
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComment on “Atmospheric chemistry of oxazole: the mechanism and kinetic studies on oxidation reaction initiated by OH radicals” by A. Shiroudi, M. A. Abdel-Rahman, A. M. El-Nahas and M. Altarawneh, New J. Chem., 2021, 45, 2237†
Tam V.-T.
Mai
 *abc and
Lam K.
Huynh
*cd
*abc and
Lam K.
Huynh
*cd
aUniversity of Science, Ho Chi Minh City, 227 Nguyen Van Cu, Ward 4, District 5, Ho Chi Minh City, Vietnam
bMolecular Science and Nano-Materials Lab, Institute for Computational Science and Technology, SBI Building, Quang Trung Software City, Tan Chanh Hiep Ward, District 12, Ho Chi Minh City, Vietnam
cVietnam National University HoChiMinh City, Quarter 6, Linh Trung Ward, Thu Duc City, Ho Chi Minh City, Vietnam
dInternational University, Block 6, Linh Trung Ward, Thu Duc City, Ho Chi Minh City, Vietnam. E-mail: hklam@hcmiu.edu.vn
First published on 21st July 2021
Abstract
In this short communication, we resolve the large discrepancy (∼750 times) in the rate constants between computational and experimental studies for the atmospheric oxidation reaction of oxazole initiated by OH radicals. This achievement is due to the use of the rigorous stochastic Rice–Ramsperger–Kassel–Marcus based master equation (RRKM-ME) rate model, which allows mimicking the dynamics of the title reaction without introducing any assumption (e.g., fast pre-equilibrium between the reactants and van der Waals pre-reactive complex). The model shows a large tunneling contribution at low temperatures and the minor role of the hindered internal rotation treatment. Detailed mechanistic insights (e.g., the NTC behaviors) are also revealed to advance related applications of the title system.
Recently, Shiroudi et al.1 (2021) theoretically studied the kinetic mechanism of the atmospheric oxidation of oxazole initiated by OH radicals, and they reported global rate constants that underestimated the measured data2,3 by a factor of ∼750 at T = 299–468 K (e.g., 1.04 × 10−14vs. 8.29 × 10−12 cm3 per molecule per s at T = 324 K and P = 100 torr). Such a significant discrepancy strongly encouraged us to reinvestigate the kinetics of the title reaction. In particular, we used a rigorous stochastic RRKM-based master equation (RRKM-ME) rate model, including the hindered internal rotation (HIR) and tunneling treatments, on the potential energy surface (PES) constructed using the same electronic structure methods (i.e., M06-2X/aug-cc-pVTZ and ROCBS-QB3//M06-2X/aug-cc-pVTZ levels of theory) suggested by Shiroudi et al.1
Fig. 1 schematically presents the detailed energy profile for the oxazole + OH → products reaction, consisting of three H-abstraction and three OH-addition channels, explored at the M06-2X/aug-cc-pVTZ level. Due to the somewhat similar systems of imidazole4/pyrrole5 + OH, the interaction between an oxazole molecule and an OH radical can form two pre-active van der Waals complexes or reactant-complexes (namely, RC1 and RC2) whose structures are presented in Fig. 2, located at −5.3 and −3.5 kcal mol−1, respectively, relative to the entrance channel. Energetically, the former complex, RC1, is well consistent with those found in imidazole4/pyrrole5 + OH (e.g., −6.8 and −4.9 kcal mol−1, respectively). Using the same electronic structure methods, Shiroudi and co-workers1 reported two complexes RC1′ and RC2′, lying at higher energies (i.e., −3.3 and −3.5 kcal mol−1, respectively). It is worth noting that different RC1/RC1′ structures and similar RC2/RC2′ structures, via different transition states, were found in Shiroudi et al.'s and our studies (cf.Fig. 2). In particular, in our study, RC1 is found to be about 2.0 kcal mol−1 lower (i.e., −5.3 vs. −3.3 kcal mol−1) due to the intramolecular hydrogen bond formed between the H atom of the OH radical and the N atom of oxazole. Also, RC1 and RC2 are found to go through transition states (TS1, TS2 & TS4) and (TS3, TS5 & TS6) while RC1′ and RC2′ proceed via (TS1 and TS4) and (TS2, TS3, TS5 & TS6), respectively.
 | ||
Fig. 1 Reaction energy profile (0 K), calculated at the M06-2X/aug-cc-pVTZ level, for the reaction of oxazole + OH → products. ZPE correction is included, and units are in kcal mol−1. Numbers in parentheses, “(![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) )”, are suggested by Shiroudi et al.1 at the same level. )”, are suggested by Shiroudi et al.1 at the same level. | ||
 | ||
| Fig. 2 Optimized structures of reactant-complexes RC1, RC1′, RC2 and RC2′ obtained at the M06-2X/aug-cc-pVTZ level of theory. Bond lengths and dihedral angles are in Å and degree (°), respectively. Values in “()” are obtained from Shiroudi et al.1 using the same method, M06-2X/aug-cc-pVTZ. ‘D’ stands for dihedral angle. | ||
From RC1, two H-abstraction (viaTS1 and TS2) and one OH-addition (viaTS4) channels can occur to form (P1 + H2O), (P2 + H2O) and I1, respectively; while the channels via transition states TS3 (direct loss-H pathway) and TS5/TS6 (OH-addition) can proceed from RC2. Note that the unimolecular decomposition of the formed adducts (i.e., I1, I2, and I3) via the β-scission of the C–H bonds is further studied to provide more insights into the reaction mechanism of the title reaction in this work. The optimized structures (reactants, complexes, TSs, intermediates, and products) are shown in Fig. S1 (ESI†). In general, our calculated numbers consistently match with those obtained from Shiroudi and co-workers1 with the mean absolute deviation (MAD) values of 0.25 and 0.35 kcal mol−1, calculated using the same methods, M06-2X/aug-cc-pVTZ and ROCBS-QB3//M06-2X/aug-cc-pVTZ, respectively (cf. Table S3, ESI†).
With the calculated equilibrium concentration ratio Keq(298 K) = [RC1]eq/[RC2]eq = 55.7 and the low intrinsic barrier (comparable with the RC2 energy) between the two complexes, RC1 is expected to dominate from both thermodynamic and kinetic points of view. In fact, two kinetic models (namely, the so-called “RC1&RC2 model” using explicitly two complexes RC1 & RC2 as described by the PES in Fig. 1 and the so-called “RC1 model” using the lowest complex RC1 as described by the PES in Fig. S2, ESI†) predict very close rate constant values with each other (i.e., the largest difference of 40.3% at high temperatures and the MAD value of ∼18.3%, cf.Fig. 4). Therefore, model RC1, in addition to model RC1&RC2, was used in some analyses to simplify the calculations (e.g., species profile simulations).
Based on the PES characterized at the M06-2X/aug-cc-pVTZ level, the predicted kinetic behaviors of the oxazole + OH → products reaction, including the time-resolved species profiles and rate coefficients, are reported thoroughly. As seen in Fig. 3 for the condition of 300 K and 760 torr, the species profiles significantly change with time; thus, at least, those species appearing in the figure (i.e., OH, RC1, I1, I3, P4 + H, P2 + H2O, P1 + H2O, P5 + H and P6 + H) should be included to characterize the dynamic/kinetic behaviors correctly. The computed total rate coefficients with the HIR and tunneling corrections, ktot, are in good agreement with those measured by Witte et al.2,3 (e.g., 8.57 × 10−12 (model RC1) vs. 8.29 × 10−12 cm3 per molecule per s at T = 324 K, cf.Fig. 4) with the MAD of less than 10%, at T = 299–468 K & P = 100 torr. In contrast, Shiroudi's numbers are much lower than the experimental values2,3 with the average factor of ∼750 (e.g., 1.04 × 10−14vs. 8.29 × 10−12 cm3 per molecule per s at T = 324 and P = 100 torr).
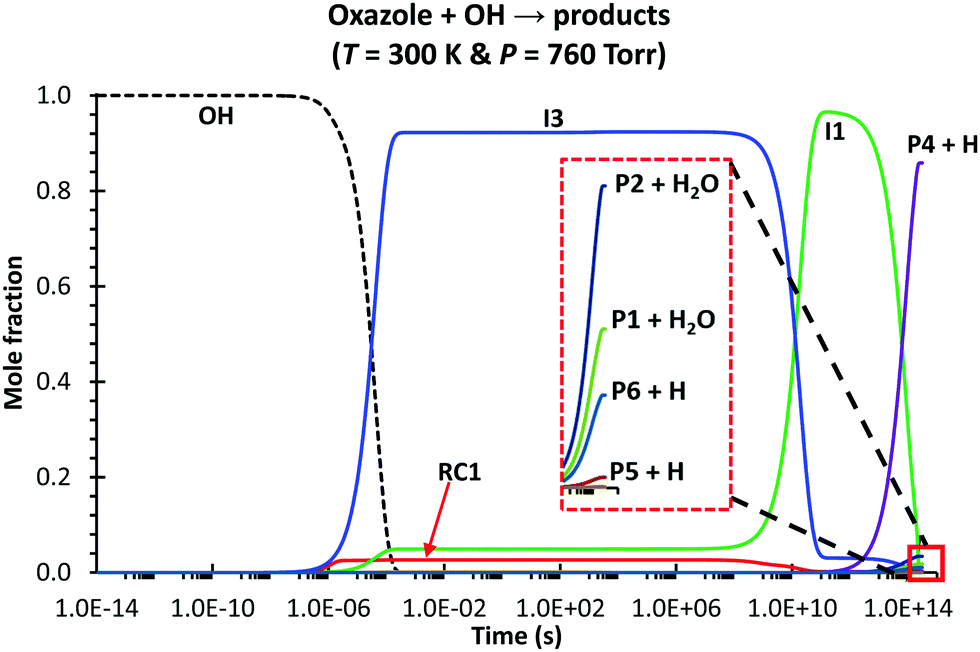 | ||
| Fig. 3 Time-dependent species profiles for the oxazole + OH reaction, simulated under the atmospheric conditions using the stochastic approach (108 trials) with [oxazole]/[Ar] = 10−4 and [oxazole]0 ≫ [OH]0 (Ar is the bath gas). The calculations were carried out using model RC1 (see the main text for the model details). Time is in a logarithm scale. | ||
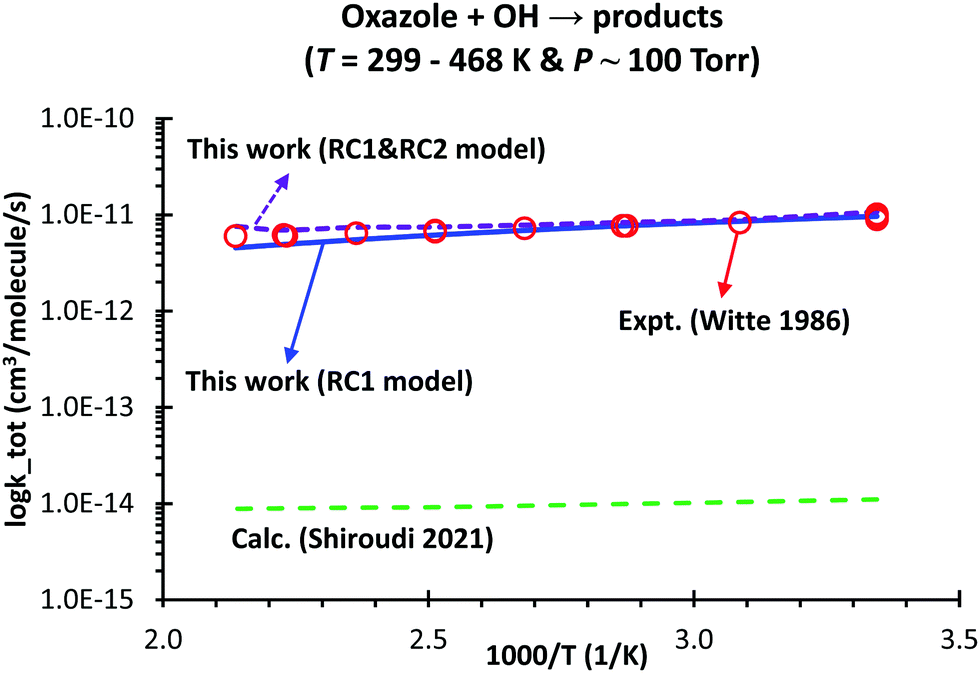 | ||
| Fig. 4 Comparison between our predicted and literature overall rate constants, ktot, of the oxazole + OH → products reaction at P = 100 torr as a function of temperature. Laboratory/computational data are collected from the works of Witte et al. (“Expt. (Witte 1986)”)2,3 and Shiroudi et al. (“Calc. (Shiroudi 2021)”),1 respectively. | ||
It is worth mentioning that the HIR correction plays a minor role in obtaining the reliable rate constants (e.g., it speeds up the calculated total rate by a factor of ∼1.5 in the temperature range of 299–468 K and P = 100 torr, cf. Table S4, ESI†). On the other hand, the tunneling treatment is found to play a more important role in kinetics and to be more relevant to the H-abstraction channels than OH-addition ones (e.g., the tunneling effect increases the individual rate constants by a factor of 30.0, 18.7, 16.9, 1.3, 1.3, 1.1, 3.6, 4.4 and 1.9 for channels viaTSx (x = 1–9), respectively, at T = 299 K, cf. Table S5, ESI†). Interestingly, a negative temperature dependence (NTD) manner, consistent with the experimental observation,2,3 of ktot(T) is also predicted for the title reaction in the considered conditions. Such behavior is similar to that of imidazole4/pyrrole5 + OH and mainly due to the faster re-dissociation of the reactant-complexes to the reactants at higher temperatures (cf. Fig. S3, ESI† for Keq(T) = kforward(T)/kreverse(T) plot as a function of temperature for the oxazole + OH → RC1 channel).
A comparison between our predicted ktot(T) and those calculated by Shiroudi and co-workers was carried out at P = 750 torr (∼1 bar), shown in Fig. 5. The calculated ktot(T) values at different temperatures and pressures (T = 299–468 K & P = 0.76–76000 torr) are tabulated in Table S2 (ESI†). Our values are found to be higher than those derived from the TST and RRKM models by Shiroudi and co-workers with an average factor of 3.1 and 125.3, respectively (cf.Fig. 5). Consequently, our calculated species branching ratios are significantly different from those suggested by Shiroudi et al. (cf.Fig. 6). Our model reveals that the production of adduct I3 (viaTS6) dominates the stabilization of I1 (viaTS4) and the bimolecular product formation from three H-abstraction channels P1/P2/P3 + H2O is insignificant in the considered conditions (T = 299–468 K & P = 100 torr). It is also observed that the yield of the main adduct I3 gradually decreases with the temperature increase (e.g., 94.9 and 92.4% at 299 and 468 K, respectively, at P = 100 torr).
 | ||
| Fig. 5 Comparison between our predicted and Shiroudi's total rate coefficients, ktot, of the oxazole + OH → products reaction as a function of temperature at 750 torr (1 bar). The calculations were carried out using model RC1 (see the main text for the model details). | ||
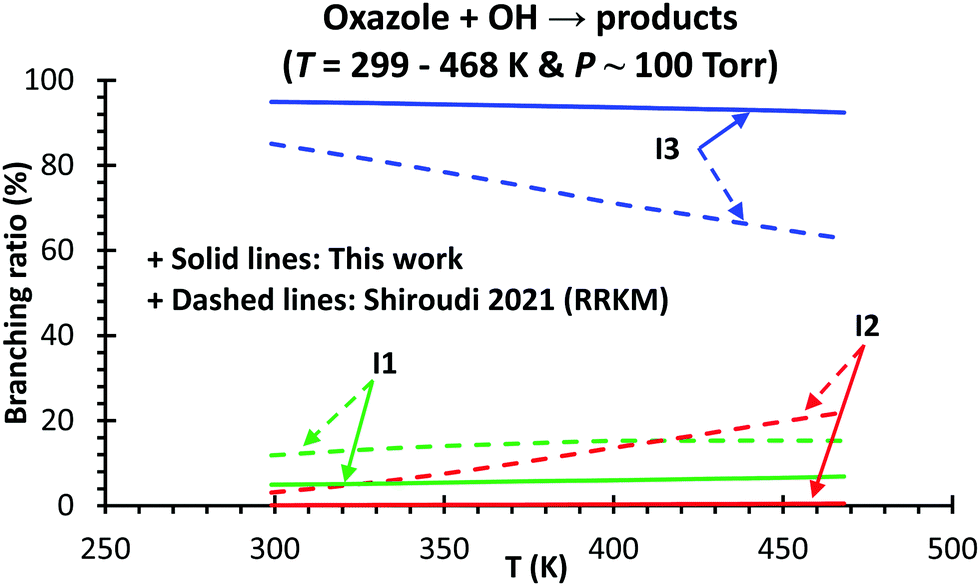 | ||
| Fig. 6 Comparison between our predicted (solid lines) and Shiroudi's (dashed lines) branching ratios as a function of temperature at P = 100 torr for the title reaction, oxazole + OH → products. The calculations were carried out using the RC1 model (see the main text for the model details). | ||
With the PES explored at the same M06-2X/aug-cc-pVTZ level, the difference between our numbers and the values suggested by Shiroudi et al. (cf.Fig. 4) is mainly due to the use of two different kinetic models. Specifically, our models (i.e., models RC1&RC2 and RC1) explicitly include all species involved to mimic the dynamics of the title reaction without introducing the hypothesis of a fast pre-equilibrium between the reactants and the van der Waals pre-reactive complexes (e.g., 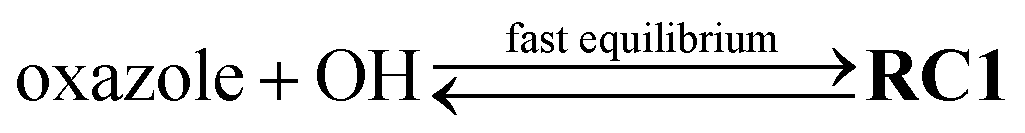 ) as introduced in Shiroudi et al.'s model (as in the reaction scheme earlier proposed by Singleton et al.6). Such a fast equilibrium hypothesis is found to be invalid for this title system. For example, at T = 300 K, the [RC1]t/[OH]t × [oxazole]t ratio does not reach its equilibrium value until 10−8 seconds (cf.Fig. 7). Therefore, the model proposed by Shiroudi et al. is likely to be compromised, at least under the considered conditions. Note that the exclusion of RC1 certainly affects the detailed reaction mechanism; for example, the negative temperature dependence of the rate constants is not observed if the reaction complexes are excluded from our kinetic models.
) as introduced in Shiroudi et al.'s model (as in the reaction scheme earlier proposed by Singleton et al.6). Such a fast equilibrium hypothesis is found to be invalid for this title system. For example, at T = 300 K, the [RC1]t/[OH]t × [oxazole]t ratio does not reach its equilibrium value until 10−8 seconds (cf.Fig. 7). Therefore, the model proposed by Shiroudi et al. is likely to be compromised, at least under the considered conditions. Note that the exclusion of RC1 certainly affects the detailed reaction mechanism; for example, the negative temperature dependence of the rate constants is not observed if the reaction complexes are excluded from our kinetic models.
 | ||
| Fig. 7 Plot of [RC1]t/[OH]t × [oxazole]t as a function of time at T = 300 K (in logarithm scale) and 760 torr, using the stochastic approach (108 trials) with [oxazole]0/[Ar] = 10−4 and [oxazole]0 ≫ [OH]0 (Ar is the bath gas). The simulation was carried out using the RC1 model (see the main text). Time is in the logarithm scale. | ||
The overall rate constants, ktot(T), based on two electronic structure methods (i.e., M06-2X/aug-cc-pVTZ and ROCBS-QB3//M06-2X/aug-cc-pVTZ) are shown in Fig. 8. Surprisingly, the calculated ktot(T) using the PES constructed at the M06-2X/aug-cc-pVTZ level is better than at the ROCBS-QB3//M06-2X/aug-cc-pVTZ method when compared to the experimental data with MADs of 9.6 and 83.7%, respectively. Since transition state TS6 (the main channel, cf.Fig. 1 and 3) is found at −1.6 and −2.2 kcal mol−1 (cf. Table S3, ESI†) at the M06-2X/aug-cc-pVTZ and ROCBS-QB3//M06-2X/aug-cc-pVTZ level, respectively, the calculated ktot(T) with the former method is slower than with the later one by an average factor of ∼2.0 in the range of T = 299–468 K & P = 100 torr. It is worth noting that the uncertainty of a kinetic model comes from different sources such as the PES and RRKM-ME model (including any hypothesis introduced); therefore, using a low level of theory might provide better kinetic data if the errors compensate the missing physics in such a model. Within this context, the M06-2X/aug-cc-pVTZ level is found to be suitable for investigating the kinetics of similar reactions (e.g., imidazole4/pyrrole5/aniline7 + OH).
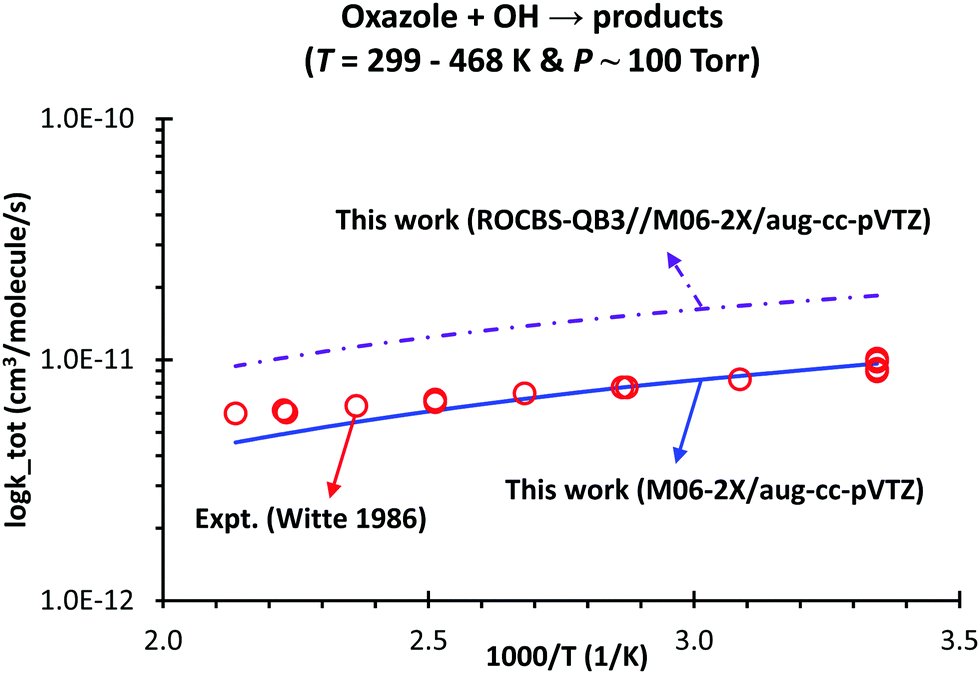 | ||
| Fig. 8 Comparison between the experimental values and our predicted total rate coefficients, ktot, of the oxazole + OH → products reaction as a function of temperature and at P = 100 torr using the M06-2X/aug-cc-pVTZ and ROCSB-QB3//M06-2X/aug-cc-pVTZ methods. The simulation was carried out using the RC1 model (see the main text). | ||
In conclusion, the use of the rigorous rate model (i.e., the stochastic RRKM-based ME statistical model with the corrections of the HIR and Eckart tunneling treatments) on the M06-2X/aug-cc-pVTZ potential energy surface helps to resolve the large kinetic discrepancy between the theoretical1 and experimental2,3 studies for the title reaction without introducing any assumption as in the previous calculations. Such an achievement confidently reveals detailed mechanistic insights via the model to advance atmospheric applications related to the title reaction.
Computational details
All electronic structures were calculated using the Gaussian 09 program.8 Optimization and frequency calculations were obtained using the M06-2X9,10/aug-cc-pVTZ11 level of theory, which is found to be reliable in studying the detailed kinetics of similar systems (e.g., imidazole4/pyrrole5/aniline7 + OH). ROCBS-QB312 single-point energy calculations were carried out for all optimized structures at the M06-2X/aug-cc-pVTZ level (denoted as ROCBS-QB3//M06-2X/aug-cc-pVTZ) to assess the kinetic/thermodynamic findings of the DFT method of M06-2X. Note that the zero-point vibrational energy (ZPE) corrections were scaled by a factor of 0.97113 prior to calculating the thermal corrections for enthalpy and entropy.The hindered internal rotation (HIR) corrections were included in thermodynamic/kinetic analyses. The M06-2X/cc-pVDZ level was used to obtain the hindrance potentials of the rotation around the C–O and O–H “single” bonds (cf. Fig. S4, ESI†) via relaxed surface scans. The HIR parameters were determined with the use of the Multi-Species Multi-Channels (MSMC) graphical user interface.14,15 The detailed procedure of the HIR treatment was presented in our previous studies.16 The tunneling correction using the Eckart tunneling model17 was included in the rate constant calculations for all elementary reaction channels. Note that the spin–orbit splitting correction (∼139.7 cm−1)18 was also considered in calculating the partition function of OH radicals.
The microcanonical rate constants k(E) of the barrierless channels (e.g., the association of oxazole and OH radicals to form the reactant-complexes, RCs, cf.Fig. 1) were treated using the Inverse Laplace Transform (ILT) technique19 with the high-pressure-limit rate constant, k∞(T), of 4.0 × 10−10 cm3 per molecule per s, with the assumed temperature-independence based on the long-range transition state (LR-TST).20 It has been shown that this approach was successfully used to capture the rate coefficients for similar systems.4,5,7 The temperature-dependent expression of 〈ΔEdown〉 = 260.0 × (T/298)0.8 cm−1 was used for the bath gas of Ar4,5 regarding the energy-transfer manner. The Lennard-Jones parameters of ε/kB = 113.5 K and σ = 3.465 Å were used for Ar21 whereas ε/kB = 467.4 K and σ = 4.6 Å were adopted for the [oxazole–OH] complex and its adducts.1 The stochastic22,23 RRKM-based ME24 statistical rate model (with the trial number of 108), including the corrections of HIR and tunneling treatments, was used for the kinetic analyses. The MSMC code25 was used for all thermodynamic and kinetic calculations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by Vietnam National University HoChiMinh City (VNU-HCM) under the grant number B2018-28-03. TVTM was funded by Vingroup Joint Stock Company and supported by the Domestic PhD Scholarship Programme of Vingroup Innovation Foundation (VINIF), Vingroup Big Data Institute (VINBIGDATA) (VINIF.2020.TS.96).References
- A. Shiroudi, M. Abdel-Rahman, A. El-Nahas and M. Altarawneh, New J. Chem., 2021, 45, 2237–2248 RSC.
- F. Witte and C. Zetzsch, 9th International Symposium on Gas Kinetics, University of Bordeaux, Bordeaux, France, 1986 Search PubMed.
- R. Atkinson, J. Phys. Chem. Ref. Data, Monogr., 1989, 1, 1–246 Search PubMed.
- T. V. Mai and L. K. Huynh, Phys. Chem. Chem. Phys., 2019, 21, 21162–21165 RSC.
- T. V. T. Mai, H. T. Nguyen and L. K. Huynh, Chemosphere, 2020, 263, 127850 CrossRef PubMed.
- D. L. Singleton and R. J. Cvetanovic, J. Am. Chem. Soc., 1976, 98, 6812–6819 CrossRef CAS.
- T. V.-T. Mai, T. T.-D. Nguyen, H. T. Nguyen, T. T. Nguyen and L. K. Huynh, Environ. Sci. Technol., 2021, 55, 7858–7868 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- G. P. Wood, L. Radom, G. A. Petersson, E. C. Barnes, M. J. Frisch and J. A. Montgomery, Jr., J. Chem. Phys., 2006, 125, 094106 CrossRef PubMed.
- I. M. Alecu, J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2872–2887 CrossRef CAS PubMed.
- T. H. M. Le, S. T. Do and L. K. Huynh, Comput. Theor. Chem., 2017, 1100, 61–69 CrossRef CAS.
- T. H. M. Le, T. T. Tran and L. K. Huynh, Chemom. Intell. Lab. Syst., 2018, 172, 10–16 CrossRef CAS.
- T. V. T. Mai, M. v. Duong, X. T. Le, L. K. Huynh and A. Ratkiewicz, Struct. Chem., 2014, 25, 1495–1503 CrossRef CAS.
- C. Eckart, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- J. A. Coxon and S. C. Foster, J. Mol. Spectrosc., 1982, 91, 243–254 CrossRef CAS.
- S. H. Robertson, M. J. Pilling, D. L. Baulch and N. J. B. Green, J. Phys. Chem., 1995, 99, 13452–13460 CrossRef CAS.
- Y. Georgievskii and S. J. Klippenstein, J. Chem. Phys., 2005, 122, 194103 CrossRef PubMed.
- H. Hippler, J. Chem. Phys., 1983, 78, 6709 CrossRef CAS.
- D. T. Gillespie, J. Comput. Phys., 1976, 22, 403–434 CrossRef CAS.
- D. T. Gillespie, A. Hellander and L. R. Petzold, J. Chem. Phys., 2013, 138, 170901 CrossRef PubMed.
- J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2006, 110, 10528–10544 CrossRef CAS PubMed.
- M. v. Duong, H. T. Nguyen, N. Truong, T. N. M. Le and L. K. Huynh, Int. J. Chem. Kinet., 2015, 47, 564–575 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nj01020d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |