
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-small FeS2 nanoparticles for highly efficient chemoselective transfer hydrogenation of nitroarenes†
Jamie P.
Southouse
a,
Laura
Lazzarini
 b,
Alex O.
Ibhadon
a and
M. Grazia
Francesconi
*c
b,
Alex O.
Ibhadon
a and
M. Grazia
Francesconi
*c
aDepartment of Chemical Engineering, University of Hull, Cottingham Road, Hull, HU6 7RX, UK
bIMEM-CNR, Parco Area delle Scienze 37/A 43124, Parma, Italy
cDepartment of Chemistry, University of Hull, Cottingham Road, Hull, HU6 7RX, UK. E-mail: m.g.francesconi@hull.ac.uk
First published on 10th August 2021
Abstract
Ultra-small FeS2 nanoparticles were prepared and used as catalysts in a hydrogen transfer reaction for the synthesis of substituted anilines. The catalytic performance is superior to current systems across all reactions, within the timeframe of two hours (100% conversion, 99.9% selectivity and activation energy 26.8 kJ mol−1). The superior catalytic performance was consistent across hydrogen transfer reactions with a number of different nitroarene substrates. Ultra-small FeS2 nanocatalyst, with size 3–6 nm, were prepared via solvothermal method, utilizing oleylamine as a capping agent and a combination of milder temperature (160 °C) and longer reaction time than those reported to date. Tests on recyclability confirmed that the FeS2 nanocatalyst remains highly chemoselective, up to four cycles.
Introduction
The industrial market for substituted anilines is large as they are used as intermediate/precursor compounds in the synthesis of various pharmaceuticals, agrochemicals, dyes and many other compounds pivotal to modern life. The industrial market share for anilines is expected to surpass 19 billion USD by 2024, making them hugely important for industrial supply.1 The chemical reaction to obtain substituted anilines involves the reduction of nitro compounds to amines.2–4 Specifically, the reaction consists of a hydrogenation of substituted aromatic nitro compounds with the substituting group constituting the functionality of the end aniline, therefore, giving it the desired properties tailored to the application. The hydrogenation reaction converts the nitro group into the amino group; however, the substituting group is also susceptible to reduction. Giving that the desired outcome is the synthesis of a substituted aniline, the hydrogenation needs to target only the nitro group.5 To avoid the undesirable outcome of a removed functionality, heterogeneous catalysts are used to direct the reaction selectively towards the reduction of the nitro group.Traditionally hydrogenation reactions involve the use of platinum group metals (PGMs) as catalyst in the activation of molecular H2 gas.6,7 Many noble metal heterogeneous catalysts have been deployed for the hydrogenation of nitrobenzenes to anilines, such as Pd/Al2O3,2,8 Pd/TiO2,6 PtZn/SiO29 and Au/TiO2.6,10
Noble metal catalysts are attractive because of the high activity towards hydrogen activation. However, platinum group metals are energy intensive in their extraction and carry possible risk of dispersion in the environment.11,12 In addition to their energy intensive extraction, these metals tend to be expensive with potential supply interruption, with Pd and Pt, costing 73.36 and 28.30 USD per gram at the time of writing, according to the ‘Study on the review of the list of Critical Raw Materials’ by the European Commission.12
The use of high pressure H2 requires specialised equipment to contain such pressure, with constant maintenance to ensure it is gas tight with no leaks.13–15 Therefore, there is the need to find more affordable routes towards substituted aniline synthesis in order to benefit a wider portion private sector and scientific research.
First row transition metals possess a wide variety of compounds, which have been demonstrated to show a wide range of catalytic properties.16–19 Transfer hydrogenation is being investigated as an alternative approach to the use of high pressure molecular hydrogen in hydrogenation reactions.20,21 Transfer hydrogenation reactions involve the use of a chemical substance that acts as hydrogen donor, such as NaBH4, alcohols, 1,2,3,4-tetrahydronaphthalene and many others.20,22,23 Hydrazine monohydrate (N2H4.H2O) has been reported to be a potent reducing agent in hydrogen transfer for the reduction of nitroarenes to anilines.13,14 J. Wang et al. showed that hydrazine monohydrate could be used to reduce nitroarenes in the presence of a surface enriched MoS2 catalyst, using 4-chloronitrobenze.14 Highly chemoselective reduction of nitroarenes was obtained by hydrogen transfer from hydrazine monohydrate using an oxygen enriched MoS2 catalyst.24 Additionally, the reduction of nitrophenol to amino phenol was obtained using hydrazine monohydrate as a hydrogen transfer agent over a Rh–Fe3O4-g-C3N4–N catalyst at 110 °C.25 Even though hydrazine monohydrate is known to be toxic, appropriate handling procedures are well established in both academia and the private sector. Furthermore, when active as a hydrogen donor it only decomposes into H2 and N2.21,26
Iron Pyrite (FeS2) has been demonstrate to be a highly chemoselective catalyst in the hydrogenation of nitroarenes under high pressure H2.17,27 It is a naturally occurring mineral that has been well studied.16,28–30 It is found commonly in iron, nickel and cobalt ore deposits. Pyrite deposits have been found globally in abundance.16,17 Iron pyrite is also a waste product from iron ore mining and for this reason it is encountering interest in upcycling as a material for energy conversion and catalysis.16,17,27,28,31 FeS2 catalysed the activation of molecular hydrogen gas at lower than usual pressure (0.5 MPa) for the chemoselective hydrogenation of nitrobenzenes.27 Bulk FeS2 pyrite, has been shown to activate molecular hydrogen for the chemoselective hydrogenation of nitroarenes at 5 MPa of hydrogen.17
The mechanism behind the selective hydrogenation of functionalised nitroarenes is somewhat mysterious.32 Across the literature there is a debate on the actual mechanism of the reduction, with the debate stemming from the direct synthesis route via forming the hydroxylamine intermediate or the indirect synthesis route via forming the hydroxylamine which condenses and is then further reduced forming the azo product which is reduced to the amine product.33,34 A graphical display of the direct synthesis vs. indirect synthesis is shown in Fig. 2.
The direct route in the reduction of functionalised nitrobenzene is seen as the more efficient route for the synthesis of functionalised anilines, as it requires fewer chemical transformations. However, the indirect route is more energetically efficient, as the azo produce represents a low energy stable intermediate, whereas in the direct route total conversion of the nitroso group to the unstable hydroxylamine intermediate.34 The total conversion of the nitroso to the hydroxylamine intermediate requires a greater energy input than the condensation step, seen in the indirect step, however the energy gap between the hydroxylamine and aniline product is substantially reduced compared to the hydrazo product seen in the indirect route. As such, the energy gap between the two routes is negligible and they can be seen as equivalent in energy.
This equivalence in energy between the two systems can be differentiated when a catalyst is introduced. By stabilizing intermediates in either route, a catalyst causes a defined difference in the energy level, thus one route shows prevalence over the other.
In this work, we set up a new catalytic system, by combining the use of hydrazine monohydrate and FeS2 nanoparticles as the catalyst. We used 4-chloronitrobenzene as the model nitro compound in which chlorine constitutes the substituting group, (Fig. 1). 4-Chloronitrobenzene was selected as the model reaction due to its prevalence making it easy to acquire in addition to its ease of analysis using our analytical equipment. 4-Chloronitrobenzene is reduced to 4-chloroaniline via selective hydrogenation reactions. Other substituted nitrobenzenes were then tested to confirm the general applicability of our catalytic system. The catalytic performance is superior to current systems across all reactions (100% conversion, 99.9% selectivity and activation energy 26.8 kJ mol−1).
 | ||
| Fig. 1 Schematic diagram of the chemoselective reduction of 4-chloronitrobenzene to 4-chloroaniline, using the catalytic system as defined in this work. | ||
 | ||
| Fig. 2 Proposed reaction scheme in the synthesis of aniline from nitrobenzene, via a direct route and an indirect route.32,34 | ||
Experimental
Materials
Anhydrous iron(II) acetate (Fe 29.5% min), sulfur powder 325 mesh 99.5%, 1-chloro-4-nitrobenzene 98+ %, 1-fluoro-4-nitrobenzne 99%, 1-bromo-4-nitrobenzene 98%, 1-iodo-4-nitrobenzene 98+ %, 1-chloro-3-nitrobenzene 98%, 1-chloro-2-nitrobenzene 99%, 4-chloroaniline, 4-fluoroaniline 99%, 4-bromoaniline 98+ %, 4-iodoaniline 99%, 3-chloroaniline 99%, 2-chloroaniline 98+ %, aniline 99+ %, nitrobenzene 99+ %, hydrazine monohydrate monohydrate 98+ %, hypophosphorus acid 50% w/w aq. solution, propan-2-ol 99.5%, glycerol 99+ %, potassium hydroxide pellets 85%, formic acid 97%, acetic acid (glacial) 99%, was purchased from Alfa Aesar.Oleylamine C-18 content 80–90% was purchased from Fisher Scientific.
Absolute ethanol, hexane (analytical grade) and toluene (analytical grade) were obtained from Honeywell Lab Chemicals.
Preparation of the FeS2 catalyst
The preparation of FeS2 nanoparticles as a hydrogenation catalyst was carried out using a modified version of the method described by C. Guo et al. and later improved for catalytic purposes by B. Ma et al.27,28 Fe(CH3COO)2 (0.48 g, 2.5 mmol) and elemental sulfur (0.48 g, 15 mmol) was weighed into a Teflon lined stainless steel autoclave. 20 mL of ethanol and 10 mL of oleylamine were added to the reagents in the Teflon vessel. The mixture was then sonicated to ensure all solid was suspended before sealing the Teflon vessel tightly in the autoclave. The autoclave was then placed a in a thermostatic oven at 160 °C for 24 h. The oven heating rate was 100 °C min−1 and its cooling rate was 0.5 °C min−1.Once cooled to room temperature the vessel was removed from the oven and opened to reveal a dark brown liquid with a pungent odour of sulfur. The black FeS2 (pyrite) nanoparticles were separated from the liquid by centrifugation. The FeS2 nanoparticles were repeatedly washed by dispersion in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ethanol and hexane. The nanoparticles were then dried under vacuum overnight at room temperature in a desiccator and stored under vacuum until usage.
:1 mixture of ethanol and hexane. The nanoparticles were then dried under vacuum overnight at room temperature in a desiccator and stored under vacuum until usage.
Characterisation of FeS2 catalyst nanoparticles
The FeS2 catalyst nanoparticles were characterised using powder X-ray diffraction (PXRD) carried out using a PANalytical Empyrean X-ray diffractometer with inline PIXcel detector. The monochromatic Cu Kα1 radiation was used and the diffractograms were obtained in the 2θ range of 20–80° over 4 hour collection time, the collection time at each point being 1521 seconds and a step of 0.02626°. A 1° divergence slit was used for all samples and a Ni anti-scatter filter, to minimise the fluorescence of iron under Cu Kα1 radiation.FTIR spectroscopy was carried out on the FeS2 catalyst material pre and post catalysis. FTIR analysis was carried out using a Thermoscientific NicoletiS5 spectrometer with a Pike Miracle diamond ATR attachment carried out from 800 cm−1 to 4000 cm−1.
Transmission Electron Microscopy (TEM) studies were performed in an ultra high-resolution (UHR) (0.18 nm) field emission JEOL 2200FS microscope operating at 200 kV, equipped with an in-column Ω energy filter, two High-Angle Annular Dark Field (HAADF) detectors for the so-called ‘Z-contrast’ imaging and an Energy Dispersive X-ray Spectrometer (EDX) for collecting X-ray spectra and X-ray mapping. The nanostructures were dispersed on holey carbon grids for the observation.
Catalytic testing
The general procedure for carrying out tests on the new catalytic system was as follows: 0.05 M substituted nitrobenzene substrate (4-chloronitrobenzene, 4-iodonitrobenzene, 4-bromonitrobenzene, 4-fluoronitrobenzene, 3-chloronitrobenzene, 2-chloronitrobenzene) with 0.1 M toluene internal standard was added to ethanol, according to the catalytic test carried out. 25 mg of FeS2 catalyst was added to the batch reactor with 50 mL of the chloronitrobenzene solution. The solution would then be purged with nitrogen 3 times until all oxygen had been removed from the reaction vessel.The solution was left to equilibrate under the nitrogen atmosphere for 30 minutes before hydrogen donor compound (hydrazine monohydrate, isopropanol, glycerol, formic acid, acetic acid, hypophosphorus acid) was injected into the reaction. Reaction was stirred rapidly at 800 rpm to avoid mass transfer mixing limitations.
After this point 250 μL aliquots of the solution were extracted at 0, 10, 20, 40, 60, 80, 100 and 120 minutes reaction time, these times were selected to obtain a full reaction profile across the reaction, opposed to a single end point. Each aliquot was analysed using a Varian 430 gas chromatograph equipped with a 30 m Stabilwax® capillary column (Restek), each aliquot was injected in triplicate to obtain the standard deviation and uncertainty in the measurements.
Results and discussion
Preparation and characterisation of FeS2 catalyst
Hydrogen transfer reactions have become promising alternatives to the more traditional molecular hydrogen activation for nitroarene reduction. In fact, hydrogen transfer reactions do not require the use of high-pressure hydrogen and can use catalysts that are more sustainable than noble metals. An environmentally friendly compound that has recently shown promise as a heterogeneous catalyst is iron pyrite (FeS2). Iron sulfide is normally used as a nanocatalyst hence many studies have reported methods for the synthesis of small particles.35 We set out to prepare FeS2 as ultra-small nanoparticles and to test their behaviour as catalysts for the hydrogen transfer reduction of nitroarenes. We designed a methodology that uses milder conditions than those previously reported and has the added advantage of giving ultra-small nanoparticles of narrow size distribution (3–6 nm).The reason behind the choice to synthesise ultra-small nanoparticles was to maximise the performance of the catalyst in the transfer hydrogenation reaction, as B. Ma et al. showed a link between the size of the FeS2 particles and the conversion of the nitrobenzene substrate, approximately the larger the size the lower the conversion.27 Therefore, we set out to produce ultra-small nanoparticles to maximise the catalytic performance. T. Li et al. reported that lowering the synthesis temperature, in the synthesis of FeS2 nanoparticles from 260 °C to 180 °C, results in a drastic decrease in the size and distribution of sizes of the obtained nanoparticles, from 18 nm to 3.5 nm.36 Thus, in this work we merged the two methodologies from Ma and Li.27,28 Specifically, we used the reaction mixtures from B. Ma et al. and C. Guo et al., Fe(CH3COO)2 and elemental sulfur suspended in ethanol and oleylamine (Table 1), because of better environmental credentials and easier experimental handling. However, our reaction temperature was lowered to precipitate the smallest possible nanoparticles. A comparison of the reaction conditions previously published for the synthesis of FeS2 nanoparticles are shown in Table 1.
| Fe Source | S Source | Solvent and capping agent | Reaction temperature (°C) | Particle size (nm) | Ref. |
|---|---|---|---|---|---|
| β-FeOOH | Elemental sulfur | Octadecylamine | 180 | 50–75 | 37 |
| FeCl2·4H2O Fe(CH3COO)2 | Elemental sulfur | Ethanol/oleylamine | 220 | 30–50 | 27,28 |
| Fe(acac)2 | Sulfur diphenyl ether | Dodecanethiol | 180–260 | 3.5–18 | 36 |
| Fe(CH3COO)2 | Elemental sulfur | Ethanol/oleylamine | 160 | 3–6 | This work |
Hence, we used a lower temperature of 160 °C for our reaction, but that lead to an amorphous product. We, therefore, extended the reaction time parameter from ten hours to twenty-four hours and this led to a single phase FeS2. The powder X-ray diffraction pattern (PXRD) (Fig. 3a) shows a single phase cubic FeS2. The diffraction peaks were indexed using the crystal structure for FeS2 reported by Finklea et al. (space group Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , a = 5.4281 Å).29 The crystal structure unit cell of FeS2 is shown in Fig. 3b.29 Using the Scherrer equation we obtained an average particle size of 4.7 nm.
, a = 5.4281 Å).29 The crystal structure unit cell of FeS2 is shown in Fig. 3b.29 Using the Scherrer equation we obtained an average particle size of 4.7 nm.
 | ||
| Fig. 3 (a) PXRD pattern of FeS2 nanoparticles with peaks indexed. (b) Crystal structure of FeS2 from model.29 The yellow spheres represent the sulfur anions and the brown octahedral the Fe–S octahedral. The crystal structure was drawn using the software VESTA.38 | ||
TEM imaging shows that the dispersed nanoparticles tend to form agglomerates while maintaining their individuality, both in the STEM-HAADF and HREM modes (Fig. 4a and b respectively). Most of our particles present anisotropic shape, being twice or more, longer than wide. Visual examination suggests uniformity in size. Indeed, their average size (length) has been measured to be between 3 and 6 nm, with a narrow size distribution (Fig. 4b). This value agrees reasonably well with the PXRD evaluation. The SAED pattern (Fig. 4c) showed that the sample displays a cubic unit cell, in agreement with the PXRD pattern and the literature for FeS2 pyrite.
 | ||
| Fig. 4 (a) HAADF Scanning TEM image of nanoparticle dispersion. (b) High resolution image of some nanoparticles showing their elongated shape, the red contours serve as guide for the eye. (c) Selected Area Electron Diffraction (SAED) taken on a large agglomerate of FeS2 nanoparticles (see 3a), with diffraction rings indexed. (d) HAADF Scanning TEM of large aggregate of nanoparticles. (e) Fe k line signal mapped onto image in Fig. 3d. (f) S k line signal mapped onto image in Fig. 3d. | ||
EDS analysis was used to confirm the chemical composition of the material. Analyses have been performed in many different regions and identified everywhere 67% of sulfur and 33% iron by atomic percentage, in line with the expected values for FeS2. The elemental mapping of the FeS2 material (Fig. 3d–f) showed homogeneous distribution of Fe and S within the catalyst material. The zones with highest intensity are the same in the HAADF picture and in the maps and correspond to regions where more particles are present. Isolated pockets of iron, potentially detrimental for catalytic performance, were not detected.18,39
The FTIR spectrum of the catalyst material was carried out to determine whether carbon species were present on the surface from the use of oleylamine as a capping agent. The presence of carbon species on the surface could block active sites, thereby be detrimental to the catalytic performance. The FTIR spectrum (Fig. S1, ESI†) shows only bands assigned to sulfur species (∼1400 cm−1 and ∼2800 cm−1) which may derive from surface oxidation of the catalyst or from residual elemental sulfur from the synthesis.40 These sulfur species are not reported to be of hinderance to the catalytic performance.
The role of the capping agent, oleylamine, showed a dramatic effect on the size and particle aggregation of the obtained FeS2 nanoparticles as shown in Fig. S2a–c (ESI†). The lack of oleylamine did not affect the FeS2 crystal structure or phase purity yet particles were shown to be larger and more aggregated Fig. 5.
 | ||
| Fig. 5 Trends of decreasing concentration for 4-CNB (blue graph) and increasing concentration for 4-CAN (orange graph) in time, during the FeS2 catalysed hydride transfer hydrogenation in ethanol, averaged across 3 test reactions. Reactions carried out using 0.05 M 4-CNB solution in ethanol at 60 °C, under N2, stirred at 800 rpm, 25 mg FeS2 catalyst loading. 1 mL, 20 mmol, of hydrazine monohydrate used as hydrogen donor in all reactions. | ||
Selective hydrogenation of substituted nitroarenes
The chemoselective transfer hydrogenation of nitroarenes was evaluated on a number of nitroarene substrates using freshly prepared FeS2 nanocatalyst, as shown by the reaction scheme in Fig. 1. However, prior to the catalytic testing on a wide range of substrates, 4-chloronitrobenzene was studied as a model compound for the hydrogen transfer reaction. Several reaction parameters were studied and optimised on the test reaction, 4-chloronitrobenzene reduction, these included reaction solvent, temperature and catalyst loading. A summary of these results is shown in Table S1 (ESI†) and an example reaction profile is shown in Fig. 6. It was found that ethanol was the ideal solvent for the reaction, at 60 °C with 25 mg of FeS2 nanocatalyst, to obtain a balance between catalytic performance and environmental considerations. | ||
| Fig. 6 Recyclability of FeS2 nanoparticles for the hydrogen transfer hydrogenation of 4-chloronitrobenzene, conversion of 4-chloronitrobenzene at 2 hours (blue) and selectivity towards 4-chloroaniline (orange). Reactions carried out using 0.05 M 4-CNB solution in ethanol at 60 °C, under N2, stirred at 800 rpm. 1 mL, 20 mmol, of hydrazine monohydrate used as hydrogen donor in all reactions. After each reaction, the catalyst was collected via centrifugation, washed using aliquots of ethanol and dried under vacuum before reuse in subsequent reactions. | ||
Once properly optimised for 4-chloronitrobenzene, for catalytic performance and environmental considerations, the hydrogen transfer reaction using ultra-small FeS2 was applied to a range of nitroarene substrates. As substrates we selected nitrobenzenes with halogen substituents, from fluorine to iodine. The properties of the halogens vary from electron withdrawing (fluorine) to electron donating (iodine). We correlated these electronic effects to the conversion and selectivity of the hydrogen transfer reaction (Table 2). The halogenated nitrobenzenes were all hydrogenated to their corresponding substituted aniline using ultra-small FeS2 nanoparticles. For all substrates, a selectivity greater than 99.8% and conversion of >99.9% were achieved within a two-hour time frame. The effect of the position of the halogen on the aromatic ring on the conversion and selectivity was also considered. Specifically, we concentrated on chloronitrobenzene with substituents in either ortho, para or meta positions on the aromatic ring. In general, there was no observable effect of the substituted aniline regardless of the electron donating/withdrawing properties or position of the halogen on the aromatic ring, in relation to the conversion or selectivity.
| Substrate | Product | Conversion (%) | Selectivity (%) |
|---|---|---|---|

|

|
99.9 | 100 |

|

|
99.9 | 100 |

|

|
100 | 99.9 |

|

|
100 | 99.8 |

|

|
99.9 | 100 |

|

|
100 | 100 |
It can therefore be concluded that ultra-small FeS2 nanoparticles are of general applicability as catalysts for the high conversion and chemoselectivity observed in the hydrogen transfer reduction of 4-chloronitrobenzene, are reproduced with other substrates. FeS2 shows advantages in the catalytic hydrogen transfer reduction of nitroarenes compared to other iron chalcogenide catalysts such as Fe2O3, specifically the γ-Fe2O3 polymorph, in that the reaction times are reduced, yet selectivity is higher under comparable reaction conditions. This was demonstrated by Ai et al. where γ-Fe2O3 was used as a catalyst in the selective hydrogen transfer using hydrazine as a donor solvent.41 They demonstrated that reduction of nitroarenes can be achieved using this method, however, reaction times are extended to 3 hours at higher temperature, 100 °C. Additional work by Sonavane et al. and Jagadeesh et al. showed that γ-Fe2O3, can be used in the selective hydrogenation of nitroarenes.42,43 Sonavane et al. showed that isopropanol can be used in the selective hydrogenation, yet, this reaction requires elevated temperatures, >80 °C, and longer reaction times, up to 6 hours, to convert the substrate using this catalyst. Jagadeesh et al. demonstrated the activation of molecular hydrogen by γ-Fe2O3, their work showed that high pressures of 50 bar were required to convert the nitroarene substrate within a 12 hour reaction.43 These studies show the advantages of using FeS2 in the selective hydrogen transfer of nitroarenes using hydrazine as a donor, where >99.9% conversion of the substrate can be achieved at relatively low temperature, 60 °C, within 2 hours at ambient pressure.
From these studies using other hydrogen sources, it was decided that other hydrogen donors should be studied to determine the relative susceptibility of these hydrogen donors in comparison to hydrazine. These hydrogen donors are summarised in Table 2. Conversion of 4-chloronitrobenzene was shown to minimal in comparison to hydrazine for all other hydrogen sources Table 3.
| Reducing agent | Conversion of p-CNB (%) | Selectivity towards p-CAN (%) |
|---|---|---|

|
99.9 | 100 |
| H2 | 0 | N/A |
| NaBH4 | 0 | N/A |

|
6.1 | 100 |

|
<1.0 | 99.2 |

|
0 | N/A |

|
0 | N/A |

|
2.0 | 100 |

|
0 | N/A |
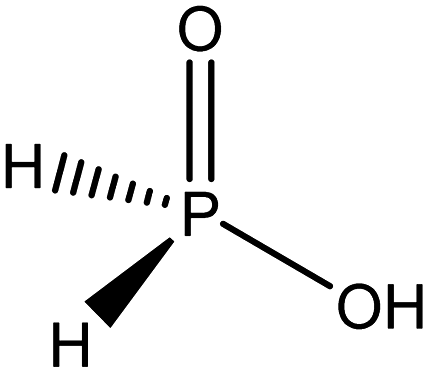
|
0 | N/A |
Alcohol derived hydrogen donors were studied in further tests in the presence of a KOH co-catalyst as literature sources show that this can be of use in developing effective hydrogen transfer catalysis system based on these donors.23,44 In the case of FeS2 it was shown that the presence of a co-catalyst has no impact on the hydrogenation of the substrate.
From the studies of the hydrogen donors, it was clear that hydrazine monohydrate was the hydrogen donor of choice for the selective hydrogen transfer of 4-chloronitrobenzene on an FeS2 catalyst. Hydrogen transfer using shows >99.9% conversion of the substrate whilst maintaining 100% selectivity towards 4-chloroaniline within 2 hours of reaction at standard pressure and 60 °C, with other hydrogen donors showing <10% conversion under the same reaction conditions.
The activation energy for the transfer hydrogenation of the model reaction, 4-chloronitrobenzene, was calculated using a range of reaction temperatures, from 25 °C to 80 °C, and using Arrhenius theory, as shown in Fig. S3 (ESI†). The activation energy for the transfer hydrogenation reaction was found to be 28.36 kJ mol−1. The activation energy was determined to be far lower than previous studies on the reduction of nitroarenes by activation of molecular hydrogen (∼90 kJ mol−1) and the activation energy of comparable nitroarene reductions based on hydrogen transfer (∼75 kJ mol−1).45
The lower activation energy, and in general superior catalytic activity, can be attributed to FeS2, which easily promotes the decomposition of hydrazine by an electron donation. This electron derives from the change in oxidation state between Fe(II) and Fe(III) on the FeS2 surface and is donated to the hydrazine molecule which releases the reactive H* and N2 gas. The generated H* is then transferred to the substrate molecule.24,32,46
Recyclability of FeS2 catalyst
The recyclability of a catalyst is a vital property of a given catalyst. Deactivation over time and repeated use of the catalyst can lead to inefficiencies in the hydrogenation reaction or a lack of selectivity in the desired product distribution.4The recyclability of the ultra-small FeS2 nanoparticles was studied, taking into account that pyrite is known to decay into iron sulfate and iron oxide over time.17,27 Structural and chemical changes in the catalyst during the reaction can influence the catalytic performance, therefore, post reaction characterisation is key.31,47 Ultra-small FeS2 nanocatalyst was used in four subsequent CNB to CAN hydrogen transfer reactions, with intermediate centrifugation and drying, then analysed via PXRD and FTIR after each reaction to detect signs of decomposition. The recyclability of the nanocatalyst is shown in Fig. 6.
FeS2 proved to be recyclable across four reaction cycles, with conversion of 4-chloronitrobenzene reducing to less than 99% only after the third cycle (Fig. 6). This reduction may not necessarily be linked to decomposition of FeS2 but may be due to the loss of catalyst after each cycle during extraction and centrifugation. Selectivity towards 4-chloroaniline was maintained throughout the reaction cycles with >99.9% of the products obtained being 4-chloroaniline.
Thus the ultra-small FeS2 nanocatalyst can be considered to be highly recyclable across at least four cycles, maintaining rapid rate of CNB conversion and complete chemoselectivity towards the CAN product. PXRD and FTIR did not show noticable changes to the FeS2 material after several reaction cycles (ESI† Fig. S4 and S5)
Conclusions
We present a highly efficient approach to the chemoselective transfer hydrogenation of substituted nitroarenes to their corresponding aniline using ultra-small FeS2 nanoparticles as a catalyst. Using a combination of lower temperature and longer reaction times, ultra-small FeS2 nanoparticles were obtained, with a narrow size distribution and anisotropic morphology. In fact, it was reported in the literature that, in general, the smaller the size of the catalyst particles an increase activity catalyst for hydrogenation catalysis. We aimed at applying this concept to hydrogen transfer reactions, an alternative to hydrogenation. Ultra-small FeS2 nanoparticles were used as catalysts for the chemoselective transfer hydrogenation of six halogenated nitroarene substrates. The heterogeneous hydrogen transfer was carried out within a two-hour reaction timeframe, using ethanol as a solvent, hydrazine monohydrate as a hydrogen donor, a catalyst loading of 25 mg and a reaction temperature of 60 °C. These parameters were optimised using 4-chloronitrobenzene as a test substrate, with a view of reaching the best compromise between reaction performance and sustainability. Across all reactions from halogenated nitroarenes to halogenated anilines, the catalytic performance was found to be superior compared to the state if the art with conversion >99.9% and selectivity >99.9%. Furthermore, the activation energy was calculated for the transfer hydrogenation of 4-chloronitrobenzene (28.36 kJ mol−1) and proved to be lower than that within comparable systems. FeS2 nanocatalyst was shown to be highly recyclable, with minimal changes to its crystal structure and surface properties after four reactions. We believe that this is the first reported instance of the use of ultra-small FeS2 nanoparticles as catalysts for a selective hydrogen transfer reaction.Conflicts of interest
There are no conflicts to declare.References
- S. Chakraborty, K. Pulindindi, ed., Global Market Insights, GMInsights, 2017, Vol. 1, pp. 1–160 Search PubMed.
- J. Zhang, L. Wang, Y. Shao, Y. Wang, B. C. Gates and F.-S. Xiao, Angew. Chem., Int. Ed., 2017, 56, 9747–9751 CrossRef CAS PubMed.
- H. Huang, X. Wang, X. Li, C. Chen, X. Zou, W. Dinga and X. Lu, Green Chem., 2017, 19, 809–815 RSC.
- T. B. Nguyen, C. P. Huang and R. Doong, Appl. Catal., B, 2019, 240, 337–347 CrossRef CAS.
- P. Sangeetha, K. Shanthi, K. S. Rama Rao, B. Viswanathan and P. Selvam, Appl. Catal., A, 2009, 353, 160–165 CrossRef CAS; M. Turáková, M. Králik, P. Lehocký, L. U. Pikna, M. Smrcová, D. Remeteiová and A. Hudák, Appl. Catal., A, 2014, 476, 103–112 CrossRef.
- Y.-W. Chen and D.-S. Lee, Mod. Res. Catal., 2013, 2, 25–34 CrossRef CAS.
- S. K. Johnston, N. Cherkasov, E. Pérez-Barrado, A. Aho, D. Y. Murzin, A. O. Ibhadon and M. G. Francesconi, Appl. Catal., A, 2017, 544, 40–45 CrossRef CAS; S. K. Johnston, T. A. Bryant, J. Strong, L. Lazzarini, A. O. Ibhadon and M. G. Francesconi, ChemCatChem, 2019, 11, 2909–2918 CrossRef; M. J. Taylor, L. J. Durndell, M. A. Isaacs, C. M. A. Parlett, K. Wilson, A. F. Lee and G. Kyriakou, Appl. Catal., B, 2016, 180, 580–585 CrossRef.
- W. Wang, W. Xu, K. B. Thapa, X. Yang, J. Liang, L. Zhu and J. I. Zhu, Catalysts, 2017, 7, 292–305 CrossRef.
- S. Iihama, S. Furukawa and T. Komatsu, ACS Catal., 2016, 6, 742–746 CrossRef CAS.
- D. Lamey, O. Beswick, F. Cárdenas-Lizana, P. J. Dyson, E. Sulman and L. Kiwi-Minsker, Appl. Catal., A, 2017, 542, 182–190 CrossRef CAS.
- B. d. R. G. e. Minières, B. G. Survey, TNO, D. Sustainability, Vol. 4 (Ed.: D. Sustainability), EU Publications, Europa, 2017, pp. 1–517.
- E. Commission, European Commission, Publications Office of the European Union, 2017, 476.
- C. Zhang, J. Lu, M. Li, Y. Wang, Z. Zhang, H. Chen and F. Wang, Green Chem., 2016, 18, 2435–2442 RSC.
- J. Wang, Y. Zhang, J. Diao, J. Zhang, H. Liu and D. Su, Chin. J. Catal., 2018, 39, 79–87 CrossRef CAS.
- W. M. N. Ratnayake, J. S. Grosserf and R. G. Ackman, J. Am. Oil Chem. Soc., 1990, 67, 940–946 CrossRef CAS.
- M. S. Faber, M. A. Lukowski, Q. Ding, N. S. Kaiser and S. Jin, J. Phys. Chem. C, 2014, 118, 21347–21356 CrossRef CAS PubMed.
- J. R. Morse, J. F. Callejas, A. J. Darling and R. E. Schaak, Chem. Commun., 2017, 53, 4807–4810 RSC.
- R. V. Jagadeesh, K. Natte, H. Junge and M. Beller, ACS Catal., 2015, 5, 1526–1529 CrossRef CAS.
- S. F. Adil, M. E. Assal, M. Kuniyil, M. Khan, M. R. Shaik, A. Alwarthan, J. P. Labis and M. R. H. Saddidui, Mater. Express, 2017, 7, 79–92 CrossRef CAS.
- J. Wang, Z. Ge, L. Pei, P. Kong, R. Wang, P. Zhu, M. Liu, X. Gu and Z. Zheng, Catal. Sci. Technol., 2019, 9, 6681–6690 RSC.
- A. Furst, R. C. Berlo and S. Hooton, Chem. Rev., 1965, 65, 51–68 CrossRef CAS.
- K. Zhang, J. M. Suh, J.-W. Choi, H. W. Jang, M. Shokouhimehr and R. S. Varma, ACS Omega, 2019, 4, 483–495 CrossRef CAS PubMed; M. A. Aramendia, V. Borau, C. Jiménez, J. M. Marinas and J. A. Pajares, J. Catal., 1982, 78, 188–196 CrossRef; K. W. Cheah, S. Yusup, G. Kyriakou, M. Ameen, M. J. Taylor, D. J. Nowakowski, A. V. Bridgewater and Y. Uemura, Int. J. Hydrogen Energy, 2019, 44, 20678–20689 CrossRef; K. W. Cheah, M. J. Taylor, A. Osatiashtiani, S. K. Beaumont, D. J. Nowakowski, S. Yusup, A. V. Bridgewater and G. Kyriakou, Catal. Today, 2019, 324, 115–125 CrossRef.
- D. Tavor, I. Gefen, C. Dlugy and A. Wolfson, Synth. Commun., 2011, 41, 3409–3416 CrossRef CAS.
- C. Zhang, Z. Zhang, X. Wang, M. Li, J. Lu, R. Si and F. Wang, Appl. Catal., A, 2016, 525, 85–93 CrossRef CAS.
- Z. Hu, J. Zhou, Y. Ai, L. Liu, L. Qi, R. Jiang, H. Bao, J. Wang, J. Hu, H. Sun and Q. Liang, J. Catal., 2018, 368, 20–30 CrossRef CAS.
- D.-X. Zhang, H. Yin, H.-F. Zhong, L.-Y. Gan and P. Wang, Int. J. Hydrogen Energy, 2020, 45, 16114–16121 CrossRef CAS; H. W. Lucien, J. Chem. Eng. Data, 1961, 6, 584–586 CrossRef.
- B. Ma, X. Tong, C. Guo, X. Guo, X. Guo and F. J. Keilc, RSC Adv., 2016, 6, 55220–55224 RSC.
- C. Guo, X. Tong and X. Guo, Mater. Lett., 2015, 161, 220–223 CrossRef CAS.
- S. Finklea, L. Cathey and E. L. Amma, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 529–537 CrossRef.
- E. Nowack, D. Schwarzenbach and T. Hahn, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 650–659 CrossRef.
- I. Temprano, T. Liu and S. J. Jenkins, Catal. Today, 2017, 286, 101–113 CrossRef CAS.
- A. Mahata, R. K. Rai, I. Choudhuri, S. K. Singh and B. Pathak, Phys. Chem. Chem. Phys., 2014, 16, 26365–26374 RSC.
- O. A. Stasyuk, H. Szatylowicz, T. M. Krygowiski and C. Fonseca Guerra, Phys. Chem. Chem. Phys., 2016, 18, 11624–11633 RSC.
- H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- D.-W. Wang, Q.-H. Wang and T.-M. Wang, CrystEngComm, 2010, 12, 755–761 RSC.
- T. Li, Z. Guo, X. Li, Z. Wu, K. Zhang, H. Liu, H. Sun, Y. Liu and H. Zhang, RSC Adv., 2015, 5, 98967–98970 RSC.
- M. Adhikari, A. Singh, E. Echeverria, D. N. McIlroy and Y. Vasquez, ACS Omega, 2020, 5, 14104–14110 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- K. J. Datta, A. K. Rathi, M. B. Gawande, V. Ranc, G. Zoppellaro, R. S. Varma and R. Zboril, ChemCatChem, 2016, 8, 2351–2355 CrossRef CAS.
- D. Rickard and G. W. Luther, Chem. Rev., 2007, 107, 514–562 CrossRef CAS PubMed; S. Phimsen, W. Kiatkittipong, H. Yamada, T. Tagawa, K. Kiatkittipong, N. Laosiripojana and S. Assabuumrungrat, Energy Convers. Manage., 2017, 151, 324–333 CrossRef.
- Y. Ai, M. He, Q. Lv, L. Liu, H. Sun, M. Ding and Q. Liang, Chem. – Asian J., 2017, 13, 89–98 CrossRef PubMed.
- S. U. Sonavane, M. B. Gawande, S. S. Deshpande, A. Vankataraman and R. V. Jayaram, Catal. Commun., 2007, 8, 1803–1806 CrossRef CAS.
- R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2017, 342, 1073–1076 CrossRef PubMed.
- M. B. Gawande, A. K. Rathi, P. S. Branco, I. D. Nogueira, A. Velhinho, J. J. Shrikhande, U. U. Indulkar, R. V. Jayaram, C. A. A. Ghumman, N. Bundaleski and O. M. N. D. Teodoro, Chem. – Eur. J., 2012, 18, 12628–12632 CrossRef CAS PubMed.
- Z. Hu, S. Tan, R. Mi, X. Li, D. Li and B. Yang, Catal. Lett., 2018, 148, 1490–1498 CrossRef CAS; J. B. Santos, G. P. Valenca and J. A. J. Rodrigues, J. Catal., 2002, 210, 1–6 CrossRef; M. Lauwiner, P. Rys and J. Wissmann, Acta Crystallogr., Sect. A: Found. Crystallogr., 1998, 172, 141–148 Search PubMed.
- M. E. Castillo, T. A. Gad-Allah, M. E. M. Ali, A. H. Salem, A. S. G. Khalil, M. G. Francesconi and D. C. Lupascu, Proceedings of the 4th World Congress on New Technologies, 2018, 1, 1–6 Search PubMed.
- R. Sun, M. K. Y. Chan and G. Ceder, Phys. Rev., 2011, 83, 235311–235312 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nj03297f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |