
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards more accurate solubility measurements with real time monitoring: a carvedilol case study†
Dóra
Csicsák
a,
Enikő
Borbás
 *b,
Szabina
Kádár
b,
Petra
Tőzsér
b,
Péter
Bagi
b,
Hajnalka
Pataki
b,
Bálint
Sinkó
c,
Krisztina
Takács-Novák
a and
Gergely
Völgyi
a
*b,
Szabina
Kádár
b,
Petra
Tőzsér
b,
Péter
Bagi
b,
Hajnalka
Pataki
b,
Bálint
Sinkó
c,
Krisztina
Takács-Novák
a and
Gergely
Völgyi
a
aDepartment of Pharmaceutical Chemistry, Semmelweis University, Hőgyes Endre u. 9., H-1092 Budapest, Hungary
bDepartment of Organic Chemistry and Technology, Budapest University of Technology and Economics, Műegyetem rakpart 3., H-1111, Budapest, Hungary. E-mail: eniko.jaksaneborbas@edu.bme.hu
cPion Inc. Billerica, 10 Cook Street, Massachusetts 01821, USA
First published on 10th June 2021
Abstract
The aim of this study was to draw attention to the challenges of solubility measurement and to present new techniques and detection methods that provide more accurate results. We investigated the effect of crystal structure, pH and buffer composition on thermodynamic equilibrium solubility using two polymorphs of carvedilol (Form I and Form II) as model compounds. Measuring the solubility of carvedilol is challenging, as the literature data show extremely high standard deviation. Therefore standardized measurements were performed in the pH range 3–10, in two solutions at 25 °C: Britton–Robinson (BR) and BR with added KCl. Solid phase analysis was performed by X-ray powder diffraction and Raman spectroscopy. The measured SpH data were compared to the theoretical Henderson–Hasselbalch (HH) curve and a perfect fit was found in the pH range 7–10. The salt formation could be observed in the acidic pH range. The counter-ion and solubility of the salt were found to be different in various buffer solutions. In situ fiber optic UV probes were used to monitor the dissolution of carvedilol polymorphs in real-time. The results showed significantly different dissolution kinetics for the polymorphs depending on the pH and the buffer solution. From the dissolution profile, the time required to reach the equilibrium was determined. In most cases, it was more than 24 hours, therefore using the standard protocol (6 hours agitation, 18 hours sedimentation) would have caused significant inaccuracy in results. In extreme cases the measured concentration after 24 hours was found to be 5 or 6 times higher than the real equilibrium solubility.
1. Introduction
Solubility is one of the most important physicochemical properties of a drug for the pharmaceutical industry. It can highly influence the absorption of a compound and consequently affect bioavailability. It is also a key parameter in the Biopharmaceutics Classification System (BCS).1 Solubility is influenced by the lattice structure of a compound. Because of the different lattice energies, the dissolution requires more or less solvation energy.2 Polymorphism is the ability of the compounds to exist in two or more crystalline structures, consequently it can affect their solubility or dissolution.3 As a rule of thumb, the polymorph having the lower lattice-free energy is the more stable form and it has lower solubility while the one with higher energy is less stable but tends to dissolve faster and has higher solubility.4 Polymorphism can be observed at least for one-third of the active pharmaceutical ingredients. The topic received increasing attention in the past decades, as more and more cases with unexplored polymorphism caused serious safety problems.5 Cases with the highest publicity (chloramphenicol palmitate, oxytetracycline, enalapril, ritonavir) are discussed in detail in the literature.6–9 As the result, the discovery and investigation of possible polymorphs (i.e. polymorph screening) is now an inevitable requirement in drug development. Solubility difference of polymorphs will affect the bioavailability/bioequivalence of a drug product if the solubility is the rate-limiting factor upon absorption (for BCS II and IV drugs).Equilibrium (or thermodynamic) solubility is the concentration of the compound in a saturated solution when the solid and solution phases are at equilibrium. This parameter can be measured by several methods, however, the “gold standard” is still the saturation shake-flask (SSF) technique.10 The kinetic solubility (not a thermodynamic constant) is the concentration when precipitation first appears in the (saturated or supersaturated) solution. It can be registered with an in situ device, such as fiber optic UV probes. The kinetic solubility is generally higher than the measured equilibrium solubility.11 In the case of ionisable molecules, solubility depends on the pH of the medium: acidic molecules have poorer solubility at acidic pH; basic molecules at alkaline pH. For amphoteric molecules, the lowest solubility can be observed at their isoelectric point.12–14 The solubility of the unionized form of the compound can be described by the intrinsic solubility value (So).
Solubility measurement of polymorphs is often challenging since in the presence of solvent changes in the crystalline structure of the compound can be observed: the less stable form can be converted to the more stable form; depending on the solvent, a hydrate or solvate may be formed. Because of these phenomena, the solubility measurements of polymorphs must always be complemented by solid-phase analysis to identify the form present in the equilibrium.15
In this work, carvedilol was used as a model compound to study the possible behaviour of polymorphs in solubility experiments. Carvedilol (CAS No. 72956-09-3) is a non-selective beta-adrenergic receptor blocker and an alpha-adrenergic receptor blocker, that acts on the beta-1 receptors of the heart to produce negative inotropic and chronotropic effects. Therefore it is primarily used to treat hypertension. However, due to its non-selective nature, it can cause bronchoconstriction on the beta-2 receptors in the lungs, which limits its applicability.16 Carvedilol is known to have many polymorphic forms, in this study Form I and II were used.17–19 Several studies published solubility data of carvedilol, but the intrinsic solubility values show poor reproducibility. The average value is log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) So [M]: −5.35 ± 0.47 (in-ADME Research).20 In the literature different methods (CheqSol, SSF method), buffers (NaOH/HCl solution, phosphate, acetate buffers, FaSSIF/FeSSIF media) and phase separation techniques (centrifugation, sedimentation, filtration) were described.21–25 These circumstances can greatly influence the measured equilibrium solubility, so this work tried to avoid as many potential errors as possible.26 Therefore real-time monitoring was used via immersed UV probes, because it needed no phase-separation and the sensitivity of UV detection is appropriate to measure the concentration of carvedilol solutions.
So [M]: −5.35 ± 0.47 (in-ADME Research).20 In the literature different methods (CheqSol, SSF method), buffers (NaOH/HCl solution, phosphate, acetate buffers, FaSSIF/FeSSIF media) and phase separation techniques (centrifugation, sedimentation, filtration) were described.21–25 These circumstances can greatly influence the measured equilibrium solubility, so this work tried to avoid as many potential errors as possible.26 Therefore real-time monitoring was used via immersed UV probes, because it needed no phase-separation and the sensitivity of UV detection is appropriate to measure the concentration of carvedilol solutions.
This study aimed to follow the dissolution of the two polymorphs of the model compound in different buffer solutions using real-time monitoring, to determine the equilibrium solubility over a wide pH range and to obtain detailed information about supersaturation and the time needed to reach the equilibrium. Furthermore, the reason for the poor reproducibility of the intrinsic solubility data found in the literature was also investigated.
2. Experimental section
2.1 Materials
The structure of the model compound is presented in Fig. 1. Carvedilol was purchased from Merck KGaA. (Darmstadt, Germany). From the commercially available Form II, the other polymorphic form (Form I) was prepared in-house based on patents and literature data17–19 and was verified by X-Ray powder diffraction (XRPD) and differential scanning calorimetry (DSC) measurements. Distilled water of Ph. Eur. grade was used. All other reagents were of analytical grade.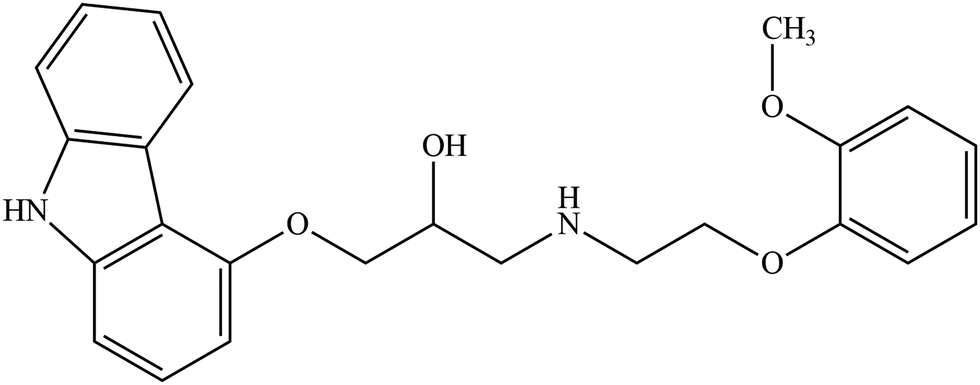 | ||
| Fig. 1 The structure of carvedilol. | ||
As solvent, two different buffer solutions were used: Britton–Robinson (BR) buffer stock solution (a mixture of acetic acid, phosphoric acid, and boric acid, each at 0.04 M) and modified BR buffer, where 0.15 M KCl was added to the solution (BR + KCl). The required amount of 0.2 M NaOH or 1 M NaOH was added to obtain the pH specified for the solubility experiments in the pH range 3–10.
2.2 Solubility measurements
2.3 Solid-phase analysis
2.4 Statistical analyses
Concentrations were expressed as means ± SD, and were compared using the “two-sample” Student's t-test. Differences were considered statistically significant when p < 0.05.3. Results and discussion
Carvedilol is a non-selective β-receptor blocker drug used mainly as an antihypertensive agent in immediate-release (IR) tablets as a racemate of the free base form and in retard tablets as that of its phosphate salt, respectively. The compound belongs to the BCS II group having low solubility and high permeability.30 Carvedilol is known to have many polymorphic forms, in this study Form I and II (nomenclature according to Pataki et al.17,18) were used. In recent literature, Form I is referred as Form III.31 Although Form I is more stable (melting point: 123–126 °C), in marketed formulations Form II (melting point: 114–115 °C) is used. Form I and Form II are monotropic forms.18 Carvedilol is a monoprotic base, (pKa: 8.06), its solubility is pH-dependent in the biorelevant pH range.3.1 Real-time dissolution monitoring by μDISS Profiler
The dissolution profile of the examined carvedilol polymorphs was followed in real-time via UV fiber-optic probes in BR or BR + KCl buffers. The dissolution kinetics was found highly different depending on the pH and the buffer solution. In Fig. 2 the dissolution kinetics of the two polymorphs in BR and BR + KCl buffer solutions can be seen at pH 4, which is representative of the acidic pH region. In the BR buffer, practically no difference could be observed, within 3 hours both forms reached the equilibrium solubility of the phosphate salt, which formed in the acidic range (pH 3–5). No change was observed until the end of the measurements (Fig. 2, A panel). On the other hand in the BR + KCl buffer solution, Form II reached a higher supersaturation at 310 μg mL−1 and held it a bit longer before reaching the equilibrium solubility of the hydrochloride salt (Fig. 2, B/2 panel). From the dissolution profile, the time required to reach the equilibrium can be determined. It was less than 24 hours in the acidic pH region, so in this case, the use of the standard protocol (18 hours agitation time, 6 hours sedimentation) was found to be appropriate.10,12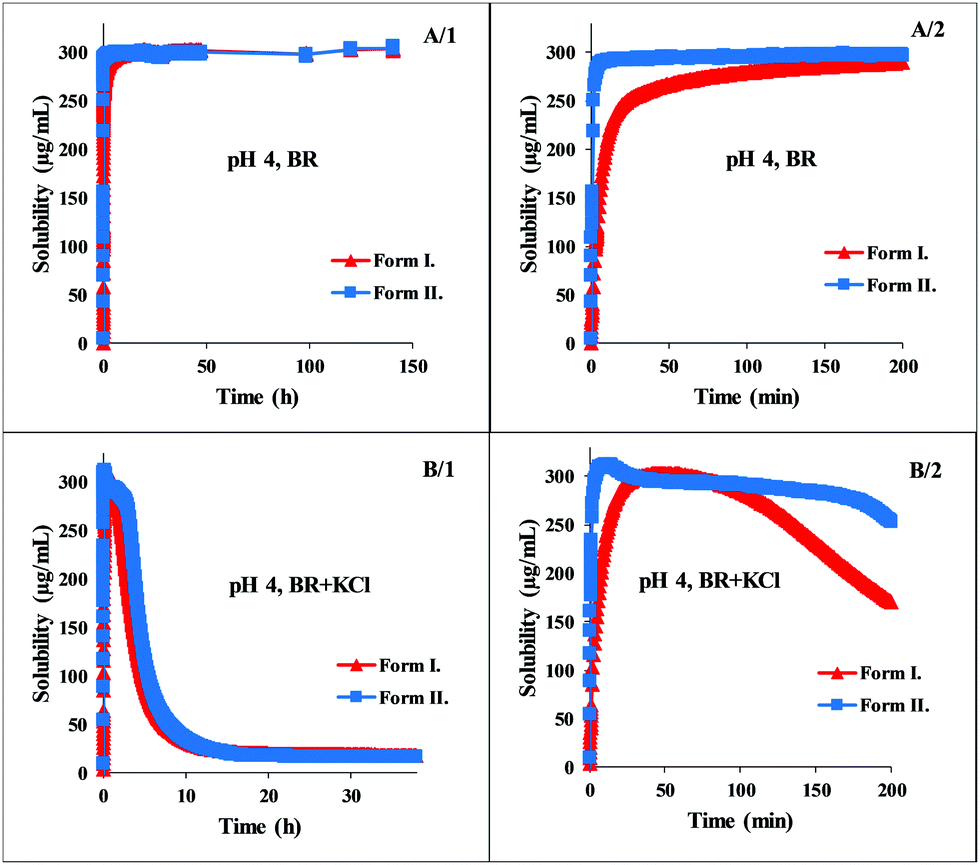 | ||
| Fig. 2 Concentration–time profile of carvedilol polymorphs at pH 4 in BR and BR + KCl buffers. | ||
At pH 8 (which represents the basic pH region), both Form I and Form II exhibit immediate dissolution with no or minimal supersaturation. Equilibrium solubility concentrations can be seen on the first panel of Fig. 3. The second panel shows the deviation between the dissolution of the two polymorphs in the first 15 hours. Not only lower equilibrium solubility, but also slower dissolution can be observed in the case of Form I. Both Form I and II needed more than one day to reach equilibrium solubility, therefore measuring the concentration after 24 hours can lead to false solubility data. After one day, the concentration of the solution of Form I was 2.96 ± 0.03 μg mL−1, which is 14% higher than the measured concentration at the real equilibrium. In contrast, Form II slowly reached the equilibrium state without supersaturation, and the concentration obtained after 24 hours was 30% lower than the equilibrium solubility. In this basic region (pH 7–10) no salt formation could be observed.
 | ||
| Fig. 3 Concentration–time profile of carvedilol polymorphs at pH 8 in the BR buffer. | ||
The greatest difference between the behavior of the two polymorphs can be observed at pH 6. Fig. 4 shows that Form II exhibits immediate dissolution and reaches supersaturation with 135 μg mL−1, at ca. 40 min. Form I dissolves much slower, producing a lower supersaturation at ca. 400 min (Fig. 4, A/2 panel). The total solubility-time profile (Fig. 4, A/1 panel) indicates that it precipitates quickly from the saturated solution but the system reaches the real solubility equilibrium state of the phosphate salt very slowly, during ca. 3 days. Reaching the equilibrium state is even slower in the presence of KCl, where hydrochloric salt is forming at the end of the measurement. However, the kinetics in the first few hours is very similar to the dissolution profile in BR buffer solution (Fig. 4, B/2 panel). Form I reached the equilibrium in BR + KCl at ca. 5 days, in contrast, Form II needed even more time, approx. 9 days (Fig. 4, B/1 panel). This slow equilibrium can cause inaccuracy if the standard 24 hours protocol is applied. In the BR buffer solution, the concentration of Form I after 24 hours is 85.95 ± 3.74 μg mL−1, which is 20.9% higher than the real equilibrium concentration. This distortion is only 7.6% in the case of Form II. In BR + KCl, this deviation is significantly higher: the concentration of Form I after 24 hours is 112.01 ± 5.81 μg mL−1 and the concentration of Form II is 91.56 ± 0.71 μg mL−1. It causes a more than 600% increase compared to the actual equilibrium solubility in the case of Form I and more than 500% in the case of Form II.
 | ||
| Fig. 4 Concentration–time profile of carvedilol polymorphs at pH 6 in BR and BR + KCl buffers. | ||
3.2 Solubility-pH profile of carvedilol polymorphs
The pH-dependent equilibrium solubility of carvedilol Form I and Form II was studied at 25 °C, in the pH 3–10 range, in BR buffer solutions (chloride ion free, I = 0.089 M) and also in pH 3–6.5 domain in BR buffer solutions using external salt (KCl) to adjust the ionic strength to 0.15 M. The average values of the solubility results in μg mL−1 unit are collected in Table 1.| Sample | pH | S pH ± SD (μg mL−1) BR | pH | S pH ± SD (μg mL−1) BR + KCl | Sample | pH | S pH ± SD (μg mL−1) BR | pH | S pH ± SD (μg mL−1) BR + KCl |
|---|---|---|---|---|---|---|---|---|---|
| Form I | 3.38 | 437.84 ± 32.64 | 3.08 | 13.54 ± 0.45 | Form II | 3.30 | 491.42 ± 18.79 | 3.12 | 13.01 ± 1.10 |
| 4.16 | 342.37 ± 2.68 | 4.09 | 14.54 ± 1.22 | 4.17 | 334.77 ± 6.96 | 4.10 | 14.09 ± 0.71 | ||
| 5.08 | 165.49 ± 8.54 | 5.05 | 12.76 ± 1.34 | 5.12 | 158.22 ± 7.34 | 5.08 | 15.19 ± 1.96 | ||
| 6.05 | 71.06 ± 2.45 | 6.06 | 18.05 ± 1.84 | 6.12 | 68.19 ± 2.50 | 6.06 | 18.01 ± 1.82 | ||
| 6.57 | 35.63 ± 1.84 | 6.62 | 46.91 ± 0.68 | ||||||
| 6.99 | 11.75 ± 0.20 | 6.99 | 22.04 ± 0.21 | ||||||
| 8.02 | 2.59 ± 0.19 | 8.06 | 4.61 ± 0.55 | ||||||
| 8.95 | 1.23 ± 0.15 | 8.96 | 2.38 ± 0.85 | ||||||
| 9.98 | 1.21 ± 0.06 | 9.84 | 2.23 ± 0.38 |
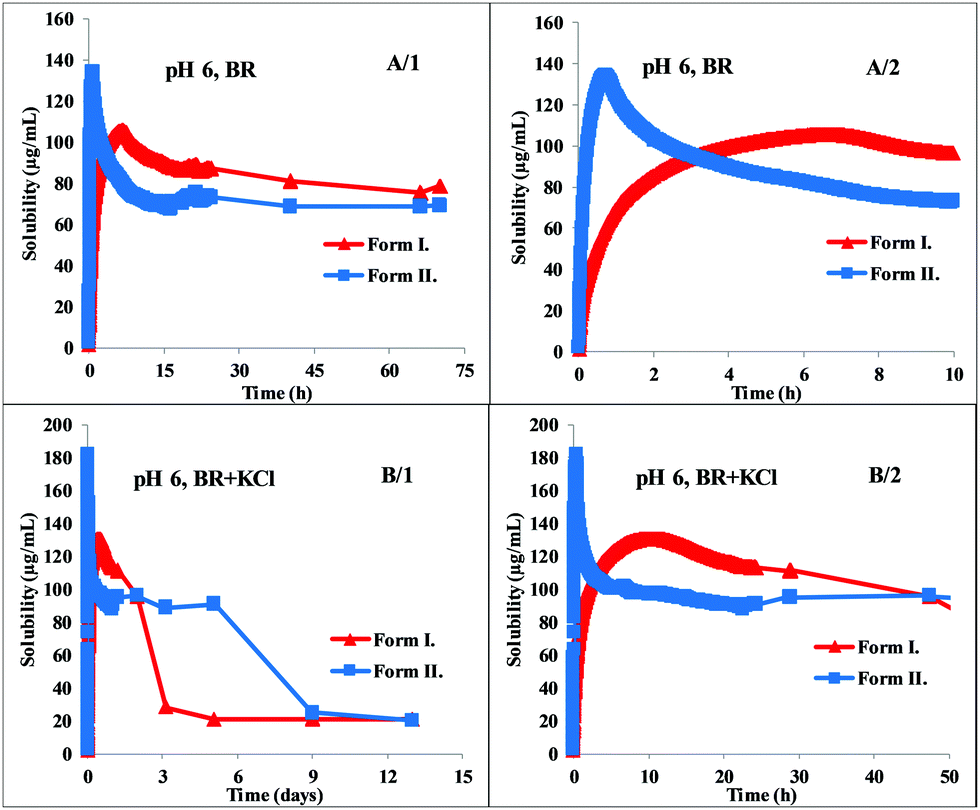
The standard deviation was found to be in the range of 1–35%, the average was 8%. A significant difference in equilibrium solubility of the two polymorphs was found in the pH range 7–10, Form II reached twice as high equilibrium concentration as Form I. This solubility ratio of polymorphs is in agreement with the general behavior of polymorphs reported in the literature.32 The experimental data showed a perfect fit to the theoretical HH curve (logS [M] vs. pH, Fig. 5) between pH 7–10. At about pH 6.5, the measured solubility starts to deviate from the HH curve; in the presence of KCl, a constant salt solubility can be observed at pH 3–6. This is a typical phenomenon when a salt starts to precipitate from the solution. In contrast, the equilibrium concentrations measured in the pure BR buffer at pH 3–6 show a continually increasing tendency and reach no constant value. Solid-phase analysis methods helped to identify which salt is formed in the different buffer media.
 | ||
| Fig. 5 Solubility-pH profile of carvedilol Form I and Form II in BR and BR + KCl buffers (solid line represents the theoretical HH curve, points represent the experimental data). | ||
3.3 Characterization of the undissolved solid phase isolated from solubility measurements
In the pH range 7–10, the Raman spectra of the solid phase isolated from the solubility suspension after equilibration showed consentaneous characteristic peaks with the starting materials (Fig. 6). This supports that during the solubility experiments, the transition did not happen and the measured solubility belongs to the specified polymorph.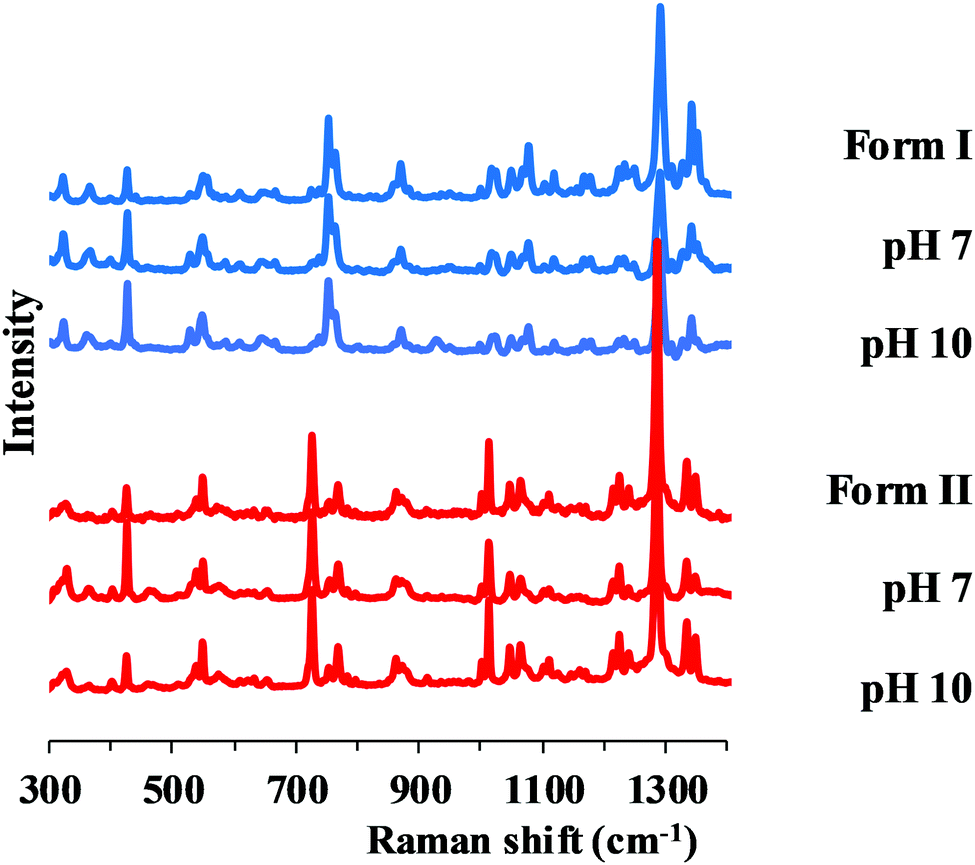 | ||
| Fig. 6 Raman spectra of original Form I, Form II and the solid phase of carvedilol polymorphs isolated from solubility suspensions from the measurements at pH 7 and 10. | ||
In the pH range 3–5, salt formation can be observed regardless of the starting polymorph, but in a different way in the two buffers used. Hydrochloride salt (Fig. 7) was identified from the BR + KCl buffer, while a phosphate salt from pure BR buffer, which was identified as carvedilol dihydrogen phosphate anhydrate (later referred as phosphate salt) based on information found in international patents.33,34
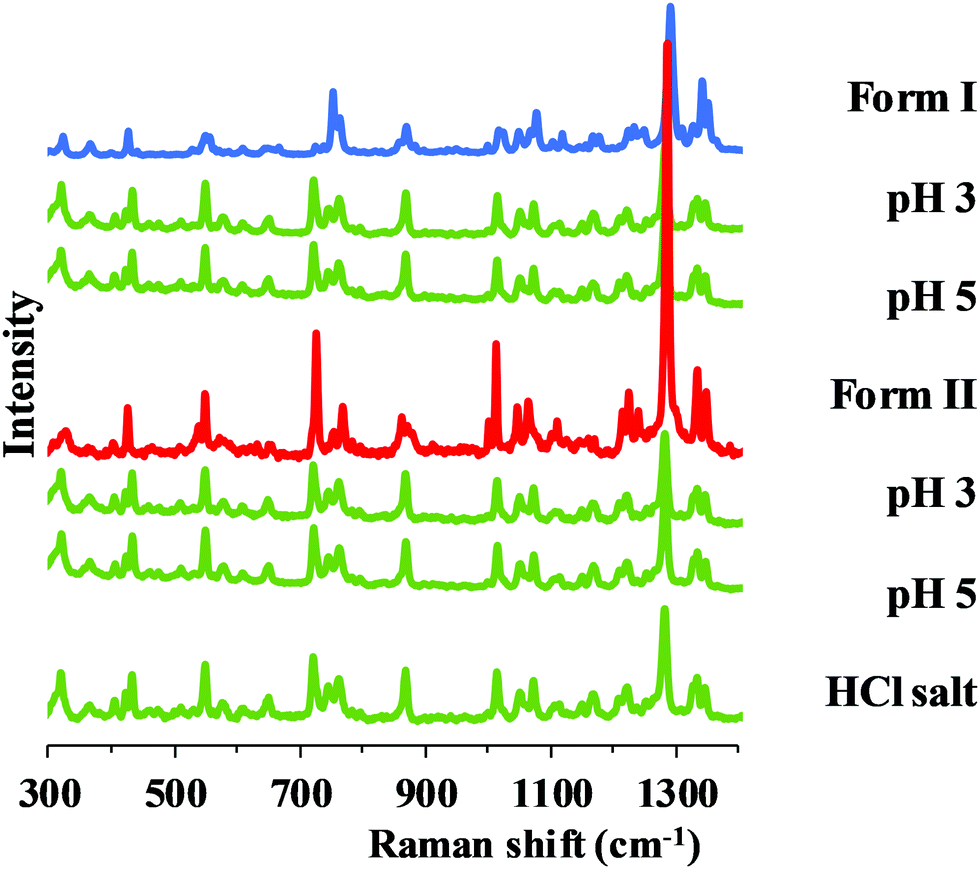 | ||
| Fig. 7 Raman spectra of carvedilol hydrochloride and the solid phase of carvedilol polymorphs isolated from solubility suspensions from the measurements at pH 3 and 5 in BR + KCl. | ||
Since XRPD and Raman spectroscopy verified the identical composition of the precipitate from pH 3–5 BR solutions (Fig. 8), the increasing solubility with decreasing pH in this region (see Table 1 and Fig. 5) can be explained by the formation of solute aggregates or complexes with a buffer component.
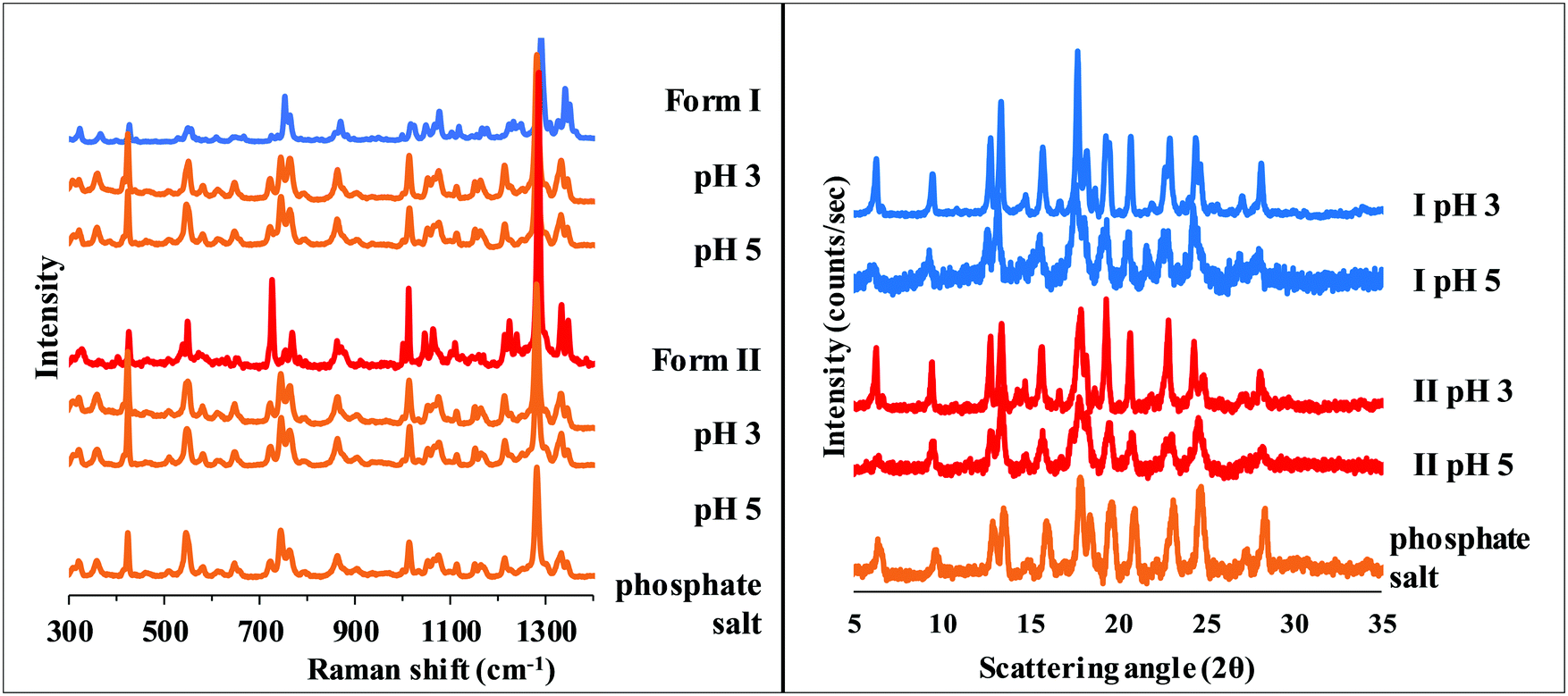 | ||
| Fig. 8 Raman spectra (left) and XRPD diffractogram (right) of carvedilol dihydrogen phosphate anhydrate and the solid phase of carvedilol polymorphs isolated from solubility suspensions from the measurements at pH 3 and 5 in BR. | ||
At pH 6.5, the two polymorphs showed different behavior at the end of the solubility measurements in the BR buffer solution. Form II formed a phosphate salt. It can be observed that the Raman spectra and the XRPD diffractogram differ from that of the original Form II and show characteristic peaks with the phosphate salt. In contrast, Raman spectra and XRPD diffractogram of the sample from the end of the solubility measurements of Form I shows a good agreement with the original form, except one peak, which can be observed between 700 and 750 cm−1 at the Raman spectra and between 17 and 19 2Θ at the XRPD diffractogram (Fig. 9), and is characteristic to the phosphate salt.
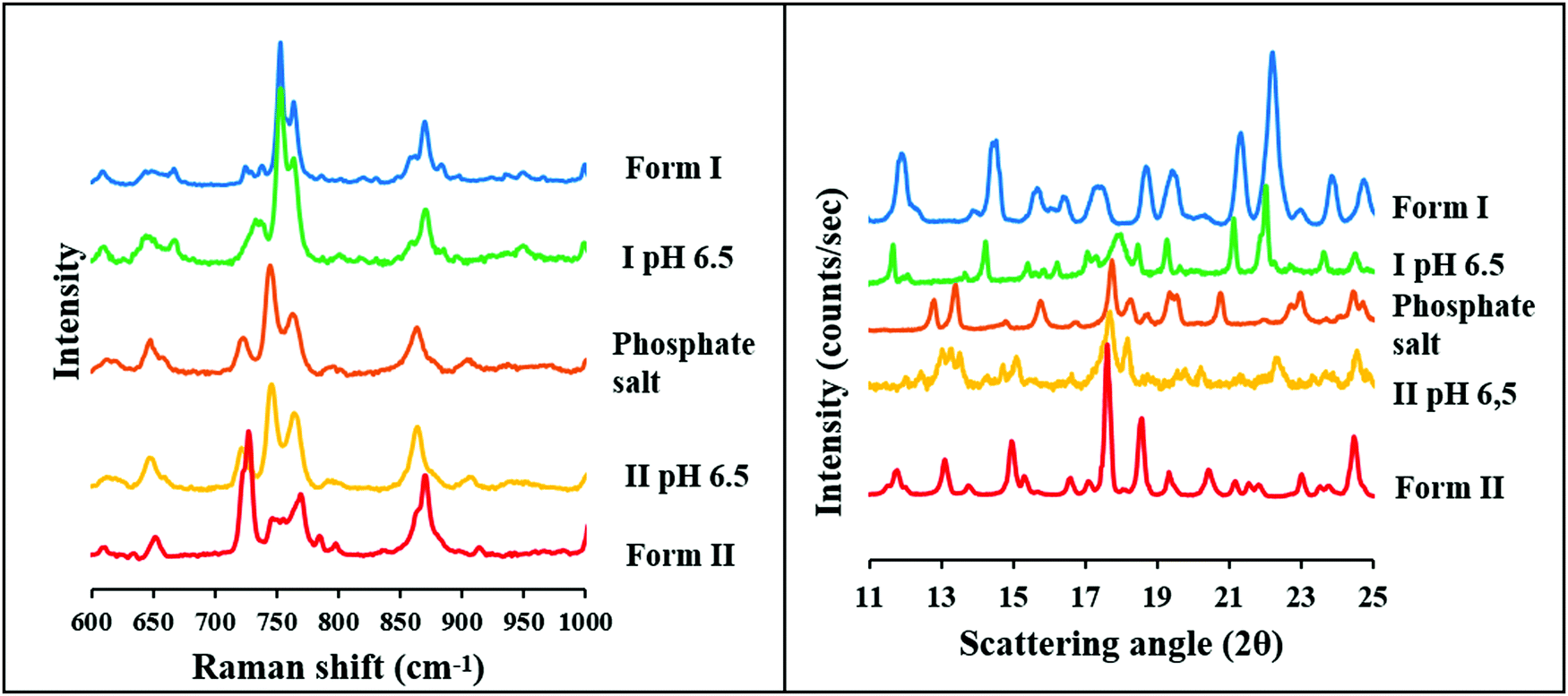 | ||
| Fig. 9 Raman spectra (left) and XRPD diffractogram (right) of carvedilol dihydrogen phosphate anhydrate, Form I, Form II and the solid phase of carvedilol polymorphs isolated from solubility suspensions from the measurements at pH 6.5 in BR. | ||
In conclusion it can be stated, based on solubility and solid-phase measurements, that the solubility of Form I and carvedilol dihydrogen phosphate anhydrate is so similar, that the original compound cannot be completely transformed into the salt at pH 6.5. It also means that the pHmax is slightly different in the case of the two polymorphs. The pHmax refers to the pH where both the phosphate salt and the carvedilol free base have equal solubilities. This phenomenon can be observed only in the BR buffer solution because in BR + KCl the solubility of the hydrochloride salt is much lower at this pH.
4. Conclusion
In this study, we have demonstrated that the poorly water-soluble carvedilol provides a good example that BCS II classified drugs may need more than 24 hours to reach their equilibrium solubility. Factors like the type of polymorph, pH, and even the buffer composition can greatly influence the equilibration time. This can explain the different equilibrium solubility values of carvedilol published in the literature. In the case of carvedilol, real-time monitoring of the dissolution was found to be essential in order to measure the solubility at the true equilibrium.The use of in situ UV probes enabled the measurement of concentration in real-time without sampling and sample preparation and provided a more in-depth knowledge of how the equilibrium is reached in case of different polymorphic forms. In most cases, Form II showed a faster dissolution and reached a higher extent of supersaturation. The faster dissolution of Form II at biorelevant pHs explains the application of the less stable polymorph in solid formulations.
Author contributions
Dóra Csicsák: investigation, formal analysis, visualization, writing – original draft. Enikő Borbás: conceptualization, supervision, writing – review & editing. Szabina Kádár: supervision, project administration, formal analysis. Petra Tőzsér: project administration, visualization. Péter Bagi: investigation, formal analysis, visualization. Hajnalka Pataki: investigation. Bálint Sinkó: supervision, project administration. Krisztina Takács-Novák: conceptualization, methodology, supervision. Gergely Völgyi: methodology, supervision, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank Dr Alex Avdeef (in-ADME Research, NY, US) for providing carvedilol literature solubility data from the Wiki-pSo Database and for the valuable discussion on the topic. Hajnalka Pataki is thankful for the János Bolyai Research Scholarship of the Hungarian Academy of Sciences. This work was financially supported by the National Research, Development, and Innovation Office of Hungary (OTKA PD-121143). The research was also supported by the ÚNKP-20-5 and ÚNKP-21-4 New National Excellence Program of the Ministry for Innovation and Technology from the source of the National Research, Development, and Innovation Fund.Notes and references
- G. L. Amidon, H. Lennernäs, V. P. Shah and J. R. Crison, Pharm. Res., 1995, 12, 413–420 CrossRef CAS PubMed.
- A. Avdeef, Absorption and Drug Development: Solubility, Permeability, and Charge State, 2012 Search PubMed.
- T. L. Threlfall, Analyst, 1995, 120, 2435–2460 RSC.
- H. G. Brittain, Polymorphism in Pharmaceutical Solids, 2nd edn, 2016 Search PubMed.
- P. A. Pangarkar, A. M. Tayade, S. G. Uttarwar and R. S. Wanare, Int. J. Pharm. Technol., 2013, 5, 2374–2402 CAS.
- R. Eyjolfsson, Pharmazie, 2002, 57, 347–348 CAS.
- D. Tempfli, E. Borbás, H. Pataki, D. Csicsák, G. Völgyi, B. Sinkó and K. Takács-Novák, Eur. J. Pharm. Sci., 2020, 149, 105328 CrossRef CAS PubMed.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Pharm. Res., 2001, 18, 859–866 CrossRef CAS PubMed.
- A. J. Aguiar, J. Krc, A. W. Kinkel and J. C. Samyn, J. Pharm. Sci., 1967, 56, 847–853 CrossRef CAS PubMed.
- E. Baka, J. E. A. Comer and K. Takács-Novák, J. Pharm. Biomed. Anal., 2008, 46, 335–341 CrossRef CAS PubMed.
- K. J. Box, G. Völgyi, E. Baka, M. Stuart, K. Takács-Novák and J. E. A. Comer, J. Pharm. Sci., 2006, 95, 1298–1307 CrossRef CAS PubMed.
- A. Avdeef, E. Fuguet, A. Llinàs, C. Ràfols, E. Bosch, G. Völgyi, T. Verbic, E. Boldyreva and K. Takács-Novák, ADMET DMPK, 2016, 4, 117–178 CrossRef.
- E. Shoghi, E. Fuguet, E. Bosch and C. Ràfols, Eur. J. Pharm. Sci., 2013, 48, 291–300 CrossRef CAS PubMed.
- A. Pobudkowska and U. Domańska, Chem. Ind. Chem. Eng. Q., 2014, 20, 115–126 CrossRef CAS.
- M. Saifee, N. Inamdar, D. L. Dhamecha and A. A. Rathi, Int. J. Health Res., 2009, 2, 291–306 CAS.
- D. McTavish, D. Campoli-Richards and E. M. Sorkin, Drugs, 1993, 45, 232–258 CrossRef CAS PubMed.
- H. Pataki, I. Csontos, Z. K. Nagy, B. Vajna, M. Molnár, L. Katona and G. Marosi, Org. Process Res. Dev., 2013, 17, 493–499 CrossRef CAS.
- H. Pataki, I. Markovits, B. Vajna, Z. K. Nagy and G. Marosi, Cryst. Growth Des., 2012, 12, 5621–5628 CrossRef CAS.
- A. G. Bubendorf, R.-D. Gabel, M. Henning, S. Krimmer, G. Neugebauer, W. Preis, A. Wirl, Eur. Pat., 2001, EP1432681B1, 2001 Search PubMed.
- Wiki-pSo Database. in-ADME Research, New York, USA.
- D. Schönherr, U. Wollatz, D. Haznar-Garbacz, U. Hanke, K. J. Box, R. Taylor, R. Ruiz, S. Beato, D. Becker and W. Weitschies, Eur. J. Pharm. Biopharm., 2015, 92, 155–170 CrossRef PubMed.
- C. A. S. Bergström, C. M. Wassvik, K. Johansson and I. Hubatsch, J. Med. Chem., 2007, 50, 5858–5862 CrossRef PubMed.
- Y. L. Hsieh, G. A. Ilevbare, B. Van Eerdenbrugh, K. J. Box, M. V. Sanchez-Felix and L. S. Taylor, Pharm. Res., 2012, 29, 2738–2753 CrossRef CAS PubMed.
- R. Hamed, A. Awadallah, S. Sunoqrot, O. Tarawneh, S. Nazzal, T. AlBaraghthi, J. Al Sayyad and A. Abbas, AAPS PharmSciTech, 2016, 17, 418–426 CrossRef CAS PubMed.
- J. Ashok Kumar, S. Ramkanth, S. Lakshmana Prabu and V. Gopal, Int. J. Curr. Pharm. Rev. Res, 2015, 6, 269–273 CrossRef.
- G. Völgyi, D. Csicsák and K. Takács-Novák, Eur. J. Pharm. Sci., 2018, 123, 98–105 CrossRef PubMed.
- V. Bijlani, D. Yuonayel, S. Katpally, B. N. Chukwumezie and M. C. Adeyeye, AAPS PharmSciTech, 2007, 8, 6–9 CrossRef PubMed.
- K. Bynum, K. Roinestad, A. Kassis, J. Pocreva, L. Gehrlein, F. Cheng and P. Palermo, Dissolution Technol., 2001, 8, 1–8 CrossRef.
- V. A. Gray, Dissolution Technol., 2003, 10, 33–36 CrossRef.
- E. Borbás, P. Tőzsér, K. Tsinman, O. Tsinman, K. Takács-Novák, G. Völgyi, B. Sinkó and Z. K. Nagy, Mol. Pharmaceutics, 2018, 15, 3308–3317 CrossRef PubMed.
- L. D. Prado, H. V. A. Rocha, J. A. L. C. Resende, G. B. Ferreira and A. M. R. De Figuereido Teixeira, CrystEngComm, 2014, 16, 3168–3179 RSC.
- M. Pudipeddi and A. T. M. Serajuddin, J. Pharm. Sci., 2005, 94, 929–939 CrossRef CAS PubMed.
- R. K. Thaper, M. D. Prabhvat, A. V. Mouneshwarachar, Y. D. Pawar, P. P. Daramwar and P. R. Upadhyay, WO2008093350A1, 2007.
- J. Hildesheim, S. Finogueev, J. Aronhime, B.-Z. Dolitzky, S. Ben-Valid, I. Kor, US Pat., US7056942B2, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nj01349a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |