
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTopologically diverse polycyclic aromatic hydrocarbons from pericyclic reactions with polyaromatic phospholes†
Réka
Mokrai
 abc,
Rózsa
Szűcs
ab,
Matthew P.
Duffy
a,
Vincent
Dorcet
a,
Thierry
Roisnel
a,
Zoltán
Benkő
*b,
László
Nyulászi
*bc,
Pierre-Antoine
Bouit
*a and
Muriel
Hissler
*a
abc,
Rózsa
Szűcs
ab,
Matthew P.
Duffy
a,
Vincent
Dorcet
a,
Thierry
Roisnel
a,
Zoltán
Benkő
*b,
László
Nyulászi
*bc,
Pierre-Antoine
Bouit
*a and
Muriel
Hissler
*a
aUniv Rennes, CNRS, ISCR - UMR 6226, F-35000 Rennes, France. E-mail: muriel.hissler@univ-rennes1.fr; pierre-antoine.bouit@univ-rennes1.fr
bDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics Szt. Gellért tér 4 H-1111 Budapest, Hungary. E-mail: zbenko@mail.bme.hu
cMTA-BME Computation Driven Chemistry Research Group. Gellért tér 4 H-1111 Budapest, Hungary. E-mail: nyulaszi@mail.bme.hu
First published on 14th April 2021
Abstract
Polycyclic Aromatic Hydrocarbons (PAHs) with planar, twisted and negatively curved topologies were obtained from polycyclic phospholes using pericyclic reactions. Deviation from planarity is due to steric interactions between the PAH core and the ester substituents. These structural effects on the optical and redox properties were studied and rationalized through DFT calculations. This synthetic approach thus allows the preparation of topologically diverse PAHs allowing fine-tuning their electronic properties, with potential applications in organic electronics.
Introduction
Polycyclic Aromatic Hydrocarbons (PAHs) are bi-dimensional π-conjugated systems consisting of sp2-hybridized carbon atoms. This particular chemical structure confers them appealing electronic properties and makes them suitable components for applications in optoelectronic devices (field-effect transistors, solar cells…).1 The properties of PAHs are intrinsically determined by their structure at the molecular level. Indeed, the optical/redox properties of these planar frameworks (such as I, Fig. 1a) can be fine-tuned by extension of their π-conjugated system, introduction of substituents or heteroatoms, or modification of their edge structures (armchair, zigzag…). | ||
| Fig. 1 (a) Representative examples of planar (I), twisted (II) and negatively curved PAHs (III). (b) Synthetic strategy devised in this article. | ||
The introduction of twist or curvature into the π-conjugated framework affording the so-called contorted PAHs, such as twisted II or negatively Gaussian curved III, (Fig. 1a)2 also appeared as an efficient way to fine-tune their electronic properties. To achieve this, two main strategies are used.3 One is the endo-skeletal approach, in which non-six membered rings are included in polybenzenoid frameworks. The other is the exo-skeletal strategy, whereby one or more cove and/or fjord regions of the fused π-system is contorted by bulky substituents.4 The development of such PAHs is highly challenging since, in the non-planar structure, the stability of the π-system has to be reduced. Thus, the formation of contorted PAHs directly depends on the efficiency of synthetic approaches to control shape, size, curvature and, therefore, to achieve well-tailored properties. In our hypothesis, the energy demand of the distortion of aromaticity can be covered by the use of weakly anti-aromatic λ5,σ4-phosphole derivatives as starting materials.5 The generation of six-membered rings stabilized by aromaticity via cycloaddition-type reactions6 provides the necessary driving force allowing to overcome the energy need of the steric repulsion. We envisage extending this approach to polycyclic phospholes7 to prepare new PAHs, whose structural characteristics will be controlled through the phosphole-based precursor (Fig. 1b). It is worth mentioning that cycloadditions have already been used to prepare PAHs.8 For example, Scott et al. selectively performed cycloadditions on the bay position of perylenes and bisanthenes using diethyl acetylenedicarboxylate.9 Heterocycles such as furanes have also been used to as diene to prepare PAHs.8 However, the rich reactivity of phospholes in cycloaddition prompt us to explore this strategy to prepare novel PAHs.
Herein, we report a selective bottom-up synthesis of topologically diverse PAHs employing pericyclic reactions with polycyclic phospholes and the investigation of their optical and redox properties based on experimental and computational studies.
Result and discussion
As a proof of concept, our strategy was first tested on the newly accessed polycyclic phosphole 1 (Scheme 1). Compound 1 was synthesized in excellent yield (88%) using Scholl conditions (AlCl3, NaCl) from binaphthyl-fused oxophosphole (see Scheme 1 for X-ray structure).10 This novel polyaromatic phosphole is fully air, moisture and temperature stable (Td10 = 363 °C). As a dienophile for the pericyclic reaction, we have chosen dimethyl acetylenedicarboxylate (DMAD). This compound provides sterically demanding ester functional groups and can allow further chemical modifications by replacing the methoxy groups with other functional units. Refluxing 1 during 24 h with a large excess of DMAD afforded the planar benzo[ghi]perylene 2 (vide infra) as a major product. These reaction conditions allowed us to introduce selectively the six-membered ring in place of the phosphole leaving untouched the bay-region on the opposite side. Based on similar reactions carried out by Mathey et al. on 3,4-dimethylphospholes,6 we assume that PAH 2 is accessed from a [4 + 2] cycloaddition between the dienic part of the P-heterocycle and the activated alkyne, followed by aromatization. Attempts to observe the elusive phosphanorbornadiene intermediate failed. 2 was fully characterized by multinuclear NMR and X-ray diffraction and our data clearly fit those reported by Hirayama et al.11 (see ESI†). Furthermore, the study of the nucleus independent chemical shift (NICS(0)) at the B3LYP/6-311 + G*//B3LYP/6-31 + G* level (see Fig. 2 and ESI†) shows that the perylene part of 1 displays a similar pattern as the perylene (P) itself and the phosphole moiety (ring A) is non-aromatic/slightly antiaromatic with a diene-like character, making this part of the molecule an ideal target for a pericyclic reaction. In contrast, the newly formed ring A together with the rings C and C′ in 2 are the most aromatic ones, forming a Clar aromaticity pattern12 (Fig. 2), providing altogether the expected driving force for the formation of PAH 2.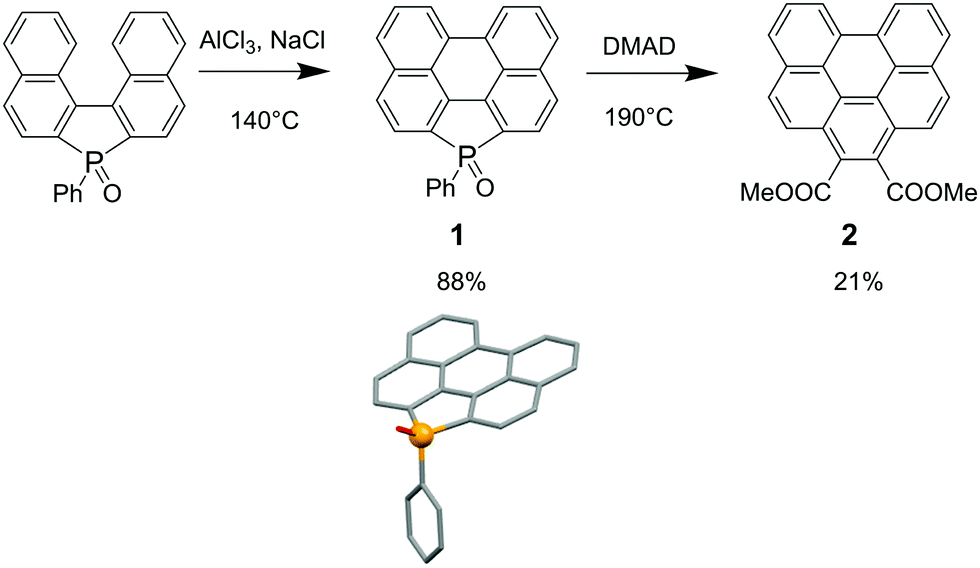 | ||
| Scheme 1 Synthesis of 2 and crystallographic structure of 1. | ||
 | ||
| Fig. 2 Visualization of the NICS(0) values (B3LYP/6-311 + G*//B3LYP/6-31 + G*) of compounds P, 1, 2 and 6–8. The NICS(0) values (which can be defined unambiguously also in the curved molecules) were presented, nevertheless the NICS(1) values show the same tendencies, see Table S18 (ESI†). | ||
To extend this approach for achieving structural diversity, we decided to engage a more sterically demanding polyaromatic thioxo-phospholes7 in pericyclic reactions with DMAD (Scheme 2). In similar conditions as for the formation of 2, polycyclic thioxophospholes 3–5, featuring respectively 4, 6 and 8 fused rings,7 were converted into Clar-type benzenoid-based PAHs 6–8 in moderate yields after purification (Scheme 2). These moderate yields come from purification difficulties since the conversion appears quantitative as observed by NMR spectroscopies (see Fig. S8b, ESI†). Following the strategy of Li et al.,13 microwave irradiation was also investigated and afforded better isolated yields in the case of 6. Importantly, all starting phospholes are fully stable in the reaction conditions in absence of dienophile (Fig S8a, ESI†). Again, the phosphanorbornadiene intermediate remained elusive.
 | ||
| Scheme 2 Synthesis of 6–8. | ||
However, as an indirect proof for the mechanism, the reaction between 3 and p-benzoquinone as a dienophile resulted in the phosphanorbornene 9 (Scheme 3 and ESI†) because the aromatization is hampered by the two sp3 carbon atoms. The newly formed PAHs can be further functionalized using simple transformations of esters, as exemplified on 6 to afford its bis-imide analogue 10 (Scheme 3 and ESI†). All compounds were unambiguously characterized by multinuclear NMR, HRMS and by X-ray diffraction performed on single crystals (vide infra).
 | ||
| Scheme 3 Synthesis of compounds 9 and 10. | ||
Importantly, the regioselectivity of the pericyclic reactions is exclusively on the dienic system of the P-ring of 1,3–5 and not on the bay position as it has been reported by Scott et al. on various PAHs.9 This clearly emphasizes the role of the P-heterocycle in the regioselectivity. It also unambiguously confirms that the P(S)Ph loss does not occur before the cycloaddition. For 6–8, the replacement of the phosphole ring by a benzenoid unit results in analogous changes as in case of 2, yielding again a Clar aromatic arrangement of the all-fused 8. Significantly, regardless of the distortion from planarity (for the similar NICS pattern of the structures calculated under planarity constraint see ESI†), the aromaticity pattern in these PAHs nicely agrees with Clar's criterion (see the rings marked with white circles in Fig. 2). This observation is further bolstered by the measured bond distances. The bonds of six-membered rings A, C and E are within the typical range of 1.38–1.42 Å, revealing benzenoid character, while in rings B and D the distances between benzenoid rings are significantly longer (1.47–1.48 Å).
Further analysis of the structural data (Fig. 3 and Table S1, ESI†) of 6 and 8 reveals that their polycyclic backbone is highly distorted in both cases in order to accommodate the two ester groups on the central phenyl ring. In addition, different curvatures are observed in these two compounds. Hence, 6 displays a twisted π-backbone with angles between the central ring (in blue, Fig. 3) and the phenyl moiety of the bay-like position (in purple, Fig. 3) of 31.4°. On the contrary, 8 possesses a negative Gaussian curvature (saddle shape) with angles between the methoxy substituted peripheral benzene ring (in green, Fig. 3) and the central ring (in blue, Fig. 3) of 25.9° and 29.4° and angles between the two central rings (blue and red) of 15.1° in the opposite direction. The structure of 7 can also be classified as negatively curved, however the X-ray data did not allow for reliable structural parameter determination (Fig. S11, ESI†). At the intermolecular level, no clear interactions are observed in the crystalline packing of 6, probably because of its highly distorted backbone. However, 8 crystallizes as isolated π-dimers (d = 3.35 Å, Fig. S15, ESI†) by overlapping of the biphenyl moieties indicating that the solid-state structure may be finally determined by the crystal packing effects.
 | ||
| Fig. 3 Crystallographic structure of 211 (top (a) and side view (b)) 6 (top (c) and side view (d)) and 8 (top (e) and side view (f)). | ||
To gain insight into the conformational stability of these compounds, we attempted to optimize further conformations from several different starting geometries at the B3LYP/6-31 + G* and B3LYP-D3/6-31 + G* levels. For 6 and 7, only the twisted and negatively curved structures, respectively, were found, which are similar to the ones obtained from the X-ray diffraction. For 8, apart from the negatively curved structure, a twisted isomer was also obtained, and the energy difference between the two conformers is only 0.5 kcal mol−1 (Table S3 and Fig. S32–S33, ESI†). We have also calculated the energy of planarization, by optimizing the PAH part under planarity constraint. For 6, 7 and 8, we have obtained the non-negligible 9.6 kcal mol−1, 10.4 kcal mol−1 and 11.6 kcal mol−1 planarization energies, respectively. Altogether, these data indicate the flexibility of the molecule in the gas phase and likely in solution. Importantly the gas phase structures clearly show significant deviation from planarity, which indicates that the curvature of these molecules is not caused by the crystal packing, but indeed results from the steric repulsion of the substituents.
The analysis of the X-ray structures thus revealed that our synthetic methodology can lead to the formation of planar, twisted or negatively curved PAHs in the solid-state, depending on the nature of the substituents. In precursor 3–5, the five-membered ring with a pyramidal phosphorus atom does not perturb the framework of the sp2-carbon atoms. On the contrary, the introduction of a six-membered ring functionalized with ester groups introduces a torsion of the carbon domain depending on the steric extent of the environment around the bay regions, which had accommodated the phosphorus-centered moiety.
To establish structure–property relationships for these structurally versatile PAHs 2 and 6–8, their UV-vis absorption and fluorescence spectra were recorded in DCM (Fig. 4) at room temperature and their cyclic voltammetry was measured in DCM using Bu4NPF6 as the electrolyte (Table 1). Furthermore, TD-DFT calculations were performed at the B3LYP/6-31 + G* level of theory. The replacement of the phosphole ring, known to have a stabilized LUMO level, by a benzenoid unit strongly influences the redox properties. While the oxidation potentials remain almost unchanged for 1 and 2, the reduction potential decreases significantly for 2, characteristic of the formation of the Clar aromatic pattern. 6 displays irreversible oxidation and reduction processes (Table 1) at rather high potentials. The extension of the π-systems in the series 6–8 leads to a decrease of the oxidation potentials and an increase of the reduction potentials (Table 1). The stability of the reduced and oxidized forms also increases since the redox potentials of 8 become reversible. Accordingly, the LUMO and HOMO levels decrease and increase, respectively (Table 1).
 | ||
| Fig. 4 UV-vis absorption (top) and emission (down) of 2 (yellow), 6 (blue), 7 (red) and 8 (green) in diluted DCM. (all compounds are excited at λmax except 2 (λexc = 386 nm)). | ||
| λ abs (nm) | log ε | λ em (nm) | Φb % | E ox (V) | E red (V) | HOMOe (eV) | LUMOe (eV) | |
|---|---|---|---|---|---|---|---|---|
| a In CH2Cl2 (10−5M). Calculated values shown in parentheses (TD-B3LYP/6-31+G*). b Measured relative to quinine sulfate (H2SO4, 0.1 M), ϕ = 55%. c In CH2Cl2 with Bu4N+PF6− (0.2 M) at a scan rate of 100 mVs−1. Eox(Ered) = Epc (Epa). Potentials vs ferrocene/ferrocenium. d Quasi-reversible processes. e At the B3LYP/6-31 + G* level. f Negatively curved structure. g Twisted structure. | ||||||||
| 1 | 486 | 4.2 | 509 | 69 | +0.83d | −1.80d | −5.53 | −2.66 |
| (424) | (575) | |||||||
| 2 11 | 386, 424 | 4.1, 3.3 | 463 | 64 | +0.87d | −2.25d | −5.65 | −2.19 |
| 6 | 277 | 4.3 | 411 | <1% | +1.15 | −2.38 | −5.98 | −1.68 |
| (310) | (472) | |||||||
| 7 | 325, 374 | 4.5, 3.9 | 496 | 6 | +0.85 | −2.28 | −5.62 | −1.85 |
| (334, 378) | (602) | |||||||
| 8 | 341, 385 | 4.3, 3.7 | 505 | 15 | +0.67 | −2.04d | −5.41f | −1.96f |
| (347, 415)e | (521)f | −5.43g | −1.78g | |||||
| (345, 399)f | (426)g | |||||||
The absorption spectrum of the planar phosphole-based PAH 1 shows structured transitions (λabsmax = 486 nm) belonging to the HOMO → LUMO π–π* transition and corresponding to vibrational fine structures characteristic of polyaromatic molecules (Fig. S35, ESI†). In DCM, 1 shows intense and structured emission which is nearly the perfect mirror image of the absorption spectrum with a respective maximum at 509 nm (Fig. S16, ESI†). The insertion of the six–membered ring in place of the phosphole ring resulting in 2, induces a blue shift of the absorption bands. The spectrum itself, (as previously described)11 is dominated by a 386 nm structured band, accompanied by a low-intensity band centered at 424 nm. Both ADC(2) and TD-B3LYP calculations reproduce this intensity pattern. According to the TD-DFT calculations, the 424 nm (λcalc= 398 nm) band belongs mainly to the HOMO → LUMO+1 transition, while the more intense band at 386 nm (λcalc= 392 nm) is the HOMO → LUMO transition. Both bands are mainly π–π* transitions since HOMO, LUMO and LUMO+1 are localized at the planar PAH framework. However, LUMO+1 has a significant contribution of the rather floppy ester functionalities (Fig. S26, ESI†) leading to the presence of the vibronic structure only at the 386 nm band.
6–8 also exhibit significantly blue-shifted absorption bands compared to their phosphole precursors, which are in agreement with the increasing aromaticity upon replacement of the phosphorus-based unit by a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C fragment. Furthermore, no vibrational fine structures in the absorption bands characteristic for planar PAH appear for 6–8 exhibiting contorted backbone (Fig. 4). The absorption spectra of the PAHs 6, 7 and 8 show a gradual bathochromic shift in the series in accordance with the increase in π-conjugation and the above discussed variation of their FMO energies. Furthermore, TD-DFT calculations (Tables S8, S10 and S12, ESI†) show that the tailing features of these spectra can be explained by low energy transitions of rather low intensity (two excitations for 6 and three for 7 and 8). In all these transitions, several electron configurations are mixed (Tables S8, S10 and S12), but the HOMO → LUMO being the main contributor for the first excitations of 7 and 8. The curvature of the molecules and the presence of flexible substituents lead to the lack of vibronic structures. Furthermore, in case of 8, the possible contribution from the twisted form in the solution acts also broadening.
C fragment. Furthermore, no vibrational fine structures in the absorption bands characteristic for planar PAH appear for 6–8 exhibiting contorted backbone (Fig. 4). The absorption spectra of the PAHs 6, 7 and 8 show a gradual bathochromic shift in the series in accordance with the increase in π-conjugation and the above discussed variation of their FMO energies. Furthermore, TD-DFT calculations (Tables S8, S10 and S12, ESI†) show that the tailing features of these spectra can be explained by low energy transitions of rather low intensity (two excitations for 6 and three for 7 and 8). In all these transitions, several electron configurations are mixed (Tables S8, S10 and S12), but the HOMO → LUMO being the main contributor for the first excitations of 7 and 8. The curvature of the molecules and the presence of flexible substituents lead to the lack of vibronic structures. Furthermore, in case of 8, the possible contribution from the twisted form in the solution acts also broadening.
All these compounds (1, 2, 6–8) are fluorescent in solution. A bathochromic shift of emission wavelengths is observed in the series 6–8, together with an increase in the fluorescence quantum yield. The low fluorescence quantum yield of 6 is in accordance with the low oscillator strength of the absorption and the flexibility of the molecule. Although their quantum yield does not reach the high value of the perfectly planar 2,11 going from the poorly luminescent 6 to the good fluorophore 8, the rigidity of their structure increases alongside with their quantum yield.
Conclusion
In conclusion, we described the use of pericyclic reactions from polycyclic phospholes to prepare planar 2, twisted 6 and negatively curved 7 and 8 PAHs. Notably, the planar framework of the phosphole starting materials can be converted into contorted PAHs. This can be attributed to two steric factors (i) introduction of a six-membered ring in place of a five-membered ring, (ii) sterically demanding ester functionalities. The impact of these structural modifications on the UV-vis absorption/emission, redox properties and aromaticity were studied experimentally and computationally. The introduction of these novel PAHs in electronic devices such as a field-effect transistor is currently under investigation.Experimental section
General. All experiments were performed under an atmosphere of dry argon using standard Schlenk techniques. Commercially available reagents were used as received without further purification. Solvents were freshly purified using MBRAUN SPS-800 drying columns. Separations were performed by gravity column chromatography on basic alumina (Aldrich, Type 5016A, 150 mesh, 58 Å) or silica gel (Merck Geduran 60, 0.063–0.200 mm). General procedure for purification with recycling preparative HPLC: Compounds were solubilized in HPLC grade chloroform (stabilized with ethanol). Prior to injection, the solution was filtered through a 0.45 μm PTFE filter. Purification was performed on a LC-9160 II NEXT system from the Japan Analytical Industry Co., Ltd. (JAI) equipped with coupled UV-vis 4C h NEXT through a set of two JAIGEL-2H columns at an elution rate of 10 mL min−1 (CHCl3).Reactions under microwave irradiations were realized in the Anton Paar Monowave 300® microwave reactor (Anton Paar France) of the S2 Wave platform, ScanMAT UMS CNRS 2001, Rennes. Microwave irradiation reactions were realized in borosilicate glass vials of 10 ml equipped with snap caps (at the end of the irradiation, cooling reaction was realized by compressed air) with the Monowave® 300 reactor. The microwave instrument consists of a continuous focused microwave power output from 0 to 800W for the Anton Paar Monowave 300® apparatus. All the experiments in this microwave reactor were performed using stirring option. The target temperature was reached with a ramp of 2–5 minutes and the chosen microwave power stayed constant to hold the mixture at this temperature. The reaction temperature was monitored using calibrated infrared sensor and the reaction time included the ramp period. The microwave irradiation parameters (power and temperature) were monitored by the internal software package for the Monowave® 300 Anton–Paar. All the other reactions were, carried out in closed Schlenk and the temperature indicated were measured inside the oil bath.
1H, 13C, and 31P NMR spectra were recorded on Bruker AV III 400 MHz NMR and 300 MHz spectrometers equiped with BBO or BBFO probeheads. Assignment of proton and carbon atoms is based on COSY, NOESY, edited-HSQC and HMBC experiments. 1H and 13C NMR chemical shifts were reported in parts per million (ppm) using residual solvent signal as reference. High-resolution mass spectra were obtained on a Varian MAT 311 or ZabSpec TOF Micromass instrument at Scanmat-UMS 2001, University of Rennes 1 and Maxis 4G. Elemental analyses were performed by Scanmat-UMS 2001, University of Rennes 1. Compounds 2–5 and 7-phenyldinaphtho[2,1-6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1′,2′-d]oxophosphole were synthesized according to published procedures.7,10 UV-Visible spectra were recorded at room temperature on a JASCO V-630 spectrophotometer. The UV-vis-NIR emission and excitation spectra measurements were recorded on a FL 920 Edimburgh Instrument equipped with a Hamamatsu R5509-73 photomultiplier for the NIR domain (300–1700 nm) and corrected for the response of the photomultiplier. Quantum yields were calculated relative to quinine sulfate (ϕ = 0.55 in H2SO4 0.1M). The electrochemical studies were carried out under argon using an Eco Chemie Autolab PGSTAT 30 potentiostat for cyclic voltammetry with the three-electrode configuration: the working electrode was a platinum disk, the reference electrode was a saturated calomel electrode and the counter-electrode was a platinum wire. All potentials were internally referenced to the ferrocene/ferrocenium couple. For the measurements, concentrations of 10−3 M of the electroactive species were used in freshly distilled and degassed dichloromethane and 0.2 M tetrabutylammonium hexafluorophosphate.
:1′,2′-d]oxophosphole were synthesized according to published procedures.7,10 UV-Visible spectra were recorded at room temperature on a JASCO V-630 spectrophotometer. The UV-vis-NIR emission and excitation spectra measurements were recorded on a FL 920 Edimburgh Instrument equipped with a Hamamatsu R5509-73 photomultiplier for the NIR domain (300–1700 nm) and corrected for the response of the photomultiplier. Quantum yields were calculated relative to quinine sulfate (ϕ = 0.55 in H2SO4 0.1M). The electrochemical studies were carried out under argon using an Eco Chemie Autolab PGSTAT 30 potentiostat for cyclic voltammetry with the three-electrode configuration: the working electrode was a platinum disk, the reference electrode was a saturated calomel electrode and the counter-electrode was a platinum wire. All potentials were internally referenced to the ferrocene/ferrocenium couple. For the measurements, concentrations of 10−3 M of the electroactive species were used in freshly distilled and degassed dichloromethane and 0.2 M tetrabutylammonium hexafluorophosphate.
Synthesis of 7-Phenyldinaphtho[2,1-6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1′,2′-d]oxophosphole
:1′,2′-d]oxophosphole
2,2′-Dibromo-1,1′-binaphthyl (1 g, 2.43 mmol) is dissolved in 70 mL of dry THF. 2.1 eq. of n-Buli (1.9 M in hexane) are slowly added at −80 °C. The solution is stirred for 1h at −80 °C and 1.3 eq of dichlorophenylphosphine are added. The solution is stirred at room temperature overnight and the solvent are evaporated. The crude mixture is dissolved in DCM (30 mL) and 1 mL of hydrogen peroxide 30% are added. After 2h of stirring, the solution is extracted with water. The solution is dried over anhydrous MgSO4 and the solvent is evaporated. The crude is purified with silica gel chromatography (dichloromethane/acetone, v/v 100/0 → 85/15) to afford 7-phenyldinaphtho[2,1-6:1′,2′-d]oxophosphole (550 mg, 60%). NMR data fit to the reported ones.10
Synthesis of 1
7-Phenyldinaphtho[2,1-6:1′,2′-d]oxophosphole (0.402 g, 1.068 mmol), aluminium chloride (1.709 g. 12.82 mmol, 10 eq) and sodium chloride (0.375 g, 6.409 mmol, 6 eq) are heated to 140 °C in an oil bath and stirred for 2.5 hours. The mixture is cooled to room temperature, then to 0 °C. Some 10% aqueous HCl solution is added and the mixture is stirred for 30 minutes. The crude is extracted with dichloromethane. The solution is dried over anhydrous MgSO4, the solvent is evaporated. The crude mixture is reacted with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (0.485 g, 2.136 mmol, 2 eq) in 160 ml of toluene. The reaction mixture is heated to 60 °C in an oil bath and stirred for 2.5 hours. The reaction is quenched with saturated NaHCO3 aqueous solution and extracted with saturated NaHCO3 solution then water. The solution is dried over anhydrous MgSO4 and the solvent is evaporated. The crude is purified with silica gel chromatography (dichloromethane/acetone, v/v 100/0 → 90/10) to afford 1 as an orange solid (349.6 mg, 88%). 1H (400 MHz, CD2Cl2): δ 8.14 (d, 2H, 3J = 7 Hz, CH); 7.79–7.88 (m, 2H, CH); 7.67–7.74 (m, 4H, CH); 7.54–7.65 (m, 5H, CH); 7.40–7.47 (m, 2H, CH). 13C{1H} NMR (100 MHz, CD2Cl2): δ 138.4 (d, J(P, C) = 24 Hz, Cq); 136.4 (d, J(P, C) = 2 Hz, Cq); 132.4 (d, J(P, C) = 2 Hz, CH); 131.9 (Cq); 130.9 (d, J(P, C) = 11 Hz, CH); 130.0 (d, J(P, C) = 100 Hz, Cq); 129.6 (CH); 128.9 (d, J(P, C) = 12 Hz, CH); 128.4 (d, J(P, C) = 11 Hz, CH); 127.8 (d, J(P, C) = 11 Hz, Cq); 127.7 (CH); 126.1 (d, J(P, C) = 11 Hz, Cq); 125.1 (d, J(P, C) = 11 Hz, CH); 121.9 (CH). 31P{1H}{13C} (162 MHz, CD2Cl2): δ +37.1 (s). Anal. Calcd. for C26H15OP: (C, 83.41, H, 4.04); Found: C, 80.91, H, 4.36. HR MS (ESI, CH2Cl2/MeOH, 1/9, m/z): [M + Na]+ calcd for C26H15ONaP: 397.0753, Found: 397.0755.
Synthesis of 2
1 (0.050 g, 0.134 mmol) and dimethyl acetylenedicarboxylate (0.835 g, 5.882 mmol, 44 eq) are stirred at 190 °C in an oil bath for 24 hours. The mixture is cooled to room temperature and the crude is purified with silica gel chromatography (dichloromethane) to afford 2 as a yellow solid (10.8 mg, 21%). 1H NMR (300 MHz, CD2Cl2): δ 9.10 (d, 2H, 3J = 7 Hz, CH); 8.42 (d, 2H, 3J = 9 Hz, CH); 8.19–8.32 (m, 4H, CH); 8.14 (t, 2H, 3J = 8 Hz, CH); 4.17 (s, 6H, CH3). 13C{1H} NMR (75 MHz, CD2Cl2): δ 168.7 (CO); 132.2 (Cq); 130.0 (Cq); 129.0 (CH); 128.0 (Cq); 127.5 (CH); 127.1 (CH); 125.4 (Cq); 125.2 (Cq); 124.8 (Cq); 124.3 (CH); 121.5 (CH); 53.0 (CH3). HR MS (ESI, CH2Cl2/MeOH, 1/9, m/z): [M + Na]+ calcd for C26H16O8Na: 415.0941, Found: 415.0941.
Synthesis of dimethyl 1,4-bis(3,5-dimethoxyphenyl)triphenylene-2,3-dicarboxylate (6)
1,3-Bis(3,5-dimethoxyphenyl)-2-phenyldibenzo [e,g] isophosphindole 2-sulfide (3) (50 mg, 0.084 mmol) and dimethyl acetylenedicarboxylate (231 mg, 1.629 mmol, 20 eq) are dissolved in 2.6 ml of chlorobenzene and placed in the Anton Paar Monowave 300® microwave cavity. The stirred mixture was irradiated at 190 °C for 40 min. The solvent is then evaporated. The product is purified with silica gel chromatography (n-heptane/ethyl acetate, v/v 90/10 → 75/25) to afford 6 as a white solid (29 mg, 58%). 1H NMR (400 MHz, CD2Cl2,): δ 8.49 (dd, 2H, 3J = 7 Hz, 4J = 1.2 Hz, CH); 7.78 (dd, 2H, 3J = 7 Hz, 4J = 1.2 Hz, CH); 7.54 (ddd, 2H, 3J = 7Hz, 3J = 7 Hz, 4J = 1.2 Hz, CH); 7.18 (ddd, 2H, 3J = 7Hz, 3J = 7 Hz, 4J = 1.2 Hz, CH); 6.55 (m, 6H, CH); 3.76 (s, 12H, CH3); 3.65 (s, 6H, CH3). 13C{1H} NMR (100 MHz, CD2Cl2): δ 168.9 (CO); 161.4 (C-OMe); 143.0 (Cq); 136.0 (ql); 132.6 (Cq); 131.8 (Cq); 131.1 (Cq); 129.6 (CH); 129.5 (Cq); 127.6 (CH); 125.8 (CH); 123.2 (CH); 108.1 (CH); 100.1 (CH); 55.4 (OCH3); 52.4 (OCH3). HR MS (ESI, CH2Cl2/MeOH, 1/9, m/z): [M + Na]+ calcd for C38H32O8Na: 639.19894, Found: 639.1991. Anal. Calcd. for C38H32O8 (C, 74.0, H, 5.2); Found: C, 72.5, H, 5.1.
Synthesis of 7
4 (0.100 g, 0.163 mmol) and dimethyl acetylenedicarboxylate (0.232 g, 1.634 mmol, 10 eq) are dissolved in 5 ml of chlorobenzene. The mixture is stirred at 160 °C in an oil bath for 4 days. The solvent is evaporated and the crude is purified with silica gel chromatography (n-heptane/ethyl acetate, v/v 90/10 → 75/25) and SEC HPLC with CHCl3 eluent to afford 7 as a yellow solid (65 mg, 65%). 1H NMR (300 MHz, CDCl3,): δ 9.51 (d, 1H, 3J = 7 Hz, CH); 8.69 (d, 1H, 3J = 8 Hz, CH); 8.60 (d, 1H, 3J = 8 Hz, CH); 7.99 (t, 1H, 3J = 8 Hz, CH); 7.88 (d, 1H, 3J = 8 Hz, CH); 7.56 (t, 1H, 3J = 2 Hz, CH); 7.40 (d, 1H, 4J = 2 Hz, CH); 7.17 (dd, 1H, 3J = 7 Hz, 4J = 7 Hz, CH); 6.88 (d, 1H, 4J = 2 Hz, CH); 6.64 (d, 2H, 4J = 2 Hz, CH); 6.57 (t, 1H, 4J = 2 Hz, CH); 4.14 (s, 3H, CH3); 3.96 (s, 3H, CH3); 3.86 (s, 3H, CH3); 3.77 (s, 6H, CH3); 3.70 (s, 3H, CH3). 13C{1H} NMR (75 MHz, CDCl3): δ 171.3 (CO); 169.1 (CO); 160.8 (C–O); 159.2 (C–O); 158.7 (C–O); 142.1 (Cq); 135.1 (Cq); 133.8 (Cq); 132.4 (Cq); 132.2 (Cq); 129.7 (CH); 129.0 (Cq); 128.8 (Cq); 128.7 (Cq); 128.3 (Cq); 128.1 (Cq); 127.8 (CH); 127.2 (CH); 127.0 (CH); 126.7 (Cq); 125.6 (CH); 123.8 (Cq); 123.7 (CH); 120.6 (CH); 115.5 (Cq); 109.0 (CH); 102.3 (CH); 100.8 (CH); 100.7 (CH); 56.0 (OCH3); 55.5 (OCH3); 53.0 (OCH3); 52.4 (OCH3). 1 Cq is missing due to overlapping. HR MS (ESI, CH2Cl2/MeOH, 1/9, m/z): [M + Na]+ calcd for C38H30O8Na: 637.1833, Found: 637.1838.
Synthesis of 8
(5) (43 mg, 0.070 mmol) and dimethyl acetylenedicarboxylate (99 mg, 0.70 mmol, 10 eq) in chlorobenzene (4 ml) are heated at 150 °C in an oil bath for 2 days. Solvent is evaporated. The product is purified with silica gel chromatography (n-heptane/ethyl acetate, v/v 90/10→ 80/20) to afford 8 as a yellow solid (21 mg, 48%). 1H NMR (400 MHz, CDCl3,): δ 9.78 (d, 2H, 3J = 7Hz, CH); 8.96 (d, 2H, 3J = 7Hz, CH); 8.07 (dd, 2H, 3J = 7Hz, 3J = 7 Hz, CH); 7.86 (d, 2H, 4J = 2 Hz, CH); 6.92 (d, 2H, 4J = 2 Hz, CH); 4.19 (s, 6H, CH3); 4.05 (s, 6H, CH3); 3.92 (s, 6H, CH3). 13C{1H} NMR (100 MHz, CDCl3): δ 171.3 (CO); 159.8 (Cq–O); 158.9 (Cq–O); 133.1 (Cq); 129.9 (Cq); 129.7 (Cq); 128.0(Cq); 127.4 (CH); 126.7 (CH); 126.1 (Cq); 125.5 (Cq); 122.9 (Cq); 121.2 (CH); 115.9 (Cq); 102.3 (CH); 100.8 (CH); 56.0 (OCH3); 55.7 (OCH3); 53.2 (OCH3). HR MS (ESI, CH2Cl2/MeOH, 1/9, m/z): [M + Na]+ calcd for C38H28O8Na: 635.1676, Found: 635.1680.
Synthesis of 9
3 (100 mg, 0.163 mmol) and 1,4-Benzoquinone (18 mg, 0.163 mmol, 1 eq) in chlorobenzene are heated 30h at 150 °C in an oil bath. The solvent is then evaporated. The product is purified with silica gel chromatography (dichloromethane/ethanol, v/v 99,8/0,2) to afford 9 as a yellow solid (62 mg, 53%). 1H NMR (300 MHz, CDCl3): δ 8,79 (d, 2H, 3J = 8.2 Hz, CH); 7.7–7.6 (m, 4H, CH); 7.44 (m, 2H, CH); 7.33 (m, 3H, CH); 7.05 (m, 2H, CH); 6.38 (m, 2H, CH); 5.82 (s, 2H, CH); 5.76 (m, 2H, CH); 5.34 (d, 2H, 3J (H,P) = 2.1 Hz, CH); 5.32 (s, 2H, CH); 3.94 (s, 6H, CH3); 2.87 (s, 6H, OCH3). 13C{1H} NMR (75 MHz, CDCl3): δ 195.8 (d, J(P, C) = 13.8 Hz, CO); 160.4 (d, J(P, C) = 2.7 Hz, C–O); 159.1 (d, J(P, C) = 3.4 Hz, C–O); 141.5 (d, J(P, C) = 2.8 Hz, CH); 134.3 (d, J(P, C) = 5.8 Hz, Cq); 133.7 (d, J(P, C) = 8.1 Hz, Cq); 132.4 (d, J(P, C) = 3.0 Hz, CH); 131.4 (Cq); 128.3 (d, J(P, C) = 11.6 Hz, CH); 127.9 (CH); 127.5 (CH); 126.9 (d, J(P, C) = 6.9 Hz, Cq); 126.1 (CH); 125.1 (Cq); 123.1 (CH); 109.1 (d, J(P, C) = 5.3 Hz, CH); 106.6 (d, J(P, C) = 4.8 Hz, CH); 100.5 (d, J(P, C) = 3.1 Hz CH); 66.6 (d, J(P, C) = 45.1 Hz); 55.4 (CH3); 54.6 (CH3); 52.0 (d, J(P, C) = 13.0 Hz, CH). 31P{1H}{13C} NMR (121 MHz, CDCl3): δ 109.3 (s). 1 C signal is missing due to overlapping. HR MS (ESI, CH2Cl2/MeOH, 9/1, m/z): [M + Na]+ calcd for C44H35O6NaPS: 745.1784, Found: 745.1787.
Synthesis of 10
6 (0.119 g, 0.193 mmol), 3-Aminopentane (0.197 g, 2.268 mmol, 11.7 eq), imidazole (5.17 g, 75.982 mmol, 392 eq) and 20 ml dihlorobenzene are stirred at 195 °C in an oil bath for 18 hours. The mixture is cooled to room temperature and some methanol is added. All of the volatiles are evaporated and the solid is dissolved in methanol, 10% aqueous HCl solution is added. The mixture is filtrated and the solid compound from the filter paper is dissolved in dichloromethane. The crude is purified with silica gel chromatography (dichloromethane) to afford 10 as yellow solid (76.3 mg, 62%). 1H NMR (300 MHz, CDCl3): δ 8.45 (d, 2H, 3J = 7 Hz, CH); 7.93 (dd, 2H, 3J = 8 Hz, 4J = 1 Hz, CH); 7.53 (td, 2H, 3J = 8 Hz, 4J = 1 Hz, CH); 7.17 (ddd, 2H, 3J = 7 Hz, 4J = 1 Hz, CH); 6.61 (s, 6H, CH); 3.99–4.12 (m, 1H, CH); 3.78 (s, 12H, CH3); 1.96–2.17 (m, 2H, CH2); 1.67–1.85(m, 2H, CH2); 0.91 (t, 6H, 3J = 7 Hz, CH3). 13C{1H} NMR (75 MHz, CDCl3): 167.1 (CO); 160.8 (C–O); 139.8 (Cq); 137.0 (Cq); 136.3 (Cq); 132.0 (Cq); 130.0 (Cq);. 129.7 (CH); 128.1 (CH); 126.2 (Cq); 126.1 (CH); 123.2 (CH); 109.1 (CH); 100.5 (CH); 55.4 (CH3); 25.1 (CH2); 11.3 (CH3); HR MS (ESI, CH2Cl2/MeOH, 1/9, m/z): [M + Na]+ calcd for C41H37NO6Na: 662.2531, Found: 662.2512. Anal. Calcd. for C41H37NO6 (C, 77.0, H, 5.8); Found: C, 73.9, H, 5.8.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the MESR, the CNRS, the Région Bretagne, Campus France, ANR (ANR Heterographene ANR-16-CE05-0003-01), PD 116329, Varga József Alapítvány, Pro Progressio Alapítvány, Tempus Közalapítvány, a János Bolyai Research Fellowship, an ÚNKP-20-5-BME-317 grant, TÉT_16-1-2016-0128, an NRDI Fund (TKP2020 IES,Grant No. BME-IE-NAT), PICS SmartPAH (08062)-MTA NKM-44/2019, China-French AIL in “Functional Organophosphorus Materials”. L. Scott and K. Itami are thanked for providing the cdx file of III.Notes and references
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef CAS PubMed; L. Chen, Y. Hernandez, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2012, 51, 7640 CrossRef PubMed; M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479 CrossRef PubMed.
- M. A. Majewski and M. Stępień, Angew. Chem., Int. Ed., 2019, 58, 86 CrossRef CAS PubMed; K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739 CrossRef PubMed; J. M. Fernández-García, P. J. Evans, S. Medina Rivero, I. Fernández, D. García-Fresnadillo, J. Perles, J. Casado and N. Martín, J. Am. Chem. Soc., 2018, 140, 17188 CrossRef PubMed; S. Ho Pun and Q. Miao, Acc. Chem. Res., 2018, 51, 1630 CrossRef PubMed; F. H. Herbstein, Acta Crystallogr., 1979, B35, 1661 CrossRef; R. R Kimura, H. Kuramochi, P. Liu, T. Yamakado, T. Osuka, T. Tahara and S. Saito, Angew. Chem., Int. Ed., 2020, 59, 16430–16435 CrossRef PubMed; T. Liu, J. Yang, F. Geyer, F. S. Conrad-Burton, R. Hernandez Sanchez, H. Li, X. Zhu, C. P. Nuckolls, M. L. Steigerwald and S. Xiao, Angew. Chem., Int. Ed., 2020, 59, 14303–14307 CrossRef PubMed.
- T. Fujikawa, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2016, 138, 3587 CrossRef CAS PubMed.
- R. A. Pascal, Chem. Rev., 2006, 106, 4809 CrossRef CAS PubMed; M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267 CrossRef PubMed.
- L. Nyulászi, O. Hollóczki, C. Lescop, M. Hissler and R. Réau, Org. Biomol. Chem., 2006, 4, 996 RSC.
- A. Marinetti, F. Mathey, J. Fischer and A. Mitschler, J. Chem. Soc., Chem. Commun., 1982, 667 RSC; A. Marinetti and F. Mathey, J. Am. Chem. Soc., 1982, 104, 4484 CrossRef CAS; T. Moller, P. Wonneberger, N. Kretzschmar and E. Hey-Hawkins, Chem. Commun., 2014, 50, 5826 RSC.
- P.-A. Bouit, A. Escande, R. Szűcs, D. Szieberth, C. Lescop, L. Nyulászi, M. Hissler and R. Réau, J. Am. Chem. Soc., 2012, 134, 6524 CrossRef CAS PubMed; F. Riobé, R. Szűcs, P.-A. Bouit, D. Tondelier, B. Geffroy, F. Aparicio, J. Buendía, L. Sánchez, R. Réau, L. Nyulászi and M. Hissler, Chem. – Eur. J., 2015, 21, 6547 CrossRef PubMed; R. Szűcs, P.-A. Bouit, L. Nyulászi and M. Hissler, ChemPhysChem, 2017, 18, 2618 CrossRef PubMed.
- For a review on pericyclic reactions toward PAH, see: O. T. Dyan, G. I. Borodkin and P. A. Zaikin, Eur. J. Org. Chem., 2019, 7271–7306 CrossRef CAS Other example: B. A. Alameddine, R. S. Anju, F. Al-Sagheer and T. A. Jenny, New J. Chem., 2016, 40, 10363 RSC.
- E. H. Fort, P. M. Donovan and L. T. Scott, J. Am. Chem. Soc., 2009, 131, 16006 CrossRef CAS PubMed.
- S. Gladiali, A. Dore, D. Fabbri, O. De Lucchi and G. Valle, J. Org. Chem., 1994, 59, 6363 CrossRef CAS.
- S. Hirayama, H. Sakai, Y. Araki, M. Tanaka, M. Imakawa, T. Wada, T. Takenobu and T. Hasobe, Chem. – Eur. J., 2014, 20, 9081 CrossRef CAS PubMed.
- E. Clar, Polycyclic Hydrocarbons, Springer, 1964 Search PubMed; E. Clar, The aromatic sextet, Wiley, 1972 Search PubMed.
- B. Yang, Y. Li and M. G. Xie, Chin. Chem. Lett., 2003, 14, 783–785 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1973069, 1895041, 1956170, 1895043, 1895042 and 1973065. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1nj01194d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |