
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis of biologically active indolocarbazole natural products
George E.
Chambers
 a,
A. Emre
Sayan
b and
Richard C. D.
Brown
*a
a,
A. Emre
Sayan
b and
Richard C. D.
Brown
*a
aSchool of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: rcb1@soton.ac.uk
bUniversity of Southampton Cancer Sciences Division, University of Southampton, Southampton SO17 1BJ, UK
First published on 5th March 2021
Abstract
Covering: up to 2020
The indolocarbazoles, in particular indolo[2,3-a]pyrrolo[3,4-c]carbazole derivatives, are an important class of natural products that exhibit a wide range of biological activities. There has been a plethora of synthetic approaches to this family of natural products, leading to advances in chemical methodology, as well as affording access to molecular scaffolds central to protein kinase drug discovery programmes. In this review, we compile and summarise the synthetic approaches to the indolo[2,3-a]pyrrolo[3,4-c]carbazole derivatives, spanning the period from their isolation in 1980 up to 2020. The selected natural products include indolocarbazoles not functionalised at indolic nitrogen, pyranosylated indolocarbazoles, furanosylated indolocarbazoles and disaccharideindolocarbazoles.
 George E. Chambers | George Chambers grew up in south–east England and studied chemistry at the University of Southampton, where he received a first class MChem in 2018. During his studies, he spent 6 months at the University of Sydney working in the group of Associate Prof. Christopher McErlean on the synthesis of trehalose-6-phosphate analogues. He then joined the group of Prof. Richard Brown for his PhD where he is currently working on the total synthesis of indolocarbazole alkaloids. |
 A. Emre Sayan | Emre Sayan was born in Ankara, Turkey, in 1972. He completed his BSc degree in Bogazici University, followed by MSc and PhD degrees (2002) in Bilkent University, department of Molecular Biology and Genetics. He worked in INSERM (Paris), MRC Toxicology Unit (Leicester, UK) and University of Leicester (UK) as postdoctoral and career development fellow between 2002–2010. He was appointed as a Lecturer in University of Southampton, UK (2010). His research group's interests include biomarker discovery and drug development for metastatic cancers. He has a fascination for natural products and he always takes lessons from nature. |
 Richard C. D. Brown | Richard Brown was born in Canterbury, UK, in 1968. Following undergraduate and postgraduate studies at the University of Southampton, UK (PhD 1994, supervisor Prof. Philip J. Kocienski), he took up a NATO postdoctoral fellowship at UC Berkeley (Prof. Clayton H. Heathcock). He returned to the University of Southampton in 1996 as a Royal Society University Research Fellow, and was promoted to Professor in 2010. His group's research includes natural product total synthesis, oxidative cyclisation methodology and electro-organic synthesis using flow reactors. Natural products provide a major inspiration for his research, with current synthetic targets including bioactive alkaloids. |
1 Introduction
1.1 The indolocarbazoles
The indolocarbazoles are a family of heterocyclic natural products consisting of a carbazole core fused to an indole system through its nitrogen-containing ring (Fig. 1). There are several indolocarbazole isomers, of which the five most commonly isolated are the 11,12-dihydroindolo[2,3-a]-carbazole (1), 5,7-dihydroindolo[2,3-b]carbazole (2), 5,8-dihydroindolo[2,3-c]carbazole (3), 5,12-dihydroindolo[3,2-a]carbazole (4) and the 5,11-dihydroindolo[3,2-b]carbazole (5). The indolocarbazoles have been the subject of several extensive reviews covering their chemistry, properties and biological activity.1–5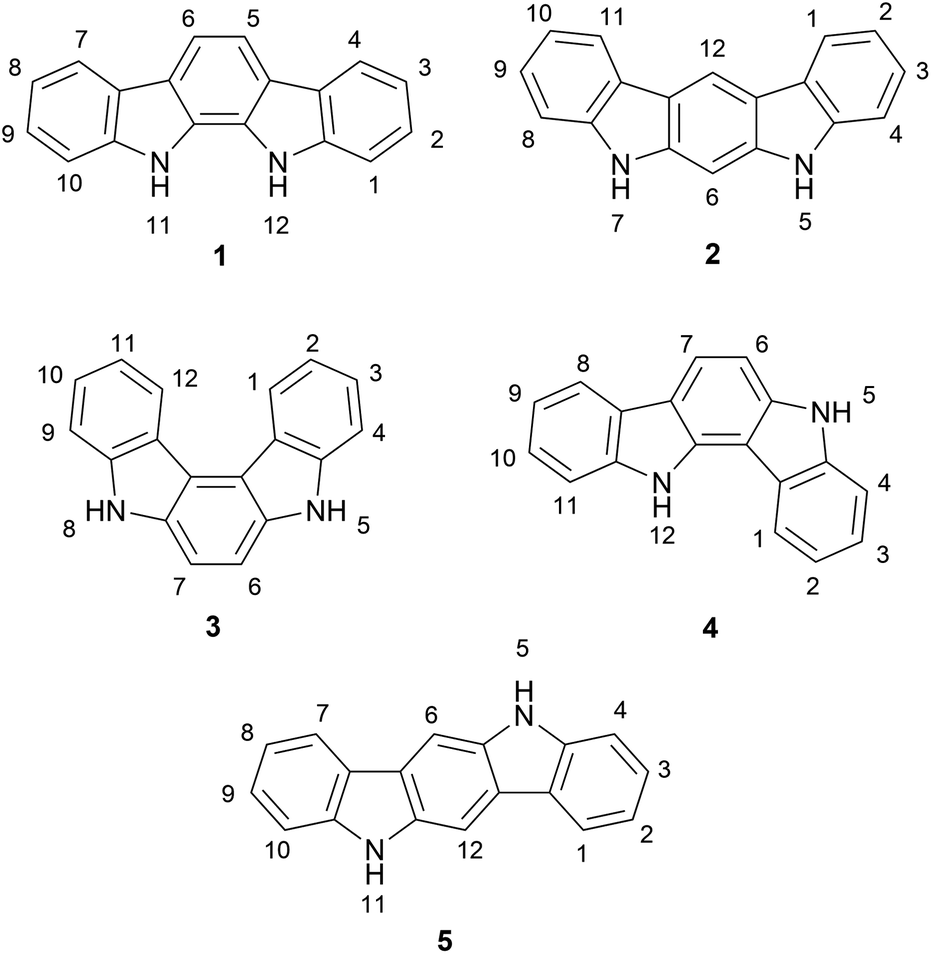 | ||
| Fig. 1 Indolocarbazole isomers. | ||
Of the indolocarbazole isomers described, the most rigorously studied is 11,12-dihydroindolo[2,3-a]-carbazole (1). In particular, a sub-family known as the indolo[2,3-a]pyrrolo[3,4-c]carbazole derivatives (6) ascended to prominence in the early 90 s after discovery of interesting biological activities associated with these compounds, in particular their nanomolar inhibition of protein kinase C (PKC) (Fig. 2). This broad sub-family of natural products includes compounds both with and without indolic functionalisation, and for convenience throughout this review they will be referred to as indolocarbazole natural products. These natural products have been reviewed previously from a broader perspective,6–8 as well as on more specific aspects such as their biological activity,4,5,9 biosynthesis10 and therapeutic application.7,11,12 The primary focus of the present review is on synthetic approaches to the indolocarbazole natural products.
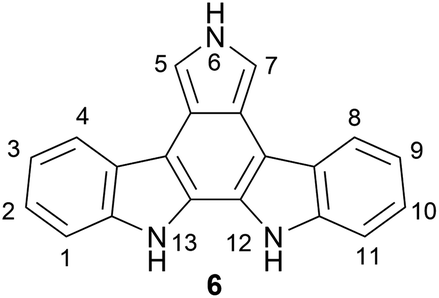 | ||
| Fig. 2 An isomer of indolo[2,3-a]pyrrolo[3,4-c]carbazole. | ||
1.2 The indolocarbazole alkaloids and their discovery
In 1977, Omura and co-workers isolated a novel alkaloid they named AM-2282, from the bacterium Streptomyces staurosporeus.13 Preliminary testing of the alkaloid revealed strong hypotensive activity as well as antimicrobial activity against fungi and yeast.13 Much effort was devoted to elucidate the alkaloid's structure, which was later confirmed by X-ray crystallography, as an indolocarbazole core bearing two glycosyl linkages at the indolic nitrogens. AM-2282 was subsequently renamed staurosporine (7, Fig. 3).14,15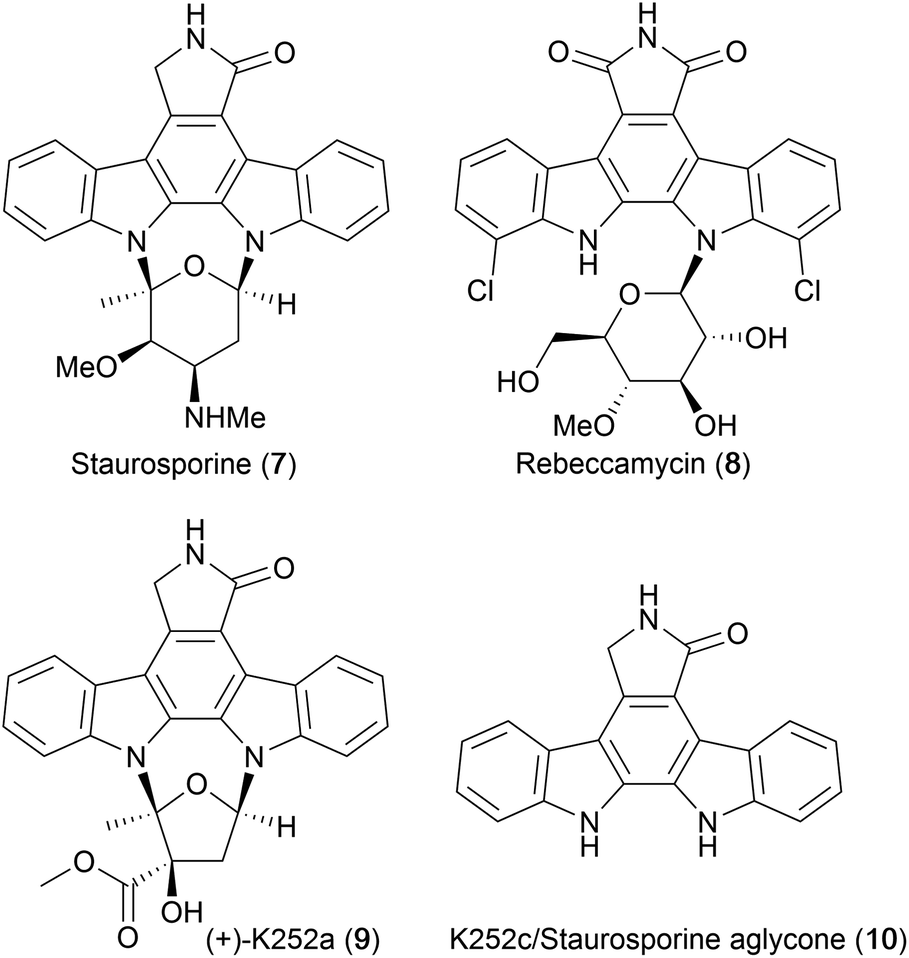 | ||
| Fig. 3 Structures of indolocarbazole alkaloids. | ||
In 1985, Clardy and co-workers isolated rebeccamycin (8),16 and later that year Sezaki and co-workers reported the discovery of another important indolocarbazole alkaloid that subsequently became known as (+)-K252a (9) when Kase and co-workers isolated several more compounds with the indolocarbazole core including the staurosporine aglycone (10).17–19 An excellent account of the discovery and isolation of members of the indolocarbazole family can be found in Knölker and Reddy's review.20
A pivotal development in the indolocarbazole alkaloid story was realisation of their potential as therapeutic agents, or as leads to develop more selective drugs. In particular, their high potency as inhibitors of protein kinase C (PKC) caused great excitement;18,21,22 K252a (9) exhibited nanomolar inhibition of PKC (IC50 = 32 nM) whilst staurosporine (7) displayed the greatest inhibitory power (IC50 = 2.7 nM).4,18,23 The PKC family of enzymes has an important role in regulation of cellular responses, which include gene expression, cell proliferation, and inflammatory response. Consequently, PKC's have been under intense investigation over many decades in order to understand their structure, molecular mechanisms and impact upon cellular function. The discovery that inhibition of PKC can lead to cancer cell apoptosis stimulated huge interest in the search for new classes of anti-cancer drugs.6,23,24 This exciting biological activity clearly heightened interest in indolocarbazoles, stimulating many synthetic endeavours that extend over four decades.
2 Syntheses of indolocarbazoles not functionalised at indolic nitrogen
2.1 Staurosporine aglycone (staurosporinone, 10)
The first indolocarbazoles covered herein are the natural products devoid of functionalisation at indolic nitrogen. One of the most important is the staurosporine aglycone (10), also commonly referred to as K252c and staurosporinone. The first synthesis was described in 1983 by Winterfeldt and co-workers (Scheme 1).25 The synthesis began with acylation of tryptamine (12) to give amide 13. Oxidation of both benzylic positions with DDQ followed by a chemoselective reduction of the α-ketoamide 14 gave hydroxyketone 15. Acylation was accompanied with cyclisation to the pentaacetate 16, which only underwent reductive elimination to the desired lactam 17 in appreciable yields when treated with TiCl3.26 Subsequent deacylation and 6π photochemical cyclisation, with oxidative aromatisation occurring under the reaction conditions, furnished aglycone 10 in moderate yield. Winterfeldt's application of the photocyclisation approach is significant, as it paved the way for many subsequent synthetic endeavours.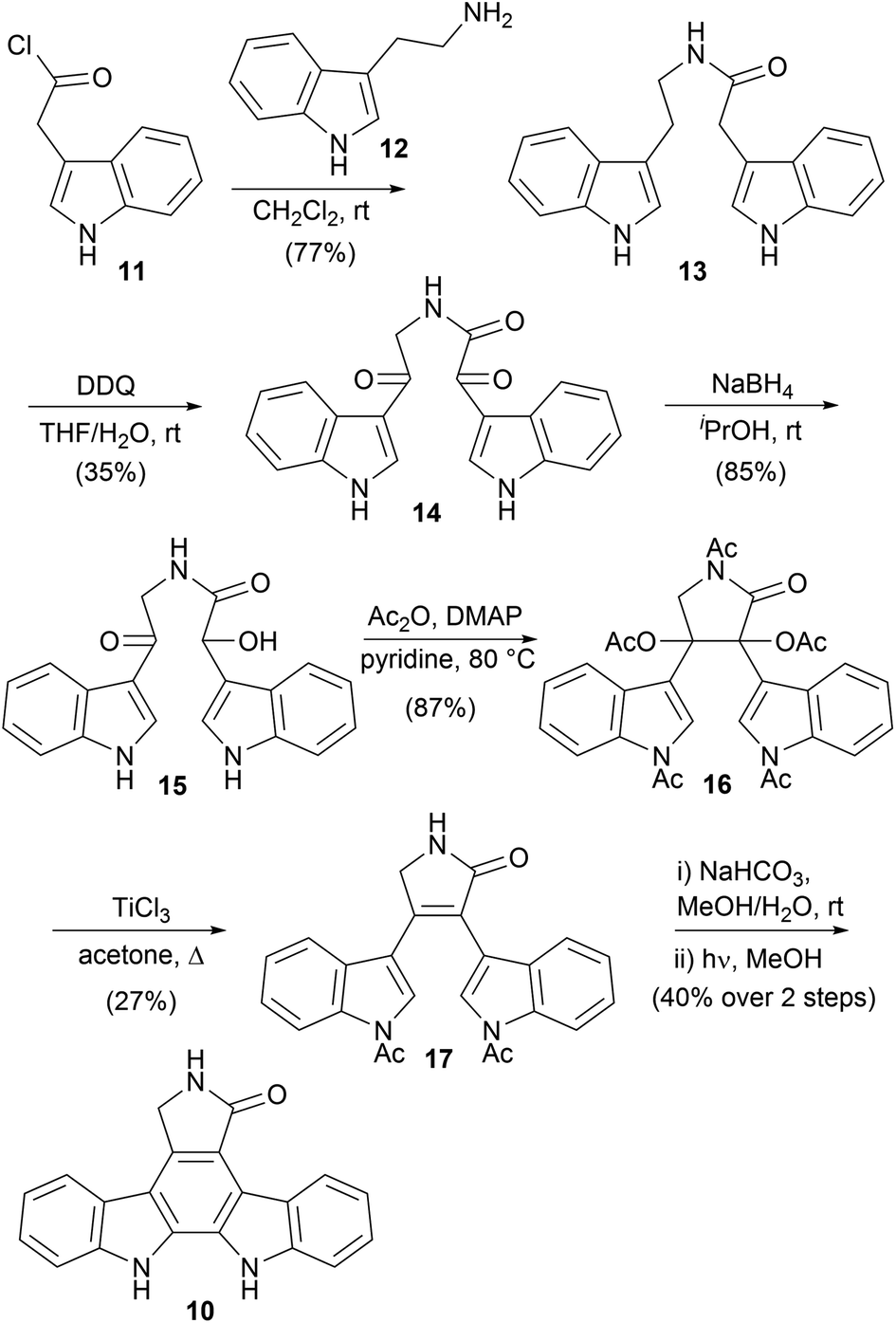 | ||
| Scheme 1 Winterfeldt and co-workers' synthesis of staurosporine aglycone (10). | ||
Discovery of important biological activity exhibited by these natural products sparked a dramatic upsurge in synthetic interest.6 In 1983, Raphael and co-workers developed a Diels–Alder/nitrene insertion approach to aglycone 10 (Scheme 2). Frustratingly, an initial attempt was thwarted by failure to cleave N-benzyl protection from the aglycone.27 However, a synthesis of the unprotected aglycone 10 was ultimately published in 1990,28 commencing with a Wittig reaction between 2-nitrocinnamaldehyde (18) and phosphonium salt 19 to give butadiene 20. The ensuing Diels–Alder reaction with dimethyl acetylenedicarboxylate gave 1,4-cyclohexadiene 21, which was subjected to dehydrogenative aromatisation and annulation to give phthalic anhydride 22. Aqueous ammonia treatment of 22 afforded maleimide 23, before two-step reduction to lactam 24.29–31 The lactam N–H was then THP protected before application of Cadogan's deoxygenative nitrene insertion, a strategy subsequently exploited in many indolocarbazole syntheses, secured aglycone 10 after acid cleavage of the lactam protection.32
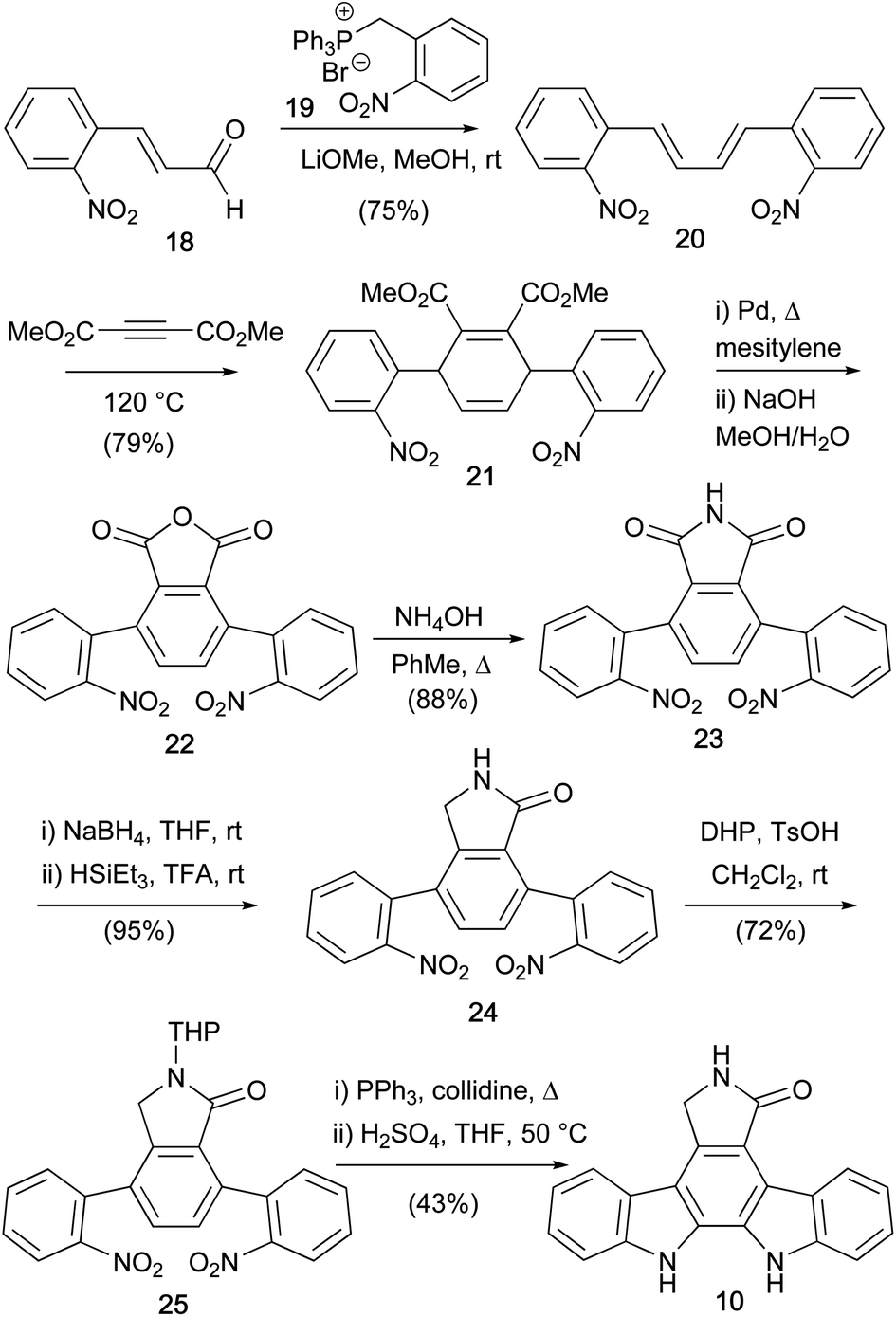 | ||
| Scheme 2 Raphael and co-workers' synthesis of staurosporine aglycone (10). | ||
Magnus and co-workers reported the synthesis of a selectively mono-protected staurosporine aglycone 34 in 1984, which is of significance as it has the potential to allow regiocontrolled functionalisation (Scheme 3).33 Phthalimido tryptamine derivative 26 was protected at its indole nitrogen,34 followed by formylation of C2 to afford 2-formyl indole 28. Condensation with 2-aminostyrene preceded the key intramolecular Diels–Alder cycloaddition, which secured pentacyclic carbamate 30. Subsequent oxidative aromatisation using DDQ gave indolocarbazole 31. The phthalimido protecting group was removed using hydrazine hydrate before chloroformylation/cyclisation with phosgene gave protected indolocarbazole 33. Finally, reductive cleavage of the p-methoxyphenylsulfonyl group under Birch conditions delivered a mono-protected staurosporine aglycone 34.
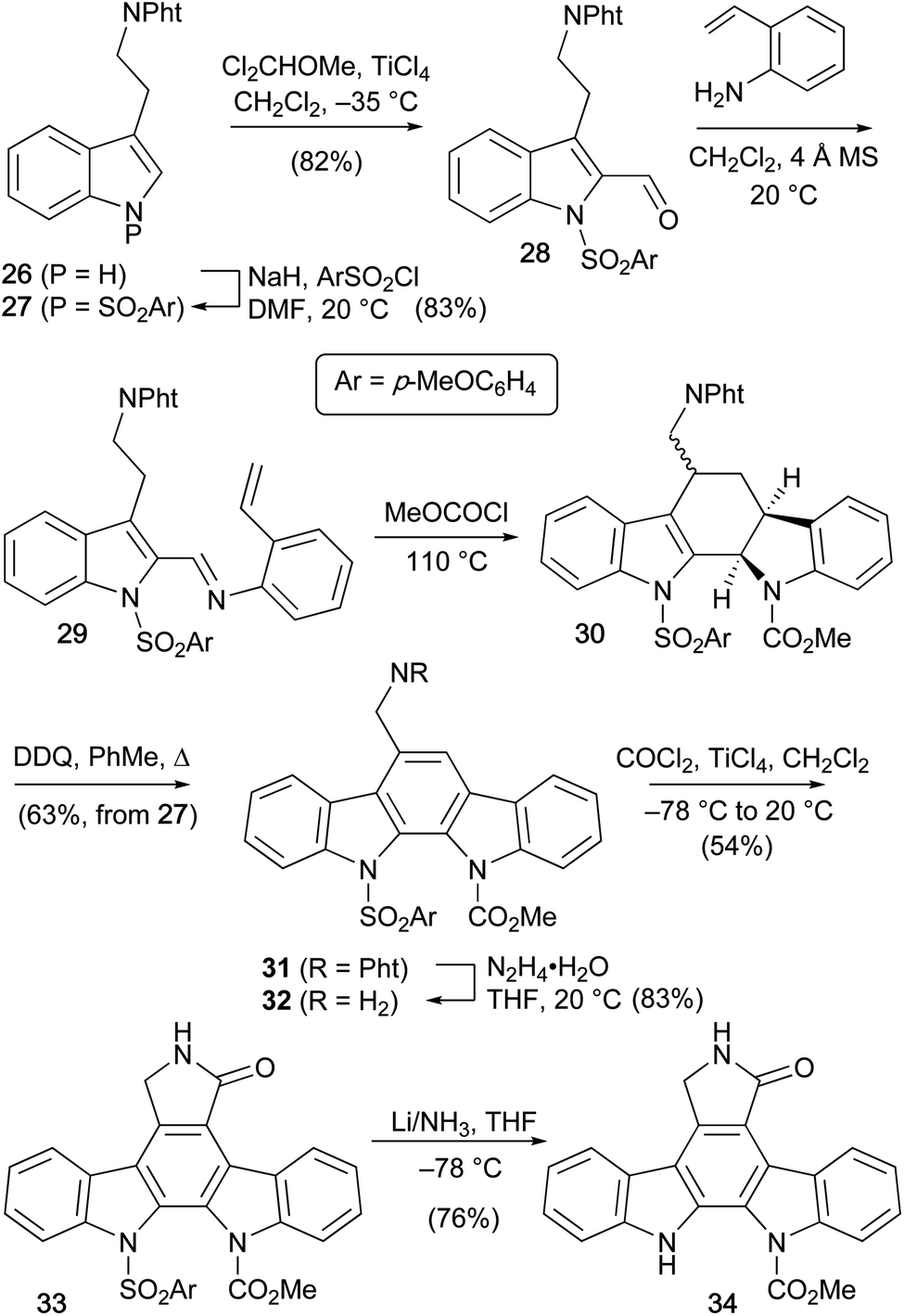 | ||
| Scheme 3 Magnus and co-workers' synthesis of a protected staurosporine aglycone 34. | ||
In 1987, as part of investigations towards the aglycone and monosaccharide moieties of staurosporine (7), Weinreb and co-workers utilised a modification of methodology developed by Steglich and co-workers to access N-benzyl staurosporine aglycone 38 (Scheme 4).35–37 Dibromomaleimide (35) was N-benzylated, then treatment with an excess of magnesiated indole gave N-benzyl bisindolylmaleimide (36). Oxidative cyclisation using TsOH/DDQ and subsequent Clemmensen reduction afforded the targeted N-benzyl staurosporine aglycone 38. The approach to the protected aglycone is notable in its brevity (4 steps from 35) and efficiency, and provided the basis for further optimisation by others.
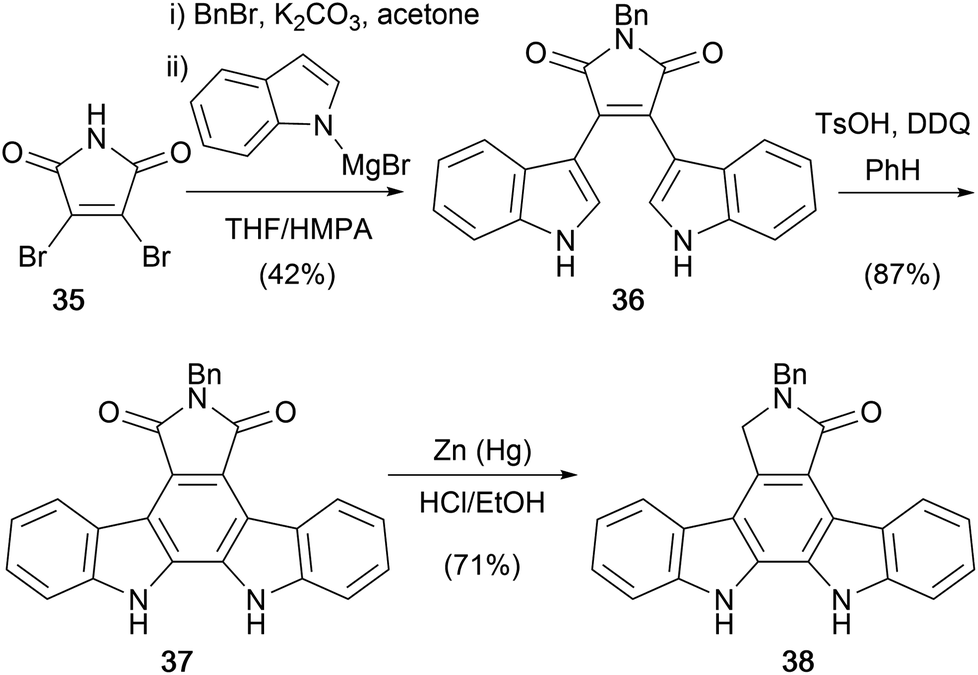 | ||
| Scheme 4 Weinreb and co-workers' synthesis of protected staurosporine aglycone 38. | ||
The first protecting group free synthesis of the aglycone 10, was completed by Moody and co-workers in 1990 (Scheme 5).38,39 The approach centred on intramolecular Diels–Alder decarboxylation sequence from pyranone 42, prepared in 6 steps from the commercially available enal 18.40,41 Allylic amine fragment 39 was prepared by Luche reduction of 18, then coupling of the resulting cinnamyl alcohol with phthalimide under Mitsonobu conditions and hydrazinolysis. Friedel–Crafts acylation of indole 40 with oxalyl chloride, and trapping with allylic amine 39, provided α-ketoamide 41. Ester hydrolysis and cyclisation using acetic anhydride gave the desired pyranone 42, which upon refluxing in BnBr open to air, forged the carbazole system in 43. Adoption of Raphael's deoxygenative nitrene insertion approach completed the staurosporine aglycone (10). The approach (8% over 9 steps from enal 18), presents carbazole 43 as a suitable intermediate for regioselective functionalisation towards staurosporine (7). However, some limitations may arise due to the rather forcing thermal conditions of nitrene insertion.
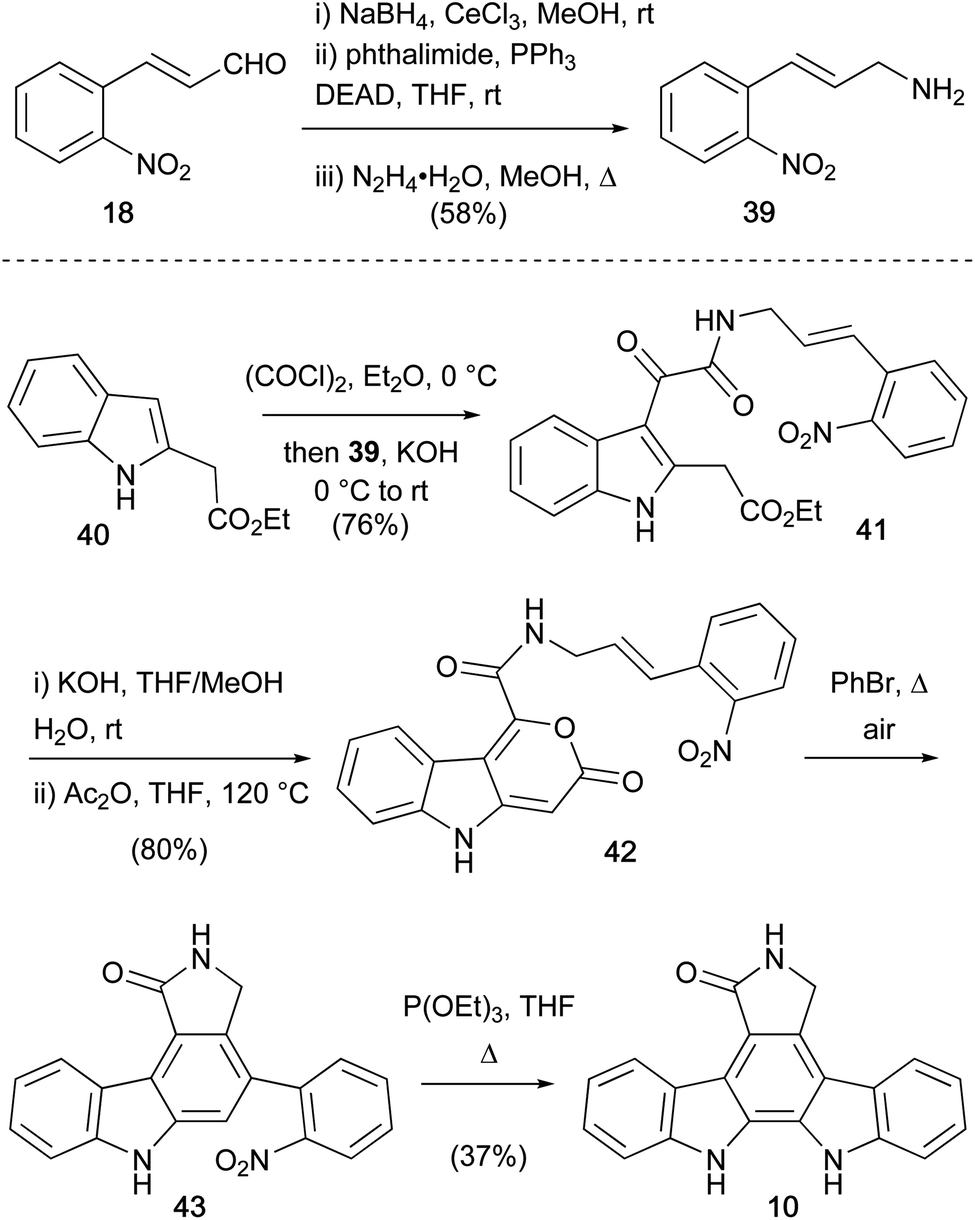 | ||
| Scheme 5 Moody and co-workers' synthesis of the staurosporine aglycone (10). | ||
Hill and co-workers from Roche, built upon methodology developed by Steglich and later Weinreb, coupling dibromomaleimide (35) with magnesiated indole to obtain bisindolylmaleimide (44) (Scheme 6).42 Cyclisation of 44 using TsOH/DDQ proved challenging, while a novel Pd(OAc)2 mediated oxidative cyclisation gave arcyriaflavin A (45). Reduction of the imide to hydroxylactam 46, and subsequent hydrogenolysis gave staurosporine aglycone (10) in only 4 synthetic operations, and without recourse to N-protection, constituting the shortest route at the time of publication.
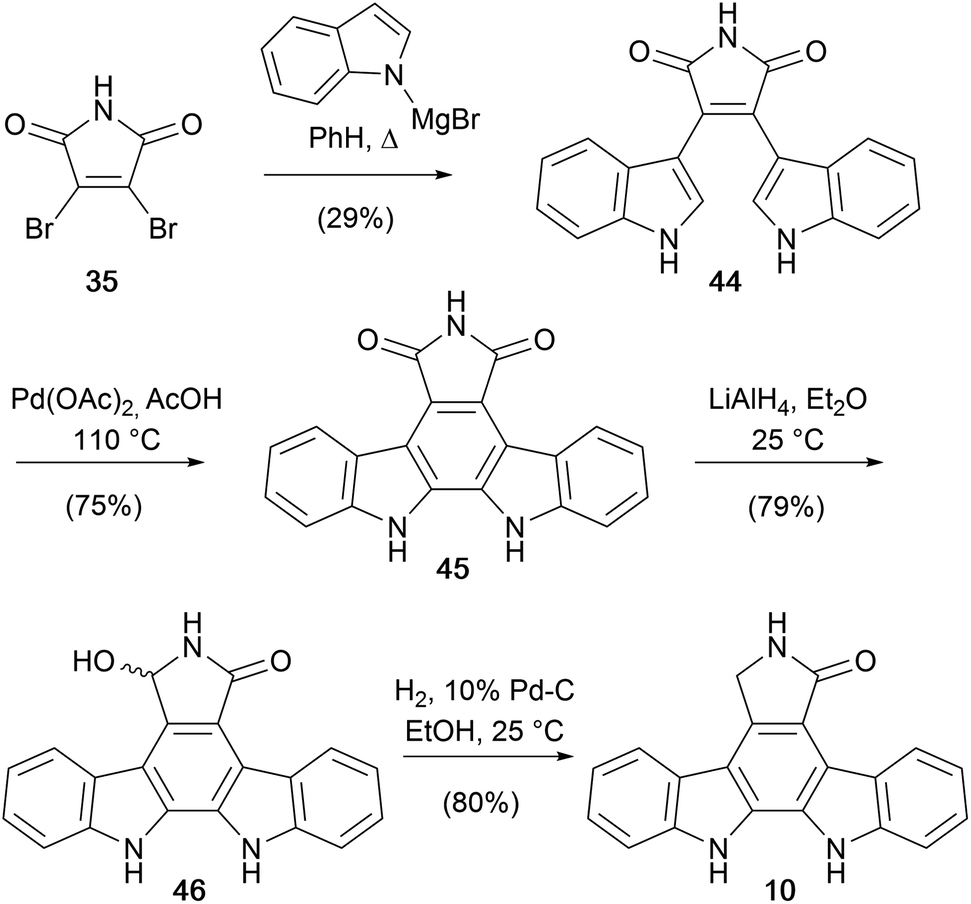 | ||
| Scheme 6 Hill and co-workers' synthesis of staurosporine aglycone (10). | ||
Lown and co-workers went on to disclose a further adaptation of the dibromomaleimide route in 1994, introducing an oxidative photocyclization step to form the pentacycle (Scheme 7).43N-Benzyl dibromomaleimide (48) was obtained from diacid 47, under peptide coupling conditions. The issue of N-debenzylation that thwarted earlier efforts was resolved, albeit indirectly, by conversion of the maleimide to anhydride 50 prior to oxidative photocyclisation to the indolocarbazole anhydride 51.44,4551 then delivered arcyriaflavin A (45) by aminolysis, and then Clemmensen reduction afforded aglycone 10.
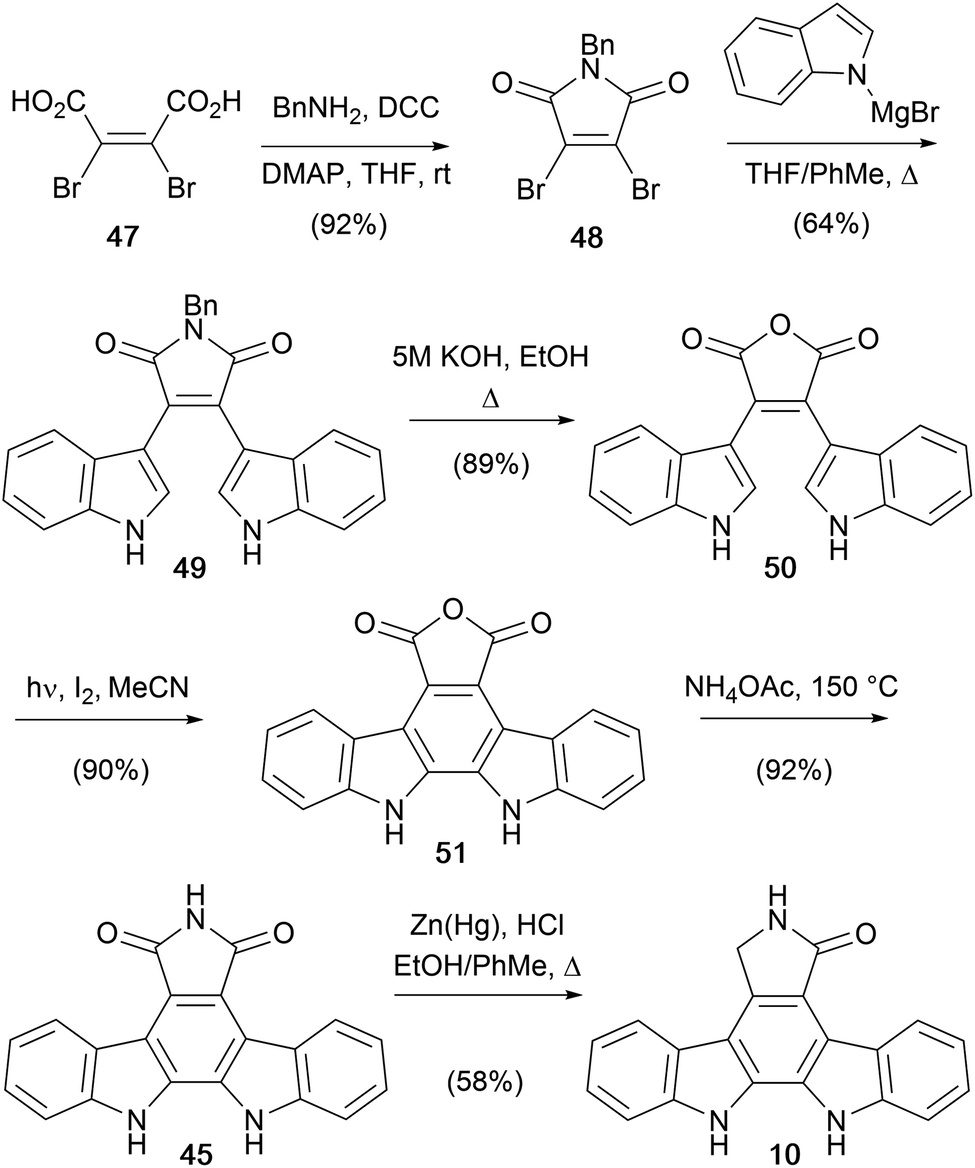 | ||
| Scheme 7 Lown and co-workers' synthesis of the staurosporine aglycone (10). | ||
With a focus directed towards scaling up the synthesis of bisindolylmaleimide (44) to meet the need of PKC-β inhibition studies,46,47 Faul and co-workers reported optimisation of Hills's route substituting 35 with dichloromaleimide.48 Using this concise and highly efficient 4 step approach, multi-gramme synthesis of staurosporine aglycone (10) was realised in 34% overall yield.
In 2000, Burtin and co-workers isolated 6-methylstaurosporinone utilising a similar approach to Faul and co-workers in 1993 (not shown).49 The route began from dichloro-N-methylmaleimide and indolylmagnesium bromide and gave the 6-methylstaurosporinone in 36% yield over 4 steps.
During expansive studies into the indolocarbazole natural product family, in 1995 Wood and co-workers disclosed a novel synthesis of staurosporine aglycone (10), which was extended to a series of protected derivatives 38, 57b–d (Scheme 8).50,51 The approach centred upon the addition of diazo-lactams 56a–e to 2,2′-biindole (53), which was prepared by double-Madelung cyclisation of diamide 46.52 The diazotetramic acids were prepared using a 4 step protocol from the N-substituted glycine esters 54a–e.53,54 Amide coupling of the esters with ethyl hydrogen malonate followed by Dieckmann cyclisation gave lactams 55a–e. The lactams were then subjected to a single pot decarboxylative diazo-transfer reaction to give diazotetramic acids 56a–e. Finally, a novel diazo-addition of lactams 56a–e into C3 of 2,2′-biindole (53) followed by a cycloaromatisation at elevated temperatures provided the staurosporine aglycone (10), and protected derivatives 38, 57b–d.
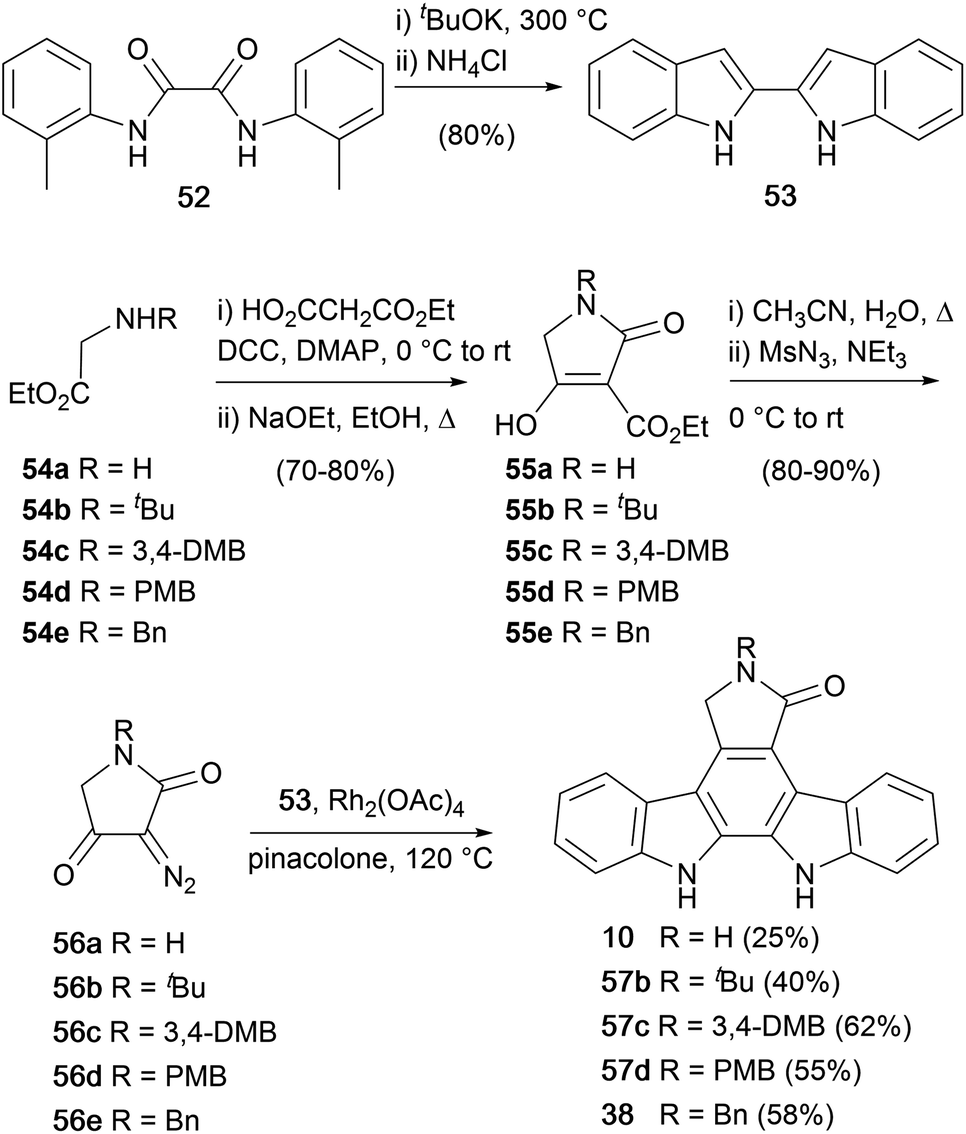 | ||
| Scheme 8 Wood and co-workers' synthesis of staurosporine aglycone (10) and protected derivatives. | ||
Faul and co-workers described their second approach to the staurosporine aglycone (10) in 1998 (Scheme 9).55 The key step was the coupling to access amide 61 and ensuing Perkin-type condensation to bisindolylmaleimide (44). This was then subjected to the oxidative cyclisation, reduction, hydrogenation sequence described by Hill and co-workers to give staurosporine aglycone (10). This synthesis represents the highest yielding (36% over 5 steps) synthesis of the aglycone 10 to date, and became a popular approach to obtain bisindolylmaleimide and arcyriaflavin A analogues.
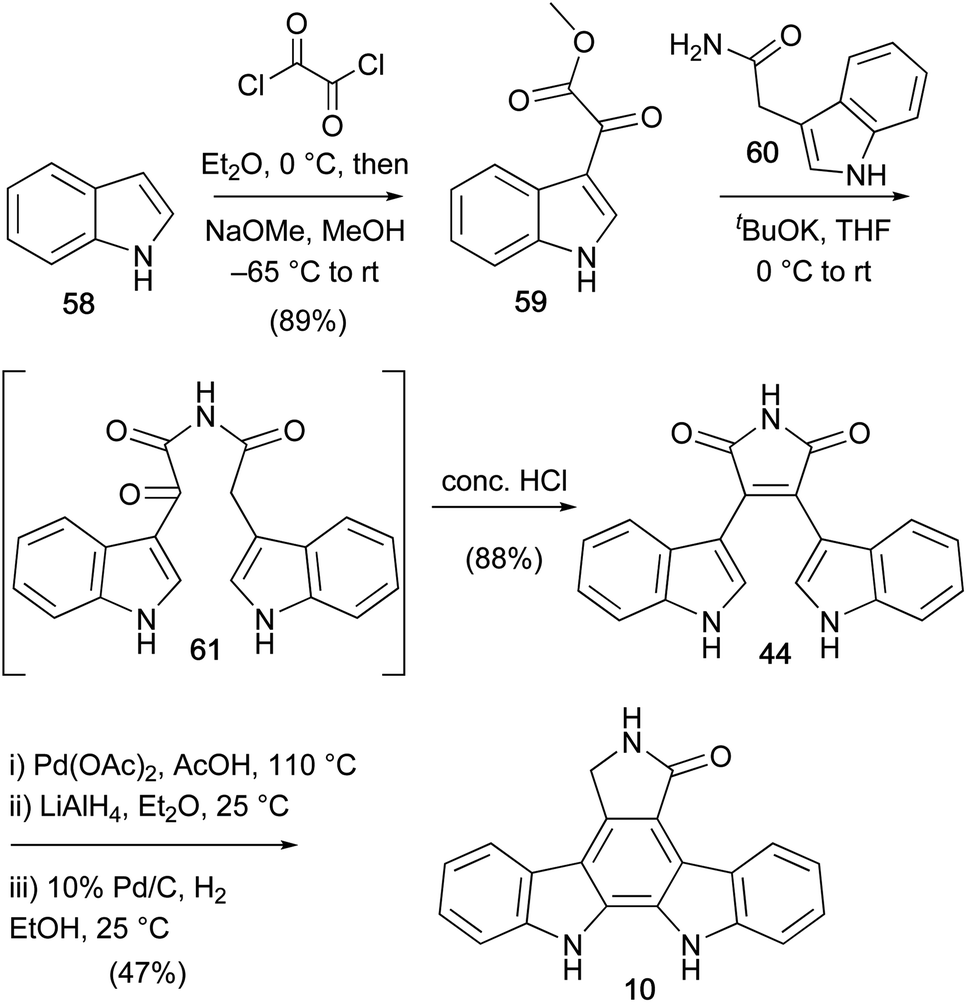 | ||
| Scheme 9 Faul and co-workers' approach to the staurosporine aglycone (10). | ||
In 1998, Beccalli and co-workers disclosed an original synthesis of staurosporine aglycone (10) from a readily accessible indole derivative 62 (Scheme 10).56,57 Treatment with ethyl chloroformate gave the protected indole derivative 63 which was selectively deprotected to give enol 64. This was converted to the corresponding vinyl triflate 65 and subsequent Stille coupling with stannane 66 secured bisindole 67.58–60 Deprotection of both indole moieties was followed by an oxidative photocyclisation to give indolocarbazole 69. Finally, chemoselective reduction of the cyano group and annulation was achieved with NaBH4–CoCl2 to give the aglycone (10).
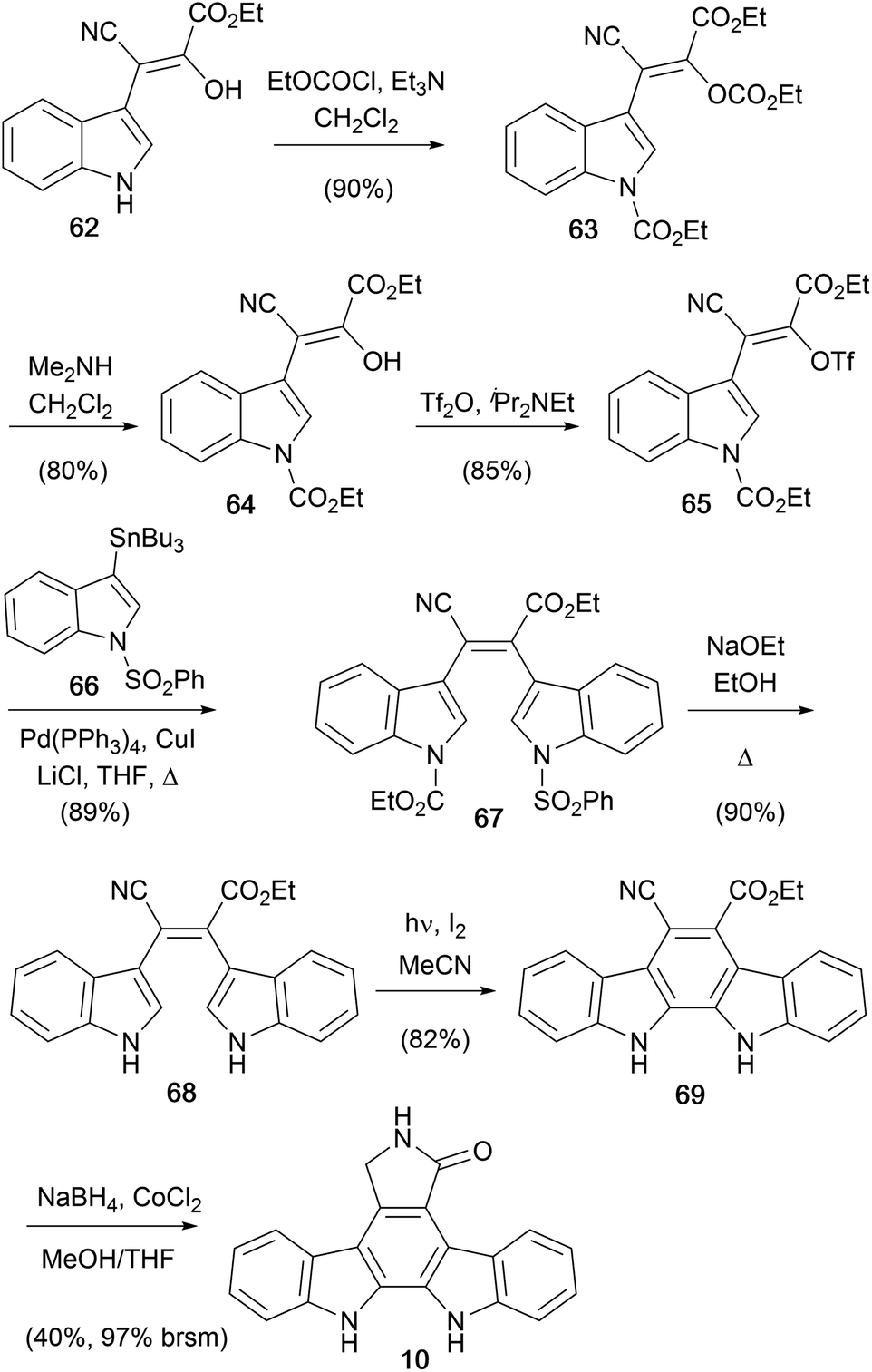 | ||
| Scheme 10 Beccalli and co-workers' synthesis of the staurosporine aglycone (10). | ||
In 1999, Mahboobi and co-workers disclosed a concise approach to the staurosporine aglycone (10) utilising a Michael addition to nitro-olefin 70 (Scheme 11).61–64 This key step provided nitrobutanoate 72 in a modest yield of 10%. Subsequent catalytic hydrogenation/lactamization furnished lactam 73 which was cyclised under oxidative conditions to the staurosporine aglycone (10).
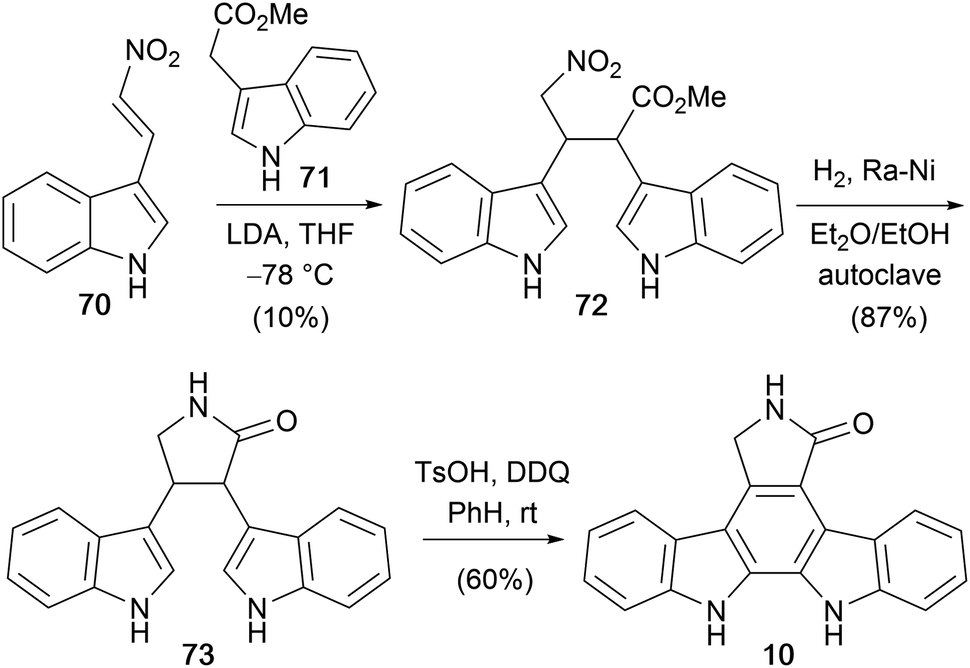 | ||
| Scheme 11 Mahboobi and co-workers' synthesis of the staurosporine aglycone (10). | ||
Inverse electron-demand Diels–Alder cycloaddition–cycloreversion reactions were at the heart of Snyder and co-workers 2001 synthesis of protected aglycone derivative 82 (Scheme 12).65 Diels–Alder cycloaddition–reversion of N-tosyl indole (74) and tetrazine 75 gave the electron deficient indole system 76.66–68
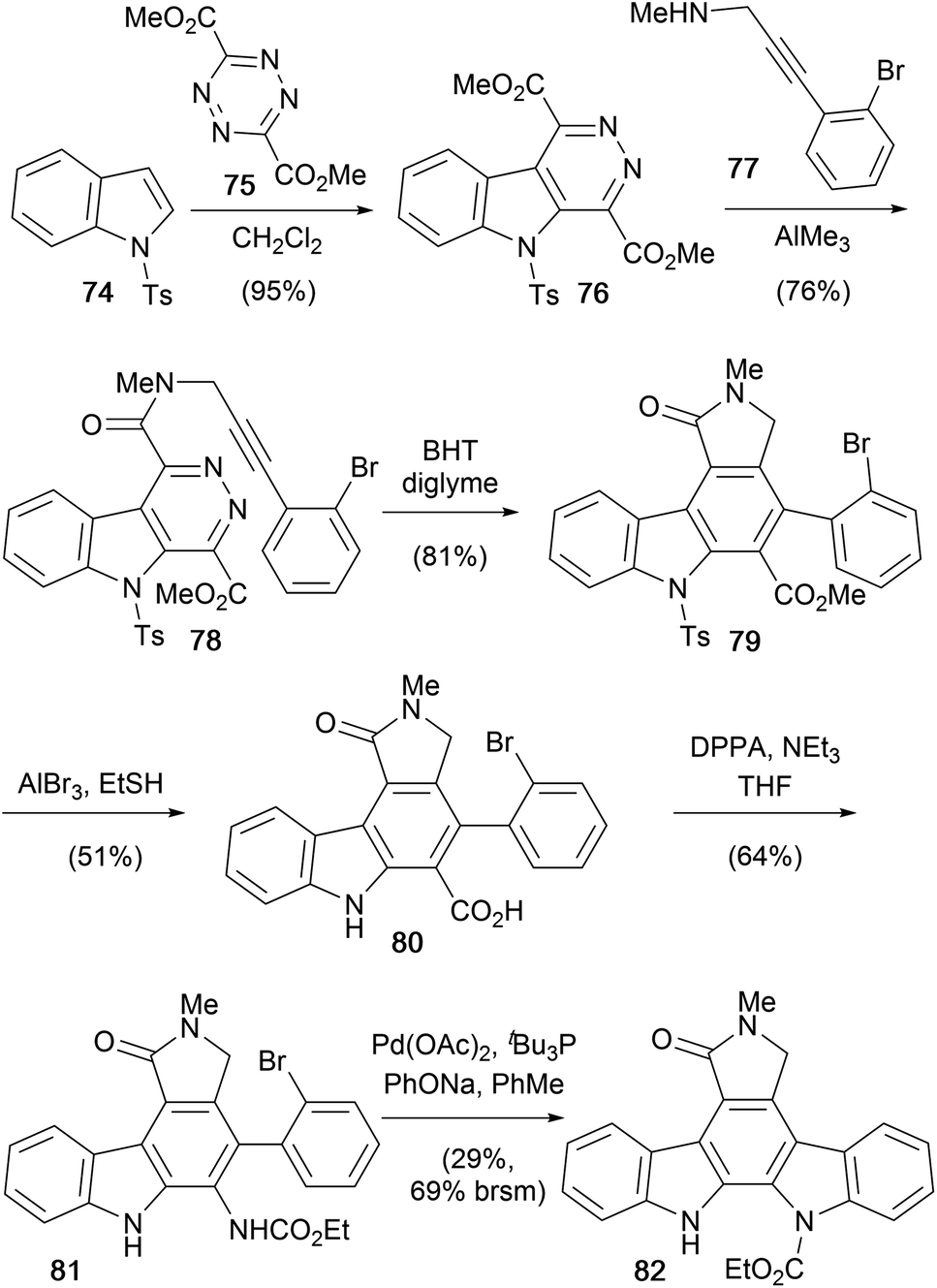 | ||
| Scheme 12 Snyder and co-workers' synthesis of protected staurosporine aglycone 82. | ||
Introduction of the acetylenic dienophile 77 was achieved by regioselective amidation of the less encumbered and more electrophilic ester, promoted by AlMe3, to give amide 78.69 Subsequent intramolecular annulation gave carbazole 79 and ensuing demethylation and carbazole deprotection secured carboxylic acid 80. Finally, Curtius rearrangement of acid 80 and Pd-catalysed amination realised protected aglycone 82.70
In 2003, Prabhakar and co-workers disclosed a short approach to staurosporine aglycone (10, Scheme 13).71 Tetramic acid 86 was prepared in two-steps from hippuric acid (83) and Meldrum's acid (84) as described by Sandris.72 2,2′-Biindole (53) was prepared according to the procedure described by Bergman (see Scheme 8), and the two were coupled to give lactam 87.52 Oxidative photocyclisation of this intermediate yielded staurosporine aglycone (10). The group subsequently published a modified approach to protected derivatives.73
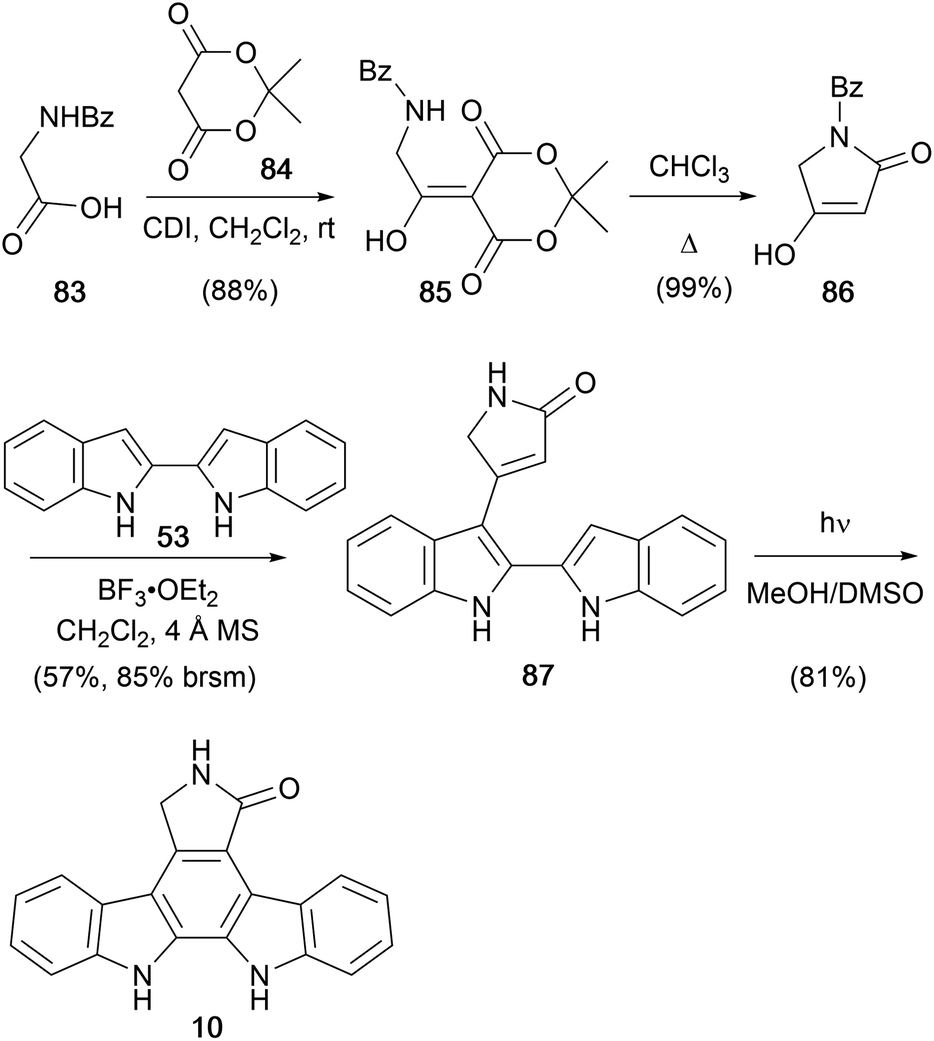 | ||
| Scheme 13 Prabhakar and co-workers' synthesis of the staurosporine aglycone (10). | ||
In the same year, Uang and co-workers reported optimisation of the photocyclisation of halogenated and methoxylated bisindolylmaleimide derivatives. Photocyclisation was performed in THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN (1:1) with catalytic I2, and was applied to the synthesis of aglycone (10).74 Electron-withdrawing groups were found to reduce irradiation times, however on multi-gram scale (5 g), 96 h of irradiation with two 400 W mercury lamps was required for full conversion of bisindolylmaleimide (44) to arcyriaflavin A (45).
:MeCN (1:1) with catalytic I2, and was applied to the synthesis of aglycone (10).74 Electron-withdrawing groups were found to reduce irradiation times, however on multi-gram scale (5 g), 96 h of irradiation with two 400 W mercury lamps was required for full conversion of bisindolylmaleimide (44) to arcyriaflavin A (45).
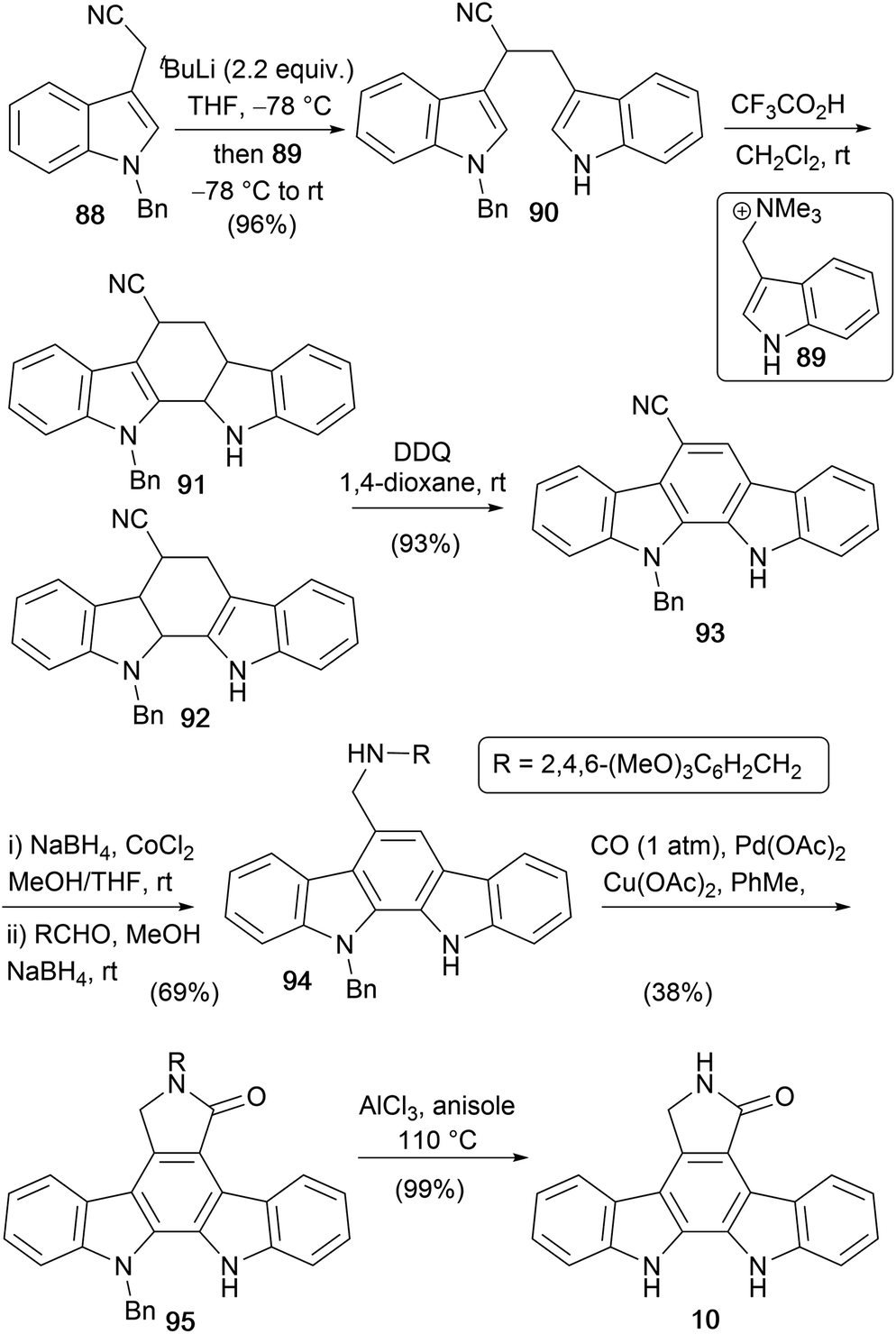
In 2007, Orito and co-workers described a new route to aglycone 10 from 3-(N-benzylindolyl)acetonitrile (88, Scheme 14).75 Coupling 88 with the carbocation precursor ammonium salt 89 gave bisindole 90, which was subjected to acid-mediated cyclisation and subsequent DDQ oxidation to give indolocarbazole 93.76–79 The cyano group was reduced before a 2-step reductive alkylation with 2,4,6-trimethoxybenzaldehyde gave amine 94.80 This was subjected to Pd-catalysed carbonylation, before AlCl3 deprotection of the two benzyl-type protecting groups gave staurosporine aglycone (10).
 | ||
| Scheme 14 Orito and co-workers' synthesis of the staurosporine aglycone (10). | ||
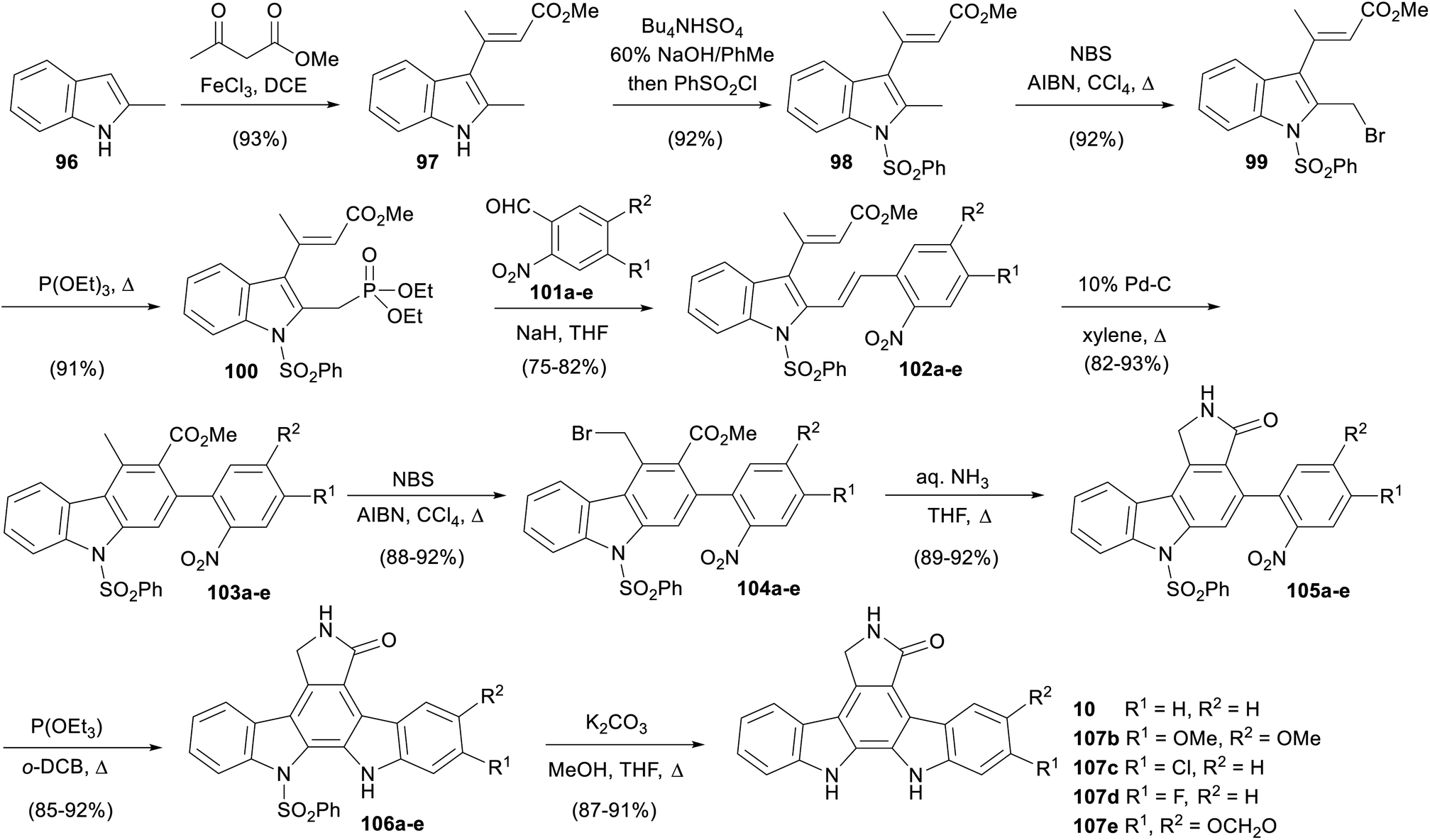
In 2011, Mohanakrishnan and co-workers published a route to various aglycone derivatives starting from 2-methylindole (96, Scheme 15).81 Condensation of indole 96 with methyl acetoacetate gave vinylindole 97 then N-sulfonylation afforded protected indole 98.82 Benzylic bromination preceded the Michaelis–Arbuzov reaction with P(OEt)3 to give phosphonate ester 100 upon workup. This was subjected to a Wittig–Horner reaction with a variety of 2-nitroarylaldehydes 101a–e to give divinylindoles 102a–e, which underwent electrocyclisation/aromatisation with Pd–C to afford carbazoles 103a–e.83 Benzylic bromination and subsequent reaction with aqueous ammonia gave lactams 105a–e. Triethyl phosphite mediated nitrene insertion effected the final ring closure, before deprotection of the phenylsulfonyl group secured staurosporine aglycone (10), as well as derivatives 107b–e. The same group subsequently reported extending the approach to synthesise a variety of aglycone analogues.84
 | ||
| Scheme 15 Mohanakrishnan and co-workers' synthesis of staurosporine aglycone derivatives. | ||
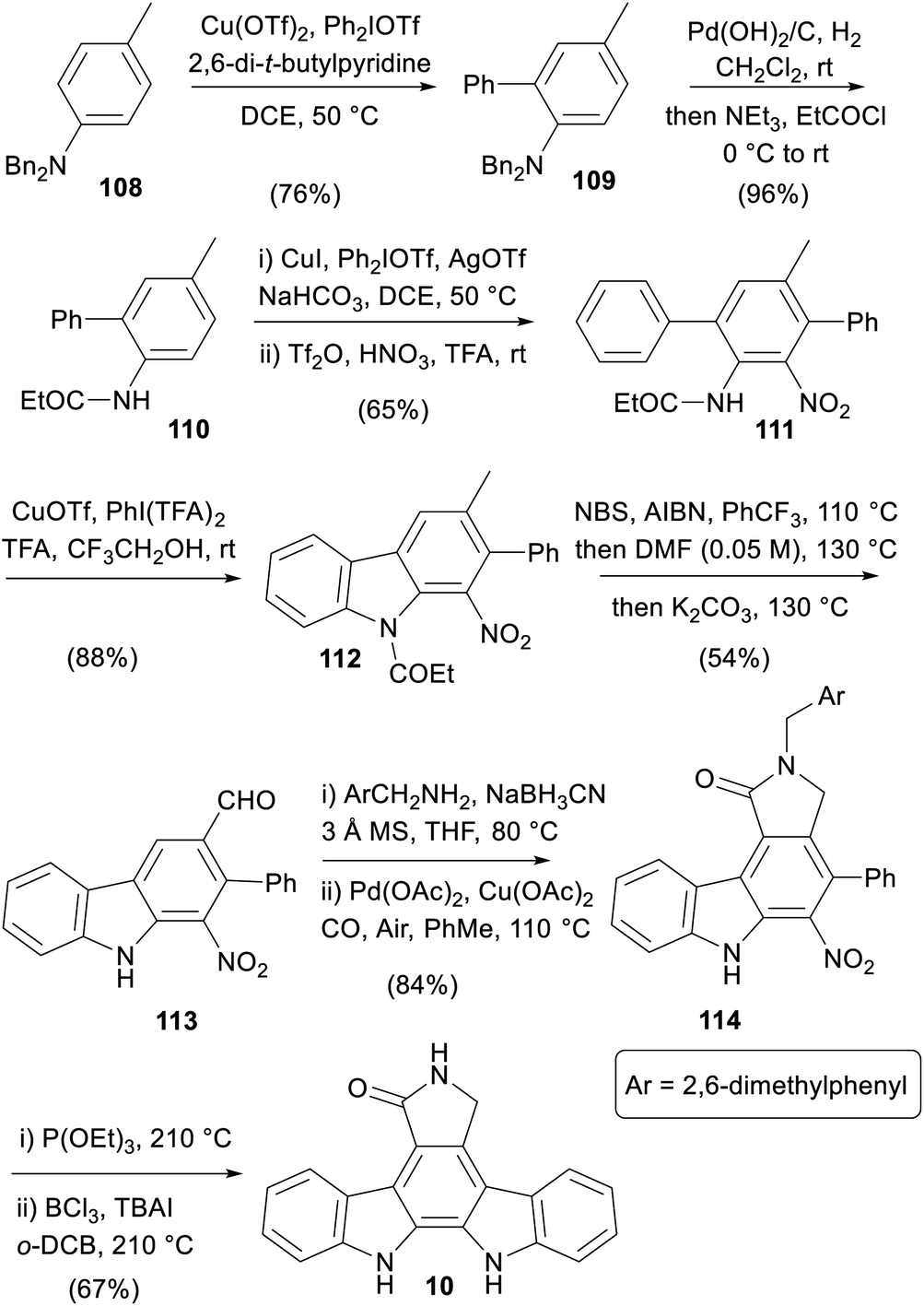
An approach centred on strategic C–H functionalisations was published in 2016 by Gaunt and co-workers, which exploited the amine moiety in the central ring to direct four out of the seven key transformations (Scheme 16).85
 | ||
| Scheme 16 Gaunt and co-workers' synthesis of the staurosporine aglycone (10). | ||
Ortho-arylation of aniline 108 was followed by hydrogenolysis and carbamoylation to switch the directing nature of the group and give anilide 110.86,87 A subsequent meta-arylation followed by nitration gave the nitro-arene 111, which underwent an oxidative cyclisation to carbazole 112.88 A one-pot radical bis-bromination, formylation and concomitant cleavage of the carbazole N-protection secured aldehyde 113.89 Reductive amination, and C–H carbonylation protocol gave γ-lactam 114, which was subjected to Cadogan nitrene insertion then N-debenzylation to form staurosporine aglycone (10).
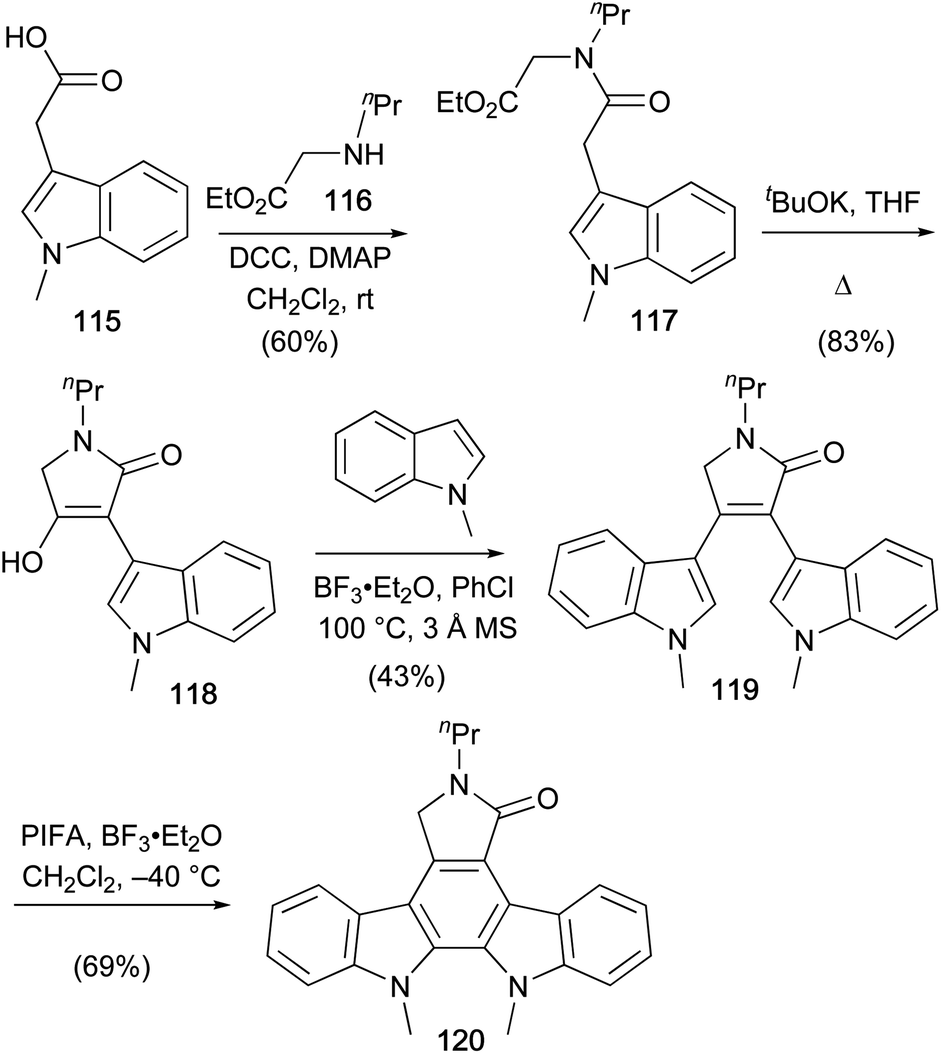
A recent synthesis of a staurosporine aglycone derivative was described by Pelkey and co-workers in 2016 (Scheme 17).90N-Methylindole-3-acetic acid (115) was coupled with amine 116 and the resulting amide cyclised to tetramic acid 118.71,91 Lewis-acid mediated arylation with N-methylindole gave lactam 119 which was cyclised to staurosporine aglycone derivative 120 using Scholl-type oxidative conditions.92
 | ||
| Scheme 17 Pelkey and co-workers' synthesis of the protected aglycone 120. | ||
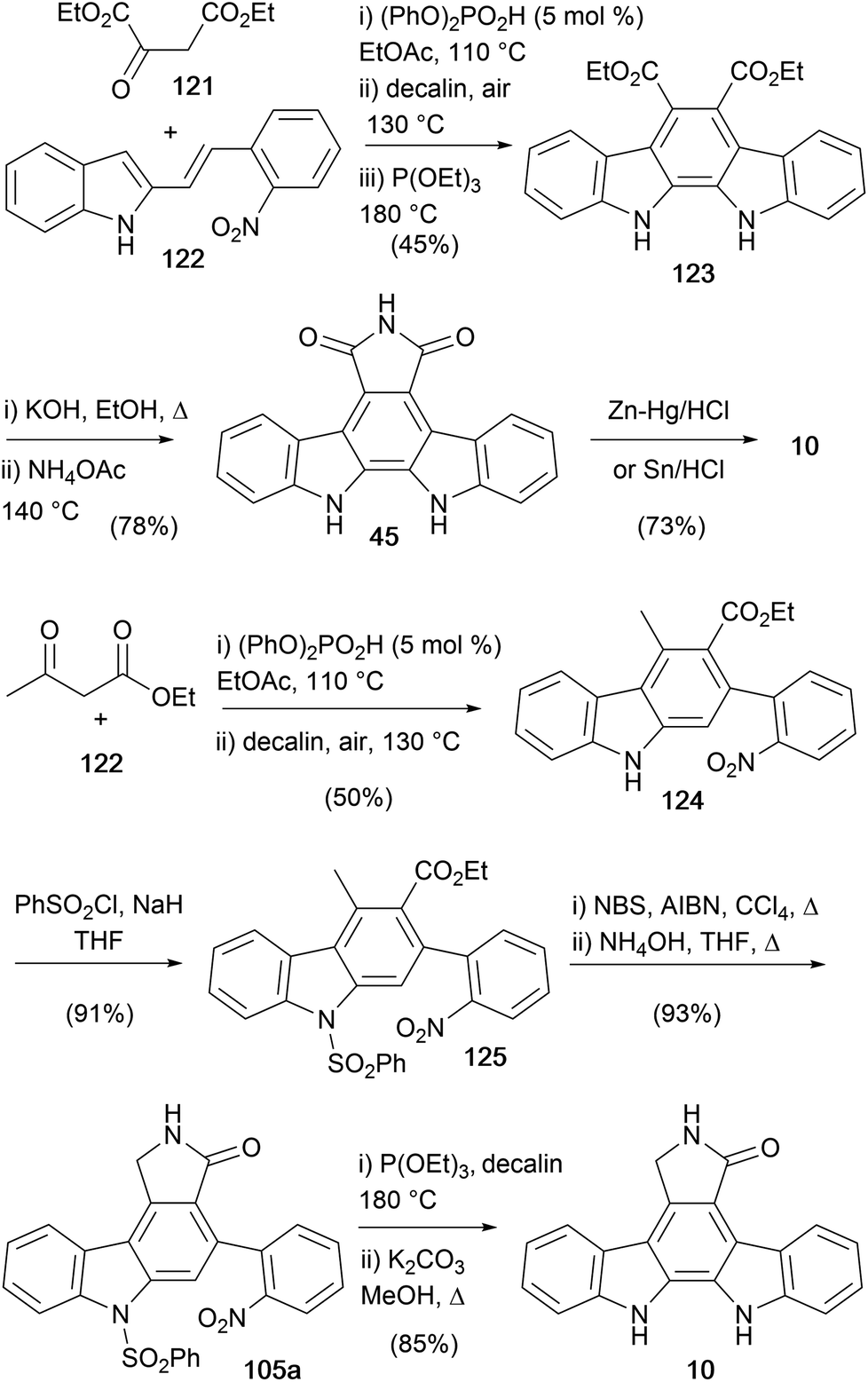
In 2018, Maji and co-workers described a route to the indolocarbazole core, leading to the synthesis of both staurosporine aglycone (10) and arcyriaflavin A (45, Scheme 18).93 The route was designed around the Brønsted acid catalysed one-pot benzannulation of 2-alkenylindoles.94–96 The first generation synthesis began by coupling diethyl 2-oxosuccinate (121) with alkenylindole 122 in the presence of phosphoric acid diphenyl ester before oxidation and Cadogan cyclisation all in one-pot gave the diester 123.32,97 This was hydrolysed before annulation with ammonium acetate gave arcyriaflavin A (45). Imide 45 was reduced via a Clemmensen reduction as described previously by Raphael and co-workers (see Scheme 4).28 In addition to this approach, the same group developed an alternate route from a similarly derived carbazole 124.93 Phenylsulfonyl protection of the indole N–H gave the intermediate carbazole 125, which was converted to staurosporine aglycone (10) using the benzylic bromination/lactamization/Cadogan cyclisation/hydrolysis process reported by Mohanakrishnan and co-workers.81
 | ||
| Scheme 18 Maji and co-workers' syntheses of the staurosporine aglycone (10). | ||
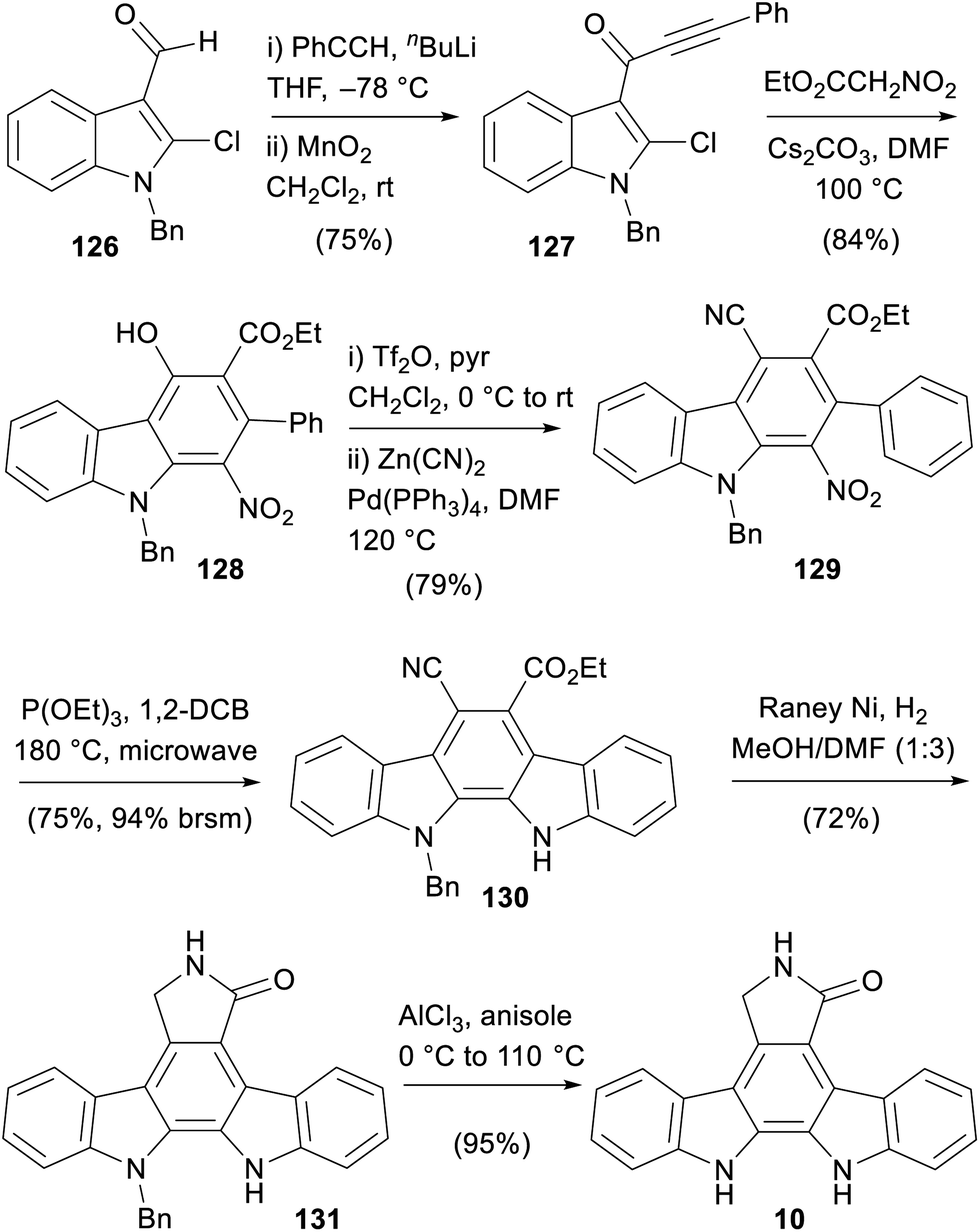
The most recent synthesis of staurosporine aglycone (9) was disclosed by Pabbaraja and co-workers in 2019 (Scheme 19).98 The synthesis began with addition of phenyl acetylene to aldehyde 126 before oxidation to ynone 127. This then underwent the key cascade upon treatment with ethyl nitroacetate to give carbazole 128.99 This was proposed to occur via initial Michael-addition and subsequent intramolecular aldol-type addition to a strained cyclobutene intermediate. A retro-nitroaldol followed by generation of an indolic iminium ion led to the final C–C bond formation with concomitant aromatisation to give the key carbazole core 128. Formation of the triflate was followed by Negishi-coupling to give cyanated nitrocarbazole 129.100,101 Cadogan cyclisation was followed by reduction to the protected aglycone 131, which was debenzylated using AlCl3 to give staurosporine aglycone (10).
 | ||
| Scheme 19 Pabbaraja and co-workers' synthesis of the staurosporine aglycone (10). | ||
2.2 Arcyriaflavin A (45)
Another sub-family of indolocarbazole natural products that don't bear any indolic nitrogen functionalisation are the arcyriaflavins. Molecularly similar to the staurosporine aglycone (10), these feature a distinctive maleimide moiety fused to the indolocarbazole core. The first of these natural products isolated were arcyriaflavin's A–C by Steglich and co-workers from the Arcyria nutans and Arcyria denudata slime moulds.35,102–104 Arcyriaflavin D was isolated from a separate slime mould Dictydiaethalium plumbeum, whilst arcyriaflavin E was isolated years later from bacterium Streptomyces cinnamoneus NBRC 13823 by Abe and co-workers (Fig. 4).105 Interest in these natural products, as with other indolocarbazoles, stems from their moderate antibiotic and antifungal properties.35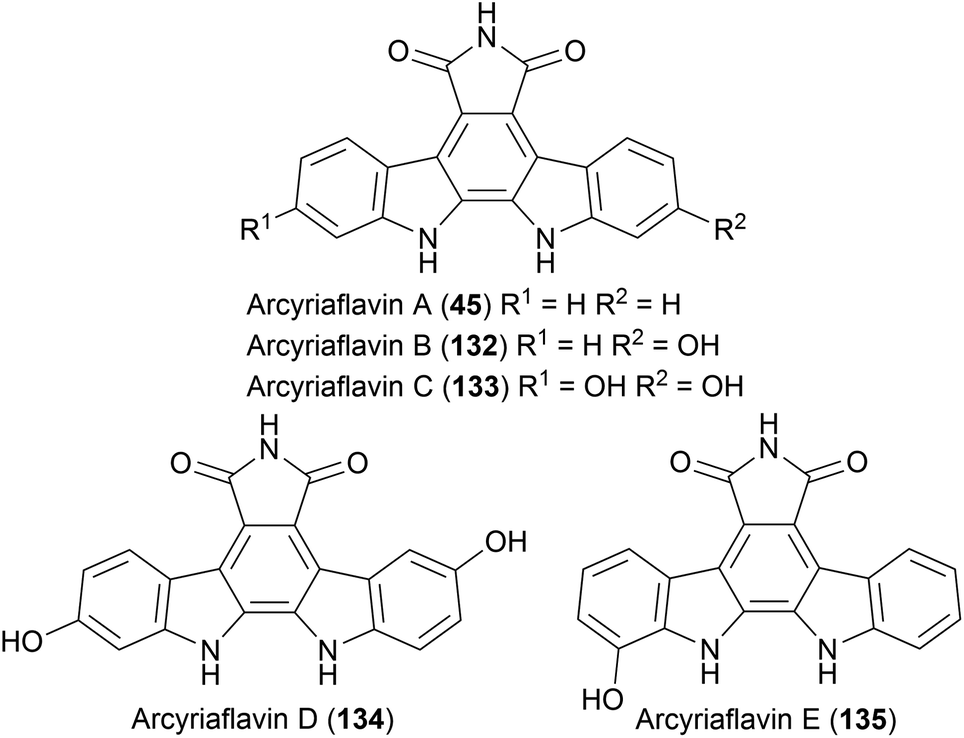 | ||
| Fig. 4 The structures of arcyriaflavins A–E. | ||
Due to the decreased molecular complexity relative to many other indolocarbazole natural products, there has been a plethora of synthetic approaches to this sub-group. In particular, many groups have utilised arcyriaflavin intermediates, particularly arcyriaflavin A (45), in the synthesis of more complex indolocarbazoles. These syntheses are covered elsewhere in this review.
The first synthesis of an arcyriaflavin A derivative was reported by Bergman and co-workers in 1987 (Scheme 20).106 The key step in their approach was oxidative coupling of the methyl indol-3-ylacetate dianon to give diester 136 as a mixture of dl and meso forms, which were easily separated by crystallisation and chromatography.107–109 Their initial approach was centred on formation of the indol-3-ylacetate trianion utilising similar chemistry, however it proved more fruitful to proceed via the dianion. The diester 136 was then subjected to imidation with benzylamine before an oxidative cyclisation gave N-benzyl protected arcyriaflavin A 37.
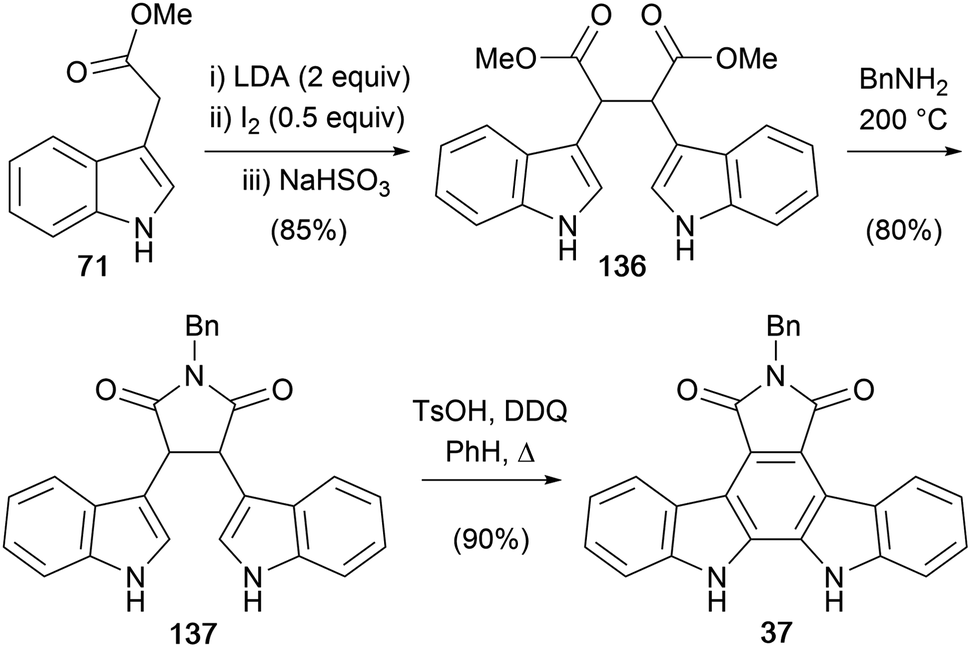 | ||
| Scheme 20 Bergman and co-workers' synthesis of the arcyriaflavin A derivative 37. | ||
In 1989, the same group developed an alternate route to a variety of arcyriaflavin derivatives, including arcyriaflavin A (45, Scheme 21).110 The approach began with a Diels–Alder cycloaddition between maleimide and butadiene 138 to access olefin 139. Condensation of the olefin 139 with 3 equiv. of phenylhydrazine gave bis(phenylhydrazone) 140. This was subjected to the key double Fischer indolisation using polyphosphoric acid trimethylsilyl ester (PPSE)111,112 before dehydrogenation of the crude mixture gave arcyriaflavin A (45).
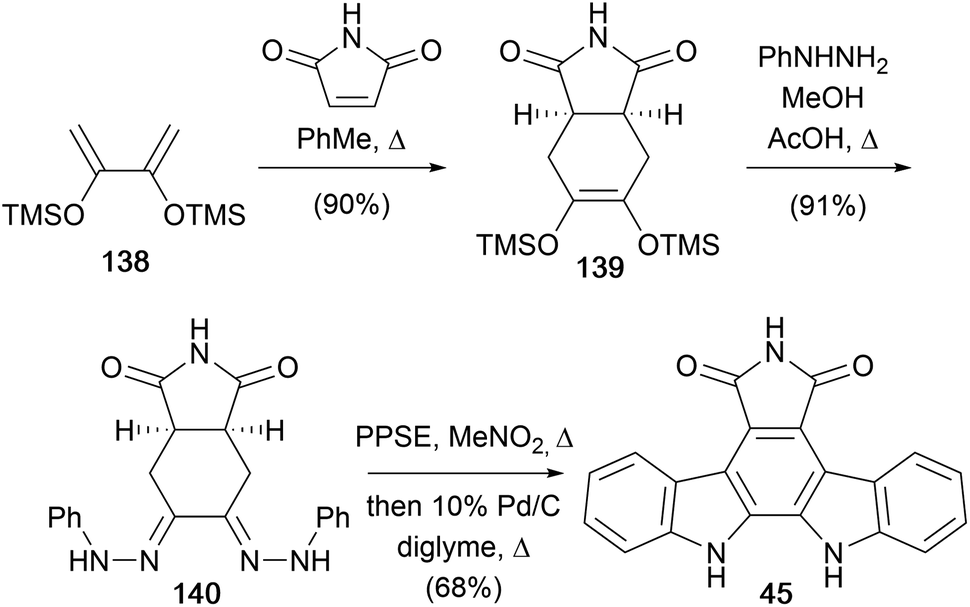 | ||
| Scheme 21 Bergman and co-workers' synthesis of arcyriaflavin (45). | ||
In 1992, Gribble and co-workers described a related approach to N-methylarcyriaflavin A (147), again utilising a Diels–Alder cycloaddition between diene 138 and N-methylmaleimide (not shown).113 Oxidation of the derived olefin with m-CPBA was followed by condensation with phenylhydrazine and the Fisher indolisation/dehydrogenation protocol as previously described by Bergman to access N-methylarcyriaflavin A (147).114
The same group developed an alternate approach starting from the commercially available cyclohexenes 141 and 142 (Scheme 22).113 The synthesis began with ozonolysis to give 1,6-dialdehydes 143 and 144, which were immediately condensed in situ with phenylhydrazine to afford the hydrazones 145 and 146 respectively. Due to instability and decomposition on attempted purification, these hydrazones were treated with PPSE without delay, giving direct access to arcyriaflavin A (45) and N-methylarcyriaflavin A (147).78 Although yields reported are modest, they represent a 3-step synthesis from commercial compounds without purification of intermediates.
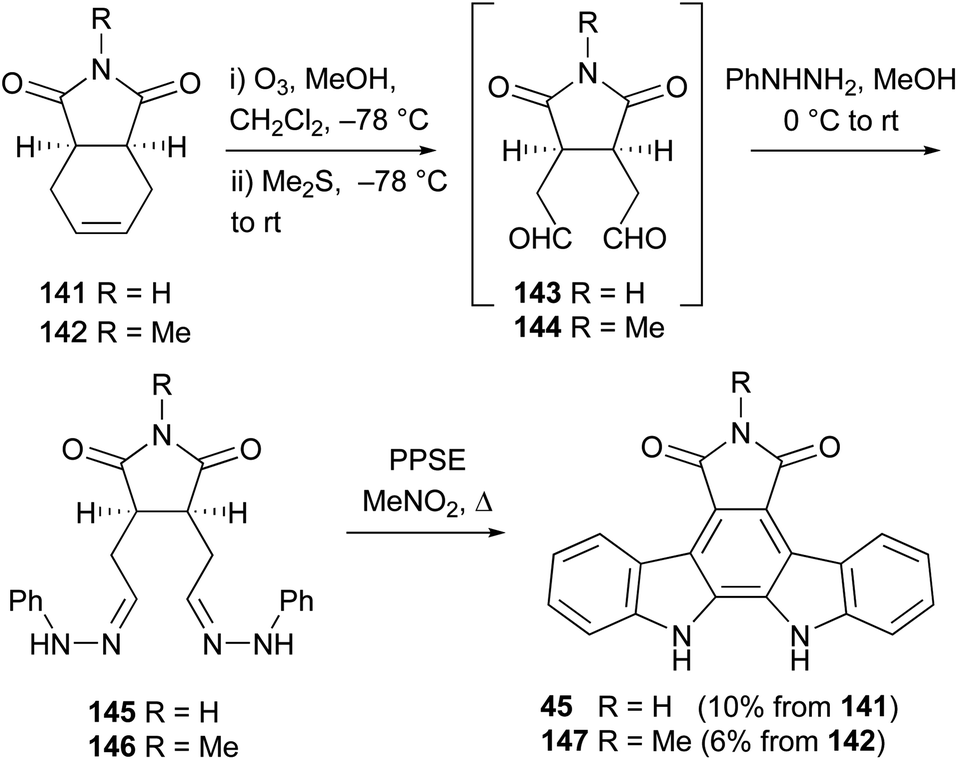 | ||
| Scheme 22 Gribble and co-workers' synthesis of arcyriaflavin A (45). | ||
A short synthesis of N-phenylarcyriaflavin A (150) was published by Somei and co-workers in 1992 (Scheme 23).115Ortho-lithiation of N-methoxyindole (148) followed by copper-mediated oxidative coupling under ultrasonic irradiation gave biindole 149. Hydrogenolysis of N–O bonds gave 2,2′-biindole (53) which underwent the final Diels–Alder cycloaddition–dehydrogenation with N-phenylmaleimide in the presence of a catalytic 10% Pd/C to afford N-phenylarcyriaflavin A (150).
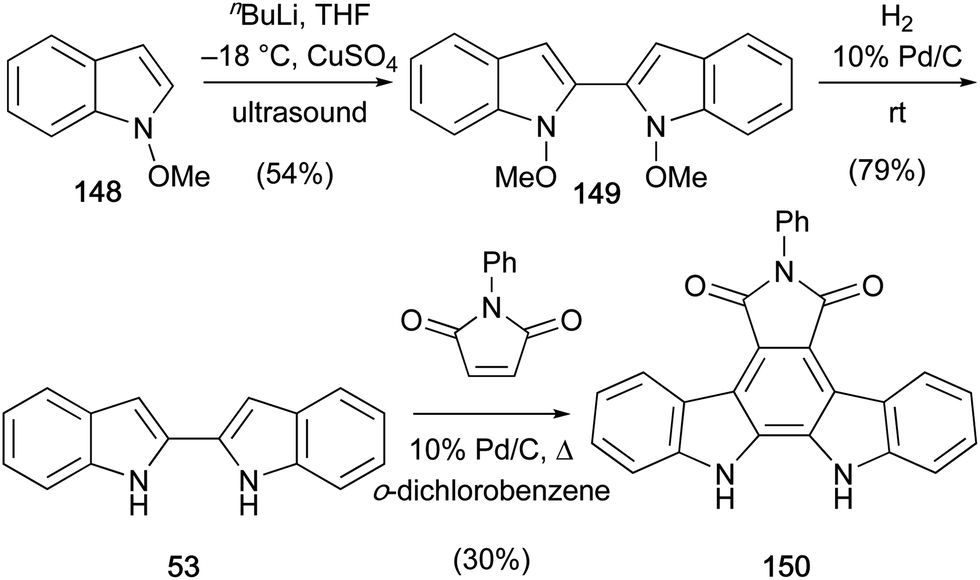 | ||
| Scheme 23 Somei and co-workers' synthesis of N-phenylarcyriaflavin A (150). | ||
Wallace and co-workers later improved the yield of the cycloaddition step to 58% using diethyl oxalate as solvent (not shown).116 In addition to this, N-methylmaleimide was also coupled with the 2,2′-biindole (53) to give N-methylarcyriaflavin A (147).
Somei and co-workers published alternative routes to protected derivatives of arcyriaflavin A (45) and staurosporine aglycone (10) later in 1995 and 2002 (not shown).117,118 The routes again proceed via 2,2′-biindole (53), with a Diels–Alder cycloaddition reaction with dimethyl acetylenedicarboxylate or N-phenylmaleimide and dehydrogenation used to form the central indolocarbazole core.
The oxidative coupling of indolyl anions using copper(II) salts was further explored by Pindur and co-workers, publishing a synthesis of N-benzylarcyriaflavin A derivative (154) in 1995 (Scheme 24).119 Lithiation of N-methylindole (151) followed by oxidative coupling gave the biindole 152.120 Interestingly, the 1,4-adduct 153 resulted from Lewis-acid treatment of biindole 152 and dimethyl acetylenedicarboxylate. Thermolysis in BnNH2 effected 6π-annulation, imidation and aromatisation together in one pot, securing arcyriaflavin A derivative 154.8
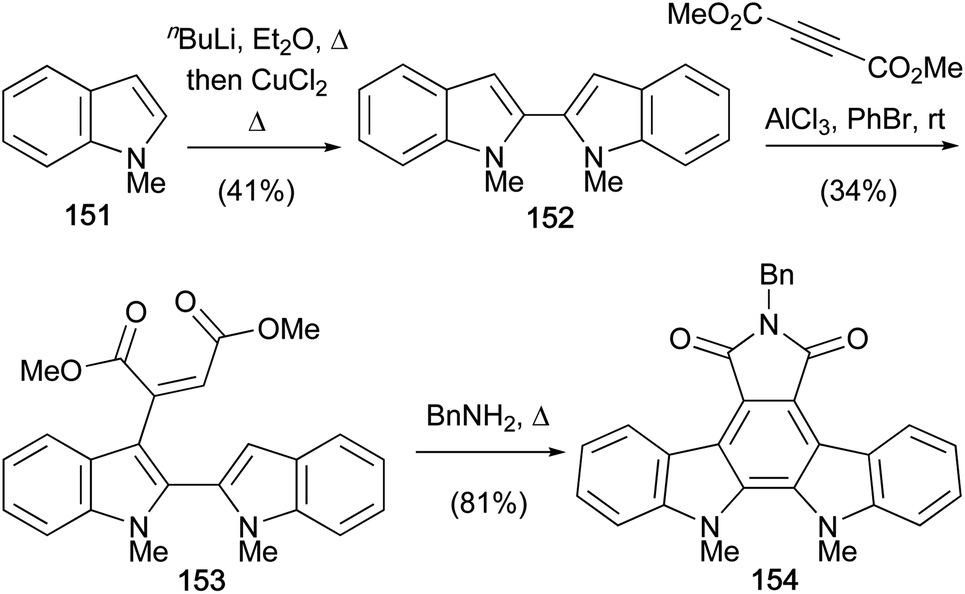 | ||
| Scheme 24 Pindur and co-workers' synthesis of arcyriaflavin A derivative 154. | ||
A Pd(0) catalysed reaction of diyne 156 and dibromomaleimide 48 was reported by Saulnier and co-workers in 1995 (Scheme 25),121 affording N-benzylarcyriaflavin A (37). After bis-trifluoroacetylation of the known diamine 155,122,123 a remarkable polyannulation cascade ensued to realise N-benzylarcyriaflavin A (37) in 52% yield.
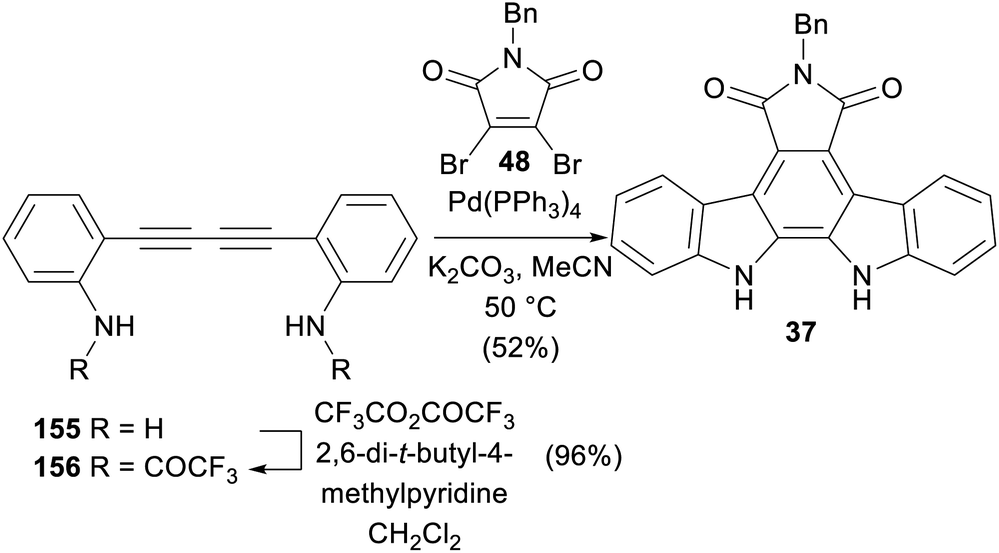 | ||
| Scheme 25 Saulnier and co-workers' synthesis of N-benzylarcyriaflavin A (37). | ||
In 1995, Lobo disclosed a novel approach to arcyriaflavin A (52) utilising a sulphur-extrusion strategy (Scheme 26).124 Coupling of readily accessible thiol 157 and dichloromaleimide (158) gave bis-sulphide 159, which when treated with PdCl2 and Hünig's base at elevated temperatures, followed by reductive workup afforded arcyriaflavin A (45).125 The mechanism proposed by the group involved formation of the C2–C2′ bond via an organopalladium intermediate, an intramolecular Diels–Alder followed by sulphur extrusion.
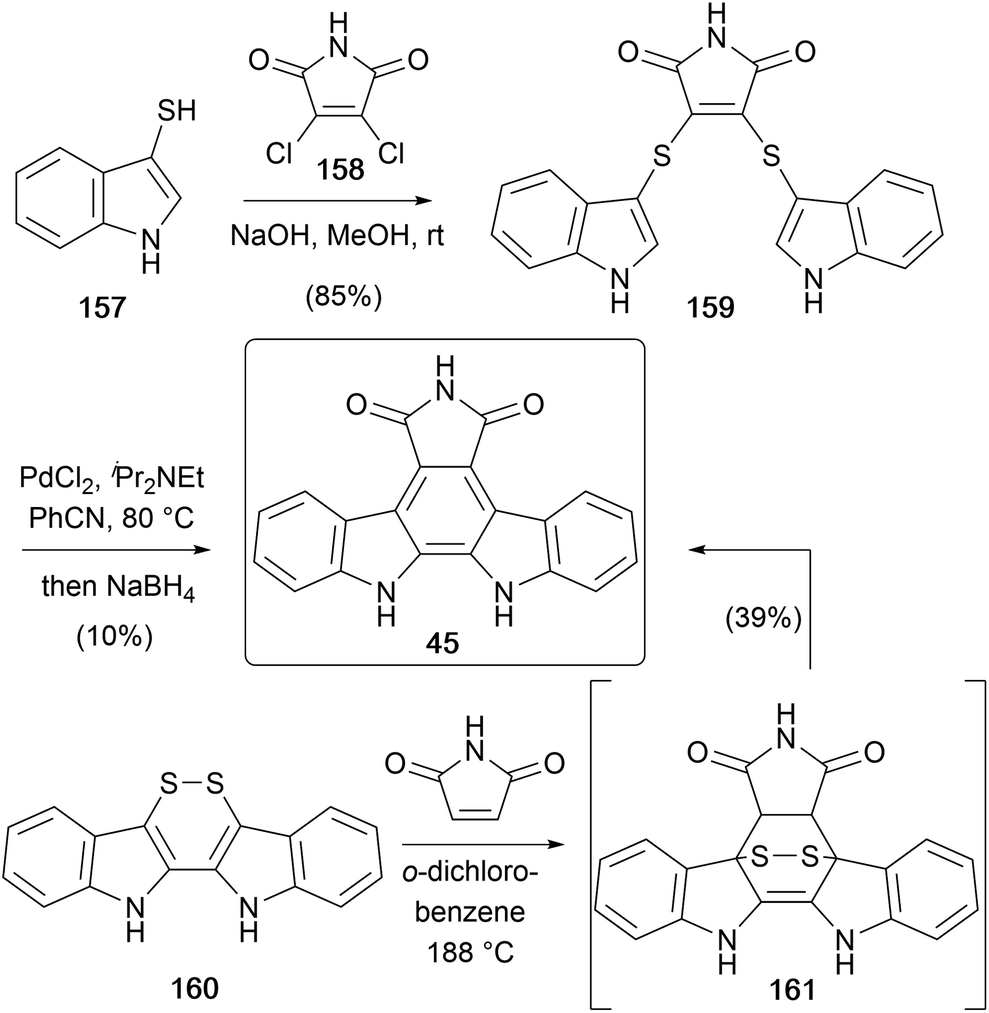 | ||
| Scheme 26 Lobo and co-workers' syntheses of arcyriaflavin A (45). | ||
The same group developed a modified approach a few years later,126 this time from disulphide 160.127 Diels–Alder cycloaddition with maleimide gave arcyriaflavin A (45) directly in 39% yield (Scheme 26). They propose the reaction proceeds via the [4+2] adduct 161, which then undergoes a retro-Diels–Alder and oxidation, likely from the extruded S2 or dissolved O2.128
In 2000, Tomé and co-workers reported a hybrid approach to arcyriaflavin A (45), wherein classical Fischer synthesis and Cadogan nitrene insertion were applied to indole ring constructions (Scheme 27).129 From commercial 2-nitrobenzaldehyde (162), Wittig olefination gave ketone 163, which was converted to the diene 164 upon treatment with TBSOTf/NEt3. Diels–Alder cycloaddition reaction with maleimides 165a or 165b gave the endo-cycloadducts 166a/b, which were deprotected to give ketones 167a/b.130 These intermediates were subjected to Fischer indole conditions with hydrazines168a/b to give indoles 169a/b in moderate yield. The corresponding carbazoles were obtained by DDQ oxidation before a final Cadogan cyclisation gave arcyriaflavin A (45) and analogue 171.
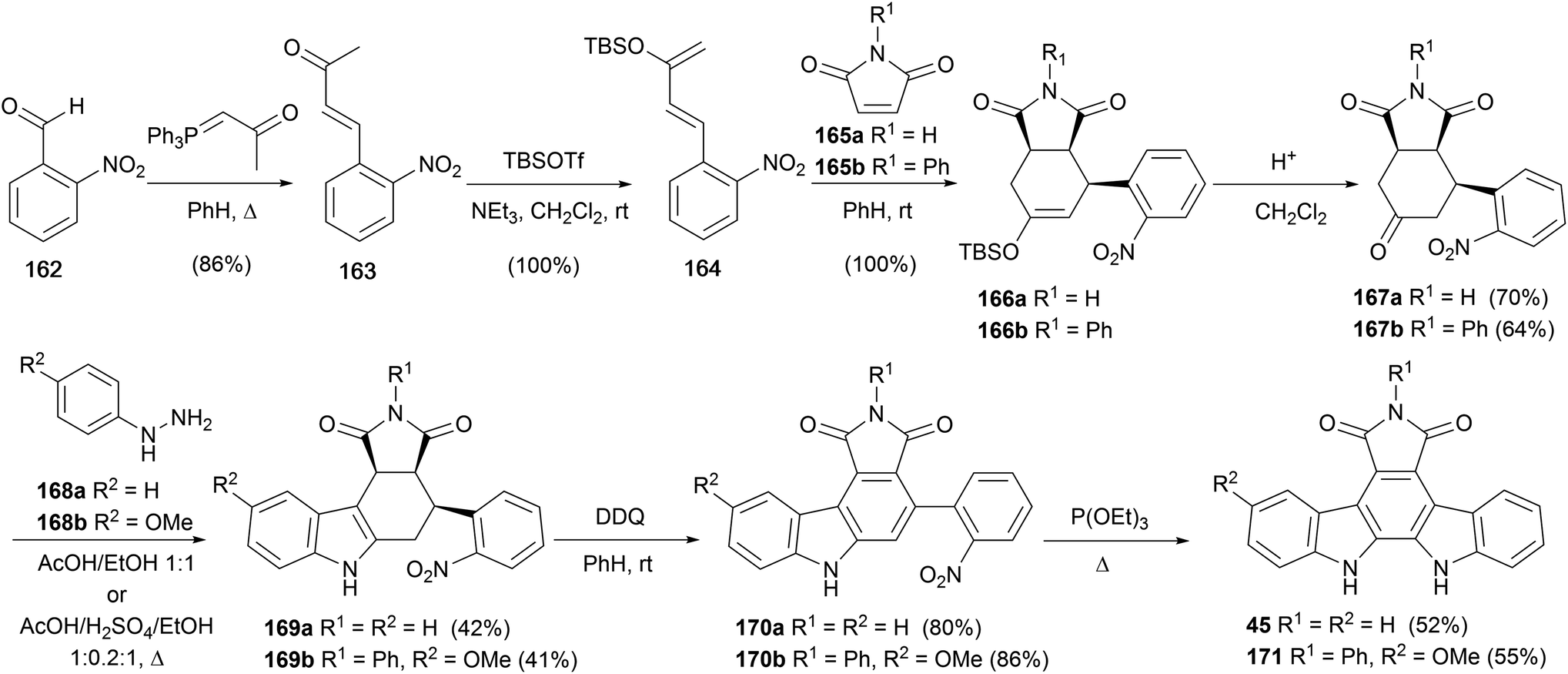 | ||
| Scheme 27 Tomé and co-workers' synthesis of arcyriaflavin A (45). | ||
Liu and co-workers adapted the Pd-catalysed oxidative cyclisation approach to produce a range of arcyriaflavin derivatives 173a–i (Table 1).131–134 Bisindolylmaleimide derivatives 172a–i were isolated as previously described from the appropriately substituted indolylmagnesium bromide with N-substituted 3,4-dihalomaleimides.48 This methodology was further developed by Liu and co-workers, using Pd(TFA)2 and Cu(OAc)2, accessing further arcyriaflavin analogues.135
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Product | R1; R1 | R2 | T (°C) | Time (h) | Yield (%) |
| 1 | 173a | 1-Cl; 11-Cl | p-tBu–Bn | 120 | 12 | 88 |
| 2 | 173b | 3-F; 9-F | p-tBu–Bn | 120 | 16 | 81 |
| 3 | 173c | 3-Br; 9-Br | p-tBu–Bn | 120 | 10 | 74 |
| 4 | 173d | 3-F; 9-F | H | 120 | 8 | 78 |
| 5 | 173e | 2-F, 3-F; 9-F, 10-F | p-tBu–Bn | 120 | 16 | 82 |
| 6 | 173f | 3-OMe; 9-OMe | p-tBu–Bn | 90 | 4 | 86 |
| 7 | 173g | H; H | p-tBu–Bn | 90 | 8 | 79 |
| 8 | 173h | 3-F; 9-F | t Bu | 90 | 6 | 62 |
| 9 | 173i | 3-F; 9-F | DMB | 90 | 5 | 58 |
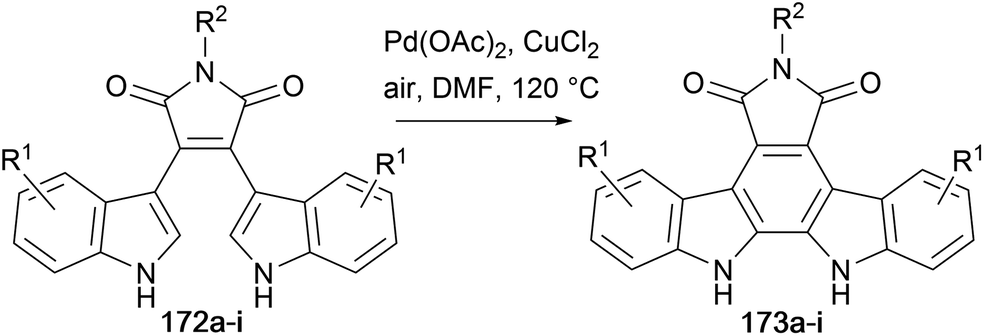
The same group developed a synthesis of arcyriaflavin A (45) using Suzuki coupling of 3-indolylboronic acid (175) with N-methyldibromomaleimide (Scheme 28),136 rather than the well-established addition–elimination (e.g. see Scheme 4). Synthesis of the boronic acid proceeded via an organomercury intermediate 174, by borylation with BH3·THF and aqueous work-up. Suzuki cross-coupling and hydrolysis of the tosyl groups gave bisindolylmaleimide 176. Formation of the free maleimide required the usual hydrolysis to anhydride 50,137 followed by re-imidation with HMDS to obtain bisindolylmaleimide (44). Finally, cyclisation in presence of a Pd(II)-catalyst provided arcyriaflavin A (45).42
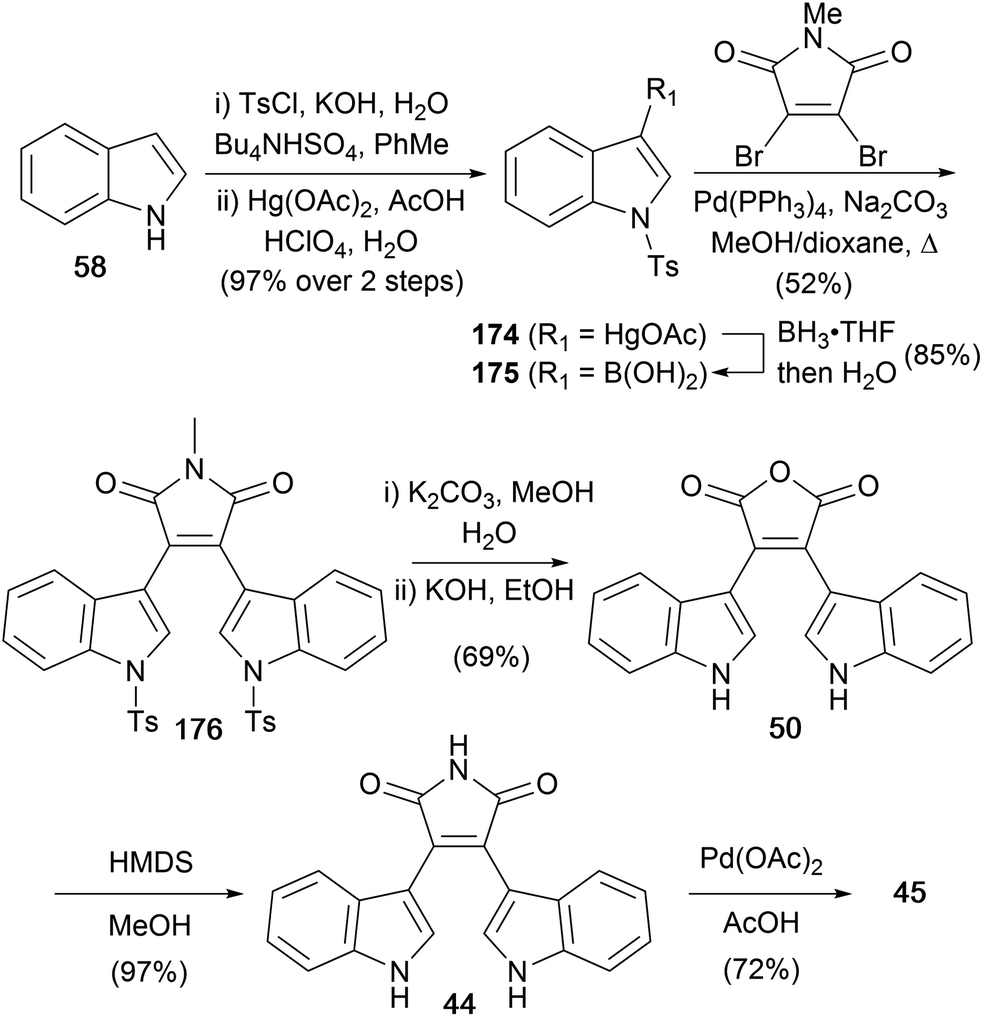 | ||
| Scheme 28 Liu and co-workers' synthesis of arcyriaflavin A (45). | ||
Starting from 1H-indole-2-methanol (177), Tilve and co-workers synthesised arcyriaflavin A (45),138 incorporating a curious graphite-promoted 1,4-addition to dimethyl acetylenedicarboxylate (Scheme 29). The resulting mixture of isomeric alkene adducts 178 (E:Z, 88:12) was subjected to one pot oxidation with MnO2 followed by a Wittig olefination to afford a mixture of four stereoisomeric trienes 179. Electrocyclisation of the mixture gave nitro-phenyl substituted carbazole 180, which was subjected to Cadogan cyclisation followed by hydrolysis to afford anhydride 51.32,139 Arcyriaflavin A (45) was finally secured in 72% yield through treatment of 51 with NH4OAc.
 | ||
| Scheme 29 Tilve and co-workers' synthesis of arcyriaflavin A (45). | ||
A gold-catalysed indolisation was applied by Aponick and co-workers in their synthesis of protected arcyriaflavin A 187 (Scheme 30).140,141 The key intermediate 184 for the Au-catalysed diene formation was prepared from Boc-protected 2-aminobenzyl alcohol 181 over four steps.142 Conversion of 181 to the terminal alkyne 183 was achieved via bromination, Pd-catalysed cross-coupling with tris(trimethylsilylacetylene)–indium and deprotection of the TMS group.143 Addition of the dianion prepared from alkyne 183 to 2-nitrobenzaldehyde gave alkyne 184, which was set-up for the Au-catalysed dehydrative cyclisation reaction to secure 2-vinylindole 185. Subsequent Diels–Alder cycloaddition with maleimide and oxidation gave carbazole 186, which was converted to protected arcyriaflavin A (187) using a Mo-variant of the Cadogan cyclisation, which allowed cyclisation at a lower temperature.144,145
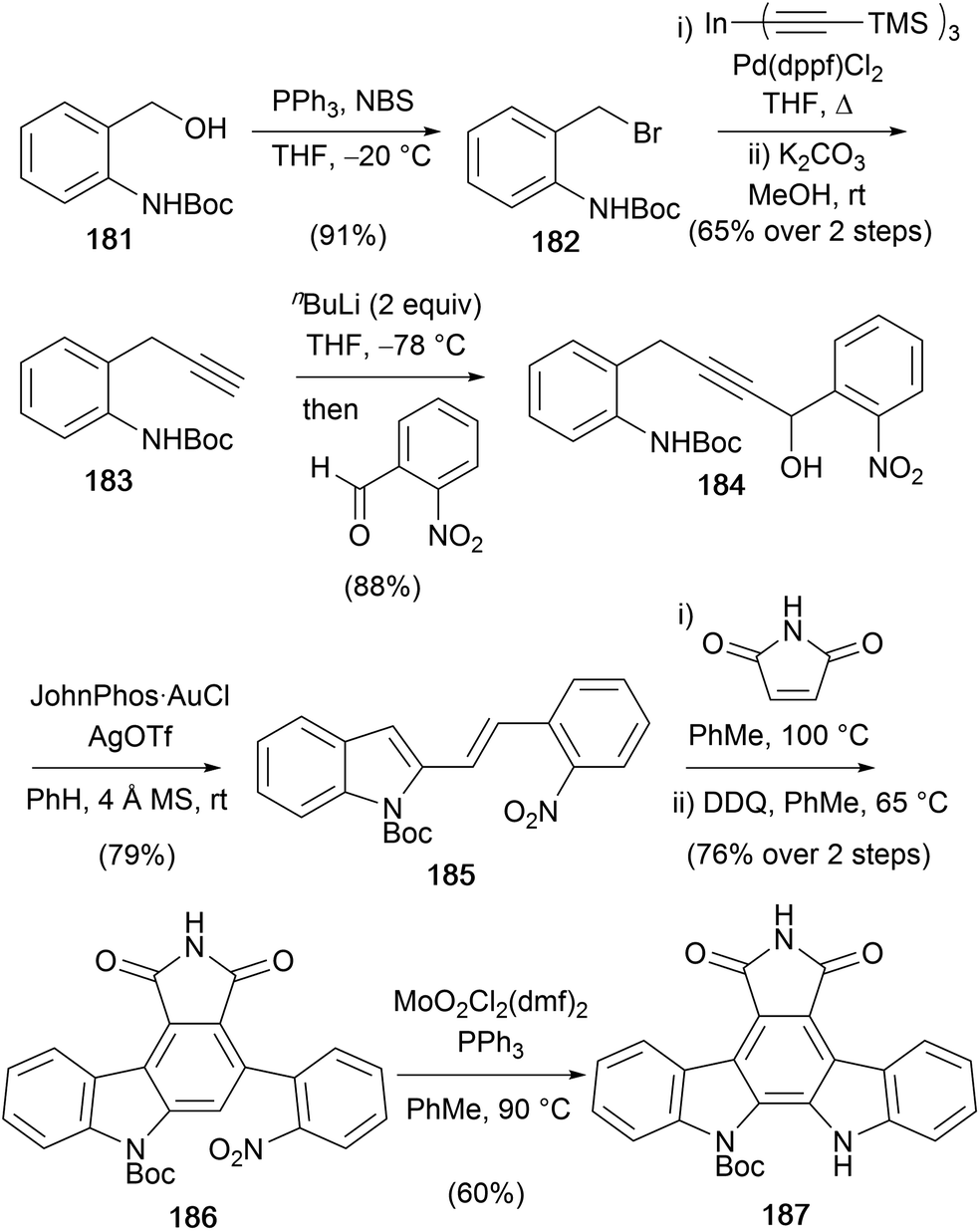 | ||
| Scheme 30 Aponick and co-workers' synthesis of protected arcyriaflavin A (187). | ||
A synthesis of arcyriaflavin A (45) was detailed by Cheon and co-workers in 2017 (Scheme 31).146 The synthesis began by accessing 2,2′-biindole 190via an imino-Stetter approach from 1-benzyl-1H-indole-2-carbaldehyde (188).147,148 Protection of the free indolic nitrogen with BnBr gave biindole 191. Reaction with ethyl glyoxylate followed by oxidation of the resulting alcohol gave diester 192. Treatment with tBuOK was immediately followed by hydrolysis and maleimide formation to afford protected arcyriaflavin A derivative 193. Finally, Lewis-acid cleavage of the benzyl groups delivered arcyriaflavin A (45).
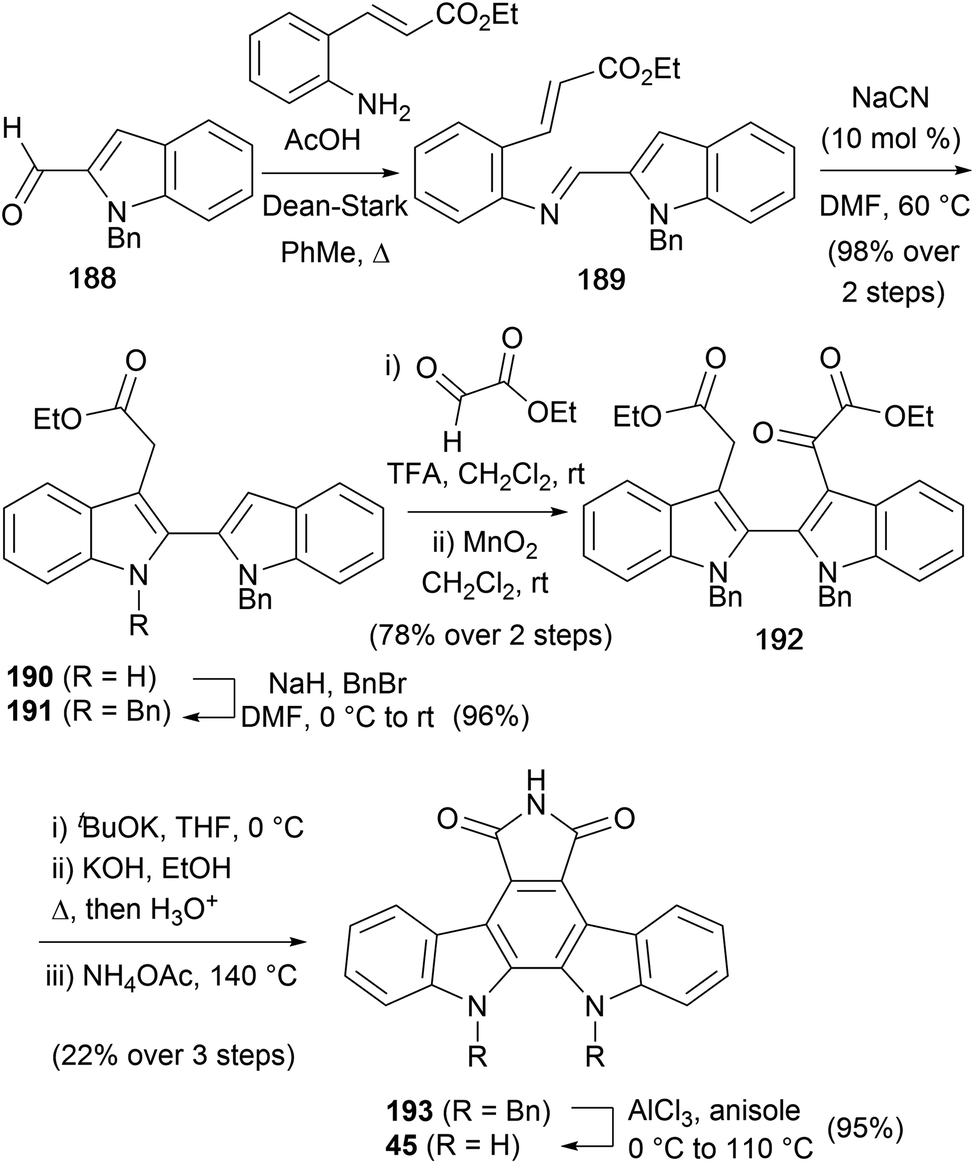 | ||
| Scheme 31 Cheon and co-workers' synthesis of arcyriaflavin A (45). | ||
The most recently disclosed synthesis of arcyriaflavin A (45) was by Tang and co-workers in 2018 (Scheme 32),149 applying a formal [1+2+3] annulation of alkenyl aryisocyanides and α,β-unsaturated ketones to the synthesis of the carbazole system. 3-(2-Nitrophenol)-2-oxobut-3-enoate (195), prepared according to known procedures, underwent formal [1+2+3] annulation reaction with aryisocyanide 194 to give diester 196. The well-established imide formation and Cadogan cyclisation ultimately secured arcyriaflavin A (45).
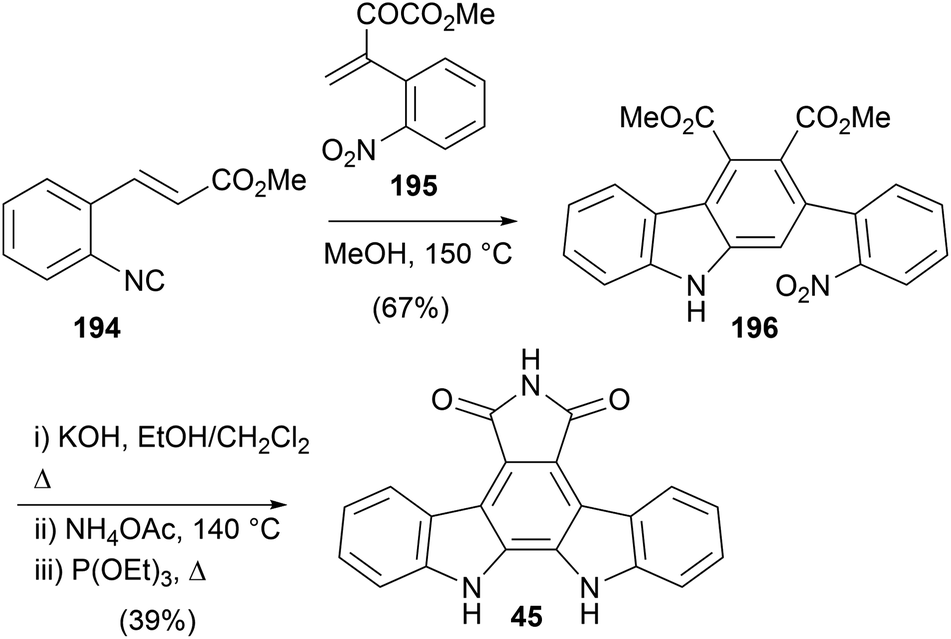 | ||
| Scheme 32 Tang and co-workers' synthesis of arcyriaflavin A (45). | ||
2.3 Arcyriaflavins B–D
As part of their pioneering work on indolocarbazole natural products Raphael and co-workers reported a synthesis of arcyriaflavin B in 1983 (Scheme 33).27 The synthesis was similar to their approach to the staurosporine aglycone (10), and began with Wittig olefination of aldehyde 18 with ylide 197 before iodine-mediated cis–trans isomerism gave diene 198. Diels–Alder cycloaddition with maleimide and DDQ oxidation secured dinitro derivative 199, which afforded arcyriaflavin B (132) after Cadogan cyclisation and demethylation of indolocarbazole 200.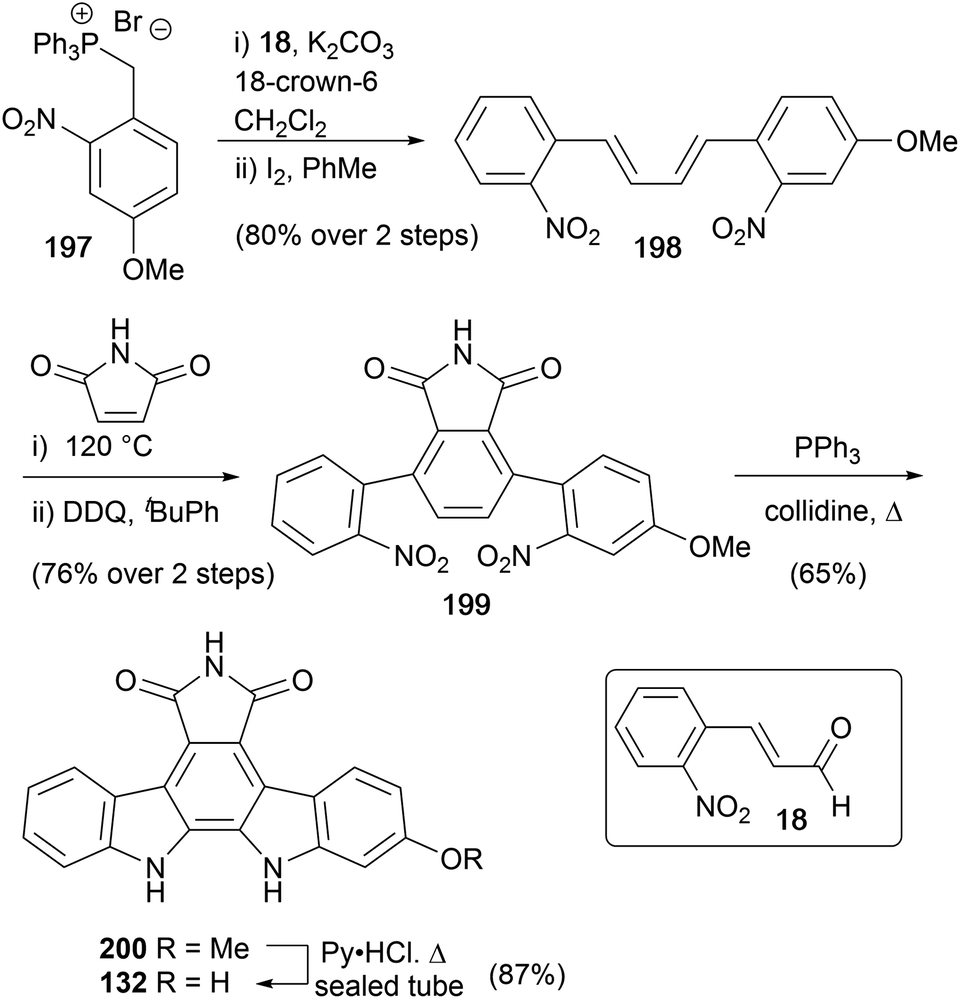 | ||
| Scheme 33 Raphael and co-workers' synthesis of arcyriaflavin B (132). | ||
Ohkubo and co-workers described a synthetic approach to arcyriaflavins B (132), C (133) and D (134) that exploited sequential substitutions of 2,3-dibromo-N-methylmaleimide (201) with indolyl nucleophiles (Scheme 34).150 Substituted indoles 58, 202 and 205, prepared from the corresponding nitrophenols,151 were lithiated and selectively coupled following a modification of Steglich and co-workers procedure yielding unsymmetrically substituted bisindolylmaleimides 206a–c.35 Cleavage of the Boc group preceded cyclisation and oxidation to indolocarbazoles 208a–c, which were converted to arcyriaflavins B (132), C (133) and D (134) utilising the protocol described by Lown and co-workers.43
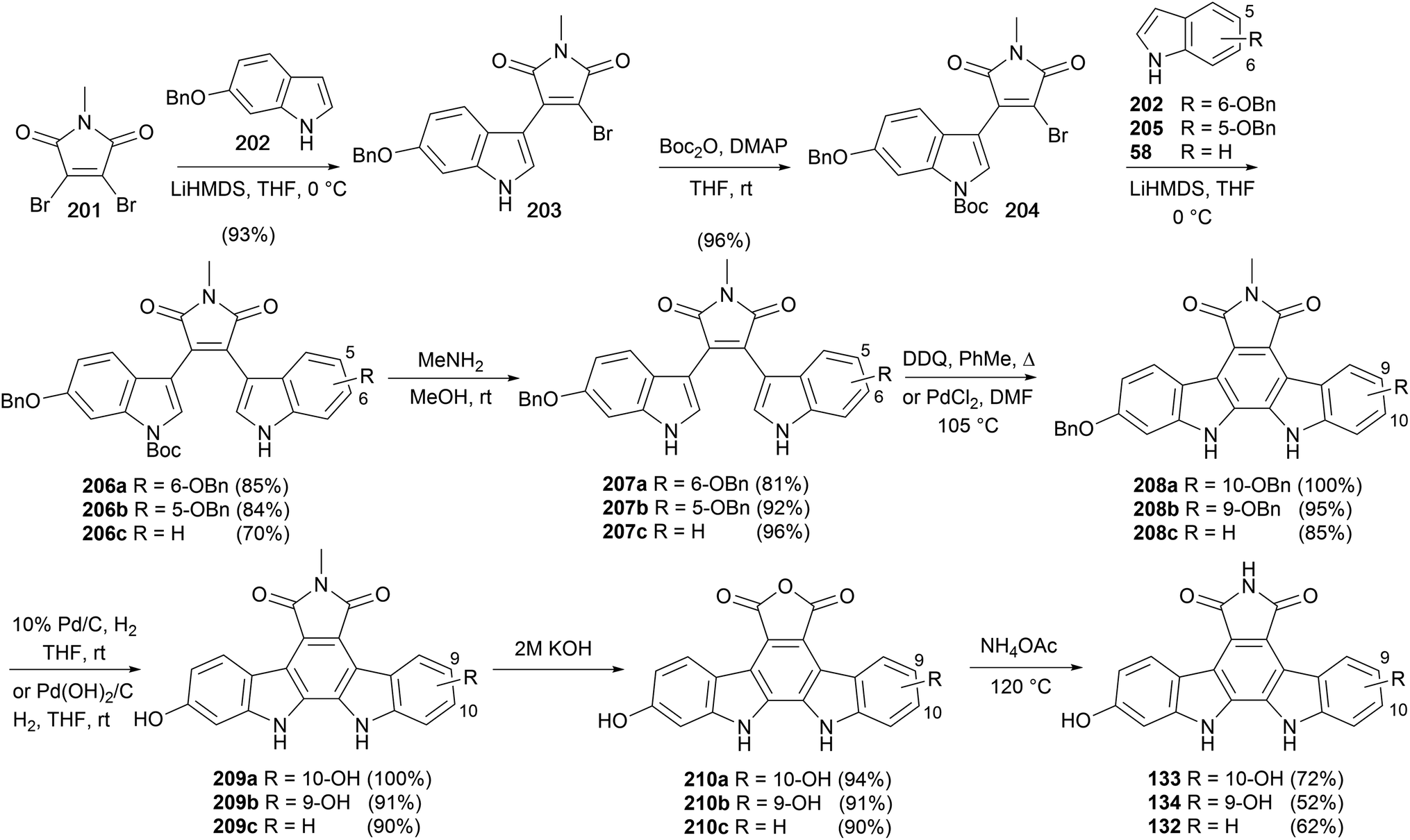 | ||
| Scheme 34 Ohkubo and co-workers' synthesis of arcyriaflavin B (132), C (133) and D (134). | ||
3 Pyranosylated indolocarbazoles
3.1 Staurosporine (7)
Due to its position as one of the most potent PKC inhibitors to date, staurosporine has been an important lead compound in the discovery of viable pharmaceutical derivatives. One such example is midostaurin, which was approved in 2017 by the FDA for the treatment of acute myeloid leukaemia.152 However, despite impressive biological activity, there have only been two total syntheses of staurosporine (7) to date.Danishefsky and co-workers completed the first total synthesis of 7 starting from BOM-protected maleimide 211 (Scheme 35).153–157 Sequential addition of the magnesiated indoles provided access to bisindolylmaleimide 214 with orthogonal protection of nitrogens. The pyranose fragment 215 was prepared in 7 steps from L-glucal derivative 228, first forming the corresponding bis-(trichloroacetimidate) followed by N-cyclisation onto the allylic trichloroacetimidate and hydrolysis of the obtained oxazoline gave trichloroacetamide 229. Oxazolidinone annulation and protecting group manipulations, before final epoxidation with DMDO gave a 5.5:1 mixture of α and β epoxides (major 215), setting the stage for indole glycosylation. The mixture of epoxides was treated with the sodiated indole 214, providing alcohol 216, which was deoxygenated under Barton conditions to give bisindolylmaleimide 217. Removal of PMB and SEM protecting groups afforded primary alcohol 219, which underwent an oxidative photocyclisation and conversion of the primary alcohol to the iodide with molecular iodine secured arcyriaflavin A derivative 220. The staurosporine framework was established in 221 by elimination of HI from 220 and iodocyclisation of the resulting exocyclic enol ether. From 221 radical deiodination, hydrogenolysis of both BOM groups, selective oxazolidinone protection (Boc), reinstallation of the maleimide BOM protection, and final treatment with Cs2CO3 in MeOH returned alcohol 226. Methylation of the amino and hydroxyl residues was followed by BOM deprotection and maleimide reduction without regiocontrol, realising a 1:1 mixture of staurosporine (7) and iso-staurosporine (227).
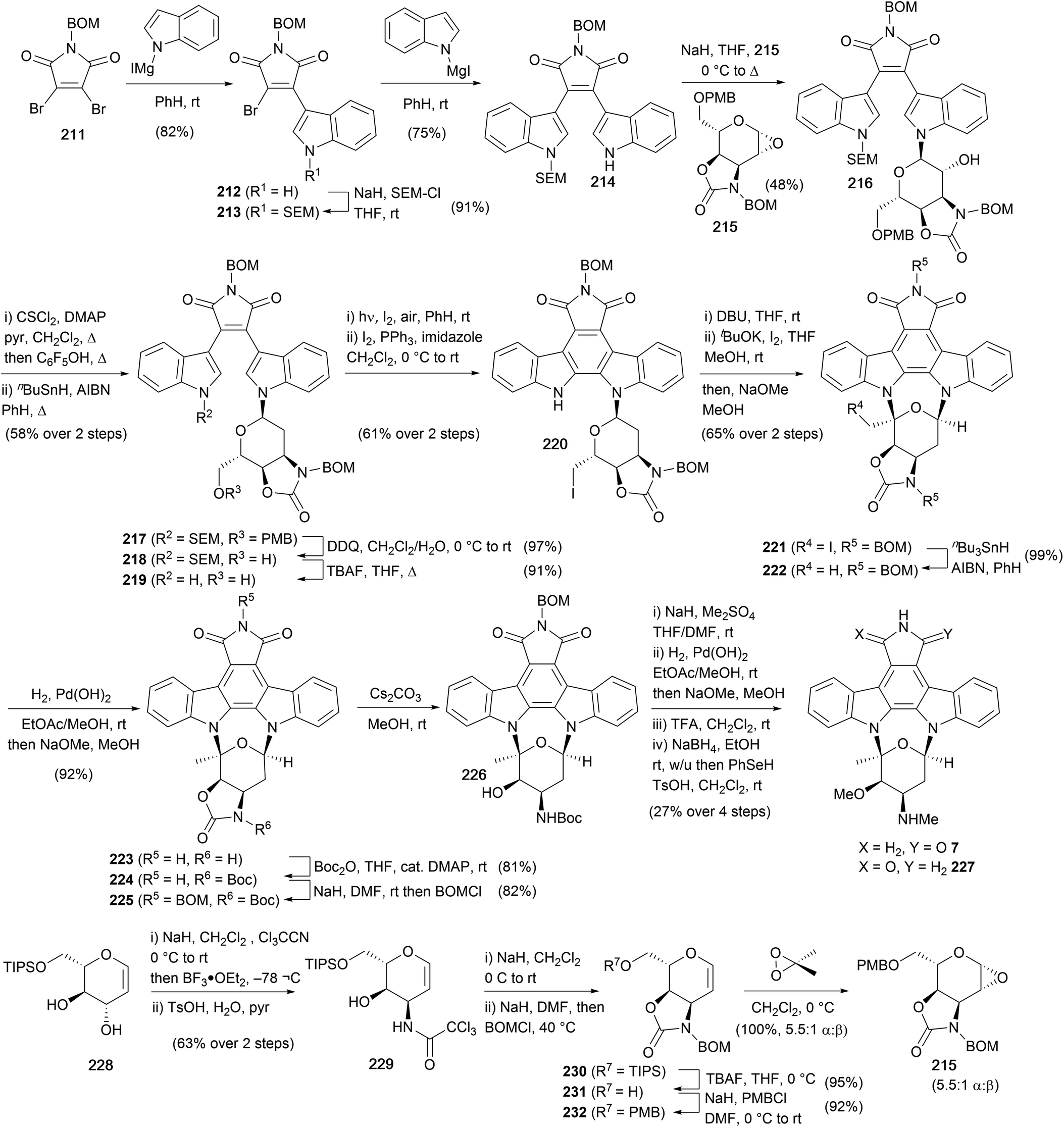 | ||
| Scheme 35 Danishefsky and co-workers' synthesis of staurosporine (7) (1993). | ||
A subsequent synthesis of staurosporine (7), was reported in 1996 by Wood and co-workers (Scheme 36).50,51,158 Their approach encompassed syntheses of several indolocarbazole natural products, with initial focus on coupling a furanose fragment to access K252a (9, see Section 4.1), with ring expansion giving access to staurosporine. The protected aglycone 57c was prepared from biindole 53 using a Rh-catalysed coupling–rearrangement–elimination sequence. The furanose fragment 242 was obtained in short order from diazoester 238, commencing with Rh-catalysed O–H insertion into secondary alcohol R-(−)-239 and ensuing [3,3]-sigmatropic rearrangement providing alcohol 240. Exposure of the α-ketol 240 to BF3·Et2O resulted in diastereoselective [1,2]-migration to afford β-ketoester 241. Ozonolysis of the alkene and acid-mediated cyclisation delivered furanose 242, which underwent an acid-catalysed double-glycosidation of DMB-protected staurosporine aglycone 57c in the presence of CSA to give a 2:1 mixture of regioisomers 233 and 234. Interestingly, a mixture containing two regioisomeric pairs of open chain monoamino acetal diastereoisomers was initially observed, which upon prolonged heating induced cycloglycosylation, converging on two of the four possible isomers, 233 and 234. After separation of the desired major isomer 233, ester reduction and reoxidation gave aldehyde 235. Which upon treatment with BF3·Et2O, underwent another α-ketol rearrangement/ring-expansion to afford the ketone 236, elegantly accessing both the pyranosylated (see later section) and furanosylated natural products from a common intermediate. In addition, a copper mediated ring-contraction of ketone 236 to ester 233 was also achieved, highlighting the potential for a biomimetic link. To complete the synthesis, oximation of ketone 236 followed by bis-methylation yielded methyl oxime 237, before a stereoselective reduction, reductive methylation and DMB-deprotection sequence delivered (+)-staurosporine (7).
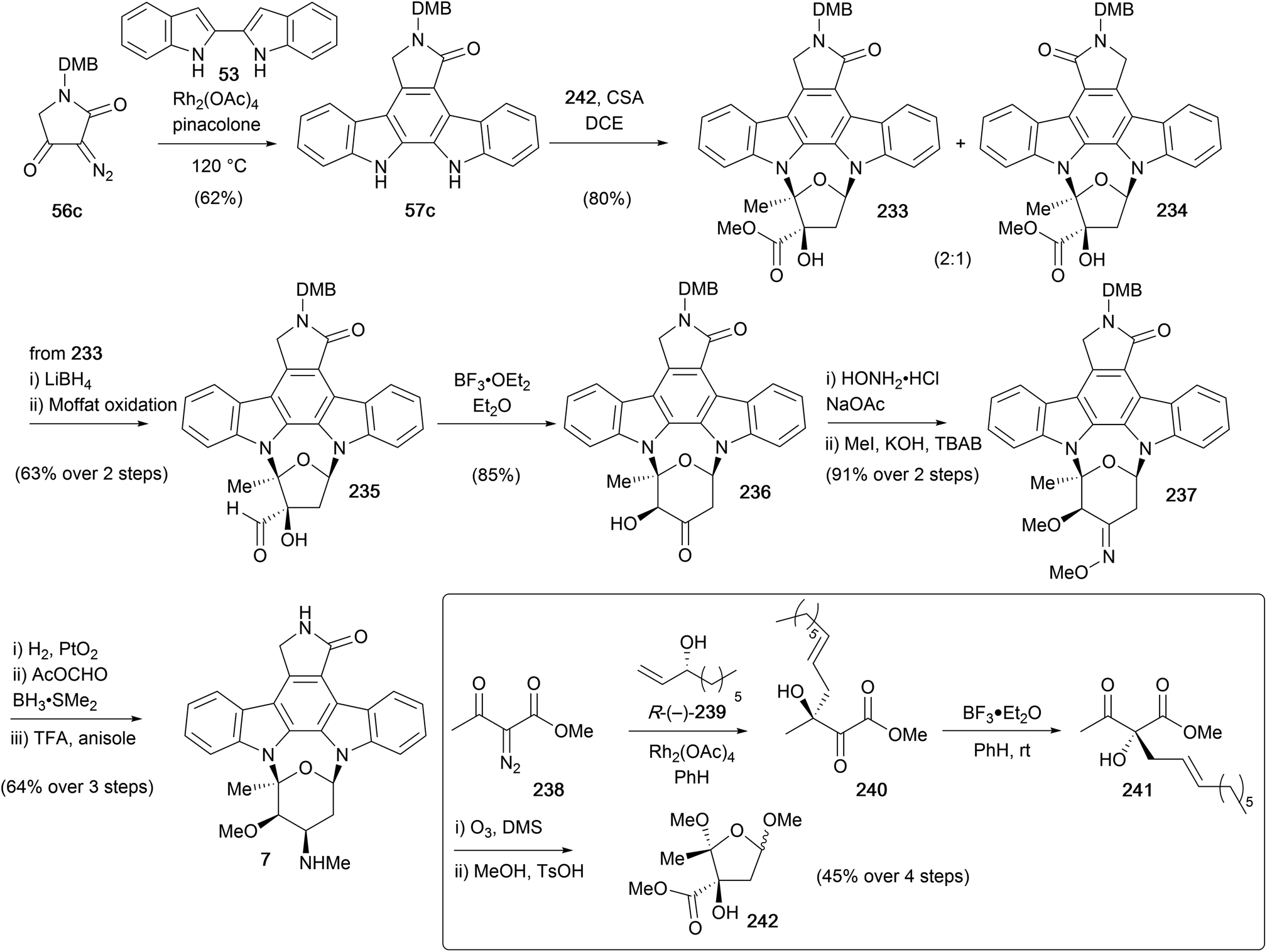 | ||
| Scheme 36 Wood and co-workers' synthesis of staurosporine (7). | ||
3.2 Rebeccamycin (8)
As the most potent indolocarbazole antitumor agent, rebeccamycin (8) has been the focus of both total synthesis and synthetic analogues studies.159–161 In contrast to the staurosporine class of indolocarbazole glycosides, rebeccamycin only has a single glycosidic linkage. However, many of this sub-family of indolocarbazole natural products exhibit extraordinary activity as poisons of DNA topoisomerase I. This is shown to lead to cell apoptosis, and rebeccamycin derivatives reached phase II clinical trials for the treatment of neoplasias including renal cell carcinoma and stage IIIB or IV breast cancer.159,162,163First isolated by Clardy and co-workers in 1985 from the actinomycete strain Nocardia aerocoligenes,16 the rebeccamycin structure was elucidated during synthetic approaches carried out by the same group in 1985.164 The synthesis began from 7-chloroindole, which was prepared according to the procedure described by Sugasawa and co-workers (Scheme 37).165 This could then be coupled with N-benzyloxymethyl-2,3-dibromomaleimide (243) using Steglich's magnesiated indole coupling methodology to give bisindolylmaleimide 244.35 Cyclisation of 244 could be achieved photochemically, however a one-pot cyclisation–oxidation and Koenigs–Knorr glycosidation with pyranosyl bromide 246 was developed to give indolocarbazole 247. Pyranosyl donor 246 was prepared according to the method of Bouveng.166 Finally hydrogenolysis of the BOM group followed by ammonolysis of the acetyl groups gave rebeccamycin (8).
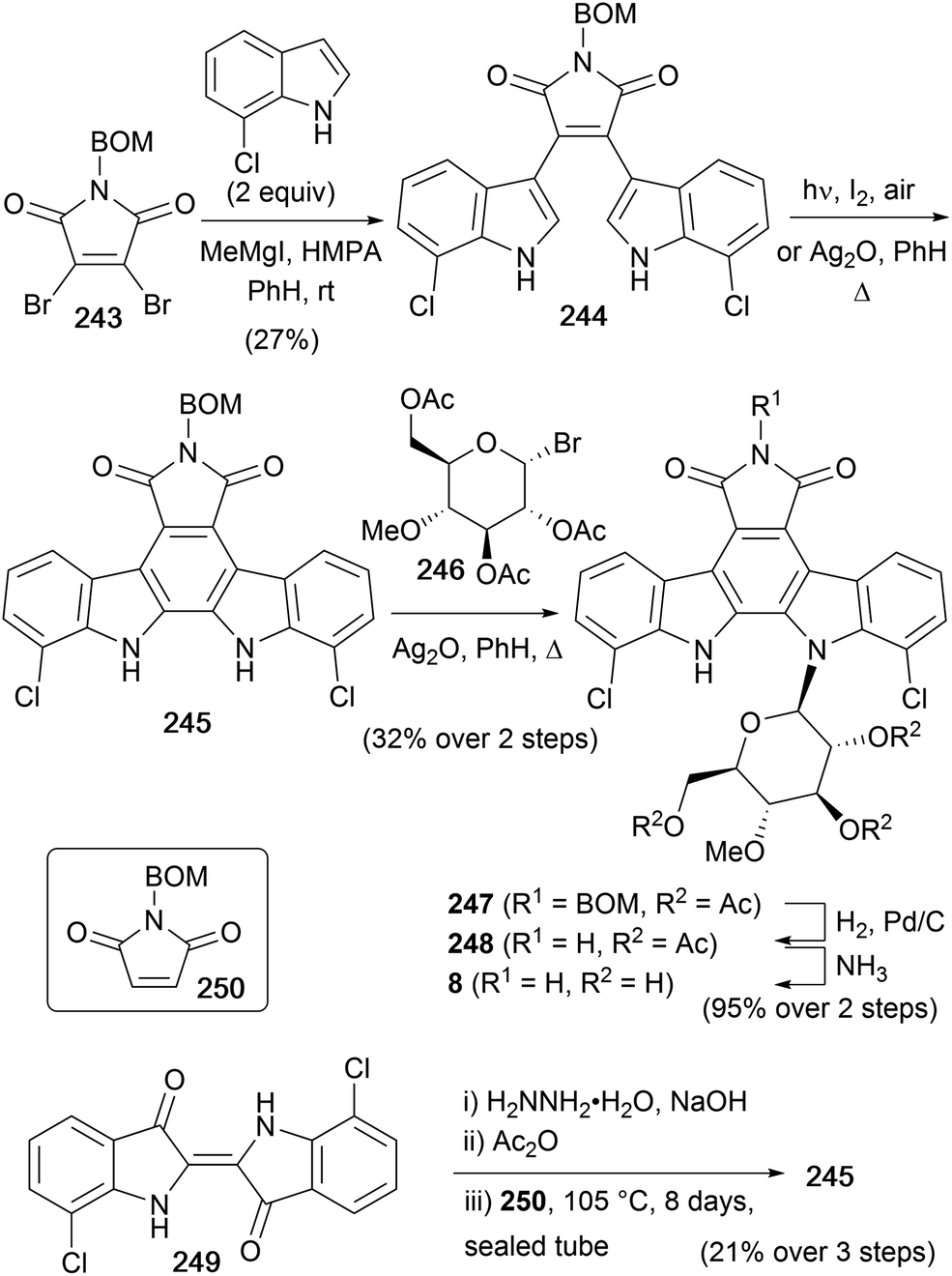 | ||
| Scheme 37 Clardy and co-workers' syntheses of rebeccamycin (8). | ||
The same group also demonstrated an alternative method to access rebeccamycin aglycone 245 (Scheme 37).164 Wolff–Kishner reduction of 7,7′-dichloroindigo (249) followed by acetylation gave a 2,2′-biindole derivative.167,168 Finally, Diels–Alder cycloaddition with N-benzyloxymethyl maleimide (250) gave N-benzyloxymethyl aglycone 245.
In 1993, Danishefsky and co-workers also completed a synthesis of rebeccamycin (8, Scheme 38).156 The synthesis began in similar vein to their synthesis of staurosporine (7) using sequential additions of indole magnesium reagents to give the asymmetric, mono-protected bisindolylmaleimide 251. The required sugar 252 was prepared from the commercially available tri-O-acetyl-D-glucan (256). Removal of the acetate protecting groups was followed by selective 3,6-O-dibenzylation using stannylene chemistry, methylation of the 4-hydroxyl and dioxirane promoted epoxidation gave glucan 252 as a 15:1 mixture of anomers.169 The anomeric mixture was then subjected to the pivotal glycosidation with bisindolylmaleimide 251 to deliver a major β-maleimide 253 in 48% and the α-anomer in 8% yield. The 5:1 ratio of 253versus the 15:1 ratio in 252 is attributed to the greater reactivity of the minor β epoxide. SEM deprotection, followed by photocyclisation gave indolocarbazole 254, and finally, hydrogenolytic benzyl cleavage and ammonolysis secured rebeccamycin (8).
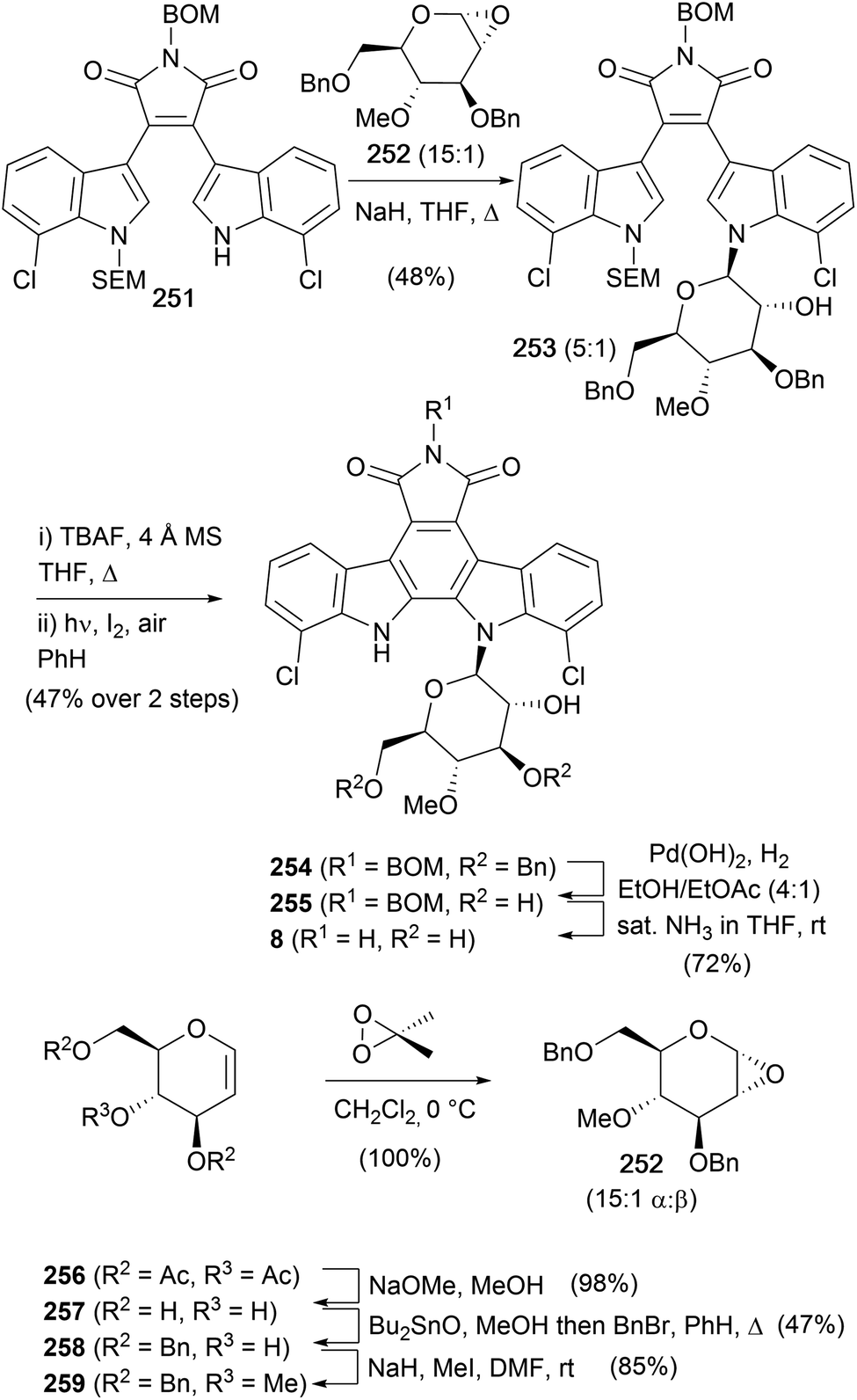 | ||
| Scheme 38 Danishefsky and co-workers' synthesis of rebeccamycin (8). | ||
The most recent synthesis of rebeccamycin (7), to date, was disclosed by Faul and co-workers in 1999 (Scheme 39).170 The synthesis began aminomethylation of 7-chloroindole (260), then homologation using cyanation and hydrolysis of the obtained nitrile gave amide 263. Glycosylation of 263 with epoxide 252, prepared according to the method of Danishefsky (see Scheme 38),156 secured glycosylated indole 264, which was treated with KOtBu and acetate 265 before addition of acid and heating promoted an intramolecular Perkin-type condensation to give the corresponding bisindolylmaleimide. The synthesis was completed with oxidative-cyclisation using Pd(OTf)2 followed by debenzylation with Pearlman's catalyst to conclude a concise synthesis of rebeccamycin (8) in 12 steps and 12% overall yield.
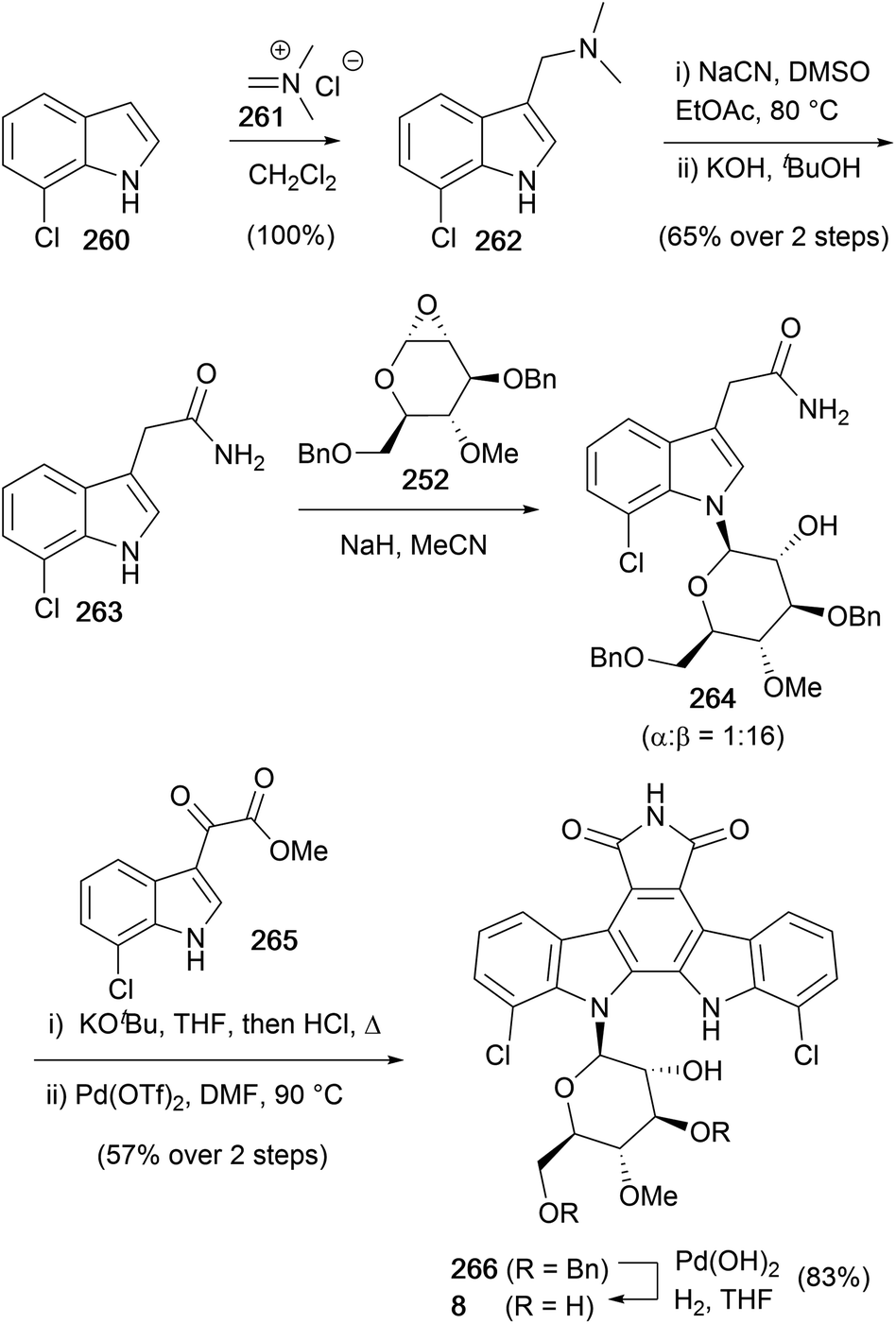 | ||
| Scheme 39 Faul and co-workers' synthesis of rebeccamycin (8). | ||
Despite having only been the focus of three synthetic approaches, there has been a great deal of interest in isolating synthetic analogues of rebeccamycin (8) as potential therapeutics. For lead references to the preparation of analogues, see recent reviews published by Kirsanov and co-workers171 and Panov and co-workers.172
3.3 Syntheses of (+)-RK-286c (268), (−)-TAN-1030a (271) and (+)-MLR-52 (272)
To date, the only syntheses of (+)-RK-286c (268), (−)-TAN-1030a (271) and (+)-MLR-52 (272) have emerged from the Wood group (Scheme 40, see also Schemes 8 and 36).51,158 These syntheses share a common ketone intermediate 236, also employed during their synthesis of staurosporine (7, Scheme 36). To access (+)-RK-286c (268), ketone 236 was reduced stereoselectively and then regioselective mono-methylation of the resulting vicinal diol gave methyl ether 267 in excellent yield. The highly selective methylation was attributed to the aglycone providing considerably different steric environments for the equatorial [C(3′)] and axial [C(4′)] hydroxyls. Finally, deprotection of the DMB group gave (+)-RK-286c (268). To complete the synthesis of (−)-TAN-1030a (271), ketone 236 was subjected to oximation with BnONH2·HCl, followed by methylation under phase-transfer conditions to give ether 269. Finally, removal of the DMB and Bn protecting groups furnished (−)-TAN-1030a (271). The third natural product obtained using this methodology was (+)-MLR-52 (272). Alcohol 267 was dehydrated with Martin's sulfurane, in preparation for stereoselective dihydroxylation of the resulting olefin and deprotection of the DMB group to secure (+)-MLR-52 (272).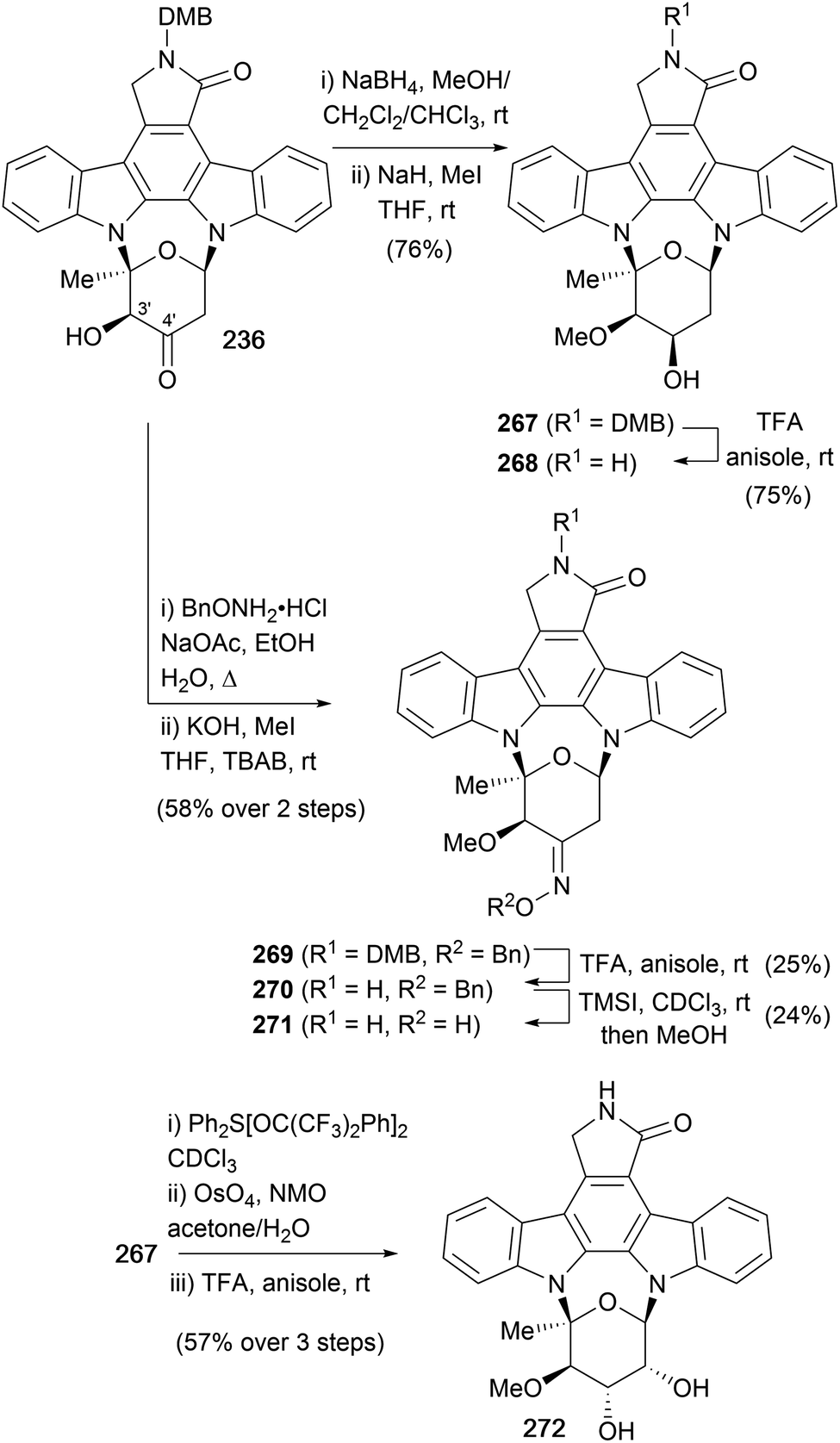 | ||
| Scheme 40 Wood and co-workers' syntheses of (+)-RK-286c (268), (−)-TAN-1030a (271) and (+)-MLR-52 (272). | ||
4 Furanosylated indolocarbazoles
4.1 K252a (9)
Another group of indolocarbazole natural products are those bearing furanose substitution on the indolic nitrogens, specifically K252a (9) is discussed here. The first synthesis of K252a was completed by Wood and co-workers in 1995 as part of their broader work discussed in this review (Scheme 41, see also Schemes 8, 36 and 40).50 After coupling N-DMB staurosporine aglycone (57c) and furanose 242 to give a 2:1 mixture of regioisomeric indolocarbazoles 233 and 234 respectively (see Scheme 36), separation of the mixture and deprotection of the DMB group provided K252a (9).50
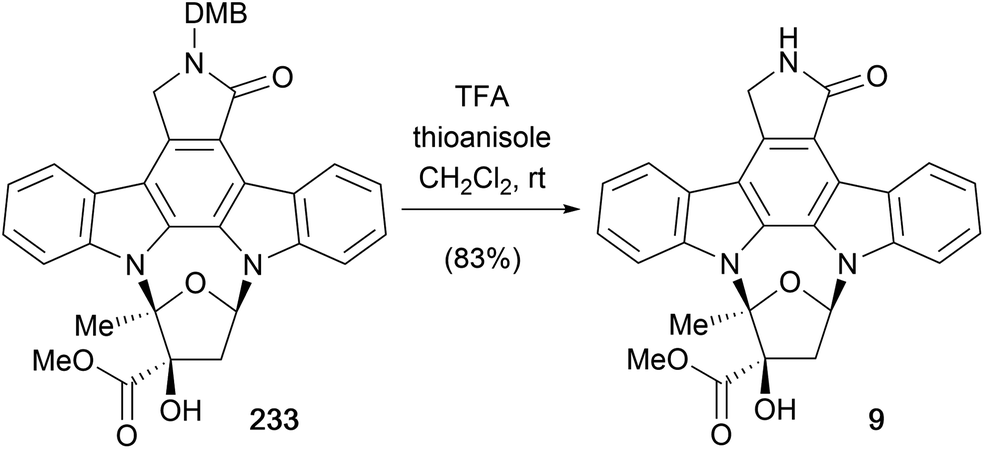 | ||
| Scheme 41 Wood and co-workers' synthesis of (+)-K252a (9). | ||
Later the same year, Lowinger and co-workers also detailed a synthesis of (±)-K252a (9, Scheme 42).173 The synthesis of the carbohydrate moiety of K252a began with allylation of methyl acetoacetate (273) and formation of the silyl-enol ether followed by chemoselective epoxidation and desilylation to access olefin 275. Ozonolysis and quenching with Me2S gave the intermediate aldehyde which cyclised to glycoside 276 using CSA. The PMB-protected aglycone 57d was prepared according to a modification of Raphael's route from aldehyde 18.28 The final stages of the synthesis mirrored Wood's approach, coupling the aglycone 57d and glycoside 276 to give a 2:1 regioisomeric mixture of 278 and 279, respectively, isomer separation, and PMB deprotection delivered (±)-K252a (9).173
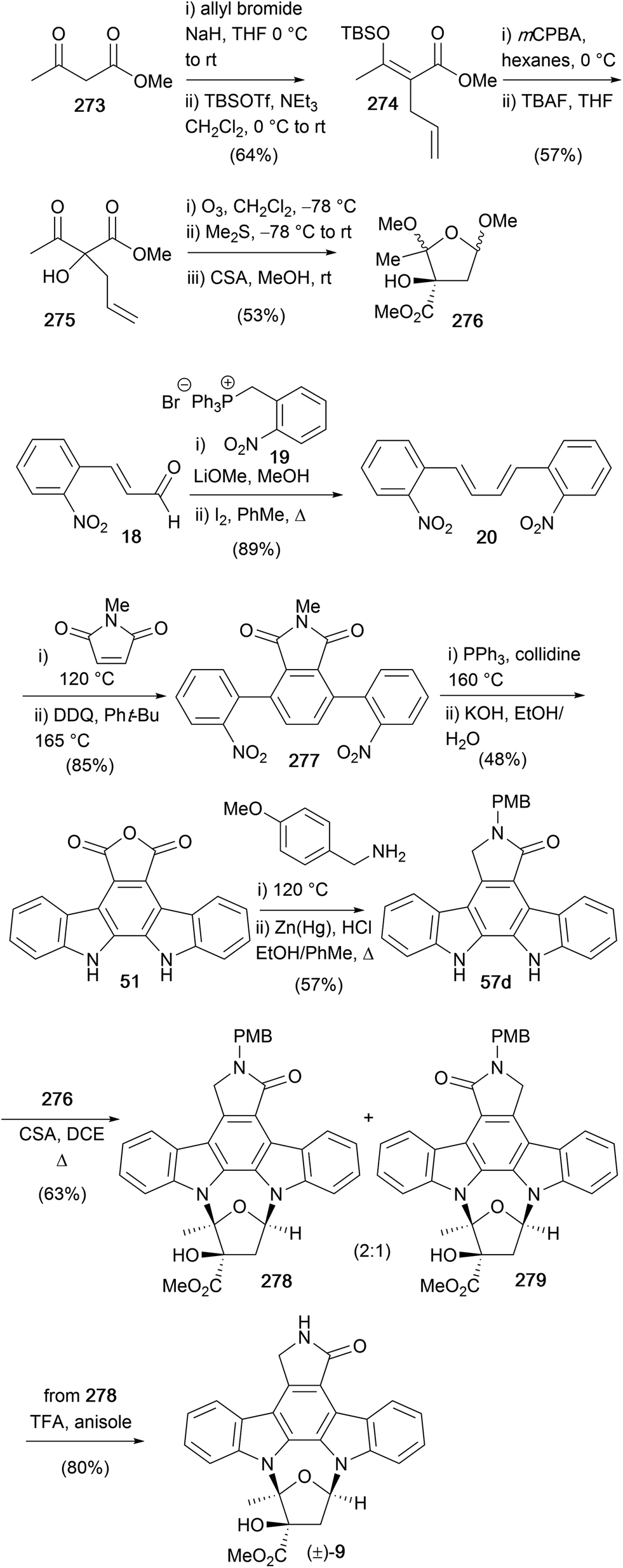 | ||
| Scheme 42 Lowinger and co-workers' synthesis of (±)-K252a ((±)-9). | ||
The most recent synthesis of (+)-K252a (9) was completed by Fukuyama and co-workers in 1999 (Scheme 43),174 focusing on solving regiochemical issues associated with previous furanose annulations reported by Wood and Lowinger. Starting with the commercially available indole acetic acid (280), protection as the allyl ester and regioselective bromination gave indole 282. N-Glycosidation was carried out with NaH and the readily available furanosyl chloride 283 to afford indole 284.175 Deprotection of the allyl ester and amide coupling with tryptamine (12) was followed by benzylic oxidation using DDQ and acetylation of the indole and amide nitrogens to give bisindole 285. Intramolecular condensation using DBU gave unsaturated lactam 286 as a 1:1 mixture of atropisomers before nonoxidative photocyclisation, hydrolysis of the acyl and toluoyl groups and selective Appel reaction of the primary alcohol gave iodide 289. Conversion of 289 to the exocyclic olefin 290 proved not to be trivial, requiring a three-step sequence involving selenation, followed by protection of the secondary alcohol, oxidation to the selenoxide with elimination providing the desired olefin 290. NEt3 and DHP present in the selenoxide elimination prevented oxidative addition of phenylselenous acid to the enol ether formed. Iodoglycosidation of the enol ether using I2/KI under basic conditions afforded cycloglycoside 291, which underwent radical deiodination, methanolysis of the acetyl group and oxidation of the secondary alcohol 293 to the corresponding ketone in excellent overall yield.153 Hydrocyanation secured the kinetic cyanohydrin which was subsequently acetylated to cyanohydrin acetate 294. Treatment with HCl in HCO2H gave the primary amide, which was converted to (+)-K252a (9) via hydrolysis and esterification with diazomethane. The overall sequence provided (+)-K252a (9) in 10% overall yield and in 23 synthetic operations.
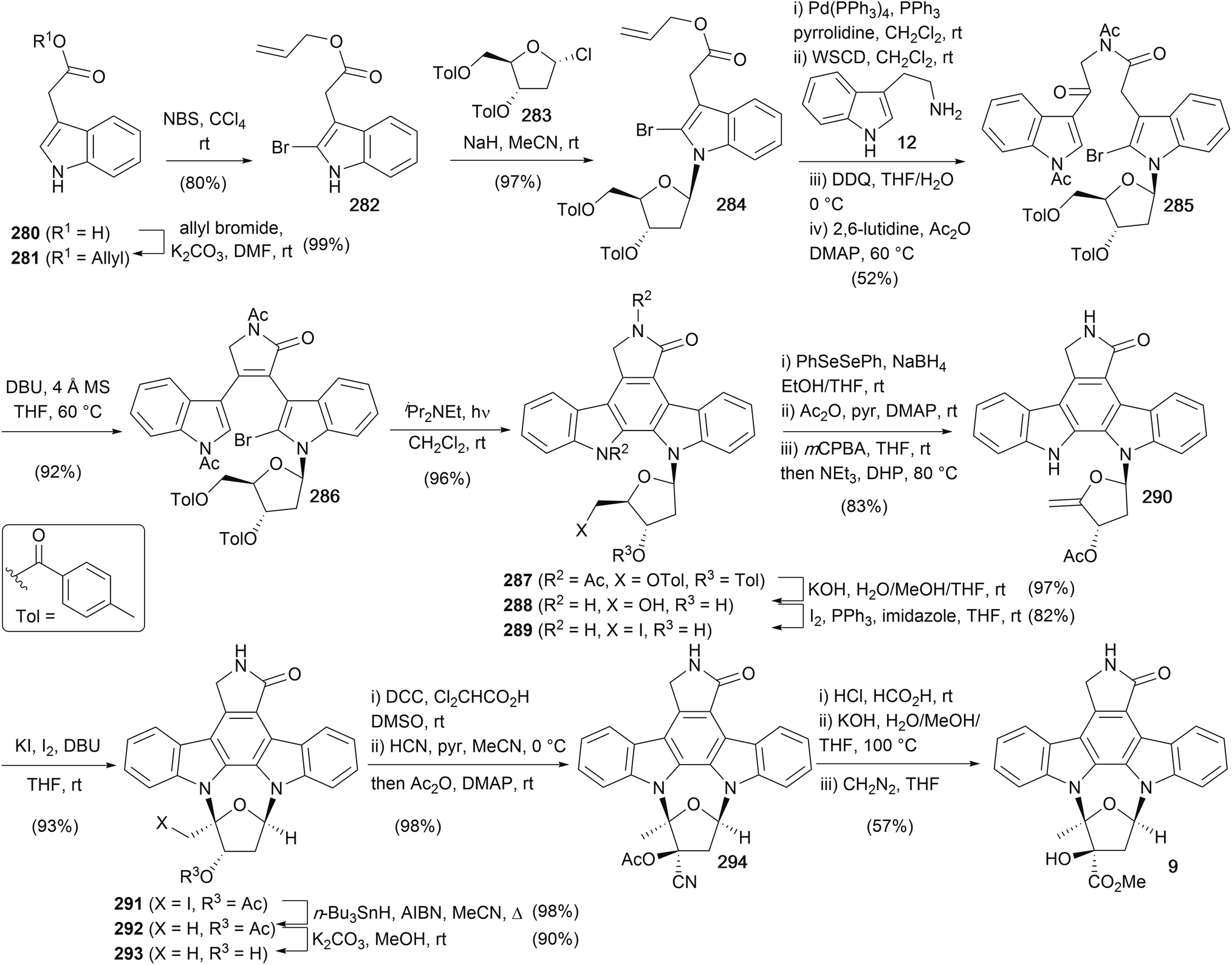 | ||
| Scheme 43 Fukuyama and co-workers' synthesis of (+)-K252a (9). | ||
5 Disaccharide indolocarbazoles
An intriguing family of indolocarbazoles contain a disaccharide unit bonded to the indolocarbazole unit. Isolated in 1989 by Matson and co-workers, these natural products fit into a similar sub-family to the rebeccamycin class of indolocarbazole glycosides, containing a single N-glycosidic linkage.176,177 They have demonstrated activity in the poisoning of DNA topoisomerase I and ultimately in cancer cell apoptosis. In addition to this, they are structurally impressive, particularly in the case of AT2433-A1 (295) and AT2433-A2 (296), which contain an asymmetric aglycon coupled to the disaccharide unit. This again presents interesting regioselectivity challenges for coupling of the sugar and aglycone fragments (Fig. 5).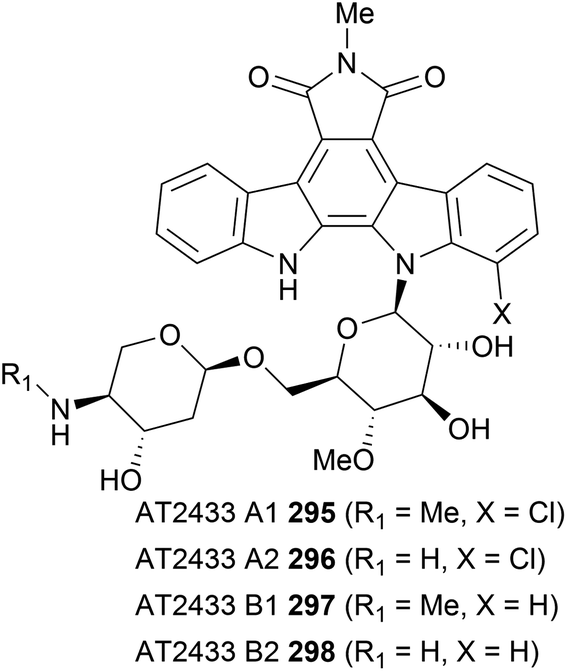 | ||
| Fig. 5 Structures of AT2433 A1 (295), A2 (296), B1 (297) and B2 (298). | ||
The first, and only synthesis of the AT2433 natural products to date, was carried out by Van Vranken and co-workers in 2000,159 utilising Mannich cyclisations to indolylindolines (Scheme 44).79,178,179 Their synthesis began with the preparation of asymmetric succinimide 299via Faul and co-workers adaption of Steglich's methodology with sequential addition of magnesiated indole and 7-chloroindole.48 Hydrogenolysis of the obtained maleimide gave succinimide 299, which was subjected to the key intramolecular Mannich cyclisation delivering indolene 300. Of particular note is the excellent control achieved in the Mannich cyclisation for either isomeric indolene through careful choice of acid used. Refluxing in neat TFA realised the thermodynamic indolene, where the indolene is formed in the more electron rich indole ring, whilst MsOH provided the desired kinetic isomer 300. Rationalisation based on the Curtin–Hammett principle, proposed that the electron-withdrawing effect of the chloro group during C–C bond formation destabilises the intermediate indolenium ion, leading to faster, irreversible cyclisation. In contrast, refluxing TFA led to the more stable indolenium ion, likely reversibly, however the group quote a diminished yield due to partial decomposition. The required disaccharide 301 was isolated via an 8-step sequence from homoallyl alcohol 304. Homoallyl alcohol 304 was prepared by Roush and Hunt's procedure from (R)-Garners's aldehyde as an 88:12 mixture of diastereoisomers.180 Benzylation of homoallyl alcohol 304 was followed by unveiling the masked aldehyde via OsO4 mediated dihydroxylation and periodate cleavage of the diol. Removal of the acetonide and concurrent cyclisation provided glycol 307. Formation of the glycosyl fluoride 308 was achieved with DAST, before coupling with primary alcohol 303 was effected under Mukaiyama–Nicolaou conditions and reductive cleavage of the Boc group provided disaccharide 309 as a mixture of α:β anomers (∼1:1). Protection of the methylamine moiety as the 2-(trimethylsilyl)ethyl carbamate 310 preceded debenzylation to give disaccharide 301. Indolene 300, was then glycosylated chemoselectively with disaccharide 301 in the presence of catalytic amounts of CSA before oxidation of the 1:1 mixture of diastereoisomers gave indolocarbazole 302. Finally, removal of the teoc group with TBAF concluded the first synthesis of AT2433-A1 (295).
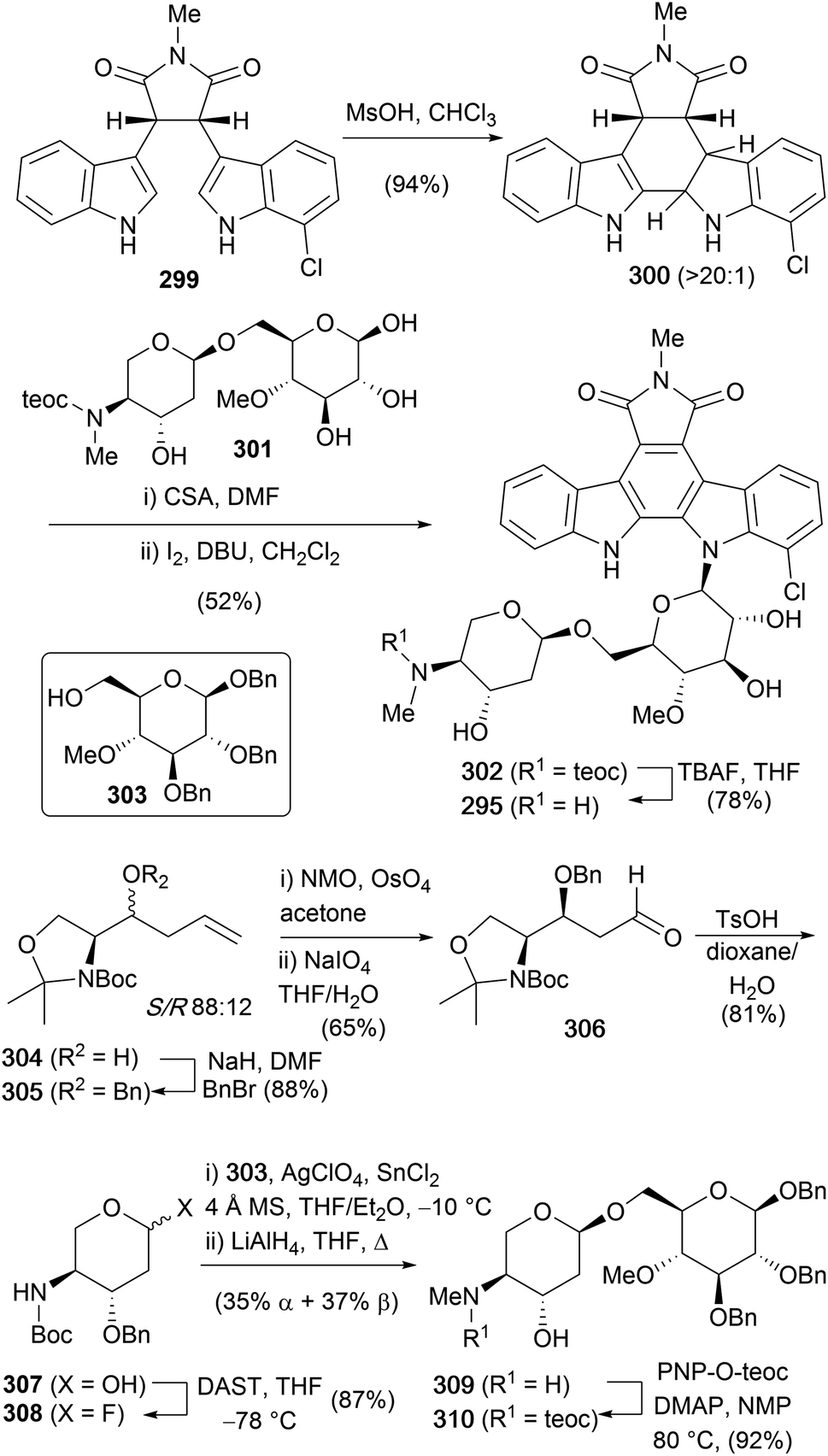 | ||
| Scheme 44 Van Vranken and co-workers' synthesis of AT2433 A1 (295). | ||
In addition, the same group also completed the first total synthesis of AT2433-B1 (297) (Scheme 45).159 Indolene 311 was prepared in an analogous fashion to chloroindolene 300, before coupling with disaccharide 301 and DDQ oxidation of the mixture of diastereoisomers provided indolocarbazole 312 in excellent yield. Removal of the teoc group was achieved with TBAF to complete the first synthesis of AT2433-B1 (297).
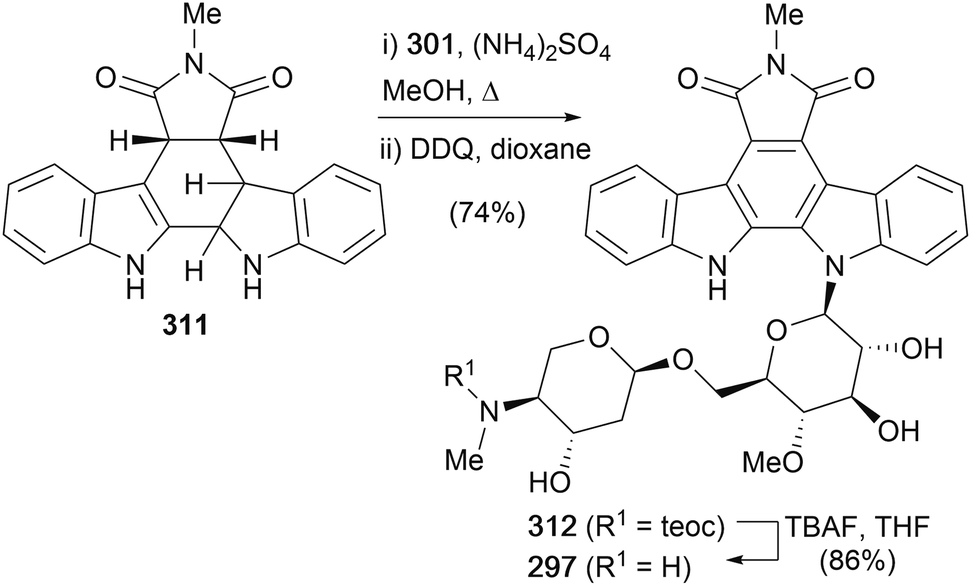 | ||
| Scheme 45 Van Vranken and co-workers' synthesis of AT2433 B1 (297). | ||
6 Conclusions
Total synthesis continues to be one of the most exciting areas of research, with papers published in the field regarded as some of the most-read manuscripts in chemistry focussed journals.181 In addition to providing a proving ground for the generality and utility of synthetic methodology, it offers access to bioactive compounds, key to advancing biological and medical science. The indolocarbazoles are prime examples, displaying extraordinary bioactivity and molecular complexity. As evident in this review, this has led to a plethora of synthetic endeavours towards indolocarbazoles. Realisation of the important bioactivities exhibited by many of these compounds spurred a greater emphasis on isolating large quantities of these materials, particularly the staurosporine aglycone (10) and arcyriaflavin A (45). The synthesis of the aglycone developed by process department at Eli Lily is a great example;55 concise, efficient and scalable, whilst also allowing access bisindolylmaleimide (44) and arcyriaflavin A (45), and their derivatives.Indolocarbazole natural products bearing glycosidic linkages present a different synthetic challenge, where regioselectivity is hampered by the remote nature of the asymmetry in the aglycone and sugar fragments. Wood and co-workers diazo-insertion approach and Danishefsky and co-workers epoxide-glycosylation have enabled the only syntheses of staurosporine (7) to date, however regioselectivity issues were not fully addressed during these remarkable approaches. Subsequent synthetic endevours, namely by Fukuyama and co-workers on K252a (9) and Van Vranken and co-workers on AT2433-A1 (295) have worked to resolve regioselectivity issues. Yet to date, a synthesis of staurosporine, that would enable isolation of large quantities of both the natural compound as well as interesting pharmaceutically relevant derivatives remains elusive. As the cost of the natural product is high, its utility as starting point for SAR studies and optimisation is restricted, and thus, total synthesis remains an attractive option.
The FDA approval of Midostaurin in 2017 for the treatment of acute myeloid leukemia (AML), highlights the remarkable bioactivity of indolocarbazoles and enduring and evolving interest. The current understanding of cancer therapy dictates a targeted strategy where a key cellular pathway is identified, signalling cascade biologically dissected and one target enzyme is selectively inhibited. This allows patient stratification according to a biomarker and applying a relatively specific drug to treat a condition. In fact, the use of Midostaurin in AML has been approved due to its inhibitory activity on FLT3 kinase, not PKC. However, in the relevant clinical trials, Midostaurin showed clinical efficacy in FLT3 wild type (non-mutated) and mutant subgroups suggesting its clinical benefit extends beyond the aimed association with a biomarker but still allowed clinical translation.182 Similar observations were made with other staurosporine derivatives such as 7-hydroxystaurosporine (UCN-01) during early phase clinical trials in which unstratified patient groups were used.183 Therefore the anti-cancer activity of Midostaurin and UCN-01 PKC inhibitors are not in question but their association with a biological condition is missing at the moment.
In recent years characterisation of cancer stem cells have paved the way for specific drugs to inhibit cancer metastasis. PKC inhibitors were identified to specifically inhibit cancer stem cell formation both in vitro and in vivo.184,185 Indolocarbazole derivatives may have an important role in these studies, and may even lead to future therapies. Synthetic approaches to these natural products and derivatives will be paramount to advancements, and this review is intended as a useful tool to aid access to these exciting scaffolds. In conclusion, indolocarbazole natural products have an enduring potential as leads for development of new medicines, and inspiration for new synthetic methodologies and approaches. From a biological perspective, interest indolocarbazoles is likely to be maintained well into the future as more biological pathways and patient treatment regimens are revealed.
7 Author contributions
All authors were involved in conceptualisation and writing (review & editing) of the manuscript. GEC was additionally responsible for writing the original draft.8 Conflicts of interest
There are no conflicts to declare.9 Acknowledgements
The authors gratefully acknowledge the European France–(Manche)–England cross-border cooperation program INTERREG V A “LABFACT”, co-financed by ERDF, and EPSRC (EP/R513325/1, DTP Research Studentship for GEC) for financial support.10 Notes and references
- J. Bergman, T. Janosik and N. Wahlström, Adv. Heterocycl. Chem., 2001, 80, 1–71 Search PubMed.
- T. Janosik, N. Wahlström and J. Bergman, Tetrahedron, 2008, 64, 9159–9180 Search PubMed.
- T. Janosik, A. Rannug, U. Rannug, N. Wahlström, J. Slätt and J. Bergman, Chem. Rev., 2018, 118, 9058–9128 Search PubMed.
- C. Sánchez, C. Méndez and J. A. Salas, Nat. Prod. Rep., 2006, 23, 1007–1045 Search PubMed.
- H. Nakano and S. Omura, J. Antibiot., 2009, 62, 17–26 Search PubMed.
- G. Gribble and S. Berthel, Studies in Natural Products Chemistry, Elsevier Science Publishers, New York, vol. 72, 1993 Search PubMed.
- M. Prudhomme, Curr. Pharm. Des., 1997, 3, 265–290 Search PubMed.
- U. Pindur, Y. S. Kim and F. Mehrabani, Curr. Med. Chem., 1999, 6, 26–69 Search PubMed.
- M. Prudhomme, Curr. Med. Chem.: Anti-Cancer Agents, 2004, 4, 509–521 Search PubMed.
- J. A. Salas and C. Méndez, Curr. Opin. Chem. Biol., 2009, 13, 152–160 Search PubMed.
- M. Prudhomme, M. Sancelme, A. Bonnefoy, D. Fabbro and T. Meyer, Eur. J. Med. Chem., 1999, 34, 161–165 Search PubMed.
- M. Prudhomme, Eur. J. Med. Chem., 2003, 38, 123–140 Search PubMed.
- S. Omura, Y. Iwai, A. Hirano, A. Nakagawa, J. Awaya, H. Tsuchya, Y. Takahashi and R. Masuma, J. Antibiot., 1977, 30, 275–282 Search PubMed.
- N. Funato, H. Takayanagi, Y. Konda, Y. Toda, Y. Harigaya, Y. Iwai and S. Omura, Tetrahedron Lett., 1994, 35, 1251–1254 Search PubMed.
- S. Omura, Y. Sasaki, Y. Iwai and H. Takeshima, J. Antibiot., 1994, 48, 535–548 Search PubMed.
- D. E. Nettleton, T. W. Doyle, B. Krishnan, G. K. Matsumoto and J. Clardy, Tetrahedron Lett., 1985, 26, 4011–4014 Search PubMed.
- M. Sezaki, T. Sasaki, T. Nakazawa, U. Takeda, M. Iwata, T. Watanabe, M. Koyama, F. Kai, T. Shomura and M. Kojima, J. Antibiot., 1985, 38, 1437–1439 Search PubMed.
- H. Kase, K. Iwahashi and Y. Matsuda, J. Antibiot., 1986, 39, 1059–1065 Search PubMed.
- S. Nakanishi, Y. Matsuda, K. Iwahashi and H. Kase, J. Antibiot., 1986, 39, 1066–1071 Search PubMed.
- H. J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4427 Search PubMed.
- H. Kase, K. Iwahashi, S. Nakanishi, Y. Matsuda, K. Yamada, M. Takahashi, C. Murakata, A. Sato and M. Kaneko, Biochem. Biophys. Res. Commun., 1987, 142, 436–440 Search PubMed.
- J. Budworth and A. Gescher, FEBS Lett., 1995, 362, 139–142 Search PubMed.
- T. Tamaoki, H. Nomoto, I. Takahashi, Y. Kato, M. Morimoto and F. Tomita, Biochem. Biophys. Res. Commun., 1986, 135, 397–402 Search PubMed.
- X. D. Zhang, S. K. Gillespie and P. Hersey, Mol. Cancer Ther., 2004, 3, 187–197 Search PubMed.
- B. Sarstedt and E. Winterfeldt, Heterocycles, 1983, 20, 469–476 Search PubMed.
- J. E. McMurry, Acc. Chem. Res., 1974, 7, 281–286 Search PubMed.
- I. Hughes and R. A. Raphael, Tetrahedron Lett., 1983, 24, 1441–1444 Search PubMed.
- I. Hughes, W. P. Nolan and R. A. Raphael, J. Chem. Soc., Perkin Trans. 1, 1990, 2475–2480 Search PubMed.
- J. H. Brewster, A. M. Fusco, L. E. Carosino and B. G. Corman, J. Org. Chem., 1963, 28, 498–501 Search PubMed.
- J. C. Hubert, J. B. P. A. Wijnberg and W. N. Speckamp, Tetrahedron, 1975, 31, 1437–1441 Search PubMed.
- J. Auerbach, M. Zamore and S. M. Weinreb, J. Org. Chem., 1976, 41, 725–726 Search PubMed.
- J. I. G. Cadogan, Synthesis, 1969, 11–17 Search PubMed.
- P. D. Magnus and N. L. Sear, Tetrahedron, 1984, 40, 2795–2797 Search PubMed.
- S. O. De Silva and V. Snieckus, Can. J. Chem., 1978, 56, 1621–1627 Search PubMed.
- W. Steglich, B. Steffan, L. Kopanski and G. Eckhardt, Angew. Chem., Int. Ed., 1980, 19, 459–460 Search PubMed.
- M. Brenner, H. Rexhausen, B. Steffan and W. Steglich, Tetrahedron, 1988, 44, 2887–2892 Search PubMed.
- R. P. Joyce, J. A. Gainor and S. M. Weinreb, J. Org. Chem., 1987, 52, 1177–1185 Search PubMed.
- C. J. Moody and K. F. Rahimtoola, J. Chem. Soc., Chem. Commun., 1990, 1667–1668 Search PubMed.
- C. J. Moody, K. F. Rahimtoola, B. Porter and B. C. Ross, J. Org. Chem., 1992, 57, 2105–2114 Search PubMed.
- C. J. Moody, P. Shah and P. Knowles, J. Chem. Soc., Perkin Trans. 1, 1988, 3249–3254 Search PubMed.
- C. J. Moody and K. F. Rahimtoola, J. Chem. Soc., Perkin Trans. 1, 1990, 673–679 Search PubMed.
- W. Harris, C. H. Hill, E. Keech and P. Malsher, Tetrahedron Lett., 1993, 34, 8361–8364 Search PubMed.
- G. Xie and J. William Lown, Tetrahedron Lett., 1994, 35, 5555–5558 Search PubMed.
- S. W. McCombie, R. W. Bishop, D. Carr, E. Dobek, M. P. Kirkup, P. Kirschmeier, S. I. Lin, J. Petrin, K. Rosinski and B. B. Shankar, et al. , Bioorg. Med. Chem. Lett., 1993, 3, 1537–1542 Search PubMed.
- S. Fabre, M. Prudhomme and M. Rapp, Bioorg. Med. Chem., 1993, 1, 193–196 Search PubMed.
- H. Ishii, M. R. Jirousek, D. Koya, C. Takagi, P. Xia, A. Clermont, S. E. Bursell, T. S. Kern, L. M. Ballas and W. F. Heath, et al. , Science, 1996, 272, 728–731 Search PubMed.
- M. R. Jirousek, J. R. Gillig, C. M. Gonzalez, W. F. Heath, J. H. McDonald, D. A. Neel, C. J. Rito, U. Singh, L. E. Stramm and A. Melikian-Badalian, et al. , J. Med. Chem., 1996, 39, 2664–2671 Search PubMed.
- M. M. Faul, K. A. Sullivan and L. L. Winneroski, Synthesis, 1995, 1511–1516 Search PubMed.
- G. E. Burtin, D. J. Madge and D. L. Selwood, Heterocycles, 2000, 53, 2119–2122 Search PubMed.
- J. L. Wood, B. M. Stoltz and H. J. Dietrich, J. Am. Chem. Soc., 1995, 117, 10413–10414 Search PubMed.
- J. L. Wood, B. M. Stoltz, S. N. Goodman and K. Onwueme, J. Am. Chem. Soc., 1997, 119, 9652–9661 Search PubMed.
- J. Bergman, E. Koch and B. Pelcman, Tetrahedron, 1995, 51, 5631–5642 Search PubMed.
- G. Lowe and H. W. Yeung, J. Chem. Soc., Perkin Trans. 1, 1973, 2907–2910 Search PubMed.
- V. J. Lee, A. R. Branfman, T. R. Herrin and K. L. Rinehart, J. Am. Chem. Soc., 1978, 100, 4225–4236 Search PubMed.
- M. M. Faul, L. L. Winneroski and C. A. Krumrich, J. Org. Chem., 1998, 63, 6053–6058 Search PubMed.
- E. M. Beccalli, M. L. Gelmi and A. Marchesini, Tetrahedron, 1998, 54, 6909–6918 Search PubMed.
- E. M. Beccalli, A. Marchesini and T. Pilati, Tetrahedron, 1993, 49, 4741–4758 Search PubMed.
- S. Achab, Tetrahedron Lett., 1996, 37, 5503–5506 Search PubMed.
- L. S. Liebeskind and R. W. Fengl, J. Org. Chem., 1990, 55, 5359–5364 Search PubMed.
- V. Farina, Pure Appl. Chem., 1996, 68, 73–78 Search PubMed.
- S. Mahboobi, E. Eibler, M. Koller, S. Kumar KC, A. Popp and D. Schollmeyer, J. Org. Chem., 1999, 64, 4697–4704 Search PubMed.
- S. Mahboobi, G. Grothus and E. von Angerer, Arch. Pharm., 1994, 327, 481–492 Search PubMed.
- S. Mahboobi, A. Popp and D. Schollmeyer, Tetrahedron: Asymmetry, 1998, 9, 2369–2376 Search PubMed.
- G. Büchi and C. P. Mak, J. Org. Chem., 1977, 42, 1784–1786 Search PubMed.
- R. Nomak and J. K. Snyder, Tetrahedron Lett., 2001, 42, 7929–7933 Search PubMed.
- D. L. Boger, R. S. Coleman, J. S. Panek and D. Yohannes, J. Org. Chem., 1984, 49, 4405–4409 Search PubMed.
- S. C. Benson, C. A. Palabrica and J. K. Snyder, J. Org. Chem., 1987, 52, 4610–4614 Search PubMed.
- K. Daly, R. Nomak and J. K. Snyder, Tetrahedron Lett., 1997, 38, 8611–8614 Search PubMed.
- A. Basha, M. Lipton and S. M. Weinreb, Tetrahedron Lett., 1977, 18, 4171–4172 Search PubMed.
- J. F. Hartwig, M. Kawatsura, S. I. Hauck, K. H. Shaughnessy and L. M. Alcazar-Roman, J. Org. Chem., 1999, 64, 5575–5580 Search PubMed.
- S. P. Gaudêncio, M. M. M. Santos, A. M. Lobo and S. Prabhakar, Tetrahedron Lett., 2003, 44, 2577–2578 Search PubMed.
- S. Hamilakis, D. Kontonassios and C. Sandris, J. Heterocycl. Chem., 1996, 33, 825–829 Search PubMed.
- M. M. M. Santos, A. M. Lobo, S. Prabhakar and M. M. B. Marques, Tetrahedron Lett., 2004, 45, 2347–2349 Search PubMed.
- G. M. Reddy, S. Y. Chen and B. J. Uang, Synthesis, 2003, 497–500 Search PubMed.
- Y. Wada, H. Nagasaki, M. Tokuda and K. Orito, J. Org. Chem., 2007, 72, 2008–2014 Search PubMed.
- J. W. Lown and G. L. Weir, Can. J. Chem., 1978, 56, 249–257 Search PubMed.
- M. Iwao and O. Motoi, Tetrahedron Lett., 1995, 36, 5929–5932 Search PubMed.
- G. W. Gribble and B. Pelcman, J. Org. Chem., 1992, 57, 3636–3642 Search PubMed.
- E. J. Gilbert and D. L. Van Vranken, J. Am. Chem. Soc., 1996, 118, 5500–5501 Search PubMed.
- T. Satoh, S. Suzuki, Y. Suzuki, Y. Miyaji and Z. Imai, Tetrahedron Lett., 1969, 10, 4555–4558 Search PubMed.
- G. G. Rajeshwaran and A. K. Mohanakrishnan, Org. Lett., 2011, 13, 1418–1421 Search PubMed.
- J. S. Yadav, B. V. S. Reddy and K. Praneeth, Tetrahedron Lett., 2008, 49, 199–202 Search PubMed.
- E. T. Pelkey and G. W. Gribble, Tetrahedron Lett., 1997, 38, 5603–5606 Search PubMed.
- P. Raju, G. G. Rajeshwaran and A. K. Mohanakrishnan, Eur. J. Org. Chem., 2015, 7131–7145 Search PubMed.
- J. C. Fox, R. E. Gilligan, A. K. Pitts, H. R. Bennett and M. J. Gaunt, Chem. Sci., 2016, 7, 2706–2710 Search PubMed.
- C. L. Ciana, R. J. Phipps, J. R. Brandt, F. M. Meyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 458–462 Search PubMed.
- R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593–1597 Search PubMed.
- C. L. Coon, W. G. Blucher and M. E. Hill, J. Org. Chem., 1973, 38, 4243–4248 Search PubMed.
- S. Mataka, G. B. Liu, T. Sawada, M. Kurisu and M. Tashiro, Bull. Chem. Soc. Jpn., 1994, 67, 1113–1119 Search PubMed.
- N. J. Truax, F. Banales Mejia, D. O. Kwansare, M. M. Lafferty, M. H. Kean and E. T. Pelkey, J. Org. Chem., 2016, 81, 6808–6815 Search PubMed.
- A. Mallinger, B. Nadal, N. Chopin and T. Le Gall, Eur. J. Org. Chem., 2010, 1142–1148 Search PubMed.
- M. M. Faul and K. A. Sullivan, Tetrahedron Lett., 2001, 42, 3271–3273 Search PubMed.
- S. Saha, A. Banerjee and M. S. Maji, Org. Lett., 2018, 20, 6920–6924 Search PubMed.
- K. Ozaki, H. Zhang, H. Ito, A. Lei and K. Itami, Chem. Sci., 2013, 4, 3416–3420 Search PubMed.
- S. Sahu, A. Banerjee and M. S. Maji, Org. Lett., 2017, 19, 464–467 Search PubMed.
- A. Banerjee, S. Sahu and M. S. Maji, Adv. Synth. Catal., 2017, 359, 1860–1866 Search PubMed.
- J. I. G. Cadogan, M. Cameron-Wood, R. K. Mackie and R. J. G. Searle, J. Chem. Soc., 1965, 4831–4837 Search PubMed.
- S. Singh, R. Samineni, S. Pabbaraja and G. Mehta, Org. Lett., 2019, 21, 3372–3376 Search PubMed.
- S. Singh, R. Samineni, S. Pabbaraja and G. Mehta, Angew. Chem., Int. Ed., 2018, 57, 16847–16851 Search PubMed.
- H. G. Selnick, G. R. Smith and A. J. Tebben, Synth. Commun., 1995, 25, 3255–3261 Search PubMed.
- R. L. Hudkins and C. H. Park, J. Heterocycl. Chem., 2003, 40, 135–142 Search PubMed.
- W. Steglich, Pure Appl. Chem., 1981, 53, 1233–1240 Search PubMed.
- M. Gill and W. Steglich, Progress in the Chemistry of Organic Natural Products, Springer-Verlag/Wien, 1987, vol. 51 Search PubMed.
- W. Steglich, Pure Appl. Chem., 1989, 61, 281–288 Search PubMed.
- S. Hoshino, L. Zhang, T. Awakawa, T. Wakimoto, H. Onaka and I. Abe, J. Antibiot., 2015, 68, 342–344 Search PubMed.
- J. Bergman and B. Pelcman, Tetrahedron Lett., 1987, 28, 4441–4444 Search PubMed.
- J. L. Belletire, E. G. Spletzer and A. R. Pinhas, Tetrahedron Lett., 1984, 25, 5969–5972 Search PubMed.
- J. L. Belletire and S. L. Fremont, Tetrahedron Lett., 1986, 27, 127–130 Search PubMed.
- J. L. Belletire and E. G. Spletzer, Tetrahedron Lett., 1986, 27, 131–134 Search PubMed.
- J. Bergman and B. Pelcman, J. Org. Chem., 1989, 54, 824–828 Search PubMed.
- K. Yamamoto and H. Watanabe, Chem. Lett., 1982, 11, 1225–1228 Search PubMed.
- P. Magnus, T. Gallagher, J. Schultz, Y. S. Or and T. P. Ananthanarayan, J. Am. Chem. Soc., 1987, 109, 2706–2711 Search PubMed.
- G. W. Gribble and S. J. Berthel, Tetrahedron, 1992, 48, 8869–8880 Search PubMed.
- S. C. Conway and G. W. Gribble, Heterocycles, 1992, 34, 2095–2108 Search PubMed.
- M. Somei and A. Kodama, Heterocycles, 1992, 34, 1285–1288 Search PubMed.
- J. F. Barry, T. W. Wallace and N. D. A. Walshe, Tetrahedron, 1995, 51, 12797–12806 Search PubMed.
- M. Somei, H. Hayashi, T. Izumi and S. Ohmoto, Heterocycles, 1995, 41, 2161–2164 Search PubMed.
- M. Somei, F. Yamada, J. Kato, Y. Suzuki and Y. Ueda, Heterocycles, 2002, 56, 81–84 Search PubMed.
- U. Pindur, Y. S. Kim and D. Schollmeyer, J. Heterocycl. Chem., 1995, 32, 1335–1339 Search PubMed.
- U. Pindur and M. H. Kim, Arch. Pharm., 1992, 325, 353–356 Search PubMed.
- M. G. Saulnier, D. B. Frennesson, M. S. Deshpande and D. M. Vyas, Tetrahedron Lett., 1995, 36, 7841–7844 Search PubMed.
- A. Arcadi, S. Cacchi and F. Marinelli, Tetrahedron Lett., 1989, 30, 2581–2584 Search PubMed.
- T. M. Cresp, J. Ojima and F. Sondheimer, J. Org. Chem., 1977, 42, 2130–2134 Search PubMed.
- A. P. Fonseca, A. M. Lobo and S. Prabhakar, Tetrahedron Lett., 1995, 36, 2689–2692 Search PubMed.
- R. L. N. Harris, Tetrahedron Lett., 1969, 10, 4465–4466 Search PubMed.
- M. M. B. Marques, A. M. Lobo, S. Prabhakar and P. S. Branco, Tetrahedron Lett., 1999, 40, 3795–3796 Search PubMed.
- J. Bergman and C. Stlhandske, Tetrahedron Lett., 1994, 35, 5279–5282 Search PubMed.
- M. M. B. Marques, M. M. M. Santos, A. M. Lobo and S. Prabhakar, Tetrahedron Lett., 2000, 41, 9835–9838 Search PubMed.
- M. Adeva, F. Buono, E. Caballero, M. Medarde and F. Tomé, Synlett, 2000, 832–834 Search PubMed.
- M. Adeva, E. Caballero, F. García, M. Medarde, H. Sahagún and F. Tomé, Tetrahedron Lett., 1997, 38, 6893–6896 Search PubMed.
- J. Wang, M. Rosingana, D. J. Watson, E. D. Dowdy, R. P. Discordia, N. Soundarajan and W. S. Li, Tetrahedron Lett., 2001, 42, 8935–8937 Search PubMed.
- H. J. Knölker and W. Fröhner, J. Chem. Soc., Perkin Trans. 1, 1998, 173–176 Search PubMed.
- H. J. Knölker, K. R. Reddy and A. Wagner, Tetrahedron Lett., 1998, 39, 8267–8270 Search PubMed.
- H. J. Knölker and N. O’Sullivan, Tetrahedron, 1994, 50, 10893–10908 Search PubMed.
- X. Pan, K. Wang, B. H. Guan and Z. Z. Liu, J. Heterocycl. Chem., 2015, 52, 911–913 Search PubMed.
- K. Wang and Z. Liu, Synth. Commun., 2009, 40, 144–150 Search PubMed.
- P. D. Davis and R. A. Bit, Tetrahedron Lett., 1990, 31, 5201–5204 Search PubMed.
- P. Torney, R. Shirsat and S. Tilve, Synlett, 2014, 25, 2121–2126 Search PubMed.
- P. T. Parvatkar, P. S. Parameswaran and S. G. Tilve, Chem.–Eur. J., 2012, 18, 5460–5489 Search PubMed.
- A. Aponick, C. Y. Li and J. A. Palmes, Org. Lett., 2009, 11, 121–124 Search PubMed.
- P. H. S. Paioti, J. M. Ketcham and A. Aponick, Org. Lett., 2014, 16, 5320–5323 Search PubMed.
- T. Kline, M. Fromhold, T. E. McKennon, S. Cai, J. Treiberg, N. Ihle, D. Sherman, W. Schwan, M. J. Hickey and P. Warrener, et al. , Bioorg. Med. Chem., 2000, 8, 73–93 Search PubMed.
- I. Pérez, J. P. Sestelo and L. A. Sarandeses, J. Am. Chem. Soc., 2001, 123, 4155–4160 Search PubMed.
- R. Sanz, J. Escribano, M. R. Pedrosa, R. Aguado and F. Arnáiz, Adv. Synth. Catal., 2007, 349, 713–718 Search PubMed.
- N. V. Borrero, L. G. DeRatt, L. Ferreira Barbosa, K. A. Abboud and A. Aponick, Org. Lett., 2015, 17, 1754–1757 Search PubMed.
- S. Lee, K. H. Kim and C. H. Cheon, Org. Lett., 2017, 19, 2785–2788 Search PubMed.
- S. J. Lee, H. A. Seo and C. H. Cheon, Adv. Synth. Catal., 2016, 358, 1566–1570 Search PubMed.
- H. A. Seo and C. H. Cheon, J. Org. Chem., 2016, 81, 7917–7923 Search PubMed.
- Y. Men, Z. Hu, J. Dong, X. Xu and B. Tang, Org. Lett., 2018, 20, 5348–5352 Search PubMed.
- M. Ohkubo, T. Nishimura, H. Jona, T. Honma and H. Morishima, Tetrahedron, 1996, 52, 8099–8112 Search PubMed.
- D. H. Lloyd and D. E. Nichols, J. Org. Chem., 1986, 51, 4294–4295 Search PubMed.
- E. S. Kim, Drugs, 2017, 77, 1251–1259 Search PubMed.
- J. T. Link, S. Raghavan and S. J. Danishefsky, J. Am. Chem. Soc., 1995, 117, 552–553 Search PubMed.
- J. T. Link, S. Raghavan, M. Gallant, S. J. Danishefsky, T. C. Chou and L. M. Ballas, J. Am. Chem. Soc., 1996, 118, 2825–2842 Search PubMed.
- J. T. Link, M. Gallant, S. J. Danishefsky and S. Huber, J. Am. Chem. Soc., 1993, 115, 3782–3783 Search PubMed.
- M. Gallant, J. T. Link and S. J. Danishefsky, J. Org. Chem., 1993, 58, 343–349 Search PubMed.
- J. T. Link and S. J. Danishefsky, Tetrahedron Lett., 1994, 35, 9135–9138 Search PubMed.
- J. L. Wood, B. M. Stoltz and S. N. Goodman, J. Am. Chem. Soc., 1996, 118, 10656–10657 Search PubMed.
- J. D. Chisholm and D. L. Van Vranken, J. Org. Chem., 2000, 65, 7541–7553 Search PubMed.
- G. Zhang, J. Shen, H. Cheng, L. Zhu, L. Fang, S. Luo, M. T. Muller, G. E. Lee, L. Wei and Y. Du, et al. , J. Med. Chem., 2005, 48, 2600–2611 Search PubMed.
- P. Moreau, F. Anizon, M. Sancelme, M. Prudhomme, C. Bailly, D. Sevère, J. F. Riou, D. Fabbro, T. Meyer and A. M. Aubertin, J. Med. Chem., 1999, 42, 584–592 Search PubMed.
- Y. Yamashita, N. Fujii, C. Murakata, T. Ashizawa, M. Okabe and H. Nakano, Biochemistry, 1992, 31, 12069–12075 Search PubMed.
- F. Anizon, L. Belin, P. Moreau, M. Sancelme, A. Voldoire, M. Prudhomme, M. Ollier, D. Sevère, J. F. Riou and C. Bailly, et al. , J. Med. Chem., 1997, 40, 3456–3465 Search PubMed.
- T. Kaneko, H. Wong, K. T. Okamoto and J. Clardy, Tetrahedron Lett., 1985, 26, 4015–4018 Search PubMed.
- T. Sugasawa, M. Adachi, K. Sasakura and A. Kitagawa, J. Org. Chem., 1979, 44, 578–586 Search PubMed.
- H. O. Bouveng, B. Lindberg and O. Theander, Acta Chem. Scand., 1957, 11, 1788–1789 Search PubMed.
- P. W. Sadler and R. L. Warren, J. Am. Chem. Soc., 1956, 78, 1251–1255 Search PubMed.
- J. Bergman and N. Eklund, Tetrahedron, 1980, 36, 1439–1443 Search PubMed.
- C. Cruzado, M. Bernabé and M. Martín-Lomas, J. Org. Chem., 1989, 54, 465–469 Search PubMed.
- M. M. Faul, L. L. Winneroski and C. A. Krumrich, J. Org. Chem., 1999, 64, 2465–2470 Search PubMed.
- R. G. Zenkov, L. V. Ektova, O. A. Vlasova, G. A. Belitskiy, M. G. Yakubovskaya and K. I. Kirsanov, Chem. Heterocycl. Compd., 2020, 56, 644–658 Search PubMed.
- A. A. Panov, A. Y. Simonov, S. N. Lavrenov, S. A. Lakatosh and A. S. Trenin, Chem. Heterocycl. Compd., 2018, 54, 103–113 Search PubMed.
- T. B. Lowinger, J. Chu and P. L. Spence, Tetrahedron Lett., 1995, 36, 8383–8386 Search PubMed.
- Y. Kobayashi, T. Fujimoto and T. Fukuyama, J. Am. Chem. Soc., 1999, 121, 6501–6502 Search PubMed.
- N. S. Girgis, H. B. Cottam and R. K. Robins, J. Heterocycl. Chem., 1988, 25, 361–366 Search PubMed.
- J. A. Matson, C. Claridge, J. A. Bush, J. Titus, W. T. Bradner, T. W. Doyle, A. C. Horan and M. Patel, J. Antibiot., 1989, 42, 1547–1555 Search PubMed.
- J. Golik, T. W. Doyle, B. Krishnan, G. Dubay and J. A. Matson, J. Antibiot., 1989, 42, 1784–1789 Search PubMed.
- J. D. Chisholm and D. L. Van Vranken, J. Org. Chem., 1995, 60, 6672–6673 Search PubMed.
- D. S. Carter and D. L. Van Vranken, Tetrahedron Lett., 1996, 37, 5629–5632 Search PubMed.
- W. R. Roush and J. A. Hunt, J. Org. Chem., 1995, 60, 798–806 Search PubMed.
- P. S. Baran, J. Am. Chem. Soc., 2018, 140, 4751–4755 Search PubMed.
- R. M. Stone, S. J. Mandrekar, B. L. Sanford, K. Laumann, S. Geyer, C. D. Bloomfield, C. Thiede, T. W. Prior, K. Döhner and G. Marcucci, et al. , N. Engl. J. Med., 2017, 377, 454–464 Search PubMed.
- P. N. Lara, P. C. Mack, T. Synold, P. Frankel, J. Longmate, P. H. Gumerlock, J. H. Doroshow and D. R. Gandara, Clin. Cancer Res., 2005, 11, 4444–4450 Search PubMed.
- W. L. Tam, H. Lu, J. Buikhuisen, B. S. Soh, E. Lim, F. Reinhardt, Z. J. Wu, J. A. Krall, B. Bierie and W. Guo, et al. , Cancer Cell, 2013, 24, 347–364 Search PubMed.
- R. Sreekumar, M. Emaduddin, H. Al-Saihati, K. Moutasim, J. Chan, M. Spampinato, R. Bhome, H. M. Yuen, C. Mescoli and A. Vitale, et al. , Cell Death Dis., 2019, 10, 703 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |