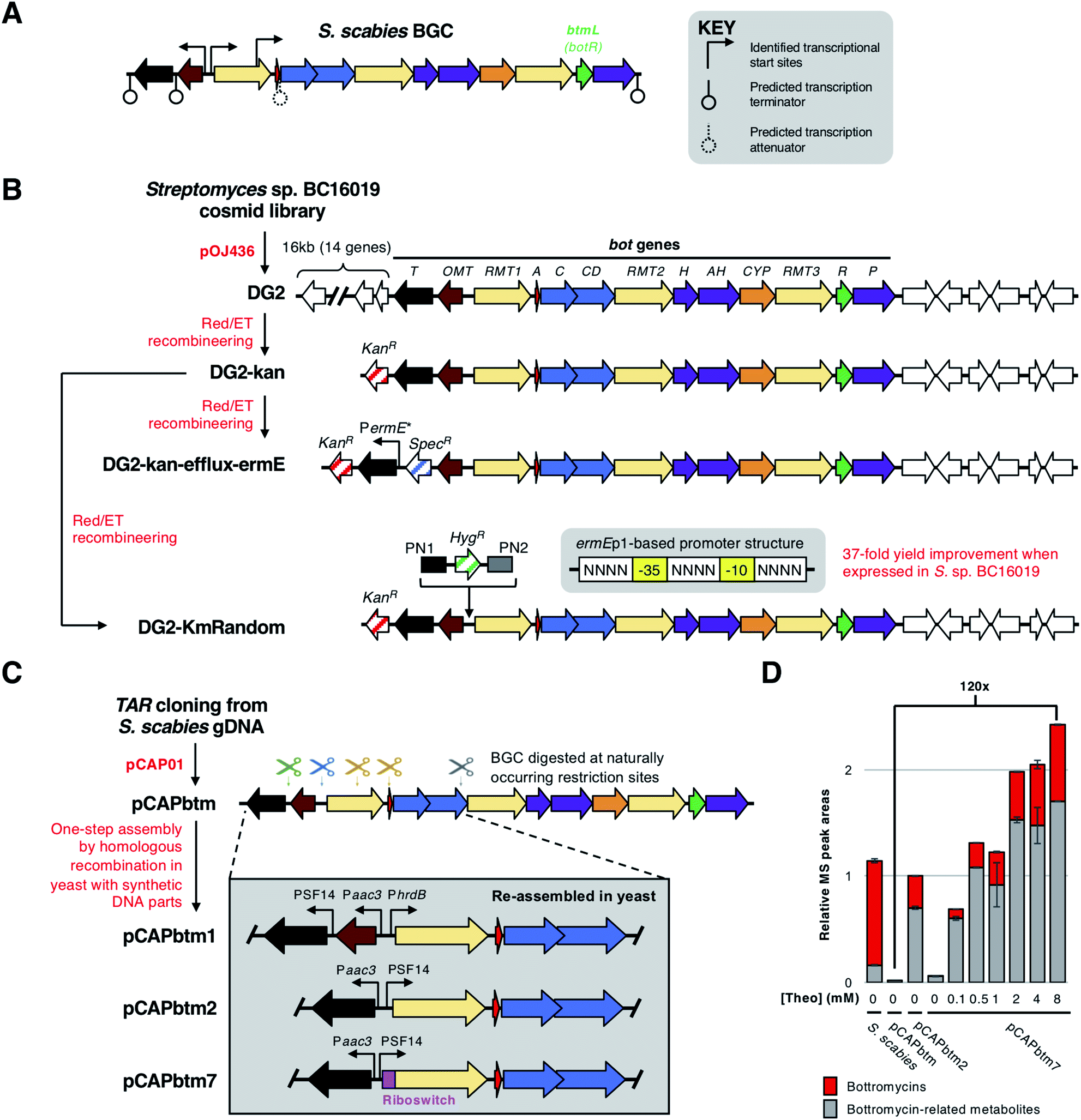 | ||
| Fig. 12 Overview of bottromycin BGC regulation and methods used genetically engineer the BGC. (A) Natural transcriptional start sites and predicted terminators determined by Vior et al.7 The gene encoding the pathway-situated transcriptional modulator BtmL (BotR) is highlighted. (B) Overview of cloning and engineering the Streptomyces sp. BC16019 BGC by Huo et al.8 and Horbal et al.9 (C) Overview of cloning and engineering the Streptomyces scabies BGC by Eyles et al.14 showing selected examples of engineered BGCs. (D) Bottromycin production chart adapted from Eyles et al.14 [Theo] = theophylline induction concentration. | ||
7.2 Heterologous expression of the bot BGC
Huo et al. were the first to express a bottromycin BGC in a heterologous host.8 Here, a pOJ436-based124 cosmid library from Streptomyces sp. BC16019 was screened for the intact bot BGC. The resulting DG2 construct was then expressed in Streptomyces albus J1074 and Streptomyces coelicolor A3(2) to successfully produce bottromycin A2, although estimated production levels (1–4 μg L−1) were much lower than from the native producer. This large construct was simplified by removing a 16 kb fragment of non-bot genes from the 5′-end of the BGC. This was replaced with a kanamycin resistance gene via Red/ET recombineering125 to generate cosmid DG2-kan (Fig. 12B).To increase production levels from DG2-kan, an approach pioneered by Ochi and colleagues126,127 was employed. This strategy involved obtaining rifampicin-resistant isolates that contain mutations in the rpoB gene, which encodes the RNA polymerase β-subunit. These rpoB mutations can enhance levels of antibiotic production in Streptomyces without affecting growth. Accordingly, bottromycin production was increased by about 10-fold in rifampicin-resistant mutants of S. coelicolor-DG2-kan. Huo et al. hypothesised that a limiting factor preventing higher yields was a lack of bottromycin resistance in S. coelicolor. The most likely self-immunity gene in the cluster is botT, which encodes a major facilitator superfamily (MFS) transporter.128 Therefore, to increase botT expression, the region preceding the botT gene was replaced with the strong PermE* promoter using Red/ET recombineering. This further increased bottromycin production levels two-fold compared to the rifampicin-resistant mutants.
Given that 11 bot biosynthetic genes (botRMT1-botP) are present on a polycistronic operon,7 it should be possible to engineer pathway regulation by simply changing the promoter preceding botRMT1 and optionally modifying the promoter(s) for botOMT and botT. Truman and colleagues used transformation-associated recombination (TAR) cloning129,130 in Saccharomyces cerevisiae (yeast) to directly capture the bottromycin BGC from the genomic DNA of S. scabies DSM 41658.14 This used the yeast/E. coli shuttle vector pCAP01,129 which can also integrate into actinobacterial genomes via the ϕC31 attachment site. The resulting vector, pCAPbtm, was introduced into S. coelicolor M1146,131 but bottromycin was produced in negligible amounts. Therefore, it was hypothesised that the bottromycin BGC could be efficiently engineered to improve productivity by use of homologous recombination in yeast. This strategy involved introducing double-strand breaks in pCAPbtm via restriction sites naturally found in the bottromycin BGC. This fragmented BGC was then repaired using a combination of double- and single-stranded DNA fragments to introduce new genetic features in a marker-free way (Fig. 12C).14
This approach was used to generate a series of modified pCAPbtm-derived plasmids that contained a variety of strong promoters (PSF14, PhrdB, Paac3, PermE*) in front of botRMT1 and botT, as well as rearranged the botA and botRMT1 genes. In an effort to limit the potential toxicity of bottromycin overproduction, the botOMT gene was removed, as previous work had shown that BotOMT-catalysed O-methylation is important for bottromycin activity.2 To fully understand the metabolic consequences of engineering regulation, the total productivity of the pathway was assessed using LC-MS-based metabolomics to detect multiple peptides derived from the bottromycin pathway that likely resulted from incomplete biosynthesis and subsequent hydrolysis of modified BotA. One surprising challenge was that BotRMT1 was inactive in all conditions tested, which resulted in all molecules lacking a β-methyl group on phenylalanine. Sequencing revealed no mutations to the gene cluster in any of the constructed vectors, so to further control expression levels, an inducible theophylline-dependent riboswitch132 was incorporated upstream of botRMT1. While this did not lead to active BotRMT1, theophylline induction did lead to the most productive heterologous expression system tested in this study, which was 120 times more productive than heterologous expression of the wild type BGC, as well as producing higher levels of bottromycin-related metabolites than the native S. scabies producer (Fig. 12D).
The DG2-kan cosmid generated by Huo et al.8 was used as the basis for BGC engineering by Luzhetskyy and colleagues.9 DG2-kan contains the bot BGC from Streptomyces sp. BC16019, and this cosmid was expressed in Streptomyces lividans TK24 to yield 0.23 mg L−1 bottromycin A2. To improve pathway productivity, the BGC was initially engineered to replace the native promoters of botOMT and botRMT1 with strong promoters from a promoter library previously generated by the Luzhetskyy group.133 The selection of strong promoters initially led to S. lividans growth problems, potentially caused by toxicity of the pathway to the host. This was overcome by selecting for bottromycin-resistant S. lividans mutants, which led to bottromycin A2 production levels of up to 3-fold higher than a control strain.
As it can be difficult to predict the precise relationship between promoter strength and pathway productivity, a ‘random rational strategy’ was employed to further boost yields from DG2-kan. Here, a library of bot BGCs was generated that featured random synthetic promoters inserted between botOMT and botRMT1. These random promoters were created based on the consensus −35 and −10 sequences of the ermEp1134 promoter (Fig. 12B). Degenerate primers were then used to randomise the sequences upstream, between and downstream of these consensus sequences. These were then introduced into the BGC using Red/ET cloning, and the mutated cosmids were conjugated into S. lividans TK24. Screening of 100 randomly selected strains harbouring mutated BGCs (DG2-KmRandom, Fig. 12B) revealed that 10% produced 5–50 fold more bottromycin A2 than a control strain harbouring unmodified DG2-kan. Quantitative RT-PCR (RT-qPCR) revealed that the transcription from both promoters had increased in the best producer, but that the strength for each promoter was very different (1:59 ratio), which emphasises the benefit of screening a promoter library. Pathway productivity was further increased by introducing this mutated BGC into the native producer, Streptomyces sp. BC16019, which therefore contained one copy each of the wild type and mutated bot BGCs. This led to a 37-fold increase in bottromycin production in relation to wild type Streptomyces sp. BC16019.
There is a lack of natural diversity within the core peptides of known bottromycins (Fig. 4B), which is in contrast to most other RiPP families.15 RiPPs are uniquely suited to pathway engineering to generate derivatives, as mutations to the core peptide lead to predictable changes to the final chemical structure. The lack of diversity amongst natural bottromycins means that there is a lot of chemical space to explore for bottromycin-like molecules, which could be important in the context of antibiotic discovery. Therefore, Luzhetskyy and colleagues generated DG2-kan cosmids with mutated botA genes,9 and then introduced these into Streptomyces sp. BC16019 ΔbotA, which is unable to make the wild type BotA precursor peptide. 12 mutants were generated, although most led to no detectable bottromycin-like metabolites, indicating a lack of pathway tolerance to these unnatural substrates, which is unlike many other RiPP pathways.135 Based on these data, the position most tolerant of modifications was Val3 of the core peptide (Fig. 13). Peptides with isoleucine and methionine residues at this position were successfully converted into bottromycin derivatives containing all expected post-translational modifications. Fig. 13 shows methionine-containing bottromycin M. Position 3 of the core peptide is effectively the only amino acid that is not post-translationally modified during biosynthesis and is the only position where natural variation has been observed, as bottromycin D from Streptomyces sp. WMMB272 features an alanine at this position.12 In this strain, an Ala3Val mutant was tolerated and therefore generated bottromycin A2. In contrast, no products were detected from three different mutants of Asp7 in this strain (Fig. 13).
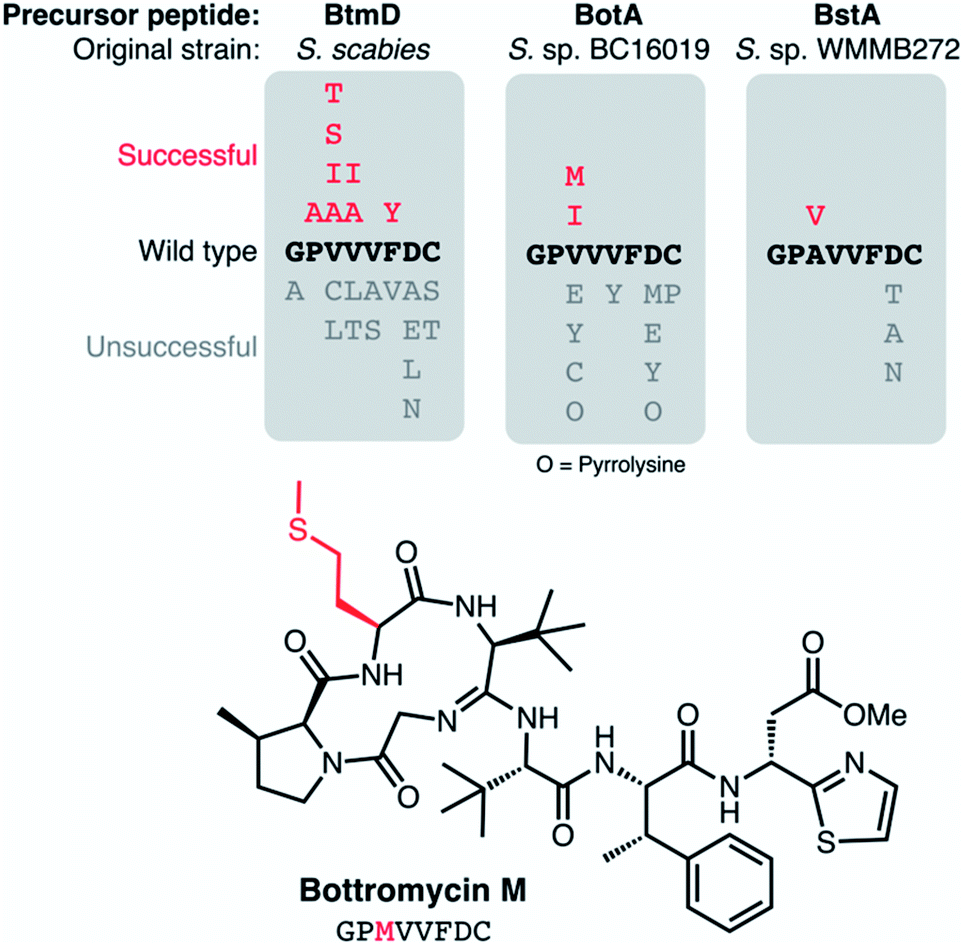 | ||
| Fig. 13 Mutations made to precursor peptide BotA and homologues in other bottromycin BGCs.2,9,136 The characterised structure of bottromycin M is shown. | ||
Comparable mutation results were obtained by Crone et al.136 Here, 22 botA mutants were generated using the precursor peptide complementation strategy previously reported for S. scabies ΔbtmD6 (the botA orthologue in this strain). As with Streptomyces sp. BC16019, Val3 of the core peptide was most tolerant of mutations, where mutations to Ala, Ile, Ser and Thr residues all led to bottromycin analogs with expected masses and tandem MS fragmentation patterns (Fig. 13). As with the other mutant studies, no bottromycin analogs were produced by mutations to Asp7, which was targeted due to the reported importance of this residue for activity.
8 Conclusions
Over 60 years after their original discovery bottromycins continue to capture the attention of research groups and provide riddles to be solved. Given the developing antibiotic crisis and the effectiveness of bottromycins against (multi) drug-resistant pathogens, the recent advances in bottromycin research should be capitalized on: through chemical synthesis and a combination of biochemistry/biotechnology new chemical space has become available to explore a more comprehensive SAR and address the in vivo instability of the compounds. In addition, the actual target within the A-site of the prokaryotic 50S ribosome has remained elusive and the MICs of some derivatives suggest an alternative, unknown target. We therefore think that despite its long history, the story of bottromycins may be just beginning.9 Conflicts of interest
There are no conflicts to declare.10 Acknowledgements
We thank Prof. Shuichi Hirono and Prof. Hiroaki Gouda for providing the coordinate file of the NMR structure of bottromycin A2 in CDCl3.11 References
- J. M. Waisvisz, M. G. van der Hoeven, J. van Peppen and W. C. M. Zwennis, Bottromycin. I. A New Sulfur-containing Antibiotic, J. Am. Chem. Soc., 1957, 79(16), 4520–4521 CrossRef CAS.
- Y. Kobayashi, M. Ichioka, T. Hirose, K. Nagai, A. Matsumoto, H. Matsui, H. Hanaki, R. Masuma, Y. Takahashi, S. Ōmura and T. Sunazuka, Bottromycin derivatives: efficient chemical modifications of the ester moiety and evaluation of anti-MRSA and anti-VRE activities, Bioorg. Med. Chem. Lett., 2010, 20(20), 6116–6120 CrossRef CAS.
- T. Otaka and A. Kaji, Mode of action of bottromycin A2. Release of aminoacyl or peptidyl tRNA from ribosomes, J. Biol. Chem., 1976, 251, 2299–2306 CrossRef CAS.
- T. Otaka and A. Kaji, Mode of action of bottromycin A2: Effect of bottromycin A2 on polysomes, FEBS Lett., 1983, 153(1), 53–59 CrossRef CAS.
- T. Otaka and A. Kaji, Mode of action of bottromycin A2: effect on peptide bond formation, FEBS Lett., 1981, 123(2), 173–176 CrossRef CAS.
- W. J. K. Crone, F. J. Leeper and A. W. Truman, Identification and characterisation of the gene cluster for the anti-MRSA antibiotic bottromycin: expanding the biosynthetic diversity of ribosomal peptides, Chem. Sci., 2012, 3(12), 3516–3521 RSC.
- N. M. Vior, E. Cea-Torrescassana, T. H. Eyles, G. Chandra and A. W. Truman, Regulation of Bottromycin Biosynthesis Involves an Internal Transcriptional Start Site and a Cluster-Situated Modulator, Front. Microbiol., 2020, 11, 495 CrossRef.
- L. Huo, S. Rachid, M. Stadler, S. C. Wenzel and R. Müller, Synthetic Biotechnology to Study and Engineer Ribosomal Bottromycin Biosynthesis, Chem. Biol., 2012, 19(10), 1278–1287 CrossRef CAS.
- L. Horbal, F. Marques, S. Nadmid, M. V. Mendes and A. Luzhetskyy, Secondary metabolites overproduction through transcriptional gene cluster refactoring, Metab. Eng., 2018, 49, 299–315 CrossRef CAS.
- H. Shimamura, H. Gouda, K. Nagai, T. Hirose, M. Ichioka, Y. Furuya, Y. Kobayashi, S. Hirono, T. Sunazuka and S. Ōmura, Structure determination and total synthesis of bottromycin A2:a potent antibiotic against MRSA and VRE, Angew. Chem., Int. Ed., 2009, 48(5), 914–917 CrossRef CAS.
- J. P. Gomez-Escribano, L. Song, M. J. Bibb and G. L. Challis, Posttranslational β-methylation and macrolactamidination in the biosynthesis of the bottromycin complex of ribosomal peptide antibiotics, Chem. Sci., 2012, 3(12), 3522–3525 RSC.
- Y. Hou, M. D. B. Tianero, J. C. Kwan, T. P. Wyche, C. R. Michel, G. A. Ellis, E. Vazquez-Rivera, D. R. Braun, W. E. Rose, E. W. Schmidt and T. S. Bugni, Structure and Biosynthesis of the Antibiotic Bottromycin D, Org. Lett., 2012, 14(19), 5050–5053 CrossRef CAS.
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H.-G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J.-K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, New developments in RiPP discovery, enzymology and engineering, Nat. Prod. Rep., 2020, 38, 130–239 RSC.
- T. H. Eyles, N. M. Vior and A. W. Truman, Rapid and Robust Yeast-Mediated Pathway Refactoring Generates Multiple New Bottromycin-Related Metabolites, ACS Synth. Biol., 2018, 7(5), 1211–1218 CrossRef CAS.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature, Nat. Prod. Rep., 2013, 30(1), 108–160 RSC.
- J. N. Elgersma, N. Rusting and J. H. van Berne, Werkwijze voor de bereiding van een antibiotische stof met behulp van actinomyceten van het geslacht Streptomyces 79749, 1955 Search PubMed.
- J. M. Waisvisz and M. G. van der Hoeven, The Chemistry and Partial Structure of Bottromycin. IV, J. Am. Chem. Soc., 1958, 80, 383–385 CrossRef CAS.
- J. M. Waisvisz, M. G. Vanderhoeven and B. T. Nijenhuis, The Structure of The Sulfur-Containing Moiety of Bottromycin, J. Am. Chem. Soc., 1957, 79(16), 4524–4527 CrossRef CAS.
- J. M. Waisvisz, M. G. van der Hoeven, J. F. Hölscher and B. te Nijenhuis, Bottromycin. II. Preliminary Degradation Studies, J. Am. Chem. Soc., 1957, 79, 4522–4524 CrossRef CAS.
- S. Nakamura, T. Chikaike, K. Karasawa, N. Tanaka, H. Yonehara and H. Umezawa, Isolation and characterization of bottromycins A and B, J. Antibiot., Ser. A, 1965, 18, 47–52 CAS.
- S. Nakamura, T. Chikaike, H. Yonehara and H. Umezawa, Structures of Bottromycins A and B, J. Antibiot., Ser. A, 1965,(18), 60–61 CAS.
- S. Nakamura, T. Yajima, Y. Lin and H. Umezawa, Isolation and characterization of bottromycins A2, B2, C2, J. Antibiot., 1967, 20(1), 1–5 CAS.
- S. Nakamura, N. Tanaka and H. Umezawa, Bottromycin A1, A2 and their structures, J. Antibiot., 1966, 19(1), 10–12 CAS.
- T. Yamada, T. Miyazawa, S. Kuwata and H. Watanabe, Studies of unusual amino acids and their peptides. VIII. The syntheses of an iminohexapeptide as a model of bottromycin and its related iminopeptides, Bull. Chem. Soc. Jpn., 1977, 50(7), 1827–1830 CrossRef CAS.
- Y. Takahashi, H. Naganawa, T. Takita, H. Umezawa and S. Nakamura, The revised structure of bottromycin A2, J. Antibiot., 1976, 29(10), 1120–1123 CrossRef CAS.
- D. Schipper, The Revised Structure of Bottromycin-A2, J. Antibiot., 1983, 36(8), 1076–1077 CrossRef CAS.
- H. Gouda, Y. Kobayashi, T. Yamada, T. Ideguchi, A. Sugawara, T. Hirose, S. Ōmura, T. Sunazuka and S. Hirono, Three-Dimensional Solution Structure of Bottromycin A2: A Potent Antibiotic Active against Methicillin-Resistant Staphylococcus aureus and Vancomycin-Resistant Enterococci, Chem. Pharm. Bull., 2012, 60(2), 169–171 CrossRef CAS.
- N. Tanaka, T. Nishimura, S. Nakamura and H. Umezawa, Activity of bottromycin against Mycoplasma gallisepticum, J. Antibiot., 1968, 21(1), 75–76 CrossRef CAS.
- S.-B. Park, I. Lee, J.-W. Suh, J.-G. Kim and C. Lee, Screening and identification of antimicrobial compounds from Streptomyces bottropensis suppressing rice bacterial blight, J. Microbiol. Biotechnol., 2011, 21, 1236–1242 CrossRef CAS.
- N. Tanaka, T. Nishimura, S. Nakamura and H. Umezawa, Biological studies on bottromycin A and its hydrazide, J. Antibiot., Ser. A, 1966, 19(4), 149–154 CAS.
- F. J. Wolf and W. J. Miller, Amides of methobottromycin, US3860703A, 1972.
- W. J. Miller, L. Chaiet, G. Rasmussen, B. Christensen, J. Hannah, A. K. Miller, F. J. Wolf and F. J. Wolf, Bottromycin. Separation of biologically active compounds and preparation and testing of amide derivatives, J. Med. Chem., 1968, 11, 746–749 CrossRef CAS.
- N. Tanaka, K. Sashikata, H. Yamaguchi and H. Umezawa, Inhibition of Protein Synthesis by Bottromycin A2 and Its Hydrazide, J. Biochem., 1966, 60(4), 405–410 CrossRef CAS.
- Y.-C. Lin and N. Tanaka, Mechanism of Action of Bottromycin in Polypeptide Biosynthesis, J. Biochem., 1968, 63(1), 1–7 CrossRef CAS.
- Y.-C. Lin, T. Kinoshita and N. Tanaka, Mechanism of protein synthesis inhibition by bottromycin A2: studies with puromycin, J. Antibiot., 1968, 21(8), 471–476 CrossRef CAS.
- N. Tanaka, Y.-C. Lin and A. Okuyama, Studies on translocation of F-met-tRNA and peptidyl-tRNA with antibiotics, Biochem. Biophys. Res. Commun., 1971, 44(2), 477–483 CrossRef CAS.
- T. Kinoshita and N. Tanaka, On the site of action of bottromycin A2, J. Antibiot., 1970, 23(6), 311–312 CrossRef CAS.
- S. Pestka and N. Brot, Studies on the Formation of Transfer Ribonucleic Acid-Ribosome Complexes: XV. Effect of Antibiotics on Steps of Bacterial Protien Synthesis: Some New Ribosomal Inhibitors of Translocation, J. Biol. Chem., 1971, 246(24), 7715–7722 CrossRef CAS.
- D. E. Brodersen, W. M. Clemons, A. P. Carter, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, The Structural Basis for the Action of the Antibiotics Tetracycline, Pactamycin, and Hygromycin B on the 30S Ribosomal Subunit, Cell, 2000, 103(7), 1143–1154 CrossRef CAS.
- Y. S. Polikanov, T. Szal, F. Jiang, P. Gupta, R. Matsuda, M. Shiozuka, T. A. Steitz, N. Vázquez-Laslop and A. S. Mankin, Negamycin interferes with decoding and translocation by simultaneous interaction with rRNA and tRNA, Mol. Cell, 2014, 56(4), 541–550 CrossRef CAS.
- N. B. Olivier, R. B. Altman, J. Noeske, G. S. Basarab, E. Code, A. D. Ferguson, N. Gao, J. Huang, M. F. Juette, S. Livchak, M. D. Miller, D. B. Prince, J. H. D. Cate, E. T. Buurman and S. C. Blanchard, Negamycin induces translational stalling and miscoding by binding to the small subunit head domain of the Escherichia coli ribosome, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(46), 16274–16279 CrossRef CAS.
- Y. S. Polikanov, N. A. Aleksashin, B. Beckert and D. N. Wilson, The Mechanisms of Action of Ribosome-Targeting Peptide Antibiotics, Front. Mol. Biosci., 2018, 5(48) CAS.
- J. Lin, D. Zhou, T. A. Steitz, Y. S. Polikanov and M. G. Gagnon, Ribosome-Targeting Antibiotics: Modes of Action, Mechanisms of Resistance, and Implications for Drug Design, Annu. Rev. Biochem., 2018, 87(1), 451–478 CrossRef CAS.
- U. Kazmaier, The Long, Long Way to Bottromycin, Isr. J. Chem., 2020 DOI:10.1002/ijch.202000068.
- S. L. Titouani, J. P. Lavergne, P. Viallefont and R. Jacquier, New syntheses of L-amino acids. I. Stereospecific synthesis of L-proline, and of the cis- and trans-3- and 4-methyl L-prolines, Tetrahedron, 1980, 36(20–21), 2961–2965 CrossRef CAS.
- C. Herdeis, H. P. Hubmann and H. Lotter, Synthesis of homochiral 3-substituted glutamic acids and prolines from pyroglutamic acid, Tetrahedron: Asymmetry, 1994, 5(3), 351–354 CrossRef CAS.
- P. Karoyan and G. Chassaing, New strategy for the synthesis of 3-substituted prolines, Tetrahedron Lett., 1997, 38(1), 85–88 CrossRef CAS.
- T. M. Kamenecka, Y.-J. Park, L. S. Lin, T. Lanza and W. K. Hagmann, Enantioselective approach to 3-substituted prolines, Tetrahedron Lett., 2001, 42(49), 8571–8573 CrossRef CAS.
- C. Flamant-Robin, Q. Wang, A. Chiaroni and N. A. Sasaki, An efficient method for the stereoselective synthesis of cis-3-substituted prolines: conformationally constrained α-amino acids, Tetrahedron, 2002, 58(52), 10475–10484 CrossRef CAS.
- R. Sharma and W. D. Lubell, Regioselective enolization and alkylation of 4-oxo-N-(9-phenylfluoren-9-yl)proline: Synthesis of enantiopure proline-valine and hydroxyproline-valine chimeras, J. Org. Chem., 1996, 61(1), 202–209 CrossRef CAS.
- J. R. Medina, C. W. Blackledge, K. F. Erhard, J. M. Axten and W. H. Miller, Benzyl 2-cyano-3,3-dimethyl-1-pyrrolidinecarboxylate, a versatile intermediate for the synthesis of 3,3-dimethylproline derivatives, J. Org. Chem., 2008, 73(10), 3946–3949 CrossRef CAS.
- T. M. Bott, J. A. Vanecko and F. G. West, One-Carbon Ring Expansion of Azetidines via Ammonium Ylide 1,2 -Shifts: A Simple Route to Substituted Pyrrolidines, J. Org. Chem., 2009, 74(7), 2832–2836 CrossRef CAS.
- S. Fuse, H. Koinuma, A. Kimbara, M. Izumikawa, Y. Mifune, H. He, K. Shin-ya, T. Takahashi and T. Doi, Total Synthesis and Stereochemistry Revision of Mannopeptimycin Aglycone, J. Am. Chem. Soc., 2014, 136(34), 12011–12017 CrossRef CAS.
- C. S. V. Houge-Frydrych, M. L. Gilpin, P. W. Skett and J. W. Tyler, SB-203207 and SB-203208, two novel isoleucyl tRNA synthetase inhibitors from a Streptomyces sp. II. Structure determination, J. Antibiot., 2000, 53(4), 364–372 CrossRef CAS.
- Y. Kataoka, Y. Seto, M. Yamamoto, T. Yamada, S. Kuwata and H. Watanabe, Studies of Unusual Amino Acids and Their Peptides. VI. The Syntheses and the Optical Resolutions of β-Methylphenylalanine and Its Dipeptide Present in Bottromycin, Bull. Chem. Soc. Jpn., 1976, 49(4), 1081–1084 CrossRef CAS.
- M. Alias, M. P. Lopez and C. Cativiela, An efficient and stereodivergent synthesis of threo- and erythro-beta-methylphenylalanine. Resolution of each racemic pair by semipreparative HPLC, Tetrahedron, 2004, 60(4), 885–891 CrossRef CAS.
- N. Grobuschek, M. G. Schmid, C. Tuscher, M. Ivanova and G. Gubitz, Chiral separation of beta-methyl-amino acids by ligand exchange using capillary electrophoresis and HPLC, J. Pharm. Biomed. Anal., 2002, 27(3–4), 599–605 CrossRef CAS.
- L. M. Nogle, C. W. Mann, W. L. Watts and Y. R. Zhang, Preparative separation and identification of derivatized beta-methylphenylalanine enantiomers by chiral SFC, HPLC and NMR for development of new peptide ligand mimetics in drug discovery, J. Pharm. Biomed. Anal., 2006, 40(4), 901–909 CrossRef CAS.
- A. Peter, G. Toth, G. Torok and D. Tourwe, Separation of enantiomeric beta-methyl amino acids and of beta-methyl amino acid containing peptides, J. Chromatogr. A, 1996, 728(1–2), 455–465 CrossRef CAS.
- G. Torok, A. Peter, D. W. Armstrong, D. Tourwe, G. Toth and J. Sapi, Direct chiral separation of unnatural amino acids by high-performance liquid chromatography on a ristocetin A-bonded stationary phase, Chirality, 2001, 13(10), 648–656 CrossRef CAS.
- E. Vekes, G. Torok, A. Peter, J. Sapi and D. Tourwe, Indirect high-performance liquid chromatographic separation of stereoisomers of beta-alkyl-substituted amino acids by the application of (S)-N-(4-nitrophenoxycarbonyl)phenylalanine methoxyethyl ester as chiral derivatizing agent, J. Chromatogr. A, 2002, 949(1–2), 125–139 CrossRef CAS.
- I. Ilisz, G. Fodor, R. Ivanyi, L. Szente, G. Toth and A. Peter, Enantioseparation of β-methyl-substituted amino acids with cyclodextrins by capillary zone electrophoresis, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 875(1), 273–279 CrossRef CAS.
- C. Jiang, D. W. Armstrong, A. W. Lantz, A. Peter and G. Toth, Enantiomeric separation of synthetic amino acids using capillary zone electrophoresis, J. Liq. Chromatogr. Relat. Technol., 2007, 30(9–12), 1421–1436 CrossRef CAS.
- R. Torok, R. Berkecz and A. Peter, Enantioseparation of phenylalanine analogs on a quinine-based anion-exchanger chiral stationary phase: Structure and temperature effects, J. Sep. Sci., 2006, 29(16), 2523–2532 CrossRef CAS.
- J. Ogawa, A. Ryono, S. X. Xie, R. M. Vohra, R. Indrati, M. Akamatsu, H. Miyagawa, T. Ueno and S. Shimizu, Separative preparation of the four stereoisomers of β-methylphenylalanine with N-carbamoyl amino acid amidohydrolases, J. Mol. Catal. B: Enzym., 2001, 12(1–6), 71–75 CrossRef CAS.
- G. Tsuchihashi, S. Mitamura and K. Ogura, Synthesis of (S,S)-2-Amino-3-phenylbutyric acid, Bull. Chem. Soc. Jpn., 1979, 52(7), 2167–2168 CrossRef CAS.
- R. Dharanipragada, E. Nicolas, G. Toth and V. J. Hruby, Asymmetric synthesis of unusual amino acids: An efficient synthesis of optically pure isomers of β-methylphenylalanine, Tetrahedron Lett., 1989, 30(49), 6841–6844 CrossRef CAS.
- R. Dharanipragada, K. Vanhulle, A. Bannister, S. Bear, L. Kennedy and V. J. Hruby, Asymmetric-Synthesis of Unsual Amino-acids – An Efficient Synthesis of Optically Pure Isomers of beta-Methylphenylalanine, Tetrahedron, 1992, 48(23), 4733–4748 CrossRef CAS.
- W. Oppolzer and R. Moretti, Enantioselective systheses of α-amino acids from 10-sulfonamido-isobornyl esters and di-t-butyl azodicarboxylate, Tetrahedron, 1988, 44(17), 5541–5552 CrossRef CAS.
- E. Medina, A. Moyano, M. A. Pericas and A. Riera, Synthesis of N-Boc-β-Aryl Alanines and of N-Boc-β-Methyl-β-aryl Alanines by Regioselective Ring-Opening of Enantiomerically Pure N-Boc-Aziridines, J. Org. Chem., 1998, 63(23), 8574–8578 CrossRef CAS.
- M. J. O'Donnell, J. T. Cooper and M. M. Mader, Acyclic Stereoselective Boron Alkylation Reactions for the Asymmetric Synthesis of β-Substituted α-Amino Acid Derivatives, J. Am. Chem. Soc., 2003, 125(9), 2370–2371 CrossRef.
- A. Enright, F.-R. Alexandre, G. Roff, I. G. Fotheringham, M. J. Dawson and N. J. Turner, Stereoinversion of β- and γ-substituted α-amino acids using a chemo-enzymatic oxidation–reduction procedure, Chem. Commun., 2003,(20), 2636–2637 RSC.
- T. Ooi, D. Kato, K. Inamura, K. Ohmatsu and K. Maruoka, Practical Stereoselective Synthesis of β-Branched α-Amino Acids through Efficient Kinetic Resolution in the Phase-Transfer-Catalyzed Asymmetric Alkylations, Org. Lett., 2007, 9(20), 3945–3948 CrossRef CAS.
- S. Nakamura, T. Chikaike, H. Yonehara and H. Umezawa, Isolation, Characterization and Structural Elucidadtion of New Amino Acids from Bottromycin A, Chem. Pharm. Bull., 1965, 13(5), 599–602 CrossRef CAS.
- Y. Seto, K. Torii, K. Bori, K. Inabata, S. Kuwata and H. Watanabe, Studies of Unusual Amino Acids and Their Peptides. VI. The Syntheses and the Optical Resolutions of β-Methylphenylalanine and Its Dipeptide Present in Bottromycin, Bull. Chem. Soc. Jpn., 1974, 47(1), 151–155 CrossRef CAS.
- F. A. Davis, R. E. Reddy and J. M. Szewczyk, Asymmetric-Synthesis of (R)-(+)-beta-Phenylalanine from (S)-(+)-Benzylidene-p-Toluenesulfinamide – Regeneration of the Sulfiniamine Precursor, J. Org. Chem., 1995, 60(21), 7037–7039 CrossRef CAS.
- T. P. Tang and J. A. Ellman, The tert-butanesulfinyl group: An ideal chiral directing group and boc-surrogate for the asymmetric synthesis and applications of beta-amino acids, J. Org. Chem., 1999, 64(1), 12–13 CrossRef CAS.
- T. Yamada, M. Yagita, Y. Kobayashi, G. Sennari, H. Shimamura, H. Matsui, Y. Horimatsu, H. Hanaki, T. Hirose, S. Ōmura and T. Sunazuka, Synthesis and Evaluation of Antibacterial Activity of Bottromycins, J. Org. Chem., 2018, 83(13), 7135–7149 CrossRef CAS.
- T. Yamada, K. Suegane, S. Kuwata and H. Watanabe, Studies of unusual amino acids and their peptides. VII. The syntheses and the reactions of iminopeptides, Bull. Chem. Soc. Jpn., 1977, 50(5), 1088–1093 CrossRef CAS.
- S. Nakamura, T. Chikaike, H. Yonehara and H. Umezawa, Structures of Bottromycins A and B, J. Antibiot., 1965, 18, 60–61 CAS.
- S. Nakamura and H. Umezawa, The structure of bottromycin A2, a new component of bottromycins, Chem. Pharm. Bull., 1966, 14(9), 981–986 CrossRef CAS.
- M. Kaneda, Studies on bottromycins. I. 1H and 13C NMR assignments of bottromycin A2, the main component of the complex, J. Antibiot., 1992, 45(5), 792–796 CrossRef CAS.
- S. Ackermann, H.-G. Lerchen, D. Haebich, A. Ullrich and U. Kazmaier, Synthetic studies towards bottromycin, Beilstein J. Org. Chem., 2012, 8, 1652–1656 CrossRef CAS.
- U. Kazmaier and C. Hebach, Peptide syntheses via Ugi reactions with ammonia, Synlett, 2003, 11, 1591–1594 CrossRef.
- R. Pick, M. Bauer, U. Kazmaier and C. Hebach, Ammonia in Ugi reactions - Four-component versus six-component couplings, Synlett, 2005, 5, 757–760 Search PubMed.
- M. Bauer, F. Maurer, S. M. Hoffmann and U. Kazmaier, Hydroxyalkyl Thiazolines, a New Class of Highly Efficient Ligands for Carbonyl Additions, Synlett, 2008,(20), 3203–3207 CAS.
- U. Kazmaier and S. Ackermann, A straightforward approach towards thiazoles and endothiopeptides via Ugi reaction, Org. Biomol. Chem., 2005, 3(17), 3184–3187 RSC.
- U. Kazmaier and A. Persch, A straightforward approach towards 5-substituted thiazolylpeptides via the thio-Ugi-reaction, Org. Biomol. Chem., 2010, 8(23), 5442–5447 RSC.
- F. Hata, A. Matsumae, K. Abe, Y. Sano, M. Otani and S. Ōmura, Fermentative preparation of bottromycin, JP47010036B, 1972.
- H.-G. Lerchen, G. Schiffer, H. Broetz-Oesterhelt, A. Mayer-Bartschmid, S. Eckermann, C. Freiberg, R. Endermann, J. Schuhmacher, H. Meier, N. Svenstrup, S. Seip, M. Gehling and D. HaebichFermentative and chemical synthesis of bottromycin derivative analogs for use in treatment or prevention of bacterial infections in humans or animals. WO2006103010A1, 2006.
- J. Kellenberger, On the Stereochemical Course of the C-methylation Steps in Bottromycin Biosynthesis, PhD thesis, ETH Switzerland, 1997.
- H. Mosimann and B. Kräutler, Methylcorrinoids Methylate Radicals-Their Second Biological Mode of Action?, Angew. Chem., Int. Ed. Engl., 2000, 39(2), 393–395 CrossRef CAS.
- M. F. Freeman, C. Gurgui, M. J. Helf, B. I. Morinaka, A. R. Uria, N. J. Oldham, H.-G. Sahl, S. Matsunaga and J. Piel, Metagenome Mining Reveals Polytheonamides as Posttranslationally Modified Ribosomal Peptides, Science, 2012, 338(6105), 387–390 CrossRef CAS.
- C. Mahlert, F. Kopp, J. Thirlway, J. Micklefield and M. A. Marahiel, Stereospecific Enzymatic Transformation of α-Ketoglutarate to (2S,3R)-3-Methyl Glutamate during Acidic Lipopeptide Biosynthesis, J. Am. Chem. Soc., 2007, 129(39), 12011–12018 CrossRef CAS.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W.-T. Liu, M. Crüsemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderón, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C.-C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C.-C. Liaw, Y.-L. Yang, H.-U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. Boya P, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodríguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P.-M. Allard, P. Phapale, L.-F. Nothias, T. Alexandrov, M. Litaudon, J.-L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D.-T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Müller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. Ø. Palsson, K. Pogliano, R. G. Linington, M. Gutiérrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking, Nat. Biotechnol., 2016, 34(8), 828–837 CrossRef CAS.
- W. J. K. Crone, N. M. Vior, J. Santos-Aberturas, L. G. Schmitz, F. J. Leeper and A. W. Truman, Dissecting Bottromycin Biosynthesis Using Comparative Untargeted Metabolomics, Angew. Chem., Int. Ed., 2016, 55(33), 9639–9643 CrossRef CAS.
- M. Matsui, J. H. Fowler and L. L. Walling, Leucine aminopeptidases: diversity in structure and function, Biol. Chem., 2006, 387(12), 1535–1544 CAS.
- S. Dorus, E. C. Wilkin and T. L. Karr, Expansion and functional diversification of a leucyl aminopeptidase family that encodes the major protein constituents of Drosophila sperm, BMC Genomics, 2011, 12, 177 CrossRef CAS.
- F. Frottin, A. Martinez, P. Peynot, S. Mitra, R. C. Holz, C. Giglione and T. Meinnel, The proteomics of N-terminal methionine cleavage, Mol. Cell. Proteomics, 2006, 5(12), 2336–2349 CrossRef CAS.
- G. Mann, L. Huo, S. Adam, B. Nardone, J. Vendome, N. J. Westwood, R. Müller and J. Koehnke, Structure and Substrate Recognition of the Bottromycin Maturation Enzyme BotP, ChemBioChem, 2016, 17(23), 2286–2292 CrossRef CAS.
- T. J. Oman and W. A. van der Donk, Follow the leader: the use of leader peptides to guide natural product biosynthesis, Nat. Chem. Biol., 2010, 6(1), 9–18 CrossRef CAS.
- S. W. Lee, D. A. Mitchell, A. L. Markley, M. E. Hensler, D. Gonzalez, A. Wohlrab, P. C. Dorrestein, V. Nizet and J. E. Dixon, Discovery of a widely distributed toxin biosynthetic gene cluster, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(15), 5879–5884 CrossRef CAS.
- L. C. Wieland Brown, M. G. Acker, J. Clardy, C. T. Walsh and M. A. Fischbach, Thirteen posttranslational modifications convert a 14-residue peptide into the antibiotic thiocillin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(8), 2549–2553 CrossRef CAS.
- E. W. Schmidt, J. T. Nelson, D. A. Rasko, S. Sudek, J. A. Eisen, M. G. Haygood and J. Ravel, Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(20), 7315–7320 CrossRef CAS.
- Y. Goto and H. Suga, In Vitro Biosynthesis of Peptides Containing Exotic Azoline Analogues, ChemBioChem, 2020, 21(1–2), 84–87 CrossRef CAS.
- J. Koehnke, F. Morawitz, A. F. Bent, W. E. Houssen, S. L. Shirran, M. A. Fuszard, I. A. Smellie, C. H. Botting, M. C. M. Smith, M. Jaspars and J. H. Naismith, An Enzymatic Route to Selenazolines, ChemBioChem, 2013, 14(5), 564–567 CrossRef CAS.
- C. J. Schwalen, G. A. Hudson, S. Kosol, N. Mahanta, G. L. Challis and D. A. Mitchell, In Vitro Biosynthetic Studies of Bottromycin Expand the Enzymatic Capabilities of the YcaO Superfamily, J. Am. Chem. Soc., 2017, 139(50), 18154–18157 CrossRef CAS.
- L. Franz, S. Adam, J. Santos-Aberturas, A. W. Truman and J. Koehnke, Macroamidine Formation in Bottromycins Is Catalyzed by a Divergent YcaO Enzyme, J. Am. Chem. Soc., 2017, 139(50), 18158–18161 CrossRef CAS.
- A. Sikandar, L. Franz, O. Melse, I. Antes and J. Koehnke, Thiazoline-Specific Amidohydrolase PurAH Is the Gatekeeper of Bottromycin Biosynthesis, J. Am. Chem. Soc., 2019, 141(25), 9748–9752 CrossRef CAS.
- J. T. Mindrebo, C. M. Nartey, Y. Seto, M. D. Burkart and J. P. Noel, Unveiling the functional diversity of the alpha/beta hydrolase superfamily in the plant kingdom, Curr. Opin. Struct. Biol., 2016, 41, 233–246 CrossRef CAS.
- A. Rauwerdink and R. J. Kazlauskas, How the Same Core Catalytic Machinery Catalyzes 17 Different Reactions: the Serine-Histidine-Aspartate Catalytic Triad of α/β-Hydrolase Fold Enzymes, ACS Catal., 2015, 5(10), 6153–6176 CrossRef CAS.
- A. Sikandar, L. Franz, S. Adam, J. Santos-Aberturas, L. Horbal, A. Luzhetskyy, A. W. Truman, O. V. Kalinina and J. Koehnke, The bottromycin epimerase BotH defines a group of atypical α/β-hydrolase-fold enzymes, Nat. Chem. Biol., 2020, 16, 1013–1018 CrossRef.
- B. I. Morinaka, A. L. Vagstad, M. J. Helf, M. Gugger, C. Kegler, M. F. Freeman, H. B. Bode and J. Piel, Radical S-Adenosyl Methionine Epimerases: Regioselective Introduction of Diverse D-Amino Acid Patterns into Peptide Natural Products, Angew. Chem., Int. Ed., 2014, 53(32), 8503–8507 CrossRef CAS.
- A. Benjdia, A. Guillot, P. Ruffié, J. Leprince and O. Berteau, Post-translational modification of ribosomally synthesized peptides by a radical SAM epimerase in Bacillus subtilis, Nat. Chem., 2017, 9(7), 698–707 CrossRef CAS.
- X. Yang and W. A. van der Donk, Post-translational Introduction of D-Alanine into Ribosomally Synthesized Peptides by the Dehydroalanine Reductase NpnJ, J. Am. Chem. Soc., 2015, 137(39), 12426–12429 CrossRef CAS.
- L. Huo and W. A. van der Donk, Discovery and Characterization of Bicereucin, an Unusual D-Amino Acid-Containing Mixed Two-Component Lantibiotic, J. Am. Chem. Soc., 2016, 138(16), 5254–5257 CrossRef CAS.
- S. Adam, L. Franz, M. Milhim, R. Bernhardt, O. V. Kalinina and J. Koehnke, Characterization of the Stereoselective P450 Enzyme BotCYP Enables the In Vitro Biosynthesis of the Bottromycin Core Scaffold, J. Am. Chem. Soc., 2020, 142(49), 20560–20565 CrossRef CAS.
- G. A. Hudson, Z. Zhang, J. I. Tietz, D. A. Mitchell and W. A. van der Donk, In Vitro Biosynthesis of the Core Scaffold of the Thiopeptide Thiomuracin, J. Am. Chem. Soc., 2015, 137(51), 16012–16015 CrossRef CAS.
- Z. Zhang, G. A. Hudson, N. Mahanta, J. I. Tietz, W. A. van der Donk and D. A. Mitchell, Biosynthetic Timing and Substrate Specificity for the Thiopeptide Thiomuracin, J. Am. Chem. Soc., 2016, 138(48), 15511–15514 CrossRef CAS.
- Q. Zheng, Q. Wang, S. Wang, J. Wu, Q. Gao and W. Liu, Thiopeptide Antibiotics Exhibit a Dual Mode of Action against Intracellular Pathogens by Affecting Both Host and Microbe, Chem. Biol., 2015, 22(8), 1002–1007 CrossRef CAS.
- X. Bai, H. Guo, D. Chen, Q. Yang, J. Tao and W. Liu, Isolation and structure determination of two new nosiheptide-type compounds provide insights into the function of the cytochrome P450 oxygenase NocV in nocathiacin biosynthesis, Org. Chem. Front., 2020, 7(3), 584–589 RSC.
- T. S. Young and C. T. Walsh, Identification of the thiazolyl peptide GE37468 gene cluster from Streptomyces ATCC 55365 and heterologous expression in Streptomyces lividans, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(32), 13053–13058 CrossRef CAS.
- J. J. Hug, J. Dastbaz, S. Adam, O. Revermann, J. Koehnke, D. Krug and R. Müller, Biosynthesis of Cittilins, Unusual Ribosomally Synthesized and Post-translationally Modified Peptides from Myxococcus xanthus, ACS Chem. Biol., 2020, 15(8), 2221–2231 CrossRef CAS.
- M. Bierman, R. Logan, K. O'Brien, E. T. Seno, R. Nagaraja Rao and B. E. Schoner, Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp., Gene, 1992, 116(1), 43–49 CrossRef CAS.
- B. Gust, G. L. Challis, K. Fowler, T. Kieser and K. F. Chater, PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(4), 1541–1546 CrossRef CAS.
- H. Hu and K. Ochi, Novel Approach for Improving the Productivity of Antibiotic-Producing Strains by Inducing Combined Resistant Mutations, Appl. Environ. Microbiol., 2001, 67(4), 1885–1892 CrossRef CAS.
- H. Hu, Q. Zhang and K. Ochi, Activation of Antibiotic Biosynthesis by Specified Mutations in the rpoB Gene (Encoding the RNA Polymerase β Subunit) of Streptomyces lividans, J. Bacteriol., 2002, 184(14), 3984–3991 CrossRef CAS.
- E. M. Quistgaard, C. Löw, F. Guettou and P. Nordlund, Understanding transport by the major facilitator superfamily (MFS): structures pave the way, Nat. Rev. Mol. Cell Biol., 2016, 17(2), 123–132 CrossRef CAS.
- K. Yamanaka, K. A. Reynolds, R. D. Kersten, K. S. Ryan, D. J. Gonzalez, V. Nizet, P. C. Dorrestein and B. S. Moore, Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(5), 1957–1962 CrossRef CAS.
- V. Noskov, N. Kouprina, S.-H. Leem, M. Koriabine, J. C. Barrett and V. Larionov, A genetic system for direct selection of gene-positive clones during recombinational cloning in yeast, Nucleic Acids Res., 2002, 30(2), e8 CrossRef.
- J. P. Gomez-Escribano and M. J. Bibb, Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters, Microb. Biotechnol., 2011, 4(2), 207–215 CrossRef CAS.
- M. M. Rudolph, M.-P. Vockenhuber and B. Suess, Synthetic riboswitches for the conditional control of gene expression in Streptomyces coelicolor, Microbiology, 2013, 159, 1416–1422 CrossRef CAS.
- T. Siegl, B. Tokovenko, M. Myronovskyi and A. Luzhetskyy, Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes, Metab. Eng., 2013, 19, 98–106 CrossRef CAS.
- W. R. Strohl, Compilation and analysis of DNA sequences associated with apparent streptomycete promoters, Nucleic Acids Res., 1992, 20(5), 961–974 CrossRef CAS.
- G. A. Hudson and D. A. Mitchell, RiPP antibiotics: biosynthesis and engineering potential, Curr. Opin. Microbiol., 2018, 45, 61–69 CrossRef CAS.
- W. J. K. Crone, Investigations into the biosynthesis of bottromycin, University of Cambridge Cambridge, 2015 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |