
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-doped mesoporous ZrO2 catalyzed chemoselective synthesis of allylic alcohols from Meerwein–Ponndorf–Verley reduction of α,β-unsaturated aldehydes†
Christianah Aarinola
Akinnawo
,
Ndzondelelo
Bingwa
and
Reinout
Meijboom
 *
*
Research Centre for Synthesis and Catalysis, Department of Chemical Sciences, University of Johannesburg, P. O. Box 524, Auckland Park, Johannesburg 2006, South Africa. E-mail: rmeijboom@uj.ac.za; Fax: +27 11 559 2819; Tel: +27 11 559 2367
First published on 12th April 2021
Abstract
Meerwein–Ponndorf–Verley reduction (MPVr) is a sustainable route for the chemoselective transformation of α,β-unsaturated aldehydes. However, tailoring ZrO2 catalysts for improved surface-active sites and maximum performance in the MPV reaction is still a challenge. Here, we synthesized mesoporous zirconia (ZrO2) and metal-doped zirconia (M_ZrO2, M = Cr, Mn, Fe, and Ni). The incorporation of metal dopants into zirconia's crystal framework alters its physico-chemical properties such as surface area and total acidity-basicity. The prepared catalysts were evaluated in the MPVr using 2-propanol as a hydrogen donor under mild reaction conditions. The catalysts' remarkable reactivity depends mainly on their surface mesostructure's intrinsic properties rather than the specific surface area. Cr_ZrO2, which is stable and sustainable, presented superior activity and 100% selectivity to unsaturated alcohols. The synergistic effect between Cr and Zr species in the binary oxide facilitated the Lewis acidity-induced performance of the Cr_ZrO2 catalyst. Our work presents the first innovative application of a well-designed mesoporous Cr_ZrO2 in the green synthesis of unsaturated alcohols with exceptional reactivity.
Introduction
The chemoselective reduction of α,β-unsaturated aldehydes to their corresponding allylic alcohols is one of the significant chemical transformations in synthetic organic chemistry. The unsaturated allylic alcohol (UAA) produced through this process is widely utilized as the primary feedstock in food and perfumery industries and intermediates in pharmaceutical industries.1,2 However, the reaction is classically carried out using gaseous hydrogen in the presence of noble metals as catalysts with significant limitations such as high-pressure requirements and low selectivity to UAA.3 The high-pressure involved in such a chemical process requires expensive equipment and an elaborate experimental set-up with associated safety risks.4,5 The problems associated with these classical methods could be avoided in the Meerwein–Ponndorf–Verley (MPV) reduction.The MPV is an alternative means of selective reduction of unsaturated carbonyls at atmospheric pressure using safe and readily available secondary alcohol as a hydrogen donor instead of high-pressure molecular hydrogen or hazardous reduction reagents such as LiAlH4 and NaBH4.6 The MPV among several reduction routes for functional group conversion has the following prevailing advantages: (i) easy to handle hydrogen donor without the requirement of heavy gas containment, (ii) cheap and environmentally friendly source of hydrogen (iii) enhanced selectivity under mild reaction conditions (iv) safer process (v) minimized waste (vi) reduced cost, e.g., maintenance, separation, and other production logistics (vii) economical and environmentally sustainable process.7,8 However, to force the equilibrium reaction towards the formation of UAA, the MPV reduction requires excess sacrificial H-donating molecule,9 which generates by-product.10,11 The product separation through distillation is a tall task owing to the close boiling points of the α,β-unsaturated aldehydes or ketones, the UAAs, and the sacrificial alcohol. Hence, efforts should be directed towards attaining 100% selectivity and product yield and recycling the by-product generated from the sacrificial hydride donor.11
The MPV reaction is highly chemoselective to the reduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in the α,β-unsaturated aldehydes but, the hydrogenation of conjugated CC is preferentially favorable thermodynamically and kinetically over the CO bond.3,12 There is an ongoing research effort to develop an efficient catalyst system mainly for the chemoselective hydrogenation of CO in the liquid phase at atmospheric pressure. The MPV reaction's generally accepted reaction mechanism involves a cyclic six-membered transition state (a determining step in the reaction rate) when both reactants simultaneously coordinate to the same Lewis acidic sites (Scheme 2). Various metal oxides exhibiting either Lewis acidic or Brønsted acidic sites, or both acidic sites are active in the MPV reaction. However, designing and developing a selective and efficient catalyst with desirable concentration and strength of acid sites has become an essential issue in selective hydrogenation of α,β-unsaturated aldehydes. Despite several studies, hydrogenation of α,β-unsaturated aldehydes in the absence of additives and/or co-solvent, with complete selectivity to UAA at atmospheric pressure, remains a challenge and requires more research attention.4,13
O bond in the α,β-unsaturated aldehydes but, the hydrogenation of conjugated CC is preferentially favorable thermodynamically and kinetically over the CO bond.3,12 There is an ongoing research effort to develop an efficient catalyst system mainly for the chemoselective hydrogenation of CO in the liquid phase at atmospheric pressure. The MPV reaction's generally accepted reaction mechanism involves a cyclic six-membered transition state (a determining step in the reaction rate) when both reactants simultaneously coordinate to the same Lewis acidic sites (Scheme 2). Various metal oxides exhibiting either Lewis acidic or Brønsted acidic sites, or both acidic sites are active in the MPV reaction. However, designing and developing a selective and efficient catalyst with desirable concentration and strength of acid sites has become an essential issue in selective hydrogenation of α,β-unsaturated aldehydes. Despite several studies, hydrogenation of α,β-unsaturated aldehydes in the absence of additives and/or co-solvent, with complete selectivity to UAA at atmospheric pressure, remains a challenge and requires more research attention.4,13
Conventionally, the MPV reaction is carried out in the homogeneous system using metal (Al, Zr) alkoxides as catalysts.14,15 They suffer deactivation, which is peculiar to homogeneous catalysts due to the difficulty separating them from the end products; hence, they are non-reusable. Besides, most homogeneous systems require base additives for their activation.11 These challenges encountered during the utilization of homogeneous catalysts have paved the way for increased research to design sustainable heterogeneous catalysts. Heterogeneous catalysts offer the advantage of the easy recovery of product and catalyst as well as possible reuse of the catalyst system, among which mesoporous metal oxides were preferred.
Metal oxide such as ZrO2,16–18 CeO2–ZrO2,4 MgO–ZrO2,19 Al2O3, Ga2O3 and In2O3 modified ZrO2,16 SiO2, ZrO2 and ZrO2/SBA-15,20,21 ZrO2–La2O3/MCM-41,22 γ-Al2O3, TiO2, and TiO2/γ-Al2O323 as an active phase has shown excellent catalytic activities in MPV reactions. The catalytic performance of metal oxides depends on the surface's structural features or environments, influencing their adsorption strength and activation of the adsorbates.24,25 Among a wide range of heterogeneous catalytic species utilized in MPV reduction, zirconia showed the most promising performance.26 The potential catalytic activity of zirconia has increased its application in catalysis. It is known for its acidic properties,27 high thermal stability, and corrosion resistance.7,28 To further emphasize this fact, Alvarez-Rodriguez et al. pointed out that the zirconia catalyst is more effective than other conventional catalysts such as alumina or silica to produce unsaturated allylic alcohol from citral and cinnamaldehyde.29 The surface active sites of zirconia has been verified to improve by addition of dopant.30 Xie et al. prepared Cr–ZrO2 using acid–base pair pathway with evaporation-induced self-assembly.31 They found out that the catalytic performance in dehydrogenation of propane with CO2 was influenced by the enhanced surface morphology, acidity and redox property of the Cr–ZrO2 catalysts with different Cr doping percentage. Also, in the report of Wu et al. the catalytic activity of the Cr2O3–ZrO2 prepared hydrothermally was attributed to the presence of Cr6+ species.32
From the above literature survey coupled with the crucial need for sustainable and environmentally benign catalytic hydrogenation processes,11 it would be interesting to develop novel mesoporous zirconia-based catalyst systems with increased acidic sites for the transfer hydrogenation process under mild reaction conditions with high selectivity to unsaturated allylic alcohol. Also, a stable catalytic MPV process without the use of additives is imperative for the green and clean synthesis of UAA. The surface properties of zirconia, a major determining factor of its catalytic activity, may depend on the method of synthesis.33 Several methods of preparing mesoporous zirconia have been reported, but the inverse micelles soft-templated technique is poorly represented. The inverse surfactant micelle approach enables the control of the surfactant-transition metal interactions, hydrolysis, and condensation of inorganic sols.34,35 Transition metal oxides prepared using the inverse micelles approach are known for their exceptional structural properties such as large surface area (SBET), porosity, and crystallinity. The large surface area avails the active sites for better catalytic activity than other porous materials with low SBET.
Hence, in this study, a triblock copolymer P-123 was employed as a structure-directing agent in the inverse micelles approach34,35 for the synthesis of mesoporous zirconia and metal-doped zirconia. The mesoporous zirconia-based materials' catalytic potential was investigated as active phases in the MPV reduction of selected aromatic aldehydes to their corresponding alcohols (Scheme 1) without any additives or co-solvent. We optimized the MPV process variables. A deep insight into the effect of cation dopant on the physicochemical properties of ZrO2, including crystal phases, structural morphology, surface area, and acidity-basicity, was evaluated. Furthermore, the relativity of these properties to its catalytic performance in the MPV process was also revealed. Elucidation of the activity was based on the observed pseudo-first-order rate constants (kobs) and calculated conversion Equations S1-3. Also, we show that all the synthesized catalysts exhibit excellent selectivity to the unsaturated allylic alcohol in 2-propanol as H-donor at 80 °C, 450 rpm, and atmospheric pressure. To the best of our knowledge, this is the first work that applied mesoporous Cr_ZrO2 prepared via inverse micelle for the MPV process. The mesoporous Cr_ZrO2 showed considerable conversion with 100% selectivity to the unsaturated alcohols in reducing citral as a model reaction and other aldehydes. The synthesized Cr_ZrO2 is a sustainable catalyst for the Meerwein–Ponndorf–Verley process.
 | ||
| Scheme 1 MPV reduction of α,β-unsaturated aldehydes to their corresponding unsaturated allylic alcohol over the mesoporous zirconia-based catalyst. | ||
2. Experimental
2.1. Materials
Nitric acid (HNO3) (69–70%) was purchased from Rochelle Chemicals (RSA). 1-Butanol (99.8%), ethanol (99.9%), poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (PEO20-PPO70-PEO20 or Pluronic P-123), zirconium(IV) butoxide solution (80% in 1-butanol), manganese(II) nitrate tetrahydrate (97%), nickel(II) nitrate hexahydrate (99%), cinnamaldehyde (99%), citral (mixture of cis- and trans-isomers) (96%), benzaldehyde (99%), crotonaldehyde (99%), furfural (99%), 2-propanol (99.5%), decane (99%) were all purchased from Sigma-Aldrich. Ferric nitrate nonahydrate was purchased from SRL chemicals, and chromic(III) nitrate nonahydrate (98%) was purchased from UNIVAR, SAAR CHEM pty. All chemicals were of analytical grade and used as received without further purification.2.2. Synthesis of catalysts
The procedure already reported was followed for the synthesis of ZrO2.34 Briefly, 15.34 g (0.040 mol) of zirconium butoxide was added to a solution containing 25.04 g (0.336 mol) 1-butanol, 4.0 g (6.8 × 10−4 mol) of P-123, and 4.0 g (0.064 mol) HNO3. The mixture was stirred overnight, and the clear gel was dried in an oven at 120 °C for 4 h. The yellow glassy thin flakes obtained were calcined in air at 350 °C for 5 h with a heating rate of 2 °C min−1. The metal-doped zirconia was synthesized by adding the metal dopant (Cr, Mn, Fe, and Ni) precursor to zirconium butoxide in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. As in the case of pure ZrO2, the same thermal treatment was applied (Scheme S1, ESI†). The samples are tagged M_ZrO2, M = metal dopant.
:4. As in the case of pure ZrO2, the same thermal treatment was applied (Scheme S1, ESI†). The samples are tagged M_ZrO2, M = metal dopant.
2.3. Characterization of catalysts
Powder X-ray diffraction (p-XRD) analyses were carried out on a Philips XPERT-PRO diffractometer system operating with Cu Kα1, Kα2, and Ni Kβ radiation (λ = 1.5406, 1.54443 and 1.39225 Å, respectively) at 25 °C. Both low and wide 2θ range (i.e., 4–90°) angle diffraction patterns with a step of 0.170° were measured. The Debye–Scherrer equation (eqn (S1), ESI†) was used to calculate the mean crystallite size. Micrometric ASAP 2460 sorption system gave the nitrogen sorption measurements. The samples were firstly degassed under flowing nitrogen at 100 °C for 18 h and under vacuum for 10 h at the same temperature before the experiments to remove any physisorbed moisture. The surface areas were calculated using the Brunauer–Emmett–Teller (BET) method. Transmission electron microscopy (TEM) for the confirmation of the mesostructure was achieved on a JEOL Jem-2100F electron microscope with an accelerating voltage of 200 kV. The pore diameter was measured using ImageJ software. A 10 mg of the catalyst was sonicated in 1 ml of methanol for 1 h, and a drop of the suspension was placed on a carbon-coated Cu-grid then allowed to dry before the TEM analysis. The prepared samples' surface morphology was identified on a Tescan Vega 3 LMH scanning electron microscope (SEM) using a scattering electron detector with a high voltage of 20.0 kV. Prior to analysis, the samples were placed on an aluminum stub and carbon-coated in an Agar Turbo carbon coater. The quantity of dopant Mn+ was verified with energy-disperse X-ray spectroscopy (EDX). The distribution of the metal species was identified by elemental mapping on SEM. Also, the dopant content in the solid samples was measured using a Spectro Acros ICP-OES spectrometer. The Fourier transform infrared spectroscopy (FTIR) spectra of the samples were recorded on a Bruker FTIR Alpha spectrometer in the 4000–400 cm−1 region. The samples were mixed with KBr, and the analysis was performed in the transmission mode under ambient conditions. The NH3/CO2 temperature-programmed desorption (TPD) studies to determine the materials' acidity or basicity were performed on a Micromeritics AutoChem II. About 0.2 g of the sample was loaded in a quartz tube reactor. The loaded sample surface was degassed in a He gas flow at 200 °C for 1 h before the TPD measurement. We used a mixture of NH3 or CO2 and helium in the ratio of 10:90 as the probe gas at a flow rate of 50 ml min−1. Measurements were performed in the temperature range of 30–550 °C at a temperature ramp of 10 °C min−1 and 3 °C min−1 for TPD-NH3 and TPD-CO2, respectively. For identifying the Lewis and Brønsted acid sites on the samples, adsorbed pyridine FTIR analysis was carried out. Before the analysis, ∼0.03 g of the sample was activated by degassing under gaseous nitrogen at 300 °C for an hour and cooled to room temperature. After that, the activated catalyst was contacted with pyridine (200 μl) at 120 °C for 30 min. Subsequently, the physisorbed pyridine was evacuated under vacuum at ambient temperature for an hour,36 and the sample was analyzed on a Bruker FTIR Alpha spectrometer. The H2-TPR (hydrogen-temperature programmed reduction) analysis was conducted on the same Micromeritics Autochem II. Approximately 30 mg of the catalyst was loaded in the quartz tube reactor and pretreated under Argon flow at 200 °C for 1 h to ensure the catalyst surface is clean before each test. After the pretreatment, H2/Ar (10:90) was passed over the catalyst at a 50 ml min−1 flow rate. The measurements were performed within the ambient temperature to 800 °C with a 10 °C min−1 ramping rate. The prepared samples' thermal stability test was performed on a PerkinElmer STA 6000 thermogravimetric analyzer (TGA). The degradation study temperature was varied from 25–900 °C with a ramping rate of 10 °C min−1 under air at a 20 ml min−1 flow rate. The UV-vis spectra of the samples were obtained on a microplate reader (PowerWave HT.Biotek microplate reader). Before obtaining the UV-vis absorption spectra, about 30 mg of the solid sample was sonicated in 2 ml methanol and decanted. After that, the supernatant was analyzed using a 24-well plate.
2.4. Evaluation of catalytic performance
The liquid phase MPV reduction experiments were performed on a carousel reaction station multi-reactor (Radley Discovery Technologies) with twelve 50 ml vials. The 50 ml reactor vial was charged with 0.4 g M-ZrO2, 2.50 mmol of aldehyde, 1.00 mmol (200 μl) decane as an internal standard, and 130 mmol (10 ml) 2-propanol. Followed by reflux at 80 °C and stirring at 450 rpm with a 16.5 mm crossbar stirrer. After filtration, the filtrate was analyzed on a Shimadzu GC-2010 with flame ionization detector (FID) using a capillary column (Restek RTX-5; 30 m, 0.25 mm ID, thickness 0.25 μm) in N2 carrier gas. The injection port and FID temperature were maintained at 200 °C and 350 °C, respectively. The products were further confirmed by a Shimadzu GC-MS QP-2010 using the same capillary column with the injection temperature at 200 °C. The ion source and interface temperatures were 200 °C and 250 °C, respectively. For the GC FID and MS, the column oven temperature program started at 40 °C (hold 2 min), then programmed at 20 °C min−1 to 280 °C (hold 5 min); the total analytical time was 19 min (details in Section S1.3.2, ESI†). The catalysts were screened with the MPV reduction of citral as a model reaction. The catalyst exhibiting the best activity was chosen for the transfer-hydrogenation of selected α,β-unsaturated aldehydes. The substrate conversion, product selectivity, and normalized activity were calculated (eqn (S2)–(S5), ESI†). Furthermore, the observed kobs for each experiment were calculated using Kinetic studio version 2.08 software. For the recyclability study, the catalyst was pretreated by calcining at 350 °C/5 h before reuse.3. Results
3.1. Surface properties of the M_ZrO2 catalysts
Fig. 1 illustrates the wide-angle XRD patterns of pure ZrO2 and M_ZrO2 samples. The diffractogram of pure ZrO2 correspond to a tetragonal crystal phase t-ZrO2 (JCPDS-89-7710), indexed at 2θ = 30.4° (101), 35.3° (110), 50.4° (112), 60.1° (211) and 73.8° (220). No extra peaks were detected, which confirms the purity of the synthesized t-ZrO2. Upon doping t-ZrO2 with a metal atom, an isomorphous substitution was observed. Suggesting that some surface Zr atoms in the M_ZrO2 samples are substituted with the dopant atoms and the formation of a homogeneous solid solution of binary MxOy–ZrO2. This claim is supported by the gradual shift of the peak at 30.4° (101) of ZrO2 towards a higher 2θ degree. No identifiable peak is associated with the dopants, which is an indication that the dopants are well incorporated into the ZrO2 matrix and high dispersion of the dopant species. The incorporation of cation into the crystal framework of zirconia significantly influenced its crystallinity. The tetragonal structure with reduced peak intensity remains in the presence of Mn and Fe, while we observed crystal distortion in the case of Ni and Cr dopants. The dopant species weakened the t-ZrO2 peaks in the case of Mn and Fe and were significantly destroyed in Ni and Cr, forming disordered ZrO2. This observation implies the degree of dopant incorporation and distribution and Mn+–Zr4+ interaction. The observed broader and weaker diffraction peaks in Mn_ZrO2 and Fe_ZrO2 explain the occurrence of higher surface area in correlation with the host ZrO2. Also, the crystallite size of t-ZrO2 (5.50 nm) decreased upon doping with Mn_ZrO2 (1.37 nm) and Fe_ZrO2 (2.59 nm). The decrease in the crystallite size possibly contributed to expanding the surface area, as depicted in Table 1. | ||
| Fig. 1 Wide-angle pXRD patterns of the pure ZrO2 and M_ZrO2 catalysts. | ||
| Entry | Meso | Dopant content (wt%) | BET surface area (m2 g−1) | Pore diameter (nm) | Pore volume (cm3 g−1) | Crystallite size (nm) | Crystal structure | ||
|---|---|---|---|---|---|---|---|---|---|
| a | b | c | d | ||||||
| Dopant content determined by EDX = a, ICP-OES = b, pore diameter determined by N2 sorption analysis = c, TEM = d, NA: not applicable. | |||||||||
| 1 | ZrO2 | — | — | 206 | 2.53 | 2.57 | 0.10 | 5.50 | Tetragonal |
| 2 | Cr_ZrO2 | 7.1 | 8.2 | 190 | 3.04 | 3.06 | 0.14 | NA | Amorphous |
| 3 | Mn_ZrO2 | 8.3 | 7.4 | 221 | 3.63 | 3.62 | 0.26 | 1.37 | Tetragonal |
| 4 | Fe_ZrO2 | 6.8 | 6.9 | 223 | 2.73 | 2.64 | 0.14 | 2.59 | Tetragonal |
| 5 | Ni_ZrO2 | 9.7 | 6.1 | 193 | 2.79 | 2.59 | 0.14 | NA | Amorphous |
| 6 | Spent Cr_ZrO2 | — | 8.1 | 133 | 2.89 | 2.48 | 0.10 | NA | — |
The experiments carried out to confirm the surface structure and porosity of pure and doped ZrO2 using nitrogen sorption analysis shown in Table 1 revealed that the pure ZrO2 exhibited pores with an average diameter of 2.53 nm within the meso range with a large BET surface area (SBET) 206 m2 g−1. The BET surface area of the M_ZrO2 catalysts depends on the final catalyst's crystallinity, which is a function of the dopant's nature. The SBET of pure ZrO2 (206 m2 g−1) increased when modified with Mn (221 m2 g−1) and Fe (223 m2 g−1) but decreased in the case of Cr (190 m2 g−1) and Ni (193 m2 g−1) dopants. The pore sizes are within the range of 2.5–3.6 nm, a characteristic feature of mesoporous oxides. The metal dopants' addition distinctly enlarged the pore size 2.5–3.6 nm and the pore volume 0.1 to 0.26 cm3 g−1. The mesostructuring is associated with the metal–metal grain boundary adhesion/expansion during the formation of oxo-metal clusters at the stage of sol condensation, diffusion of volatile species, and subsequent removal of the surfactant template. The degree of grain segregation is directly proportional to the porosity of the material.37–39 Also, the larger the porosity, the slower the grain growth, as depicted in the correlation of the pore size and the crystallite size (Table 1). The observed physisorption isotherms for all the catalysts in Fig. 2a are typical of Type IV compared with the IUPAC classification reported by Thommes et al.40,41 This further indicates that all the catalysts are mesoporous materials with thin capillary pores. The ZrO2 catalysts exhibited hysteresis loops P/P0 typical of H2 type indicating the occurrence of cavitation controlled evaporation; this depicts that the materials possess a heterogeneous pore network with the neck size (W) distribution much more narrow than the size distribution of the cavities (Wc), that is, W < Wc.40–42 The pore size distribution of pure zirconia and M_ZrO2 are shown in Fig. 2b. All the catalysts showed unimodal pore size distribution, with the M_ZrO2 exhibiting a narrower pore distribution compared to the pure ZrO2 catalyst. Significantly, the corresponding BET surface area, pore-size distribution, and pore volume is an indication that the catalysts are mesoporous with high surface area, and the surface mesostructure of ZrO2 could be tailored by adding foreign atomic specie.
 | ||
| Fig. 2 (a) N2 adsorption–desorption isotherms (b) pore size distribution of pure ZrO2 and M_ZrO2 catalysts. | ||
Moreover, the transmission electron microscopy (TEM) and high transmission (HRTEM) images of the zirconia systems as displayed in Fig. 3a, b, d, e and Fig. S1, S2 (ESI†) reveal that they are made of nanosized particles with intraparticle voids that were preserved upon the addition of different metal ions. The TEM images (Fig. 3a, d and Fig. S1, S2, ESI†) show that the pores are well distributed in the ZrO2 matrix, supporting the evidence of the presence of pore and the pore enlargement upon doping as presented by the N2 sorption experiment (Table 1 and Fig. 2b). A similar observation was reported in the work of Xie et al.31
 | ||
| Fig. 3 (a–c) TEM, HRTEM, and SEM images of the meso-ZrO2, respectively; (d–f) TEM, HRTEM, and SEM images of the Cr_ZrO2, respectively; (g) EDX mapping of Cr_ZrO2 showing the metal distribution, and (h) EDX of Cr_ZrO2 indicating the weight percentage of the Cr content. | ||
The surface morphologies of the catalysts are shown in Fig. 3c, f, and Fig. S3 (ESI†). Modification of the t-ZrO2 surface morphology upon the introduction of dopants is insignificant; this is due to the homogeneity of the M_ZrO2 solid structures, as observed in the XRD patterns. The EDX mapping (Fig. 3g and Fig. S4, ESI†) reveals the uniform distribution of the dopant species in the ZrO2 matrix. The SEM-EDX analyses (Fig. 3h and Fig. S5, ESI†) confirm the metal constituents of the synthesized ZrO2 systems and indicate the actual weight percentage of the dopants incorporated.
The thermal stability of the catalysts (Fig. 4a) suggests that the catalysts are thermally stable. The dopant species enhance the thermal stability of the host ZrO2 (8%), except Mn_ZrO2, which shows an approximately similar weight loss of 8.3%. The 5% degradation between 30–257 °C is attributed to the removal of adsorbed surface and bulk water molecules, while above 257 °C could be classified as degradation due to the decomposition of the organic surfactant residue. Above 683 °C, the spectra seemingly flatten out, suggesting minimal or no decomposition and the inorganic material's stability. The samples are also stable in the catalytic characterization and application temperature within this study's scope.
 | ||
| Fig. 4 (a) TGA spectra, (b) H2-TPR profiles of the ZrO2, and M_ZrO2 catalysts. | ||
The catalysts' hydrogen consumption temperatures (Fig. 4b) and the minimum temperatures (Table 2) suggest the catalysts' reducibility. The pure zirconia showed a poor hydrogen uptake with a small peak around 652 °C, corresponding to the reduction of bulk lattice oxygen of zirconia. We found that doping enhanced the reducibility of ZrO2, with a significant shift in its reduction peak to lower temperatures; this depicts the rate of the redox reaction. However, the degree of reducibility is dependent on the kind of doping species. The reduction peaks between 261–360 °C likely represent the reduction of the metal dopant species: Cr3+ → Cr2+, Fe3+ → Fe2+, Mn2+ → Mn0, Ni2+ → Ni0. The reduction peaks demonstrated in the region of 430–486 °C and above 600 °C are typical of surface and bulk reduction of lattice oxygen of zirconia, respectively. The Cr_ZrO2 demonstrated the superior reduction capacity of surface interaction with hydrogen at the lowest temperature of 261 °C. This is likely due to the strong synergistic interaction between the Cr3+ and Zr4+ (CrxOy–ZrO2 solid solution). The H2-TPR data supported the superior catalytic activity of Cr–Zr active phase species for the H abstraction-release mechanism in the MPV dehydrogenation–hydrogenation reaction.
| Entry | Catalyst | Total acidity (mmolNH3 g−1) | Acidic site density (μmol m−2) | Total basicity (mmolCO2 g−1) | Basic site density (μmol m−2) | Ratio acid:base |
T H2 consumption (°C) |
|---|---|---|---|---|---|---|---|
| 1 | ZrO2 | 0.32 | 0.0016 | 0.86 | 0.0042 | 0.37 | 652 |
| 2 | Cr_ZrO2 | 0.66 | 0.0035 | 0.26 | 0.0014 | 2.54 | 261 |
| 3 | Mn_ZrO2 | 0.47 | 0.0021 | 1.12 | 0.0051 | 0.42 | 270 |
| 4 | Fe_ZrO2 | 0.41 | 0.0018 | 0.93 | 0.0042 | 0.44 | 331 |
| 5 | Ni_ZrO2 | 0.52 | 0.0027 | 0.95 | 0.0049 | 0.55 | 360 |
We investigated the surface acid–base properties of the prepared catalysts by NH3- and CO2-TPD analyses. The spectra are depicted in Fig. 5 and 6, respectively. The total acidity and basicity, along with their density, are summarized in Table 2. The total acidity and basicity were obtained from the peak area of NH3 and CO2 desorption, respectively. The acid or base sites density was derived by dividing the total acidity or basicity by the surface area (Table 1). The NH3/CO2 desorption peak around 200 °C represents the acid/base sites of a weak strength, from 200 °C to 350 °C depicts medium strength acid/base sites, and above 400 °C corresponds to strong acid/base sites.43
 | ||
| Fig. 5 CO2-TPD profiles of the ZrO2 and M_ZrO2 catalysts. | ||
 | ||
| Fig. 6 NH3-TPD profiles of the ZrO2 and M_ZrO2 catalysts. | ||
The surface basic properties of all the catalysts are depicted in Table 2 and Fig. 5. A similar chair-like CO2-TPD profile was reported for ZrO244 and Cu/ZrO2/CaO.45 The base concentrations of the prepared catalysts ranged from 0.3–1.1 mmolCO2 g−1. The basic sites distribution of the catalysts illustrated in Fig. 5 depicted that all the catalysts exhibited basic sites of both weak and strong strength, although with different peak intensities. The base concentration of the pure ZrO2 (0.9 mmolCO2 g−1) was approximately similar in the presence of Fe (0.9 mmolCO2 g−1) but slightly increased upon doping with Mn (1.1 mmolCO2 g−1) and Ni (1.0 mmolCO2 g−1). An exception occurred in the case of Cr_ZrO2; the Cr species significantly decreased the base concentration of pure ZrO2 to 0.3 mmolCO2 g−1. The catalysts' basic density showed a similar trend, which was also confirmed by the reduction in the peak intensity representing the weak strength basic sites on the pure ZrO2 in the case of Cr_ ZrO2.
Fig. 6 shows the distribution of the acidic sites of weak to strong strength in the meso-ZrO2. Upon doping, the acidic sites underwent modulation. In the presence of Mn, Fe, and Ni, only two broad peaks representing weak and medium strength acid sites were observed. Whereas the Cr_ZrO2 appears unique, which gave a more prominent shoulder desorption peak on the high-temperature side at ca 436 °C, suggesting a stronger surface acidic site. A similar trend of NH3 desorption over Cr doped ZrO2 was reported.31 The Cr_ZrO2 (0.7 mmolNH3 g−1) possessed the highest concentration of acidity among the catalysts (Table 2). The NH3 desorption results suggest the proton-donating capacity of the surface acid site on the catalysts. The stronger the proton-donor tendency, the more strongly it binds with the base (NH3), and the higher the required NH3 desorption temperature. Hence, Cr_ZrO2 possesses stronger electrophilic active sites (acid sites) needed for the selective adsorption of citral via the CO bond.
Comparatively, the NH3- and CO2-TPD data revealed that all the catalysts exhibit both surface acidic and basic sites. However, these active sites are not equivalent, as observed in the data derived from the acid to base ratio (Table 2); the dominance in terms of strength, total concentration, and density depends on the metal–metal interaction nature. Meanwhile, Cr_ZrO2 presents more acid sites density and strength than the pure ZrO2, which possibly originates from the interaction of the Cr–Zr species at the atomic level. The tuning of the surface acid–base character of ZrO2 was achieved by incorporating metal ions, which facilitated the understanding of the effect of acid/base sites of the catalysts on the MPV reduction of aldehydes.
To further understand the surface components, types, and structures of the acid sites of the catalytic materials, investigation of the surface functionality and nature of acid sites was achieved through FTIR (Fig. 7a and b) and pyridine-adsorbed FTIR (Fig. 7c and d) spectroscopy methods, respectively. In Fig. 7a, the broad absorption band around 500–823 cm−1 is typical of Zr–O–Zr vibration in the tetragonal structure, and 1534 cm−1 suggests the stretching vibration of the Zr–O bond. The strong bands around 1394 and 3143 cm−1 are ascribed to the C–O–C and C–H stretching vibrations of P-123, respectively.46 This agrees well with the TG analysis (Fig. 4a), indicating residual surfactant is present in the catalysts. The IR peak at 1622 cm−1 suggests the bending hydroxyl group vibrations, while the broad and strong peak at 3435 cm−1 is a reflection of physically adsorbed moisture on the surface, hence showing the O–H stretching of water.47
 | ||
| Fig. 7 FTIR spectra of the (a) ZrO2 and M_ZrO2, (b) fresh and spent Cr_ZrO2, and Pyridine-adsorbed FTIR spectra of the distribution of the acid sites of (c) ZrO2 and M_ZrO2, (d) fresh and spent Cr_ZrO2, recorded at room temperature. L (Lewis acid site), B (Brønsted acid site), and L + B (Lewis + Brønsted acid site). | ||
Upon doping, the peak intensity at 3435 cm−1 decreased, indicating a decline in hydroxy groups and hydrophilic property of ZrO2.48 Like pXRD patterns (Fig. 1), the peak intensity at 500–823 cm−1 representing the t-ZrO2 decreased upon the substitution of Mn+ into ZrO2. Besides, the shifting of the bands towards the lower wavenumber was observed (for instance, 591 to 519 cm−1), which is most significant in Cr_ZrO2. This shifting is due to variation in the bond length when Mn+ ions replace Zr4+ ions. Hence, it confirmed the successful incorporation of the metal ions into the ZrO2 lattice. In Cr_ZrO2 catalyst, small peaks at 1210 and 1740 cm−1 are observed, attributed to the formation of CrxOy,49 due to the strong interaction between Cr–Zr. This could be responsible for the generation of more active sites on the surface of Cr_ZrO2. It is observed in Fig. 7b that the peak at 1740 cm−1 corresponding to the Cr species disappeared after five catalytic cycles, which suggests that the Cr_ZrO2 perhaps undergoes a surface structural transformation in the course of further thermal pretreatments during reuse.
Pyridine is a sensitive probe molecule for the classification of Lewis acid and Brønsted acid sites. As depicted in Fig. S5.7c (ESI†), the pyridine-IR bands at 957, 1400, and 1615 cm−1 are typical of the Lewis acid site. Two different acidic strengths due to the Lewis acid sites are shown in ZrO2, Fe_ZrO2, and Mn_ZrO2, whereas Lewis acidity of three different strengths was observed for both Ni_ZrO2 and Cr_ZrO2. The higher the assumed frequency of the IR bands, the stronger the acidity of the sites.50 The IR band at 1538 cm−1 suggests Brønsted acid site while 1485 cm−1 indicates C–C oscillation of pyridine aromatic ring chemisorbed on both Brønsted and Lewis acid sites.51 The ZrO2 catalyst showed no peak typical of the Brønsted acid site, but two characteristic bands (1400 and 1615 cm−1) associated with pyridinium ions coordinately bonded to Lewis acid sites.52 These two adsorption bands were retained with increased intensity upon the addition of Mn and Fe species. More broad bands for Lewis acid, Brønsted acid, and a combination of Lewis acid and Brønsted acid sites were found when ZrO2 was doped with Ni and Cr, accompanied by an increased intensity on Cr_ZrO2 catalyst. The pyridine-adsorbed IR reveals that the total acidity obtained from the NH3-TPD data has a larger Lewis to Brønsted acid ratio, which is highest in the Cr_ZrO2 and the main factor that governs its catalytic activity in this study. The results indicate the possibility of tuning the active sites of ZrO2 by adding foreign atomic species.
The UV-vis absorption spectra of undoped and metal-doped ZrO2 are shown in Fig. 8. The results indicate that the undoped ZrO2 and M_ZrO2 (M = Fe, Mn, and Ni) have no absorption peaks in the visible wavelength region of 300 to 700 nm. However, after Cr doping, a new absorption peak appears at around 360 nm; this is attributed to the band-gap transition of ZrO2 due to Cr3+ ions.53,54 The Cr-3d electronic configuration results in the appearance of some localized states in the host band-gap and makes electron transfer easier than the undoped system. This agrees well with the H2-TPR data (Fig. 4b) of Cr_ZrO2. All these issues explain the reason for the generation of more Lewis acid sites on Cr_ZrO2 compared to the undoped system.
 | ||
| Fig. 8 UV-vis spectra of the mesoporous ZrO2 and M_ZrO2. | ||
3.2. Surface performance of the M_ZrO2 catalysts in MPV reduction of citral
| Entry | Catalyst | Conversion (%) | Selectivity (%) | Amount converted (mmol) | Activitynormalized* (mmol g−1) × 102 | k obs (h−1) |
|---|---|---|---|---|---|---|
| Reaction condition: 2-propanol:citral = 52 molar ratio, 1.0 mmol decane, 0.4 g catalyst, stirring rate = 450 rpm, T = 80 °C, t = 10 h. * Activitynormalized obtained by normalizing the amount converted at 10 h to the mass of the catalyst used in each run. | ||||||
| 1 | ZrO2 | 62.6 | >99 | 1.57 | 393 | 0.24 ± 0.03 |
| 2 | Cr_ZrO2 | 76.4 | >99 | 1.91 | 478 | 0.40 ± 0.04 |
| 3 | Mn_ZrO2 | 55.8 | >99 | 1.39 | 348 | 0.23 ± 0.02 |
| 4 | Fe_ZrO2 | 41.1 | >99 | 1.03 | 258 | 0.20 ± 0.03 |
| 5 | Ni_ZrO2 | 26.8 | >99 | 0.67 | 168 | 0.06 ± 0.05 |
 | ||
| Fig. 9 Correlation of the surface properties with catalytic activity in MPV reduction of citral. | ||
 | ||
| Fig. 10 (a) Leaching test and (b) recyclability of Cr_ZrO2 catalyst in MPV reduction of citral. Reaction condition: 2-propanol:citral = 52 molar ratio, 1.0 mmol decane, 0.4 g catalyst, stirring rate = 450 rpm, T = 80 °C, t = 6 h. | ||
Nevertheless, the TEM image (Fig. S7c and d, ESI†) shows the stability of the mesostructure after 5 consecutive catalytic cycles. Moreover, the comparison of the Cr content before (8.2 wt%) and after five use (8.1%) as quantified on the ICP/OES showed no leached Cr species. The leaching test (Fig. 10a) and the ICP results (Table 1) confirmed the homogeneity of the Cr_ZrO2 solid structure. Hence, the mesoporous Cr_ZrO2 is catalytically stable and reusable with retained activity.
3.3. Substrate scope of mesoporous Cr_ZrO2 catalyst
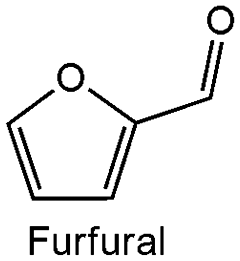
The chromium doped zirconia exhibited the best catalytic activity in the reduction of citral with 76.4% conversion, Table 3. Hence, the catalytic scope of Cr_ZrO2 in MPV reduction is extended to some unsaturated aldehydes to form their corresponding unsaturated alcohols Table 4. The Cr_ZrO2 is most active in the MPV reduction of furfural. The appreciable reactivity suggests that the mesoporous Cr_ZrO2 acid catalyst is highly active in the MPV process and 100% selective to unsaturated allylic alcohols.| Entry | Substrate | k obs (h−1) | Activitynormalized* (mmol m−2 g−1) × 10−5 | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|---|
| Reaction condition: 2.5 mmol substrate, 1.0 mmol decane, 130 mmol 2-propanol, 0.4 g catalyst, stirring rate 450 rpm, temperature 80 °C, and reaction time 10 h. *Activitynormalized obtained by normalizing the amount converted at 10 h to the SBET. | |||||
| 1 |
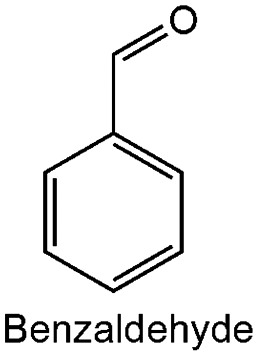
|
0.05 ± 0.05 | 741 | 56.31 | 100 |
| 2 |
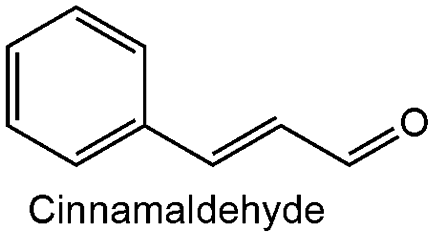
|
0.28 ± 0.06 | 790 | 60.09 | 100 |
| 3 |
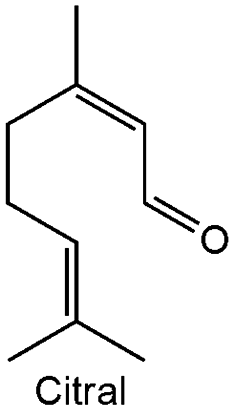
|
0.40 ± 0.04 | 1006 | 76.40 | 100 |
| 4 |
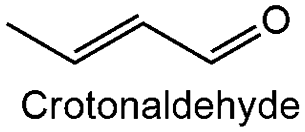
|
0.24 ± 0.07 | 581 | 44.14 | 100 |
| 5 |
|
0.49 ± 0.02 | 1292 | 98.17 | 100 |
4. Discussion
Herein, a series of transition metal-doped mesoporous ZrO2 (M_ZrO2, M = Cr, Mn, Fe, and Ni) catalysts were designed for the MPV reduction of aldehydes. Interestingly, the tunability of the surface properties of the resulting catalysts is governed by the metal dopant's nature. The SBET decreases from 206 to 189 and 193 m2 g−1 upon doping with Cr and Ni, respectively. An improvement in SBET from 206–223 m2 g−1 was observed when doped with Mn and Fe (221 and 223 m2 g−1, respectively). Also, upon doping, there was an enlargement of pore diameter from 2.53–3.63 nm and increased pore volume from 0.10–0.26 cm3 g−1 (Table 1). The mesostructure properties of the synthesized pure zirconia ZrO2 and the metal-doped zirconia M_ZrO2 are typical of type IV hysteresis loops as shown by the BET isotherms (Fig. 2a). This indicates the successful design of a mesopore structure of the materials via a sol–gel approach. This study took advantage of the structure-directing ability of P-123 in the inverse micelles system, which serves as the nanoreactors. Also, a control condensation of the oxo-clusters was achieved by forming NOx species from the nitric acid's thermal decomposition.34The increase in the porosity (pore diameter and pore volume) upon doping and SBET in the case of Mn_ZrO2 and Fe_ZrO2 could be due to the metal–metal interactions during condensation of the inorganic sols and the condition of the reaction media. A similar phenomenon was explained by Grosso et al.,55 that the mesostructuring occurs during the formation of surfactant-templated inorganic materials by evaporation. The chemical composition of the film governs the meso-organization. Also, it depends on relative vapor pressure in the environment, the evaporation conditions, and the chemical conditions in the initial solution. The changes in the structure and crystallography of ZrO2 resulting from doping species modified the concentration of active phases involved in the catalyzed reaction on the surface of the ZrO2 based catalysts.
The NH3-TPD and CO2-TPD experiments showed that the synthesized catalysts possess acid–base properties that could be tuned. The acid/base strength and density are related to the nature of the metal dopant. The acid density decreases in this order Cr_ZrO2 > Ni_ZrO2 > Mn_ZrO2 > Fe_ZrO2 > ZrO2. On the other hand, the basicity of the M_ZrO2 gave this trend Mn_ZrO2 > Ni_ZrO2 > Fe_ZrO2 > ZrO2 > Cr_ZrO2, which could be related to neither the electron density nor ionic charge. These results show that the presence of metal dopant in the ZrO2 framework possibly tunes the active acid–base sites, resulting mainly from the metal–metal synergy between the metal dopant and the host ZrO2. It was revealed that the acid:base in ratio 3:1 is required for the chemoselective transfer hydrogenation of citral via the MPV system. To further understand the kinds of acid sites on the solid catalysts, the pyridine-adsorbed experiments indicated that the metal–metal synergy generated both surface Lewis and Brønsted acid sites with a higher concentration of Lewis acid sites having weak, medium, and strong strength. The Lewis acid sites are possibly generated by the concerted metal ions (Cr3+ and Zr4+) acting as the electron-acceptor. Also, the metal–metal synergy influenced the hydrogen consumption, with the Cr–Zr catalyst showing superior reducibility (H-abstraction capacity).
According to Stavale et al., the electronic structure and chemical properties of oxide materials, and their catalytic activities, could be tailored by doping with metal.56 The approach takes advantage of the metal dopants' tendency to exchange electrons with the host oxide and surface-bound adsorbates. It has also been reported that dopant-modified metal oxides exhibit improved catalytic performance than their pure oxides.57 In our case, the MPV process is catalytic driven; no activity was observed in the blank reaction. The catalytic activity of the materials in the MPV reduction of α,β-unsaturated aldehydes is dependent on the concentration of the surface acidic sites. Among the transition metal dopants incorporated, the catalytic activity of pure ZrO2 (62.6%) in terms of citral conversion in the model reaction was only improved by Cr_ZrO2 (76.4%). The observed linear relationship between the surface acidity and activity of the synthesized catalysts suggests that the MPV reduction reaction is perhaps governed by the extent of acidity induced by the electronic interaction between Cr and Zr. The experiment performed with pure chromium oxide showed no activity after 24 h, this suggests that the synergistic interaction might be responsible for the enhanced activity in Cr_ZrO2. Also, the acid character of the Cr_ZrO2 catalyst with the polarity of the citral molecule made it possible for citral to preferably adsorb through the carbonyl group. Hence, the transfer hydrogenation of the carbonyl to produce UAA is favored. A similar scenario in which the adsorption of citral on the Lewis acid site is via the carbonyl was reported.58 The local structure of the Lewis acid sites on zirconia catalyst was also reported.59,60 In view of these and the findings of this study, a possible mechanism for MPV reduction of citral on the Lewis acid sites of M_ZrO2 is proposed (Scheme 2).
 | ||
| Scheme 2 Proposed mechanism for the chemoselective MPV reduction of α,β-unsaturated aldehydes to their corresponding unsaturated allylic alcohol over the Lewis acid sites on mesoporous M_ZrO2 catalyst. | ||
Distinctively, despite the high acidic properties of the catalytic system, no secondary products were formed. Acidic catalysts frequently favor secondary reactions as either dehydration of alcohol or aldehyde condensation.61,62 All the prepared catalysts in this work exhibited excellent selectivity to unsaturated allylic alcohol as evidenced in the GC spectra (Fig. S8 and S9, ESI†) compared to their previously reported counterparts in Table 4; this is paramount to a sustainable catalytic process. Moreover, the MPV process in this work was carried out under milder reaction conditions in the absence of additives and gaseous hydrogen. The reactivity retained after five consecutive runs evidence the sustainability of the Cr_ZrO2 catalyst.
Furthermore, the synthesized Cr_ZrO2 in this work showed considerable reactivity compared to its counterparts in literature Table 5. Our catalyst gave a maximum selectivity of 100% to the UAA under milder reaction conditions in the absence of H2 gas pressure. For instance, it is more reactive than the ZrSr-PN catalyst in the MPV reduction of cinnamaldehyde; the ZrSr-PN gave 24.0% conversion of cinnamaldehyde in 24 h while our Cr_ZrO2 gave 60% conversion in 10 h. Also, in the MPV reduction of furfural, our Cr_ZrO2 showed higher activity of 85% conversion in 4 h and 98.2% in 10 h at 80 °C than P-Zr 200 (55.3%), ME-Zr-200UW (67.6%), and Zr-SBA-15 (54%) after 24, 24 and 6 h, respectively. However, Pt/ZrO2 synthesized by Wei et al.4 gave better activity than our catalyst in cinnamaldehyde reduction and Ru/ZrO2 in citral reduction but, this is due to the H2 gas pressure used in their catalytic system.
| Entry | Catalysts | S BET (m2 g−1) | Substrate | Time (h) | Conversion (%) | Selectivity UAA % | Reaction conditions | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Pt/ZrO2 | 42 | Crotonaldehyde | 8 | 37 | 4 | 30 °C, 0.414 MPa | 33 |
| 2 | ZrO2/Gaim 300 | 175 | Crotonaldehyde | 8 | 26 | 86 | 130 °C, atm | 16 |
| 3 | Zr-BEA | 470 | Crotonaldehyde | — | 15 | 72 | 200 °C, atm | 26 |
| 4 | Zr-SBA-15 | 553 | Furfural | 6 | 54 | 41 | 90 °C, atm | 21 |
| 5 | P-Zr 200 | 230 | Furfural | 24 | 55.3 | 79.9 | 100 °C, atm, 1000 rpm | 18 |
| 6 | ME-Zr-200UW | 210 | Furfural | 24 | 67.6 | 83.6 | 100 °C, atm 1000 rpm | 18 |
| 7 | ZrO2 | 154 | Benzaldehyde | 8 | 33.7 | 99.2 | 82 °C, atm | 63 |
| 8 | 5%ZrO2/Si-MCM-41 | 776 | Benzaldehyde | 8 | 88.5 | 99.1 | 82 °C, atm | 63 |
| 9 | Zr-beta zeolite | 490 | Citral | 5 | 91 | 74 | 82 °C, atm | 64 |
| 10 | Ru/ZrO2 | 102 | Citral | 3.3 | 90 | 30 | 50 °C, 5 MPa, 500 rpm | 29 |
| 11 | ZrSr-PN | 191 | Cinnamaldehyde | 24 | 24 | 98 | 82 °C, atm 1000 rpm | 7 |
| 12 | Pt/ZrO2 | 274 | Cinnamaldehyde | 0.5 | 59 | 99 | 60 °C, 1.0 MPa | 4 |
| 13 | Cr–ZrO2 | 189 | Benzaldehyde | 4 | 24 | 100 | 80 °C, atm 450 rpm | This work |
| 14 | Cr–ZrO2 | 189 | Cinnamaldehyde | 4 | 42 | 100 | 80 °C, atm 450 rpm | |
| 15 | Cr–ZrO2 | 189 | Citral | 4 | 63 | 100 | 80 °C, atm 450 rpm | |
| 16 | Cr–ZrO2 | 189 | Crotonaldehyde | 4 | 30 | 100 | 80 °C, atm 450 rpm | |
| 17 | Cr–ZrO2 | 189 | Furfural | 4 | 85 | 100 | 80 °C, atm 450 rpm |
5. Conclusion
Herein, we have demonstrated that incorporating metal dopants into zirconia's crystal framework alters its physico-chemical properties such as surface area, mesopore structure, crystallinity, basicity, acidity, reducibility, and thermal stability. The reducibility and the strength of the Lewis acid sites govern the activity of ZrO2 based catalysts in the MPV reduction of citral. Specifically, the Cr dopant weakens the crystallinity of ZrO2. However, it improves the reducibility, acidity, and catalytic reactivity for MPV reduction of aldehydes. All the prepared zirconia-based catalysts in this work showed a remarkable selectivity of 100% to UAA. The surface-induced performance of the Cr_ZrO2 is due to the enhanced active centers generated from the synergistic electronic interaction between CrOx and ZrO2. Hence, this work unveils that the reactivity of ZrO2 depends solely on the intrinsic properties of its' surface structure rather than the specific surface expanse. Also, the Cr_ZrO2 exhibited good stability and recyclability for at least five reaction cycles. We proposed a plausible mechanism of the MPV transformation over the Lewis acidic sites. This work presents the first application of a well-designed mesoporous Cr_ZrO2via an inverse micelle approach in the MPV reduction of aldehydes with exceptional reactivity and efficient reusability. The green production of UAA was successfully achieved under mild reaction conditions without pressurized hydrogen gas. The Cr_ZrO2 is proposed to be a potential sustainable catalyst for industrial applications.Conflicts of interest
The authors declare no competing interest.Acknowledgements
We profoundly acknowledge the financial support by the National Research Foundation/the World Academy of Science NRF/TWAS UID: 105463, NRF (UID: 121039), and the University of Johannesburg. Also, Shimadzu company for their instruments and Spectrum UJ for the use of SEM and TEM instruments.References
- D. Das, K. Pal, J. Llorca, M. Dominguez, S. Colussi, A. Trovarelli and A. Gayen, React. Kinet., Mech. Catal., 2017, 122, 135–153 CrossRef CAS.
- X. Lan and T. Wang, ACS Catal., 2020, 10, 2764–2790 CrossRef CAS.
- M. S. Ide, B. Hao, M. Neurock and R. J. Davis, ACS Catal., 2012, 2, 671–683 CrossRef CAS.
- S. Wei, Y. Zhao, G. Fan, L. Yang and F. Li, Chem. Eng. J., 2017, 322, 234–245 CrossRef CAS.
- K. Maresz, A. Ciemięga and J. Mrowiec-Białoń, Catalysts, 2018, 8, 221 CrossRef.
- S. Shylesh, M. P. Kapoor, L. R. Juneja, P. P. Samuel, C. Srilakshmi and A. P. Singh, J. Mol. Catal. A: Chem., 2009, 301, 118–126 CrossRef CAS.
- F. J. Urbano, M. A. Aramendía, A. Marinas and J. M. Marinas, J. Catal., 2009, 268, 79–88 CrossRef CAS.
- G. Wang, X. Deng, D. Gu, K. Chen, H. Tüysüz, B. Spliethoff, H. Bongard, C. Weidenthaler, W. Schmidt and F. Schüth, Angew. Chem., Int. Ed., 2016, 55, 11101–11105 CrossRef CAS PubMed.
- E. Plessers, D. E. De Vos and M. B. J. Roeffaers, J. Catal., 2016, 340, 136–143 CrossRef CAS.
- Y. Leng, L. Shi, S. Du, J. Jiang and P. Jiang, Green Chem., 2020, 22, 180–186 RSC.
- R. A. Farrar-Tobar, A. Dell’Acqua, S. Tin and J. G. de Vries, Green Chem., 2020, 22, 3323–3357 RSC.
- Z. Hu, S. Shi, L. Wang, S. Chen, Y. You, S. Wang and C. Chen, Appl. Surf. Sci., 2020, 146992–147002 CrossRef CAS.
- E. V. Ramos-Fernández, B. Samaranch, P. R. de la Piscina, N. Homs, J. L. G. Fierro, F. Rodríguez-Reinoso and A. Sepúlveda-Escribano, Appl. Catal., A, 2008, 349, 165–169 CrossRef.
- E. J. Creyghton, J. Huskens, J. C. Van der Waal and H. Van Bekkum, Studies in Surface Science and Catalysis, Elsevier, 1997, 108, pp. 531–537 Search PubMed.
- S. H. Liu, S. Jaenicke and G. K. Chuah, J. Catal., 2002, 206, 321–330 CrossRef CAS.
- J. F. Miñambres, M. A. Aramendía, A. Marinas, J. M. Marinas and F. J. Urbano, J. Mol. Catal. A: Chem., 2011, 338, 121–129 Search PubMed.
- S. Axpuac, M. A. Aramendía, J. Hidalgo-Carrillo, A. Marinas, J. M. Marinas, V. Montes-Jiménez, F. J. Urbano and V. Borau, Catal. Today, 2012, 187, 183–190 CrossRef CAS.
- V. Montes, J. F. Miñambres, A. N. Khalilov, M. Boutonnet, J. M. Marinas, F. J. Urbano, A. M. Maharramov and A. Marinas, Catal. Today, 2018, 306, 89–95 CrossRef CAS.
- M. B. Gawande, A. K. Rathi, P. S. Branco, T. M. Potewar, A. Velhinho, I. D. Nogueira, A. Tolstogouzov, C. A. A. Ghumman and O. M. N. D. Teodoro, RSC Adv., 2013, 3, 3611 RSC.
- Y. Kuwahara, W. Kaburagi, Y. Osada, T. Fujitani and H. Yamashita, Catal. Today, 2017, 281, 418–428 CrossRef CAS.
- J. Iglesias, J. Melero, G. Morales, J. Moreno, Y. Segura, M. Paniagua, A. Cambra and B. Hernández, Catalysts, 2015, 5, 1911–1927 CrossRef CAS.
- B. Zhang, F. Xie, J. Yuan, L. Wang and B. Deng, Catal. Commun., 2017, 92, 46–50 CrossRef.
- W. Cai, J. Yang, H. Sun, Y. Wang, T. Ling, X. Guo, L. Peng and W. Ding, Chin. J. Catal., 2017, 38, 1330–1337 CrossRef CAS.
- A. Taguchi and F. Schüth, Microporous Mesoporous Mater., 2005, 77, 1–45 CrossRef CAS.
- J. Védrine, Catalysts, 2017, 7, 341 CrossRef.
- V. L. Sushkevich, I. I. Ivanova, S. Tolborg and E. Taarning, J. Catal., 2014, 316, 121–129 CrossRef CAS.
- D. Kaucký, Z. Sobalík, J. M. Hidalgo, R. Černý and O. Bortnovský, J. Mol. Catal. A: Chem., 2016, 420, 107–114 CrossRef.
- F. Alonso, P. Riente and M. Yus, Tetrahedron, 2008, 64, 1847–1852 CrossRef CAS.
- J. Álvarez-Rodríguez, I. Rodríguez-Ramos, A. Guerrero-Ruiz, E. Gallegos-Suarez and A. Arcoya, Chem. Eng. J., 2012, 204, 169–178 CrossRef.
- N. J. A. Rahman, A. Ramli, K. Jumbri and Y. Uemura, Sci. Rep., 2019, 9, 1–12 Search PubMed.
- Z. Xie, Y. Ren, J. Li, Z. Zhao, X. Fan, B. Liu, W. Song, L. Kong, X. Xiao and J. Liu, J. Catal., 2019, 372, 206–216 CrossRef CAS.
- R. Wu, P. Xie, Y. Cheng, Y. Yue, S. Gu, W. Yang, C. Miao, W. Hua and Z. Gao, Catal. Commun., 2013, 39, 20–23 CrossRef CAS.
- J. Hidalgo-Carrillo, M. A. Aramendía, A. Marinas, J. M. Marinas and F. J. Urbano, Appl. Catal., A, 2010, 385, 190–200 CrossRef CAS.
- A. S. Poyraz, C.-H. Kuo, S. Biswas, C. K. King’ondu and S. L. Suib, Nat. Commun., 2013, 4, 1–10 Search PubMed.
- C. A. Akinnawo, N. Bingwa and R. Meijboom, Catal. Commun., 2020, 106115 CrossRef CAS.
- N. P. Rajan, G. S. Rao, V. Pavankumar and K. V. R. Chary, Catal. Sci. Technol., 2014, 4, 81–92 RSC.
- Z. Y. Xun, Y. T. Yu and W. B. Liu, Acta Metall. Sin., 2020, 1–18 Search PubMed.
- T. Lu, C. Liu, Z. Li, Q. Wu, J. Wang, T. Xu, J. Liu, H. Wang and S. Ma, J. Alloys Compd., 2020, 817, 153334 CrossRef CAS.
- T. Uhlířová and W. Pabst, J. Eur. Ceram. Soc., 2020, 3181–3190 CrossRef.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 1051–1069 CAS.
- M. Thommes, Chem. Ing. Tech., 2010, 82, 1059–1073 CrossRef CAS.
- K. S. W. Sing and R. T. Williams, Adsorpt. Sci. Technol., 2004, 22, 773–782 CrossRef CAS.
- Y. Li, L. Wang, R. Yan, J. Han and S. Zhang, Catal. Sci. Technol., 2015, 5, 3682–3692 RSC.
- C. A. Akinnawo, N. Bingwa and R. Meijboom, Microporous Mesoporous Mater., 2020, 110693–110704 Search PubMed.
- W. Wisaijorn, P. Praserthdam, S. Assabumrungrat and S. Soisuwan, Energy Procedia, 2015, 79, 685–690 CrossRef CAS.
- C. Suwanchawalit and S. Wongnawa, J. Nanopart. Res., 2010, 12, 2895–2906 CrossRef CAS.
- K. Anandan, K. Rajesh, K. Gayathri, S. V. Sharma, S. G. M. Hussain and V. Rajendran, Phys. E, 2020, 114342–114356 CrossRef CAS.
- S. Bhaskar, E. W. Awin, K. C. H. Kumar, A. Lale, S. Bernard and R. Kumar, Sci. Rep., 2020, 10, 1–13 CrossRef PubMed.
- N. H. R. Annuar, S. Triwahyono, A. A. Jalil, N. Basar, T. A. T. Abdullah and A. Ahmad, Appl. Catal., A, 2017, 541, 77–86 CrossRef CAS.
- M. I. Zaki, M. A. Hasan, F. A. Al-Sagheer and L. Pasupulety, Colloids Surf., A, 2001, 190, 261–274 CrossRef CAS.
- W. Gac, M. Greluk, G. Słowik, Y. Millot, L. Valentin and S. Dzwigaj, Appl. Catal., B, 2018, 237, 94–109 CrossRef CAS.
- Y. Zhou, L. Zhang and S. Tao, ACS Appl. Nano Mater., 2019, 2, 5125–5131 CrossRef CAS.
- S. Chang and R. Doong, J. Phys. Chem. B, 2004, 108, 18098–18103 CrossRef CAS.
- J. Singh, R. Kumar, V. Verma and R. Kumar, Ceram. Int., 2020, 46, 24071–24082 CrossRef CAS.
- D. Grosso, F. Cagnol, G. J. de, A. A. Soler-Illia, E. L. Crepaldi, H. Amenitsch, A. Brunet-Bruneau, A. Bourgeois and C. Sanchez, Adv. Funct. Mater., 2004, 14, 309–322 CrossRef CAS.
- F. Stavale, X. Shao, N. Nilius, H.-J. Freund, S. Prada, L. Giordano and G. Pacchioni, J. Am. Chem. Soc., 2012, 134, 11380–11383 CrossRef CAS PubMed.
- M. E. Assal, M. Kuniyil, M. Khan, M. R. Shaik, A. Al-Warthan, M. R. H. Siddiqui, J. P. Labis and S. F. Adil, Adv. Mater. Sci. Eng., 2017, 2017, 3958319 Search PubMed.
- S. Santiago-Pedro, V. Tamayo-Galván and T. Viveros-García, Catal. Today, 2013, 213, 101–108 CrossRef CAS.
- X. Tang, L. Hu, Y. Sun, G. Zhao, W. Hao and L. Lin, RSC Adv., 2013, 3, 10277–10284 RSC.
- J. Zhang, K. Dong, W. Luo and H. Guan, ACS Omega, 2018, 3, 6206–6216 CrossRef CAS PubMed.
- V. A. Ivanov, J. Bachelier, F. Audry and J. C. Lavalley, J. Mol. Catal., 1994, 91, 45–59 CrossRef CAS.
- Z. Xiao, Mol. Catal., 2017, 436, 1–9 CrossRef CAS.
- B. Zhang, T. Minhui, Y. Jian and W. U. Lei, Chin. J. Catal., 2012, 33, 914–922 CrossRef CAS.
- Y. Zhu, S. Liu, S. Jaenicke and G. Chuah, Catal. Today, 2004, 97, 249–255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nj00936b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |