
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electro-oxidative depolymerisation of technical lignin in water using platinum, nickel oxide hydroxide and graphite electrodes
Nicola
Di Fidio
 a,
Johan W.
Timmermans
b,
Claudia
Antonetti
a,
Anna Maria
Raspolli Galletti
a,
Richard J. A.
Gosselink
b,
Roel J. M.
Bisselink
*b and
Ted M.
Slaghek
*b
a,
Johan W.
Timmermans
b,
Claudia
Antonetti
a,
Anna Maria
Raspolli Galletti
a,
Richard J. A.
Gosselink
b,
Roel J. M.
Bisselink
*b and
Ted M.
Slaghek
*b
aDepartment of Chemistry and Industrial Chemistry, University of Pisa, Via G. Moruzzi 13, 56124 Pisa, Italy
bWageningen Food and Biobased Research, Wageningen University & Research, Bornse Weilanden 9, 6708 WG Wageningen, The Netherlands. E-mail: roel.bisselink@wur.nl; ted.slaghek@wur.nl; Tel: +31 317483871 Tel: +31 317481977
First published on 27th April 2021
Abstract
In order to improve the lignin exploitation to added-value bioproducts, a mild chemical conversion route based on electrochemistry was investigated. For the first time, soda lignin Protobind™ 1000 (technical lignin from the pulp & paper industry) was studied by cyclic voltammetry to preliminarily investigate the effect of the main reaction parameters, such as the type of electrode material (platinum, nickel oxide hydroxide, graphite), the pH (12, 13, 14), the scan rate (10, 50, 100, 250 mV s−1), the substrate concentration (2, 20 g L−1) and the oxidation/reduction potential (from −0.8 to +0.8 V). Under the optimal reaction conditions among those tested (NiOOH electrode, pH 14, lignin 20 g L−1, 0.4 V), the electro-oxidative depolymerisation of lignin by electrolysis was performed in a divided cell. The reaction products were identified and quantified by ultra-pressure liquid chromatography coupled with mass spectrometry. The main products were sinapic acid, vanillin, vanillic acid, and acetovanillone. The obtained preliminary results demonstrated the potential feasibility of this innovative electrochemical route for lignin valorisation for the production of bio-aromatic chemicals.
1. Introduction
Among renewable resources, low-cost and abundantly available lignocellulosic biomass plays a fundamental role in the bio-based and circular economy. Lignocellulosic biomass is mainly composed of three biopolymers: cellulose, hemicellulose and lignin. Cellulose is the most abundant biopolymer in the world. The second one is lignin which represents a promising and renewable source of aromatic compounds.1 In order to promote the economic sustainability and profitability of biorefinery models, the full exploitation of the starting feedstock should be pursued by the conversion of all its components into added-value molecules and/or materials.2In this perspective, the valorisation of technical lignins, which represent one side-stream of the existing industrial-scale biorefineries and paper industry, is a strategic approach to enhance the biorefinery and paper industry sustainability.3 Side-streams of pulp & paper industry are rich in technical lignin, due to the almost complete valorisation of hemicellulose and cellulose components. In the present preliminary investigation, the soda technical lignin Protobind™ 1000 (P1000) was adopted as starting material. P1000 lignin is produced on an industrial scale by the company GreenValue, starting from a mix of wheat straw and Sarkanda grass. It is obtained by alkaline extraction from biomass with sodium hydroxide.4 In the literature, numerous studies propose several interesting approaches for lignin upgrading, such as pyrolysis, enzymatic or chemical depolymerisation and surface functionalisation.5–9 However, most of them require harsh reaction conditions, such as high temperature, high pressure, expensive and hazardous catalysts which make the process not economically viable on a larger scale.10 The electrochemical depolymerisation of lignin, especially if powered by renewable electricity, is a promising technology compared to conventional chemical oxidation because it can operate under mild, safe and eco-friendly reaction conditions, such as room temperature and atmospheric pressure.11 Among electrochemical approaches, the electro-oxidation of lignin at the anode is the most common one studied.12 Even if electrocatalytic approaches similar to that adopted in the present study were reported in the literature for other combinations of lignins and working electrodes,13–15 investigations regarding the electrochemical conversion of P1000 lignin, up to now, are absent. Only a few studies are available concerning its chemical valorisation through inorganic catalysts, such as CuMgAlOx or NiMo sulphide, in organic solvents under harsh reaction conditions.16–18
During the electrochemical oxidation of lignin, the surface functionalisation (α-carbonylation) and the cleavage of C–C/C–O bonds are the two main competing reactions.19 In particular, the cleavage of β-O-4 linkages is considered the rate-determining step in lignin depolymerisation.20,21 Mechanistic studies demonstrated that the C–O bond of the β-O-4 aryl ether linkage and Cα–Cβ bonds could be cleaved by electrocatalysts.21,22
In the literature, the direct electro-oxidation of technical lignins was performed using Ni, Pb/PbO2, Ti/SnO2, Sb2O3, RuO2–IrO2/Ti electrodes as catalysts, namely as working electrode (anode) or as an immobilised coating on the surface of the electrode.23–26 The present preliminary investigation, for the first time, is aimed to assess the performances of Pt, Ni/NiOOH and graphite as electrode materials for the P1000 lignin electro-oxidative depolymerisation. One of the main limitations in the scaling-up of electrochemical approaches could be the high cost of metal electrodes or electrode coatings, if for example Pt-based electrodes are involved.27 Thus, a challenging effort is represented by the development of electrochemical processes based on low-cost electrocatalysts,12 justifying the choice of graphite and nickel in the present work. In detail, a preliminary investigation of the performances of three different electrode materials (Pt, Ni/NiOOH and graphite) for the P1000 lignin electro-oxidative depolymerisation was performed by cyclic voltammetry adopting different reactions conditions, such as pH 12, 13 and 14, lignin concentration 2 and 20 g L−1, scan rates 10, 50, 100 and 250 mV s−1. Moreover, in order to validate the catalytic performances of the selected electrodes, the cyclic voltammetry study was also performed on guaiacol, considered as a model compound of a prominent lignin structural unit. Finally, the most efficient electrode and the optimal reaction conditions among those tested were then adopted in the electrolysis of soda P1000 lignin into added-value aromatic compounds.
2. Materials and methods
2.1 Chemicals and materials
The commercially available soda lignin Protobind™ 1000 (P1000) was provided by GreenValue S.A. (Switzerland). It has been obtained from a mixture of wheat straw and Sarkanda grass. A detailed characterisation of P1000 lignin is described in previous work,4 which has the chemical composition of (wt% on dry matter): glucan 0.5 ± 0.2, xylan 1.5 ± 0.1, galactan 0.2 ± 0.0, arabinan 0.2 ± 0.2, mannan < 0.1, rhamnan < 0.1, acid-insoluble lignin 85.1 ± 0.7, acid-soluble lignin 5.4 ± 0.0, ash 2.5 ± 0.0, other compounds 4.6 ± 1.2.HPLC-grade water and acetonitrile were products of Brunschwig Chemie (Amsterdam, The Netherlands). Formic acid (98–100%) was purchased from Riedel-de Haën (Seelze, Germany). The phenolics were obtained from Sigma-Aldrich (St. Louis, MO, USA). Milli-Q water was used throughout for preparation of all eluents and standard solutions. All other reagents and compounds were of the available highest purity.
Three working electrode materials were tested in the present investigation: Pt wire (geometric area = 1.0 cm2, Metrohm), Ni/NiOOH wire (geometric area = 0.37 cm2, Sigma Aldrich), graphite rod (geometric area = 1.0 cm2, Alfa Aesar).
2.2 NiOOH electrode preparation
A layer of NiOOH was formed on a nickel substrate in an electrochemical cell having a three-electrode configuration. The working electrode was a Ni wire (l = 1.15 cm, d = 0.1 cm, geometric area = 0.37 cm2), which was washed with ethanol, acetone and demi-water prior to the electrochemical treatment. As counter electrode a platinum wire (l = 20 cm, d = 0.025 cm, geometric area = 1.57 cm2) was used. An Ag/AgCl electrode (Radiometer Analytical REF201) was used as a reference electrode.The electrolyte solution was composed of 0.05 M NiSO4, 0.1 M CH3COONa and 0.005 M NaOH at room temperature. The thickness of the deposited oxides was controlled via applied current. Thus, to get a layer of around 0.4 × 10−6 g, six consecutive potentiometric cycles or 12 steps (0.5 mA cm−2, 60 s) were applied.28 After the preparation, the Ni electrode was rinsed with ethanol and demi-water.
2.3 Cyclic voltammetry
Cyclic voltammetry (CV) was used in order to examine electrocatalytic properties of various electrode materials for the electro-oxidative depolymerisation of P1000 lignin. The experimental setup consisted of an undivided electrochemical cell linked to a potentiostat (Ivium Technologies, the Netherlands), Pt wire as a counter electrode and Ag/AgCl electrode (Radiometer Analytical REF201) as a reference electrode. The following materials were tested as working electrode: Pt wire, Ni/NiOOH wire and graphite rod. All electrodes were used as purchased, except Ni/NiOOH, which was pretreated to deposit NiOOH layer on the electrode (see Section 2.2). Investigated electrolyte solutions were composed of 0.01 M NaOH + 0.99 M NaClO4 (pH 12), 0.10 M NaOH + 0.90 M NaClO4 (pH 13), 1.0 M NaOH (pH 14). The electrochemical cell contained 50 mL of solution. Argon gas was purged in the electrolyte solution before the measurements in order to remove oxygen. The CV measurements were performed at room temperature (20 °C). All the currents reported in this work were normalised with respect to the geometric area of the electrodes. Different scan rates (10, 50, 100, 250 mV s−1) were investigated modulating the potentiostat. Eight full cycles were performed for the scan rate of 10 mV s−1, fifteen full cycles were performed for the scan rate of 50 and 100 mV s−1, while twenty-five full cycles were performed for the scan rate of 250 mV s−1. In order to investigate the effect of the substrate concentration on the electrocatalytic properties, two values of lignin concentration, 2 and 20 g L−1, were tested.2.4 Electro-oxidative depolymerisation
Electrolysis of P1000 lignin was performed in a custom-made double-walled divided electrochemical glass cell in a three-electrode configuration under galvanostatic conditions (6.4 mA cm−2) using a potentiostat (Fig. 1). The current was measured through the potentiostat obtaining the value of 150 mA. Then it was correlated to the geometric area of the NiOOH working electrode (23.55 cm2) used in the electrolysis. Based on this calculation, the current density was 6.4 mA cm−2.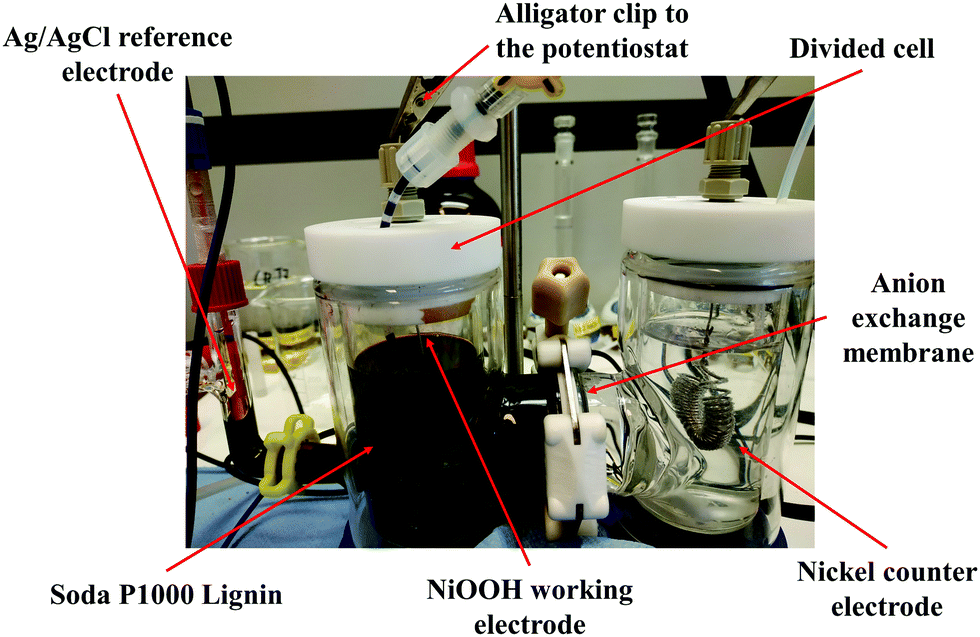 | ||
| Fig. 1 Picture of the custom-made double-walled divided electrochemical glass cell in a three-electrode configuration used for the electrolysis of soda P1000 lignin under the optimal reaction conditions. | ||
The electrochemical cell contained 150 mL catholyte (1.0 M NaOH, pH 14), 150 mL anolyte (1.0 M NaOH, pH 14, 20 g L−1 P1000 lignin). It was divided by an anion exchange membrane, with a dry thickness of 130 μm (Fumasep® FAA-3-PK-130, Fumatech). Prior to use, the membrane was immersed in an aqueous solution of 1 M NaOH for 24 h at room temperature, in order to exchange the bromide (Br−) counter ions into hydroxyl (OH−). The presence of an anion exchange membrane ensured the migration of OH− ions from the catholyte to the anolyte. Argon gas was purged in anolyte and catholyte solutions prior to and during the electrochemical measurements in order to completely remove oxygen in the electrochemical cell thus avoiding the involvement of the atmospheric oxygen in the investigated reaction. Both compartments were stirred and electrolysis was performed at room temperature. The three-electrode configuration consisted of the Ni counter electrode (l = 75 cm, d = 0.1 cm, geometric area = 23.55 cm2), the Ag/AgCl reference electrode (Radiometer Analytical REF201) via a Luggin capillary and the Ni/NiOOH working electrode (l = 75 cm, d = 0.1 cm, geometric area = 23.55 cm2). Counter and working electrodes were spirally wound to fit into the electrochemical cell. Around 4 mL of the catholyte was used to fill the Luggin capillary and reservoir into which the Ag/AgCl reference electrode was placed. Electrolysis was carried out at 0.4 V vs. Ag/AgCl for 4 h. The anolyte and catholyte solutions were separately collected after electrolysis. The lignin-containing solution was then analysed by UPLC/MS analysis.
The theoretical electrochemical conversion (%) was calculated as the ratio between the consumed moles of electrons during electrolysis and the theoretical amount of moles of electrons required for the complete electrolysis of the starting lignin (3 g). For the calculation of this last factor, the monomer average molecular weight of 195.2 g mol−1 and 4 electrons consumed per mole were considered according to the literature.29
2.5 UPLC/MS analysis
Samples from the electrochemical divided cell were neutralised with formic acid and centrifuged in order to separate the unreacted lignin and the insoluble high molecular weight oligomers. Then, the liquid fraction was recovered by microfiltration (0.22 μm filter). The UPLC/MS analysis was performed by a Dionex RSLC system with an UltiMate 3000 Rapid Separation pump and auto-sampler. The detector was a Dionex Ultimate 3000 RS Diode Array Detector (280 nm) in combination with a Thermo Scientific™ LCQ Fleet Ion Trap Mass spectrometer. The separating column was a Waters Acquity UPLC BEH C18 reversed-phase column (1.7 μm particle size, 2.1 × 150 mm) with a sample loop of 100 μL. The guard column was a Waters VanGuard Acquity UPLC BEH C18 guard column (1.7 μm particle size, 2.1 × 5 mm).The column temperature was maintained at 40 °C. Eluent A consisted of Biosolve ULC/MS grade water with 1 mL L−1 formic acid (MS for positive and negative modus). Eluent B consisted of Biosolve ULC/MS grade acetonitrile. Elution was performed at a flow rate of 0.35 mL min−1, using the following gradient (expressed as solvent B, while solvent A is the complementary part): initial composition 4.0% B; 0.0–1.0 min 4.0% B; 1.0–17.0 min 56.0% B; 17.0–20.0 min 70.0% B; 20.0–24.0 min 100% B; 24.0–30.0 min 4.0% B. Heated electrospray ionization (HESI) mass spectrometry was performed in both positive and negative modes. The LCQ mass spectrometer was operated with the HESI set on 150 °C and the capillary temperature at 235 °C, sheath gas at 20 arbitrary units, the auxiliary gas at 5 arbitrary units and the sweep gas at 4 arbitrary units. The electrospray voltage was set to 5.0 kV. In the positive modus, the capillary voltage was set at 11.0 V and the tube lens offset at 45.0 V. In the negative modus the capillary voltage was set at −1.0 V and the tube lens offset at −44.9 V. The injection time was 100 ms. Mass spectra were recorded from m/z 70–500 at a unit mass resolution without in-source fragmentation. For sequential MS/MS experiments the normalised collision energy was 35%, with wideband activation turned off.
Standard (stock) solutions were obtained by weighing the phenolics of interest (with analytical precision, on an analytical balance) in a volumetric flask (50 or 100 mL) and subsequently adding/dissolving the phenolics in a mixture of methanol and Milli-Q-water (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v). trans-Cinnamic acid (for phenolic acids) and 1-methyl-napthalin (for phenolics) were used as internal standards (I.S.). For sample preparation, 500 μL sample (or standard solution) was mixed with 500 μL I.S., mixed and transferred into a 1.0 mL Dionex vial, to be ready for analysis. All the structural identifications were confirmed by using authentic aromatic standards. Retention times, UV-vis spectra, and MS/MS spectra of the compounds were matched with those of the corresponding commercial standards.
:50 v/v). trans-Cinnamic acid (for phenolic acids) and 1-methyl-napthalin (for phenolics) were used as internal standards (I.S.). For sample preparation, 500 μL sample (or standard solution) was mixed with 500 μL I.S., mixed and transferred into a 1.0 mL Dionex vial, to be ready for analysis. All the structural identifications were confirmed by using authentic aromatic standards. Retention times, UV-vis spectra, and MS/MS spectra of the compounds were matched with those of the corresponding commercial standards.
3. Results and discussion
3.1 Cyclic voltammetry study – effect of the electrocatalyst
In electrochemical reactions based on direct electro-oxidation of lignin, the anode material and structure, especially in terms of surface features, play a crucial role in the process performance.30 The electrocatalyst should be both stable towards anodic corrosion and passivation, and catalytically effective for lignin depolymerisation. A preliminary investigation on the Pt, NiOOH and graphite electrodes was performed by cyclic voltammetry in order to identify the oxidation potential of P1000 lignin in the electrolyte as a function of pH and the kind of electrocatalyst and to evaluate the optimal reaction conditions in terms of the electrode, pH and lignin concentration.Eight full cycles were performed with the scan rate of 10 mV s−1 for the three electrodes, as showed in Fig. 2. In all the cases the last and the second cycles were similar, thus confirming limited electrode passivation during the cyclic voltammetry experiments. The same behaviour of nickel and graphite electrodes in the cyclic voltammetry of Kraft lignin was observed by Di Marino et al.14 In the first cycle on Pt and graphite electrodes two oxidation peaks were observed at ca. 0.2 and 0.5 V. According to Milczarek,31 in the first anodic scan the second oxidation peak at higher potential value is related to the irreversible oxidation.
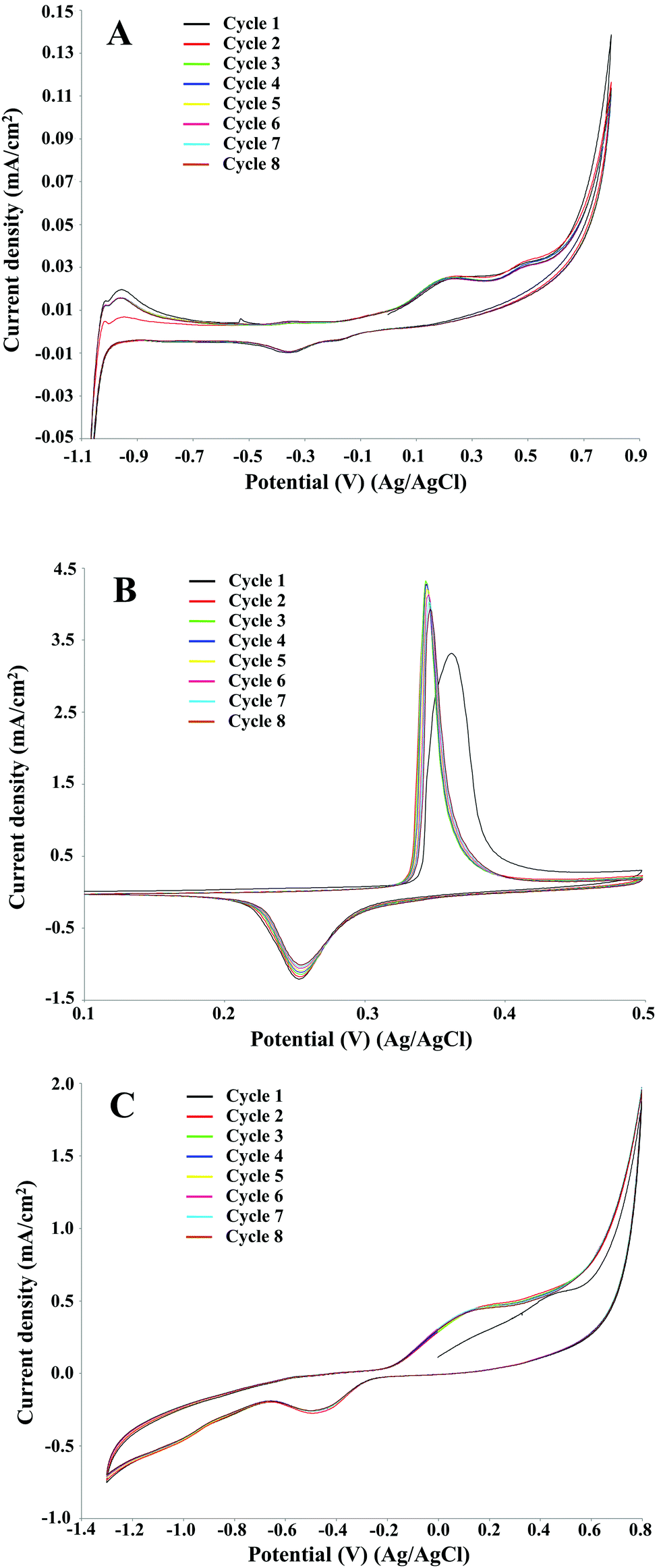 | ||
| Fig. 2 Cyclic voltammetry of 2 g L−1 P1000 lignin in 1 M NaOH (pH 14). Voltammograms recorded at 10 mV s−1 on platinum (A), NiOOH (B), and graphite (C) electrodes at room temperature. | ||
Fig. 3 compares the results obtained by cyclic voltammetry of soda P1000 lignin (2 g L−1) on Pt, NiOOH and graphite electrodes at pH 14.
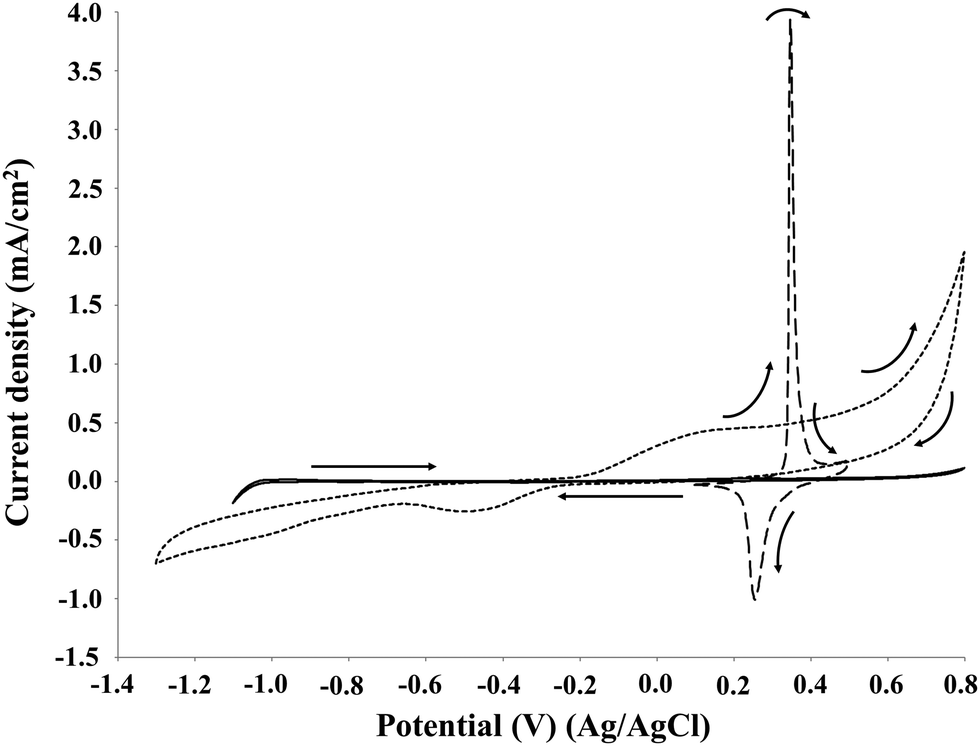 | ||
| Fig. 3 Cyclic voltammetry of 2 g L−1 P1000 lignin in 1 M NaOH (pH 14). Voltammograms recorded at 10 mV s−1 at room temperature. Solid line: on platinum electrode; dashed line: on NiOOH electrode; dotted line: on graphite electrode. | ||
The profiles obtained on platinum, nickel oxide hydroxide and graphite electrodes differ considerably. In all the cases one or two oxidation peaks were observed between 0.2 and 0.5 V. Similar results were obtained by Parpot et al.32 for the electro-oxidation of Kraft lignin on Ni and Pt electrodes and by Movil-Cabrera et al.33 on Co core/Pt partial shell nanoparticle alloy electrocatalyst. Moreover, the potential value of 0.45 V was obtained by Caravaca et al. for the electro-oxidation of Kraft lignin on bimetallic Pt–Ru anode.34
For each electrode material, the current density increased significantly compared to the control (no lignin) as can be seen in Fig. 4. In particular, on Pt electrode (Fig. 4A) two oxidation peaks were observed at 0.2 and 0.5 V. Differently, on NiOOH (Fig. 4B) and graphite (Fig. 4C) electrodes only one oxidation peak was ascertained: in the first case it was around 0.35 V, while in the second case it was around 0.2 V. Regarding the lignin reactivity, on Pt electrode the current density increased to around 23 μA cm−2 at 0.2 V and around 28 μA cm−2 at 0.5 V with respect to the control test. On NiOOH electrode the current density increased to around 2 mA cm−2 at 0.35 V, while on graphite electrode it increased to around 0.5 mA cm−2 at 0.2 V. Moreover, in the presence of lignin, on NiOOH electrode the charge density, namely the supplied charge related to the electrode area, of the oxidation and reduction peaks were 9.8 and 6.1 mC cm−2, respectively. In the absence of lignin, namely in the control test, the charge density of the oxidation and reduction peaks were 6.3 and 5.5 mC cm−2, respectively. Thus, the net charge density in the oxidation sweep was 3.5 mC cm−2, corresponding to an increase of about 56% respect to the control test. The net charge density in the reduction sweep was only 0.6 mC cm−2, corresponding to an increase of about 10% respect to the control test. On this basis, the electrochemical oxidation of lignin in alkaline medium resulted an irreversible reaction.
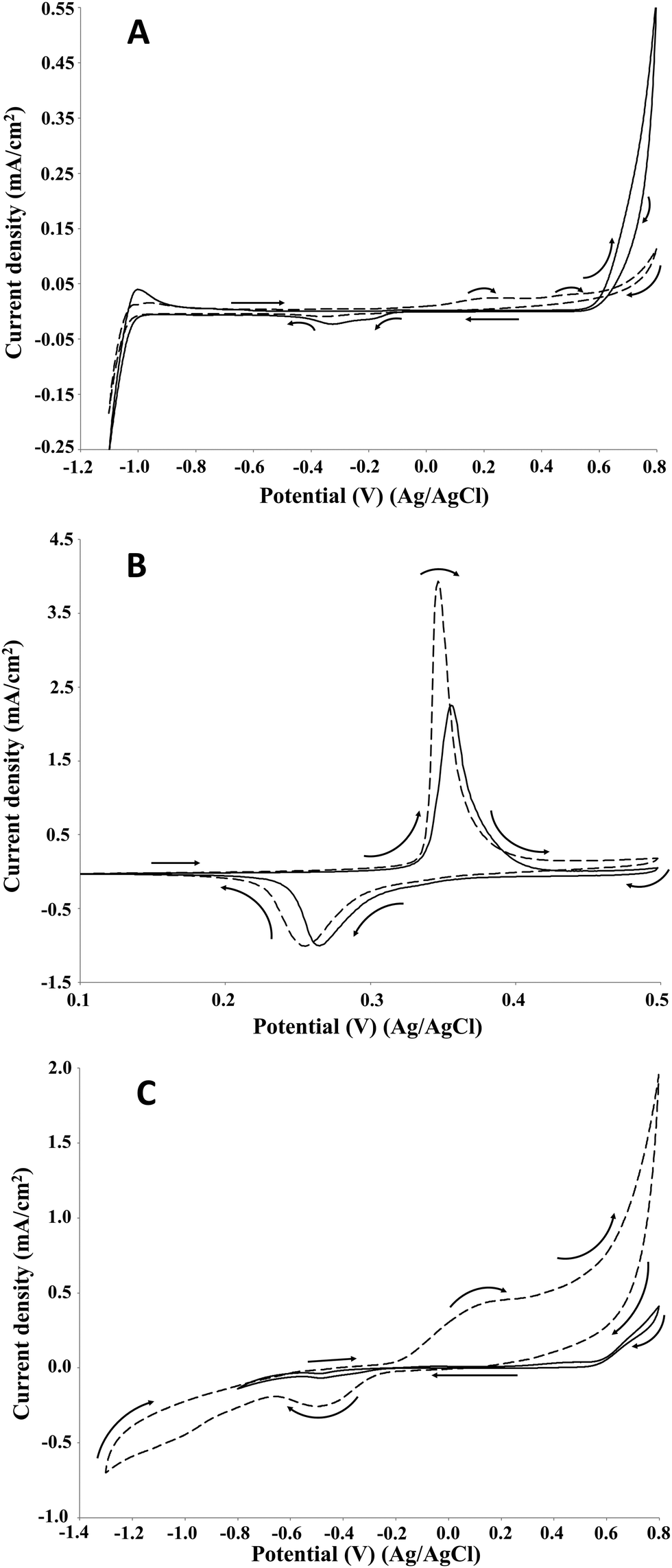 | ||
| Fig. 4 Cyclic voltammetry of 2 g L−1 P1000 lignin in 1 M NaOH (pH 14). Voltammograms recorded at 10 mV s−1 on platinum (A), NiOOH (B), and graphite (C) electrodes at room temperature. Solid line: control; dashed line: lignin. | ||
By comparing the results obtained for the three electrode materials, the NiOOH electrode showed the maximum current density in the electro-oxidation of P1000 lignin.
In order to confirm the attribution of the increase in the current density of the oxidation peak to the lignin oxidation on the NiOOH, the same cyclic voltammetry study was performed on guaiacol, which is one of the main structural units of lignin. Guaiacol, being a monomer, is not involved in the oxidative depolymerisation process of lignin but its use can provide useful information on the performances of several electrode materials in terms of electrode stability, current density and reproducibility of the electrochemical reaction. Moreover, guaiacol represents a common model compound used for the study of the oxidation of phenolic groups which are typical of the lignin structure.32 Electrochemical oxidation of guaiacol on Pt, Au, Ti/Sb–SnO2, Ti/Pb3O4, Ni, vitreous carbon and oxides of cobalt electrodes has been investigated in previous studies.32,35,36 According to the literature, the cyclic voltammetry of guaiacol is characterised by a first irreversible discharge involving one or two electrons leading to the formation of a radical species. In particular, in the presence of an acidic medium, the electrochemical mechanism involves two electrons, while in an alkaline medium a one-electron discharge is favoured.32,35Fig. 5 shows the voltammograms acquired for each electrode of the present investigation.
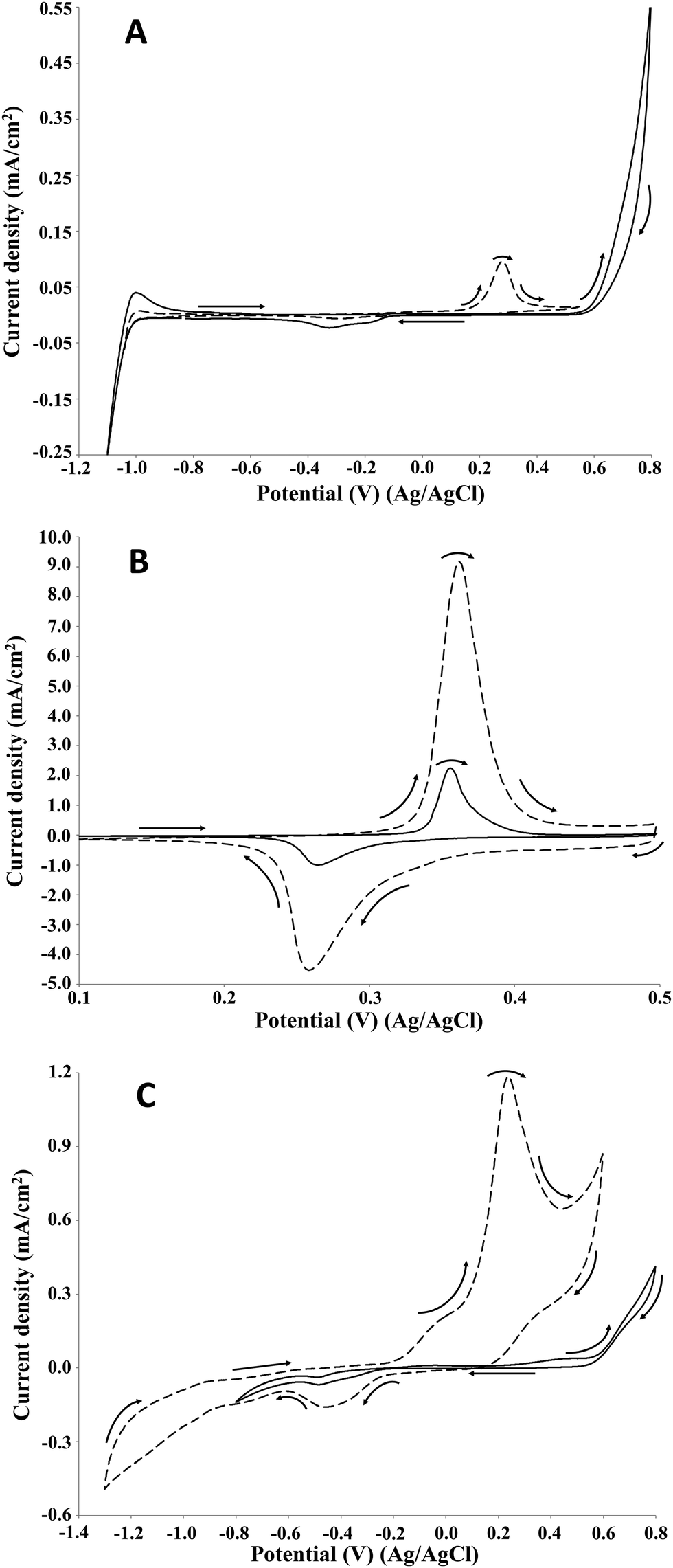 | ||
| Fig. 5 Cyclic voltammetry of 2 g L−1 guaiacol in 1 M NaOH (pH 14). Voltammograms recorded at 10 mV s−1 on platinum (A), NiOOH (B), and graphite (C) electrodes at room temperature. Solid line: control; dashed line: guaiacol. | ||
Differently from lignin, on Pt electrode only one oxidation peak was observed at around 0.3 V, which corresponded to the first peak registered for lignin. The obtained voltammogram agreed with the information reported in the literature for the Pt electrode in alkaline medium.37 The net current density was around 0.1 mA cm−2, namely significantly higher respect to the values obtained for the lignin oxidation. On the NiOOH electrode, the presence of one oxidation peak at around 0.4 V was confirmed. Similarly to the lignin oxidation, the current density of the oxidation peak of guaiacol was significantly higher than the control test. For the guaiacol electro-oxidation, the net current density was 7 mA cm−2, namely 3.5-folds higher than the value registered for the lignin oxidation (2 mA cm−2). Moreover, the net charge density of the oxidation and reduction peaks were 0.8 and 0.4 mC cm−2, respectively. Also on the graphite electrode, one oxidation peak was observed at around 0.3 V, according to the behaviour of Pt and NiOOH electrocatalysts. Moreover, the potential agreed with the value acquired for the lignin oxidation. The net current density of the peak was around 1.2 mA cm−2, namely 3-folds higher than the value registered for the lignin oxidation (0.5 mA cm−2). The cyclic voltammetry study on the guaiacol electro-oxidation confirmed the NiOOH as the best electrodes among those tested in terms of current density.
3.2 Cyclic voltammetry study – effect of scan rate
In order to establish if the electro-oxidation of P1000 lignin is mass transfer controlled, the effect of the scan rate of the three electrodes at pH 14 in the presence of 2 g L−1 lignin was investigated. For all the electrodes a linear relation was obtained by relating the current density of the lignin oxidation peak with the squared root of the scan rate (Fig. 6). This linear relationship is an indication that the investigated process is mass transfer controlled, according to the Randles-Sevcik equation:38| Ip = 0.4463·z·F·A·C·[(z·F·v·D)/(R·T)]1/2 |
| Ip = 2.686 × 105·z3/2·A·D1/2·C·v1/2 |
485 C mol−1), A is the area of the electrode (cm2), C is the initial concentration of the analyte (mol cm−3), v is the potential scan rate (V s−1), D is the diffusion coefficient of the analyte (cm2 s−1), R is the gas constant (J mol−1 K−1) and T is the temperature (K).
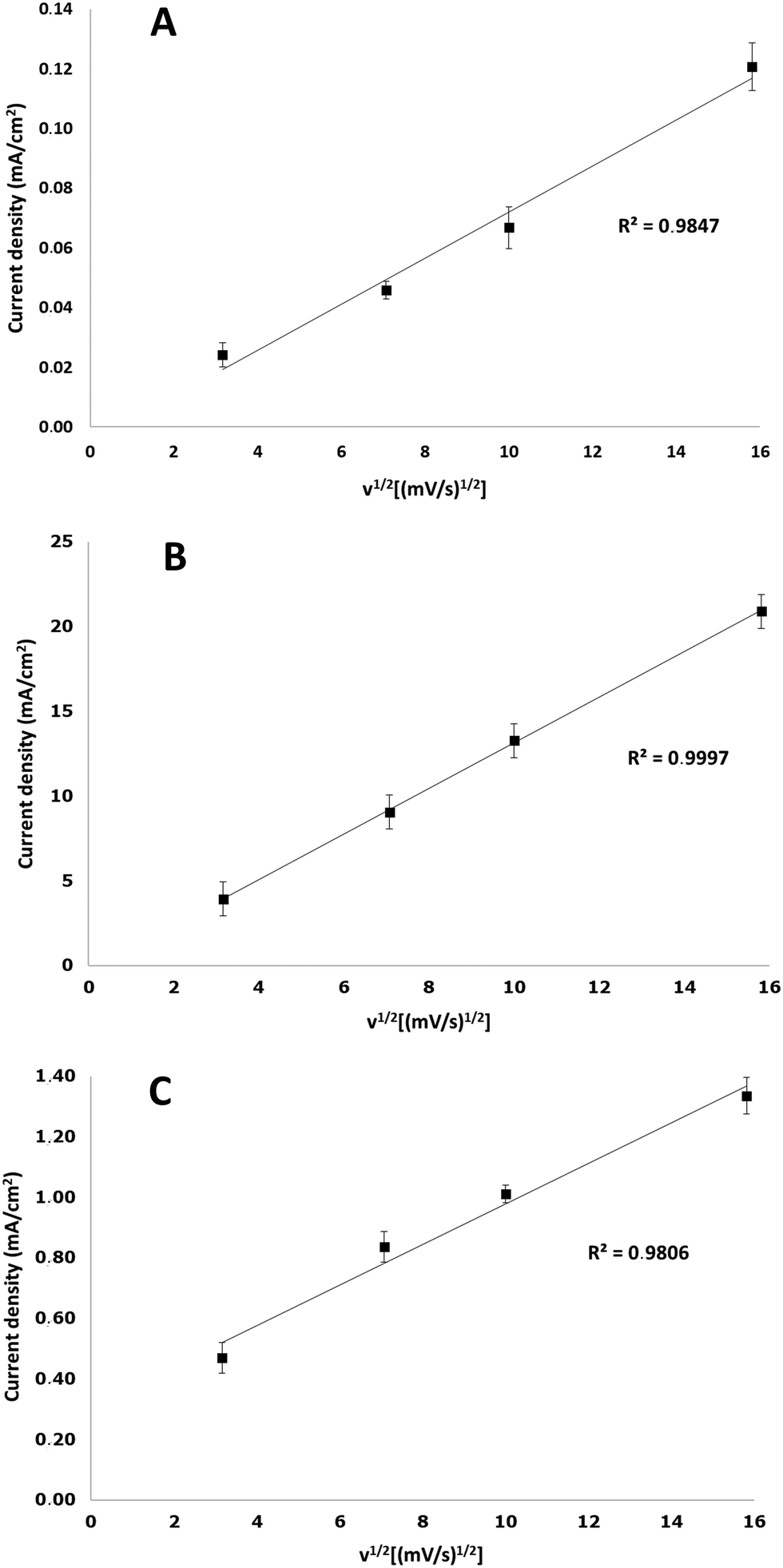 | ||
| Fig. 6 Current density (mA cm−2) as a function of the square root of scan rate in cyclic voltammetry measurements of 2 g L−1 P1000 lignin at pH 14 on platinum (A), NiOOH (B), and graphite (C) electrodes at room temperature. | ||
3.3 Cyclic voltammetry study – effect of lignin concentration
The effect of the increase in the lignin concentration on the current density of the oxidation peak was investigated by cyclic voltammetry on the NiOOH electrode at pH 14 (Fig. 7).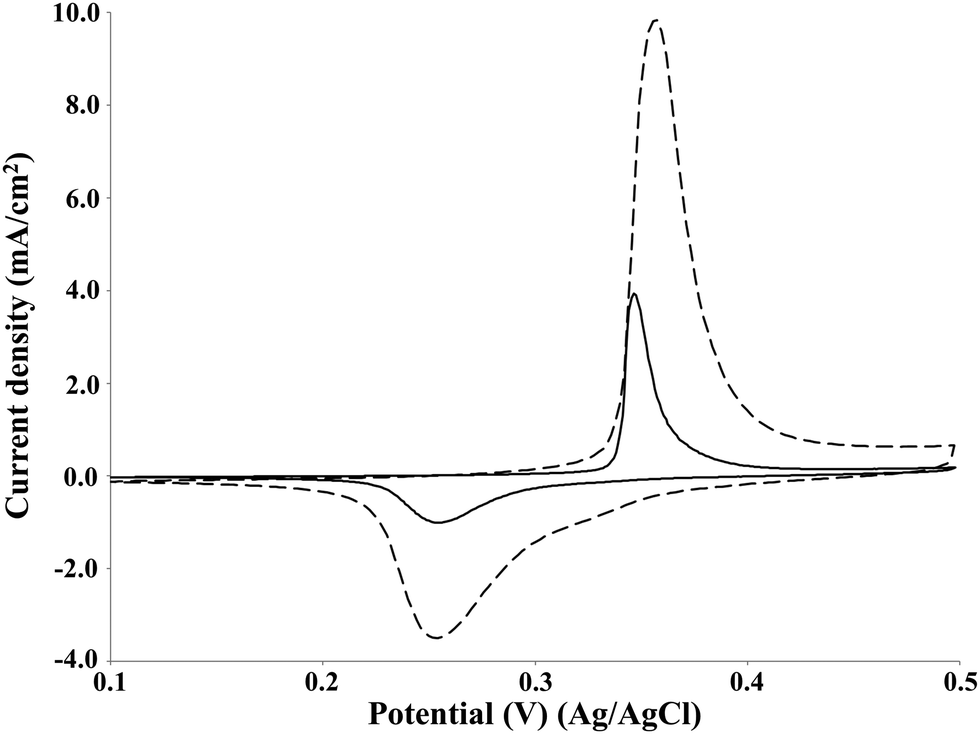 | ||
| Fig. 7 Cyclic voltammetry of P1000 lignin at the concentration of 2 g L−1 (solid line) and 20 g L−1 (dashed line). Voltammograms recorded at 10 mV s−1 on NiOOH electrode at room temperature. | ||
The increase of lignin concentration from 2 to 20 g L−1 resulted in a 2.5-folds increase of the current density of the oxidation peak of P1000 lignin from 4 to around 10 mA cm−2. A similar increase of the electrode activity with increasing of lignin concentration was obtained by Cai et al. in the cyclic voltammetry, adopting different concentrations in the range 20–40 g L−1 on Pb/PbO2 electrode in alkali solution.23 Moreover, the same phenomenon is reported in the literature for the electrochemical oxidation of guaiacol35 and urea on the NiOOH electrode in alkaline media.39
3.4 Cyclic voltammetry study – effect of pH
The effect of pH on the oxidative potential of P1000 lignin was investigated by cyclic voltammetry on the NiOOH electrocatalyst (Fig. 8).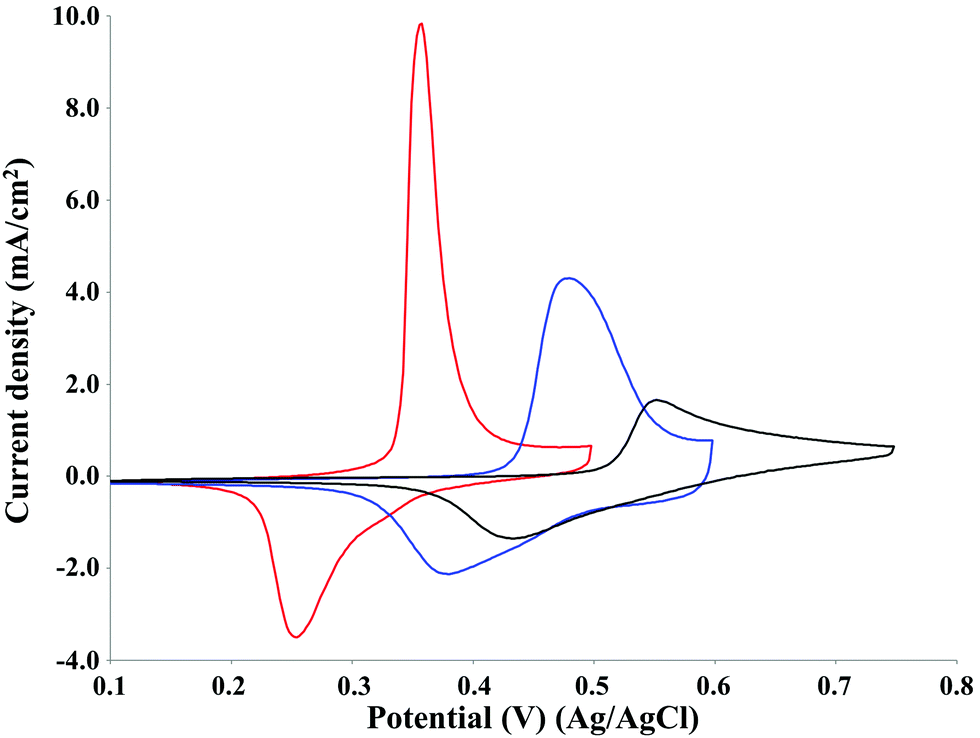 | ||
| Fig. 8 Cyclic voltammetry of 2 g L−1 P1000 lignin at pH 12 (black line), 13 (blue line) and 14 (red line). Voltammograms recorded at 50 mV s−1 on NiOOH electrode at room temperature. | ||
The pH increase resulted in a decrease in the potential of the lignin oxidation peak according to the Nernst equation.40,41 It was around 0.55 V at pH 12, around 0.45 V at pH 13 and around 0.35 V at pH 14. Similar results were obtained by Vedharathinam and Botte for the electrochemical oxidation of urea on NiOOH electrode in alkaline media.39 Moreover, the increase in pH resulted in an increase of the peak current density which is related to the kinetics of the oxidation current for Ni(OH)2/NiOOH according to the literature.42,43 In particular, since the lignin oxidation is catalysed by NiOOH species and the formation of this last one on the electrode surface is strongly affected by the OH− activity, the increase of pH, namely the increase of OH− concentration, increases the anodic current density.39 In fact, it was around 1 mA cm−2 at pH 12, around 4 mA cm−2 at pH 13 and around 10 mA cm−2 at pH 14. The net charge density of the oxidation peaks at pH 12, 13 and 14 were 3.1, 5.3 and 5.3 mC cm−2, respectively. The net charge density of the reduction peaks at pH 12, 13 and 14 were 2.8, 3.8 and 3.6 mC cm−2, respectively. The increase of pH from 12 to 13 or 14 determined an increase of 71% of the charge density of the oxidation peak, corresponding to 2.2 mC cm−2. The same effect of the pH increase on the current density was observed by Cai et al. in the cyclic voltammetry of Pb/PbO2 in alkali solution for the commercial corn stover lignin.23 Based on the results obtained, pH 14 was selected as the optimal reaction condition for P1000 lignin electrolysis. This reaction condition agreed with the information reported in the literature for the electro-oxidative depolymerisation of other technical lignins.13,23,32
Considering the performances of Pt, NiOOH and graphite electrodes and the results obtained by the preliminary cyclic voltammetry study, the following reaction conditions were selected for the electrolysis of P1000 lignin: NiOOH electrode, pH 14, 0.4 V, 20 g L−1 lignin. NiOOH was selected as the most efficient electrocatalyst based on the highest net current density registered for the lignin oxidation peak in the cyclic voltammetry.
3.5 Electrolysis of lignin
The initial cyclic voltammetry study aimed at a preliminary screening for determining the optimal reaction conditions for the electrolysis of soda P1000 lignin in order to produce added-value aromatic compounds. NiOOH electrode showed the maximum current density in the electro-oxidation of P1000 lignin during the cyclic voltammetry study. In particular, by comparing the current density at the maximum oxidative potential value for each electrode, on NiOOH electrode the current density increased to around 2 mA cm−2 at 0.35 V, while on graphite and platinum electrodes it increased to around 0.5 mA cm−2 and 23 μA cm−2 at 0.2 V, respectively. Besides, for all the three materials, the electro-oxidation of P1000 lignin resulted mass transfer controlled. Finally, nickel, as well as graphite, represents a cheaper material than platinum and it is characterised by better mechanical properties than graphite, especially in terms of brittleness. Based on all these results and considerations, NiOOH was selected as the preferred electrode material for the following constant-potential lignin electrolysis in water. Alkaline solutions are typically used to dissolve lignin favouring its oxidative depolymerisation.34,44 In the literature, several hypotheses on the oxidation mechanism of lignin to give valuable aromatics are reported.22,32,45 In the case of electro-oxidative depolymerisation, the R–O–R ether linkages, which are thermally and oxidatively more labile with respect to the C–C linkages, are firstly involved in the mechanism.46 Mechanistic studies found the C–O bond of the abundant β-O-4 aryl ether linkage is cleaved by the electrocatalyst.22 Subsequently, the same electrode material is able to cleave Cα–Cβ bonds in the electro-oxidation of β-O-4 linkages.21 Moreover, the cleavage of Cα–Cβ bonds competes with the Cα-carbonylation reaction.19 The ether bonds breaking leads to the formation of oxygenated aromatics such as vanillin, vanillic acid, vanillin acetate and guaiacol. Moreover, during electrolysis in alkaline solution, different substituted phenolates are produced, which are involved in a direct electron transfer leading to the synthesis of radical species which take part in the lignin depolymerisation.32 Recently, Chen et al. studied the electrochemical oxidation mechanisms for the C–O and C–C cleavages of β-O-4 linkages in lignin model monomers and dimers such as 2-phenoxy-1-phenethanol, 2-phenoxyacetophenone and 2-phenoxy-1-phenylethane.47 Authors demonstrated that the conversions of aryl ring in the lignin model dimers are involved in the starting single electron transfer oxidations, forming their benzene radical cation intermediates. Then, these last ones process further chemical conversion on their β-O-4 bonds due to electron delocalisation, resulting in cationic products and the phenolic radical. The cationic products are finally transformed into alcohol products.47Fig. 9 shows the results of the P1000 lignin electro-oxidative depolymerisation under the optimal reaction conditions.
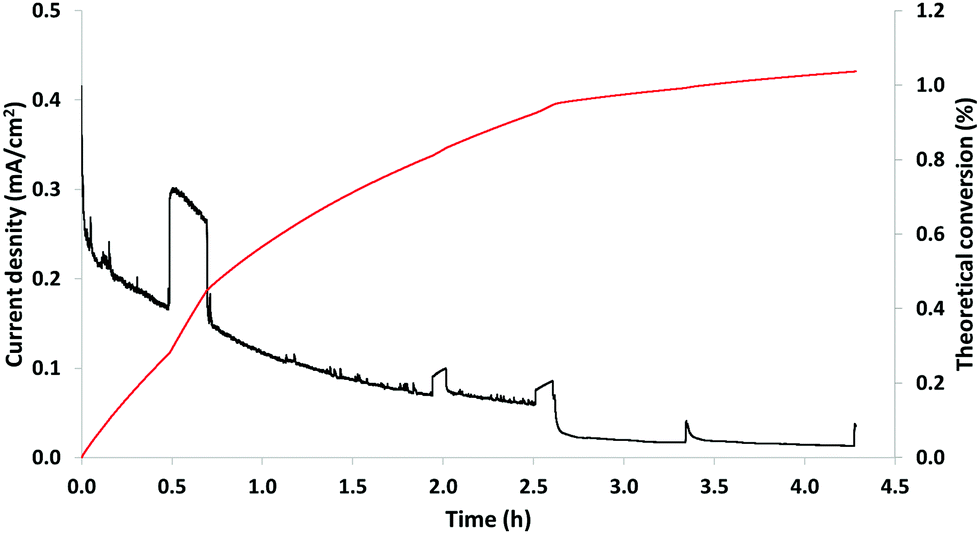 | ||
| Fig. 9 Constant potential electrolysis of 20 g L−1 P1000 lignin on NiOOH electrode at pH 14, 0.4 V and room temperature. Black line: current density (mA cm−2); red line: theoretical conversion (%). | ||
In particular, Fig. 9 shows the variation of the current density (mA cm−2), black line, and the theoretical conversion (%), red line, as a function of the reaction time. At the end of the reaction, the theoretical conversion was 1.0%, which represented the oxidation degree of the implemented process. The decreasing current density observed in Fig. 9 might be attributed to the passivation of the electrode. This phenomenon might be the result of lignin (or its degradation products) adsorbed on the active sites of the electrode. In addition, the flat-topped peaks in Fig. 9 of the black line are considered artefacts, which are related to sampling. Possibly, sampling generated a (partial) de-passivation of the electrode resulting in a temporarily increase of the current density followed by re-passivation of the surface.
Table 1 reports the list of the identified aromatic compounds and their concentrations obtained after the P1000 electrolysis.
| Number | Compound | Structure | Concentration (mg L−1) | Yielda (wt%) |
|---|---|---|---|---|
| a Products yield was calculated with respect to the lignin loading in the electrochemical cell. | ||||
| 1 | Vanillic acid |
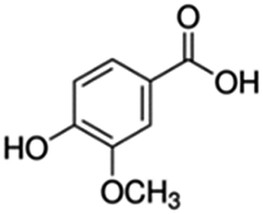
|
23.4 | 0.12 |
| 2 | 4-Hydroxybenzaldehyde |
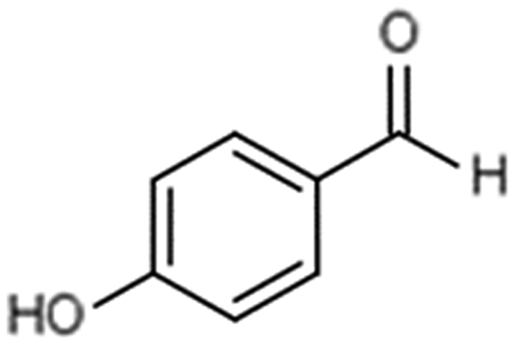
|
17.3 | 0.09 |
| 3 | Syringic acid |
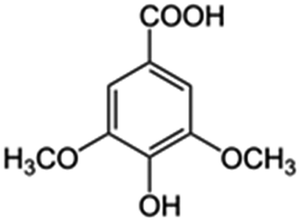
|
20.6 | 0.10 |
| 4 | 3,5-Dimethoxy-4-hydroxyphenylacetic acid |
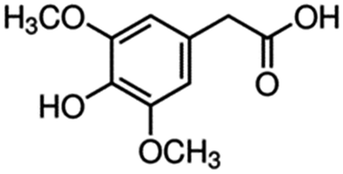
|
2.8 | 0.01 |
| 5 | Vanillin |
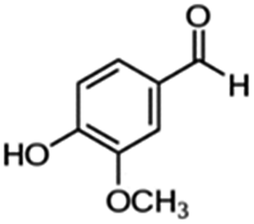
|
23.8 | 0.12 |
| 6 | p-Coumaric acid |
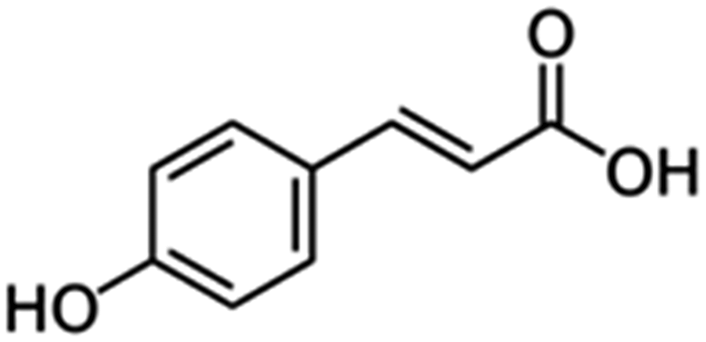
|
18.2 | 0.09 |
| 7 | 3,5-Dimethoxy-4-hydroxybenzaldehyde |
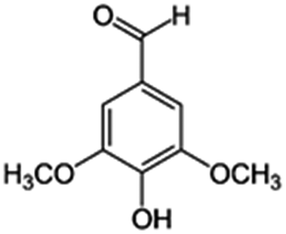
|
13.3 | 0.07 |
| 8 | Acetovanillone |
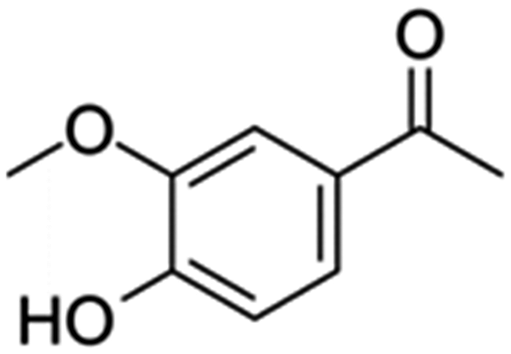
|
30.2 | 0.15 |
| 9 | Sinapic acid |
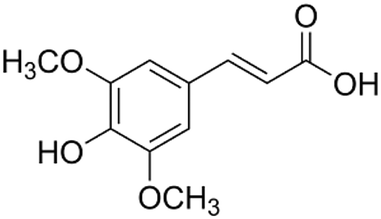
|
64.3 | 0.32 |
| 10 | 3,5-Dimethoxy-4-hydroxyacetophenone |
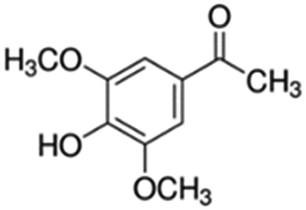
|
9.9 | 0.05 |
| 11 | 2,6-Dimethoxyphenol |
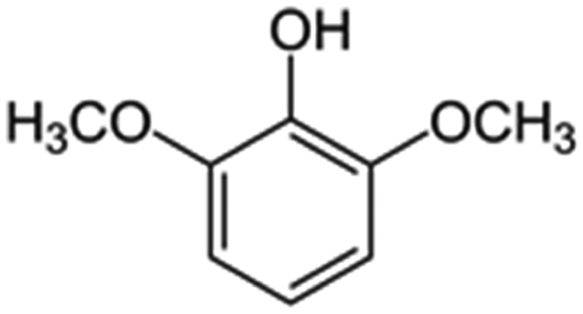
|
14.1 | 0.07 |
| 12 | 2,4-Dihydroxyacetophenone |
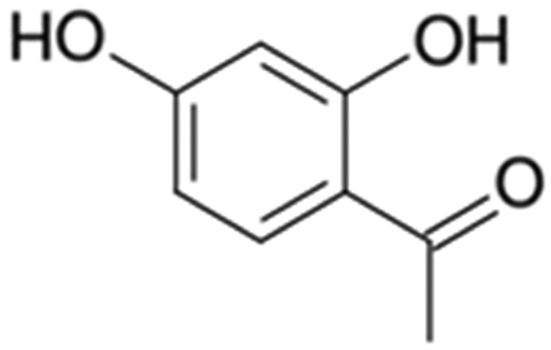
|
0.1 | 0.00 |
| 13 | 4-Ethylcatechol |
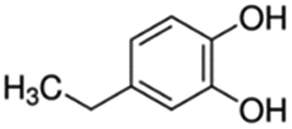
|
5.1 | 0.03 |
| 14 | 2-Hydroxy-4-methoxyacetophenone |
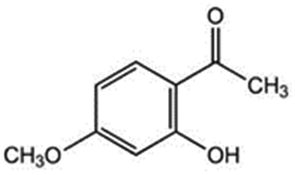
|
2.6 | 0.01 |
14 added-value bioproducts were identified (Table 1). The main products were sinapic acid (64.3 mg L−1, 0.32 wt%), acetovanillone (30.2 mg L−1, 0.15 wt%), vanillin (23.8 mg L−1, 0.12 wt%) and vanillic acid (23.4 mg L−1, 0.12 wt%). The sum of aromatics concentrations resulted 245.7 mg L−1, corresponding to a production of around 1.2 kg of aromatics from 100 kg of P1000 lignin. This yield value of 1.23 wt% is in line with the literature. In fact, according to Weber and Ramasamy, mass yields are typically ≤2 wt%.46 For example, Di Marino et al. obtained 14 aromatic compounds with an overall yield of 2.0 wt% respect to the starting lignin,25 while Ghahremani and Staser identified 4 products with an overall yield of 0.2 wt% respect to the starting lignin.48 Moreover, similar aromatic bioproducts were obtained in the study of Long et al.49 on the chemical depolymerisation of organosolv pine lignin and in the study of Yang et al.50 on the enzymatic depolymerisation of Kraft lignin. The amount of products corresponds with the estimated maximum conversion and therefore indicates a high coulombic efficiency for the depolymerisation of P1000 lignin.
4. Conclusions
In the present study, for the first time, the electro-oxidative depolymerisation of soda P1000 lignin into added-value aromatic compounds was preliminarily investigated. In particular, three anode materials were tested, Pt, NiOOH and graphite, as well as three pH values, 12, 13 and 14 and two lignin concentrations, 2 and 20 g L−1. The preliminary cyclic voltammetry study allowed us to identify the optimal reactions conditions for the lignin electrolysis. NiOOH, pH 14, 20 g L−1 lignin and 0.4 V resulted the best reaction conditions among those tested. Adopting these parameters, the constant potential electrolysis of P1000 lignin was performed into a divided cell in the presence of an anion exchange membrane. 14 main aromatic compounds were identified and quantified by UPLC-MS. The main products were sinapic acid, acetovanillone, vanillin and vanillic acid, achieving the overall oxidation degree of 1%. The present research aids to future research involving elucidation of solutions aiming to avoid the electrode passivation and to increase the oxidation of soda P1000 lignin in order to raise the aromatics yield.Author contributions
Nicola Di Fidio: methodology, investigation, data curation, formal analysis, writing – original draft. Johan W. Timmermans: methodology, formal analysis, writing – review & editing, supervision. Claudia Antonetti: writing – review & editing, formal analysis. Anna Maria Raspolli Galletti: writing – review & editing, formal analysis, supervision. Richard J. A. Gosselink: conceptualization, writing – review & editing, supervision, funding acquisition, resources. Roel J. M. Bisselink: conceptualization, methodology, formal analysis, writing – review & editing, supervision. Ted M. Slaghek: conceptualization, writing – review & editing, supervision, funding acquisition, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The contribution of COST Action LignoCOST (CA17128), supported by COST (European Cooperation in Science and Technology), in promoting interaction, exchange of knowledge and collaborations in the field of lignin valorisation, is gratefully acknowledged. The contribution of COST Action LignoCOST in supporting the present work by a Short-Term Scientific Mission (Call No. 1, 2nd Grant Period) is acknowledged. Federico Maria Vivaldi of the Department of Chemistry and Industrial Chemistry of the University of Pisa is gratefully acknowledged for the fruitful discussion and his suggestions about the data elaboration.References
- I. De Bari, D. Cuna and N. Di Fidio, in Biofuels Production and Processing Technology, ed. M. Riazi and D. Chiaramonti, CRC Press, Boca Raton, 2017, ch. 19, pp. 533–561 DOI:10.1201/9781315155067.
- N. Di Fidio, S. Fulignati, I. De Bari, C. Antonetti and A. M. Raspolli Galletti, Bioresour. Technol., 2020, 313, 123650 CrossRef CAS PubMed.
- V. K. Garlapati, A. K. Chandel, S. J. Kumar, S. Sharma, S. Sevda, A. P. Ingle and D. Pant, Renewable Sustainable Energy Rev., 2020, 130, 109977 CrossRef CAS.
- S. Constant, H. L. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. Van Es, R. J. Grisel, B. M. Weckhuysen, W. J. Huijgen and R. J. A. Gosselink, Green Chem., 2016, 18, 2651–2665 RSC.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed.
- T. Li, H. Ma, S. Wu and Y. Yin, Energy Convers. Manage., 2020, 207, 112551 CrossRef CAS.
- J. Dillies, C. Vivien, M. Chevalier, A. Rulence, G. Châtaigné, C. Flahaut, V. Senez and R. Froidevaux, Biotechnol. Appl. Biochem., 2020, 67, 774–782 CrossRef CAS PubMed.
- P. J. De Wild, W. J. Huijgen and R. J. A. Gosselink, Biofuels, Bioprod. Biorefin., 2014, 8, 645–657 CrossRef CAS.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- F. Bateni, R. Ghahremani and J. A. Staser, J. Appl. Electrochem., 2021, 51, 65–78 CrossRef CAS.
- X. Du, H. Zhang, K. P. Sullivan, P. Gogoi and Y. Deng, ChemSusChem, 2020, 13, 4318–4343 CrossRef CAS PubMed.
- S. Singh and H. R. Ghatak, J. Wood Chem. Technol., 2017, 37, 407–422 CrossRef CAS.
- D. Di Marino, V. Aniko, A. Stocco, S. Kriescher and M. Wessling, Green Chem., 2017, 19, 4778–4784 RSC.
- S. Singh and H. R. Ghatak, Holzforschung, 2018, 72, 187–199 CAS.
- B. Joffres, C. Lorentz, M. Vidalie, D. Laurenti, A. A. Quoineaud, N. Charon, A. Daudin, A. Quignard and C. Geantet, Appl. Catal., B, 2014, 145, 167–176 CrossRef CAS.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS PubMed.
- J. Pu, D. Laurenti, C. Geantet, M. Tayakout-Fayolle and I. Pitault, Chem. Eng. J., 2020, 386, 122067 CrossRef CAS.
- T. Shiraishi, T. Takano, H. Kamitakahara and F. Nakatsubo, Holzforschung, 2012, 66, 303–309 CAS.
- J. Gierer and I. Norén, Holzforschung, 1980, 34, 197–200 CrossRef CAS.
- E. Baciocchi, M. Bietti and O. Lanzalunga, Acc. Chem. Res., 2000, 33, 243–251 CrossRef CAS PubMed.
- V. L. Pardini, C. Z. Smith, J. H. Utley, R. R. Vargas and H. Viertler, J. Org. Chem., 1991, 56, 7305–7313 CrossRef CAS.
- P. Cai, H. Fan, S. Cao, J. Qi, S. Zhang and G. Li, Electrochim. Acta, 2018, 264, 128–139 CrossRef CAS.
- Y. Jia, Y. Wen, X. Han, J. Qi, Z. Liu, S. Zhang and G. Li, Catal. Sci. Technol., 2018, 8, 4665–4677 RSC.
- D. Di Marino, D. Stöckmann, S. Kriescher, S. Stiefel and M. Wessling, Green Chem., 2016, 18, 6021–6028 RSC.
- H. Zhu, Y. Chen, T. Qin, L. Wang, Y. Tang, Y. Sun and P. Wan, RSC Adv., 2014, 4, 6232–6238 RSC.
- M. Shestakova and M. Sillanpää, Rev. Environ. Sci. Bio/Technol., 2017, 16, 223–238 CrossRef CAS.
- R. Latsuzbaia, R. Bisselink, A. Anastasopol, H. Van der Meer, R. Van Heck, M. Segurola Yagüe, M. Zijlstra, M. Roelands, M. Crockatt and E. Goetheer, J. Appl. Electrochem., 2018, 48, 611–626 CrossRef CAS.
- T. K. Dier, D. Rauber, D. Durneata, R. Hempelmann and D. A. Volmer, Sci. Rep., 2017, 7, 1–12 CrossRef CAS PubMed.
- S. Stiefel, A. Schmitz, J. Peters, D. Di Marino and M. Wessling, Green Chem., 2016, 18, 4999–5007 RSC.
- G. Milczarek, Electroanalysis, 2007, 19, 1411–1414 CrossRef CAS.
- P. Parpot, A. Bettencourt, A. Carvalho and E. Belgsir, J. Appl. Electrochem., 2000, 30, 727–731 CrossRef CAS.
- O. Movil-Cabrera, A. Rodriguez-Silva, C. Arroyo-Torres and J. A. Staser, Biomass Bioenergy, 2016, 88, 89–96 CrossRef CAS.
- A. Caravaca, W. E. Garcia-Lorefice, S. Gil, A. de Lucas-Consuegra and P. Vernoux, Electrochem. Commun., 2019, 100, 43–47 CrossRef CAS.
- Y. Samet, R. Abdelhedi and A. Savall, Phys. Chem. News, 2002, 8, 89–99 CAS.
- D. Shao, W. Chu, X. Li, W. Yan and H. Xu, RSC Adv., 2016, 6, 4858–4866 RSC.
- S. J. Reddy and V. Krishnan, Indian J. Chem., 1978, 16, 684–687 Search PubMed.
- J. J. Van Benschoten, J. Y. Lewis, W. R. Heineman, D. A. Roston and P. T. Kissinger, J. Chem. Educ., 1983, 60, 772 CrossRef CAS.
- V. Vedharathinam and G. G. Botte, Electrochim. Acta, 2012, 81, 292–300 CrossRef CAS.
- M. M. Walczak, D. A. Dryer, D. D. Jacobson, M. G. Foss and N. T. Flynn, J. Chem. Educ., 1997, 74, 1195 CrossRef CAS.
- F. Vivaldi, D. Santalucia, N. Poma, A. Bonini, P. Salvo, L. Del Noce, B. Melai, A. Kirchhain, V. Kolivoška, R. Sokolova, M. Hromadovà and F. Di Francesco, Sens. Actuators, B, 2020, 322, 128650 CrossRef CAS.
- P. Robertson, J. Electroanal. Chem. Interfacial Electrochem., 1980, 111, 97–104 CrossRef CAS.
- J. Kaulen and H. J. Schäfer, Tetrahedron, 1982, 38, 3299–3308 CrossRef CAS.
- M. Zirbes, D. Schmitt, N. Beiser, D. Pitton, T. Hoffmann and S. R. Waldvogel, ChemElectroChem, 2019, 6, 155–161 CrossRef CAS.
- V. Tarabanko, D. Petukhov and G. Selyutin, Kinet. Catal., 2004, 45, 569–577 CrossRef CAS.
- R. S. Weber and K. K. Ramasamy, ACS Omega, 2020, 5, 27735–27740 CrossRef CAS PubMed.
- J. Chen, H. Yang, H. Fu, H. He, Q. Zeng and X. Li, Phys. Chem. Chem. Phys., 2020, 22, 11508–11518 RSC.
- R. Ghahremani and J. A. Staser, Holzforschung, 2018, 72, 951–960 CrossRef CAS.
- J. Long, Y. Xu, T. Wang, Z. Yuan, R. Shu, Q. Zhang and L. Ma, Appl. Energy, 2015, 141, 70–79 CrossRef CAS.
- Y. Yang, W. Y. Song, H. G. Hur, T. Y. Kim and S. Ghatge, Int. J. Biol. Macromol., 2019, 124, 200–208 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |