
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the antioxidant activity of thiaflavan compounds: a quantum chemical study
Douniazed
Hannachi
 *ab,
Nour
El Houda Amrane
b,
Lynda
Merzoud
c and
Henry
Chermette
*c
*ab,
Nour
El Houda Amrane
b,
Lynda
Merzoud
c and
Henry
Chermette
*c
aDépartement de Chimie, Faculté des Sciences, Université de Setif-1, El Bez, Setif, Algeria. E-mail: h_douniazed@yahoo.fr
bLaboratoire d’Electrochimie, d’Ingénierie Moléculaire et de Catalyse Redox (LEIMCR), Faculté de Technologie, Université Sétif-1, 19000, Sétif, Algeria
cUniversité de Lyon, Université Claude Bernard Lyon 1, CNRS UMR 5280, Institut des Sciences Analytiques, 69622 Villeurbanne Cedex, France. E-mail: henry.chermette@univ-lyon1.fr
First published on 17th June 2021
Abstract
Density functional theory calculations at the B3LYP level are performed to theoretically investigate the antioxidant properties of 30 thiaflavan compounds. The main theoretical parameters, such as the bond dissociation enthalpy, ionization potential, proton dissociation enthalpy, proton affinity, electron transfer enthalpy, aromaticity index and spin density of O-atoms in the gas, water and benzene phases, have been determined. On the basis of our calculations, the OH groups in the A-ring are the main contributors of the antioxidative activities of thiaflavan comparatively to the B-ring. The reactivity of the thiaflavan compounds with the DNA bases is determined and the thymine moiety in DNA is found to be the primary target for reduction. Among the 30 compounds theoretically studied, the ones containing a ferrocene moiety at ring C′ of thiaflavan are the most promising for future applications in the pharmacology field and the food industry.
Introduction
Free radicals are highly reactive and unstable compounds, as they can induce oxidative damage in biological systems such as DNA, RNA, proteins and lipids. They play an important role in various diseases such as cancer, aging, arthritis, cardiovascular disease and neurodegenerative disease.1–3 This destructive reaction can be inhibited by antioxidant compounds capable of scavenging free radicals through electron or proton transfer mechanisms. In the literature, three important mechanisms describe antioxidant reactions4,5 as shown in Scheme 1.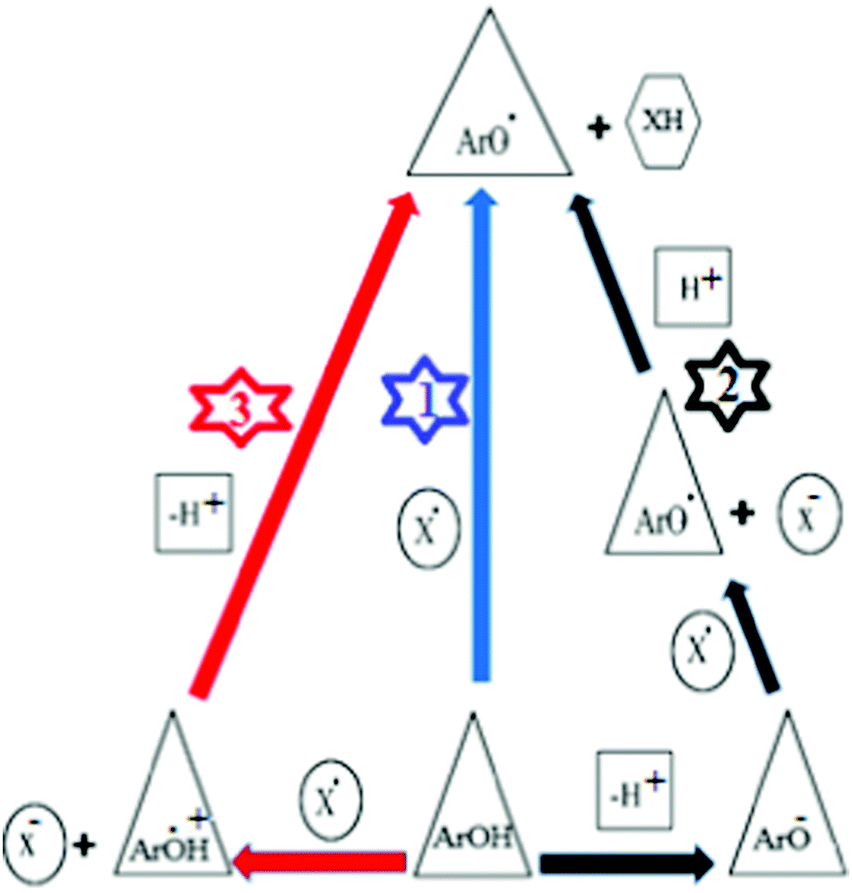 | ||
| Scheme 1 Antioxidant mechanisms: (1) HAT, (2) SPLET, and (3) SETPT. (ArOH): antioxidant, (ArO˙): radical, (ArOH+): radical cation, (ArO−): anion, (X): free radical. | ||
(1): Hydrogen atom transfer mechanism (HAT)
According to this mechanism the free radical removes one hydrogen atom from the antioxidant (ArOH), so that the antioxidant compound itself becomes a radical. In this mechanism the BDE (Bond Dissociation Enthalpy) is generally the numerical parameter for estimating the antioxidant activity, see (1) in Scheme 1.(2): Sequential proton loss electron transfer (SPLET)
SPLET is a two-step reaction (see (2) in Scheme 1). In this reaction, the antioxidant dissociates into an anion (ArO−) and a proton. The created ion reacts with the free radical and therefore becomes itself a radical (ArO˙). The numerical parameters related with the SPLET mechanism are: PA (Proton Affinity) for the first step and ETE (Electron Transfer Enthalpy) for the second step.(3): Single-electron transfer followed by proton transfer (SET-PT)
This mechanism also consists of a two-step reaction (see (3) in Scheme 1). In the SET mechanism, the antioxidant provides an electron to the free radical and then becomes itself a radical cation (ArOH+). A numerical parameter related to this mechanism is the IP (Ionization Potential). In the second step, the antioxidant cationic radical decomposes into a radical (ArO˙) and a proton. In this mechanism, the proton dissociation enthalpy (PDE) of the antioxidant is the energetic factor for evaluating the antioxidant action.In 2014 Lai et al.6 synthesized and studied the antioxidant properties of several thiaflavan compounds (T2, T5, T6, T8, T9, T10, T13, T14, and T16, see Scheme 2) and concluded that:
 | ||
| Scheme 2 Structures of the thiaflavans Ti (i = 1 to 24) | ||
✓ The (ArO˙) radicals are stable like those of curcumin and flavonoid antioxidant derivatives.
✓ The ferrocene moiety at ring B is beneficial for enhancing the abilities of thiaflavans to trap radicals and to inhibit DNA oxidation.
✓ The antioxidant effectiveness generated by hydroxyl groups or the ferrocene moiety at ring B is enhanced by the electron-donating group attached to ring A.
✓ The sulfur and oxygen atoms on ring C induce different effects on the antioxidant property of the hydroxyl group located at the para positions in ring A. The hydroxyl group at the para position of the oxygen atom (position 3) exhibits a higher antioxidant effectiveness than that at the para position of the sulfur atom (position 4). The sulfur atom is beneficial for the hydroxyl group in ring A to quench radicals, while the oxygen atom is beneficial for the hydroxyl group in ring A to inhibit DNA oxidation.
The inhibition effect on DNA oxidation and the ability to scavenge radicals are characteristic properties of antioxidant compounds (see e.g.ref. 7). Accordingly, the aim of this work is to perform a quantum chemical investigation to clarify the effects of hydroxyl groups at ring A and B, a ferrocene moiety at ring B, and ring C′ on the antioxidant effectiveness of the 30 thiaflavans gathered in Schemes 2 and 3. One can notice that the difference between {T7 and T7′}, {T8 and T8′}, {T15 and T15′}, {T19 and T19′} and {T25 and T25′} is only the orientation of H atoms in the clockwise or anti-clockwise direction. In the present work, we have attempted to study the interaction between the thiaflavan compounds and DNA bases with the goal to better understand the antioxidant effects on the DNA molecule. To this end, we assumed that the DNA oxidation inhibition is related to the calculated charge transfer between the thiaflavan molecules and the DNA bases using the ΔN method. The values of ΔN let us estimate the electron donor and electron acceptor characters.
The present paper is then organized as follows: in Section 1, some computational details and definitions are given; in Section 2, the interaction between the molecules and DNA is studied; in Section 3, the antioxidant parameters are discussed; and the paper ends with some concluding remarks.
Theory and computational details
Theory
Conceptual Density Functional Theory (DFT) supplies many reactivity indices to chemist researchers, which can successfully be used for describing and predicting chemical reactivity.8,9 Global descriptors such as electronegativity (χ), chemical potential (μ), chemical hardness (η), and the electrophilicity index (ω)10–12 are now widely used.The electronic chemical potential is the tendency of electrons to escape from the equilibrium state of an atom or molecule. It is approximated by:
 | (1) |
The hardness (η) expresses the resistance of a molecular system to changing its number of electrons.
| η = εL − εH | (2) |
In these equations, εH and εL are the energies of the highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO), respectively.
The global electrophilicity index (ω) introduced by Parr et al.13 expresses the ability of a molecule to accept electrons from the surroundings, and it is given by the following equation:
 | (3) |
The fraction of transferred electrons (ΔN) from the nucleophile (Nu) to the electrophile (E) can be estimated according to Pearson and co-workers.8,14
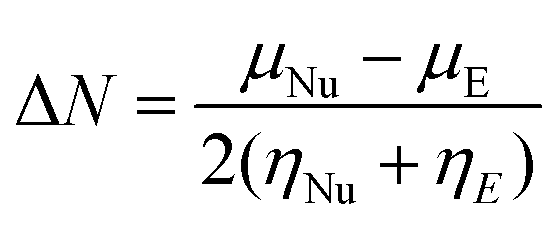 | (4) |
Computational details
All DFT calculations were carried out using the Gaussian 09 program package developed by Frisch and co-workers.15 The water, benzene and gas phase geometries of the neutral compounds (ArOH) and the corresponding radical (ArO˙), ionic (ArO−) and radical cationic (ArOH˙+) structures were optimized using the Becke's three-parameter Lee–Yang–Parr hybrid functional (B3LYP)16 without any constraints. Stuttgart–Dresden basis sets and pseudopotentials17 were applied to Fe and the Pople-style basis 6-311G++ was applied to H, C, O and S atoms.Solvent effects were modeled by employing the CPCM18 continuum solvent model based on the optimized gas-phase geometries. The harmonic vibrational frequencies are calculated at the same level of theory for the four states (neutral compounds (ArOH), radical (ArO˙), ionic ((ArO−)) and radical cationic (ArOH˙+)) to confirm that the optimized geometries correctly correspond to local minima and to estimate the free energy.
The reaction enthalpies related to the HAT, SET-PT and SPLET mechanisms are usually denoted as follows:19,20
The BDE (bond dissociation enthalpy) is used to estimate the reactivity of ArOH in HAT. The calculated equation for the BDE is:
| BDE = H(ArO˙) + H(H˙) − H(ArOH) | (5) |
The ionization potential (IP) and proton dissociation enthalpy (PDE) are calculated to describe the SETPT mechanism. The corresponding equations for the IP and PDE are:
| IP = H(ArOH+˙) + H(e−) − H(ArOH) | (6) |
| PDE = H(ArO˙) + H(H+) − H(ArOH+˙) | (7) |
The proton affinity (PA) of ArO− anions is used to characterize the reaction enthalpy of the first step of SPLET. It is given by:
| PA = H(ArO−) + H(H+) − H(ArOH) | (8) |
The electron transfer enthalpy (ETE) is related to the second step of SPLET. It is obtained by:
| ETE = H(ArO˙) + H(e−) − H(ArO−) | (9) |
The calculated enthalpies for electrons (e), protons (H+) and hydrogen atoms (H˙) in the gas phase are 0.75, 1.484 and −312.28 kcal mol−1, respectively.21 Antioxidant molecules are inside the body and have to be functional in a biological membrane, where there are unsaturated lipids (non-polar media), as well as in physiological liquids (polar media). To study the antioxidant mechanism correctly, we will calculate the antioxidant parameters in water (polar) and non-polar environments such as benzene.22,23 The calculated enthalpies in water for electrons (e), protons (H+) and hydrogen atoms (H˙) are −3.608, −250.96 and −313.24 kcal mol−1, respectively. The calculated enthalpies for e, H+ and H˙ in the benzene phase are −1.577, −208.08 and −311.09 kcal mol−1, respectively.21
Results and discussion
Reactivity
The values of the dipole moment, the chemical potential (μ, eV), the chemical hardness (η, eV) and the electrophilicity index (ω, eV), calculated for each of the thiaflavans T1 to T24 and for the DNA bases in the gas and solvent phases, are given in Table 1.| Gas | Water | Benzene | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D | μ | η | ω | D | μ | η | ω | D | μ | η | ω | |
| A | 2.556 | –3.785 | 5.283 | 1.356 | 3.522 | –3.895 | 5.286 | 1.435 | 3.027 | –3.839 | 5.286 | 1.394 |
| C | 7.456 | –4.038 | 5.155 | 1.581 | 10.598 | –4.132 | 5.41 | 1.578 | 8.951 | –4.084 | 5.282 | 1.579 |
| G | 7.719 | –3.515 | 5.208 | 1.186 | 10.378 | –3.644 | 5.176 | 1.282 | 8.803 | –3.534 | 5.285 | 1.181 |
| T | 4.988 | –4.402 | 5.285 | 1.833 | 6.853 | –4.284 | 5.258 | 1.745 | 5.881 | –4.338 | 5.274 | 1.784 |
| T1 | 2.447 | –3.311 | 5.051 | 1.085 | 3.149 | –3.407 | 5.159 | 1.125 | 2.800 | –3.355 | 5.102 | 1.103 |
| T2 | 1.124 | –3.282 | 4.974 | 1.083 | 1.809 | –3.387 | 5.073 | 1.13 | 1.445 | –3.331 | 5.027 | 1.103 |
| T3 | 1.827 | –3.169 | 4.970 | 1.01 | 2.779 | –3.29 | 5.135 | 1.054 | 2.247 | –3.223 | 5.064 | 1.026 |
| T4 | 2.193 | –3.542 | 4.994 | 0.945 | 2.841 | –3.456 | 5.238 | 1.063 | 2.410 | –3.288 | 5.233 | 1.038 |
| T5 | 2.784 | –3.284 | 5.111 | 1.055 | 3.283 | –3.37 | 5.212 | 1.089 | 3.027 | –3.312 | 5.179 | 1.059 |
| T6 | 2.302 | –3.239 | 5.044 | 1.04 | 3.247 | –3.324 | 5.179 | 1.066 | 2.750 | –3.271 | 5.13 | 1.043 |
| T7 | 2.751 | –3.223 | 4.859 | 1.069 | 4.023 | –3.316 | 4.966 | 1.107 | 3.359 | –3.266 | 4.917 | 1.085 |
| T7′ | 4.089 | –3.240 | 4.824 | 1.088 | 5.354 | –3.319 | 4.969 | 1.108 | 4.715 | –3.278 | 4.895 | 1.097 |
| T8 | 4.216 | –3.230 | 4.836 | 1.078 | 5.915 | –3.303 | 5.147 | 1.059 | 5.057 | –3.227 | 5.065 | 1.028 |
| T8′ | 2.141 | –3.241 | 5.054 | 1.039 | 3.509 | –3.309 | 5.143 | 1.064 | 2.713 | –3.258 | 5.123 | 1.036 |
| T9 | 3.345 | –3.281 | 5.088 | 1.058 | 4.856 | –3.409 | 5.156 | 1.127 | 4.101 | –3.339 | 5.13 | 1.086 |
| T10 | 2.549 | –3.279 | 4.978 | 1.079 | 3.780 | –3.337 | 5.173 | 1.076 | 3.145 | –3.289 | 5.105 | 1.059 |
| T11 | 2.836 | –3.203 | 4.968 | 1.032 | 5.063 | –3.308 | 5.024 | 1.089 | 3.812 | –3.25 | 5.005 | 1.055 |
| T12 | 3.536 | –3.228 | 4.874 | 1.069 | 5.094 | –3.337 | 4.988 | 1.116 | 4.228 | –3.277 | 4.934 | 1.088 |
| T13 | 2.299 | –3.134 | 4.998 | 0.982 | 3.315 | –3.204 | 4.908 | 1.046 | 2.759 | –3.168 | 4.96 | 1.011 |
| T14 | 0.926 | –3.113 | 5.071 | 0.955 | 1.679 | –3.169 | 4.976 | 1.009 | 1.247 | –3.125 | 5.008 | 0.975 |
| T15 | 4.146 | –3.143 | 4.882 | 1.011 | 5.378 | –3.165 | 4.971 | 1.007 | 4.750 | –3.121 | 4.999 | 0.974 |
| T15′ | 2.540 | –3.179 | 4.883 | 1.035 | 3.880 | –3.165 | 4.975 | 1.007 | 3.150 | –3.147 | 5.025 | 0.985 |
| T16 | 2.618 | –3.494 | 4.169 | 1.464 | 3.563 | –3.636 | 4.294 | 1.539 | 3.060 | –3.56 | 4.23 | 1.498 |
| T17 | 2.252 | –3.503 | 4.107 | 1.494 | 3.030 | –3.619 | 4.249 | 1.541 | 2.623 | –3.558 | 4.178 | 1.514 |
| T18 | 4.086 | –3.341 | 4.207 | 1.327 | 5.552 | –3.519 | 4.341 | 1.426 | 4.782 | –3.426 | 4.275 | 1.372 |
| T19 | 2.493 | –3.465 | 4.120 | 1.457 | 3.619 | –3.595 | 4.219 | 1.531 | 3.022 | –3.527 | 4.173 | 1.49 |
| T19' | 3.692 | –3.471 | 4.081 | 1.476 | 5.012 | –3.471 | 4.081 | 1.476 | 4.318 | –3.531 | 4.149 | 1.502 |
| T20 | 3.044 | –3.379 | 4.152 | 1.375 | 4.157 | –3.496 | 4.286 | 1.426 | 3.566 | –3.434 | 4.218 | 1.398 |
| T21 | 5.185 | –3.504 | 4.028 | 1.524 | 7.073 | –3.631 | 4.179 | 1.577 | 6.080 | –3.564 | 4.101 | 1.548 |
| T22 | 4.003 | –3.443 | 4.185 | 1.416 | 5.529 | –3.580 | 4.314 | 1.486 | 4.717 | –3.506 | 4.250 | 1.446 |
| T23 | 4.625 | –3.433 | 4.142 | 1.423 | 6.311 | –3.561 | 4.262 | 1.488 | 5.415 | –3.497 | 4.192 | 1.458 |
| T24 | 5.498 | –3.430 | 4.182 | 1.407 | 7.489 | –3.568 | 4.305 | 1.479 | 6.438 | –3.493 | 4.244 | 1.437 |
The chemical potential (μ) of the thiaflavans ranges from −3.503 to −3.113 eV, whereas for the DNA bases the values range from −4.402 to −3.515 eV and decrease following the order thymine > cytosine > adenine > guanine in the gas and solvent phases. The DFT results are in agreement with the experimental tendency.24
According to the absolute scale of global electrophilicity power (ω) proposed by Domingo et al.,25 the DNA bases can be classified as strong electrophiles (1.18 to 1.83 eV). Besides, the thiaflavan compounds T1 to T24 display a moderate electrophilicity power in the gas and solvent phases.
On the other hand, the computed hardness of the DNA bases follows the order: thymine > adenine > guanine > cytosine and the thiaflavan ones range from 4 to 5.3 eV. Among the thiaflavan compounds, T1 to T15 have the largest hardness with respect to T16 to T24 in water, benzene and the gas phase, respectively. The hardness of Ti (i = 1–24) decreases with the increase in the number of OH substitutions in the ring. We can see that T14 and T15 are more stable than T2 and T7, respectively, and T8′ than T8, T7 than T7′ and T19 than T19′.
Chattaraj, Lee, and Parr26 have established that a variety of acid–base reactions do follow the HSAB principle summarized as: “hard likes hard and soft likes soft”. From this principle, it is anticipated that the thiaflavan compounds would prefer to react with the DNA bases having the closest hardness values.
It is well-established that the oxidative damage of DNA bases is a rather complicated operation related to charge transport and reactions controlled by a combination of entropy, enthalpy, steric and other factors.27 The charge transfer (ΔN) between the T1 to T24 molecules and the DNA bases has been calculated and reported in Table 2 and Fig. 1.
| Gas | Water | Benzene | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A/Ti | C/Ti | G/Ti | T/Ti | A/Ti | C/Ti | G/Ti | T/Ti | A/Ti | C/Ti | G/Ti | T/Ti | |
| T1 | 0.022 | 0.035 | 0.009 | 0.052 | 0.023 | 0.034 | 0.011 | 0.042 | 0.023 | 0.035 | 0.008 | 0.047 |
| T2 | 0.024 | 0.037 | 0.011 | 0.054 | 0.024 | 0.035 | 0.012 | 0.043 | 0.024 | 0.036 | 0.009 | 0.048 |
| T3 | 0.030 | 0.042 | 0.016 | 0.06 | 0.029 | 0.039 | 0.017 | 0.047 | 0.029 | 0.041 | 0.014 | 0.053 |
| T4 | 0.026 | 0.038 | 0.013 | 0.055 | 0.026 | 0.037 | 0.014 | 0.044 | 0.026 | 0.037 | 0.011 | 0.050 |
| T5 | 0.024 | 0.036 | 0.011 | 0.053 | 0.024 | 0.035 | 0.013 | 0.043 | 0.025 | 0.036 | 0.010 | 0.049 |
| T6 | 0.026 | 0.039 | 0.013 | 0.056 | 0.027 | 0.038 | 0.015 | 0.046 | 0.027 | 0.039 | 0.012 | 0.051 |
| T7 | 0.027 | 0.04 | 0.014 | 0.058 | 0.028 | 0.039 | 0.016 | 0.047 | 0.028 | 0.04 | 0.013 | 0.052 |
| T7′ | 0.026 | 0.04 | 0.013 | 0.057 | 0.028 | 0.039 | 0.016 | 0.047 | 0.027 | 0.039 | 0.012 | 0.052 |
| T8 | 0.027 | 0.04 | 0.014 | 0.057 | 0.028 | 0.039 | 0.016 | 0.047 | 0.029 | 0.041 | 0.014 | 0.053 |
| T8′ | 0.026 | 0.039 | 0.013 | 0.056 | 0.028 | 0.038 | 0.016 | 0.046 | 0.027 | 0.039 | 0.013 | 0.051 |
| T9 | 0.024 | 0.036 | 0.011 | 0.054 | 0.023 | 0.034 | 0.011 | 0.042 | 0.024 | 0.035 | 0.009 | 0.048 |
| T10 | 0.024 | 0.037 | 0.011 | 0.054 | 0.026 | 0.037 | 0.014 | 0.045 | 0.026 | 0.038 | 0.011 | 0.05 |
| T11 | 0.028 | 0.041 | 0.015 | 0.058 | 0.028 | 0.039 | 0.016 | 0.047 | 0.028 | 0.04 | 0.013 | 0.052 |
| T12 | 0.027 | 0.04 | 0.014 | 0.057 | 0.027 | 0.038 | 0.015 | 0.046 | 0.027 | 0.039 | 0.012 | 0.051 |
| T13 | 0.031 | 0.044 | 0.018 | 0.061 | 0.033 | 0.044 | 0.021 | 0.053 | 0.032 | 0.044 | 0.017 | 0.057 |
| T14 | 0.032 | 0.045 | 0.019 | 0.062 | 0.035 | 0.046 | 0.023 | 0.054 | 0.034 | 0.046 | 0.019 | 0.059 |
| T15 | 0.031 | 0.044 | 0.018 | 0.061 | 0.035 | 0.046 | 0.023 | 0.054 | 0.034 | 0.046 | 0.02 | 0.059 |
| T15' | 0.029 | 0.042 | 0.016 | 0.06 | 0.035 | 0.046 | 0.023 | 0.054 | 0.033 | 0.045 | 0.018 | 0.057 |
| T16 | 0.015 | 0.029 | 0.001 | 0.047 | 0.013 | 0.025 | 0.0004 | 0.033 | 0.014 | 0.027 | −0.001 | 0.04 |
| T17 | 0.015 | 0.028 | 0.0006 | 0.047 | 0.014 | 0.026 | 0.001 | 0.034 | 0.014 | 0.027 | −0.001 | 0.041 |
| T18 | 0.023 | 0.037 | 0.009 | 0.055 | 0.019 | 0.031 | 0.006 | 0.039 | 0.021 | 0.034 | 0.005 | 0.047 |
| T19 | 0.017 | 0.03 | 0.002 | 0.049 | 0.015 | 0.027 | 0.002 | 0.036 | 0.016 | 0.029 | 0.0003 | 0.042 |
| T19' | 0.016 | 0.03 | 0.002 | 0.049 | 0.022 | 0.034 | 0.009 | 0.043 | 0.016 | 0.029 | 0.0001 | 0.042 |
| T20 | 0.021 | 0.035 | 0.007 | 0.054 | 0.02 | 0.032 | 0.007 | 0.041 | 0.021 | 0.034 | 0.005 | 0.047 |
| T21 | 0.015 | 0.029 | 0.0005 | 0.048 | 0.013 | 0.026 | 0.0006 | 0.034 | 0.014 | 0.027 | −0.001 | 0.041 |
| T22 | 0.020 | 0.033 | 0.004 | 0.047 | 0.013 | 0.026 | −0.002 | 0.039 | 0.017 | 0.030 | 0.001 | 0.043 |
| T23 | 0.021 | 0.034 | 0.005 | 0.0488 | 0.014 | 0.027 | −0.001 | 0.040 | 0.018 | 0.031 | 0.001 | 0.044 |
| T24 | 0.021 | 0.034 | 0.005 | 0.047 | 0.0141 | 0.026 | −0.001 | 0.040 | 0.018 | 0.031 | 0.002 | 0.044 |
 | ||
| Fig. 1 Calculated charge transfer between the thiaflavan Ti (i = 1 to 24) molecules and the DNA bases (A (triangles): adenine, C (squares): cytosine, G: (circles) guanine and T (stars): thymine) in the gas and solvent phases (black: gas, blue: water and red: benzene). | ||
Our results indicate that these molecules act as electron donors (ΔN > 0) in the gas and solvent phases, and thymine has the maximum accepting power to interact with the Ti compounds, with respect to cytosine, adenine and guanine (see Fig. 1). Exceptions are the cases of the interaction of the {T16, T17 and T21 in benzene} and {T22, T23 and T24 in water} compounds, which are more inclined to react with guanine. The negative ΔN indicates that these compounds act as electron acceptors only with guanine in benzene and water.
Thus, our quantum chemical calculations indicate that the thymine moiety in DNA has the largest ΔN value and guanine has the smallest value (in water, benzene and the gas phase) and hence oxidation reactions should be commonly found to occur primarily at the guanine site. This result agrees well with the experimental and calculation results of alkylating drug molecules.28,29
Antioxidant activity
The quantum chemical calculations of antioxidant parameters BDE, IP, PA, PDE and ETE for the thiaflavan compounds (T1 to T24) have been determined in the gas phase as well as in water and benzene.Ionization potential
The calculated ionization potentials (IPs) for the thiaflavan compounds are given in Tables 3 and 4. All the thiaflavans can easily donate an electron (IP > 0) and they can be sorted in the following order: T13–T15, T16–T24 and T1–T12 in all environments, namely the water, benzene and gas phases.| Ti | j-R | Gas | Experimental (k × 103 M−1 s−1) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HAT | SETPT | SPLET | |||||||
| BDE | IP | PDE | PA | ETE | ABTS˙+ | DPPH | Galvinoxyl radical | ||
| T1 | 3-OH | 75.97 | 169.00 | 221.47 | 338.99 | 51.48 | |||
| T2 | 4-OH | 77.14 | 169.13 | 222.52 | 338.55 | 53.10 | |||
| T3 | 1-OH | 79.98 | 164.41 | 230.08 | 334.74 | 59.74 | 0.168 | — | — |
| T4 | 1-OH | 79.62 | 163.54 | 230.59 | 331.35 | 62.77 | 0.202 | — | — |
| T5 | 3-OH | 75.95 | 168.45 | 222.00 | 338.71 | 51.75 | 8.92 | 0.043 | 0.025 |
| 1-OH | 80.19 | 168.45 | 226.24 | 334.04 | 60.65 | ||||
| T6 | 4-OH | 77.10 | 167.96 | 223.64 | 338.55 | 53.05 | 10.8 | 0.069 | 0.034 |
| 1-OH | 80.12 | 167.96 | 226.66 | 334.14 | 60.49 | ||||
| T7 | 3-OH | 69.79 | 165.08 | 219.21 | 329.48 | 54.81 | |||
| T7′ | 4-OH | 70.50 | 164.75 | 220.25 | 329.07 | 55.93 | |||
| T8 | 2-OH | 73.61 | 162.68 | 225.43 | 325.66 | 62.45 | |||
| T8′ | 1-OH | 74.91 | 164.69 | 224.72 | 327.54 | 61.87 | |||
| T9 | 3-OH | 75.61 | 166.68 | 223.44 | 338.63 | 51.49 | 11.7 | 1.18 | 0.52 |
| 1-OH | 73.51 | 166.68 | 221.33 | 326.58 | 61.44 | ||||
| T10 | 4-OH | 77.10 | 167.66 | 223.94 | 338.23 | 53.38 | 16.7 | 1.61 | 0.57 |
| 1-OH | 73.84 | 167.66 | 220.68 | 325.88 | 62.46 | ||||
| T11 | 1-OH | 79.83 | 163.60 | 230.73 | 334.06 | 60.27 | |||
| 4-OH | 70.48 | 163.60 | 221.38 | 328.14 | 56.84 | ||||
| T12 | 2-OH | 80.05 | 164.31 | 230.24 | 337.46 | 57.09 | |||
| 4-OH | 70.61 | 164.31 | 220.80 | 328.28 | 56.84 | ||||
| T13 | 3-OH | 75.82 | 143.77 | 246.56 | 338.79 | 51.54 | 0.16 | — | — |
| T14 | 4-OH | 77.07 | 143.00 | 248.57 | 338.14 | 53.43 | 22.9 | 0.124 | 0.084 |
| T15 | 4-OH | 69.63 | 143.53 | 240.60 | 329.32 | 54.80 | |||
| T15′ | 3-OH | 70.17 | 145.65 | 239.03 | 328.52 | 56.15 | |||
| T16 | 3-OH | 76.31 | 160.07 | 230.75 | 334.15 | 56.66 | 4.89 | 0.0071 | |
| T17 | 4-OH | 77.01 | 159.88 | 231.64 | 333.49 | 58.02 | |||
| T18 | 1-OH | 76.50 | 155.02 | 235.99 | 331.66 | 59.35 | 8.14 | 0.031 | |
| T19 | 3-OH | 69.35 | 158.02 | 225.84 | 325.16 | 58.69 | |||
| T19′ | 4-OH | 69.57 | 158.03 | 226.04 | 324.46 | 59.61 | |||
| T20 | 1-OH | 76.61 | 155.55 | 235.56 | 330.07 | 61.05 | |||
| 4-OH | 69.55 | 155.55 | 228.50 | 324.72 | 59.34 | ||||
| T21 | 2-OH | 80.75 | 158.04 | 237.22 | 335.45 | 59.80 | |||
| 4-OH | 69.53 | 158.04 | 225.99 | 324.26 | 59.78 | ||||
| T22 | 1-OH | 70.76 | 158.35 | 226.92 | 323.15 | 62.11 | |||
| T23 | 1-OH | 70.67 | 157.17 | 228.00 | 322.09 | 63.09 | |||
| 4-OH | 76.66 | 157.17 | 233.99 | 333.10 | 58.07 | ||||
| T24 | 1-OH | 70.79 | 157.31 | 227.98 | 322.81 | 62.48 | |||
| 3-OH | 76.24 | 157.31 | 233.43 | 334.06 | 56.68 | ||||
| Ti | j-R | Water | Benzene | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HAT | SET-PT | SPLET | HAT | SET-PT | SPLET | ||||||
| BDE | IP | PDE | PA | ETE | BDE | IP | PDE | PA | ETE | ||
| T1 | 3-OH | 73.80 | 128.09 | 4.38 | 36.60 | 95.87 | 76.72 | 146.01 | 32.14 | 102.57 | 75.58 |
| T2 | 4-OH | 74.76 | 127.58 | 5.85 | 36.11 | 97.32 | 77.88 | 145.87 | 33.44 | 102.11 | 77.19 |
| T3 | 1-OH | 79.08 | 125.89 | 11.86 | 35.32 | 102.44 | 81.26 | 142.89 | 39.8 | 99.78 | 82.91 |
| T4 | 1-OH | 77.88 | 126.10 | 10.45 | 35.77 | 100.78 | 81.11 | 143.17 | 39.38 | 96.51 | 85.4 |
| T5 | 3-OH | 73.77 | 127.79 | 4.65 | 36.69 | 95.75 | 76.7 | 145.73 | 32.4 | 102.53 | 75.6 |
| 1-OH | 79.08 | 132.31 | 5.44 | 35.29 | 102.46 | 81.42 | 145.73 | 37.13 | 99.43 | 83.42 | |
| T6 | 4-OH | 74.71 | 127.43 | 5.95 | 36.12 | 97.26 | 77.84 | 145.5 | 33.77 | 102.2 | 77.07 |
| 1-OH | 79.09 | 127.43 | 10.33 | 35.25 | 102.51 | 81.42 | 145.5 | 37.34 | 99.55 | 83.3 | |
| T7 | 3-OH | 69.11 | 124.38 | 3.40 | 31.35 | 96.43 | 71.2 | 142.29 | 30.33 | 95.1 | 77.53 |
| T7′ | 4-OH | 69.53 | 124.31 | 3.90 | 30.83 | 97.37 | 71.86 | 142.08 | 31.22 | 94.67 | 78.63 |
| T8 | 2-OH | 74.03 | 125.30 | 7.40 | 37.44 | 95.26 | 75.52 | 141.77 | 35.18 | 92.58 | 84.37 |
| T8′ | 1-OH | 74.54 | 125.29 | 7.92 | 30.86 | 102.35 | 76.48 | 143.03 | 34.88 | 93.8 | 84.11 |
| T9 | 3-OH | 73.66 | 128.67 | 3.66 | 36.72 | 95.62 | 76.43 | 146.24 | 31.63 | 102.38 | 75.49 |
| 1-OH | 73.45 | 128.67 | 3.45 | 31.42 | 100.70 | 75.22 | 146.24 | 30.41 | 94.9 | 81.75 | |
| T10 | 4-OH | 74.75 | 127.59 | 5.83 | 36.10 | 97.31 | 77.88 | 145.57 | 33.74 | 102.03 | 77.28 |
| 1-OH | 73.96 | 127.59 | 5.04 | 30.40 | 102.23 | 75.63 | 145.57 | 31.49 | 92.82 | 84.24 | |
| T11 | 1-OH | 78.14 | 125.03 | 11.79 | 36.24 | 100.57 | 80.83 | 141.81 | 40.45 | 99.74 | 82.53 |
| 4-OH | 69.68 | 125.03 | 3.327 | 30.69 | 97.66 | 71.86 | 141.81 | 31.48 | 94 | 79.29 | |
| T12 | 2-OH | 79.07 | 125.4 | 12.34 | 36.34 | 101.4 | 81.33 | 142.42 | 40.34 | 101.52 | 81.23 |
| 4-OH | 69.84 | 125.4 | 3.11 | 30.85 | 97.66 | 72.01 | 142.42 | 31.02 | 94.15 | 79.29 | |
| T13 | 3-OH | 73.51 | 100.92 | 31.26 | 36.61 | 95.57 | 76.56 | 120.49 | 57.5 | 102.56 | 75.43 |
| T14 | 4-OH | 74.71 | 100.29 | 33.08 | 36.22 | 97.16 | 77.8 | 120.03 | 59.19 | 102.01 | 77.21 |
| T15 | 4-OH | 68.90 | 100.33 | 27.24 | 31.50 | 96.07 | 71.69 | 141.87 | 31.25 | 94.53 | 78.59 |
| T15′ | 3-OH | 69.38 | 100.99 | 27.06 | 31.01 | 97.04 | 71.03 | 141.87 | 30.60 | 95.08 | 77.39 |
| T16 | 3-OH | 74.48 | 120.09 | 13.06 | 34.53 | 98.62 | 77.27 | 137.66 | 41.04 | 99.08 | 79.62 |
| T17 | 4-OH | 74.76 | 119.70 | 13.73 | 33.94 | 99.49 | 77.88 | 137.38 | 41.93 | 98.44 | 80.87 |
| T18 | 1-OH | 74.41 | 117.91 | 15.17 | 33.82 | 99.26 | 77.38 | 134.21 | 44.6 | 97.65 | 81.17 |
| T19 | 3-OH | 69.02 | 118.42 | 9.28 | 29.5 | 98.19 | 71.49 | 136.38 | 36.54 | 92.43 | 80.49 |
| T19′ | 4-OH | 69.06 | 118.44 | 9.29 | 28.96 | 98.78 | 71.17 | 135.84 | 36.76 | 91.31 | 81.29 |
| T20 | 1-OH | 74.44 | 117.02 | 16.1 | 33.74 | 99.37 | 77.43 | 133.96 | 44.9 | 96.77 | 82.09 |
| 4-OH | 68.93 | 117.02 | 10.59 | 29.07 | 98.53 | 71.11 | 133.96 | 38.58 | 91.53 | 81.02 | |
| T21 | 2-OH | 79.61 | 118.79 | 19.49 | 35.78 | 102.5 | 81.97 | 136.03 | 47.37 | 100.3 | 83.11 |
| 4-OH | 69.19 | 118.79 | 9.07 | 28.9 | 98.96 | 71.18 | 136.03 | 36.58 | 91.21 | 81.4 | |
| T22 | 1-OH | 70.48 | 119.67 | 9.48 | 29.08 | 100.07 | 72.38 | 136.56 | 37.25 | 90.81 | 83.00 |
| T23 | 1-OH | 70.38 | 118.46 | 10.58 | 29.02 | 100.02 | 72.30 | 135.44 | 38.29 | 90.26 | 83.47 |
| 4-OH | 74.67 | 118.46 | 14.88 | 33.96 | 99.38 | 77.61 | 135.44 | 43.59 | 98.24 | 80.80 | |
| T24 | 1-OH | 70.60 | 119.03 | 10.24 | 29.04 | 100.22 | 72.42 | 135.75 | 38.11 | 90.63 | 83.22 |
Bond dissociation enthalpy
The bond dissociation enthalpy is an important parameter related to the breaking of an O–H bond resulting in the abstraction of an H atom. The highest BDE value indicates the lowest antioxidant activity.The calculated BDEs in the gas and solvent phases are reported in Tables 3 and 4. Firstly, the calculated BDE values in the gas and solvent phases show a nice correlation with experimental results presented by Lai and co-workers6 of the rate constant (ln(k)) for thiaflavans in scavenging [ABTS]˙+ (2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonate) cationic radical), see Fig. 2. These lines provide a trend between the BDE and PDE and (available) experimental ln(k).
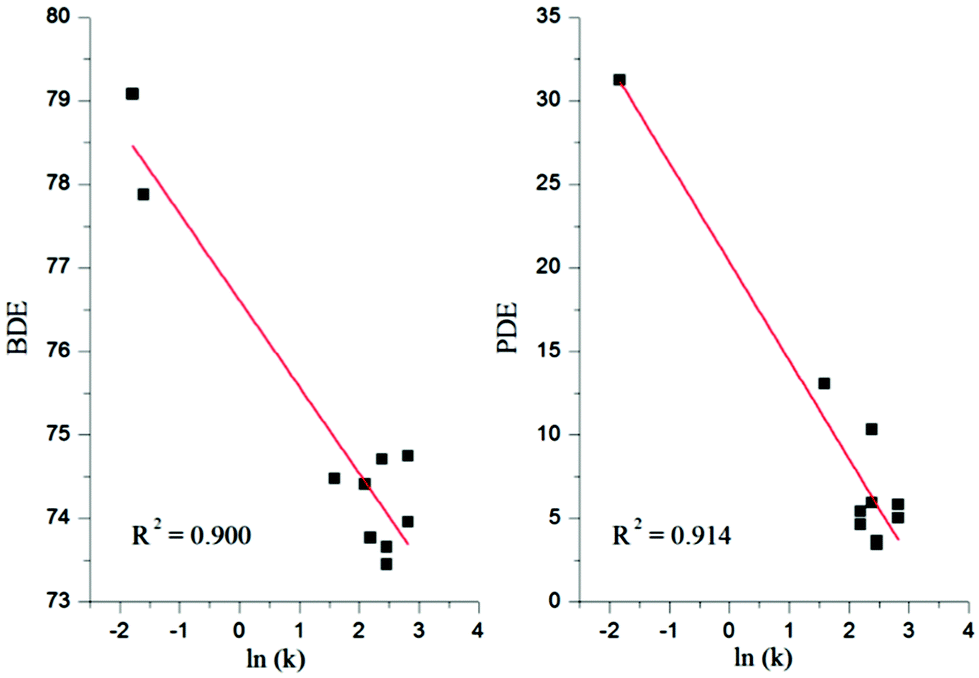 | ||
| Fig. 2 Correlation between the BDE and PDE values calculated in water with the available experimental ln(k) for thiaflavans in scavenging ABTS+˙. | ||
On the other hand, comparing between the gas, polar and non-polar phase BDE values, no dramatic differences are found (all are less than 5 kcal mol−1). However, one can notice that the O–H BDE values of the thiaflavan compounds are lower in water than in the gas phase and benzene. This indicates that polar phases facilitate the HAT mechanism. The BDE values in the gas phase are in the following order:
T21–R2 > T5–R1 ∼ T6–R1 ∼ T12–R2 ∼ T3–R1 ∼ T11–R1 ∼ T4–R1 > T2–R4 ∼ T6–R4 = T10–R4 ∼ T14–R4 ∼ T17–R4 > T23–R4 ∼ T20–R1 ∼ T18–R1 ∼ T16–R3 ∼ T24–R3 > T1–R3 ∼ T5–R3 ∼ T13–R3 ∼ T9–R3 > T8′–R1 > T10–R1 ∼ T8–R2 ∼ T9–R1 > T24–R1 ∼ T22–R1 ∼ T23–R1 ∼ T12–R4 ∼ T7′–R4 ∼ T11–R4 ∼ T15′–R3 > T7–R3 ∼ T15–R4 ∼ T19′–R4 ∼ T20–R4 ∼ T21–R4 ∼ T19–R3.
The high BDE values of the T3–R1, T4–R1, T5–R1, T6–R1, T11–R1, T12–R2 and T22–R2 compounds indicate that a single hydroxyl group at the 1 or 2-position in ring B exhibits a weak ability to donate its hydrogen atom. On the other hand, the BDE value of the T2–R4, T6–R4 and T17–R4 compounds is higher than that of the T1–R3, T5–R3 and T16–R3 compounds (∼1.14 kcal mol−1). Our calculation shows that the bond dissociation enthalpy of a single O–H group at the B ring is about 5 kcal mol−1 higher than that at the A ring.
Therefore, the hydroxyl groups at the A ring play a key role in scavenging radicals and the antioxidant effectiveness.
On the other hand, this result shows that the hydrogen atom transfer from ring A of two hydroxyl groups at the 3 and 4-positions is easier than that of the B-ring of two OH groups at the 1 and 2-positions; this clearly proves that ring A has to be considered as the primary target for radical attack in thiaflavans.
Proton dissociation enthalpies
The PDE represents the reaction enthalpy of the second step in the SETPT mechanism, which is responsible for the production of the radical cation (ArOH˙+) from the neutral (ArOH) compound by loss of one electron followed by the deprotonation of the radical cation. The PDE values let us suggest the thermodynamically preferred hydroxyl group for deprotonation from [ArOH˙+].As shown in Fig. 2, the correlation between the PDE and 10 experimental values of the rate constant (k) for thiaflavans in scavenging [ABTS˙+] is fairly good in both the gas and solvent phases (the correlation coefficient is 0.914).
To the best of our knowledge, the PDE value of thiaflavan antioxidants has not been studied previously. The average calculated PDEs of the T1 to T24 compounds in water, benzene and the gas phase are about 11, 38 and 229 kcal mol−1, respectively (see Tables 3 and 4). The calculated PDEs are dramatically lower in water and in benzene than that in the gas phase, confirming that solvation facilitates electron donation, particularly with a polar medium. We can attribute these results to the enhanced stability of the radical cation in a polar medium, as expected from the electrostatic interactions between its charge and the polar solvent. The delocalization and conjugation of the π-electrons of the Ti molecules are also larger in a polar medium.30,31 On the other hand, the dipole moments of the Ti optimized geometries are higher in water than in benzene and the gas phase (see Table 1), as expected from the polarization induced by the polar solvent.
One can see from Tables 3 and 4 that the PDE values of T13 to T15 (ferrocene moiety at ring B) are higher than all other thiaflavan compounds. Besides, the PDE values of a single hydroxyl group at the 3-position are lower than that at the 4, 1 and 2-positions, respectively, in both the gas and solvent phases.
We can conclude that the hydrogen abstraction from the hydroxyl group in the A-ring is much easier than in the B-ring and, among all the thiaflavan compounds, it is more difficult in the case of the presence of a ferrocene moiety at ring B. Generally, OH groups in the A-ring contribute greatly to the second step of the SETPT mechanism, whereas OH groups in the B-ring play a relatively little role.
Electron transfer enthalpy
The calculated ETE values in the gas phase as well as in solvents for Ti (i = 1 to 24) are gathered in Tables 3 and 4.The ETEs are higher in water (∼99 kcal mol−1) than in the corresponding benzene (∼80 kcal mol−1) and gas (∼58 kcal mol−1) phases. These results indicate that the polar medium favors the reaction less than the non-polar and gas phases in the second step of the SPLET mechanism.
The lowest ETEs in the gas phase are found for T1–R3, T5–R3, T9–R3, and T13–R3 (∼51 kcal mol−1) and for T2–R4, T6–R4, T10–R4, and T14–R4 (∼53 kcal mol−1) and the higher values are found for T4–R1, T8–R2, T9–R1, T10–R1, T22–R1, T23–R1 and T24–R1. The ETE values are lower than their corresponding IP values of the thiaflavans. Hence, the single electron transfer process from the anionic form is preferred with respect to that from the neutral form.
Proton affinities
The proton affinity of hydroxyl groups (PA OH) is a theoretical descriptor related with the SPLET antioxidant mechanism. The PA parameter describes the thermodynamic stability of O–H bonds in the antioxidant compounds. The antioxidant molecules with the lowest PA values are the most active.The computed PAs for the various Ti compounds in the gas and solvents phases are reported in Tables 3 and 4. The highest values in the gas phase of the PA for the Ti compounds in different positions of the hydroxyl group (OH = R, j = 1 to 4) are found for the molecules with a single hydroxyl group in ring A and B (T1, T2, T5–R3, T6–R4, T9–R3, T10, T12, T13, and T14, PA = 338 kcal mol−1). The lowest PA values are found in the compounds of a C′ ring and two OH groups attached to the same (A or B) ring (T23–R1 > T24–R1 > T22 > T21–R4 > T19–R4 > T20–R4, PA = 322–324 kcal mol−1). We can attribute these results to the formation of an intramolecular hydrogen bond between the hydrogen atom of a hydroxyl and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group ({C2–O–H and C1O} and {C3–O–H and C4–O}). This may also be related to the fact that ring C′ leads to a better resonance between the A–C′–B-rings (see Scheme 2), which makes these molecules yield high reactivity (also these compounds have the lowest values of η, see Table 1), and the corresponding radicals are more stable (this point is discussed in the next section – Mulliken spin density – vide infra).
O group ({C2–O–H and C1O} and {C3–O–H and C4–O}). This may also be related to the fact that ring C′ leads to a better resonance between the A–C′–B-rings (see Scheme 2), which makes these molecules yield high reactivity (also these compounds have the lowest values of η, see Table 1), and the corresponding radicals are more stable (this point is discussed in the next section – Mulliken spin density – vide infra).
On the other hand, the structure of these molecules shows a co-planarity between the A, C′ and B-rings (the torsion angle between the three rings is equal to ∼6°). The presently calculated average PA value for the Ti compounds in water, benzene and the gas phase reaches 33, 96 and 331 kcal mol−1, respectively.
Furthermore, our calculation indicates that the PA values in polar media are lower than the BDE and IP values for all thiaflavan compounds. This result shows that, in water, the SPLET mechanism should dominate, from the thermodynamic point of view. On the other hand, the BDE values are lower than the PA and IP in benzene and the gas phase, respectively, for all Ti compounds.
According to our DFT calculations, the orientation of H atoms in the {T7 and T7′}, {T8 and T8′}, {T15 and T15′}, {T19 and T19′} and {T25 and T25′} compounds plays an insignificant role in the BDE, IP, PA, PDE and ETE energy in the gas and solvent phases, as expected.
Mulliken spin density (MSD)
The stability of the formed free radical (ArO˙) plays a key role in the antioxidant activity of a molecule. The spin densities of the antioxidant free radical can provide some information about the stability of ArO˙ and the kinetics of the free radical scavenging reaction. The more delocalized the spin density in the antioxidant radical, the more easily the antioxidant radical is formed. A lower spin density corresponds to a decrease of the BDE value.32Table 5, which gathers Mulliken spin densities of the O-atom of various free radicals of the thiaflavan compounds formed after H abstraction, shows that the Mulliken spin density in water is lower than those of the benzene and gas phases, respectively.
| Ti | j-O˙ | Gas | Water | Benzene | Ti | j-O˙ | Gas | Water | Benzene |
|---|---|---|---|---|---|---|---|---|---|
| T1 | 3-O˙ | 0.454 | 0.383 | 0.421 | T13 | 3-O˙ | 0.452 | 0.381 | 0.419 |
| T2 | 4-O˙ | 0.451 | 0.369 | 0.414 | T14 | 4-O˙ | 0.447 | 0.366 | 0.410 |
| T3 | 1-O˙ | 0.502 | 0.449 | 0.476 | T15 | 4-O˙ | 0.427 | 0.347 | 0.391 |
| T4 | 1-O˙ | 0.499 | 0.446 | 0.473 | T15′ | 3-O˙ | 0.387 | 0.340 | 0.366 |
| T5 | 1-O˙ | 0.502 | 0.449 | 0.476 | T16 | 3-O˙ | 0.408 | 0.320 | 0.370 |
| 3-O˙ | 0.454 | 0.382 | 0.420 | T17 | 4-O˙ | 0.401 | 0.293 | 0.355 | |
| T6 | 4-O˙ | 0.451 | 0.368 | 0.414 | T18 | 1-O˙ | 0.398 | 0.303 | 0.360 |
| 1-O˙ | 0.503 | 0.449 | 0.477 | T19 | 3-O˙ | 0.337 | 0.277 | 0.312 | |
| T7 | 3-O˙ | 0.389 | 0.342 | 0.367 | T19′ | 4-O˙ | 0.319 | 0.252 | 0.291 |
| T7′ | 4-O˙ | 0.372 | 0.319 | 0.348 | T20 | 4-O˙ | 0.318 | 0.248 | 0.289 |
| T8 | 2-O˙ | 0.484 | 0.425 | 0.457 | 1-O˙ | 0.396 | 0.285 | 0.357 | |
| T8′ | 1-O˙ | 0.425 | 0.392 | 0.409 | T21 | 4-O˙ | 0.319 | 0.254 | 0.291 |
| T9 | 1-O˙ | 0.423 | 0.394 | 0.408 | 2-O˙ | 0.516 | 0.458 | 0.489 | |
| 3-O˙ | 0.454 | 0.387 | 0.422 | T22 | 1-O˙ | 0.356 | 0.301 | 0.334 | |
| T10 | 1-O˙ | 0.424 | 0.392 | 0.408 | T23 | 1-O˙ | 0.354 | 0.295 | 0.331 |
| 4-O˙ | 0.450 | 0.369 | 0.414 | 4-O˙ | 0.397 | 0.289 | 0.351 | ||
| T11 | 4-O˙ | 0.369 | 0.320 | 0.347 | T24 | 1-O˙ | 0.357 | 0.304 | 0.335 |
| 1-O˙ | 0.499 | 0.446 | 0.473 | 3-O˙ | 0.407 | 0.319 | 0.369 | ||
| T12 | 4-O˙ | 0.369 | 0.320 | 0.347 | |||||
| 2-O˙ | 0.505 | 0.455 | 0.481 |
The Mulliken spin density on the O-atom, calculated for the thioflavans radicals in water are ordered from largest to smallest as follow: {T21–2-O˙ > T12–2-O˙ > T3–1-O˙ > T5–1-O˙ > T6–1-O˙ > T4–1-O˙ > T11–1-O˙ > T8–2-O˙ > T9–1-O˙ > T8′–1-O˙ > T10–1-O˙ > T9–3-O˙ > T1–3-O˙ > T5–3-O˙ > T13–3-O˙} with values in the 0.458–0.381 and {T2–4-O˙ > T10–4-O˙ > T6–4-O˙ > T14–4-O˙ > T15–4-O˙ > T7–3-O˙ > T15′–3-O˙ > T11–4-O˙ > T12–4-O˙ > T16–3-O˙ > T7′–4 O˙ > T24–3-O˙ > T24–1-O˙ > T18–1-O˙ > T22–1-O˙ > T23–1-O˙ > T17–4-O˙ > T23–4-O˙ > T20–1-O˙} providing values in the 0.369–0.285 range and finally {T19–3-O˙ > T21–4-O˙ > T19′–4-O˙ > T20–4-O˙} giving values amounting to 0.277 to 0.248. This means that the formation of radicals T19–3-O˙, T21–4-O˙, T19′–4-O˙ and T20–4-O˙ is more favorable for spin density localization than the formation of the other thiaflavan radicals. This also means that the stabilization of the T19–3-O˙, T21–4-O˙, T19′–4-O˙ and T20–4-O˙ radicals is higher than the other thiaflavan radicals.
This result indicates that the Ti radicals having ring C′ and hydroxyl and CO groups on ring A (R3 and R4) are predicted to be stable compounds and consequently to be potential antioxidants. As can be seen from Tables 3 and 4, the Mulliken spin atomic densities agree well with the BDE results.
NICS aromaticity index
The nucleus independent chemical shift (NICS) index is the most popular aromaticity index.33 Since its introduction in 1996,34 the NICS index has been widely used to characterize the aromaticity and anti-aromaticity of rings,35–39 clusters,40–45 transition states46–51 and transition metal complexes.52 Originally, the NICS was calculated at the ring center and described as the negative value of the isotropic shielding constant.34,52 In this work, the NICSzz(1) index is calculated at 1 Å above the ring center giving the out-of plane component NICSz(1) for T1 to T25 of the thiaflavan antioxidant structures. NICS analysis is applied to identify the aromaticity of the structures, which may reflect their stability. Note that strongly negative values indicate aromaticity and positive values indicate anti-aromaticity.Note that aromaticity is related to molecular properties such as biological activities and behavior in various ways.53 The impact of aromatic rings on properties such as solubility and bioactivities suggests that the presence of more than three aromatic rings may not be optimal for viable drugs. All the studied molecules have aromatic 6-membered rings. Therefore, a possibility to relate the NICS aromaticity index with the antioxidative properties of the studied molecules is examined. The obtained results, presented in Table 6, show that the OH groups affect the chemical shift in the molecule. Note that the most extended conjugation leads to more stability. However, the aromaticity inside the carbon of ring A is very weak, and the A ring is anti-aromatic.
| NICSzz(1) | NICSzz(1) | ||||
|---|---|---|---|---|---|
| T1 | A | 19.37 | T13 | A | 11.55 |
| B | 7.40 | C | –3.73 | ||
| C | –5.31 | T14 | A | 18.31 | |
| T2 | A | 19.38 | C | — | |
| B | 7.93 | T15 | A | 17.43 | |
| C | –5.18 | C | –3.54 | ||
| T3 | A | 19.90 | T16 | A | 17.48 |
| B | 4.85 | B | 23.07 | ||
| C | –5.08 | C′ | –7.60 | ||
| T4 | A | 14.23 | T17 | A | 16.50 |
| B | 4.27 | B | 23.20 | ||
| C | –0.71 | C′ | –8.64 | ||
| T5 | A | 13.34 | T18 | A | 18.49 |
| B | 6.26 | B | 20.99 | ||
| C | –5.41 | C′ | –8.23 | ||
| T6 | A | 19.40 | T19 | A | 1.87 |
| B | 5.61 | B | 18.76 | ||
| C | –5.18 | C′ | –0.31 | ||
| T7 | A | 18.44 | T20 | A | 15.97 |
| B | 7.60 | B | 20.62 | ||
| C | –5.26 | C′ | –8.01 | ||
| T8 | A | 18.33 | T21 | A | 15.81 |
| B | 8.52 | B | 20.73 | ||
| T9 | A | 1.87 | C′ | –8.35 | |
| B | 18.76 | T22 | A | 18.33 | |
| C | –0.31 | B | 20.18 | ||
| T10 | A | 19.31 | C′ | –9.16 | |
| B | 9.23 | T23 | A | 16.63 | |
| C | –5.18 | B | 20.06 | ||
| T11 | A | 4.83 | C′ | –8.89 | |
| B | 14.01 | T24 | A | 16.89 | |
| C | –0.79 | B | 20.05 | ||
| T12 | A | 8.54 | C′ | –8.52 | |
| B | 12.55 | T25 | A | 12.86 | |
| C | –3.20 | C′ | –5.96 |
This result indicates that the hydrogen atom transfer from ring A of two hydroxyl groups at the 3 and 4-positions is easy. On the other hand, the chemical shift inside the carbon of ring B is also weak but less than in the A-ring, confirming that ring A should be considered the primary target for radical attack in thiaflavans. Furthermore, the NICSzz(1) results obtained for the C-ring indicate that this ring is always aromatic and, therefore, more stable. Antioxidant activity is due to the capacity to eliminate reactive oxygen species by trapping free radicals. Based on the NICS analysis, we thus obtained a relationship between the NICS and the hydroxyl groups related to antioxidant activity. Therefore, the lowest value of the enthalpy of dissociation corresponds to the decrease of the aromaticity. This information underlines the importance of aromaticity and the BDE indices, indicating the antioxidant behavior and thus the biological activities.
Design of the best thiaflavan antioxidants
Experimental results show that transition metal complexes show encouraging antioxidant activity54–57 and sometimes the ligand exhibits more potent in vitro antioxidant capacity than its complexes.58Towards the design of better thiaflavan antioxidants, we investigated the influence of the ferrocenyl group at the B-ring, a C′-ring and the position/number of OH groups in the A and B rings on the BDE, IP, PA, PDE, and ETE of the T1 to T24 compounds. Our DFT calculations show that the effectiveness of the thiaflavan antioxidants increases in the case of:
1 – The presence of two hydroxyl groups in the A-ring (3 and 4-positions).
2 – The presence of ring C′.
3 – The presence of a ferrocene moiety at ring B. The relevance of the Fe(II)/Fe(III) redox couple in ferrocene is certainly an actor in the efficiency of the T25 thiaflavan system. This has been already suggested by Zai-Qun Liu59 and Arezki et al.,60 who even suggest that the redox couple of Fe(II)/Fe(III) may control the antioxidative property of the molecule.59,60
According to this study, we can conclude that the best thiaflavan antioxidants are those shown in Scheme 3.
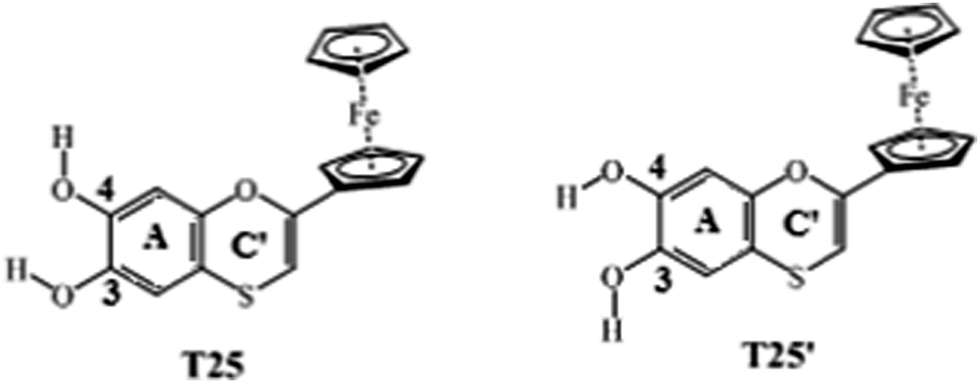 | ||
| Scheme 3 Thiaflavan antioxidants T25 and T25′. | ||
The calculated reactivity indices of these compounds (Table 7) show that T25 and T25′ have the lowest chemical potential and largest electrophilicity index (ω) among all the studied thiaflavan compounds (see Tables 7 and 1). According to the absolute scale25 of global electrophilicity power (ω), T25 and T25′ can be classified as moderate electrophiles.
| Gas | Water | Benzene | ||
|---|---|---|---|---|
| μ | T25 | −3.504 | −3.326 | −3.275 |
| T25′ | −3.501 | −3.310 | −3.269 | |
| η | T25 | 4.020 | 4.458 | 4.427 |
| T25′ | 3.989 | 4.201 | 4.311 | |
| ω | T25 | 1.524 | 1.240 | 1.211 |
| T25′ | 1.574 | 1.303 | 1.219 |
Table 8 presents the calculated BDE, IP, PA, PDE and ETE values in the gas phase as well as in solvents for T25 and T25′. By comparison, it can be deduced that the BDE values are lower than the PA and IP values of these compounds (the PA and IP are the first step of the SPLET and SETPT process, respectively) in benzene and the gas phase.
| Ti | j-OH | Gas | Water | Benzene | |
|---|---|---|---|---|---|
| BDE | T25 | 4-OH | 68.99 | 66.11 | 70.56 |
| T25′ | 3-OH | 65.16 | 63.77 | 68.95 | |
| IP | T25 | 4-OH | 142.91 | 98.62 | 121.18 |
| T25′ | 3-OH | 140.87 | 98.63 | 119.016 | |
| PA | T25 | 4-OH | 325.63 | 31.42 | 91.40 |
| T25′ | 3-OH | 325.27 | 27.4 | 92.147 | |
| PDE | T25 | 4-OH | 240.59 | 26.16 | 49.20 |
| T25′ | 3-OH | 238.80 | 23.81 | 52.97 | |
| ETE | T25 | 4-OH | 57.87 | 93.36 | 78.98 |
| T25′ | 3-OH | 54.39 | 94.97 | 79.84 |
Consequently, the HAT mechanism is predicted to be more favored in gas and non-polar media and the SPLET process in polar media (see Table 8). On the other hand, our calculation indicates that the BDE value of T25′ is lower by ∼5 kcal mol−1 than the smallest BDE value among all Ti (i = 1 to 24) compounds. Furthermore, the BDE value shows that the hydroxyl groups at the 3-position play a key role in scavenging radicals and exhibiting antioxidant effectiveness.
The Mulliken spin atomic density (see Scheme 4) appears to be slightly more delocalized for radicals issued from the 4-position (C4O4) than for the 3-position (C3O3) with values equal to 0.477 and 0.437, respectively (see Scheme 4).
 | ||
| Scheme 4 Spin atomic density distribution in thiaflavan radicals T25 and T25′ in the gas phase (black), water (blue) and benzene (red). | ||
This means that the formation of C3O3 radicals is more favorable for spin density localization than the formation of C4O4 thiaflavan radicals. The intramolecular hydrogen bond between the hydroxyl hydrogen atom and the CO group of T25′ {C4–O–H and C3O} is always slightly smaller than that of T25 {C3–O–H and C4O} in all the gas, benzene and water phases. Therefore, the BDE values are lower in the 3-position than in the 4-position.
In the end, we can conclude that thiaflavan T25′ is an attractive object for future studies of antioxidant activity.
Conclusions
In this work, for the first time the antioxidative properties of thiaflavan and thiaflavan-ferrocene in the gas, water and benzene phases have been studied from a theoretical point of view.The calculated molecular properties (chemical potential, hardness and electrophilicity index) of DNA and the thiaflavan compounds clearly confirm that the thiaflavans mostly act as electron donors in their interaction with DNA. Our results confirm that the thymine moiety in DNA is the primary target for thiaflavan drugs.
Based on the above results, three main antioxidant mechanisms, namely HAT, SET-PT and SPLET, were taken into account to analyze the antioxidative capacity of the thiaflavan compounds in the gas, water and benzene phases. The DFT calculation shows nice correlation between BDE and PDE values calculated in the gas and solvent phases and experimental data of the rate constant (ln(k)) for thiaflavans in scavenging ABTS+˙.
As for the HAT mechanism, from the magnitude of the lowest BDE values, in the gas and solvent phases, the two hydroxyl groups in the same ring A or B (3–4-position in the A ring and 1–2-position in the B ring) have higher H-atom donation ability than a single hydroxyl group in the A or B ring. Our result indicates that hydroxyl groups at the A ring play the key role in scavenging radicals and exhibiting antioxidant effectiveness.
For the SET-PT mechanism, in the studied environments, the antioxidant activity of the investigated compounds reveals that PDE values of T13 to T15 (ferrocene moiety at ring B) are the highest compared to the other thiaflavan compounds.
For the SPLET mechanism, in the studied phases, from the PA of the investigated compounds, we can conclude that rings B and C′ show the strongest antioxidant activity of all the cases.
We have seen that the HAT mechanism is more favorable than SPLET and SET-PT in the gas and benzene phases. On the other hand, the calculation carried out in polar media indicates that the SPLET mechanism is more preferable than the HAT and SET-PT mechanisms.
From the calculated results, OH groups in the A-ring contribute mainly to the antioxidative activities as compared with the B-ring. On the other hand, in thiaflavan, the dihydroxyl groups in ring A are more active sites for trapping radicals in the 3-position than in the 4-position.
Furthermore, the calculation of the aromaticity index confirms that ring A should be considered the primary target for radical attack in thiaflavans.
Our DFT calculations also reveal that the best antioxidant is T25′, which has two hydroxyl groups in the A-ring (3 and 4-positions), a C'-ring and a ferrocene moiety at ring B.
This work provides impetus and a benchmark to guide experiments for determination of the detailed properties of T25′. This compound is then an attractive object for future studies of antioxidant properties.
Author contributions
All the authors discussed the results. Douniazed Hannachi: project initiation, conceptualization, calculation supervision, data interpretation, draft writing. Nour El Houda Amrane: calculation achievements, data interpretation, draft writing. Lynda Merzoud: calculations, data interpretation, draft writing. Henry Chermette: calculations, data interpretation, draft writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge GENCI/CINES for HPC resources/computer time (Project cpt2130), and PSMN of ENS-Lyon for computing resources.Notes and references
- M. Dizdaroglu, P. Jaruga, M. Birincioglu and H. Rodriguez, Free Radical Biol. Med., 2002, 32, 1102–1115 CrossRef CAS PubMed.
- Y. Fang, S. Yang and G. Wu, Nutrition, 2002, 18, 872–879 CrossRef CAS PubMed.
- A. C. Maritim, R. A. Sanders and J. B. Watkins, J. Biochem. Mol. Toxicol., 2003, 17, 24–38 CrossRef CAS PubMed.
- N. Liang and D. D. Kitts, Molecules, 2014, 19, 19180–19208 CrossRef PubMed.
- A. Urbaniak and M. Molski, CMST, 2012, 18, 117–128 CrossRef.
- H. W. Lai and Z. Q. Liu, Eur. J. Med. Chem., 2014, 81, 227–236 CrossRef CAS PubMed.
- L. W. Wattenberg, Inhibition of Carcinogenesis by Naturally-Occurring and Synthetic Compounds in Antimutagenesis and Anticarcinogenesis Mechanisms II, Boston, MA, Springer, 1999, vol. 52, pp. 155–16 Search PubMed.
- H. Chermette, J. Comput. Chem., 1999, 20, 129–154 CrossRef CAS.
- J. Padmanabhan, R. Parthasarathi, U. Sarkar, V. Subramanian and P. K. Chattaraj, Chem. Phys. Lett., 2004, 383, 122–128 CrossRef CAS.
- P. K. Chattaraj and R. G. Parr, Struct. Bonding, 1993, 80, 151–174 CrossRef.
- P. Mondal, K. K. Hazarika and R. C. Deka, PhysChemComm, 2003, 6, 24–27 RSC.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1873 CrossRef CAS PubMed.
- R. G. Parr, L. V. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- M. J. Frisch, et al., G. 09 Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J. Thom. H. Dunning and P. J. Hay, Modern Theoretical Chemistry 3, Plenum, New York, 1977 Search PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- Y. Z. Zheng, G. Deng, R. Guo, D. F. Chen and Z. M. Fu, Int. J. Mol. Sci., 2019, 20, 1450 CrossRef PubMed.
- M. Szeląg, A. Urbaniak and H. A. R. Bluyssen, Open Chem., 2015, 13, 17–31 Search PubMed.
- J. Rimarčík, V. Lukeš, E. Klein and M. Ilčin, THEOCHEM, 2010, 952, 25–30 CrossRef.
- S. Kandasamy and K. Rathinam, Mol. Phys., 2011, 109, 839–852 CrossRef CAS.
- K. S. Kumar and R. Kumaresan, Int. J. Quantum Chem., 2011, 111, 4483–4496 CrossRef CAS.
- U. Norinder, J. Mol. Struct. THEOCHEM, 1987, 151, 259–269 CrossRef.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417–4423 CrossRef CAS.
- P. K. Chattaraj, H. Lee and R. G. Parr, J. Am. Chem. Soc., 1991, 113, 1855–1856 CrossRef CAS.
- S. Kanvah, J. Joseph, G. B. Schuster, R. N. Barnett, C. L. Cleveland and U. Z. I. Landman, Acc. Chem. Res., 2010, 43, 280–287 CrossRef CAS PubMed.
- K. Mineura, S. Fushimi, Y. Itoh and M. Kowada, Cell Biol. Int. Rep., 1987, 11, 797–802 CrossRef CAS PubMed.
- V. Kumar, G. Jain, S. Kishor and L. M. Ramaniah, Comput. Theor. Chem., 2011, 968, 18–25 CrossRef CAS.
- J. S. Wright, E. R. Johnson and G. A. DiLabio, J. Am. Chem. Soc., 2001, 123, 1173–1183 CrossRef CAS PubMed.
- Y. Z. Zheng, G. Deng, Q. Liang, D. F. Chen, R. Guo and R. C. Lai, Sci. Rep., 2017, 7, 1–11 CrossRef PubMed.
- M. Saqib, S. Iqbal, A. Mahmood and R. Akram, Int. J. Food Prop., 2016, 19, 745–751 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. von Ragué Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- P. Von Rague, C. Maerker, A. Dransfeld, H. Jiao, N. J. R. V. E. Hommes, D. Erlangen and R. V. February, Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef PubMed.
- G. Subramanian, P. von Ragué Schleyer and H. Jiao, Angew. Chem., Int. Ed. Engl., 1996, 35, 2638–2641 CrossRef.
- H. Jiao, P. V. R. Schleyer, B. R. Beno, K. N. Houk and R. Warmuth, Angew. Chem., Int. Ed. Engl., 1997, 36, 2761–2764 CrossRef CAS.
- A. A. Fokin, H. Jiao and P. V. R. Schleyer, J. Am. Chem. Soc., 1998, 120, 9364–9365 CrossRef CAS.
- T. K. Zywietz, H. Jiao, P. V. R. Schleyer and A. De Meijere, J. Org. Chem., 1998, 63, 3417–3422 CrossRef CAS.
- S. Patchkovskii and W. Thiel, J. Mol. Model., 2000, 6, 67–75 CrossRef CAS.
- M. Bühl, Chem. – Eur. J., 1998, 4, 734–739 CrossRef.
- S. M. Ferrer and J. M. Molina, J. Comput. Chem., 1999, 20, 1412–1421 CrossRef CAS.
- M. L. McKee, Z. X. Wang and P. Von Ragué Schleyer, J. Am. Chem. Soc., 2000, 122, 4781–4793 CrossRef CAS.
- A. Hirsch, Z. Chen and H. Jiao, Angew. Chem., Int. Ed., 2000, 39, 3915–3917 CrossRef CAS PubMed.
- M. Bühl and A. Hirsch, Spherical aromaticity of fullerenes, 2001, vol. 101 Search PubMed.
- A. Hirsch, Z. Chen and H. Jiao, Angew. Chem., Int. Ed., 2001, 40, 2834–2838 CrossRef CAS PubMed.
- H. Jiao and P. R. Von Schleyer, J. Phys. Org. Chem., 1998, 11, 655–662 CrossRef CAS.
- F. P. Cossío, I. Morao, H. Jiao and P. Von Ragué Schleyer, J. Am. Chem. Soc., 1999, 121, 6737–6746 CrossRef.
- D. Sawicka, S. Wilsey and K. N. Houk, J. Am. Chem. Soc., 1999, 121, 864–865 CrossRef CAS.
- M. Manoharan, F. De Proft and P. Geerlings, J. Org. Chem., 2000, 65, 7971–7976 CrossRef PubMed.
- A. R. De Lera, R. Alvarez, B. Lecea, A. Torrado and F. P. Cossío, Angew. Chem., Int. Ed., 2001, 40, 557–561 CrossRef CAS PubMed.
- S. P. Verevkin, H. D. Beckhaus, C. Rüchardt, R. Haag, S. I. Kozhushkov, T. Zywietz, A. De Meijere, H. Jiao and P. Von Ragué Schleyer, J. Am. Chem. Soc., 1998, 120, 11130–11135 CrossRef CAS.
- P. Von Ragué Schleyer, B. Kiran, D. V. Simion and T. S. Sorensen, J. Am. Chem. Soc., 2000, 122, 510–513 CrossRef.
- T. J. Ritchie and S. J. F. Macdonald, Drug Discovery Today, 2009, 14, 1011–1020 CrossRef CAS PubMed.
- A. A. Al-Amiery, A. A. H. Kadhum and A. B. Mohamad, Bioinorg. Chem. Appl., 2012, 795812 Search PubMed.
- N. K. Singh, P. Tripathi, M. K. Bharty, A. K. Srivastava, S. Singh and R. J. Butcher, Polyhedron, 2010, 29, 1939–1945 CrossRef CAS.
- A. Walcourt, M. Loyevsky, D. B. Lovejoy, V. R. Gordeuk and D. R. Richardson, Int. J. Biochem. Cell Biol., 2004, 36, 401–407 CrossRef CAS PubMed.
- D. C. Onwudiwe and A. C. Ekennia, Res. Chem. Intermed., 2017, 43, 1465–1485 CrossRef CAS.
- T. Bal-Demirci, M. Şahin, E. Kondakçi, M. Özyürek, B. Ülküseven and R. Apak, Spectrochim. Acta, Part A, 2015, 138, 866–872 CrossRef CAS PubMed.
- Z. Q. Liu, Chem. Rec., 2019, 19, 2385–2397 CrossRef CAS PubMed.
- A. Arezki, E. Brule and G. Jaouen, Organometallics, 2009, 28, 1606–1609 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |