
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA combined structural and computational investigation of aminobenzylnaphthol compounds derived from the Betti reaction using valine methyl ester†
Maria Annunziata M.
Capozzi
 *a,
Angel
Alvarez-Larena
b,
Joan F.
Piniella Febrer
b and
Cosimo
Cardellicchio
*c
*a,
Angel
Alvarez-Larena
b,
Joan F.
Piniella Febrer
b and
Cosimo
Cardellicchio
*c
aDipartimento di Chimica, Università di Bari, via Orabona 4, 70125 Bari, Italy
bUniversitat Autònoma de Barcelona, Servei de Difracció de Raigs X, 08193 Bellaterra, Cerdanyola del Vallès, Barcelona, Spain
cCNR ICCOM, Dipartimento di Chimica, Università di Bari, via Orabona 4, 70125 Bari, Italy. E-mail: cardellicchio@ba.iccom.cnr.it
First published on 14th October 2021
Abstract
The crystal structures of racemic and (S,S)-aminobenzylnaphthol compounds, obtained via the Betti reaction between 2-naphthol, aryl aldehydes and valine methyl ester, were investigated using X-ray diffraction measurements. The molecular shapes of these molecules are similar with variation in hydrogen/fluorine/chlorine, but different crystallographic assemblies were observed. It is worth pointing out that the racemic fluorinated compound crystallizes as a conglomerate. The lattice energies of the crystal structures were calculated by means of the CrystalExplorer17 program. The main weak interactions assembling the crystal structures were recognized, and their contributions were analyzed using the calculated energetic data. It is interesting to point out the situation, which has seldom been reported in the literature, that CH⋯π interactions play a role, despite the presence of potential hydrogen-bonding donors and acceptors.
Introduction
In the early years of the 20th century, the Italian scientist Mario Betti discovered a straightforward multi-component condensation reaction that has been distinguished using his name.1,2 The Betti reaction was seldom applied for many decades, until some of us brought it to the attention of the scientific community, first in 1998 and later in the following years.1,2 Since then, many research groups in the world have applied it.1 The Betti reaction uses 2-naphthol, aryl aldehydes and amines as components to yield aminobenzylnaphthols easily.1,2 Many aminobenzylnaphthols have been resolved into their enantiomers, which were later applied in asymmetric synthesis.1,2 Relevant progress was obtained when enantiopure amines were employed in condensation reactions.3–7 In fact, aminobenzylnaphthols bearing two stereogenic centers were easily produced.3–7 In a solventless and truly “green synthetic process” 2-naphthol, aryl aldehydes and (S)-1-arylethylamine reacted at 60 °C to yield the corresponding (S,S)-aminobenzylnaphthols.6,7 If small amounts of ethanol are added to the crude reaction mixture, these molecules can be obtained as crystalline solids (51–68% yields). The stereochemical preference towards the (S,S)-stereoisomers was explained by invoking an asymmetric transformation of the second kind3,6 in which a more stable crystal subtracts itself to the equilibrium. To gain further insight into this mechanism, the crystal structures of many (S,S)-aminobenzylnaphthols, also bearing halogen atoms and other substituents on the aryl groups, have been investigated via X-ray diffraction experiments.6,7 Hydrogen bonds, involving the naphthol hydroxyl group networks, have been observed, together with unusual networks of many cooperating T-shaped CH⋯π interactions.8,9Only a few papers have been reported regarding investigation of the bio-activity of the aminobenzylnaphthols synthesized so far.10–12 Thus, we reasoned that potential bio-active molecules could be obtained if an amino acid moiety is encompassed into the aminobenzylnaphthol framework via a Betti reaction involving amino acid derivatives.13
Result and discussion
General considerations
In the previous work of some of us,13 2-naphthol, (S)-valine methyl ester 1 and aryl aldehydes 2–4 were reacted at 60 °C without any solvent to yield aminobenzylnaphthols 5–7 (Scheme 1, X = H, F, Cl; Table 1).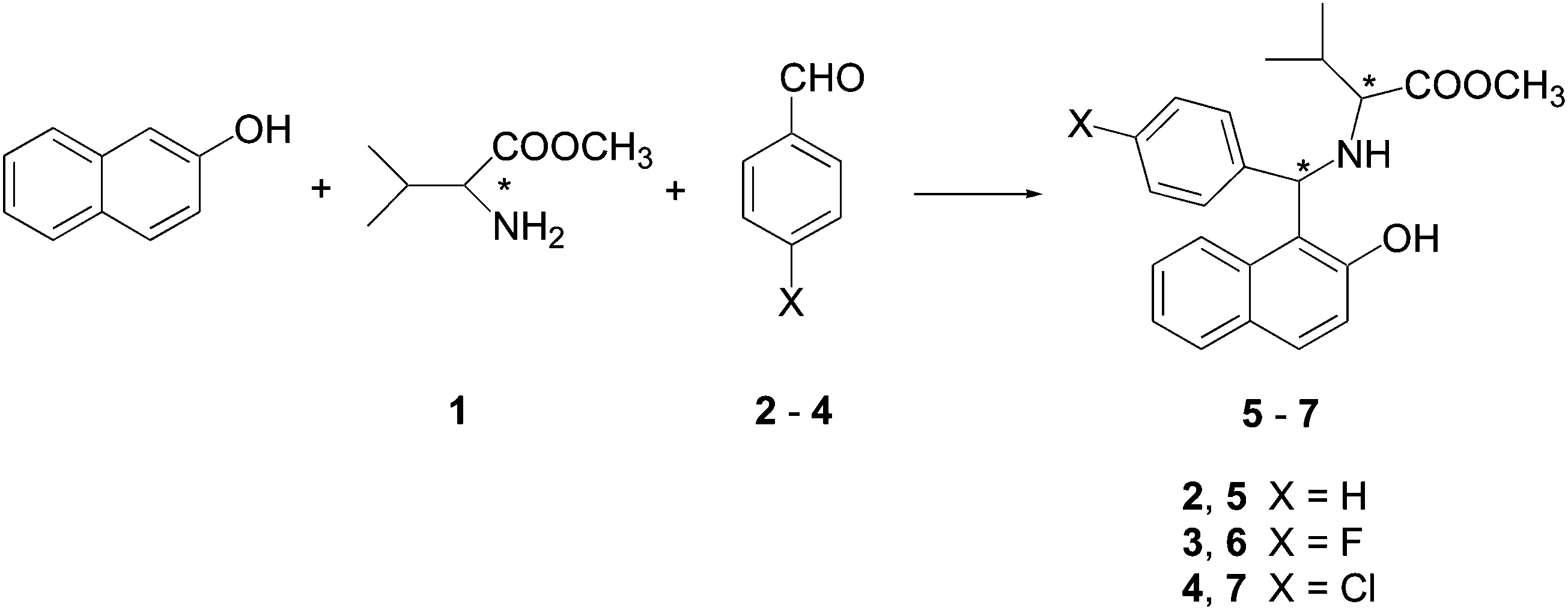 | ||
| Scheme 1 Betti reaction of 2-naphthol, valine methyl ester and aryl aldehydes. | ||
| Entries | Valine methyl ester | Aryl aldehyde | X | Products |
|---|---|---|---|---|
| a 60 °C temperature; no solvent. b Room temperature, diethyl ether as the solvent; addition of lithium perchlorate and chlorotrimethylsilane. | ||||
| 1 | (S)-1 | 2 | H | (S,S) + (R,R)-5a |
| 2 | (S)-1 | 2 | H | (S,S)-5b |
| 3 | (S)-1 | 3 | F | (S,S)-6a |
| 4 | (S) + (R)-1 | 3 | F | (S,S) + (R,R)-6a |
| 5 | (S)-1 | 4 | Cl | (S,S)-7a |
| 6 | (S) + (R)-1 | 4 | Cl | (S,S) + (R,R)-7a |
A systematic investigation of the use of amino acid derivatives in the Betti reaction was blocked by an unexpected difficulty. In fact, when benzaldehyde 2 was used under the standard reaction conditions, the stereogenic center of the valine was completely scrambled, and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of (S,S)- and (R,R)-enantiomers of 5 was obtained as the main product (Table 1, entry 1).13 This drawback was explained, and using milder reaction conditions (Table 1, entry 2) enabled the formation of the desired (S,S)-aminobenzylnaphthol 5 without any loss of chirality.13
:1 mixture of (S,S)- and (R,R)-enantiomers of 5 was obtained as the main product (Table 1, entry 1).13 This drawback was explained, and using milder reaction conditions (Table 1, entry 2) enabled the formation of the desired (S,S)-aminobenzylnaphthol 5 without any loss of chirality.13
On the other hand, when 4-fluorobenzaldehyde 3 and 4-chlorobenzaldehyde 4 were employed in the Betti process, the corresponding (S,S)-aminobenzylnaphthols 6 and 7 were obtained (Table 1, entries 3 and 5).13 In Table 1, we also reported the synthesis of racemic aminobenzylnaphthols 6 and 7, which were obtained starting from a 1:1 mixture of (R)- + (S)-valine methyl ester 1 (Table 1, entries 4 and 6).13
HPLC analysis using chiral columns confirmed the presence of a single enantiomer in macroscopic samples of (S,S)-aminobenzylnaphthols 5–7, and the presence of the enantiomer couple in the samples of racemic5–7.
In the present work, we consider it of interest to investigate the crystal structures of the six aminobenzylnaphthols 5–7, three racemic and three having the (S,S)-configuration, looking for a reshaping of the molecular assemblies due to the amino acid moiety.
Samples for the single-crystal X-ray diffraction experiments were grown by slow evaporation from ethanol. They were found to be suitable for these experiments, and some difficulties were only found for aminobenzylnaphthols (S,S)-5, which is constituted only by light atoms (C, H, N, and O).
Crystal data and structure refinement details are reported in the ESI† (Table S1–S5, ESI†), together with ORTEP plots and packing plots (Fig. S1–S10, ESI†). One molecule is present in each asymmetric unit in all the molecules but one, (S,S)-7, in which Z′ = 2.
In our previous work,13 the (S,S)-configurations of the synthesized aminobenzylnaphthols 5–7 were attributed with the aid of 1H-NMR spectra. The present X-ray diffraction investigation confirms the validity of that attribution.
As expected, equimolar amounts of (S,S)- and (R,R)-stereoisomers were found in the structures of racemic aminobenzylnaphthols 5 and 7. On the other hand, an unusual and unexpected situation occurred in the case of the fluorinated aminobenzylnaphthol 6. The single-crystal X-ray diffraction experiment of (S,S)-6 and of the believed “racemic-6” yielded the same results. Racemic-6 seems to be formed by crystals having the same chirality. This fact can be interpreted with the formation of a racemic conglomerate, that is, a mixture of separated (R,R)- and (S,S)-crystals.
A preliminary investigation on the thermal analysis of the synthesized molecules gave unsatisfactory results, because decomposition was observed around the melting points, probably due to a retro-Betti reaction1,2 that yields the original components of the Betti condensation.
At this stage, X-ray powder diffraction (XRPD) spectra of the aminobenzylnaphthols 5–7 were recorded. First, the XRPD spectrum of each (S,S)-aminobenzylnaphthol 5–7 was compared with XRPD spectrum of its racemic counterpart (Fig. S11–S13, ESI†). As expected, the patterns are clearly different for aminobenzylnaphthols 5 and 7, that do not form conglomerates. On the other hand, the two patterns are identical for the “racemic”-6 and (S,S)-aminobenzylnaphthol-6, an argument that gives support to the fact that this molecule is a racemic conglomerate.14
It is worth mentioning that a very large family of aminobenzylnaphthols has been synthesized over the years using the Betti reaction, but the present work is the first report of a conglomerate in this family. Moreover, this unexpected result occurred only through the unusual presence of a fluorine atom, whereas the almost similar hydrogen- of chlorine-containing molecules behave as usual.
A multi-disciplinary analysis of the crystal structures
The crystal structures were analyzed with the aid of the powerful CrystalExplorer17 program.15–20 This program is able also to estimate the lattice energy, starting from the experimental coordinates derived from the X-ray diffraction experiments.18–20 For a given molecule, a network of adjacent molecules within a certain radius (for example, of 18 Å) is built up around this central species.18–20 At this stage, the interaction energy of the pairs constituted by each molecule of the network with the central species is calculated. The total energy is computed by direct summation of the energy of each pair.18–20The energy calculations of each pair are based upon estimating each of the four contributions (electrostatic, polarization, dispersion and exchange-repulsion), and then adding these terms, after multiplication for suitable weights.20
In our work, the energies of the racemic and the (S,S)-crystals 5–7 were calculated using CrystalExplorer17 by expanding the network to a radius of 18 Å around the central molecule. The spreadsheets for each set of calculations are reported in the ESI† (Fig. S14–S19, ESI†). From inspection of the spreadsheets reported in the ESI,† the most relevant contributions to the final energies come from the molecules that are within an almost 10 Å distance from the central molecule. The total energies are reported in Table 2.
| Entrya | Compound | E_ele | E_pol | E_dis | E_rep | Total |
|---|---|---|---|---|---|---|
| a Data extracted from Fig. S14–S19 (ESI). b As an average value of the values reported in Fig. S17 and S18 of the pair of molecules in the asymmetric unit. | ||||||
| 1 | (S,S)-5 | −31.7 | −6.2 | −142.8 | 48.6 | −132.1 |
| 2 | racem-5 | −30.2 | −7.9 | −167.0 | 56.5 | −148.6 |
| 3 | (S,S)-6 | −39.3 | −6.2 | −150.3 | 52.5 | −143.4 |
| 4b | (S,S)-7 | −39.0 | −6.4 | −154.7 | 57.9 | −142.2 |
| 5 | racem-7 | −43.6 | −7.1 | −163.6 | 61.4 | −152.9 |
From a first inspection of the calculations, racemic aminobenzylnaphthol-5 and -7 (entries 2 and 5) are more stable in energy than the corresponding (S,S)-enantiomers 5 and 7 (entries 1 and 4), as expected. Moreover, these calculations point towards a large contribution of the dispersion energies for the stability of the crystal structures. Some weak interactions that build up the structures, such as CH⋯π interactions8,9 and halogen bonding,21 are included among the dispersion phenomena.
As a whole, the (S,S)-aminobenzylnaphthols 5–7 have a similar shape (Fig. S20, ESI†), whether the aldehyde substituent is a hydrogen, fluorine or chlorine atom, as already reported for the crystal structures of aminobenzylnaphthols6 and of some cyclic phosphonamides that are derived from them.22
However, our analysis, which will be shown in the remainder of this paper, shows unequivocally that the five crystal structures are built up by different weak interactions. Different crystal packings as a consequence of the simple halogen atom variation are reported, as demonstrated by a very recent example of the effect of the haloaryl moiety.23
First of all, in the aminobenzylnaphthols reported so far, the amino group is involved in intramolecular hydrogen bonding with the close hydrogen atom of the naphthol group and, usually, it does not participate in other interactions.6,7 In compounds 5–7, we also observed this unusual behavior. The OH⋯N distances and angles of a typical intramolecular hydrogen bond were measured and are reported in Table 3. Also in the present cases, the nitrogen atom is excluded from participating in any further interaction.
| Entry | Compound | Distance OH⋯N (Å) | OHN angle |
|---|---|---|---|
| 1 | (S,S)-5 | 1.74 | 155° |
| 2 | racem-5 | 1.84 | 151° |
| 3 | (S,S)-6 | 1.77 | 149° |
| 4 | (S,S)-7-a | 1.87 | 146° |
| 5 | (S,S)-7-b | 1.78 | 143° |
| 6 | racem-7 | 1.82 | 154° |
As far as intermolecular hydrogen bonding is concerned, after having excluded the nitrogen atom, the presence of the naphthol and the carbonyl moieties in these molecules seems to offer effective hydrogen-bonding acceptors. However, we do not see hydrogen bonding connected with the presence of the carbonyl group in compounds (S,S)-5, racemic-5 and (S,S)-7.
The situation in which there are no hydrogen bonds, despite the presence of strong donor and acceptor groups, is not common.24–26 This concerns almost 2.5% of the whole Cambridge Structural Database (CSD) structures. Most of these “anomalies” were due to steric factors,24–26 but the remaining cases are strictly connected with the presence of D–H⋯π interactions (D = carbon or nitrogen).24–26 Thus, the presence of CH⋯π interactions, as occurs in other aminobenzylnaphthol compounds, should be expected.
The energetic data that were calculated previously (Fig. S14–S19, ESI†) can provide a more quantitative analysis of these results. We extracted (Table 4) the energetic data connecting pairs of molecules of special interest.
| Entrya | Compound | Symm. operator | R (Å) | E_ele | E_pol | E_dis | E_rep | Total |
|---|---|---|---|---|---|---|---|---|
| a Data extracted from Fig. S14–S19 (ESI). b Distance between molecular centroids. | ||||||||
| 1 | (S,S)-5 | −x, +y + 1/2, −z | 9.95 | −6.1 | −0.9 | −25.0 | 16.2 | −19.0 |
| 2 | racem-5 | −x, −y, −z | 8.68 | −1.8 | −1.9 | −38.3 | 19.1 | −24.8 |
| 3 | (S,S)-6 | −x, y + 1/2, −z + 1/2 | 6.81 | −18.0 | −2.7 | −53.8 | 31.8 | −48.2 |
| 4 | (S,S)-6 | x, y, z | 9.05 | −7.7 | −1.9 | −31.7 | 15.1 | −27.9 |
| 5 | (S,S)-6 | −x + 1/2, −y, z + 1/2 | 9.48 | −5.7 | −1.4 | −24.2 | 14.1 | −19.4 |
| 6 | (S,S)-7 | 7.07 | −10.7 | −2.3 | −66.2 | 39.8 | −46.1 | |
| 7 | racem-7 | x, y, z | 9.89 | −4.4 | −1.1 | −15.8 | 9.0 | −13.7 |
| 8 | racem-7 | −x, −y, −z | 7.51 | −21.6 | −4.0 | −90.0 | 52.6 | −71.6 |
In the case of (S,S)-5, no evidence of hydrogen bonding can be found. According to the energetics data of Table 2, the most relevant contribution to the stability of the structure is due to the dispersion energy. Looking for a more directional contribution, only a weak T-shaped CH⋯π interaction8,9 between the para-hydrogen of the phenyl group and the plane of the naphthyl group (distance of the hydrogen from the plane = 2.78 Å) can be recognized. This pattern can be visualized if the electrostatic potential is drawn on the Hirshfeld surface. It is possible to point out the electrophilic light-blue area of the hydrogen atom pointing towards the electron-rich naphthyl plane (Fig. 1).
 | ||
| Fig. 1 Graphical recognition of the CH⋯π interaction in aminobenzylnaphthol (S,S)-5. Electrostatic potential map drawn on the Hirshfeld surface. | ||
According to the data in Table 4 (entry 1), the interaction between the pairs of molecules described in Fig. 1 contributes only 19 kJ mol−1 to the total energy of the crystal (132 kJ mol−1, Fig. S14, ESI†). Other types of phenomena, connected to the dispersion energy, seem to be relevant in this molecule.
In the case of racemic-5, a molecule of (S,S)-5 is bound with a molecule of (R,R)-5 by two symmetrical weak hydrogen bonds (Fig. 2) between the naphthol oxygen and one of the phenyl group's meta-hydrogen atoms (distance O⋯H 2.76 Å). This pattern, in which the couple of enantiomers is arranged around a center of symmetry, is indicated as R22(16), according to the nomenclature introduced by Bernstein et al.27 A similar but smaller pattern (that is, R22(14), based on the naphthol oxygen atom and the hydrogen in the ortho-position of the phenyl group) was reported in a different racemic crystal structure of an aminobenzylnaphthol.28
 | ||
| Fig. 2 (S,S)- and (R,R)-enantiomers paired around the center of symmetry by weak hydrogen bonding in the crystal structure of rac-aminobenzylnaphthol 5. (Bond lengths are given in Å.) | ||
The energetics data referring to this interaction are reported in Table 4 (entry 2). Even in this case, the weak hydrogen bonding that was recognized seems to offer only a small contribution to the overall stability of the structure.
However, the coupling of the two enantiomers around a center of symmetry in rac-5 looks to provide a higher energetic stability with respect to (S,S)-5 (see Table 2) as well as a higher density (1.167 Mg m−3 for the latter; 1.244 Mg m−3 for the former, see Tables S1 and S2, ESI†).
A further facility offered by the CrystalExplorer17 program is represented by so-called “fingerprint analysis”, that provides a summary of the interactions that build up the crystal structures.
In Fig. 3, the fingerprint analysis of (S,S)-5 and rac-5 is shown and compared.
 | ||
| Fig. 3 Comparison of the CrystalExplorer17 fingerprint analysis for compounds (S,S)-5 (left) and rac-5 (right). | ||
The compact patterns of these two fingerprint plots are characteristic of structures in which the dispersion forces are prevailing.15,16
The crystal structure of (S,S)-6 is governed mainly by hydrogen bonding (Fig. 4 and 5).
 | ||
| Fig. 4 Weak hydrogen bonding involving the carbonyl oxygen atom in aminobenzylnaphthol (S,S)-6. (Bond lengths are given in Å.) | ||
 | ||
| Fig. 5 Weak hydrogen bonding involving the naphthol oxygen atom in aminobenzylnaphthol (S,S)-6. (Bond lengths are given in Å.) | ||
In fact, first, the carbonyl oxygen atom is involved at the same time into two contemporaneous weak hydrogen bonds (Fig. 4), with a phenyl hydrogen atom (distance O⋯HC 2.75 Å) and a naphthyl hydrogen atom (distance O⋯HC 2.63 Å). These pairs of molecules are described in Table 4 as entries 3 and 4, respectively.
Moreover, the naphthol oxygen atom is involved in weak hydrogen bonding with a naphthyl hydrogen atom (distance O⋯H–C 2.58 Å, Fig. 5). This pair of molecules is described in Table 4, entry 5.
By inspection of the entries 3–5 in Table 4 and comparison with all of the energetics data (Fig. S16, ESI†), the interactions involving only these three pairs of molecules contribute almost 2/3 of the overall energy of the crystal structure. The dispersion components to the energy are relevant, but to a lower extent than for the other cases in which hydrogen bonding is not present.
The CrystalExplorer17 fingerprint analysis (Fig. 6, left) confirmed our analysis. The compact form of the pattern is again characteristic of structures in which the dispersion components are relevant. However, the spikes, that were revealed in the right part of Fig. 6 by filtering through the O⋯H contribution to the surface, show weak forms of hydrogen bonding.
 | ||
| Fig. 6 CrystalExplorer17 fingerprint analysis for compound (S,S)-6 (left). Analysis filtered through the O⋯H contribution to the surface (right). | ||
In the case of (S,S)-7, two molecules are found in the asymmetric unit. There is a short contact connecting the naphthol oxygen atom with a methyl hydrogen atom (distance O⋯H3C = 2.65 Å). However, this short contact cannot be qualified as a hydrogen bond, since a CH⋯O angle of only 101° was measured.9 In this situation, patterns of the CH⋯π interaction gain relevance. We observed that one phenyl hydrogen atom in the meta-position interacts with the plane of the naphthyl group of another molecule (distance of the hydrogen from the naphthyl plane 2.60 Å; Fig. 7). This pattern is repeated between another meta-hydrogen of the phenyl group with the plane of the naphthyl group of another molecule (distance of the hydrogen from the naphthyl plane 2.55 Å; Fig. 7). These distances of the hydrogen atom from the aryl plane are allocated in the lower range of distances for this interaction,8 suggesting a significant contribution to the stability of the crystal structure. The energetics data for the interaction between the two molecules in the asymmetric unit is reported in Table 4 (entry 6). Due to the short distance between the molecules of this pair, this interaction contributes significantly to the final energy.
 | ||
| Fig. 7 CH⋯π interactions in the crystal of (S,S)-aminobenzylnaphthol 7. (Bond lengths are given in Å.) | ||
The situation is different for racemic crystal-7 in which the two nucleophilic oxygen atoms (the naphthol and the carbonyl oxygen atoms) behave as hydrogen-bonding acceptors, as usually occurs. A first hydrogen bond was recognized (Fig. 8) between the carbonyl oxygen and one naphthyl hydrogen atom (distance O⋯HC = 2.63 Å). This interaction was described in Table 4 (entry 7). The dispersion components seem to offer a slightly lower contribution to the total energy.
 | ||
| Fig. 8 Weak hydrogen bonding among molecules of the same chirality in racemic-7. (Bond lengths are given in Å.) | ||
A second hydrogen bond was recognized (Fig. 8) between the naphthol oxygen and one methyl hydrogen atom (distance O⋯H3C = 2.71 Å).
The unusual aspect of these hydrogen bonds is that they connect molecules that have the same chirality (for example, one (S,S)-7 molecule with another (S,S)-7, and so on).
The pairing of molecules of different chirality (an (S,S)-7 with an (R,R)-7) around the center of symmetry of the racemic-7 compound occurs through two weak T-shaped CH⋯π interactions between the methine hydrogen of the iso-propyl group and the plane of the naphthyl moiety (distance of the hydrogen from the plane = 2.80 Å), as represented in Fig. 9.
 | ||
| Fig. 9 Pairing of (S,S)-7 with (R,R)-7 around a center of symmetry with the aid of CH⋯π interactions. (Bond lengths are given in Å.) | ||
This tight pairing of two molecules of different chirality is the first contributor to the overall energy (Fig. S19, ESI†). Dispersion components provide the most relevant energetic contribution (Table 4, entry 8) in this pair.
The fingerprint analysis is reported in Fig. 10. The wings, which are unusual in the fingerprint analysis of compound 7, should be due to the contribution of the CH⋯π interactions.15,16 In Fig. 11, the wings are revealed by filtering the pattern through the C⋯H components to the surface.
 | ||
| Fig. 10 Comparison of the CrystalExplorer17 fingerprint analysis for compounds (S,S)-7 (left) and rac-7 (right). | ||
 | ||
| Fig. 11 C⋯H components in the CrystalExplorer17 fingerprint analysis for compounds (S,S)-7 (left) and rac-7 (right). | ||
Conclusion
X-ray diffraction studies on racemic and (S,S)-aminobenzylnaphthols 5–7, changing from hydrogen to fluorine to chlorine substituent atoms, revealed new and interesting features. This halogen switch, that formally deals with a peripheral part of the synthesized molecule, yields different molecular assemblies in which CH⋯π interactions can play a role, despite the presence of potential hydrogen-bonding donors and acceptors.An uncommon and interesting situation was observed in the fluorine switch. Actually, the fluorine-substituted molecule 6 in this sequence yields a conglomerate, a behavior that has not been observed in the large family of aminobenzylnaphthol compounds synthesized so far. In this aminobenzylnaphthol 6, different from compounds 5 and 7, hydrogen bonds are predominant over other interactions and give a higher contribution to the stability of the crystal structure.
The CrystalExplorer17 release was used not only to recognize the main interactions that build up the structures but also to estimate the lattice energies of the racemic and enantiopure crystals. In these computations, dispersion energies were found to make a predominant contribution. An interesting extension of these calculations was the extraction of the data referring to particular pairs of molecules from the whole analysis, and to discuss the interactions concerning these pairs in light of the energetics data provided by the calculations.
These aminobenzylnaphthol compounds, which easily form chiral intermediates, continue to provide new and unexpected structural results.
Experimental section
For the powder X-ray diffraction experiments, data were collected with Cu Kα radiation using a Panalytical XPert Pro diffractometer. In the single-crystal X-ray diffraction experiments, data were collected with Mo Kα radiation using a Bruker SMART-APEX diffractometer. An empirical absorption correction was applied (SADABS). Structures were solved by direct methods (SHELXS-86) and refined by full-matrix least-squares methods on F2 for all reflections (SHELXL-2016).29 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions with isotropic displacement parameters fixed at 1.2 times the Ueq of the corresponding carbon atoms. Crystal data and further refinement details are collected in the ESI† (Tables S1–S5, ESI†). These data were deposited at the Cambridge Crystallographic Data Centre with the following depository codes: 2072225, 2072226, 2072227, 2072228 and 2072229.†The CrystalExplorer17 program was used for energetics calculations using B3LYP/6-31G(d, p) as a wavefunction source and 1.057, 0.54, 0.871, and 0.618 as suitable weights in the calculations.20
Author contributions
M. A. M. Capozzi: synthesis and chemical characterization of the molecules; and writing of the manuscript. A. Alvarez-Larena: X-ray diffraction experiments; resolution of the crystal structures; and writing of the manuscript. J. F. Piniella: X-ray diffraction experiments; resolution of the crystal structures; and writing of the manuscript. C. Cardellicchio: synthesis and chemical characterization of the molecules; theoretical calculations; writing of the manuscript; and coordination of the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks are due to Dr Paola Fini (CNR-IPCF Bari) for preliminary thermal analysis and to Prof. Mark A. Spackman for his suggestions on the calculations performed using CrystalExplorer17. CC thanks the Short Term Mobility program of the Italian National Research Council for staying at the UAB – Barcelona. Italian Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR) is gratefully acknowledged for support.References
- C. Cardellicchio, M. A. M. Capozzi and F. Naso, Tetrahedron: Asymmetry, 2010, 21, 507–517 CrossRef CAS.
- C. Cardellicchio, G. Ciccarella, F. Naso, E. Schingaro and F. Scordari, Tetrahedron: Asymmetry, 1998, 9, 3667–3675 CrossRef CAS; C. Cardellicchio, G. Ciccarella, F. Naso, F. Perna and P. Tortorella, Tetrahedron, 1999, 55, 14685–14692 CrossRef.
- C. Cimarelli, A. Mazzanti, G. Palmieri and E. Volpini, J. Org. Chem., 2001, 66, 4759–4765 CrossRef CAS PubMed.
- C. Boga, E. Di Martino, L. Forlani and F. Torri, J. Chem. Res. (S), 2001, 43–45 CrossRef CAS.
- D.-X. Liu, L.-C. Zhang, Q. Wang, C.-S. Da, Z.-Q. Xin, R. Wang, M. C. K. Choi and A. S. C. Chan, Org. Lett., 2001, 3, 2733–2735 CrossRef CAS PubMed.
- C. Cardellicchio, M. A. M. Capozzi, A. Alvarez-Larena, J. F. Piniella and F. Capitelli, CrystEngComm, 2012, 14, 3972–3981 RSC.
- M. A. M. Capozzi, G. Terraneo and C. Cardellicchio, Acta Crystallogr., Sect. C: Struct. Chem., 2019, C75, 189–195 CrossRef PubMed.
- M. Nishio, CrystEngComm, 2004, 6, 130–158 RSC; M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC; M. Nishio, Y. Umezawa, J. Fantini, M. S. Weiss and P. Chakraborti, Phys. Chem. Chem. Phys., 2014, 16, 12648–12683 RSC.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- M. A. M. Capozzi, C. Cardellicchio, A. Magaletti, A. Bevilacqua, M. Perricone and M. R. Corbo, Molecules, 2014, 19, 5219–5230 CrossRef PubMed.
- A. Puerta, A. R. Galán, R. Abdilla, K. Demanuele, M. X. Fernandes, G. Basica and J. M. Padrón, J. Mol. Clin. Med., 2019, 2, 35–40 CrossRef.
- N. V. Gyémánt, H. Engi, Z. Schelz, I. Szatmári, D. Tóth, F. Fülöp, J. Molnár and P. A. M. de Witte, Br. J. Cancer, 2010, 103, 178–185 CrossRef PubMed.
- M. A. M. Capozzi and C. Cardellicchio, Tetrahedron: Asymmetry, 2017, 28, 1792–1796 CrossRef CAS.
- G. Coquerel, Top. Curr. Chem., 2007, 269, 1–51 CAS.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC; J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, B60, 627–668 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017, http://hirshfeldsurface.net. Search PubMed.
- S. P. Thomas, P. R. Spackman, D. Jayatilaka and M. A. Spackman, J. Chem. Theory Comput., 2018, 14, 1614–1623 CrossRef CAS PubMed.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- M. J. Turner, S. Grabowsky, D. Jayatilaka and M. A. Spackman, J. Phys. Chem. Lett., 2014, 5, 4249–4255 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- M. A. M. Capozzi, C. Pigliacelli, G. Terraneo and C. Cardellicchio, CrystEngComm, 2019, 21, 7224–7232 RSC.
- D. Vargas-Oviedo, J. Portilla and M. A. Macias, J. Mol. Struct., 2021, 1230, 129869 CrossRef CAS.
- R. Taylor and P. A. Wood, Chem. Rev., 2019, 119, 9427–9477 CrossRef CAS PubMed.
- P. A. Wood and P. T. A. Galek, CrystEngComm, 2010, 12, 2485–2491 RSC.
- K. M. Sureshan and R. G. Gonnade, CrystEngComm, 2013, 15, 1676–1679 RSC.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 14, 1555–1573 CrossRef.
- Q. Zhao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, E68, o529 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2072225–2072229. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1nj03538j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |