
Water stable fluorescent organotin(IV) compounds: aggregation induced emission enhancement and recognition of lead ions in an aqueous system†
Kulwinder
Kaur
a,
Raghubir
Singh
 b,
Varinder
Kaur
*a and
Neena
Capalash
c
b,
Varinder
Kaur
*a and
Neena
Capalash
c
aDepartment of Chemistry, Panjab University, Sector-14, Chandigarh-160014, India. E-mail: var_ka04@yahoo.co.in
bDepartment of Chemistry, DAV College, Sector 10, Chandigarh-160011, India. E-mail: raghu_chem2006@yahoo.com
cDepartment of Biotechnology, Panjab University, Chandigarh-160014, India. E-mail: caplash@pu.ac.in
First published on 20th November 2021
Abstract
Herein, synthesis, spectroscopic studies, single-crystal X-ray diffraction, aggregation-induced emission enhancement (AIEE) and sensing application of water-stable organotin(IV) compounds (4a–6a and 4b–6b) obtained from 3-hydroxy-4H-chromen-4-one ligands are reported. All the synthesized organotin(IV) compounds were characterized using elemental analysis, FT-IR spectroscopy, multi-nuclei NMR (1H, 13C, and 119Sn) spectroscopy, UV–VIS, fluorescence spectroscopy, mass spectrometry, and single-crystal X-ray diffraction. The 119Sn NMR signal of compounds in the range of δ −144.92 to −190.68 ppm indicated the formation of hexacoordinated organotin species. The spectroscopic and single-crystal X-ray diffraction studies confirmed the formation of [L2SnR2] type compounds (where L is the bidentate ligand and R is an alkyl group) with a ‘skew-trapezoidal bipyramidal’ geometry. Furthermore, DFT calculations of compound 4b based on the DGDZVP basis set fully supported the stability of the structure where two short bonds Sn–O(C–O) acquire the cis position rather than the trans position. Single-crystal X-ray diffraction analysis of the crystals grown in the presence of water confirmed the stability of 4a in water. Moreover, the water stability of a test compound 4a was established by 119Sn NMR data and spectrofluorimetric data. The spectrofluorimetric scan at different time intervals revealed the stability and constant emission response up to 24 h. The compounds were found to be fluorescent and exhibited aggregation-induced emission enhancement in MeOH/H2O mixtures, which was confirmed by HRTEM analysis. The test compound 4a showed selective spectrofluorimetric recognition of Pb2+ ions in an aqueous medium by displaying an enhanced emission signal at 478 nm and enabled detection up to 22.66 μM. A mechanism of interaction is also proposed by spectroscopic experiments, spectrofluorimetric experiments and computational studies.
1. Introduction
The diversified structural features, hypercoordination, simple synthetic protocols, and convenient characterization procedures have established organotin(IV) compounds to be a highly versatile and interesting class of coordination compounds.1,2 Organotin(IV) compounds are well-known for their catalytic utility,3 anti-microbial properties4 (such as disinfectants, marine antifouling paints, pesticides, fungicides, antibacterial agents, and wood preservatives) and antitumor activity.5 Recently organotin(IV) compounds were mainly derived from 2-thienyl-selenoacetic acid (i),6 cyano-coumarin (ii),7 salicylaldehyde-o-aminophenol Schiff base (iii),8 thiocarbonohydrazone (iv, iv′),9 2/4-(2-hydroxynaphthylazo)-benzoic acids (v),10 and 2,6-di-tert-butylphenol derivatives (vi)11 (shown in the ESI†, Chart 1). Besides, organotin(IV) tricyclic cages with fascinating structural architectures are obtained from tripodal amine ligands.12–15Despite the wide spectrum of applications of organotin(IV) compounds, their exploration in aqueous systems is difficult due to their partial hydrolysis to form oxo/hydroxo-clusters or complete hydrolysis to form tin oxides.16 For instance, fluorescent organotin(IV) mononuclear compounds have been recently investigated for the recognition of chemical warfare agents and metal ions. However the developed protocols cannot be applied to the aqueous samples due to their insolubility or instability in water.17,18 Similarly, organotin(IV) compounds have been proved as anti-tumour agents with excellent activity, but their mode of interaction could not be developed in the aqueous system.19 Moreover, proper investigation of mechanistic aspects of organotin(IV) compounds in biological systems is also lacking due to inadequate structural data or physical properties to trace them inside the cell.20,21 Therefore, water stable and fluorescent organotin(IV) compounds (4a–6a and 4b–6b) have been synthesized in the present work. The structural attributes of the compounds are investigated so that the compounds may be further explored in the applications related to aqueous systems and biological systems.
The precursors used to obtain organotin(IV) compounds 4a–6a and 4b–6b, dialkyltin oxide and 3-hydroxy-4H-chromen-4-one compounds, were selected based on some salient features. The dialkyltin oxide (alkyl = butyl and methyl) was chosen instead of their chlorostannane analogues to avoid hydrolysis during the course of the reaction. Some biological investigations on organotin compounds revealed that the toxicity of organotin compounds increases with the increase in the number of hydrocarbyl groups, i.e. with the order R3Sn > R2Sn > RSn.22 Therefore, the usage of dialkylstannanes has been preferred over trialkylstannanes. Besides, 3-hydroxy-4H-chromen-4-one compounds as a ligand offer some advantages like these are comprised of electron-rich aromatic rings and impart fluorescence nature to the synthesized organotin(IV) compounds. These ligands consist of an easily ionizable hydroxyl group at the C-3 position of the benzopyrone moiety and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at the C-4 position. Therefore, 2
O at the C-4 position. Therefore, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 tin chelates are formed by the deprotonation of the hydroxyl group. The ligands are obtained in excellent yields via simple synthetic protocols i.e. the Algar–Flynn–Oyamada reaction and Claisen–Schmidt reaction.23,24 Although few reports on 3-hydroxy-4H-chromen-4-one derived organotin compounds (5a, 5b) have been published long back, however, their characterization techniques include elemental analysis, FTIR, and 119mSn Mössbauer only.25,26 The present report discusses actual structural attributes of all the synthesized compounds based on 1H NMR, 13C NMR, 119Sn NMR, and single-crystal X-ray diffraction analysis, which is a necessity to explore the organometallic compounds in various fields.
:1 tin chelates are formed by the deprotonation of the hydroxyl group. The ligands are obtained in excellent yields via simple synthetic protocols i.e. the Algar–Flynn–Oyamada reaction and Claisen–Schmidt reaction.23,24 Although few reports on 3-hydroxy-4H-chromen-4-one derived organotin compounds (5a, 5b) have been published long back, however, their characterization techniques include elemental analysis, FTIR, and 119mSn Mössbauer only.25,26 The present report discusses actual structural attributes of all the synthesized compounds based on 1H NMR, 13C NMR, 119Sn NMR, and single-crystal X-ray diffraction analysis, which is a necessity to explore the organometallic compounds in various fields.
The organotin(IV) compounds 4a–6a, and 4b crystallize in skew-trapezoidal bipyramidal geometry in which axially situated alkyl substituents are trans-disposed while equatorially bonded alike O-donors are coordinated in a cis-fashion. This type of coordination leads to the formation of beautiful packing structures in which aromatic skeletons form a layer and alkyl substituents protrude outwards like antennae. The hexacoordination at Sn(IV) provides stability to the compounds in aqueous systems. The layered structural packing imparts aggregation-induced emission enhancement (AIEE) in the water–methanol solvent system. This type of structural attribute leads to the emission enhancement in the presence of Pb2+ ions selectively. Therefore, a protocol is standardized to detect and quantify the lead ions in aqueous systems. The presence of lead ions is one of the major environmental concerns due to their fatal effects on the cardiovascular system, memory loss, muscle paralysis, and reproductive disorders. The present method is based on a simple technique (i.e. spectrofluorimetry) and offers the advantage of detecting the lead ions in the aqueous system without adjusting the pH of the system. Yatsimirsky and coworkers reported a similar work where the 3-hydroxyflavonediphenyltin(IV) complex (vii as shown in the ESI†, Chart 1) has been explored for the fluorometric detection of pyrophosphate by using chlorostannanes RxSnCl4−x as starting materials.27 However, this is the first report where organotin(IV) compounds are explored for the spectrofluorimetric detection of lead ions in aqueous systems.
2. Results and discussion
2.1. Synthetic aspect
The precursors, 2-hydroxychalcones 1a–3a, were synthesized by the Claisen–Schmidt reactions of 2-hydroxyacetophenone with different aromatic aldehydes viz. p-tolualdehyde, benzaldehyde, and 2-furancarboxaldehyde as per the reported methods.28,29 Then, 2-hydroxychalcone moieties were cyclized into respective 3-hydroxy-4H-chromen-4-one derivatives (3-hydroxy-benzo-γ-pyrone derivatives or chromones) 1b–3b by the Algar–Flynn–Oyamada reaction following reported methods.29,30 The 3-hydroxy-4H-chromen-4-one compounds were further reacted with dialkyltin oxide (Bu2SnO or Me2SnO) to achieve two series of hyper-coordinated organotin(IV) compounds 4a–6a (i.e. L2SnBu2) and 4b–6b (i.e. L2SnMe2), respectively (Scheme 1). The synthesized compounds were soluble in chloroform, methanol, toluene, dimethylsulfoxide, acetonitrile and tetrahydrofuran but were insoluble in water.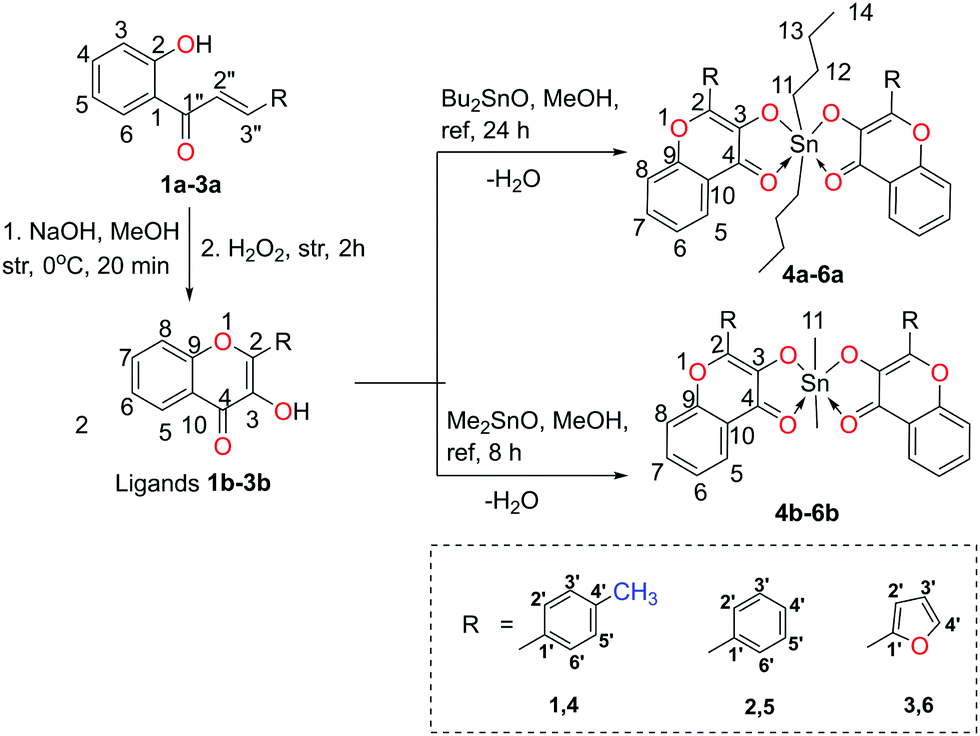 | ||
| Scheme 1 Synthetic route to obtain organotin(IV) compounds using dialkyltin oxide and 3-hydroxy-4H-chromen-4-one ligands (numbering of atoms in each structure is given to assign NMR signals). | ||
2.2. Spectroscopic aspects
The FT-IR spectra of organotin(IV) compounds 4a–6a and 4b–6b exhibited the vibrational bands for Sn–O and Sn–C bonds in the range of 470–495 cm−1 and 524–534 cm−1, respectively, which corroborated with the reported range (Sn–O: 410–470 cm−1 and Sn–C: 520–600 cm−1).26 Besides, the phenolic O–H vibrational band observed in the ligands (in the range of 3215–3276 cm−1) disappeared in the organotin(IV) compounds 4a–6a and 4b–6b suggesting the coordination via deprotonation. It was also supported by the absence of a 1H NMR singlet pertaining to the phenolic proton in the region of δ 6.92–7.04 ppm. The n-butyl and methyl protons appeared as upfield signals in ranges of δ 0.72–1.63 (in 4a–6a) and δ 0.84–0.85 ppm (in 4b–6b), respectively. The tin satellites were also observed due to spin–spin coupling 2J(1H-119Sn, H-11) and 2J(1H-117Sn, H-11) with J values in the range of 132–133 Hz and 84–92 Hz, respectively. In the 13C NMR spectra of 4a–6a and 4b–6b, the most downfield signal in the range of δ 174.63–177.17 ppm was attributed to the carbonyl carbon of the α,β-unsaturated moiety. Besides, the methyl and butyl signals resonated in the most upfield region i.e. in the range of δ 13.63–27.20 ppm in 4a–6a and δ 6.78–7.02 ppm in 4b–6b.Depending upon the nature of substituents and coordination number around the tin atom, the electron density around the tin nucleus varies, due to which the chemical shift value can adopt the range of +800 to −600 ppm in 119Sn NMR spectroscopy.1 The 119Sn NMR signal values vary with the variation of the size of the chelate rings. The general trend observed in the 119Sn NMR signal is as follows: most downfield signals in four-membered organotin(IV) chelates [for example, −148.1 ppm31 in the (Bu2Sn(O2C(CH2)3Ph)2 complex; −125 ppm in Me2Sn(benzoate)2;32 −114 ppm in Me2Sn(O2CC4H3S)233 and −141 ppm in Bu2Sn(O2CC4H3S)233)], intermediate in five-membered organotin(IV) chelates [for example, −197 ppm in Me2Sn(tropolonate)2;32 −262 ppm in Bu2Sn(oxinate)2;32 and −174 ppm in Me2Sn(kojate)234] and most upfield signal in six-membered chelates [for example, −365 ppm in Me2Sn(acetylacetonate)234]. The presence of sharp singlets in the range of δ −181.14 to −190.68 ppm for compounds 4a–6a and δ −144.92 to −154.04 ppm for 4b–6b in 119Sn NMR spectra (recorded in CDCl3) suggested the formation of hexacoordinated organotin(IV) compounds containing five-membered chelate rings.
Although 5a and 5b are reported in the literature, their spectroscopic data are not available for comparsion.25,26 The mass spectra of 4a–6a and 4b–6b followed a similar fragmentation pattern. A peak was observed at m/z 485.09, 471.08, 461.05, 401.01, 386.98, and 376.96 due to the [M–L + H]+ fragment in all the compounds. Apart from this, characteristic peaks were observed for some of the compounds. For example; peaks at m/z 679.08 and 651.06 appeared due to the loss of one butyl chain from 4a and 5a, respectively. Compounds 4b, 5b, and 6b showed molecular ion peaks as Na adducts at m/z 675.08, 647.03 and 626.98, respectively.
2.3. Structural aspects
Compounds 4a–6a and 4b were obtained in crystalline forms by evaporation of the solvent and their structures were elucidated by single-crystal X-ray diffraction. Selected bond lengths and bond angles for these compounds are presented in Table 1, and their crystallographic data and structure refinement parameters are listed in Table 2.| 4a | 5a | 6a | 4b | |
|---|---|---|---|---|
| 4a: C* = C33, C** = C37, 5a: C* = C31, C** = C35, 6a: C* = C33, C** = C29, 4b: C* = C33, C** = C34 | ||||
| Bond length (Å) | ||||
| Sn–O4 | 2.073(3) | 2.080(2) | 2.076(2) | 2.078(2) |
| Sn–O3 | 2.066(3) | 2.071(3) | 2.067(2) | 2.070(2) |
| Sn–O2 | 2.485(3) | 2.446(3) | 2.491(2) | 2.416(3) |
| Sn–O1 | 2.437(3) | 2.418(3) | 2.524(3) | 2.437(3) |
| Sn–C* | 2.133(6) | 2.135(4) | 2.107(4) | 2.102(5) |
| Sn–C** | 2.129(6) | 2.089(5) | 2.136(5) | 2.115(4) |
| Bond angles (°) | ||||
| O4 Sn O1 | 72.23(11) | 72.51(9) | 71.56(9) | 72.06(9) |
| O3 Sn O2 | 71.63(12) | 72.30(9) | 71.55(9) | 72.87(9) |
| O3 Sn O4 | 79.31(11) | 80.70(10) | 79.04(9) | 77.53(9) |
| O1 Sn O2 | 137.12(11) | 134.65(9) | 138.02(8) | 137.58(9) |
| O4 Sn C* | 108.4(2) | 106.70(13) | 108.73(14) | 105.96(17) |
| O4 Sn C** | 102.7(2) | 102.15(15) | 101.50(15) | 103.65(15) |
| O3 Sn C** | 108.3(2) | 105.30(14) | 106.04(14) | 106.54(15) |
| O3 Sn C* | 101.7(2) | 100.19(14) | 104.86(14) | 102.26(16) |
| C* Sn O2 | 83.0(2) | 82.05(13) | 83.17(13) | 82.10(17) |
| C* Sn O1 | 83.1(3) | 84.66(14) | 81.81(13) | 83.67(16) |
| C** Sn O1 | 83.1(2) | 83.98(14) | 83.70(14) | 83.75(14) |
| C** Sn O2 | 82.1(2) | 82.03(15) | 83.22(15) | 83.62(14) |
| C** Sn C* | 140.0(3) | 144.04(18) | 140.06(17) | 142.28(19) |
| Parameter | 4a | 5a | 6a | 4b |
|---|---|---|---|---|
| Empirical formula | C40H40O6Sn | C38H35O6Sn | C34H32O8Sn | C34H28O6Sn |
| Formula weight | 735.41 | 706.35 | 687.28 | 651.25 |
| T (K) | 293(2) | 293(2) | 293(2) | 293(2) |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/n | P21/n |
| a (Å) | 8.2279(2) | 11.8150(6) | 9.4660(2) | 11.5018(3) |
| b (Å) | 12.1695(2) | 11.9686(6) | 16.2902(4) | 14.9532(3) |
| c (Å) | 18.0429(4) | 12.7177(7 | 19.8338(4) | 17.5970(5) |
| α (°) | 85.696(2) | 67.247(5) | 90 | 90 |
| β (°) | 89.597(2) | 78.484(5) | 98.453(2) | 99.344(2) |
| γ (°) | 75.392(2) | 85.626(4) | 90 | 90 |
| V (Å3) | 1743.16(7) | 1625.06(16) | 3025.21(12) | 2986.31(13) |
| Z | 2 | 2 | 4 | 4 |
| ρ calc (g cm−3) | 1.401 | 1.444 | 1.509 | 1.449 |
| μ (mm−1) | 0.779 | 0.832 | 0.897 | 0.899 |
| F (000) | 756.0 | 722.0 | 1400.0 | 1320.0 |
| Crystal size (mm3) | 0.33 × 0.24 × 0.13 | 0.245 × 0.111 × 0.09 | 0.31 × 0.22 × 0.14 | 0.321 × 0.221 × 0.111 |
| Radiation | Mo-Kα (λ = 0.71073) | Mo-Kα (λ = 0.71073) | Mo-Kα (λ = 0.71073) | Mo-Kα (λ = 0.71073) |
| 2θ range | 6.792 to 54.9 | 6.616 to 54.888 | 6.502 to 54.734 | 6.354 to 54.764 |
| Index ranges | −10 ≤ h ≤ 10, −15 ≤ k ≤ 14, −23 ≤ l ≤ 22 | −15 ≤ h ≤ 15, −15 ≤ k ≤ 15, −16 ≤ l ≤ 16 | −10 ≤ h ≤ 11, −20 ≤ k ≤ 20, −25 ≤ l ≤ 24 | −14 ≤ h ≤ 12, −19 ≤ k ≤ 19, −22 ≤ l ≤ 22 |
| Reflections collected | 24253 | 22851 | 27284 | 20045 |
| Independent reflections | 7414 | 6935 | 6420 | 6180 |
| R int, Rsigma | 0.1112, 0.0784 | 0.0706, 0.0552 | 0.0946, 0.0650 | 0.0729, 0.0501 |
| θ max (°) and completeness (%) | 27.450 and 0.929 | 27.444 and 0.934 | 27.367 and 0.937 | 27.382 and 0.911 |
| Absorption correction | Multi-scan | Multi-scan | Multi-scan | Multi-scan |
| T min and Tmax | 0.783 and 0.906 | 0.822 and 0.929 | 0.769 and 0.885 | 0.761 and 0.907 |
| Data/restraints/parameters | 7414/15/428 | 6935/40/436 | 6420/11/390 | 6180/0/374 |
| GOF on F2 | 1.014 | 1.047 | 1.055 | 1.057 |
| R 1, wR2 [I > 2σ (I)] | 0.0659, 0.1742 | 0.0501, 0.1298 | 0.0491, 0.1120 | 0.0404, 0.1014 |
| R 1, wR2 [all data] | 0.0775, 0.1874 | 0.0659, 0.1421 | 0.0823, 0.1291 | 0.0531, 0.1082 |
| Largest diff. peak/hole (e Å−3) | 1.08/−1.42 | 0.75/−0.91 | 0.49/−0.41 | 0.28/−0.40 |
The compounds were formed with the general formula L2SnR2 (where R = butyl for 4a–6a and methyl for 4b–6b, and L is a bidentate ligand) in which hexacoordinated Sn was surrounded by two bidentate ligands and two alkyl substituents constructing skew-trapezoidal bipyramidal geometry.35–37 The R groups occupied axial positions and ligand donors were present at equatorial positions, therefore, the overall configuration of compounds was found to be trans to R groups. As the bidentate ligand L itself was comprised of an asymmetric environment of the donor system (i.e. a CO and a –OH), there might be two possible configurations of ligand donors (i.e. cis and trans). Interestingly, all the organotin(IV) compounds were obtained in the cis-configuration to L in which Sn–O(CO) and Sn–O(C–O) occupied adjacent positions constructing butterfly-like structures. The stability of cis-configured ligating molecules over the trans-configured orientation was supported by theoretical studies. The density-functional calculations were performed with the aid of Gaussian 03,38 with the B3LYP hybrid method.39 The DGDZVP basis set40 was used to fully optimize the geometry of the proposed structures followed by frequency calculations. For this, possible structures of compound 4b (test molecule) based on different binding modes of ligands in the SnO4 trapezoidal plane were analyzed by computational means (see Fig. S1 and Tables S1, S2, ESI†). The two geometries of compound 4b were optimized concerning Sn–O(CO) and Sn–O(C–O) bonds; (A) in the cis-configuration and (B) in the trans configuration. The theoretical approach revealed the electronic energy of optimized cis and trans configurations equal to −4887591.0 kcal mol−1 and −4887587.8 kcal mol−1, respectively. Therefore, the cis-configuration of ligands is −3.2 kcal mol−1 more stable than the trans-configuration, which is in complete agreement with the structure obtained from single-crystal X-ray diffraction analysis. The dipole moment of the trans configuration was observed as 0.52 Debye more than the cis-configuration.
Besides, Molecular Electrostatic Potential (MEP) provides information about the charge density distribution over a molecule in the form of a three-dimensional map. It helps to identify the polarity and various reactive sites in the molecule for hydrogen bonding, electrophilic and nucleophilic reactions, and other non-covalent interactions.41 The MEP surface for 4b was plotted from the optimized geometry of the molecule by DFT at the B3LYP/DGDZVP level (Fig. S2, ESI†). The electron-dense red area possessing O atoms is more prone to be attacked by an electrophile. The blue electron-deficient regions (due to C and H) are suitable for interactions with a nucleophile.
As a consequence of asymmetrical steric constraints of the bidentate ligand, each molecule attained “skew-trapezoidal bipyramidal” geometry instead of octahedral geometry.35 This geometry may be viewed as the overall distortion of the typical octahedral geometry.42 The Sn–O(CO) bond length was found to be ∼20% more than the Sn–O(C–O) bond, distinguishing the two as Sn–O coordinate and Sn–O covalent bonds, respectively. Generally, the angle between the lines joining the metal centre to the midpoints of the bidentate ligands is 180° in the case of octahedral complexes but here the two bidentate ligands were twisted towards each other in such a way that the angle was reduced to ∼120°. The metal-bound oxygen atoms of bidentate ligands remained coplanar with the Sn centre. The C–Sn–C bond angle was reclined in the range of 140.0°–145.5° instead of being collinear giving an antenna-like appearance. Irrespective of the enormous distortions, the axial and equatorial planes were still perpendicular to each other.
Compound 4a was crystallized in a triclinic crystal system with a P space group (Fig. 1A). The asymmetric unit contained one molecule and the unit cell was comprised of two molecules of compound 4a. The Sn–O(CO) bond lengths were found to be 2.437(3) Å and 2.485(3) Å whereas the Sn–O(C–O) bond lengths were found to be 2.073(3) Å and 2.066(3) Å, respectively. The C–Sn–C bond angle between two butyl groups and the Sn centre was 140.0(3)°. The stabilization of butyl groups at a reduced angle (instead of 180°) is attributed to the cis configuration of equatorial donor sets. The steric hindrance caused by the phenyl rings adjacent to Sn–O3 and Sn–O4 bonds forced the butyl chains to tilt towards the less hindered site and deviated the linearity of the C–Sn–C bond.
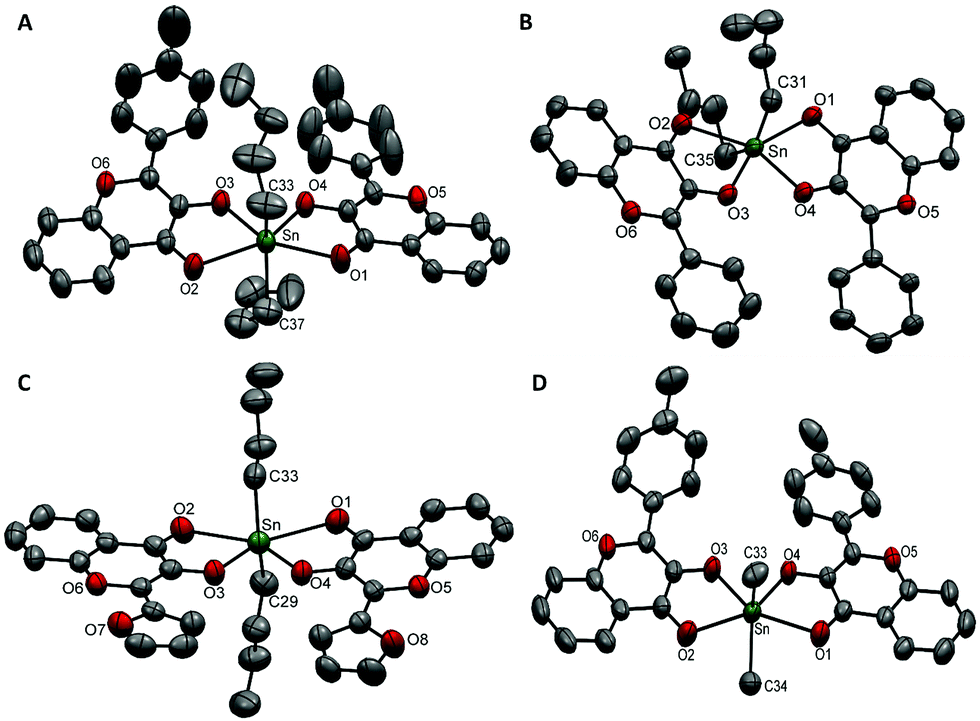 | ||
| Fig. 1 ORTEP presentation of (A) 4a, (B) 5a, (C) 6a, and (D) 4b with the partial numbering scheme (thermal ellipsoids are drawn at 40% probability and hydrogen atoms have been omitted for clarity). | ||
Compound 5a (Fig. 1B) was crystallized in a triclinic crystal system with the P space group. Its structural features were found to be similar to those of 4ai.e. a single molecule in an asymmetric unit and two molecules in the unit cell. Similarly, two Sn–O(CO) bonds were longer [i.e.5a; 2.418(3) Å/2.446(3) Å], and two Sn–O(C–O) bonds were shorter [i.e.5a; 2.071(3) Å/2.080(2) Å] accompanied by a deviation of C–Sn–C bond angle from linearity 144.04(18)°. The butyl chain composed of C-35 to C-38 showed thermal disorder and therefore carbon atoms C-36 to C-38 were located at different positions. This resulted in large sized ellipsoids which were resolved by splitting atoms C-36 to C-38 into their certain positions.
Compounds 6a (Fig. 1C) and 4b (Fig. 1D) were crystallized in a monoclinic crystal system with the P21/n space group. Their asymmetric unit consisted of one molecule of the compound and the unit cell contained four molecules.
Although the structures of all the compounds were similar, their crystalline lattices followed different packing arrangements of unit cells along the crystallographic axes. In the unit cell of 4a, two molecules were connected by non-covalent π⋯π interactions of 3.659 Å. Here, two off-set parallel displaced aromatic rings (p-tolyl) of neighbouring molecules favour the π⋯π interactions (Fig. S3, ESI†).43 The intramolecular H-bonding C–H⋯O was observed at 2.189 Å and 2.219 Å.44 The intramolecular H-bonds and intermolecular secondary interactions of the γ-pyrone ring in 5a were present as C–H⋯O (2.217, 2.233 Å) and 3.594 Å, respectively (Fig. S4, ESI†). In the lattice, edge to face C–H⋯π interactions (3.549 Å) were also observed (Fig. S4, ESI†). Non-covalent π⋯π interactions were also significant in the crystalline structure of compound 6a. The intermolecular secondary contacts were observed between two off-set parallel displaced benzene rings of the benzo-γ-pyrone moiety (3.721 Å) and intramolecular C–H⋯O interactions were present at 2.426 and 2.458 Å (Fig. S5, ESI†). In 4b, intramolecular C–H⋯O hydrogen bonds (2.220, 2.223 Å) along with edge to face C–H⋯π (3.525 Å) and π⋯π interactions (3.614 Å) hold the lattice together (Fig. S6, ESI†).
2.4. Spectrophotometric and spectrofluorimetric aspects
Like other flavonols, 2-hydroxychalcones 1a–3a and 3-hydroxy-4H-chromen-4-one 1b–3b showed two major absorption bands in the UV–Vis region (see Fig. S7A and S7B, ESI†). 2-Hydroxychalcones 1a–3a exhibited maximum absorption at 322–362 nm due to the π → π* transition (see Fig. S7A, ESI†).45 In the case of 3-hydroxy-4H-chromen-4-one 1b–3b, the absorption band located in the wavelength range of 347–357 nm (the band-I) with the shoulder at 305–319 nm corresponds to the conjugated system between ring B (cinnamoyl) and the carbonyl group of ring C (see the inset in Fig. S7B, ESI†). A less intense band centred in the range of 259–261 nm (band-II) could be assigned to the transition for the conjugated system between ring A (benzoyl) and the carbonyl of ring C.46 In organotin(IV) compounds (4a–6a, 4b–6b), band-I was slightly shifted to a longer wavelength and appeared in the region of 363–367 nm. The additional absorbance band centred in the region of 394–415 nm indicated complex formation (see Fig. S7C and D, ESI†).47To further understand the electronic transitions, Time-Dependent Density Functional Theory (TD-DFT) calculations were carried out at the B3LYP/DGDZVP level for 3-hydroxy-4H-chromen-4-one 1b and organotin compound 4b. The geometry of ligand 1b was optimized by the B3LYP/DGDZVP method (see Table S3 in the ESI†). The two main transitions for 3-hydroxy-4H-chromen-4-one 1b were HOMO → LUMO and HOMO−1 → LUMO, however, the HOMO−3 → LUMO transition was forbidden due to zero value of oscillator strength (f) (i.e. the strength of the electronic transition) as shown in Table 3. In 1b, the HOMO was more localized at rings B and C, but the LUMO was delocalized from ring B to the C1′–C2 bond, and from the C2–C3 bond to C3–C4 and C4–C10 bonds. Thus, theoretical wavelength (i.e. 353 nm) and experimental wavelength (i.e. 349 nm) for the HOMO → LUMO transition were in good agreement and correspond to band I for 1b. The HOMO−1 was more localized at rings A and B, but in the HOMO−1 → LUMO transition, charge density was shifted from ring A to bond C10–C4, thus this transition corresponds to benzoyl band II (Table S4, ESI†). The calculated value of HOMO−1 → LUMO for 1b was 298 nm, however, the experimental value was found to be 260 nm. This shift might be influenced by various factors like temperature, intermolecular interactions, solvent interactions etc. The tin compound 4b showed the first HOMO → LUMO band at 426 nm, which correlate with the experimental value of 409 nm. Since this band was mainly associated with the ligand moiety (see Fig. S8, ESI†), conceptualization of ligand-to-metal charge transfer (LMCT) is ruled out.48 But after complexation the HOMO and LUMO gap was reduced by 3.8 eV (Fig. S8, ESI†). Therefore, a red shift in the absorption spectra of tin compounds was observed.
| Compound | Excited state | Excitation energy (eV) | Wavelength (nm) | Transition (molecular contribution) | f |
|---|---|---|---|---|---|
| 1b | S1 | 3.5083 | 353.40 | HOMO → LUMO (0.69614) | 0.4453 |
| S2 | 4.0115 | 309.07 | HOMO−3 → LUMO (0.69592) | 0.0000 | |
| S3 | 4.1494 | 298.80 | HOMO−1 → LUMO (0.67295), HOMO → LUMO+1 (−0.11397) | 0.1116 | |
| 4b | S1 | 2.9104 | 426.01 | HOMO−1 → LUMO+1 (0.46333), HOMO → LUMO (0.53093) | 0.0041 |
| S2 | 2.9110 | 425.91 | HOMO−1 → LUMO (0.51173), HOMO → LUMO+1 (0.48442) | 0.0004 | |
| S3 | 3.0036 | 412.78 | HOMO−1 → LUMO (−0.48365), HOMO → LUMO+1 (0.51159) | 0.0105 |
The emission spectra of 18.54 μM methanolic solution of 3-hydroxy-4H-chromen-4-one (1b–3b) and organotin(IV) compounds (4a–6a, 4b–6b) exhibited emission maxima in the range of 529–533 nm and 455–461 nm, respectively (Fig. 2A–C). The Stokes shift i.e., the difference between emission maxima and absorption maxima for 3-hydroxy-4H-chromen-4-one (1b–3b), was found to be in the range of 176–182 nm and for organotin(IV) compounds (4a–6a, 4b–6b) it was in the range of 46–61 nm.
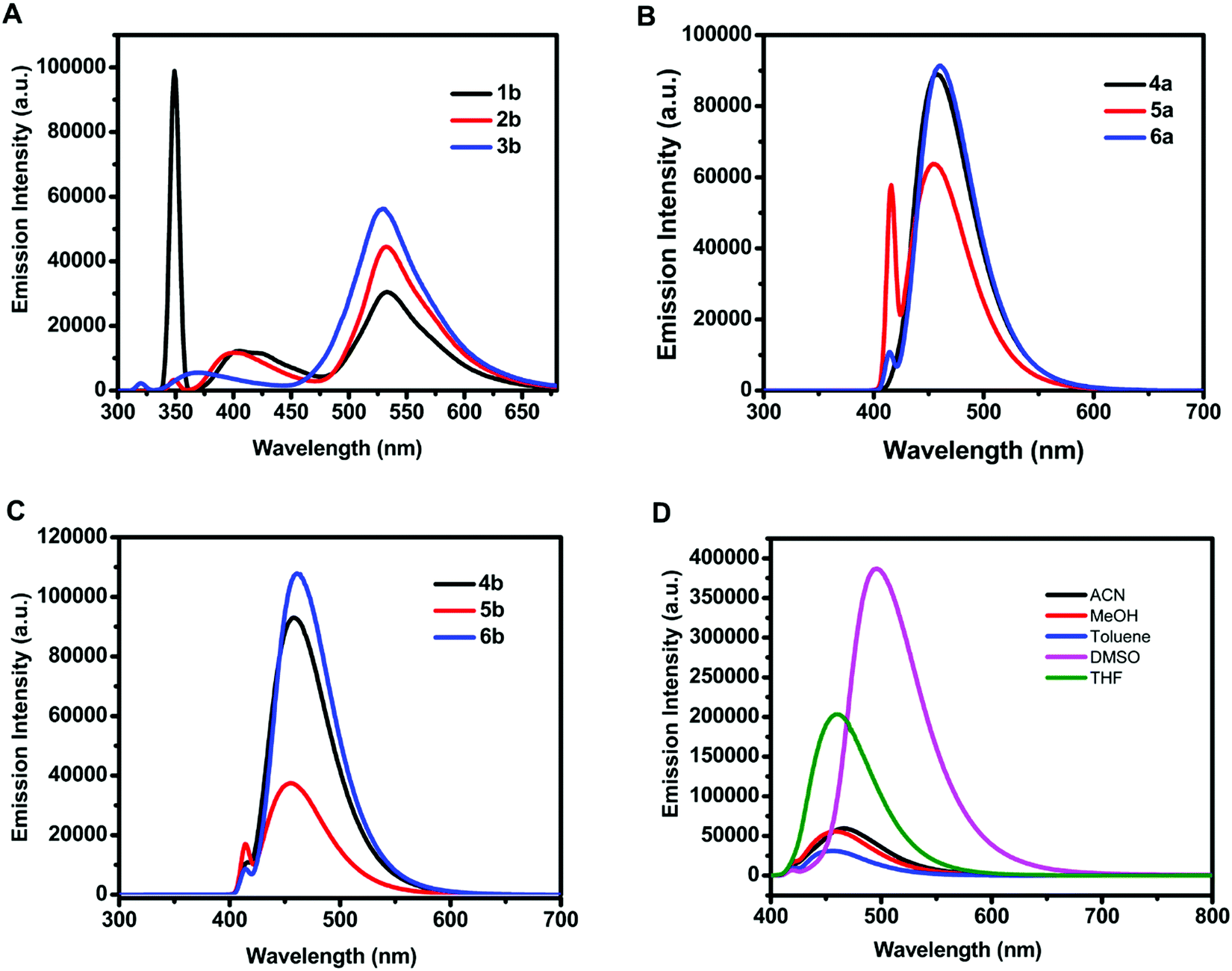 | ||
| Fig. 2 Fluorescence spectra of methanolic solution (18.54 μM) of (A) 3-hydroxy-4H-chromen-4-one (1b–3b) at λex = 350 nm, (B) organotin(IV) compounds (4a–6a) at λex = 415 nm and (C) (4b–6b) at λex = 415 nm and (D) 4a in solvents of different polarity: acetonitrile (ACN), methanol (MeOH), toluene, dimethylsulfoxide (DMSO), and tetrahydrofuran (THF). | ||
The fluorescence intensity of organotin compound 4a was enhanced as compared to that of the free ligand to 21-fold in dimethylsulfoxide and 13-fold in methanol (Fig. S9, ESI†). This significant enhancement can be attributed to the chelation-enhanced fluorescence (CHEF) effect. Mahajan et al. have given a mechanistic view of CHEF which suggests that restriction in free rotation of the CO group for adjacent rings increases the rigidity of ligand assembly after the formation of a complex, and thus a considerable emission enhancement occurs.49 Similarly, Czarnik et al. have reported that the transfer of the electron pair from the donor to the excited state of the fluorophore quenches the fluorescence signal and shows photoinduced electron transfer (or PET). However, the coordination of the donor with the metal centre restricts the electron transfer and stops PET.50 Since the X-ray crystal structure of the synthesized organotin compounds confirms the coordination of CO oxygen to the Sn atom, the applicability of the CHEF effect is valid in this case also.
2.5. Aggregation induced emission enhancement (AIEE)
Furthermore, the effect of solvents on the emission intensity of a test compound 4a (18.54 μM) was observed. The emission maximum of 4a was located at 455 nm in toluene, 457 nm in methanol, 460 nm in tetrahydrofuran, 467 nm in acetonitrile, and 495 nm in dimethylsulfoxide (Fig. 2D) revealing a red shift and maximum fluorescence intensity in aprotic polar solvents. The emission intensity and wavelength for 4a followed the order as non-polar < polar protic < aprotic polar solvents.Due to the weak fluorescence of 4a in MeOH (Fig. 2D), aggregation-induced emission enhancement was investigated in a mixture of MeOH and H2O. The emission intensity of 4a exhibited enhancement with the incremental addition of water (10 μL). It was enhanced to 2.3-fold with the addition of 160 μL of water and then attained a constant plateau (Fig. 3A). Since 4a was insoluble in water, the organotin(IV) compound showed aggregation with the increase in water fraction. The aggregate formation also led to the enhancement in quantum yield suggesting AIEE in the methanol–water mixture. The quantum yield of 4a in methanol and the MeOH/H2O mixture (with 7.4% fraction of H2O) was found to be 0.59 and 0.72, respectively.
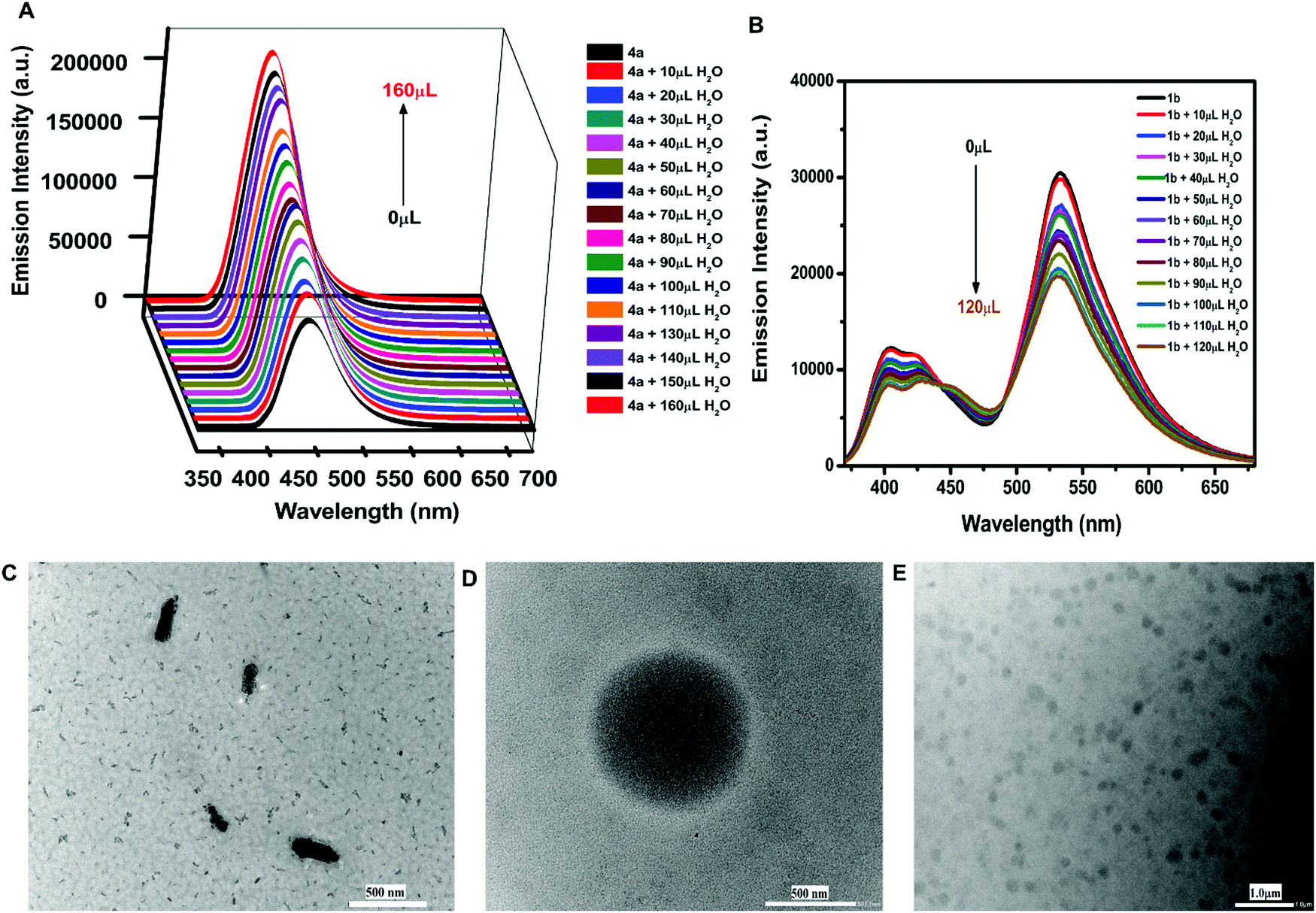 | ||
| Fig. 3 (A) and (B) Fluorescence spectra of 4a and 1b in the MeOH/H2O mixture (λex = 420 nm; 18.54 μM each), respectively; (C) HRTEM images of 4a in MeOH; and (D) and (E) HRTEM images of 4a in the MeOH/H2O (1:1 v/v) mixture at different resolution, respectively. | ||
A similar phenomenon was observed in other organotin compounds (see Fig. S10, ESI†). Thus, all the organotin compounds possessed considerable AIEE properties. The aggregation results from various types of intermolecular arrangements like J-type, H-type, and herringbone stackings. The herringbone and J-type arrangements restrict the intramolecular motions (RIM) and cause the inhibition of non-radiative decay processes producing emission enhancement.51 However, H-type stacking (face-to-face) promotes radiation-less relaxation of the excited state of the fluorophore, due to which the emission intensity decreases.52 The J-type interactions involve different types of stacking patterns like brickwork, ladder, and staircase arrangement.53 Compounds 4a and 5a followed the brickwork pattern. However, 6a and 4b possessed the herringbone arrangement of the molecules (see Fig. S11, ESI†). This type of intermolecular interaction must have provided the rigidity or spatial constraint to the molecular arrangement, which in turn have locked the intramolecular rotations or vibrations and hence caused the AIEE. To check whether free ligands were also associated with this emission enhancement property or not, their fluorescence spectra were also investigated in MeOH/H2O mixtures. However, in the case of ligands the emission intensity was quenched with increasing concentration of water (Fig. 3B and Fig. S12, ESI†). Hence, the free ligand possessed aggregation caused quenching (ACQ) properties where the fluorescence intensity is reduced after aggregation due to increased radiationless decay processes.54
The HRTEM results revealed the formation of micelle-type-micro-aggregates of 4a in the MeOH/H2O mixture (Fig. 3D and E). The aggregate formation with increasing concentration of water was also supported by FESEM studies. For FESEM, samples of aggregates of 4a were obtained from the MeOH/H2O mixture at two different water fractions i.e., at 4.76% and 33.33% water in MeOH. The aggregate formation was more at 33.33% water fraction (Fig. S13A and B, ESI†).
2.6. Water stability
The stability of 4a was checked in an aqueous medium. For this, 4a was dissolved in 5 mL of MeOH and then 1 mL of water was added and allowed to crystallize by slow evaporation of the solvent. The crystals obtained (4a′) from the mixture were analyzed by single-crystal X-ray diffraction. The crystallographic data, bond lengths, bond angles, and structure refinement parameters of 4a′ were compared with previous data of 4a obtained in the chloroform–methanol mixture (1:1) (Tables S5 and S6, ESI†). The similar structural parameters confirmed the stability of 4a in water. In addition, the solution phase stability of the compound was also investigated by performing the 119Sn NMR titration of 4a. The compound (0.030 g) was dissolved in 0.75 mL of the mixture of CDCl3 and DMSO-d6. The water was added in the fractions of 10 μL to the mixture during titration. The successive addition of water showed a negligible shift in the 119Sn NMR signal (at −189 ppm), thus confirming its stability in an aqueous–organic mixed solvent (Fig. 4A). The hexacoordination at the Sn centre and octahedral geometry must be the reason for water stability because the coordination sphere was not disturbed by the addition of water as indicated by the unaltered position of the 119Sn NMR signal.
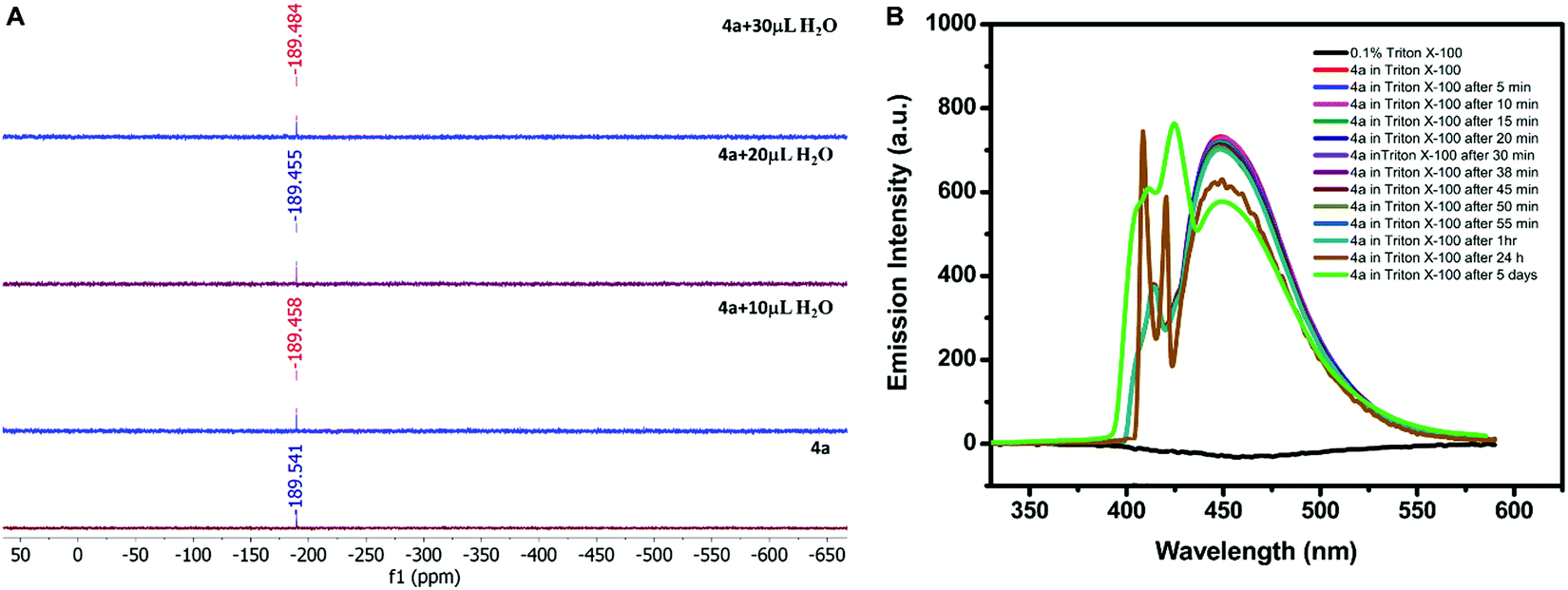 | ||
| Fig. 4 (A) 119Sn-NMR titration of 4a (0.034 g in 0.75 mL CDCl3 + DMSO-d6) with successive addition of water. (B) Emission stability of 0.2 mM 4a in 0.1% (v/v) Triton X-100 at different intervals of time. | ||
Although, 4a was found to be stable in aqueous–organic mixed systems investigation of its application in aqueous systems was again a challenge due to its insolubility in water. Therefore, it was solubilized in pure water with the help of a surfactant. Out of various surfactants i.e. cetyltrimethylammonium bromide (CTAB), sodium lauryl sulphate (SLS), and 4-octylphenol polyethoxylate (Triton X-100), 4a displayed maximum solubility in the presence of Triton X-100 (0.1% solution). So, 0.2 mM solution of 4a was prepared in 0.1% (v/v) Triton X-100. The stability of 4a in Triton X-100 was investigated by recording its emission profile at different intervals of time. The emission signal was found to be almost constant after 1 hour in the presence of Triton X-100; however, its intensity was reduced to some extent after 24 h and after 5 days as depicted in Fig. 4B. Moreover, the response was not completely diminished even after five days.
2.7. Lead detection
Due to the water stability of organotin(IV) compounds, the metal recognition property of the test compound 4a was investigated against various metal ions like Zn2+, Sm3+, Pb2+, Ni2+, Mg2+, K+, Gd3+, Fe3+, Dy3+, Cu2+, Co2+, Cd2+, and Ca2+. The 10 mM stock solution of nitrate salt of each metal ion was prepared in water. The fluorescence intensity of 4a was enhanced significantly in the presence of Pb2+ ions with a red shift of 28 nm and a quantum yield of Φ = 0.91 (Fig. 5A). The response was also observed by a colour change from yellow to blue in UV-light. There was no considerable interference of co-existing metal ions on the spectrofluorimetric response of 4a towards Pb2+ ions (except Fe3+ ions) (Fig. 5B). Furthermore, titration of 4a (0.1 mM) was carried out with every 10 μL addition of 1.5 mM Pb(NO3)2 solution (Fig. 5C). The calibration curve was plotted from the data and the limit of detection (LOD) was calculated. The LOD was found to be 22.66 μM following equation 3 σ/n (where σ = standard deviation of emission measurements taken for 4a (as shown in Table S7, ESI†) and n = slope of a plot of emission intensity of 4aversus Pb2+ ion concentration) (Fig. 5D). The time-dependent fluorescence of 4a in the presence of Pb2+ ions (Fig. 6A) confirmed the stability of the enhancement process with time. The percentage enhancement in the emission intensity of 4a as a function of time was determined by (I − I0)/I0 × 100, where I0 and I are the maximum emission intensities in the absence and presence of Pb2+ ions, respectively (Fig. 6B). A constant signal was attained immediately after the addition of Pb2+ ions and the emission enhancement efficiency of Pb2+ ions was observed to be 66.45%. A comparison with the previous fluorophores and methods reported for the detection of Pb2+ ions is summarized in Table 4.55–60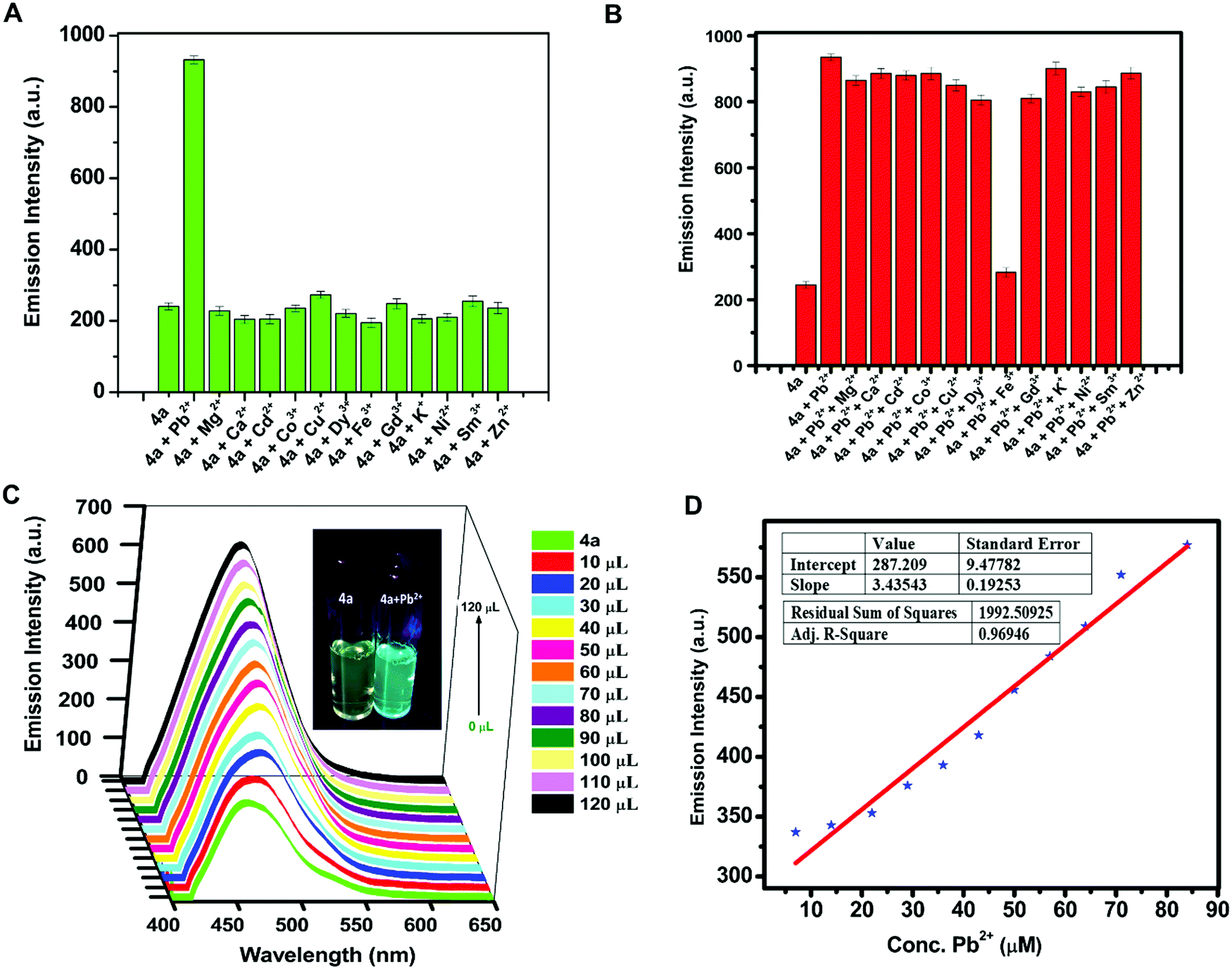 | ||
| Fig. 5 (A) 2D bar representation of the relative emission intensity of 0.2 mM 4a (λex = 410 nm) with 50 μL addition of metal nitrates (10 mM); (B) 2D bar representation of relative emission intensity of 0.2 mM 4a (λex = 410 nm) upon addition of 50 μL of Pb(NO3)2 in the presence of 50 μL of other metal nitrates (10 mM) in water; (C) 3D plot of fluorescence intensity of 0.1 mM 4a (λex = 410 nm) with successive 10 μL addition of Pb(NO3)2 (1.5 mM), inset: images of 4a and 4a+Pb2+ under 365 nm UV light; and (D) limit of the detection (LOD) plot for the recognition of Pb2+ ions. | ||
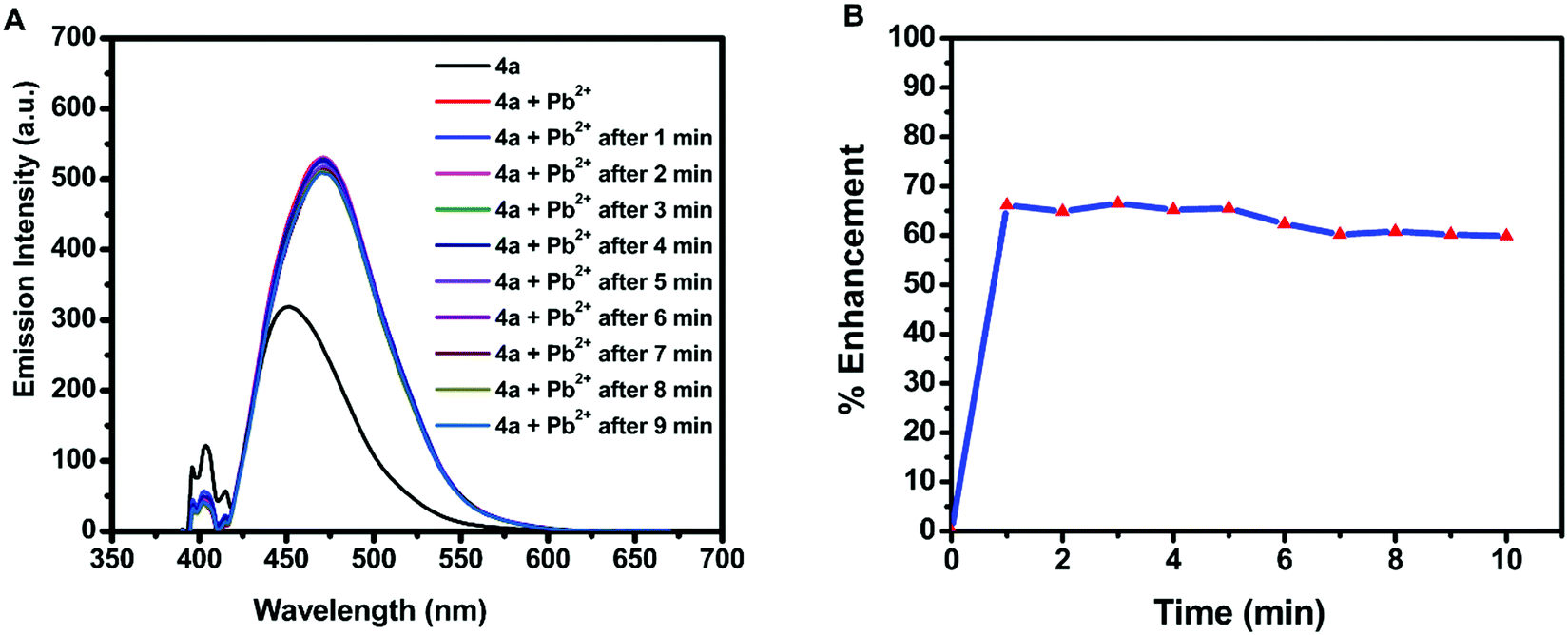 | ||
| Fig. 6 (A) Time-dependent fluorescence of 4a (λex = 410 nm) in the presence of Pb2+ ions and (B) percentage emission enhancement of 4a with Pb2+ ions as a function of time. | ||
| S.no. | Compound | Technique | Result | Solvent system | Detection limit (μM) | Ref. |
|---|---|---|---|---|---|---|
| 1 | Pseudo-crown ether PD-18C6 | Fluorescence | Signal amplification with a blue shift | Water | _ | 55 |
| DLS | Micron-aggregate formation | |||||
| 2 | Rhodamine linked triazole motif | Fluorescence | Turn-on | Semi-aqueous system [CH3CN–water (4/1, v/v; 10 mM tris HCl buffer)] | 3.9 | 56 |
| UV | Additional peak | |||||
| 3 | Rhodamine derivatives | Fluorescence | Turn-on | Ethanol/water solution (9/1, 1 mM HEPES buffer) | 0.016 | 57 |
| 4 | PSAM (Schiff based derived polymer) | Fluorescence | Turn-on | 10 mM Tris-HCl buffer solution | 0.7 | 58 |
| 5 | Calix[4]pyrrole derived aminoanthraquinone (CAAQ) | Fluorescence | Turn-off | Dimethyl sulfoxide | 10.0−10−4 | 59 |
| UV | Red shift | |||||
| 6 | [2-N,N-Bis(pyridin-2-ylmethyl)methanamine-BODIPY] | Fluorescence | Turn-on | CH3CN | 0.060 to 0.089 | 60 |
| 7 | Organotin(IV) compound | Fluorescence | Enhancement with red shift | Water | 22.66 | Present report |
| DLS | Micron-aggregate formation |
To study the mechanism of interaction between 4a and Pb2+ ions, 1H NMR spectra of 4a were recorded in the presence and absence of Pb2+ ions. The different solubility of 4a (partial solubility in DMSO, proper solubility in CHCl3) and Pb2+ ions (complete solubility in DMSO, insolubility in CHCl3) complicated the investigation. The 1H NMR signals retained their position but exhibited a broadening of signals in the presence of Pb2+ ions. This observation just confirmed the stability of the organotin(IV) compound even in the presence of Pb2+ ions but did not suggest the type of interaction (Fig. S14, ESI†).
After the 1H NMR mechanistic approach, we opted for the theoretical binding energy calculations of 4a with Pb2+. Based on the MEP diagram of 4b, the electron-rich site (observed as the red area) was proposed as a site of interaction (Fig. 7). The binding energy calculation revealed that 4a+Pb2+ is 263.8 kcal mol−1 more stable than 4a. The tolyl rings of 4a distorted from the planarity after interaction with Pb2+ ions as depicted in Fig. S15 (ESI†) in the optimized geometry of 4a (Table S8, ESI†) and 4a+Pb2+ (Table S9, ESI†).
| ΔEBinding = E4a+Pb(II) − (E4a + EPb(II)) = −8027.783973 − (−8024.761952 − 2.601709) = −0.420313 Hartree = −263.8 kcal mol−1 |
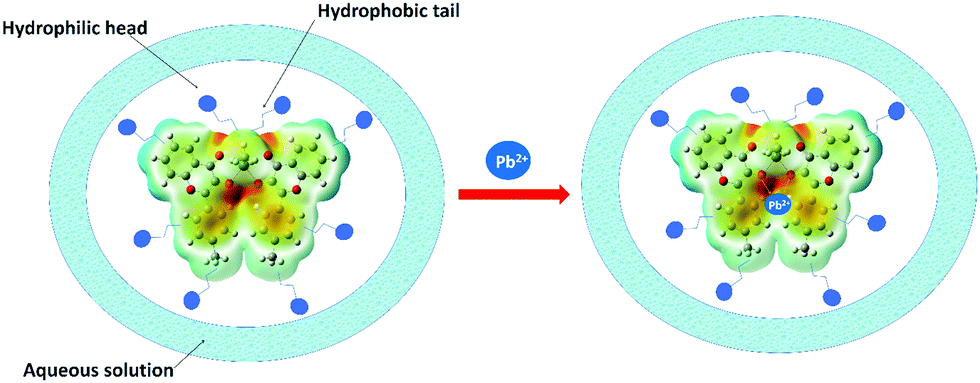 | ||
| Fig. 7 Proposed mechanism of Pb2+ binding using the MEP diagram of 4b which suggested the available electron density as the reactive site. | ||
The formation of micelle-aggregates of the Pb2+–organotin(IV) compound in surfactants resulted in the aggregation-induced emission enhancement with a red shift. It was also supported by the dynamic light scattering (DLS) measurement which displayed that nano-sized aggregates (of size ∼437.5 nm) of 4a/Triton X-100 solution enlarged to form micron-sized aggregates (∼3311.66 nm) in the presence of 10 mM Pb2+ along with the nano-aggregates (as shown in Fig. S16, ESI†). The formation of pseudo-micelle aggregates in the presence of Pb2+ ions might have provided restrictions to the molecular vibrations and rotations of the fluorophore, and thus lowered the radiationless decay processes and ultimately amplified the emission intensity.
3. Conclusions
The present work is comprised of 3-hydroxy-4H-chromen-4-one derived water-stable organotin(IV) compounds. The synthetic protocol of these compounds is very efficient, facile and convenient. The spectroscopic and structural aspects revealed the formation of hexacoordinated organotin(IV) compounds in pure form. The single-crystal X-ray diffraction analyses confirmed the skew-trapezoidal bipyramidal geometry of the synthesized compounds in which the equatorial position was occupied by four oxygen atoms of the ligand in a cis fashion and axial positions were occupied by two alkyl groups. The coordination patterns of ligands and alkyl substituents imparted a butterfly-like shape to the compounds. In all the compounds, two alkyl groups attained pseudo-axial positions around Sn, due to which the collinear C–Sn–C bonds were reduced to angle 140.0(3)°–145.5(3)°. The packing diagrams of crystals revealed the presence of non-covalent interactions such as C–H⋯O, π⋯π and C–H⋯π which fabricated the lattice arrangement of the molecules. The computational approach provided additional support for the cis alignment of Sn–O(CO) and Sn–O(C–O) bonds, thus assuring the trapezoidal arrangement of ligands around the Sn centre. 119Sn NMR, single-crystal X-ray diffraction, and spectrofluorimetric response in the presence of water revealed the stability of compounds in aqueous systems. The organotin compounds exhibited a blue shift in emission spectra for the free ligand and the fluorescence intensity enhances upon aggregation supported by HRTEM analysis in MeOH/H2O mixtures. The test compound 4a spectrofluorimetric recognizes the Pb2+ ions in an aqueous medium and offered emission enhancement with a red shift of 28 nm and a detection limit of 22.66 μM. The MEP surface diagram suggested the reactive sites on the molecule for ion binding.
4. Experimental section
4.1. Materials
All the commercially purchased solvents like methanol, ethanol, hexane, toluene, acetonitrile, dimethylsulfoxide, and tetrahydrofuran were dried before use with standard procedures. The chemicals namely 2-hydroxyacetophenone (Aldrich), sodium hydroxide (Avra), p-tolualdehyde (Aldrich), benzaldehyde (Qualigens), 2-furancarboxaldehyde (Aldrich), dibutyltin oxide (Merck), dimethyltinoxide (TCI), zinc nitrate hexahydrate (CDH), samarium nitrate hexashydrate (Merck), lead nitrate (Fisher scientific), nickel nitrate hexahydrate (CDH), magnesium nitrate hexahydrate (CDH), potassium nitrate (CDH), gadolinium nitrate hexahydrate (Merck), ferric nitrate nonahydrate (Merck), dysprosium nitrate hexahydrate (Merck), copper nitrate trihydrate (Merck), cobalt nitrate hexahydrate (Merck), cadmium nitrate tetrahydrate (Avra), calcium nitrate tetrahydrate (CDH), Triton X-100 (Merck), cetyltrimethylammonium bromide (Merck), and sodium lauryl sulphate (Merck) were used without any further purification.4.2. Physical measurements
Fourier Transform Infrared Spectra (in a neat solid-state) were obtained from Thermo-scientific NICOLET IS50 FT-IR and PerkinElmer RX-I FT-IR spectrometers. Mass spectral analyses were carried out on a VG Analytical (70-S) spectrometer (ESI† source with capillary voltage 2500 V). A FLASH-2000 organic elemental analyzer was used for C, H, and N elemental microanalyses. The NMR spectra (1H, 13C, and 119Sn) were recorded at 25 °C on a Bruker Avance II FT-NMR (AL 500 MHz) or Bruker Avance II FT NMR (AL 400 MHz) spectrometer and chemical shifts (in ppm) were noted using tetramethylsilane (TMS) as a reference for 1H and 13C, and tetramethylstannane for 119Sn NMR. The single-crystal X-ray structure analyses were carried on a RIGAKU XTA Lab SuperNova, Single source (Mo-Kα, λ = 0.71073) at offset/far, HyPix3000 diffractometer. The structures and packing diagrams were solved using Olex2 with the ShelXT structure solution program using intrinsic phasing and refined with the ShelXL refinement package using Least Squares minimization.61–63 To fix the disordered structures some restraints such as DELU, SIMU, DFIX, and ISOR were used. The wireframe representation of crystal structures was obtained by Mercury 4.2.0 software (Cambridge Structural Database, Cambridge, UK). A Jasco V-530 UV–Vis spectrophotometer was used for recording the absorption spectra. The fluorescence spectra were recorded on a Shimadzu fluorescence spectrophotometer RF-6000. The Density-Functional Theory (DFT) computations were carried out by Gaussian 03, using the B3LYP hybrid method (Becke three parameters exchange functional and the Lee_Yang_Parr correlation functional) in combination with basis set GenECP: DGDZVP for Sn, H, C, O and Lanl2DZ for Pb. The morphology of the test compound was investigated using a FESEM Zeiss Ultra Plus and HRTEM using a Hitachi SU8010 instrument and JEOL JEM 2100 Plus, respectively. The fluorescence measurements of the metal recognition part were carried out on a PerkinElmer LS-55 fluorescence spectrophotometer with a slit width of 6 nm, whose set range of emission intensity was 0 to 1000 (a.u.).4.3. Syntheses of ligands
The ligands were synthesized in two steps by the reported methods. In the first step, 2-hydroxychalcones (1a–3a) were prepared by the Claisen–Schmidt condensation reaction as reported earlier.23 In the second step, 3-hydroxy-4H-chromen-4-one ligands (1b–3b) were prepared by the Algar–Flynn–Oyamada reaction of 2-hydroxychalcones.24:1 v/v) mixture for single-crystal X-ray crystallography. A slightly modified procedure was followed for the synthesis of the second series of organotin(IV) compounds 4b–6b. For this, a solution containing 1.0 mmol ligand (the same amounts as mentioned above) and dimethyltin oxide (0.08 g, 0.5 mmol) were heated to reflux in methanol (30.0 mL) for 8 h. The solvent was evaporated to dryness which yielded a yellow solid. The products were recrystallized in the chloroform–methanol mixture (1:1 v/v) by slow evaporation.
Compound 4a. Yield: 82% (0.29 g, 0.39 mmol); m.p. 191–193 °C; FT-IR ν (cm−1): 483 (Sn–O), 535 (Sn–C), 1613 (C
O), 2908–2953 (alkyl C–H); 1H-NMR (500 MHz, CDCl3): δ (ppm) 0.72 (6H, t, 3J =7.3 Hz, H14), 1.31 (4H, sextet, 3J = 7.0 Hz, H13), 1.54–1.59 (8H, m, 2J (1H-117Sn) = 92 Hz, 2J (1H-119Sn) = 133 Hz, H11,12), 2.48 (6H, s, CH3), 7.34–7.38 (6H, m, H3′,5′,6), 7.60–7.66 (4H, m, H7,8), 8.23 (2H, d, 3J = 8.0 Hz, H5), 8.63 (4H, d, 3J = 8.0 Hz, H2′,6′); 13C-NMR (125 MHz, CDCl3): δ (ppm) 13.64 (C14), 19.17 (CH3), 26.30 (C13), 27.01 (C12), 27.19 (C11), 118.13 (C8), 120.25 (C6), 123.81 (C10), 125.40 (C7), 127.82 (C3′,5′), 129.30 (C2′,6′), 130.38 (C1′), 132.57 (C5), 139.02 (C4′), 145.69 (C9), 148.22 (C2), 154.67 (C3), 176.84 (CO). 119Sn-NMR (186 MHz, CDCl3): δ (ppm) −189.64. MS: m/z (%): 485.09 (100) [M–L + H]+, 679.08 (2.3) [M–C4H9]+. Elemental analysis: calculated for C40H40O6Sn: C, 65.32; H, 5.48; found: C, 65.29; H, 5.46.
Compound 5a. Yield: 79% (0.27 g, 0.38 mmol); m.p. 181–184 °C; FT-IR ν (cm−1): 495 (Sn–O), 524 (Sn–C), 1613 (C
O), 2855–2954 (alkyl C–H); 1H-NMR (500 MHz, CDCl3): δ (ppm) 0.73 (6H, t, 3J = 7.5 Hz, H14), 1.32 (4H, sextet, 3J = 7.0 Hz, H13), 1.52–1.63 (8H, m, 2J (1H-117Sn) = 84 Hz, 2J (1H-119Sn) = 132 Hz, H11,12), 7.38 (2H, td, 4J = 1.5, 3J = 8.5 Hz, H6), 7.46 (2H, td, 4J = 1.0, 3J = 6.5 Hz, H4′), 7.53–7.56 (4H, m, H3′,5′), 7.63 (2H, dd, 4J = 1.5, 3J = 8.5 Hz, H8), 7.67 (2H, td, 4J = 1.5, 3J = 6.5 Hz, H7), 8.24 (2H, dd, 4J = 1.5, 3J = 8.0 Hz, H5), 8.76 (4H, d, 3J = 7.5 Hz, H2′,6′); 13C-NMR (125 MHz, CDCl3): δ (ppm) 13.68 (C14), 26.36 (C13), 27.18 (C12), 27.20 (C11), 118.19 (C8), 120.15 (C6), 123.93 (C10), 125.45 (C7), 127.78 (C3′,5′), 128.52 (C4′), 129.10 (C2′,6′), 132.85 (C1′), 133.12 (C5), 146.08 (C9), 147.75 (C2), 154.75 (C3), 177.17 (CO). 119Sn-NMR (186 MHz, CDCl3): δ (ppm) −190.68. MS: m/z (%): 239.05 (45) [L + H]+, 471.08 (100) [M–L + H]+, 651.06 (5) [M–C4H9]+, 957.18 (7) [M + Bu2SnO]+, 985.18 (55) [M + Bu2SnO + CO]+. Elemental analysis: calculated for C38H36O6Sn: C, 64.52; H, 5.13; found C, 64.49; H, 5.12.
Compound 6a. Yield: 81% (0.27 g, 0.39 mmol); m.p. 210–212 °C; FT-IR ν (cm−1): 470 (Sn–O), 525 (Sn–C), 1609 (C
O), 2863–2953 (alkyl C–H); 1H-NMR (500 MHz, CDCl3): δ (ppm) 0.72 (6H, t, 3J = 7.3 Hz, H14), 1.30 (4H, sextet, 3J = 7.2 Hz, H13), 1.53–1.62 (8H, m, 2J (1H-117Sn) = 88 Hz, 2J (1H-119Sn) = 132 Hz, H11,12), 6.69 (2H, dd, 3J = 2.0, 3.5 Hz, H3′), 7.37–7.42 (2H, m, H6), 7.65–7.68 (4H, m, H7,8), 7.72 (2H, dd, 4J = 1.0, 3J = 2.0 Hz, H2′), 7.97 (2H, dd, 4J = 1.0, 3J = 3.5 Hz, H4′), 8.24 (2H, dd, 4J = 1.0, 3J = 8.0 Hz, H5); 13C-NMR (125 MHz, CDCl3): δ (ppm) 13.63 (C14), 26.22 (C13), 27.06 (C12), 27.20 (C11), 112.47 (C2′), 115.82 (C3′), 118.27 (C8), 120.55 (C6), 124.11 (C10), 125.34 (C7), 132.70 (C5), 141.99 (C4′), 143.88 (C1′), 143.98 (C9), 146.28 (C2), 154.34 (C3), 175.53 (CO). 119Sn-NMR (186 MHz, CDCl3): δ (ppm) −181.14. MS: m/z (%): 229.03 (90) [L + H]+, 461.05 (100) [M–L + H]+, 965.13 (15) [M + Bu2SnO + CO]+. Elemental analysis: calculated for C34H32O8Sn: C, 59.41; H, 4.69; found C, 59.37; H, 4.68.
Compound 4b. Yield: 80% (0.25 g, 0.38 mmol); m.p. 277–281 °C; FT-IR ν (cm−1): 484 (Sn–O), 535 (Sn–C), 1617 (C
O), 2908–2958 (alkyl C–H); 1H-NMR (400 MHz, CDCl3): δ (ppm) 0.84 (6H, s, 2J (1H-117Sn) = 88 Hz, 2J (1H-119Sn) = 133 Hz, H11), 2.48 (6H, s, CH3), 7.34–7.40 (6H, m, H6,3′,5′), 7.61–7.68 (4H, m, H7,8), 8.21 (2H, d, 3J = 8.0 Hz, H5), 8.60 (4H, d, 3J = 8.0 Hz, H2′,6′); 13C-NMR (125 MHz, CDCl3): δ (ppm) 6.78 (C11), 21.65 (CH3), 118.13 (C8), 120.21 (C6), 123.93 (C10), 125.38 (C7), 127.94 (C3′,5′), 129.31 (C2′,6′), 130.17 (C1′), 132.74 (C5), 139.25 (C4′), 145.04 (C9), 148.74 (C2), 154.70 (C3), 176.47 (CO). 119Sn-NMR (186 MHz, CDCl3): δ (ppm) −153.29. MS: m/z (%): 253.08 (58) [L + H]+, 401.01 (100) [M–L + H]+, 637.06 (5) [M–CH3]+, 675.08 (3) [M + Na]+, 845.04 (13) [M + Me2SnO + CO]+. Elemental analysis: calculated for C34H28O6Sn: C, 62.70; H, 4.33; found C, 62.67, H, 4.31.
Compound 5b. Yield: 76% (0.23 g, 0.36 mmol); m.p. 272–275 °C; FT-IR ν (cm−1): 497 (Sn–O), 525 (Sn–C), 1613 (C
O), 2982–3052 (alkyl C–H); 1H-NMR (500 MHz, CDCl3): δ (ppm) 0.85 (6H, s, 2J (1H-117Sn) = 87 Hz, 2J (1H-119Sn) = 133 Hz, H11), 7.39 (2H, td, 4J = 1.5, 3J = 7.0 Hz, H6), 7.47 (2H, td, 4J = 1.0, 3J = 7.5 Hz, H4′), 7.54–7.57 (4H, m, H3′,5′), 7.64 (2H, dd, 4J = 1.0, 3J = 8.5 Hz, H8), 7.68 (2H, td, 4J = 2.0, 3J = 7.0 Hz, H7), 8.23 (2H, dd, 4J = 1.5, 3J = 8.0 Hz, H5), 8.73 (4H, d, 3J = 7.0 Hz, H2′,6′); 13C-NMR (125 MHz, CDCl3): δ (ppm) 6.92 (C11), 118.18 (C8), 120.12 (C6), 124.49 (C10), 126.07 (C7), 127.84 (C3′,5′), 128.52 (C4′), 130.00 (C2′,6′), 132.89 (C1′), 132.99 (C5), 145.40 (C9), 148.24 (C2), 154.78 (C3), 176.78 (CO). 119Sn-NMR (186 MHz, CDCl3): δ (ppm) −154.07. MS: m/z (%): 239.05 (100) [L + H]+, 386.98 (90) [M–L + H]+, 647.03 (0.4) [M + Na]+, 816.98 (18) [M + Me2SnO + CO]+. Elemental analysis: calculated for C32H24O6Sn: C, 61.67; H, 3.88; found C, 61.63; H, 3.87.
Compound 6b. Yield: 75% (0.22 g, 0.36 mmol); m.p. 292–295 °C; FT-IR ν (cm−1): 497 (Sn–O), 525 (Sn–C), 1617 (C
O), 2998–3027 (alkyl C–H); 1H-NMR (500 MHz, CDCl3): δ (ppm) 0.85 (6H, s, 2J (1H-117Sn) = 87 Hz, 2J (1H-119Sn) = 133 Hz, H11), 6.70 (2H, dd, 3J = 2.0, 3.5 Hz, H3′), 7.38–7.41 (2H, m, H6), 7.66–7.67 (4H, m, H7,8), 7.73 (2H, dd, 4J = 0.5, 3J =2.0 Hz, H2′), 7.95 (2H, dd, 4J = 0.5, 3J = 3.5 Hz, H4′), 8.21 (2H, dd, 4J = 1.0, 3J = 8.5 Hz, H5); 13C-NMR (125 MHz, CDCl3): δ (ppm) 7.02 (C11), 112.51 (C2′), 116.00 (C3′), 118.91 (C8), 121.25 (C6), 124.29 (C10), 127.57 (C7), 132.42 (C5), 141.81 (C4′), 143.43 (C1′), 144.17 (C9), 148.65 (C2), 155.71 (C3), 174.63 (CO). 119Sn-NMR (186 MHz, CDCl3): δ (ppm) −144.92. MS: m/z (%): 229.03 (100) [L + H]+, 376.96 (83) [M–L+ H]+, 626.98 (1) [M + Na]+, 796.94 (12) [M + Me2SnO + CO]+. Elemental analysis: calculated for C28H20O8Sn: C, 55.76; H, 3.34; found C, 55.74; H, 3.33.
4.4. Photophysical studies
The absorption and emission spectra of the compounds were recorded in the range of 200–700 nm and 250–700 nm, respectively, at room temperature. Stock solutions (1 mM) of the compounds were prepared in dry methanol, which were further diluted to 0.12 mM for absorption and to 18.54 μM for emission study.For the AIEE properties, 2 mL of 18.54 μM methanolic solution of compounds were taken in the cuvette and emission spectra were recorded. Then distilled water was added in fractions (of 10 μL) to the solution and an emission profile was recorded. The fluorescence quantum yield was noted relative to the standard solution of perylene in ethanol (Φ = 0.92).64
The recognition studies were carried out by dissolving metal nitrates in the water and the test compound in a 0.1% (v/v) Triton X-100 aqueous solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge DST-INSPIRE (IF180276), DST-SERB [SPG/2021/000445] and DST-FIST for providing financial support.References
- A. G. Davies, Organotin Chemistry. John Wiley & Sons, 2006 Search PubMed.
- C. G. dos Santos and G. M. de Lima, Coord. Chem. Rev., 2020, 410, 213236 CrossRef CAS.
- M. A. da Silva, A. S. S. dos Santos, T. V. dos Santos, M. R. Meneghetti and S. M. P. Meneghetti, Catal. Sci. Technol., 2017, 7, 5750–5757 RSC.
- A. F. Hill and M. J. Fink, Advances in Organometallic Chemistry, Academic Press, 1st edn, 2008 Search PubMed.
- A. Khan, S. Parveen, A. Khalid and S. Shafi, Inorg. Chim. Acta, 2020, 505, 119464 CrossRef CAS.
- Y. Y. Zhang, R. F. Zhang, S. L. Zhang, S. Cheng, Q. L. Li and C. L. Ma, Dalton Trans., 2016, 45, 8412–8421 RSC.
- L. Hu, H. Wang, T. Xia, B. Fang, Y. Shen, Q. Zhang, X. Tian, H. Zhou, J. Wu and Y. Tian, Inorg. Chem., 2018, 57, 6340–6348 CrossRef CAS PubMed.
- Z. J. Zhang, H. T. Zeng, Y. Liu, D. Z. Kuang, F. X. Zhang, Y. X. Tan and W. J. Jiang, Inorg. Nano-Met. Chem., 2018, 48, 486–494 CrossRef CAS.
- J. Wang, Y. T. Wang, Y. Fang, Y. L. Lu and M. X. Li, Toxicol. Res., 2019, 8, 862–867 CrossRef CAS PubMed.
- P. Debnath, K. S. Singh, K. K. Singh, S. S. Singh, L. Sieroń and W. Maniukiewicz, New J. Chem., 2020, 44, 5862–5872 RSC.
- T. A. Antonenko, D. B. Shpakovsky, M. A. Vorobyov, Y. A. Gracheva, E. V. Kharitonashvili, L. G. Dubova, E. F. Shevtsova, V. A. Tafeenko, L. A. Aslanov, A. G. Iksanova, Y. G. Shtyrlin and E. R. Milaeva, Appl. Organomet. Chem., 2018, 32, e4381 CrossRef.
- N. Srivastav, K. Kumar, R. Singh and V. Kaur, New J. Chem., 2020, 44, 3168–3184 RSC.
- N. Singh, N. Srivastav, R. Singh, V. Kaur, E. Brendler, J. Wagler and E. Kroke, New J. Chem., 2018, 42, 1655–1664 RSC.
- K. Kumar, N. Srivastav, M. Khera, N. Goel, R. Singh and V. Kaur, Inorg. Chem., 2020, 59, 13098–13108 CrossRef CAS.
- T. Zöller, C. Dietz, L. Iovkova-Berends, O. Karsten, G. Bradtmöller, A. K. Wiegand and K. Jurkschat, Inorg. Chem., 2012, 51, 1041–1056 CrossRef PubMed.
- T. Zöller and K. Jurkschat, Inorg. Chem., 2013, 52, 1872–1882 CrossRef.
- N. Singh, K. Kumar, N. Srivastav, R. Singh, V. Kaur, J. P. Jasinski and R. J. Butcher, New J. Chem., 2018, 42, 8756–8764 RSC.
- R. Vinayak and H. P. Nayek, New J. Chem., 2019, 43, 7259–7268 RSC.
- S. N. Syed Annuar, N. F. Kamaludin, N. Awang and K. M. Chan, Front. Chem., 2021, 9, 657599 CrossRef.
- J. Usta and D. E. Griffiths, Biochem. Biophys. Res. Commun., 1992, 188, 365–371 CrossRef CAS.
- S. Parveen, S. Tabassum and F. Arjmand, J. Organomet. Chem., 2016, 823, 23–33 CrossRef CAS.
- R. de Carvalho Oliveira and R. E. Santelli, Talanta, 2010, 82, 9–24 CrossRef CAS.
- D. N. Dhar, The Chemistry of Chalcones and Related Compounds, Wiley, New York, 1981, 213 Search PubMed.
- T. Oyamada, Bull. Chem. Soc. Jpn., 1935, 10, 182–186 CrossRef CAS.
- S. J. Blunden and P. J. Smith, J. Organomet. Chem., 1982, 226, 157–163 CrossRef CAS.
- L. Nagy, H. Mehner, A. Christy, E. Sletten, F. Edelmann and Q. Andersen, J. Radioanal. Nucl. Chem., 1998, 227, 89–99 CrossRef CAS.
- R. Villamil-Ramos, V. Barba and A. K. Yatsimirsky, Analyst, 2012, 137, 5229–5236 RSC.
- M. Muthukumar, P. Viswanathamurthi and K. Natarajan, Spectrochim. Acta, Part A, 2008, 70, 1222–1226 CrossRef CAS.
- K. Kaur, R. Kaur, J. Tomar and M. Bansal, Photochem. Photobiol. Sci., 2017, 16, 1311–1319 CrossRef CAS.
- A. Kurzwernhart, W. Kandioller, S. Bächler, C. Bartel, S. Martic, M. Buczkowska, G. Mühlgassner, M. A. Jakupec, H. B. Kraatz, P. J. Bednarski, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, J. Med. Chem., 2012, 55, 10512–10522 CrossRef CAS.
- C. S. Rocha, B. P. de Morais, B. L. Rodrigues, C. L. Donnici, G. M. de Lima, J. D. Ardisson and R. S. Bitzer, Polyhedron, 2016, 117, 35–47 CrossRef CAS.
- J. Otera, J. Organomet. Chem., 1981, 221, 57–61 CrossRef CAS.
- C. Vatsa, V. K. Jain, T. Kesavadas and E. R. Tiekink, J. Organomet. Chem., 1991, 410, 135–142 CrossRef CAS.
- J. Otera, T. Hinoishi, Y. Kawabe and R. Okawara, Chem. Lett., 1981, 273–274 CrossRef CAS.
- T. S. Basu Baul, W. Rynjah, E. Rivarola, C. Pettinari and A. Linden, J. Organomet. Chem., 2005, 690, 1413–1421 CrossRef CAS.
- F. A. Shah, S. Sabir, K. Fatima, S. Ali, I. Qadri and C. Rizzoli, Dalton Trans., 2015, 44, 10467–10478 RSC.
- R. K. Sharma, G. Kedarnath, A. Wadawale, C. A. Betty, B. Vishwanadh and V. K. Jain, Dalton Trans., 2012, 41, 12129–12138 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- D. Martínez-Otero, B. Flores-Chávez, J. G. Alvarado-Rodríguez, N. Andrade-López, J. Cruz-Borbolla, T. Pandiyan, V. Jancik, E. González-Jiménez and C. Jardinez, Polyhedron, 2012, 40, 1–10 CrossRef.
- L. R. Domingo, Molecules, 2016, 21, 1319 CrossRef PubMed.
- S. W. Ng, C. Wei, V. G. K. Das, J. P. Charland and F. E. Smith, J. Organomet. Chem., 1989, 364, 343–351 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- B. E. Aksöz and R. Ertan, FABAD J. Pharm. Sci., 2012, 37, 205–216 Search PubMed.
- S. B. Bukhari, S. Memon, M. Mahroof-Tahir and M. I. Bhanger, Spectrochim. Acta, Part A, 2009, 71, 1901–1906 CrossRef.
- S. M. Ahmadi, G. Dehghan, M. A. Hosseinpourfeizi, J. E. N. Dolatabadi and S. Kashanian, DNA Cell Biol., 2011, 30, 517–523 CrossRef CAS.
- J. Ren, S. Meng, C. E. Lekka and E. Kaxiras, J. Phys. Chem. B, 2008, 112, 1845–1850 CrossRef CAS.
- P. G. Mahajan, D. P. Bhopate, G. B. Kolekar and S. R. Patil, Sens. Actuators, B, 2015, 220, 864–872 CrossRef CAS.
- A. W. Czarnik, Fiber Optic Medical and Fluorescent Sensors and Applications, 1992, vol. 1648, pp. 164–180 Search PubMed.
- V. S. Vyas, R. Gutzler, J. Nuss, K. Kern and B. V. Lotsch, CrystEngComm, 2014, 16, 7389–7392 RSC.
- K. Li, Y. Zhang, B. Qiao, F. Tao, T. Li, Y. Ding and Y. Cui, RSC Adv., 2017, 7, 30229–30241 RSC.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef.
- J. B. Birks, Aromatic Molecules, Wiley, New York, 1970, 704 Search PubMed.
- T. Hayashita, D. Qing, R. A. Bartsch, S. Elshani, R. E. Hanes and N. Teramae, Supramol. Chem., 2005, 17, 141–146 CrossRef CAS.
- K. Ghosh, T. Sarkar, A. Majumdar, S. K. Mandal and A. R. Khuda-Bukhsh, Anal. Methods, 2014, 6, 2648–2654 RSC.
- W. Su, S. Yuan and E. Wang, J. Fluoresc., 2017, 27, 1871–1875 CrossRef CAS PubMed.
- G. Wei, G. Zhao, N. Lin, S. Guang and H. Xu, Org. Electron., 2020, 82, 105711 CrossRef CAS.
- K. D. Bhatt, H. D. Shah, K. M. Modi, M. B. Narechania and C. Patel, Supramol. Chem., 2019, 31, 268–282 CrossRef CAS.
- X. Li, G. Tian, D. Shao, Y. Xu, Y. Wang, G. Ji and Y. A. Son, Mol. Cryst. Liq. Cryst., 2020, 706, 38–46 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2014, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2014, 71, 3–8 CrossRef.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Appendix A. Supplementary data. CCDC 2000494, 2000491, 2000497 and 2000492 contain the supplementary crystallographic data for 4a, 5a, 6a, and 4b respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1nj04612h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2022 |