
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and structure of thienyl Fischer carbene complexes of PtII for application in alkyne hydrosilylation†
Zandria
Lamprecht
 a,
Frederick P.
Malan
a,
Simon
Lotz
a and
Daniela I.
Bezuidenhout
*b
a,
Frederick P.
Malan
a,
Simon
Lotz
a and
Daniela I.
Bezuidenhout
*b
aDepartment of Chemistry, University of Pretoria, Private Bag X20, Hatfield 0028, Pretoria, South Africa
bLaboratory of Inorganic Chemistry, Environmental and Chemical Engineering, University of Oulu, P. O. Box 3000, 90014 Oulu, Finland. E-mail: daniela.bezuidenhout@oulu.fi
First published on 16th March 2021
Abstract
Transmetallation of group 6 thienylene Fischer carbene complexes to PtII precursors yielded new examples of neutral platinum(II) bisethoxycarbene complexes with either 2-thienyl (T) or 5-thieno[2,3-b]thienylene (TT) carbene substituents. The use of analogous aminocarbene group 6 precursors proceeded to give isomeric platinum(II) product mixtures where the resultant bisaminocarbene ligands displayed different orientations due to restricted rotation around the Pt–aminocarbene bond caused by the sterically demanding TT substituents. The well-defined PtII ethoxycarbene complexes were screened as catalyst precursors in the benchmark hydrosilylation reaction employing phenylacetylene and triethylsilane substrates. Marked selectivity for the β-E isomer (E)-triethyl(styryl)silane was observed, and the (pre)catalysts proved recyclable, active in solvent-free reactions, and displaying a high alkyne functional group tolerance.
Introduction
Acyclic heteroatom-stabilized carbene complexes of platinum(II) have been known since 1915,1–3 and yet the number of isolated examples are limited. In general, there are four methods available to prepare PtII Fischer carbene complexes (FCCs). Firstly, ligand modification of PtII isocyanide precursors with alcohols or primary amines produces the corresponding acyclic aminoxy- or diaminocarbene PtII complexes, respectively (Fig. 1a).4 Cationic alkoxycarbene complexes can be prepared by reacting the PtII precursors with alkynes (Fig. 1b), to yield the π-bonded intermediates that lead to carbene formation after alkyne modification,5 while neutral bisalkoxycarbene complexes are obtained from the reaction of SiMe3-substituted alkynes and H2PtCl6·6H2O (Fig. 1c).6 The first transmetallation reactions from group 6 transition metal FCCs to produce mononuclear Pt-biscarbene complexes employed [PtCl2(MeCN)2] or PtCl2 precursors (Fig. 1d) and the products were stabilized by amine-chelate formation.7 | ||
| Fig. 1 Heteroatom-stabilized FCCs of platinum(II). | ||
We have recently prepared PtII multicarbene complexes (Fig. 1e) by carbene transfer reactions of the ethoxy- and aminocarbene ligands of W(0) FCCs to a PtII centre.8 The major products obtained from the reactions with group 6 carbene precursors are neutral bisethoxycarbene complexes of PtII and cationic mononuclear trisaminocarbene complexes (Fig. 1e). In this study, PtII Fischer carbene complexes with (annulated) thiophene substituents, are synthesized for the first time employing the methodology of carbene transfer, and their use as catalysts for the alkyne hydrosilylation reaction is investigated. Hydrosilylation of terminal alkynes is one of the leading methods to produce organosilanes, and is catalysed by many transition metals including platinum.9–18 Although PtII complexes containing N-heterocyclic carbene (NHC) ligands are commonly employed as catalysts for this reaction,9,12,18 no Fischer carbene catalysts have been reported.
Results and discussion
Synthesis of platinum(II) FCCs
PtII multicarbene complexes were synthesized by transmetallation reactions of the chromium or tungsten carbonyl FCC precursors P1–P4, containing either a 2-thienyl (T) or a 5-thieno[2,3-b]thienylene (TT) substituent (see Experimental section and ESI† for the preparation of P1–P4), via a modified procedure of that reported by Sierra and co-workers (Scheme 1).7 Transmetallation occured by stirring a group 6 FCC with a platinum precursor for 24–30 h in refluxing dichloromethane (DCM). The syntheses of PtII FCCs are facile, but purification of the compounds proved challenging. The complexes are light sensitive, insoluble in most solvents (e.g. dichloromethane, chloroform, acetonitrile) and chromatographic purification processes are compromised. Four PtII biscarbene complexes were successfully isolated, namely cis-[PtCl2{C(OEt)-2-C4H3S}2] (1), cis-[PtCl2{C(OEt)-5-C6H3S2}2] (2), cis-[PtCl2{C(NH2)-5-C6H3S2}2] (3a/b) and cis-[PtCl2{C(NMe2)-5-C6H3S2}2] (4a/b) (Fig. 2). The transmetallation reactions do not run to completion, even with prolonged reaction times, and starting material is recovered from all reactions. | ||
| Scheme 1 Fischer carbene ligand transfer to a platinum(II) metal centre. | ||
 | ||
| Fig. 2 PtII Fischer carbene complexes 1–4. | ||
The cis-biscarbene complexes of 3 and 4 have three isomeric possibilities where the TT spacers and amino fragments have various orientations to yield different geometric stereoisomers (Fig. 2) due to restricted rotation enforced by the sterically demanding thieno[2,3-b]thienylene carbene substituent and the amino group with increased Ccarbene–N bond order. Evidence for the formation of two out of the possible three isomers is observed for 3 and 4. In addition, the formation of a cationic Pt triscarbene complex ([PtCl{C(NMe2)-5-C6H3S2}3]Cl, 4d) is observed for the reactions done with group 6 transition metal dimethylaminocarbene complexes. The orientation of the carbene substituents in 4d is unknown.
Compounds 3 were synthesized using P3W (see ESI,† Fig. S1) and Pt(COD)Cl2 in DCM. The cis isomer 3 was obtained with evidence for the isolation of two-out-of-three possible geometric stereoisomers that are inseparable. The two isomers are 3a (yield 64%) and 3b (yield 13%), obtained in the ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as determined by integrating the 1H NMR spectrum resonances (ESI,† Fig. S9).
:1 as determined by integrating the 1H NMR spectrum resonances (ESI,† Fig. S9).
Compounds 4 were isolated from transmetallation reactions using P4Cr/W along with Pt(COD)Cl2 or cis-[PtCl2(NCMe)2] in DCM. The preferred reaction conditions are employed by reacting P4Cr with Pt(COD)Cl2 in DCM, to produce 4 in 71% yield. Excess P4Cr and Pt(COD)Cl2 are removed by washing the reaction precipitate with minimal amounts of DCM, chloroform and ether. Two-out-of-three possible geometric stereoisomers of the cis-biscarbene complex of 4 are obtained.
The two isomers, 4a (yield 39%) and 4b (yield 8%), are accompanied by the triscarbene complex (4d, yield 24%) in the ratio 5:1:3 as determined by integrating 1H NMR spectrum resonances (ESI,† Fig. S11). Compounds 4 only dissolve in DMSO and partly in chlorinated solvents (e.g. DCM and CDCl3), hence the more insoluble 4d could be purified by washing the precipitate with a variety of solvents. In an attempt to purify 4, a short silica gel filter using acetone as eluent produced only the decomposition product (NMe2)C(O)-5-C6H3S2 (5, yield 24%).
The solubility of PtII FCCs decrease (1 > 2) with an increase in the number of thiophene units in the annulated spacer (1 < 2). A decrease in solubility is also seen when changing the heteroatom substituent from ethoxy- (2) to amino- (3, 4).
The mechanism for obtaining a PtII mononuclear biscarbene complex from Pt(COD)Cl2, requires the substitution of the η4-COD ligand, leaving two cis empty coordination sites on the metal. A stepwise mechanism is likely followed were the COD ligand remains partially coordinated (η2-COD), still bound to the metal through one of the two double bonds of cyclooctadiene, as the first carbene ligand coordinates.19,20 Coordination of the second carbene ligand, to the PtII metal, completely displaces the COD ligand. Further substitution would require that the trans effect of a carbene ligand is relatively stronger than that of a chloro ligand.8
Spectroscopic characterization
NMR spectra were recorded in CDCl3, CD2Cl2 and (CD3)2SO, depending on the solubility of the compounds. 1H NMR data are summarized in Table 1. The aromatic proton adjacent to the carbene carbon is most affected by the coordination of the carbene fragment. In 1 this is seen in the resonance of H3 and even more so in H4 of 2 (ESI,† Fig. S6 and S7). Due to conjugation in the thiophene ring of 1, H5 is also shifted significantly downfield. The methylene resonances of PtII ethoxycarbene complexes appear as broadened peaks instead of quartets. One such broad peak/unresolved quartet is observed in the 1H NMR spectrum of 1 and two in the case of 2, resonating ca. 0.6 ppm apart (Fig. S6 and S7, ESI†). Due to the bulky nature of thienylenes and the cis-orientation of the two carbene ligands in the square planar structure of the platinum complexes, the peak broadening is ascribed to restricted rotation of the OEt group around the carbene carbon, more pronounced in 2 (larger TT spacer) compared to 1 (T spacer). The broad signals of 2 are duplicated indicating different electronic environments for the ethoxy substituents. Recording the NMR spectrum of 2 in a different solvent, e.g. CD2Cl2 or at a lower temperature (2 °C) did not significantly improve the resolution of the spectra.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Complex | H3 | H4 | H5 | H4′ | H5′ | OEta | NH2/NMe2e |
| a Proton chemical shifts for the ethoxy fragment are reported with the first value being the chemical shift of the methylene group, and the second the chemical shift of the methyl group. In the case of 2 there are two values for the methylene groups. b NMR data recorded in CDCl3. c NMR data recorded in CD2Cl2. d NMR data recorded in (CD3)2SO. e Proton chemical shifts for the NH2 fragments of 3a and 3b. In the case of 4 the chemical shifts represent the methyl groups of NMe2. f Broadened resonances observed. The first values reported being the chemical shifts of the two carbene fragments opposite each other and the second the chemical shifts of the carbene fragment trans to a chloro ligand. | |||||||
| 1 | 8.69 | 7.28 | 8.09 | 5.60, 1.59 | |||
| 1 | 8.68 | 7.31 | 8.13 | 5.54, 1.59 | |||
| 2 | 9.08 | 7.35 | 7.44 | 5.77, 5.18, 1.59 | |||
| 2 | 9.03 | 7.39 | 7.49 | 5.73, 5.21, 1.58 | |||
| 3a | 8.26 | 7.40 | 7.66 | 10.77, 10.61 | |||
| 3b | 8.56, 8.46 | 7.46, 7.43 | 7.73, 7.72 | 11.42, 11.18 | |||
| 4a | 8.06 | 7.35 | 7.45 | 4.22, 3.86 | |||
| 4a | 8.00 | 7.45 | 7.73 | 4.06, 3.77 | |||
| 4b | 7.61 | 7.29 | 7.38 | 3.30 | |||
| 4b | 7.80 | 7.41 | 7.70 | 4.15, 3.65 | |||
| 4d | 7.65, 7.37 | 7.26, 6.78 | 7.37, 6.87 | 4.10, 3.63, 3.38, 3.30 | |||

In the 1H NMR spectrum of 3a, a double duplication of the carbene ligand resonances is observed in a 1:1 ratio (ESI,† Fig. S9), for the compound 3b. The two cis-biscarbene isomers are not separable. The difference between the two stereoisomers 3a and 3b can be visualized by restricting rotation around the Pt–Ccarb bond. In 3a a structure is assigned whereby each amino substituent is on the same side as the sulphurs in the adjacent TT ring, of the two carbene ligands (Fig. 2). The 1:1 ratio of the aromatic proton resonances of 3b indicates that the molecule constitutes of two different carbene ligands. In 3b one ligand has the same conformation as the TT substituents in 3a, but the other TT substituent is rotated around the Ccarb–CTT bond to take up a position where the amino substituent is on the opposite side of the sulphurs in the TT ring. The greatest difference is observed for the NH and H4 proton resonances of 3a and 3b. Two significantly different NH resonances are observed, hence H-bonding interactions are suspected. Because three geometric stereoisomers are possible for 3 (Fig. 2), 3b is expected to be one of the minor isomers. A third isomer 3c, with both amino substituents on opposite sides of the sulphurs in the TT rings, was not observed.
 | ||
| Fig. 3 The molecular structures of 1, 3b and 4a with the atomic displacement ellipsoids shown at the 50% probability level. | ||
In solution, the TT spacers may rotate and the major product (3a) is expected to be the energetically favoured compound where the amino groups are on the same side as the sulphurs of the TT-rings. This is also the favoured orientation for group 6 transition metal aminocarbene complexes.21,22 Compound 3b is presumed to be the result of both intra- and intermolecular NH interactions and H-bonding interactions with polar solvents (DMSO-d6 or THF), causing restricted rotation in the molecule. Single crystal X-ray diffraction confirms the molecular structure of this minor isomer 3b (vide infra), and is obtained due to the preferential crystallisation of this compound. The possibility of a cationic triscarbene monochlorido PtII complex for 3, similar to those reported earlier by us,8 is excluded due to the 1:1 integration of the carbene ligand's protons. Similarly, the trans-biscarbene isomer would be expected to yield only one set of duplicate signals and is therefore also ruled out as the possible product of 3b.
1H NMR spectra of mixtures of 4a, 4b and 4d in CDCl3 were recorded (ESI,† Fig. S11). DMSO was employed as a deuterated solvent for 1H NMR spectroscopy of the least soluble, purified fraction 4d (ESI,† Fig. S12). Poor resolution and peak broadening are observed for 4d, ascribed to small differences in chemical shifts of protons as more than one of the same carbene ligand is present in the macromolecule, along with the impact of bulky carbene substituents restricting rotation in the molecule. This is not uncommon especially for cationic triscarbene complexes of PtII with two carbene ligands trans to each other and one carbene ligand trans to Cl.8
Two of the three geometric isomers of the cis-biscarbene complex (4a/b, Fig. 2) are identified in the reaction mixture. The sharp, resolved signals of the biscarbene complexes are clearly distinguishable from the broadened signals of the triscarbene complex (Fig. S11, ESI†). The major cis-biscarbene isomer (4a) has the NMe2 fragments rotated away from the sulphur atoms in the TT spacers and the minor isomer (4b) has the NMe2 fragments rotated to the same side as the sulphur atoms in the TT spacers. A single crystal of 4a could be isolated to confirm the molecular structure (vide infra). Hydrogen bonding interactions closer than 2.6 Å are absent in the solid state structure of 4a and did not affect the rotations in the molecule. The third isomer 4c with two different orientations of the NMe2 fragments, would result in duplicated proton resonances in a 1:1 ratio (similar to 3b), and is not observed. The individual proton resonances of 4a are more downfield compared to 4b (Fig. S11, ESI†). Two significantly different NMe2 resonances are observed for 4a in CDCl3, ca. 0.3 ppm apart, that are more downfield compared to the single resonance of 4b at 3.30 ppm (Table 1). In (CD3)2SO both NMe2 fragments of 4a and 4b appear as two signals, respectively.
A cationic triscarbene complex structure for 4d is further supported by the poorer solubility of the complex in deuterated solvents and the upfield shifts of the TT proton resonances when compared with the corresponding biscarbene complexes. In (CD3)2SO the 2:1 ratio of the two carbene ligands trans to each other and one carbene ligand trans to Cl are observed from proton integration 2:3 (2H + 1H):2:1:1 in the aromatic region (Fig. S12, ESI†), for the three thienothienylene carbene substituents. The resonances of the two trans carbene ligands are chemically equivalent and more downfield than the third carbene ligand (trans to Cl). The three NMe2 fragments in 4d resonate as four peaks (Table 1), with the two trans NMe2 signals appearing as two broad signals (0.47 ppm apart) more downfield compared to the two broad signals of NMe2trans to Cl (0.08 ppm apart).
13C NMR data are summarized in Table 2. The carbene carbon chemical shifts of the PtII ethoxycarbene complexes (1 and 2) are at 235.2 and 233.2 ppm, respectively, comparable to the PtII ethoxy-FCC ([PtCl2{C(OEt)-p-C6H4NMe2}2], 238.5 ppm in CD2Cl2).8 Compared to the carbene carbon signals of PtCl2 bisalkoxycarbene complexes with aliphatic carbene substituents (Fig. 1c, ca. 278 ppm in CDCl3)6 and mononuclear Pt-bisethoxycarbene complexes with cyclic amine substituents (Fig. 1d, ca. 198 ppm in CDCl3),71 and 2 are shifted significantly upfield and downfield, respectively. The carbene carbon chemical shift of 2 is ca. 36 ppm downfield from the corresponding signal in the analogue aminocarbene complex 3a (196.5 ppm), measured in ((CD3)2SO); commensurate with the strong shielding effect of the amino-substituent of the carbene carbon atom, compared to an ethoxy substituent.8
| Complex | C2 | C3 | C4 | C5 | C4′ | C5′ | Ccarb | OEtb |
|---|---|---|---|---|---|---|---|---|
| a NMR data recorded in CD2Cl2. b Carbon chemical shifts for the ethoxy fragment are reported with the first value being the chemical shift of the methylene group, and the second the chemical shift of the methyl group. c Assignments could not be made unambiguously. d NMR data recorded in (CD3)2SO. | ||||||||
| 1a | 150.5 | 145.4 | 130.4 | 142.5 | 235.2 | 80.5, 14.8, 14.8 | ||
| 2a | 140.2 | 153.6 | 122.1 | 131.9 | 233.2 | 80.1, 14.9 | ||
| 3ad | 124.2 | 150.8 | 121.0 | 130.9 | 196.5 | |||
Carbon chemical shifts are very similar in 1 and 2, with the carbene carbon resonance slightly more downfield in 1. The methyl resonance in 1 is duplicated. Broad signals are obtained for the carbene carbon and C4 resonance of 2 (ESI,† Fig. S8).
Compound 3b was isolated as a mixture with 3a as the major component and no 13C NMR data were obtained for this complex, or the insoluble 4d. 2D NMR spectroscopy was employed to assign especially the aromatic resonances for the complex mixtures (ESI†).
Molecular structures
Crystallizations of the platinum(II) complexes 1, 2, 3b and 4a, precursor P4W and the oxidized ester-product 5 were carried out from saturated DCM solutions of the compounds, layered with hexane. The structures of P4W and 5 are reported in the ESI.† Due to the poor solubility of 3, the compound was crystallized from a large amount of THF layered with hexane. Single crystal X-ray diffraction studies confirmed the molecular structures (Fig. 3). Selected bond lengths, angles and torsion angles are reported in Table 3, employing the atom numbering scheme as used in the NMR assignments. The single crystal X-ray diffraction data set for 2 is of too poor quality to publish, but the molecular structure is included in the ESI,† Fig. S19 to prove atom connectivity and to use as proof of the molecule's conformation. The square planar cis-Pt dichloride fragment of 1 and 2 is attached to two thiophenes and two [2,3-b]-TTs respectively, each through an ethoxycarbene carbon.| Complex | 1 | 3b | 4a |
|---|---|---|---|
| a First set of data reported for first carbene fragment and the second for the second carbene fragment. b First mean plane drawn through C2, C3, C4 and C5, and the second through M, Ccarb and O/N. c Mean plane drawn through C2, C3, C4 and C5 of each thienylene. | |||
| Bond lengths | |||
| M–Ccarb | 1.986(7), 1.945(9) | 1.968(3), 1.964(2) | 1.993(9), 1.98(1) |
| Ccarb–O/N | 1.285(9), 1.34(1) | 1.303(4), 1.297(4) | 1.30(1), 1.29(1) |
| Pt–Cl | 2.372(3), 2.373(2) | 2.3874(7), 2.3765(8) | 2.363(3), 2.383(2) |
| Ccarb–C2/C5 | 1.42(1), 1.39(1) | 1.451(4), 1.449(4) | 1.46(1), 1.49(1) |
| C2–C3 | 1.47(1), 1.43(1) | 1.378(4), 1.383(4) | 1.39(1), 1.37(2) |
| C3–C4 | 1.44(1), 1.40(1) | 1.445(4), 1.414(4) | 1.43(1), 1.42(1) |
| C4–C5 | 1.35(1), 1.36(2) | 1.396(4), 1.375(4) | 1.37(2), 1.36(2) |
| S–C2 | 1.728(7), 1.77(1) | 1.703(3), 1.712(3) | 1.71(1), 1.73(1) |
| S–C5 | 1.685(9), 1.69(1) | 1.758(3), 1.747(3) | 1.754(8), 1.731(9) |
| Bond angles | |||
| M–Ccarb–O/N | 126.0(6), 127.5(6) | 119.1(2), 121.3(2) | 122.8(7), 125.5(7) |
| M–Ccarb–C2/C5 | 121.1(6), 123.2(6) | 122.8(2), 120.6(2) | 117.2(7), 115.8(7) |
| O/N–Ccarb–C2/C5 | 113.0(7), 109.3(7) | 118.1(3), 118.2(3) | 119.9(9), 118.6(8) |
| Torsion angles | |||
| M–Ccarb–C2/C5–C3/C4 | 14(1), 9(1) | 5.4(4), −179.0(2) | −139(1), 116(1) |
| O/N–Ccarb– C2/C5–C3/C4 | -167.5(7), −170.7(8) | −174.6(3), 1.1(5) | 46(2), −64(1) |
| Angle between two mean planesb | 10.03, 9.76 | 4.18, 2.88 | 39.54, 61.34 |
| Angle between two thienylene mean planesc | 72.75 | 87.10 | 11.15 |
In 3b and 4a, the square planar cis-Pt dichloride fragment is attached to two [2,3-b]-TT spacers, in the case of the former through aminocarbene carbons and the latter through dimethylaminecarbene carbons.
The ethoxycarbene complexes 1 and 2 have the anti-isomer arrangement in the solid state with their ethyl groups and metal moiety on the same side of the (M)Ccarb–O(Et) bonds.23–26 In both structures the ethoxy substituents appear on the same side as the thienylene spacer's adjacent sulphur atom. In the case of the aminocarbene complex 3b, the first amino group is on the same side as its thienylene spacer's adjacent sulphur atom and the second on the opposite side of its thienylene spacer's adjacent sulphur atom. For P4W, the dimethylamine group is on the same side as the sulphur atoms in the TT spacer (ESI,† Fig. S19), contradictory to what is observed for 4a (Fig. 3 and 4) where the dimethylamine groups are remote from the sulphur atoms in the TT spacers. The thienylene spacer, carbene carbon, metal and amino fragment of 3b (for both carbene fragments) are approximately in the same plane. The angle between a mean plane through C2, C3, C4 and C5 and a second through Pt, Ccarb and N confirms this (Table 3). This is not the case in 1 and 4a, and P4W and 5 (ESI†), where the angles between the two mean planes deviate from linearity as much as 10–89°. These results are also supported by the torsion angles reported in Table 3 and Table S6, ESI.† The two T and two TT fragments in 1 and 3b, respectively, are approximately perpendicular to each other.
 | ||
| Fig. 4 Angles between the thienylene fragments of 1, 3b and 4a. | ||
The angles between the fragments (mean plane drawn through C2, C3, C4 and C5 of each thienylene) are 72.75 and 87.10° individually (Fig. 4). The two carbene pπ-orbitals are also approximately perpendicular to each other and hence do not compete for the same π-interaction-orbital as the Pt center. This would explain why steric factors play such a small part in these carbene ligands. This is in contrast to the two TT fragments in 4a where the spacers are almost parallel (11.15° angle between the fragments). The averaged Pt–Ccarb bond lengths for Pt-carbene complexes in this study, 1, 3b and 4a, are the same and independent of the type of thienylene spacer or ethoxy/amino/dimethylamine group present in the molecule (1.966(8), 1.966(3) and 1.987(9) Å respectively). Compared to a PtII bisethoxycarbene complex [PtCl2{C(OEt)-p-C6H4NMe2}2] with an averaged Pt–Ccarb bond length of 1.935 (16) Å, the former is slightly longer in length.8
In Pt cationic tris-dimethylaminecarbene complexes ([PtCl{C(NMe2)-p-C6H4NMe2}3]+[W(CO)5Cl]− and [PtCl{C(NMe2)-p-C6H4NMe2}3]+PF6−) the carbene ligands trans to each other have longer averaged Pt–Ccarb bond lengths, 2.053(4) and 2.055(4) Å respectively, compared to their singular carbene ligand trans to Cl, 1.975(6) and 1.973(4) Å respectively. The Pt–Ccarb bond lengths in Pt cationic tris-dimethylaminecarbene complexes are longer compared to Pt biscarbene complexes (1, 3b, 4a and [PtCl2{C(OEt)-p-C6H4NMe2}2]).8
The Ccarb–C2/C5 bond lengths of 1 are shorter compared to 3b and 4a (Table 3), confirming that the ethoxycarbene complex is more dependent on electron density from the thienylene substituent to stabilize the carbene carbon compared to amino- and dimethylaminecarbene complexes. For 3b, NH⋯S intramolecular interactions are observed (Fig. S20, ESI†) for the sulfur of the thienylene spacer that is cis to the NH2 unit. This is not possible if the sulphurs of the thienylene spacer are trans to the NH2 unit, instead intermolecular hydrogen bonding interactions occur between TT-H4 and NH with the oxygen of a co-crystallized THF molecule (2.510 and 2.091 Å, respectively). Thus, two different orientated TT spacers are observed.
Catalytic hydrosilylation
The well-studied and documented a hydrosilylation model reaction with phenylacetylene and triethylsilane is depicted in Scheme 2.9–11,14,27,28 Six possible products have been reported for the reaction; triethyl(1-phenylvinyl)silane (α-isomer), (E)-triethyl(styryl)silane (β-E-isomer), (Z)-triethyl(styryl)silane (β-Z-isomer), hydrogenated products (styrene and ethylbenzene) and a dehydrogenative silylation product (triethyl(phenylethynyl)silane). PtII NHC catalysts display almost exclusive selectivity for the β-E or α-isomers.11,14,27,29,30 Due to the lack of solubility and the ambiguity with regards to the purity/molecular structure of the aminocarbene complexes 3 and 4, only the well-defined ethoxy-FCCs 1 and 2 were evaluated as catalyst precursors for phenylacetylene hydrosilylation with triethylsilane. The reactions in Tables 4 and 5 were performed in toluene-d8, except for the neat experiments (entries 14 and 15, Table 4). A stability test was performed for 2 (entry 1, Table 4), in the absence of any substrate. After heating the reaction tube with 2 for 6 hours at 80 °C, no decomposition indicative of a Pt-catalyzed carbene self-dimerization product is observed.31,32 | ||
| Scheme 2 Hydrosilylation of alkynes using hydrosilane.11 | ||
| Entry | Catalyst precursor | Catalyst loading (mol%) | Time (h) | Temp. (°C) | Conv.a (%) | Total% yieldb/TON/TOF (h−1) | Product distribution β-E/α/β-Z (%) |
|---|---|---|---|---|---|---|---|
| a Conversion as determined by NMR integration based on the two substrates (phenylacetylene and triethylsilane) and referenced to internal standard anisole. b Yield as determined from NMR integration based on the limiting substrate (most of the time phenylacetylene). c Experiments not performed in duplicate. d Substrate loading = 0. e An unidentified precipitate was observed. f Reactions were performed neat (no solvent). Complete conversion was observed, however no yield could be calculated. | |||||||
| 1cd | 2 | 2 | 6 | 80 | 0 | 0/0/0 | 0/0/0 |
| 2ce | — | — | 6 | 80 | 25 | 1/0/0 | 100/0/0 |
| 3e | 2 | 1 | 6 | 80 | 98 | 83/83/14 | 74/24/2 |
| 4 | 2 | 0.5 | 6 | 80 | 97 | 91/182/30 | 75/23/2 |
| 5e | 2 | 0.3 | 6 | 80 | 99 | 87/290/48 | 75/23/2 |
| 6 | 2 | 0.2 | 6 | 80 | 40 | 39/195/33 | 41/56/3 |
| 7e | 2 | 0.3 | 6 | 40 | 16 | 5/17/3 | 60/40/0 |
| 8e | 2 | 0.3 | 3 | 80 | 98 | 85/283/94 | 74/24/2 |
| 9e | 2 | 0.3 | 2 | 80 | 94 | 81/270/135 | 74/23/3 |
| 10 | 2 | 0.3 | 1 | 80 | 48 | 41/137/137 | 71/27/2 |
| 11e | 1 | 0.3 | 2 | 80 | 98 | 83/277/138 | 73/23/4 |
| 12e | K2PtCl4 | 0.3 | 2 | 80 | 13 | 0/0/0 | 0/0/0 |
| 13 | cis-[PtCl2(NCMe)2] | 0.3 | 2 | 80 | 52 | 44/147/73 | 50/45/5 |
| 14f | 2 | 0.3 | 2 | 80 | — | — | 76/22/2 |
| 15f | 1 | 0.3 | 2 | 80 | — | — | 77/22/1 |
| Entry | Substrate | Time (h) | Conv. (%) | Total % yieldb/TON/TOF (h−1) | Product distribution β-E/α/β-Z (%) |
|---|---|---|---|---|---|
| a An unidentified precipitate was observed. | |||||
| 1 |

|
2 | 99 | 94/313/157 | 76/21/3 |
| 2 |

|
2 | 100 | 100/333/167 | 99/0/1 |
| 3 |
|
2 | 99 | 94/313/157 | 61/37/2 |
| 4 |

|
2 | 99 | 98/327/163 | 88/11/1 |
| 5a |
|
2 | 100 | 87/290/145 | 81/16/3 |
| 6 |

|
2 | 6 | 0/0/0 | 0/0/0 |
| 7 |
|
2 | 73 | 73/243/122 | 69/31/0 |
| 8a |

|
2 | 13 | 0/0/0 | 0/0/0 |
| 9 |

|
1 | 98 | 95/317/317 | 86/12/2 |
| 10 |
|
1 | 86 | 86/287/287 | 99/0/1 |
| 11 |
|
1 | 38 | 36/120/120 | 72/28/0 |
| 12 |

|
1 | 99 | 96/320/320 | 89/10/1 |
Optimisation of the hydrosilylation reaction conditions were performed using 2 (entries 3–10, Table 4). The optimal reaction conditions were found to be 0.3 mol% catalyst loading at 80 °C for 2 hours (entry 9). Complete conversion was reached within this time, compared to some of the most active catalysts where 6 hours are required.9 Pronounced selectivity for the β-E-isomer was observed, as have been reported for most PtII catalysed hydrosilylation reactions.14,33 In the case of entry 9, product distribution (β-E/α/β-Z) is determined as 74/23/3 (see NMR spectrum in Fig. S22, ESI†). By-products from the reaction; styrene, ethylbenzene and triethyl(phenylethynyl)silane, are reported in Table S8, ESI.†
Styrene formed (up to 7% yield) during most of the optimization reactions, as long as the catalyst loading is high enough (>0.2 mol%) and the reaction time long enough (>1 h). Ethylbenzene was not observed and triethyl(phenylethynyl)silane is mostly observed in trace amounts (<1% yield). Changing the catalyst precursor to 1 (entry 11, Table 4) leads to a slight improvement in the % conversion and yield, but the selectivity is not influenced. The insignificantly different behaviour leads to the conclusion that there is virtually no difference between the reactivity of the T- and TT-substituted carbene ligands. However, the presence of the Fischer carbene ligand is required for catalytic activity, as determined from comparison to the performance of the platinum precursors, K2PtCl4 (entry 12) and cis-[PtCl2(NCMe)2] (entry 13).
Observation of plausible intermediates of platinum complexes containing chelated acyclic aminocarbene ligands during hydrosilylation catalysis, has been reported previously.9 These intermediates represent either products of oxidative addition of the H–Si bond of the hydrosilane to the catalyst precursor's core or a product of the 1,2-alkyne insertion into the Pt–H bond of the catalyst core, already containing the silyl fragment (see Scheme S2, ESI†). K2PtCl4 and cis-[PtCl2(NCMe)2] did not perform as well as our platinum catalyst precursors, 2 and 1, that display pronounced selectivity for one-to-two products. Performing the reactions neat (solvent free) with 2 (entry 14) and 1 (entry 15), led to reaction completion. No yield could be calculated for these reactions, but the product distribution could be determined as a ratio. Compared to entries 9 and 11, respectively, slightly more β-E-isomer and less β-Z-isomer formed. Complex 2 displayed high functional group tolerance as substrate screening was investigated (Table 5). Compared to hydrosilylation with phenylacetylene as substrate, entries 1–5 led to higher % yields of the hydrosilylation isomeric products.
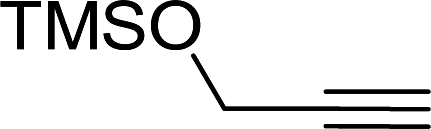
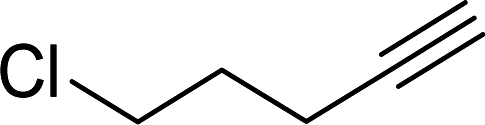
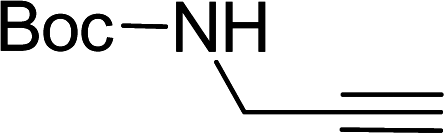
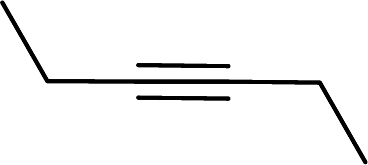
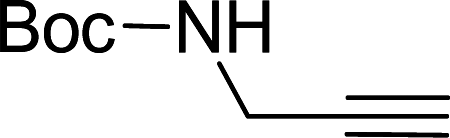
Entry 7, carried out with N-Boc-propargylamine as substrate, yielded less hydrosilylation isomeric products. No products are obtained when using propargylamine and bis(trimethylsilyl)acetylene as substrates. Because of the high % yields obtained, the reaction time of selected substrates was decreased with the aim to decrease % yields to enable comparisons. The % yields of entries 9 and 12 (Table 5) were insignificantly influenced by the time change. In the case of internal alkyne, 3-hexyne (entry 10) and amine-functionalised alkyne, N-Boc-propargylamine (entry 11), a decrease in activity is seen, more extreme in the case of the latter. Selectivity of 2 for the β-E-isomer is better in entries 1, 2, 4 and 5 (Table 5), compared to hydrosilylation with phenylacetylene as substrate. The NMR spectrum of entry 5, Table 5 is displayed in Fig. S27, ESI.† Complete selectivity for the β-E-isomer is observed when the internal alkyne 3-hexyne is used as substrate, as the expected α-isomer formation is not possible. Selectivity for the β-E-isomer is poorer in the cases where 3-TMSO-1-propyne (entry 3) and N-Boc-propargylamine (entry 7) are used as substrates (TMSO = trimethylsulfoxide, Boc = tert-butoxycarbonyl). When the reaction time of selected substrates is decreased, a slightly higher selectivity for the β-E-isomer is observed. Assignments of 1H NMR signals (Table S7, ESI†), for products obtained during hydrosilylation were made according to literature reports: phenylacetylene,14,34–37 1-hexyne,38–41 3-hexyne,42–45 3-TMSO-1-propyne,46 5-hexyn-1-ol38 and 5-chloro-1-pentyne.47 The only evidence of a dehydrogenative silylation product is observed as a byproduct in the reaction using phenylacetylene as a substrate (triethyl(phenylethynyl)silane, 1.23 (9H, t, 2J 7.9)).35
Comparison of the catalytic performance of 2 and 1 with recent examples of PtII NHC catalysts for this benchmark hydrosilylation reaction, reveals superior selectivity of the PtII FCCs 1 and 2 in the majority of cases.11,14,27,29,30 Similarly, the activity of the FCCs compares well with the state-of-the-art NHC complexes, as complete conversion is possible with 0.3 mol% catalyst loading at 80 °C for 2 hours, although some reports cite lower catalyst loading,11,14 or higher turnover numbers (TONs).9
Considering the evidence for vinylsilane product formation via the standard Chalk–Harrod mechanism, the same mechanism is proposed for 2 and 1 (Scheme S2, ESI†).9,34 Oxidative addition of the silane to the Pt metal centre is followed by chloride ligand dissociation. Alkyne migratory 1,2-insertion occurs into the Pt–H bond, followed by reductive elimination to give the β-E-isomer. The α-isomer is formed through 2,1-insertion of the alkyne into the Pt–H bond, followed by reductive elimination. As the formation of the β-Z-isomer is negligible, the possibility of alkyne insertion into the Pt–Si bond, is discarded.
Finally, the recyclability of 2 was investigated. A catalytic experiment was done in duplicate with 0.3 mol% catalyst loading at 80 °C for 2 hours in toluene-d8. After reaction completion/full conversion, substrates phenylacetylene and triethylsilane were added again, with the same equivalents as employed in the first batch reaction. This procedure was repeated four times, and the reaction still ran to completion and no decrease in activity was observed.
Conclusions
The number of carbene ligands that coordinate to the PtII metal, depends on the steric and electronic properties of the carbene ligand as determined by its substituents. In this study, mostly neutral mononuclear PtII-biscarbene complexes were obtained (1, 2, 3a/b and 4a/b) from carbene transfer via group 6 FCC precursors. In the case of both the Pt-biscarbene complexes 3 and 4, two geometric stereoisomers are observed. The formation of an insoluble cationic Pt triscarbene complex 4d was only indicated for the transmetallation reaction from dimethylaminocarbene complexes.This study reports the first utilization of Fischer carbene ligands coordinated to a PtII metal centre, as molecular catalyst precursors for alkyne hydrosilylation reactions. The PtII FCCs 1 and 2 were tested as catalyst precursors for the benchmark reaction of phenylacetylene with triethylsilane. Both (pre)catalysts exhibited good activity as complete conversion was reached in a short period of time, and a high functional group tolerance for the alkyne substrates. At optimal reaction conditions, product distribution (β-E/α/β-Z) was determined as 73/23/4 and 74/23/3, respectively. Notably, this selectivity is an improvement on that of known PtII NHC complexes, with comparable activity. The possibility of excluding solvent use in neat hydrosilylation reactions with 1/2 as catalyst precursors, proved possible with the additional advantage of improved selectivity for the β-E-isomer, while 2 can be reused for at least four batch hydrosilylation reactions, without any evidence of decomposition or decreased reactivity.
Experimental
General
All operations were carried out using standard Schlenk techniques or vacuum line techniques under an inert atmosphere of nitrogen or argon, using oven-dried glassware. Silica gel 60 (particle size 0.063–0.20 mm) was used as resin (stationary phase) for all column chromatography separations.Chemical reagents and solvents
THF and diethyl ether were distilled over sodium wire and benzophenone under N2 (g) atmosphere, hexane over sodium wire and DCM over CaH2. Other chemicals were used as they were commercially supplied by Sigma Aldrich and Strem Chemicals. The n-BuLi used in syntheses was from a stock 1.6 M solution in hexane.Triethyloxonium tetrafluoroborate was prepared according to literature procedure and stored in diethyl ether under Ar (g).48 Boron trifluoride etherate was distilled before use. Pt(COD)Cl2,49cis-[PtCl2(NCMe)2],50 and group 6 FCC precursors21,51–53 [Cr(CO)5{C(OEt)-2-C4H3S}] P1Cr, [Cr(CO)5{C(OEt)-5-C6H3S2}] P2Cr, [W(CO)5{C(OEt)-5-C6H3S2}] P2W, and [W(CO)5{C(NH2)-5-C6H3S2}] P3W were prepared according to literature procedures. The synthesis and characterization of [Cr(CO)5{C(NMe2)-5-C6H3S2}] P4Cr and [W(CO)5{C(NMe2)-5-C6H3S2}] P4W are reported in the ESI.†
Synthesis and characterization of Fischer carbene complexes
Synthesis of platinum(II) ethoxycarbene complexes of thiophene
Excess P1Cr (3.00 g, 9.0 mmol) and Pt(COD)Cl2 (1.53 g, 4.1 mmol) were dissolved in 20 mL dried DCM (degassed for 15 min) and allowed to reflux under an inert atmosphere at 50 °C for 24–30 hours. The solvent was removed under vacuum and the reaction mixture dissolved in a minimal amount of DCM. The product was cannula filtered and triturated with hexane. The product was repeatedly washed with large amounts of dry ether to separate unreacted Pt(COD)Cl2 from 1 (listed in ESI,† Table S2), before being dried under vacuum.1: δ1H(400.13 MHz; CDCl3; Me4Si) 8.09 (2 H, d, 3J5,4 4.7, H5), 7.28 (2 H, dd, 3J 4.7, 3.7, H4), 8.69 (2 H, d, 3J3,4 3.7, H3), 5.60 (4 H, s, br, CH2), 1.59 (6 H, t, J 6.8, CH3). δ1H(300.13 MHz; CD2Cl2; Me4Si) 8.13 (2 H, dd, 3J5,4 5.0, 4J5,3 0.9, H5), 7.31 (2 H, dd, 3J 5.0, 3.8, H4), 8.68 (2 H, d, 3J3,4 3.8, H3), 5.54 (4 H, s, br, CH2), 1.59 (6 H, t, J 7.1, CH3). δ13C(75.468 MHz; CD2Cl2; Me4Si) 235.2 (Ccarb), 142.5 (C5), 130.4 (C4), 145.4 (C3), 150.5 (C2), 80.5 (CH2), 14.8 and 14.8 (CH3). m/z (C14H16O2Cl2S2Pt, 546.39 g mol−1) calculated: 736.9030, found: 737.1392 (30%, [M2-{C(OEt)C4H3S}2-H]−).
Synthesis of platinum(II) ethoxycarbene complexes of [2,3-b]-TT
The same reaction method as above was applied to excess P2Cr (0.17 g, 0.4 mmol) and Pt(COD)Cl2 (0.06 g, 0.2 mmol). Instead of using large amounts of ether, the product was repeatedly washed with minimum amounts of chloroform, followed by a single ether wash. Dissolved in DCM, the product (listen in ESI,† Table S3) was dried on MgSO4, filtered and dried under vacuum.2: λmax(CH2Cl2)/nm 400 (ε/dm3 mol−1 cm−1 33140), 355 (43120). δ1H (400.13 MHz; CDCl3; Me4Si) 9.08 (2H, s, H4), 7.44 (2H, d, 3J5′,4′ 5.2, H5′), 7.35 (2H, s, br, H4′), 5.77 and 5.18 (2H + 2H, s, br, CH2), 1.59 (6H, s, br, CH3). δ1H (500.139 MHz; CD2Cl2; Me4Si) 9.03 (2H, s, H4), 7.49 (2H, d, 3J5′,4′ 5.1, H5′), 7.39 (2 H, s, br, H4′), 5.73 and 5.21 (2H + 2H, s, br, CH2), 1.58 (6H, s, br, CH3). δ1H (400.13 MHz; CD2Cl2; Me4Si at 2 °C) 9.05 (2H, s, H4), 7.49 (2H, s, br, H5′), 7.39 (2H, s, br, H4′), 5.73 and 5.14 (2H + 2H, s, br, CH2), 1.61 (6H, s, br, CH3). δ13C (125.75 MHz; CD2Cl2; Me4Si) 233.2 (br, Ccarb), 153.6 (C5), 140.2 (br, C4), 152.3 and 148.3 (C3 and C2), 131.9 (C5′), 122.1 (C4′), 80.1 (CH2), 14.9 (CH3). m/z(C18H16O2S4Cl2Pt, 658.56 g mol−1) calculated: 691.8747, found: 691.8950 (35%, [M + Cl]−).
Synthesis of platinum(II) aminocarbene complexes of [2,3-b]-TT
The same reaction method as above was applied to excess P3W (0.11 g, 0.22 mmol) and Pt(COD)Cl2 (0.03 g, 0.1 mmol). A cream brown reaction mixture was obtained when minimum amounts of DCM were added. The product was washed repeatedly with DCM and ether using cannula filtration. The product was dried under vacuum, dissolved in THF and filtered through Celite and MgSO4 before being put up for crystals. The products isolated are listed in ESI,† Table S4.3a: νNH(KBr pellet)/cm−1 3294s, br (νas) and 3210s, br (νs). δ1H (500.139 MHz; (CD3)2SO; Me4Si) 10.77 and 10.61 (2H + 2H, s, br, NH2), 8.26 (2H, s, H4), 7.66 (2H, d, 3J5′,4′ 5.3, H5′), 7.40 (2 H, d, 3J4′,5′ 5.3, H4′). δ13C(125.75 MHz; (CD3)2SO; Me4Si) 196.5 (Ccarb), 150.8 (C5), 124.2 (C4), 146.0 and 145.8 (C3 and C2), 130.9 (C5′), 121.0 (C4′). m/z (C14H10N2Cl2S4Pt, 600.49 g mol−1) calculated: 677.7935, found: 677.7938 (9%, [M + Br]−).
3b: δ1H (500.139 MHz; (CD3)2SO; Me4Si) 11.42 and 11.18 (2H + 2H, s, br, NH2), 8.56 and 8.46 (2H, s, H4), 7.73 and 7.72 (2H, d, 3J5′,4′ 5.3, H5′), 7.46 and 7.43 (2 H, d, 3J4′,5′ 5.3, H4′).
Synthesis of platinum(II) dimethylamine-carbene complexes of [2,3-b]-TT
The same reaction method was applied to excess P4Cr (0.17 g, 0.45 mmol) and Pt(COD)Cl2 (0.056 g, 0.15 mmol). The solvent was removed under vacuum and the reaction mixture washed with minimal amounts of DCM, chloroform and ether. The product (listed in ESI,† Table S5) was dried under vacuum. Filtering the product through silica gel, with acetone as eluent, causes the product to decompose to the corresponding organic ester product 5.4a: δ1H (300.13 MHz; CDCl3; Me4Si) 8.06 (2H, s, H4), 7.45 (2H, d, 3J 5.3, H5′), 7.35 (2H, d, 3J 5.3, H4′), 4.22 and 3.86 (6H + 6H, s, NCH3). δ1H (300.13 MHz; (CD3)2SO; Me4Si) 8.00 (2 H, s, H4), 7.73 (2 H, d, 3J 5.3, H5′), 7.45 (2H, d, 3J 5.3, H4′), 4.06 and 3.77 (6H + 6H, s, NMe2). m/z (C18H18N2Cl2S4Pt, 656.59 g mol−1) calculated: 688.7982, found: 688.9038 (12%, [M + Br-NMe2-H]−), calculated: 661.9028, found: 661.9876 (10%, [M + Br-2Cl-2H]−).
4b: δ1H (400.13 MHz; CDCl3; Me4Si) 7.61 (2H, s, H4), 7.38 (2H, d, 3J 5.3, H5′), 7.29 (2 H, d, 3J 5.3, H4′), 3.30 (12H, s, NMe2). δ1H (500.139 MHz; (CD3)2SO; Me4Si) 7.80 (2H, s, H4), 7.70 (2H, d, 3J 5.2, H5′), 7.41 (2H, d, 3J 5.2, H4′), 4.15 and 3.65 (6H + 6H, s, NMe2).
4d: δ1H (300.13 MHz; (CD3)2SO; Me4Si) 7.65 (2H, s, br, H4 (trans to carbene ligand)), 7.37 (3H, s, br, H5′ (trans to carbene ligand) + H4 (trans to Cl)), 7.26 (2H, s, br, H4′ (trans to carbene ligand)), 6.87 (1H, s, br, H5′ (trans to Cl)), 6.78 (1H, s, br, H4′ (trans to Cl)), 4.10 and 3.63 (6H + 6H, s, br, NMe2 (trans to carbene ligand)), 3.38 and 3.30 (3H and 3H, s, br, NCH3 (trans to Cl)).
5: νCO(hexane)/cm−1 1639m, br (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration). δ1H(300.13 MHz; CDCl3; Me4Si) 7.48 (1H, s, H4), 7.36 (1H, d, 3J5′,4′ 5.3, H5′), 7.21 (1H, d, 3J4′,5′ 5.3, H4′), 3.21 (6H, s, br, NMe2). δ13C (75.468 MHz; CDCl3; Me4Si) 164.1 ((NMe2)C(O)), 145.9 (C5), 121.8 and 121.8 (C4), 141.1 and 140.2 (C3 and C2), 128.6 (C5′), 120.3 (C4′), 38.0 (br, NMe2). m/z (C9H9NOS2, 211.3 g mol−1) calculated: 212.0204, found: 212.0264 (84%, [M + H]+), calculated: 234.0023, found: 234.0091 (100%, [M + Na]+).
O stretching vibration). δ1H(300.13 MHz; CDCl3; Me4Si) 7.48 (1H, s, H4), 7.36 (1H, d, 3J5′,4′ 5.3, H5′), 7.21 (1H, d, 3J4′,5′ 5.3, H4′), 3.21 (6H, s, br, NMe2). δ13C (75.468 MHz; CDCl3; Me4Si) 164.1 ((NMe2)C(O)), 145.9 (C5), 121.8 and 121.8 (C4), 141.1 and 140.2 (C3 and C2), 128.6 (C5′), 120.3 (C4′), 38.0 (br, NMe2). m/z (C9H9NOS2, 211.3 g mol−1) calculated: 212.0204, found: 212.0264 (84%, [M + H]+), calculated: 234.0023, found: 234.0091 (100%, [M + Na]+).
Catalytic hydrosilylation reactions
A dry high pressure NMR tube fitted with a J. Young valve, was charged with the (pre)catalyst precursor, alkyne, triethylsilane and internal standard in toluene-d8 under an atmosphere of argon, before heating. Each reaction contained triethylsilane (40.0 μL, 0.25 mmol) and 0.2 mL toluene-d8. Anisole was used as internal standard (27 μL, 0.25 mmol) and was integrated in the proton NMR spectra to represent three protons (area: 3.34–3.31 or 3.36–3.30 ppm). No triethylsilane was added in entry 1 (Table 4) and entries 14 and 15 (Table 4) were done solvent free. After a predetermined time at a certain temperature (80 °C, except entry. 7, Table 4, was performed at 40 °C), the NMR data are collected and analysed. Reaction conditions applied to individual reactions are reported in Table S8, ESI.† After the reactions were completed the solutions appeared pale yellow, indicating the presence of hydrosilylation isomeric products that formed.A descriptive example for a performed catalytic run (entry 9, Table 4) is as follows: 0.3 mol% of 2, phenylacetylene (27.5 μL, 0.25 mmol), triethylsilane (40.0 μL, 0.25 mmol), anisole (27.0 μL, 0.25 mmol) and 0.2 mL toluene-d8 was added to a high pressure NMR tube, under an atmosphere of argon. The sealed NMR tube was then placed in an oil bath set at 80 °C for 2 hours. The NMR tube is allowed to reach RT before NMR spectra were collected. Catalytic reactions were performed in duplicate, unless stated otherwise, and the averaged results reported.
Characterization and analytical techniques
Ultraviolet spectra could not be obtained for 1, 3, 4 and 5 (these compounds are yellow to colourless when diluted to 0.01 mM in 10 mL of DCM and 3 and 4 are insoluble in DCM). 4d did not ionize during mass spectrometric (MS) analysis. Dimerization of 1 through bridging chlorido ligands was observed in the MS of the sample.First-order analysis was carried out to assign signals of the 1H NMR spectra. Additional 2D [1H, 1H] COSY NMR experiments were done where confirmation of the proton assignments were required. Assigning the carbon chemical shifts, obtained from proton-decoupled 13C NMR spectra, was possible with the assistance of 2D [1H, 13C] HSQC and 2D [1H, 13C] HMBC NMR experiments (see ESI,† Section S3). Standard Bruker pulse programs were used in the experiments.
UV-Vis spectroscopy
Measurements were performed on 10 mL DCM solutions of 0.01 mM analyte concentration at 25 °C. Absorptions were measured in the range 200–1000 nm using a UV-Vis spectrophotometer Specord 200 plus. WinASPECT PLUS (version 4.2) software was used for data visualization.X-ray diffraction analysis
Single crystal diffraction data for P4W, 2, 3b and 5, were collected at 150 K on a Bruker D8 Venture diffractometer with a kappa geometry goniometer and a Photon 100 CMOS detector using a Mo-Kα IμS.micro focus source. Data were reduced and scaled using SAINT and absorption intensity corrections were performed using SADABS (APEX III control software).54 Single crystals of 1 and 4a were analyzed on a Rigaku XtaLAB Synergy R diffractometer, with a rotating-anode X-ray source and a HyPix CCD detector. Data reduction and absorption were carried out using the CrysAlisPro (version 1.171.40.23a) software package.55 X-ray diffraction measurements were all performed at 150 K (except for 4a at 293 K) using an Oxford Cryogenics Cryostat. All structures were solved by an intrinsic phasing algorithm using SHELXTS56 and were refined by full-matrix least-squares methods based on F2 using SHELXL.57 All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were placed in idealized positions and refined using riding models. The X-ray crystallographic coordinates for the structures of compounds P4W, 1, 2, 3b, 4a and 5 have been deposited at the Cambridge Crystallographic Data Centre (CCDC), with deposition numbers CSD 2061163–2061168.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the National Research Foundation, South Africa (NRF 105740; NRF 105529, NRF 87788 and NRF 9772) and Sasol Technology R&D Pty. Ltd. (South Africa) for financial support.References
- L. Chugaev and M. Skanavy-Grigorizeva, Russ. Chem. Soc., 1915, 47, 776 Search PubMed.
- L. Chugaev, M. Skanavy-Grigorizeva and A. Posniak, Z. Anorg. Allg. Chem., 1925, 148, 37 CrossRef CAS.
- W. M. Butler, J. H. Enemark, J. Parks and A. L. Balch, Inorg. Chem., 1973, 12, 451 CrossRef CAS.
- E. M. Badley, J. Chatt, R. L. Richards and G. A. Sim, J. Chem. Soc. D, 1969, 53, 1322 RSC.
- M. H. Chisholm and H. C. Clark, Inorg. Chem., 1971, 10, 1711 CrossRef.
- M. Werner, T. Lis, C. Bruhn, R. Lindner and D. Steinborn, Organometallics, 2006, 25, 5946 CrossRef CAS.
- M. P. López-Alberca, M. J. Mancheño, I. Fernández, M. Gómez-Gallego, M. A. Sierra and R. Torres, Chem. – Eur. J., 2009, 15, 3595 CrossRef PubMed.
- N. Weststrate, Structural and electronic features of tungsten(0) and platinum(II) complexes with Fischer carbene ligands, PhD thesis, University of Pretoria, Pretoria, 2017 Search PubMed.
- R. S. Chay, B. G. M. Rocha, A. J. L. Pombeiro, V. Y. Kukushkin and K. V. Luzyanin, ACS Omega, 2018, 3, 863 CrossRef CAS PubMed.
- K. Stefanowska, A. Franczyk, J. Szyling, K. Salamon, B. Marciniec and J. Walkowiak, J. Catal., 2017, 356, 206 CrossRef CAS.
- J. J. Hu, F. Li and T. S. A. Hor, Organometallics, 2009, 28, 1212 CrossRef CAS.
- G. De Bo, G. Berthon-Gelloz, B. Tinant and I. E. Markó, Organometallics, 2006, 25, 1881 CrossRef CAS.
- C. Xu, B. Huang, T. Yan and M. Cai, Green Chem., 2018, 20, 391 RSC.
- B. G. M. Rocha, E. A. Valishina, R. S. Chay, M. F. C. Guedes Da Silva, T. M. Buslaeva, A. J. L. Pombeiro, V. Y. Kukushkin and K. V. Luzyanin, J. Catal., 2014, 309, 79 CrossRef CAS.
- F. Alonso, R. Buitrago, Y. Moglie, J. Ruiz-Martínez, A. Sepúlveda-Escribano and M. Yus, J. Organomet. Chem., 2011, 696, 368 CrossRef CAS.
- M. Chauhan, B. J. Hauck, L. P. Keller and P. Boudjouk, J. Organomet. Chem., 2002, 645, 1 CrossRef CAS.
- N. Nakata, M. Sakashita, C. Komatsubara and A. Ishii, Eur. J. Inorg. Chem., 2010, 447 CrossRef CAS.
- E. Bolbat, K. Suarez-Alcantara, S. E. Canton and O. F. Wendt, Inorg. Chim. Acta, 2016, 445, 129 CrossRef CAS.
- A. M. Voutchkova, M. Feliz, E. Clot, O. Eisenstein and R. H. Crabtree, J. Am. Chem. Soc., 2007, 129, 12834 CrossRef CAS PubMed.
- F. Hanasaka, Y. Tanabe, K. I. Fujita and R. Yamaguchi, Organometallics, 2006, 25, 826 CrossRef CAS.
- Z. Lamprecht, Fischer carbene complexes of 3,3′-bithiophene and thieno[2,3-b]thiophene, MSc dissertation, University of Pretoria, Pretoria, 2015 Search PubMed.
- Z. Lamprecht, F. P. Malan, D. C. Liles, S. Lotz and D. I. Bezuidenhout, J. Organomet. Chem., 2020, 925, 121466 CrossRef CAS.
- D. M. Andrada, M. E. Zoloff Michoff, I. Fernández, A. M. Granados and M. A. Sierra, Organometallics, 2007, 26, 5854 CrossRef CAS.
- I. Fernández, F. P. Cossio, A. Arrieta, B. Lecea, M. J. Mancheño and M. A. Sierra, Organometallics, 2006, 23, 1065 CrossRef.
- N. Lugan, I. Fernández, R. Brousses, D. A. Valyaev, G. Lavigne and N. A. Ustynyuk, Dalton Trans., 2013, 42, 898 RSC.
- D. A. Valyaev, R. Brousses, N. Lugan, I. Fernández and M. A. Sierra, Chem. – Eur. J., 2011, 17, 6602 CrossRef CAS PubMed.
- M. Poyatos, A. Maisse-François, S. Bellemin-Laponnaz and L. H. Gade, Organometallics, 2006, 25, 2634 CrossRef CAS.
- L. Yong, K. Kirleis and H. Butenschön, Adv. Synth. Catal., 2006, 348, 833 CrossRef CAS.
- F. Zhang, Y. Bai, X. Yang, J. Li and J. Peng, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 1271 CrossRef CAS.
- C. Lu, S. Gu, W. Chen and H. Qiu, Dalton Trans., 2010, 39, 4198 RSC.
- M. P. López-Alberca, M. J. Mancheño, I. Fernández, M. Gómez-Gallego, M. A. Sierra and R. Torres, Org. Lett., 2007, 9, 1757 CrossRef PubMed.
- M. A. Sierra, J. C. del Amo, M. J. Mancheño and M. Gómez-Gallego, J. Am. Chem. Soc., 2001, 123, 851 CrossRef CAS PubMed.
- G. Berthon-Gelloz, J. M. Schumers, G. De Bo and I. E. Markó, J. Org. Chem., 2008, 73, 4190 CrossRef CAS PubMed.
- A. Tyagi, S. Yadav, P. Daw, C. Ravi and J. K. Bera, Polyhedron, 2019, 172, 167 CrossRef CAS.
- M. Wissing and A. Studer, Chem. – Eur. J., 2019, 25, 5870 CrossRef CAS PubMed.
- H. Liang, Y.-X. Ji, R.-H. Wang, Z.-H. Zhang and B. Zhang, Org. Lett., 2019, 21, 2750 CrossRef CAS PubMed.
- K. H. Lee, B. Lee, K. R. Lee, M. H. Yi and N. H. Hur, Chem. Commun., 2012, 48, 4414 RSC.
- X. Zhao, D. Yang, Y. Zhang, B. Wang and J. Qu, Org. Lett., 2018, 20, 5357 CrossRef CAS PubMed.
- S. V. Maifeld, M. N. Tran and D. Lee, Tetrahedron Lett., 2005, 46, 105 CrossRef CAS.
- S. Schwieger, R. Herzog, C. Wagner and D. Steinborn, J. Organomet. Chem., 2009, 694, 3548 CrossRef CAS.
- Z. Zhang, T. Xiao, H. Al-Megren, S. A. Aldrees, M. Al-Kinany, V. L. Kuznetsov, M. L. Kuznetsov and P. P. Edwards, Chem. Commun., 2017, 53, 4026 RSC.
- R. Cano, M. Yus and D. J. Ramón, ACS Catal., 2012, 2, 1070 CrossRef CAS.
- K. Jakobsson, T. Chu and G. I. Nikonov, ACS Catal., 2016, 6, 7350 CrossRef CAS.
- K. M. McWilliams and R. J. Angelici, Organometallics, 2007, 26, 5111 CrossRef CAS.
- R. J. Abraham, M. Canton and L. Griffiths, Magn. Reson. Chem., 2001, 39, 421 CrossRef CAS.
- Z. Gan, Y. Wu, L. Gao, X. Sun, J. Lei, Z. Song and L. Li, Tetrahedron, 2012, 68, 6928 CrossRef CAS.
- R. Takeuchi, S. Nitta and D. Watanabe, J. Org. Chem., 1995, 60, 3045 CrossRef CAS.
- H. Meerwein, Org. Synth., 1966, 46, 113 CrossRef CAS.
- H. C. Clark and L. E. Manzer, J. Organomet. Chem., 1973, 59, 411 CrossRef CAS.
- F. P. Fanizzi, F. P. Intini, L. Maresca and G. Natile, J. Chem. Soc., Dalton Trans., 1990, 199 RSC.
- J. A. Connor and E. M. Jones, J. Chem. Soc. A, 1971, 1974 RSC.
- Z. Lamprecht, N. A. van Jaarsveld, D. I. Bezuidenhout, D. C. Liles and S. Lotz, Dalton Trans., 2015, 44, 19218 RSC.
- L. Yang, P. Ye, W. Li, W. Zhang, Q. Guan, C. Ye, T. Dong, X. Wu, W. Zhao, X. Gu, Q. Peng, B. Tang and H. Huang, Adv. Opt. Mater., 2018, 6, 1 Search PubMed.
- APEX3. (including SAINT and SADABS). BrukerAXS Inc., Madison, WI 2016.
- Rigaku Oxford Diffraction. Rigaku Corporation: Oxford, UK 2018.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis details, NMR, crystal data collection, structure refinement, crystal packing details and hydrosilylation catalysis details. CCDC CSD 2061163–2061168. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1nj00791b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |