
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, structural analysis, electrochemical and magnetic properties of tetrachloroferrate ionic liquids†
Anham
Zafar
ab,
Imtiaz-ud-Din.
 b,
Safeer
Ahmed
b,
Dejan-Krešimir
Bučar
a,
Muhammad Nawaz
Tahir
c and
Robert G.
Palgrave
*a
b,
Safeer
Ahmed
b,
Dejan-Krešimir
Bučar
a,
Muhammad Nawaz
Tahir
c and
Robert G.
Palgrave
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1E 0AJ, UK. E-mail: r.palgrave@ucl.ac.uk
bDepartment of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan
cDepartment of Physics, University of Sargodha, Sargodha 40100, Pakistan
First published on 18th June 2021
Abstract
Eight ionic liquids have been synthesized with the tetrachloroferrate anion and varying cations with the general formula of [RA]+[FeCl4]− (R = –CH3, –CH2C6H5; A = pyridine, benzimidazole, trimethylamine, triphenylphosphine). They were characterized using spectroscopic techniques such as FT-IR, ESI-MS, Raman, and AAS along with single-crystal XRD to validate their structural motifs. The ionic conductivity, electrochemical and magnetic properties were determined to assess their possible uses in synthesis, separation, magnetic and transportation technologies, and high-energy capacitors. The diffusion coefficient value for these compounds was calculated to be around 7.55 × 10−8 cm2 s−1 and the heterogeneous rate constant value is about 3.60 × 10−3 cm s−1, which quantitatively complemented not only quasi reversibility but also moderately fast electron transfer processes. The effective magnetic moment values for the ionic liquids were determined to be similar to that expected from the spin-only approximation.
Introduction
Ionic liquids (ILs) are unique chemical materials that have attracted steeply increasing scientific interest over the past few years due to their diverse applications.1 The peculiar and distinctive characteristics of ILs offer enhanced proficiency for numerous processes and quality products that make a rationale to investigate their prospects further.2 They have a huge potential in future ground-breaking technologies, while some ILs have already made their approach from the laboratory to practical applications as well and are applied successfully in various electro-analytical,3 energy generation/storage,4 synthetic,5 and engineering applications.6 The first-row transition metal ions in di-,7 tri-8 or tetra-valent9 form exhibit interesting catalytic performances10,11 and fascinating magnetic behaviour.12,13 Metal-containing ionic liquids offer synergic effects to the materials as reports have manifested that the incorporation of iron in their anions generates significant electrochemical,14 magnetic,15 photophysical16 and catalytic properties.10,17In the present study, the electrochemical behaviour of tetrachloroferrate anions with various cations has been investigated. The sole purpose is to assess the reproducibility and sustainability of charge-transfer processes and to demonstrate the non-adsorptive nature of these compounds on the surface of the working electrode. The ionic liquids, as a class, possess high conductivities relative to conventional organic and non-aqueous solvents or electrolytes, which make them adjustable for use in various appliances.18 Some reports have manifested the active role of ILs in the development of high-energy19 and energy-storage devices.20 The presence of tetrachloroferrate (high-spin d5 iron III) anions, including their combination with several cations, is considered to be the cause of various rare properties such as magnetic separation, super-exchange magnetic interactions, gas adsorption, cellulose dissolution and environmental remediation.21 The iron-incorporated ILs cause strong sensitivity to magnetic fields, which suggests the additional benefit of their ease of recycling and recoverability.15 The magnetic susceptibilities of these magnetic ionic liquids (MILs) were measured at temperatures that may enable advanced potential application prospects in the fields of catalytic reactions and separation processes in comparison with conventional ILs.22
The designability of tetrachloroferrate as an anion and appropriate structural modification have opened a new dimension to ionic liquids and their applications in various fields. Keeping these aspects in view and as a continuation of our earlier work,23,24 we report herein the synthesis, structural elucidation of iron-incorporated MILs to further explore their utility in a diverse field of studies. Their unique characteristics, such as a wide range of physico-chemical properties, high conductivities and magnetic susceptibility data, could be exploited in electrical as well as magnetic separation techniques in addition to synthetic chemistry.
Materials and methods
All reagents and solvents were purchased from Sigma-Aldrich Chemicals and used without further purification. The NMR spectra were taken in D2O/DMSO as a solvent and recorded using a Bruker Avance III 600 spectrometer. The FTIR spectra were recorded using a Thermo Shimadzu FT-IR Spectrophotometer (750–3400 cm−1) with a resolution of 4 cm−1. A Renishaw inVia Raman microscope was used to record the Raman spectra. The acquisition time was 20 s, the excitation wavelength 785 nm (130 mW, 10%) and the laser spot 1 μm2. Electrospray mass spectrometry analyses were performed using the Vanquisher LC system connected to an Orbitrap Exactive Plus mass spectrometer operating in positive ionization mode with a HESI ion source. The metal concentration was determined using an atomic absorption spectrophotometer with an air–C2H2 flame (Shimadzu AA-670 Japan). Optimum conditions for the analysis of iron were using a hollow cathode lamp (HCL) with a current of 8 mA, slit width 0.2 and wavelength 248.3 nm. A conductometer (Art. Nr. 103635) was used to measure the conductance of the synthesized compounds. The magnetic susceptibility was measured using an Evans balance developed by Dennis Evans in 1973. All the voltammetric measurements were carried out using an Eco Chemie Autolab PGSTAT 302 potentiostat/galvanostat at an applied potential (±2 mV) and current (±0.2 A).Synthesis
The iron-containing ionic liquids (1–8) were synthesized using a reported methodology with slight modifications25 comprising of two steps. The first step involves the synthesis of the required cation through a quaternization reaction whereas the second step deals with an anion-exchange reaction that results in the preparation of ionic liquids as the target compounds. The synthesized products include methyl and benzyl substituent-based cations, and tetrachloroferrate as the anionic moiety.Methyl-based ionic liquids (1–4)
The quaternization reaction was carried out to prepare the methyl-based cation by taking stoichiometric amounts of methyl iodide with the respective amine such as pyridine, benzimidazole, trimethylamine or triphenylphosphine and stirring for around 7 hours in THF.26,27 The methyl-based iodide salts (I1–I4), thus formed, were filtered off and the filtrate was discarded as unreacted materials. The halide salt prepared must be precluded from intense light to avert decomposition. A stoichiometric amount of iron(III) chloride, dissolved in 6 M hydrochloric acid, was mixed with methyl-based iodide salts and stirred for another 4–5 hours.28 The hydrogen iodide, thus produced, was removed by washing with ethyl acetate. The methyl-based ionic liquids (1–4) were obtained and purified by recrystallization in a mixture of solvents: dichloromethane and n-hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) as enunciated in (Scheme 1).
:3) as enunciated in (Scheme 1).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (1633), νCC (1487), νHC (3070).
N (1612), νCC (1442), νHC (3093).
C (1633), νHC (3068).
N (1633), νCC (1487), νHC (3070).
N (1612), νCC (1442), νHC (3093).
C (1633), νHC (3068).
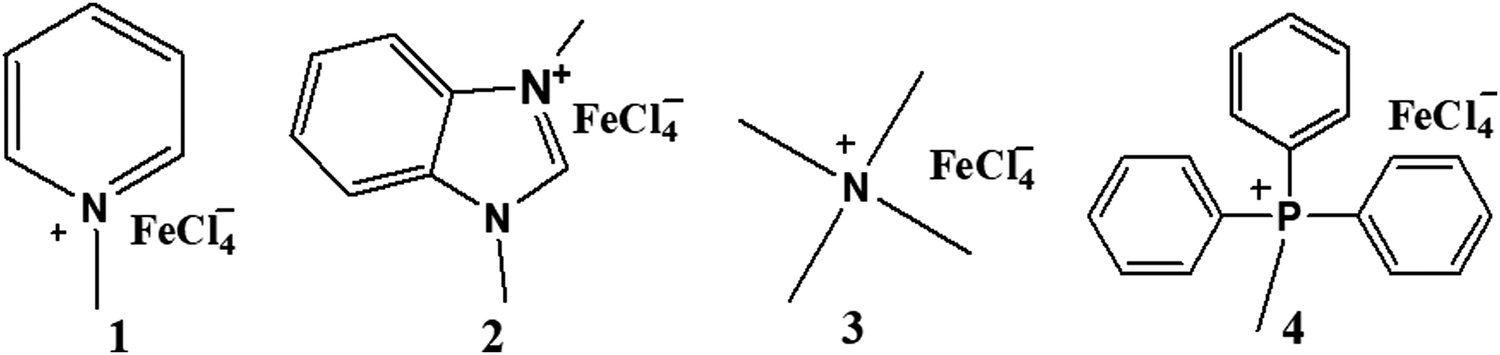 | ||
| Scheme 1 Structures of methyl-based ionic liquids. | ||
Benzyl-based ionic liquids (5–8)
The quaternization reaction was carried out to synthesize the cations of interest. Benzyl chloride and the respective amine or PPh3 in stoichiometric amounts were mixed and refluxed in THF for 48 hours.29 The benzyl-based chloride salts (I5–I8) were formed and filtered off to remove the unreacted impurities, then dried in a vacuum but not heated above 80 °C to avoid the reverse reaction. Then stoichiometric amounts of these synthesized benzyl-based chloride species and iron salts (FeCl3·6H2O) were stirred for another 4–5 hours in methanol30,31 to get the target compounds (5–8), which were subsequently purified by washing with diethyl ether (Scheme 2).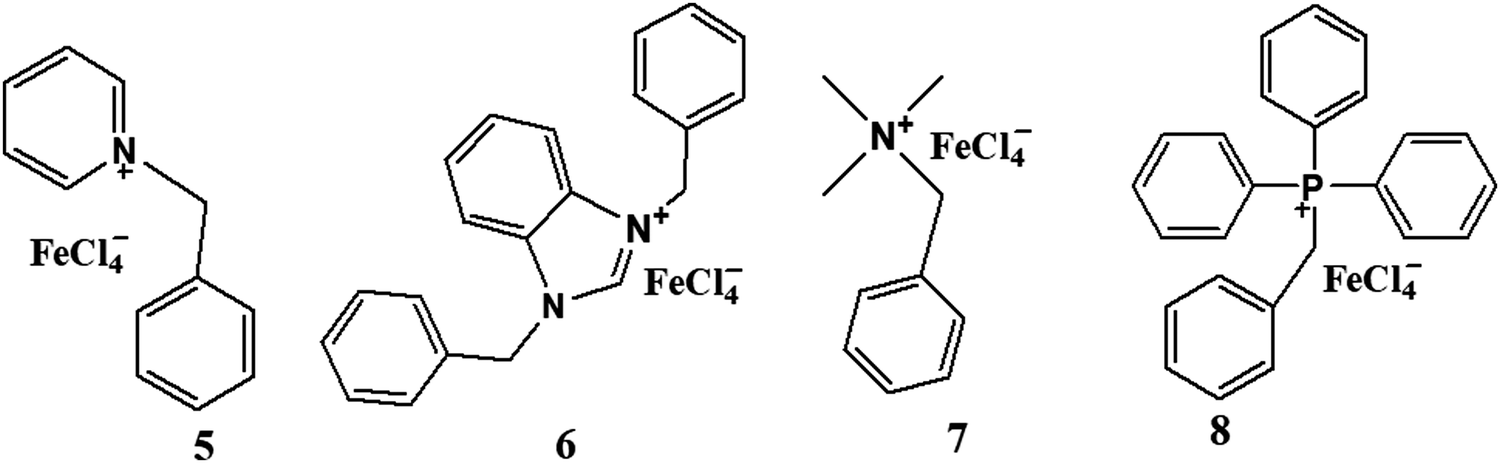 | ||
| Scheme 2 Structures of benzyl-based ionic liquids. | ||
N (1628), νCC (1486), νHC (3059).
N (1618), νCC (1434), νHC (3052).
C (1478).
C (1641), νHC (2911).
X-Ray crystallography
Single crystals of 2 and 6 were obtained via slow solvent evaporation from dichloromethane and methanol solutions, respectively.A suitable crystal for 2 was selected and mounted on a Bruker kappa APEXII CCD single-crystal X-ray diffractometer using the micro focus MoKα X-ray beam (λ = 0.71073 Å). The crystal was kept at 296 K during the data collection. The data were corrected using a Lorentz polarization absorption correction. The crystal structure was solved and refined using the ShelXS-97 and ShelXL programmes.32 Single X-ray diffraction data for compound 6 was collected using a dual-source Agilent SuperNova single-crystal X-ray diffractometer using a micro-focus Cu X-ray beam (λ = 1.54184 Å, generated at 50 kV and 0.8 mA) and a 135 mm Atlas CCD detector. The sample temperature was controlled using an Oxford Instruments Cryojet5. The data were acquired, processed and corrected using the CrysAlisPro program.33 Structure solution and refinement were accomplished using the Olex2 program suite.34 The structure was solved using SHELXT,35 while structure refinement was accomplished using SHELXL.36 All non-hydrogen atoms of 2 and 6 were refined anisotropically, while hydrogen atoms associated with the carbon atoms were refined isotropically in geometrically constrained positions [Uiso(H) = 1.2Ueq(C)]. The crystallographic and refinement parameters for 2 and 6 are shown in Table 2.
Voltammetric analysis
All the voltammetric measurements were carried out using an Eco Chemie Autolab PGSTAT 302 potentiostat/galvanostat, Utrecht, The Netherlands, having GPES 4.9. A conventional three-electrode system was employed using a Platinum disc as the working electrode with an area of 0.017 cm2, Pt wire the counter electrode and Ag wire the reference electrode calibrated against the standard Fc+/Fc couple (potential = 0.493 V vs. Ag/AgCl and 0.601 V vs. Ag wire). A control (blank scan) was run using deionized water and 0.1 M KCl as the supporting electrolyte, to ensure the electroinactivity of the medium, prior to analyzing the ionic liquids. The response of 1 mM of the ionic liquid was checked in both the absence and the presence of the supporting electrolyte. The cyclic voltammetric data were calculated at different scan rates (from 0.02 to 1.2 V s−1) at 28 °C ± 0.5.Ionic conductance
A conductometer (Art. Nr. 103635) was used to measure the ionic conductance of 1–8 for 1 mM, 0.5 mM and 0.4 mM solutions in deionized water.Magnetic susceptibility
An Evans balance was used to measure the magnetic susceptibility of the synthesized iron-containing ILs. This instrument works with the help of two permanent magnets equipped back-to-back and endorsed by the suspension strip. An optical transducer detects a deflection when the sample tube is inserted between the two magnets. One of these two magnets has a coil attached with it, through which a counteracted current is passed. The sample exerts some force on the magnet, which is proportional to the applied current to the coil to counteract the force and therefore allows for a reading to be taken.For paramagnetic materials, the gram magnetic susceptibility χg must be a positive number and it is calculated from the following equation [χg = l × CBal(R − R0)/(m × 109)] where l = length of the sample in the tube (cm), CBal = balance calibration constant, R = the measurement taken with the sample, R0 = the measurement taken without the sample, and m = sample mass in grams.
From the gram magnetic susceptibility, the molar magnetic susceptibility χm and μeff are calculated using the following relationships [χm = χg·molecular weight; μeff = 2.828(χm·T)1/2]. However, a correction must be made as the magnetic field will be weakly repelled from the pair of electrons in compounds.37
Results and discussion
Eight iron-incorporated ILs with varying cations substituted with methyl and benzyl groups, respectively, have been synthesized, and the details are enunciated in Scheme 3.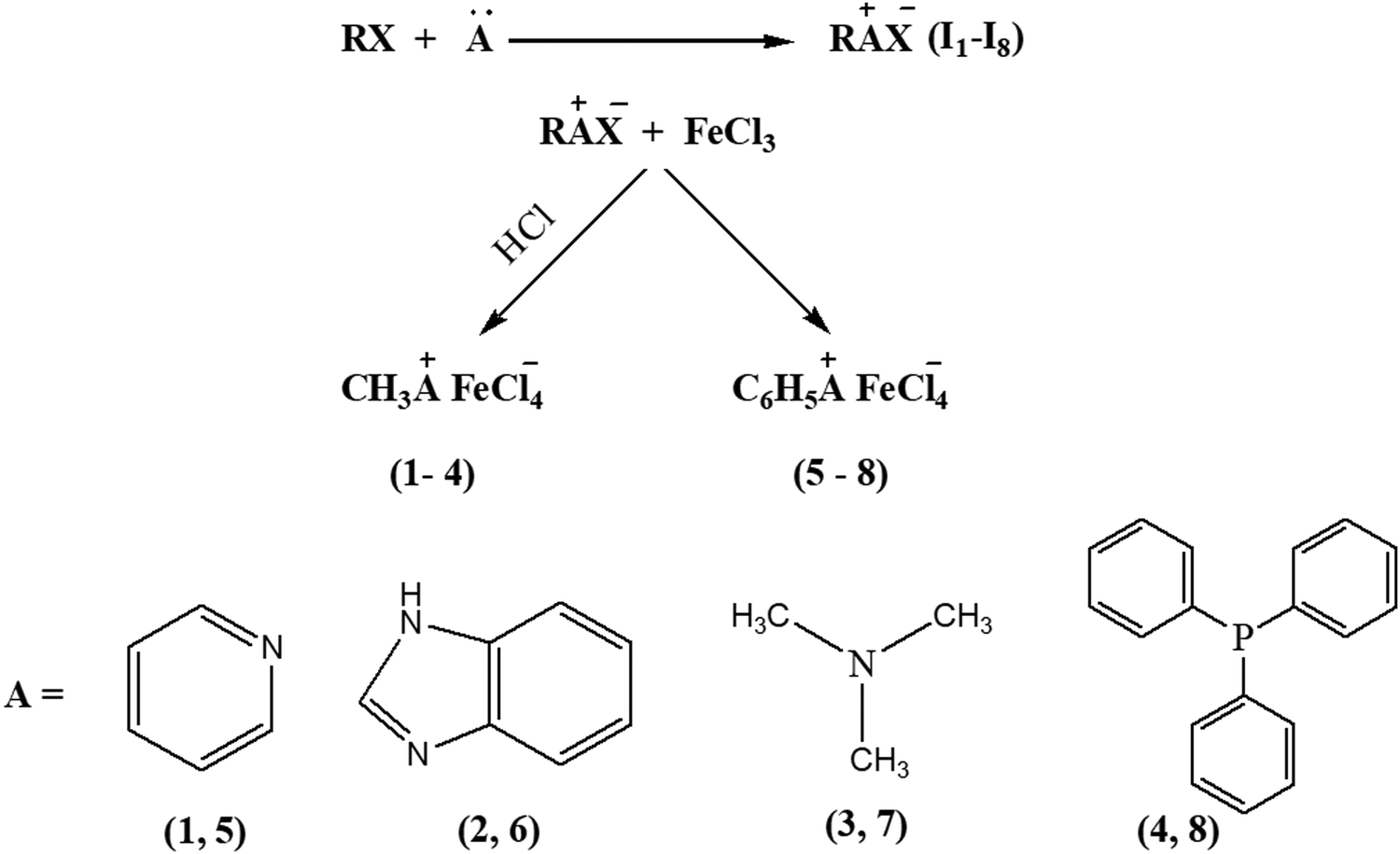 | ||
| Scheme 3 Synthetic pathway for 1–8. | ||
The synthesized compounds are stable at room temperature. They were characterized using FTIR, Raman, AAS and single-crystal X-ray analyses along with mass spectrometry. One of the significant IR absorption bands for the methyl-containing ILs (1–4) is the stretching frequency of ν(C–H), which appears around 3000 cm−1 that ensured the occurrence of methylation for all the precursors.
The absence of the stretching frequency for ν(N–H) at 3300 cm−1, present in benzimidazole, in benzimidazolium salts 2 and 6 indicates that methylation has occurred, that is, the H of the NH functionality has been replaced by a methyl group. The stretching frequency for ν(N–C) appeared in the range of 1000–1300 cm−1 for methyl-containing ionic liquids 1–3, and the stretching frequency for ν(P–C) in the case of 4 occurred at around 1438 cm−1. The IR absorption band for ν(C–H) appeared at around 3000 cm−1 in the benzyl-containing halide salts 5–8. The appearance of the absorption band in the range of 1000–1160 cm−1 indicated that the methylene of benzyl chloride is attached to the nitrogen of the precursors 5–7. The appearance of the IR absorption band around 1437 cm−1 indicates the attachment of methylene (of benzyl chloride) to the phosphine moiety as for 8.
Atomic absorption spectroscopic quantification data indicated that the calculated and observed values of iron (Fe3+) are in accordance with each other, as depicted in Table 1. The calculated and observed values of iron deviate within the range of ±0.05%.
| MILs | Calculated (%) | Observed (%) |
|---|---|---|
| 1 | 19.17 | 19.13 |
| 2 | 16.19 | 16.21 |
| 3 | 20.55 | 20.56 |
| 4 | 11.77 | 11.74 |
| 5 | 15.18 | 15.19 |
| 6 | 11.30 | 11.32 |
| 7 | 16.05 | 16.03 |
| 8 | 10.13 | 10.15 |
The electrospray analysis using mass spectrometry for 1–8 was operated in positive ionization mode with a HESI ion source, which confirms the presence of the corresponding cations. ESI-MS for 1–8 are shown as follows:
1: [C6H8N][FeCl4]. m/z (ESI-MS+): 94.06 (100%) [C6H8N]+.
2: [C9H11N2][FeCl4]. m/z (ESI-MS+): 147.09 (100%) [C9H11N2]+.
3: [C4H12N][FeCl4]. m/z (ESI-MS+): 74.09 (100%) [C4H12N]+.
4: [C19H18P][FeCl4]. m/z (ESI-MS+): 277.11 (100%) [C19H18P]+.
5: [C12H12N][FeCl4]. m/z (ESI-MS+): 170.09 (100%) [C12H12N]+.
6: [C14H13N2][FeCl4]. m/z (ESI-MS+): 299.15 (100%) [C14H13N2]+.
7: [C10H16N][FeCl4]. m/z (ESI-MS+): 150.12 (100%) [C10H16N]+.
8: [C25H22P][FeCl4]. m/z (ESI-MS+): 353.14 (100%) [C25H22P]+.
The Raman spectra of all the synthesized ILs are similar in the region 200–500 cm−1, indicating their identical anionic structure. In the literature, the band at 332 cm−1 is assigned to the totally symmetric Fe–Cl stretching vibration of FeCl4.38,39 Hence, this validated that all the synthesized ionic liquids have the same [FeCl4] anion. The Raman spectra for 1–8 are shown in Fig. S22 (ESI†).
Single-crystal X-ray analyses were carried out to authenticate the crystal structures of 2 and 6. Both salts crystallise in the monoclinic space group P21/c with one benzimidazolium cation and one tetrachloroferrate(III) anion in the asymmetric unit. The structural data of the MILs containing benzimidazole as the cation with tetrachloroferrate(III) have been studied less compared with other types of cations in spite of their extensive applications in green catalysis40 and as magnetic materials.41 So far, a very limited number of articles have appeared in the literature, showing X-ray single-crystal analysis of dialkylbenzimidazolium-based ILs with iron(III) chloride.42–44 The crystal structures of 2 and 6 are shown in Fig. 1 and their crystallographic data and selected bond lengths and angles are listed in Tables 2 and 3, respectively.
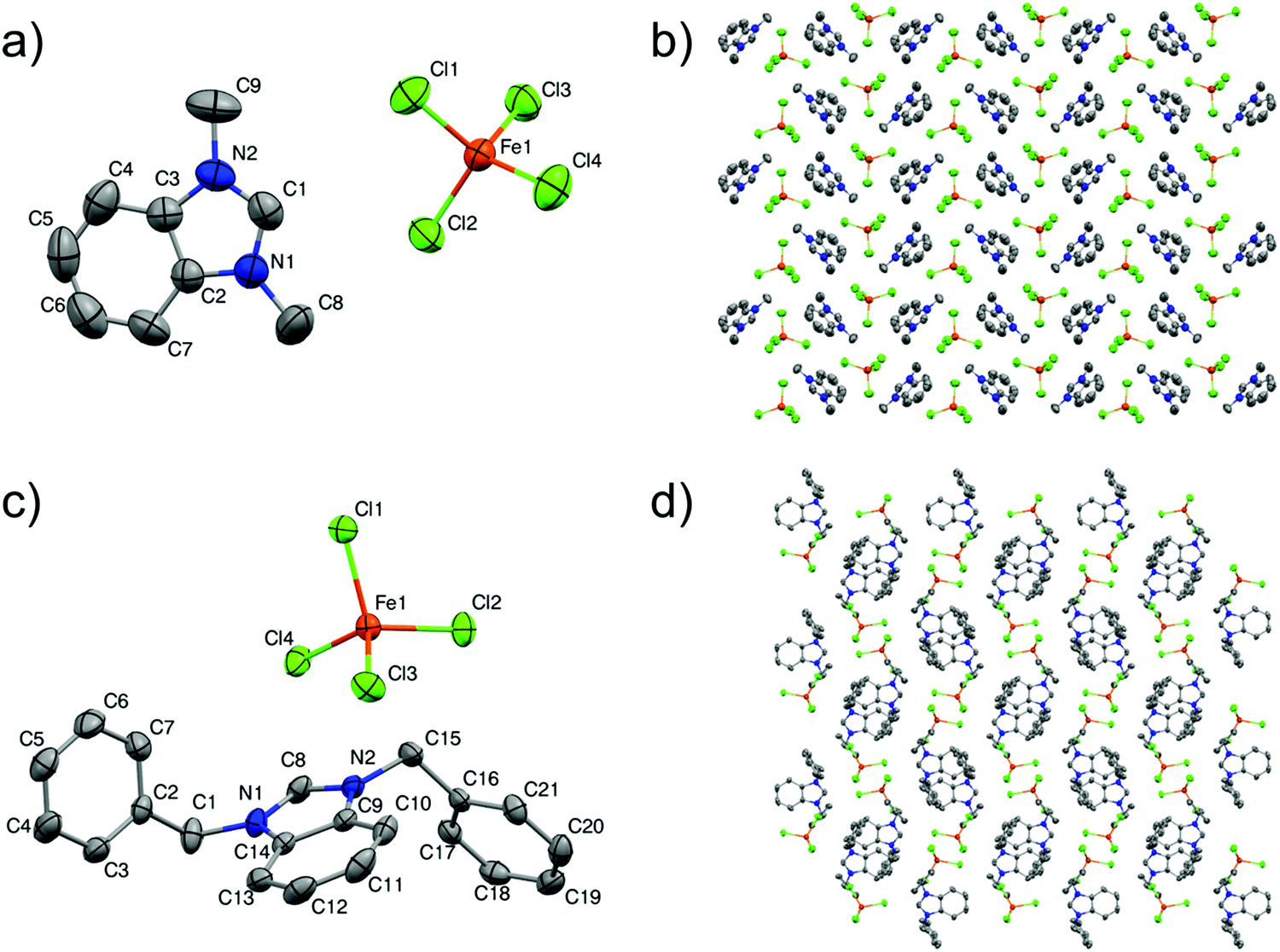 | ||
| Fig. 1 X-Ray crystal structure of 2 and 6: (a) perspective view of the asymmetric unit of 2, (b) crystal packing of 2 viewed along the crystallographic axis a, (c) perspective view of the asymmetric unit of 6, (d) crystal packing of 6 viewed along the crystallographic axis a. The thermal ellipsoids are drawn at the 50% probability level. All hydrogen atoms are omitted to enhance the clarity of the diagrams. | ||
| Identification code | 2 | 6 |
|---|---|---|
| Empirical formula | C9H11Cl4FeN2 | C21H19Cl4FeN2 |
| M r/g mol−1 | 344.85 | 497.03 |
| Crystal system | Monoclinic | Monoclinic, |
| Space group | P21/c | P21/c |
| a/Å | 7.3345(5) | 11.8089(1) |
| b/Å | 15.4892(10) | 14.1846(2) |
| c/Å | 12.8785(7) | 13.2623(2) |
| α/° | 90 | 90 |
| β/° | 100.608(2) | 96.253(1) |
| γ/° | 90 | 90 |
| V/Å3 | 1438.06(16) | 2208.28(5) |
| Z | 4 | 4 |
| ρ calc/g cm−3 | 1.593 | 1.495 |
| T/K | 296(2) | 150.0(1) |
| μ/mm−1 | 1.767 | 10.00 |
| F(000) | 692.0 | 1012 |
| Radiation | MoKα (λ = 0.71073 Å) | CuKα (λ = 1.54184 Å) |
| 2θ range for data collection/° | 5.26–3.998 | 9.158–133.12 |
| Index ranges | −9 ≤ h ≤ 9 | −14 ≤ h ≤ 14 |
| −16 ≤ k ≤ 16 | −19 ≤ k ≤ 19 | |
| −16 ≤ l ≤ 15 | −15 ≤ l ≤ 15 | |
| Number of collected reflections | 8966 | 36222 |
| Unique reflections | 3133 | 3889 |
| Number of observed reflections [I > 2σ(I)] | 2368 | 3673 |
| R int | 0.0343 | 0.0425 |
| R(F), F > 2σ(I) | 0.0336 | 0.0238 |
| wR(F2), F > 2σ(I) | 0.0826 | 0.0600 |
| R(F), all data | 0.0504 | 0.0256 |
| wR(F2), all data | 0.0925 | 0.0610 |
| Goodness-of-fit on F2 | 1.020 | 1.040 |
| Δ r(max, min)/e Å−3 | 0.315/−0.273 | 0.305/−0.243 |
| 2 | 6 | |
|---|---|---|
| Bond lengths/Å | ||
| Fe(1)–Cl(4) | 2.1853(8) | 2.1948(5) |
| Fe(1)–Cl(3) | 2.1912(8) | 2.1937(5) |
| Fe(1)–Cl(2) | 2.1938(8) | 2.1957(5) |
| Fe(1)–Cl(1) | 2.1967(7) | 2.1991(5) |
| Bond angles/° | ||
| Cl(4)–Fe(1)–Cl(3) | 110.00(3) | 110.05(2) |
| Cl(4)–Fe(1)–Cl(2) | 110.39(3) | 110.66(2) |
| Cl(3)–Fe(1)–Cl(2) | 109.12(3) | 108.31(2) |
| Cl(4)–Fe(1)–Cl(1) | 111.13(3) | 107.16(2) |
| Cl(3)–Fe(1)–Cl(1) | 110.09(3) | 111.17(2) |
| Cl(2)–Fe(1)–Cl(1) | 106.02(3) | 109.49(2) |
The cations are composed of the benzimidazolium core with methyl and benzyl chains, and the corresponding dialkylbenzimidazolium moiety, in both the structures, is not discernibly changed after reaction of the iron(III) salt, which is in accordance with previously published reports.45 The anion [FeCl4]− has a distorted tetrahedral geometry by coordinating with four chlorides. The bond lengths of Fe–Cl are found in the range of 2.185(8)–2.196(7) Å for 2 and 2.193(5)–2.199(5) Å for 6. These values are in good agreement with previous studies.46 The mean values of the Cl–Fe–Cl angles for 2 and 6 are 109.39(3)° and 109.47(2)°, respectively, which are the values for [FeCl4]− found in the literature.47
The ions interact with each other in both the structures through C–H⋯Cl, π(C–C)⋯Cl and π(C–N)⋯Cl contacts. The cations and anions are arranged in parallel layers and the interactions generated a 3-D packing network in both the structures, as illustrated in Fig. 1. Hydrogen bonding has not been observed in 6, which is analogous to previous reports having bulky imidazolium-based cations,48,49 while in the case of 2 the cations and anions have the C–H⋯Cl interaction and (four) H-bonds are formed between the acidic hydrogen at the C4-, C8- and C9-position of benzimidazole with the [FeCl4]− anion at distances between 2.84 and 2.92 Å, and the data are presented in Table 4. However, these are very weak interactions and are not categorized as typical hydrogen bonds. Thus, one can safely assume that the cations are not static and these H-bonds become weakened at room temperature.
| D | H | A | d(D–H)/Å | d(H–A)/Å | d(D–A)/Å | D–H–A/° |
|---|---|---|---|---|---|---|
| C(4) | H(4) | Cl(3) | 0.93 | 2.85 | 3.696(3) | 152.6 |
| C(8) | H(8)B | Cl(1) | 0.96 | 2.84 | 3.760(4) | 161.1 |
| C(8) | H(8)C | Cl(2) | 0.96 | 2.92 | 3.608(4) | 129.8 |
| C(9) | H(9)B | Cl(2) | 0.96 | 2.93 | 3.569(3) | 125.4 |
Furthermore, compared with the previous structural reports on tetrachloroferrate(II)-containing ILs, the anion [FeCl4]2− has a slightly flattened tetrahedral structure compared with [FeCl4]− and the average bond length is 2.292(2) Å, which is 0.11 Å more than [FeCl4]− due to the additional electron involved.50 The mean value for Cl–Fe–Cl bond angles is 110.18(9)° ranging from a high of 113.63(11)° to a low of 107.96(7)°.51
Solubility of MILs
The solubility of all MILs in different solvents is listed in Table 5. Due to the basic dipole forces between the polar organic solvent and the ILs, all the synthesized ILs have very good solubility in methanol, ethanol, acetonitrile and DMSO. They are insoluble in non-polar solvents like n-hexane, diethyl ether and benzene. The solubility pattern of these ILs is the same as their corresponding iodide and chloride salts (I1–I8). However, interestingly these salts are completely insoluble in chloroform and toluene, and on incorporating the iron moiety, they became partially soluble and imparted a yellow color to them.| MILs | H2O | MeOH | EtOH | ACN | DMSO | DCM | CCl4 | THF | DMF | EtOAc | Et2O | n-Hex |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a ++: miscible, ±: slightly miscible, −−: immiscible. | ||||||||||||
| 1 | ++ | ++ | ++ | ++ | ++ | ++ | ± | −− | −− | −− | −− | −− |
| 2 | ++ | ++ | ++ | ++ | ++ | ++ | ± | −− | −− | −− | −− | −− |
| 3 | ++ | ++ | ++ | ++ | ++ | ++ | ± | −− | −− | −− | −− | −− |
| 4 | ++ | ++ | ++ | ++ | ++ | ++ | ± | −− | −− | −− | −− | −− |
| 5 | ++ | ++ | ++ | ++ | ++ | ++ | ± | −− | −− | −− | −− | −− |
| 6 | ++ | ++ | ++ | ++ | ++ | ++ | ± | −− | −− | −− | −− | −− |
| 7 | ++ | ++ | ++ | ++ | ++ | ++ | ± | −− | −− | −− | −− | −− |
| 8 | ++ | ++ | ++ | ++ | ++ | ++ | ± | −− | −− | −− | −− | −− |
Cyclic voltammetry (CV) analysis
The electrochemical behaviour of the MILs was determined comprehensively in both positive and negative potential regions using the cyclic voltammetry technique (Fig. 2). All the ILs were found to be electro-active in the negative potential region of 0 to −1.2 V whereas 1–4 were found to be electro-active in the positive potential region of 0 to 1 V while the ILs 5–8 were apparently inactive in the positive potential region.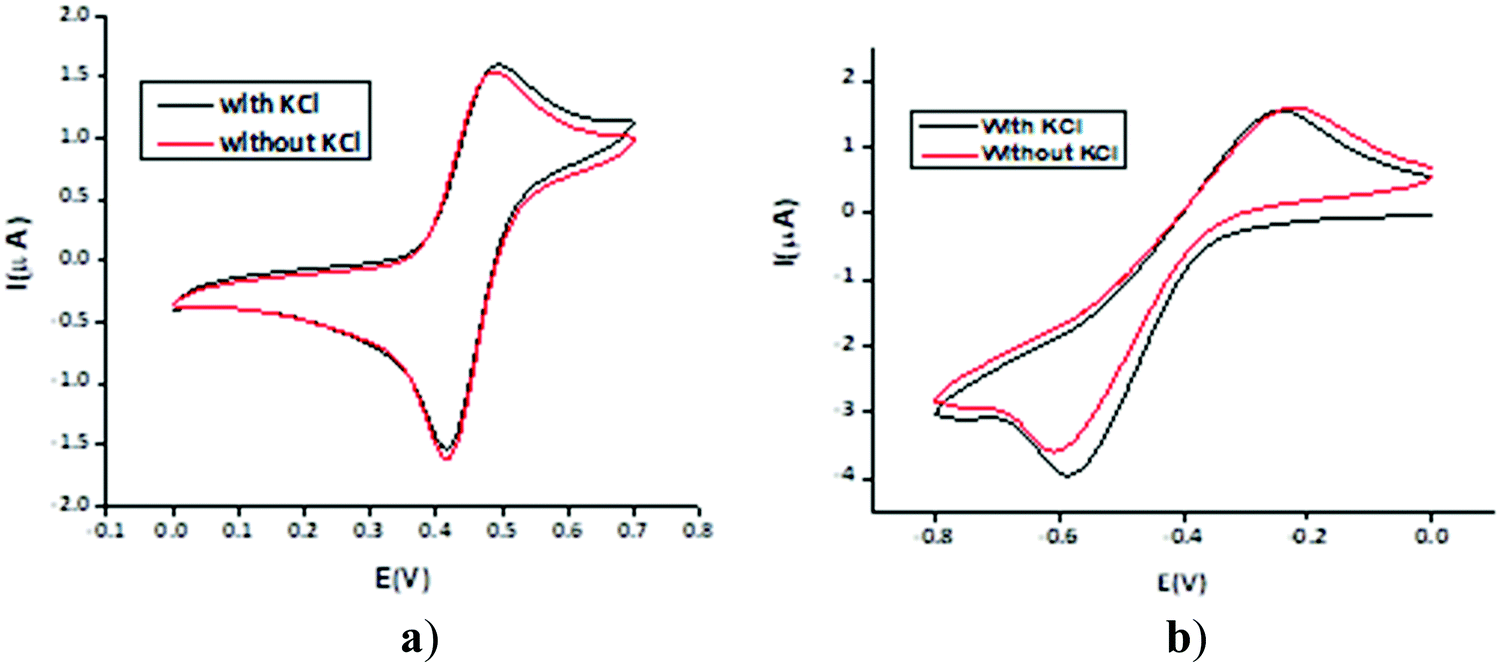 | ||
| Fig. 2 Cyclic voltammograms of a 1 mM solution of 1 with KCl, on Pt as the working electrode vs. Ag wire at 28 °C and with a scan rate of 0.05 V s−1 at (a) positive potential and at (b) negative potential. | ||
To observe the effect of the supporting electrolyte (KCl), the cyclic voltammograms of the ILs were recorded in both the presence and absence of the supporting electrolyte. The two redox peaks in the positive potential region were determined for 2 and 3. The cyclic voltammograms for the ILs were recorded both in the presence and absence of the supporting electrolyte (KCl), to observe its effect.
The representative cyclic voltammograms for 1 under different conditions are shown in Fig. 2. It is apparent that recording the signal from zero towards a positive potential gave a highly reversible voltammogram without any pre- or post-peak humps. Moreover, there is a negligible difference in the two voltammograms obtained either with or without the supporting electrolyte, indicating that the ILs can suppress the migration effect and consequently reduce the unwanted capacitive current and increase the charge-transfer current. It may be construed from these findings that these materials may be used as supporting electrolytes in various electroanalytical applications depending on their inactivity in the potential range of interest.
The quasi-reversible electrochemical signature observed in the negative potential region (around −0.6 V vs. Ag wire) is ascribed to the typical reduction of pyridinium cations.52 The quasi-reversibility on the Pt electrode is the conversion of pyridinium cations to pyridine and the production of surface hydrogen gas in an aqueous medium and the adsorbed hydrogen gas is oxidized to nascent hydrogen which protonates the pyridine back to pyridinium in the reverse scan and can be used for CO2 reduction.53 On the other hand, the reversible electro-oxidation, as discussed earlier, in the positive potential region is a less reported phenomenon. Compound 1 showed single reversible electrooxidation, while 2 and 3 showed two reversible electron-transport (ET) signatures whereas only an irreversible cathodic peak at 0.236 V was observed for 4. The rest of compounds 5–8 did not reflect clear redox waves but a hump was observed in each case between 0.4 and 0.5 V.
The apparently reversible electro-oxidation around E1/2 ≈ 0.5 V was attributed to the Fe3+/Fe2+ redox process, and the slight variation in peak positions is due to the presence of the reorganization energy of the complexes. This is quite analogous to the results attained for the reported molten salts of iron.54 The submergence of this redox peak from 5 to 8 could be argued to be due to the electron-donating effect of the benzyl group. This is also supported for 4, which has bulky phenyl groups (owing to their electron-donating nature), and as a result, the cathodic peak potential is at a less positive value. The second peaks for 2 and 3 are possibly the electrooxidation of the whole complex. The results of cyclic voltammetry scans are quite interesting for these compounds but still require further investigations for their possible applications in various domains of electrochemistry.
Stability of MILs
Cyclic voltammograms for the solutions of 1–8 with a concentration of 1 mM were recorded by taking successive scans (1–10) at a constant scan rate of 0.05 V s−1 without using KCl. The motive was to assess the reproducibility and sustainability of the charge-transfer processes in the given time scale. There is a negligible decrease in either the cathodic or anodic current for all the compounds, as exhibited in Fig. 3. This clearly demonstrates the non-adsorptive nature of the compounds on the surface of the Pt working electrode.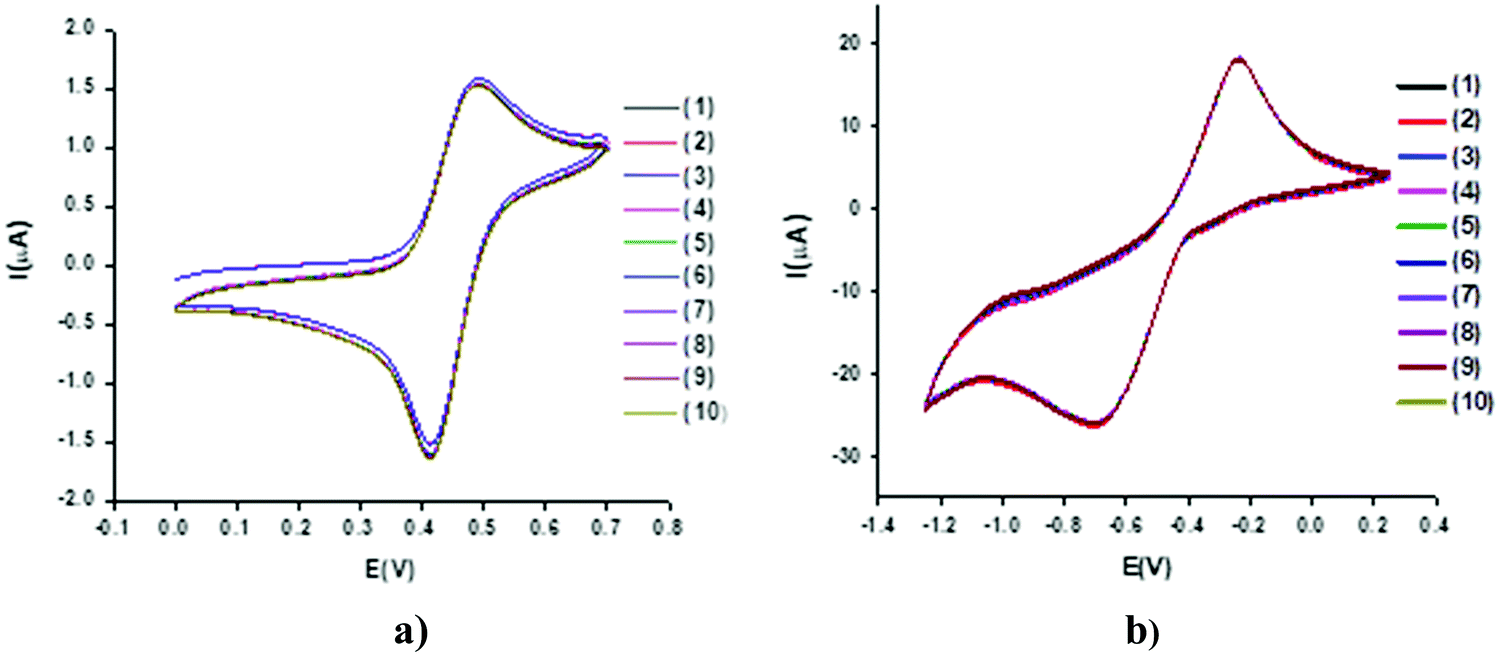 | ||
| Fig. 3 Cyclic voltammograms of a 1 mM solution of 1 in deionized water with 1–10 successive scans at 50 mV s−1 using a Pt electrode vs. Ag wire in (a) positive, and (b) negative potential regions. | ||
The essential electrochemical parameters for 1–8 are given in Tables 6 and 7. It can be inferred from the data that they exhibited their first (or the only redox wave) half wave redox potential (E1/2) around 0.5 V in the positive region and −0.4 V in the negative region. The second redox wave was determined around E1/2 of 0.8 V for (2 and 3). The first redox process in the positive potential region was found to be reversible as evinced from the peak width and current ratio values while the second redox process was quasi-reversible. Similarly, the redox process in the negative potential region was also found to be quasi-reversible for these synthesized compounds.
| MILs | a E p (V) | c E p (V) | E 1/2 (V) | ΔEp (V) | |aEp − aEp/2| (V) | |cEp − cEp/2| (V) | |aip/cip| | |
|---|---|---|---|---|---|---|---|---|
| 1 | 0.520 | 0.457 | 0.489 | 0.063 | 0.057 | −0.048 | 1.20 | |
| 2 | (i) | 0.520 | 0.469 | 0.494 | 0.050 | 0.030 | −0.045 | 0.6 |
| (ii) | 0.899 | 0.789 | 0.844 | 0.109 | 0.018 | −0.054 | 0.3 | |
| 3 | (i) | 0.534 | 0.464 | 0.499 | 0.070 | 0.026 | −0.043 | 0.60 |
| (ii) | 0.778 | 0.725 | 0.751 | 0.053 | 0.037 | −0.035 | 1.05 | |
| 4 | — | 0.236 | — | — | — | −0.146 | — | |
| 5 | — | — | — | — | — | — | — | |
| 6 | — | — | — | — | — | — | — | |
| 7 | — | — | — | — | — | — | — | |
| 8 | — | — | — | — | — | — | — | |
| MILs | a E p (V) | c E p (V) | E 1/2 (V) | ΔEp (V) | |aEp − aEp/2| (V) | |cEp − cEp/2| (V) | |aip/cip| |
|---|---|---|---|---|---|---|---|
| 1 | −0.241 | −0.564 | −0.402 | 0.323 | 0.140 | −0.097 | 1.44 |
| 2 | −0.251 | −0.583 | −0.417 | 0.337 | 0.120 | −0.044 | 1.45 |
| 3 | −0.374 | −0.459 | −0.416 | 0.832 | 0.048 | −0.046 | 1.04 |
| 4 | −0.278 | −0.549 | −0.413 | 0.271 | 0.131 | −0.090 | 1.26 |
| 5 | −0.300 | −0.551 | −0.425 | 0.251 | 0.106 | −0.080 | 1.32 |
| 6 | −0.229 | −0.710 | −0.469 | 0.481 | 0.136 | −0.085 | 1.60 |
| 7 | −0.207 | −0.651 | −0.429 | 0.444 | 0.136 | −0.143 | 1.00 |
| 8 | −0.127 | −0.986 | −0.556 | 0.859 | 0.172 | −0.055 | 3.1 |
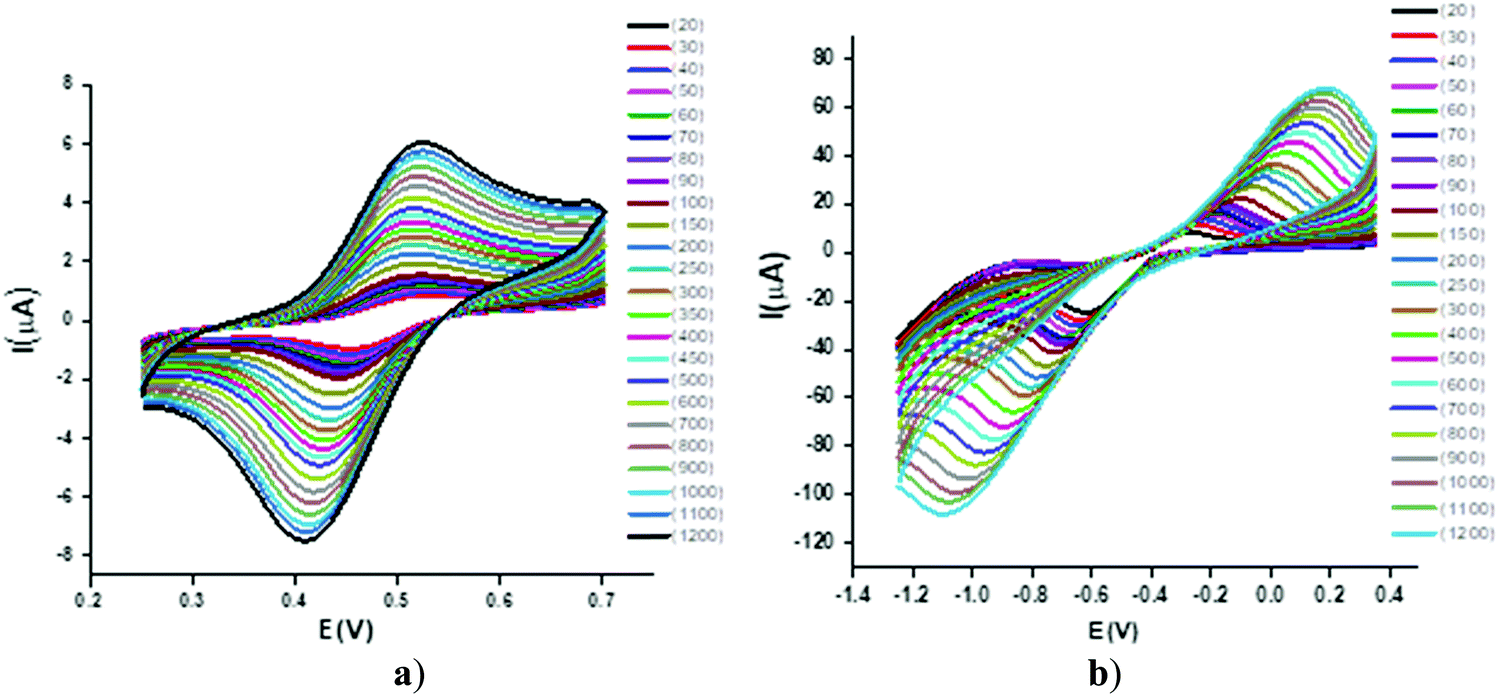 | ||
| Fig. 4 Cyclic voltammograms of a 1 mM solution of 1 in deionized water at 20–1200 mV s−1 using a Pt electrode vs. Ag wire in (a) positive and (b) negative potential regions. | ||
Effect of scan rate and heterogeneous kinetics
The CV response was studied by varying the scan rate from 0.025 to 1.200 V s−1 (Fig. 4). The changes in the voltammetric signature on the variation of the scan rate indicated the dependence of the peak potential values on the scan rate, thus confirming the quasi-reversibility of the process and that the increase in peak current was according to the Randles–Sevcik equation55 [ip = (2.99 × 105)n(αna)1/2ACD1/2v1/2], where v = scan rate (V s−1), C = concentration of electro-active species (mol cm−3), n = number of electrons, na = number of electrons exchanged in the rate determining step, A = area of electrode (cm2), α = transfer coefficient, and Do = diffusion coefficient (cm2 s−1), which was used to calculate the diffusion coefficient values and eventually the heterogeneous rate constants by employing the Nicholson method of peak separation [k° = ψ[πDonFv/RT]1/2, where ψ is a dimensionless parameter linked to peak separation, F is the Faraday constant and R and T have their usual significance. The linear relationship was shown in corresponding plots of the square root of the potential v1/2vs. the peak current ip (Fig. 5).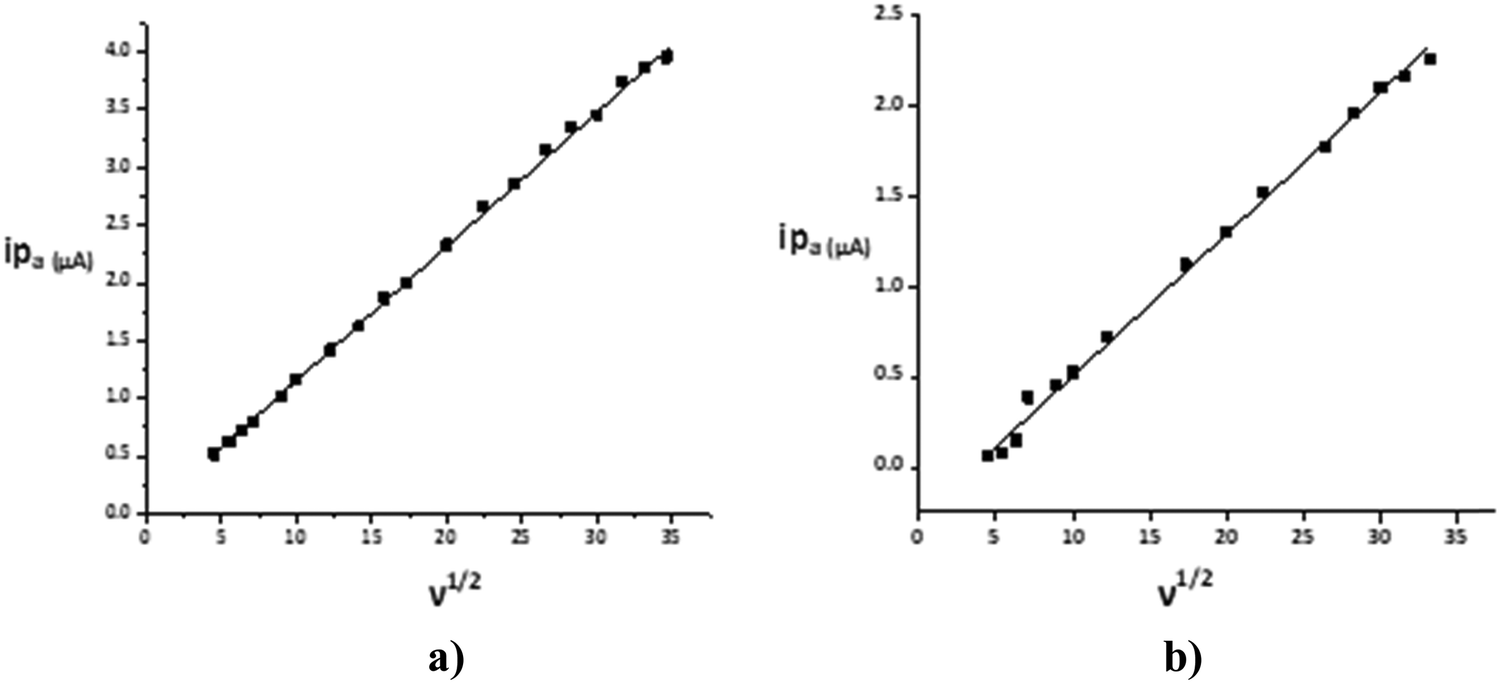 | ||
| Fig. 5 Plot of ipvs. v1/2 for a 1 mM solution for 1 in deionized water in (a) positive and (b) negative potential regions. | ||
The calculated diffusion coefficient value, 7.55 × 10−8 cm2 s−1, and the linearity of the ‘ipvs. v1/2’ plots established the diffusion-controlled nature of the electrode reaction.56 The heterogeneous rate constant value 3.60 × 10−3 cm s−1 quantitatively approved the quasi-reversible process and the moderately fast electron-transfer process.
Ionic conductivity
The molar conductance values for 1–8 at different concentrations are presented in Table 8. The ionic liquids are comprised entirely of ions, therefore, they have higher values of ionic conductance.57 The ionic liquids, thus, possess high conductivities in comparison with conventional organic and non-aqueous solvents (∼10 mS cm−1), which make them adaptable to be used as the electrolyte in various appliances.58 It is now established that the properties of the ionic liquid are highly influenced by the presence of even a very small amount of impurities such as halide ions, particularly chloride ions.59 The conductivity of ionic liquids, in general, is weakly interrelated with the type and size of ions, whereas the anionic size and type are not linked with the conductance. As illustrated from the molar conductance values, the ionic liquids with a higher cationic size show higher conductance because there is more delocalization of charge and more charge–charge separation; consequently, they conduct electricity more efficiently. This effect is more dominant over the steric hindrance, which decreases the mobility of ions owing to the larger size.60 The higher conductivity values of these compounds made them promising candidates for electrolytes and high-energy capacitors.61| MILs | 1 mM (mS cm−1) | 0.5 mM (mS cm−1) | 0.4 mM (mS cm−1) |
|---|---|---|---|
| 1 | 10.99 | 5.28 | 4.21 |
| 2 | 9.22 | 4.54 | 3.63 |
| 3 | 7.68 | 3.55 | 2.82 |
| 4 | 12.02 | 6.23 | 4.99 |
| 5 | 5.16 | 2.55 | 2.08 |
| 6 | 8.36 | 4.12 | 3.27 |
| 7 | 10.44 | 5.07 | 4.17 |
| 8 | 10.09 | 4.94 | 3.99 |
The molar conductivity values of the IL solutions increase with increasing concentration due to the increase in the effective concentration of ions as well as the ionic strength.
Magnetic studies
The ionic liquids exhibit paramagnetic behavior over a wide range of temperatures. Values of the product of molar susceptibility and temperature (χMT) for 1–8 were determined in the range of 3.49–4.50 (emu mol−1 K Oe−1) whereas the reported value is 4.37 (emu mol−1 K) for the Fe3+ system. At temperatures higher than 20 K, the magnetic susceptibility data follow the Curie–Weiss law with Weiss temperatures and the effective paramagnetic moment value was determined to be [μeff] = 5.66 μB for an Fe3+ system, which is consistent with the literature value reported for a high-spin d5 system (Table 9). The data revealed that the calculated μeff values for the synthesized MILs, owing to the FeCl4− moiety, are in good agreement with the reported values.62 The gram and molar susceptibilities of the synthesized MILs are also listed in Table S5 (ESI†).| Cation | M.p. (°C) | d (g cm−3) | η (mPa) | Yield (%) | χ M T (emu mol−1 K) | μ eff (μB) | Ref. |
|---|---|---|---|---|---|---|---|
| M.p.: melting point, d: density, η: viscosity, χMT: magnetic susceptibility, μeff: effective paramagnetic moment, —: not reported. | |||||||
| 1-Butyl-3-methylimidazolium | −8.29 | 1.38 | 340 | — | 4.11 | 5.73 | Hayashi et al., 200465 |
| 1-Decyl-3-methylimidazolium | — | — | — | — | 4.01 | 5.66 | Del Sesto et al., 200863 |
| 1-Ethyl-3-methylimidazolium | ∼11 | 1.42 | 180 | — | 4.03 | 5.67 | Yoshida et al., 200666 De Pedro et al., 201067 |
| Tricaprylylmethylammonium | — | — | — | — | 3.2 | 5.02 | D. Kogelnig et al., 201068 |
| Tetrabutylammonium | 130 | — | — | 88 | 4.27 | 5.84 | Krieger et al., 201069 |
| Methylbutylpyrrolidinium | 191 | — | — | 76 | 4.47 | 5.98 | Krieger et al., 201069 |
| Methyltributylammonium | 109 | — | — | 91 | 4.42 | 5.95 | Krieger et al., 201069 |
| Trihexyltetradecylphosphonium | ∼11 | 1.02 | 914 | — | 4.34 | 5.87 | N. Deng et al., 201170 |
| Dibutylbenzimidazolium | 70 | — | — | 76 | 3.9 | 5.56 | Wang, Ke Fei, et al., 201141 |
| Butylpyridinium | 38.4 | — | — | — | 4.31 | 5.85 | W. Zhu et al., 201371 |
| N-Decylpyridinium | 10 | 1.63 | 719.5 | 95 | — | — | Zhuravle et al., 201472 |
| 1,3-Dimethylimidazolium | 285 | — | — | 91 | 4.25 | 5.83 | García-Saiz et al., 201473 |
| 1,3-Dibenzylbenzimidazolium | 175 | — | — | 87 | — | — | LI et al., 201742 |
| 1-Hexyl-3-methylimidazolium | — | 1.30 | 450 | — | — | 5.80 | X. Li et al., 201774 |
| 1-Octyl-3-methylimidazolium | — | 1.24 | 770 | — | — | 5.85 | X. Li et al., 201774 |
| 3-Phenyl-1H-imidazolium | 90.2 | — | — | 99 | 4.85 | 6.23 | J. Chang et al., 201875 |
| 3-Methylphenyl-1H-imidazolium | 68.6 | — | — | 99 | 5.02 | 6.34 | J. Chang et al., 201875 |
| 3-Methoxyphenyl-1H-imidazolium | 83.4 | — | — | 99 | 4.01 | 5.66 | J. Chang et al., 201875 |
| 3-Nitrophenyl-1H-imidazolium | 83.7 | — | — | 99 | 4.87 | 6.24 | J. Chang et al., 201875 |
| Methylpyridinium (1) | 117 | — | — | 95 | 3.93 | 5.59 | This work |
| 1,3-Dimethylbenzimidazolium (2) | 120 | — | — | 96 | 3.49 | 5.27 | This work |
| Tetramethylammonium (3) | 123 | — | — | 93 | 3.72 | 5.44 | This work |
| Methyltriphenylphosphonium (4) | 155 | — | — | 94 | 4.50 | 5.98 | This work |
| Benzylpyridinium (5) | 113 | — | — | 93 | 3.82 | 5.51 | This work |
| 1,3-Dibenzylbenzimidazolium (6) | 119 | — | — | 95 | 3.95 | 5.60 | This work |
| Benzyltrimethylammonium (7) | 111 | — | — | 92 | 4.23 | 5.84 | This work |
| Benzyltriphenylphosphonium (8) | 112 | — | — | 93 | 4.31 | 5.86 | This work |
Various counter cations have also been examined with the transition-metal based ILs, generally with a high-spin d5 system as for the tetrachloroferrate(III) anion.59 The synthesized MILs described a strong response to the externally applied magnetic fields due to their high single-ion magnetic moments.63 The MILs are not only influenced by the applied magnetic field but are also able to separate and transport the diamagnetic materials depending upon their magnetic susceptibility and density parameters.64 The data suggest that the cationic moiety did not interfere with the unpaired electrons of the metal but may enhance its magnetic character at a wide range of temperatures. The characteristic physical and magnetic properties of the reported compounds have been critically reviewed and a comparative account has been illustrated in Table 9.
Conclusion
Eight iron-incorporated ionic liquids have been synthesized and their chemical and structural aspects were characterized using spectroscopic and single-crystal XRD techniques. Cyclic voltammetry data validated the electro-active and non-adsorptive nature of the synthesized ILs. From the diffusion coefficient and rate constant values, it may be inferred that the redox reactions of ILs are diffusion-controlled and moderately fast electron-transfer processes. They are promising candidates for electrolytic and high-energy capacitor appliances due to their higher values of conductivity. The synthesized MILs exhibited significant magnetic properties and their effective magnetic moment values demonstrated that they may find applications in material sciences as well. The literature suggests that iron-containing ILs show magnetically active properties and are attracted by an external magnetic field that may be used for the magnetic manipulation and transportation of N2 bubbles in catalytic and polymerization reactions as well as for a photo-controllable paramagnetic supramolecular system. Moreover, the intermixing of ionic liquids and the electrode materials may become beneficial for the development of environmentally friendly sophisticated storage devices.Abbreviations
| ILs | Ionic liquids |
| MILs | Magnetic ionic liquids |
| AAS | Atomic absorption spectroscopy |
| mM | Millimolar |
| THF | Tetrahydrofuran |
| DCM | dichloromethane |
| DMF | Dimethylformamide |
| ACN | Acetonitrile |
Conflicts of interest
The authors declare no competing interests.Acknowledgements
AZ acknowledges the Commonwealth Scholarship and the Franz Sondheimer Bursary received from University College London.References
- A. J. Greer and J. Jacquemin, Molecules, 2020, 25, 5207 CrossRef CAS PubMed.
- C. Kolbeck, M. Killian, F. Maier, N. Paape, P. Wasserscheid and H. P. Steinrück, Langmuir, 2008, 24, 9500–9507 CrossRef CAS PubMed.
- J. L. Anderson and D. W. Armstrong, Anal. Chem., 2006, 78, 2893–2902 CAS.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. Zheng, S. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- Suresh and J. S. Sandhu, Green Chem. Lett. Rev., 2011, 4, 289–310 CrossRef CAS.
- A. Berthod, M. J. Ruiz-Ángel and S. Carda-Broch, J. Chromatogr. A, 2008, 1184, 6–18 CrossRef CAS.
- I. Castro, M. L. Calatayud, C. Yuste, M. Castellano, R. Ruiz-García, J. Cano, J. Faus, M. Verdaguer and F. Lloret, Polyhedron, 2019, 169, 66–77 CrossRef CAS.
- E. Pardo, F. Lloret, R. Carrasco, M. C. Muñoz, T. Temporal-Sánchez and R. Ruiz-García, Inorg. Chim. Acta, 2004, 357, 2713–2720 CrossRef CAS.
- R. Ruiz, A. Aukauloo, Y. Journaux, I. Fernández, J. R. Pedro, A. L. Roselló, B. Cervera, I. Castro and M. C. Muñoz, Chem. Commun., 1998, 989–990 RSC.
- S. E. Fortea-Pérez, F. R. Moubtassim, M. L. E. I. Armentano, D. D. Munno, G. Julve and M. Stiriba, Inorg. Chem. Front., 2018, 5, 2148–2156 RSC.
- F. R. Fortea-Pérez, D. Armentano, M. Julve, G. D. Munno and S. E. Stiriba, J. Coord. Chem., 2014, 67, 4003–4015 CrossRef.
- M. Castellano, R. Ruiz-García, J. Cano, J. Ferrando-Soria, E. Pardo, F. R. Fortea-Pérez, S. E. Stiriba, M. Julve and F. Lloret, Acc. Chem. Res., 2015, 48, 510–520 CrossRef CAS PubMed.
- M. Castellano, R. Ruiz-García, J. Cano, J. Ferrando-Soria, E. Pardo, F. R. Fortea-Pérez, S. E. Stiriba, W. P. Barros, H. O. Stumpf, L. Cañadillas-Delgado, J. Pasán, C. Ruiz-Pérez, G. de Munno, D. Armentano, Y. Journaux, F. Lloret and M. Julve, Coord. Chem. Rev., 2015, 303, 110–138 CrossRef CAS.
- D. M. Cabral, P. C. Howlett and D. R. MacFarlane, Electrochim. Acta, 2016, 220, 347–353 CrossRef CAS.
- A. Joseph, G. Zyła, V. I. Thomas, P. R. Nair, A. S. Padmanabhan and S. Mathew, J. Mol. Liq., 2016, 218, 319–331 CrossRef CAS.
- R. Decadt, K. Van Hecke, D. Depla, K. Leus, D. Weinberger, I. Van Driessche, P. Van Der Voort and R. Van Deun, Inorg. Chem., 2012, 51, 11623–11634 CrossRef CAS PubMed.
- J. Wang, S. Zeng, F. Huo, D. Shang, H. He, L. Bai, X. Zhang and J. Li, J. Cleaner Prod., 2019, 206, 661–669 CrossRef CAS.
- Y. Yoshida, A. Otsuka, G. Saito, S. Natsume, E. Nishibori, M. Takata, M. Sakata, M. Takahashi and T. Yoko, Bull. Chem. Soc. Jpn., 2005, 78, 1921–1928 CrossRef CAS.
- N. Böckenfeld, S. S. Jeong, M. Winter, S. Passerini and A. Balducci, J. Power Sources, 2013, 221, 14–20 CrossRef.
- A. Brandt, S. Pohlmann, A. Varzi, A. Balducci and S. Passerini, MRS Bull., 2013, 38, 554–559 CrossRef CAS.
- B. Tobias, O. Breunig, M. Valldor, K. Merz, V. Vasylyeva, A. Mudring and D. Bochum, Cryst. Growth Des., 2011, 11, 2564–2571 CrossRef.
- K. Bica and P. Gaertner, Eur. J. Org. Chem., 2008, 3453–3456 CrossRef CAS.
- M. Mehmood, Imtiaz-ud-Din., A. Raheel, I. Haq and M. Nawaz, Heliyon, 2020, 6, e05156 CrossRef PubMed.
- T. Fatima, A. Akbar, M. Sabieh, M. T. Nawaz and Imtiaz-ud-Din., J. Mol. Struct., 2019, 1184, 462–467 CrossRef CAS.
- R. Engel, J. L. I. Rizzo, C. Rivera, M. Ramirez, M. L. Huang, D. Montenegro, C. Copodiferro, V. Behaj, M. Thomas, B. Klaritch-vrana and J. F. Engel, Chem. Phys. Lipids, 2009, 158, 61–69 CrossRef CAS PubMed.
- P. Bonĥte, A. P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef PubMed.
- J. Sun, M. Forsyth and D. R. Macfarlane, J. Phys. Chem. B, 1998, 8858–8864 CrossRef CAS.
- J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1992, 965–967 RSC.
- A. G. Böwing and A. Jess, Green Chem., 2005, 7, 230–235 RSC.
- M. S. Sitze, E. R. Schreiter, E. V. Patterson, R. G. Freeman, D. Science and E. Normal, Inorg. Chem., 2001, 2298–2304 CrossRef CAS PubMed.
- A. Elaiwi, P. B. Hitchcock, K. R. Seddon, N. Srinivasan, Y. M. Tan, T. Welton and J. A. Zora, J. Chem. Soc., Dalton Trans., 1995, 3467–3472 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- R. Naeem, M. Ali Ehsan, R. Yahya, M. Sohail, H. Khaledi and M. Mazhar, Dalton Trans., 2016, 45, 14928–14939 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2014, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2014, 3–8 Search PubMed.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- W. Jiang, W. Zhu, Y. Chang, Y. Chao, S. Yin, H. Liu, F. Zhu and H. Li, Chem. Eng. J., 2014, 250, 48–54 CrossRef CAS.
- X. Pei, Y. H. Yan, L. Yan, P. Yang, J. Wang, R. Xu and M. B. Chan-Park, Carbon, 2010, 48, 2501–2505 CrossRef CAS.
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS.
- K. F. Wang, L. Zhang, R. R. Zhuang and F. F. Jian, Transition Met. Chem., 2011, 36, 785–791 CrossRef CAS.
- Z. Li, B. Lu, H. Sun, Q. Shen and Y. Zhang, Appl. Organomet. Chem., 2017, 31, 1–7 Search PubMed.
- C. L. Xia, C. F. Xie, Y. F. Wu, H. M. Sun, Q. Shen and Y. Zhang, Org. Biomol. Chem., 2013, 11, 8135–8144 RSC.
- C. H. Yan, L. L. Wang, H. H. Gao, H. M. Sun and Q. Shen, Chin. Sci. Bull., 2012, 57, 1953–1958 CrossRef CAS.
- E. Despagnet-Ayoub, K. Miqueu, J. M. Sotiropoulos, L. M. Henling, M. W. Day, J. A. Labinger and J. E. Bercaw, Chem. Sci., 2013, 4, 2117–2121 RSC.
- T. Bäcker, O. Breunig, M. Valldor, K. Merz, V. Vasylyeva and A. V. Mudring, Cryst. Growth Des., 2011, 11, 2564–2571 CrossRef.
- T. Głowiak, E. Ďurčanská, I. Ondrejkovičová and G. Ondrejovič, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42(10), 1331–1333 CrossRef.
- A. Thirumurugan and C. N. R. Rao, Cryst. Growth Des., 2008, 8, 1640–1644 CrossRef CAS.
- H. N. Deng, Y. L. Xing, C. L. Xia, H. M. sun and Q. Shen, J. Chem. Soc., Dalton Trans., 1972, 1772 Search PubMed.
- J. W. Lauher and J. A. Ibers, Inorg. Chem., 1975, 14, 348–352 CrossRef CAS.
- C. Zhong, T. Sasaki, A. Jimbo-Kobayeshi, E. Fujiwara, A. Kobayashi, M. Tada and Y. Iwasawa, Bull. Chem. Soc. Jpn., 2007, 80, 2365–2374 CrossRef CAS.
- E. Martínez-González, H. G. Laguna, M. Sánchez-Castellanos, S. S. Rozenel, V. M. Ugalde-Saldivar and C. Amador-Bedolla, ACS Appl. Energy Mater., 2020, 3, 8833–8841 CrossRef.
- E. Lebègue, J. Agullo and D. Bélanger, ChemSusChem, 2018, 11, 219–228 CrossRef PubMed.
- Y. Castrillejo, A. M. Martínez, M. Vega, E. Barrado and G. Picard, J. Electroanal. Chem., 1995, 397, 139–147 CrossRef.
- H. L. Lord, W. Zhan and J. Pawliszyn, Comprehensive Sampling and Sample Preparation, 2012, vol. 2, pp. 677–697 Search PubMed.
- S. H. Ng, J. Wang, D. Wexler, K. Konstantinov, Z. P. Guo and H. K. Liu, Angew. Chem., Int. Ed., 2006, 45, 6896–6899 CrossRef CAS PubMed.
- S. Werner, M. Haumann and P. Wasserscheid, Annu. Rev. Chem. Biomol. Eng., 2010, 203–230 CrossRef CAS PubMed.
- C. A. Schroll, S. Chatterjee, T. G. Levitskaia, W. R. Heineman and S. A. Bryan, Anal. Chem., 2013, 85, 9924–9931 CrossRef CAS PubMed.
- Y. Yoshida and G. Saito, J. Mater. Chem., 2006, 1254–1262 RSC.
- D. R. McFarlane, J. Sun, J. Golding, P. Meakin and M. Forsyth, Electrochim. Acta, 2000, 45, 1271–1278 CrossRef CAS.
- M. Galiński, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef.
- Y. Jiang, C. Guo and H. Liu, China Particuol., 2007, 5, 130–133 CrossRef CAS.
- R. E. Del Sesto, T. M. McCleskey, A. K. Burrell, G. A. Baker, J. D. Thompson, B. L. Scott, J. S. Wilkes and P. Williams, Chem. Commun., 2008, 447–449 RSC.
- M. E. Danebrock, C. B. H. Evers and W. Jeitschko, J. Phys. Chem. Solids, 1996, 57, 381–387 CrossRef CAS.
- S. Hayashi and H. O. Hamaguchi, Chem. Lett., 2004, 33, 1590–1591 CrossRef CAS.
- Y. Yoshida and G. Saito, J. Mater. Chem., 2006, 16, 1254–1262 RSC.
- I. De Pedro, D. P. Rojas, J. Albo, P. Luis, A. Irabien, J. A. Blanco and J. R. Fernández, J. Phys.: Condens. Matter, 2010, 22, 296006 CrossRef CAS PubMed.
- D. Kogelnig, A. Stojanovic, F. V.d. Kammer, P. Terzieff, M. Galanski, F. Jirsa, R. Krachler, T. Hofmann and B. K. Keppler, Inorg. Chem. Commun., 2010, 13, 1485–1488 CrossRef CAS.
- E. W. Krieger, B. M. Lee, H. Y. Emge, T. J. Wishart and J. F. Castner, Jr., Phys. Chem. Chem. Phys., 2010, 12, 8919–8925 RSC.
- N. Deng, M. Li, L. Zhao, C. Lu, S. L. de Rooy and I. M. Warner, J. Hazard. Mater., 2011, 192, 1350–1357 CrossRef CAS PubMed.
- W. Zhu, P. Wu, L. Yang, Y. Chang, Y. Chao, H. Li, Y. Jiang, W. Jiang and S. Xun, Chem. Eng. J., 2013, 229, 250–256 CrossRef CAS.
- O. E. Zhuravlev, V. M. Nikol’Skii and L. I. Voronchikhina, Russ. J. Gen. Chem., 2014, 84, 1141–1145 CrossRef CAS.
- A. García-Saiz, P. Migowski, O. Vallcorba, J. Junquera, J. A. Blanco, J. A. González, M. T. Fernández-Díaz, J. Rius, J. Dupont, J. R. Fernández and I. D. Pedro, Chem. – Eur. J., 2014, 20, 72–76 CrossRef PubMed.
- X. Li, Q. Zhou, X. Lu and S. Zhang, J. Mol. Liq., 2017, 243, 285–292 CrossRef CAS.
- J.-C. Chang, C.-H. Yang, I.-W. Sun, W.-Y. Ho and T.-Y. Wu, Materials, 2018, 11, 2539 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1519185 and 2045996 for 2 and 6. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1nj01400e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |