
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation and application of solvent-free liquid proteins with enhanced thermal and anhydrous stabilities
Alex P. S.
Brogan

Department of Chemistry, King's College London, Britannia House, 7 Trinity Street, London, SE1 1DB, UK. E-mail: alex.brogan@kcl.ac.uk
First published on 9th March 2021
Abstract
Chemical modification of proteins to yield solvent-free liquid proteins has been demonstrated to be a robust method for stabilizing proteins against temperature, aggregation, and non-aqueous environments. This new class of biomaterial shows exceptional compatibility with ionic liquids, with enzyme architecture preserved and its activity enhanced. This review provides an overview of the work to-date on developing solvent-free liquid proteins and provides a perspective on the current move towards applications such as non-aqueous biocatalysis, therapeutic protein stabilization, and soft functional materials.
 Alex P. S. Brogan | Dr Alex P. S. Brogan received his PhD from the School of Chemistry, University of Bristol in 2012. After a short post doc there, he moved to the Department of Chemical Engineering, Imperial College London, where he was a Research Associate until 2019. In 2019, Dr Brogan established his independent career as a Lecturer in Chemistry in the Department of Chemistry, King's College London. The primary focus of his research is to make biological systems more compatible with synthetic systems, so that we can fully exploit nature's toolkit to make our own industry more sustainable and more environmentally friendly. |
Introduction
Ever since the pioneering work of Klibanov and others to show that enzymes could retain activity in organic solvents,1–4 subsequent research focussed on understanding and improving their activity in non-aqueous environments.5–10 The ultimate goal has been – and continues to be – the expansion of the repertoire of enzymes for their use in industrial biocatalysis.11–17 The drive behind using enzymes in non-aqueous environments lies in the tantalising prospect of using the vast range and high selectivity and specificity of enzymes with the ubiquity of organic solvents for the synthesis of chemical commodities. The manufacturing of many of the products required for modern living would benefit tremendously by using biocatalysts (e.g. for enantiopurity or selective transformations). However, the synthetic reactions to be catalysed are frequently very difficult to perform in aqueous solutions due to limited substrate reactivity, side reactions (e.g. unwanted hydrolysis), or low solubilities. For many industrial applications, product separation and purification from water presents an expensive problem (both energetically and financially) that severely limits applications. It would therefore be a great advantage to perform biocatalytic reactions in non-aqueous solvents: performing enzymatic conversions with improved kinetics due to optimal substrate solubilities. However, despite all that we know, the use of enzymes in non-aqueous solvents remains severely limited by their low solubilities and limited stabilities.The balance between enzyme stabilization, biocatalytic activity, and industrial usability is difficult to achieve. Traditionally, the focus has been on overcoming enzyme stability in organic solvents through protection strategies involving immobilization on, or encapsulation within, solid supports.18 Typically, the supports used for these strategies consist of either silica, polymer composites, or the enzymes themselves in the form of cross linked enzyme aggregates.11,12,18 However, the increase in solvent and temperature stability provided by the solid support often comes at the price of enzyme activity, and slow kinetics burdened by mass transfer issues severely limits application to predominately small molecule biocatalysis.
Alternatively, protein engineering strategies such as site directed mutagenesis19 and directed evolution20,21 are quickly becoming powerful tools for the development of both enzymes with new reactivities and improved stabilities. Whilst these are clearly of great importance for the field of biocatalysis, development of new enzymes is limited by the speed of verifying positive alterations and progressing through the rounds of evolution required for optimisation. As such, the focus has naturally been on introducing new reactivities as opposed to improving solvent stability. The use of synthetic biology tools to develop new enzymes has allowed for some innovative engineering solutions to effectively separate aqueous biocatalysts from non-aqueous reactions.16,22–25 However, much like immobilization strategies, these methods are ultimately limited by the scope of suitable substrates.
In light of the effort and cost required to develop new enzymes and effective stabilization strategies for industrial purposes, naturally, the focus of biocatalysis development has been predominately on high value products where alternative synthesis strategies are overburdensome. However, nature has a vast toolbox with great untapped potential particularly in diversifying chemical feedstocks away from fossil fuels and towards more sustainable resources.11 Furthermore, establishing robust stabilization strategies for a broad variety of enzymes will be invaluable for the full optimisation of industrial biocatalysis for products ranging from platform chemicals to fine chemical commodities. This will enable synergistic development of versatile enzymes alongside reaction engineering: a holistic consideration of enzyme, stabilization strategy, and solvent choice otherwise termed biocatalysis engineering.26,27
Given the apparent gulf between the potential capabilities of enzyme engineering and the currently niche role of biocatalysis in industrial settings, there remains an impetus to develop universal approaches to stabilizing proteins against non-aqueous environments. There have been many exciting innovations in the development of chemoenzymatic cascades,28–31 unnatural photoenzymatic pathways,32–34 and electrobiocatalysis.35,36 The co-deployment of enzymes and abiological reactions will ultimately have greater applicability in non-aqueous solvents. As such, a platform technology that will allow proteins and enzymes to be used in a much wider range of environments, could open avenues for introducing new functionalities and reactivities for enzymes, broader stabilities, and new soft materials for biointerfacing.
Solvent-free liquid proteins
The alternative to bulk immobilization or encapsulation of enzymes is stabilizing individual proteins. The benefits of stabilization through the construction of single particle constructs lie in the potential for both maintaining reactivities for a broad range of substrates (small and large) and the application to a greater variety of proteins (with respect to size and activity). Additionally, single protein constructs will have advantages in facing industrial application as purity will allow for improved reproducibility when scaled up. The traditional approach for producing monodisperse single particles, taking a cue from colloidal science, is the synthesis of protein–polymer conjugates. This approach relies on the tethering of a polymer to the surface of a protein, typically via: conjugation of a pre-made polymer to the protein surface (“grafting-to”),37–42 growing polymers from a protein surface (“grafting-from”),43–45 or more recently; in synthesizing polymers that maximise noncovalent interactions with protein surfaces.46,47 However, the often bespoke chemistries required and sometimes marginal increases in protein stability has meant that the focus for these protein–polymer conjugates frequently lies in improving the pharmacokinetics of therapeutic biomolecules, and thus versatility of the approach to a broad range of proteins has yet to be demonstrated.We decided to take an alternative approach, derived from a materials science perspective: could we create hybrid materials that incorporated the intricacies of proteins but confer additional properties onto that base architecture. Inspired by the work of Giannelis and others on nanoparticle fluids,48–50 we set out to investigate whether proteins could become a functional component of a “solvent” in its own right. This would be achieved with a reductionist approach of building the new class of material in the simplest way possible, in a bid to maximize its versatility. The result was what we termed solvent-free liquid proteins, or liquid proteins for short.51 These distinct liquid-like materials are made by a straightforward 3-step process (Fig. 1a). The first step is charge homogenization of the protein surface through EDC mediated coupling of a diamine to surface orientated acidic residues, resulting in a cationized protein. The second step follows with electrostatic complexation by a stoichiometric (by charge) quantity of anionic polymer surfactant molecules to form the nanoconjugate. Finally, the nanoconjugate is freezedried to yield a protein that is liquid at room temperature in the absence of any solvent, and critically, the absence of any water molecules beyond those tightly bound within the protein structure.52 Critical in providing the intermolecular interactions to sustain a liquid phase in the absence of solvent, was a complete corona of surfactant molecules surrounding the protein. The importance of which has been discussed previously in a review by Perriman and Mann.51
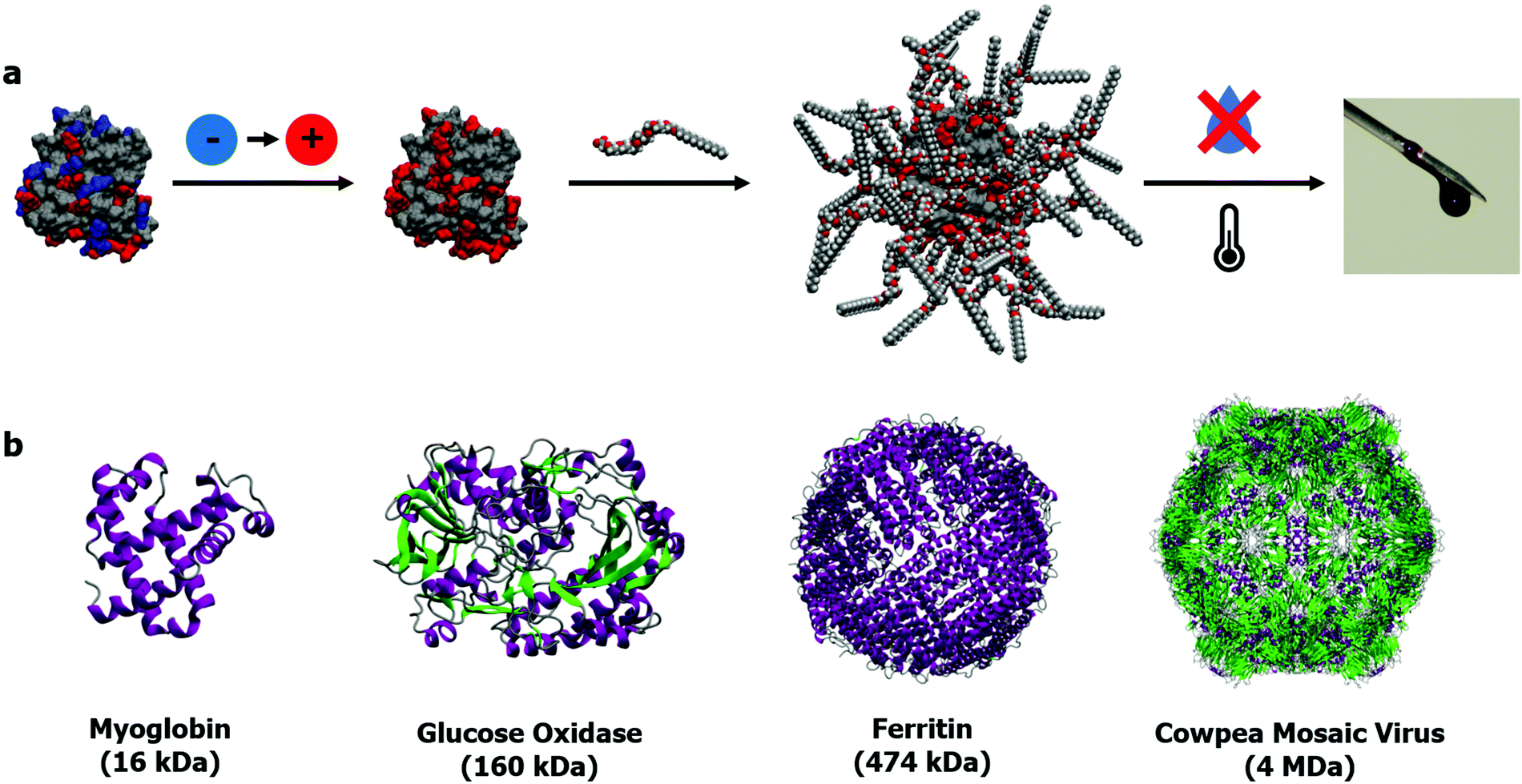 | ||
| Fig. 1 (a) 3-Step synthesis of solvent-free liquid proteins: (1) protein cationization, (2) polymer–surfactant conjugation to form stoichiometric nanoconjugate, (3) dehydration and thermal annealing to yield solvent-free liquid protein. (b) Selection of proteins (structures from PDB entries: 1MBD, 3QVP,631MFR,64 and 1NY765) to demonstrate the versatile size range of solvent-free liquid proteins: from small globular proteins to complex self-assembled structures such as ferritin and viral capsids. | ||
Importantly, with respect to potential applications of this biomaterial, not only is the protein structure maintained under anhydrous conditions, but thermal stability is drastically improved up to 100 °C greater than observed in aqueous solutions.53 Additionally, retained structure allowed for retained biological function at unprecedented temperatures up to and including 150 °C.52,54 The protective surfactant corona coating the protein surface also prevents the aggregation of reactive intermediates on the unfolding pathway.55 Elastic incoherent neutron scattering56 and molecular dynamics simulations57 revealed that these extraordinary properties stemmed entirely from the stoichiometric surface modification, which dynamically replaced water whilst also forming the protective corona. The simplicity of the approach provides a versatile modification strategy for proteins of all sizes (Fig. 1b), ranging from small globular proteins such as myoglobin52,53,58 and lysozyme,55 large multimeric proteins and enzymes such as glucose oxidase59 and hemoglobin,60 and intact viruses such as the cowpea mosaic virus61 and the M13 bacteriophage.62
Thermal stability and anhydrous activity
The defining features of solvent-free liquid proteins is the extreme thermal stability of the protein coupled with retained function in the absence of water. Using synchrotron radiation circular dichroism, we were able to probe the secondary structure of proteins in the solvent-free liquid up to 250 °C (Fig. 2a). Initial experiments on solvent-free myoglobin demonstrated a half denaturation temperature (Tm) of 160.1 °C, an almost 90 °C increase in thermal stability compared to its aqueous counterpart (Tm = 70.5 °C).53 Similarly, solvent-free lipases (from Rhizomucor miehei and Thermomyces lanuginosus) were thermally stable up to 100 °C greater (Tm range of 168–180 °C) than what was achievable in water.54 Modified lysozyme also showed a comparative increase in thermal stability, although interestingly, in the absence of water the thermal unfolding shifted from a 2-state to a 3-state mechanism.55 In this case, an otherwise reactive intermediate on the aggregation pathway remained stable up to 178 °C (Fig. 2b). This indicated that the solvent-free liquid not only protected the protein from thermal denaturation, but also against aggregation.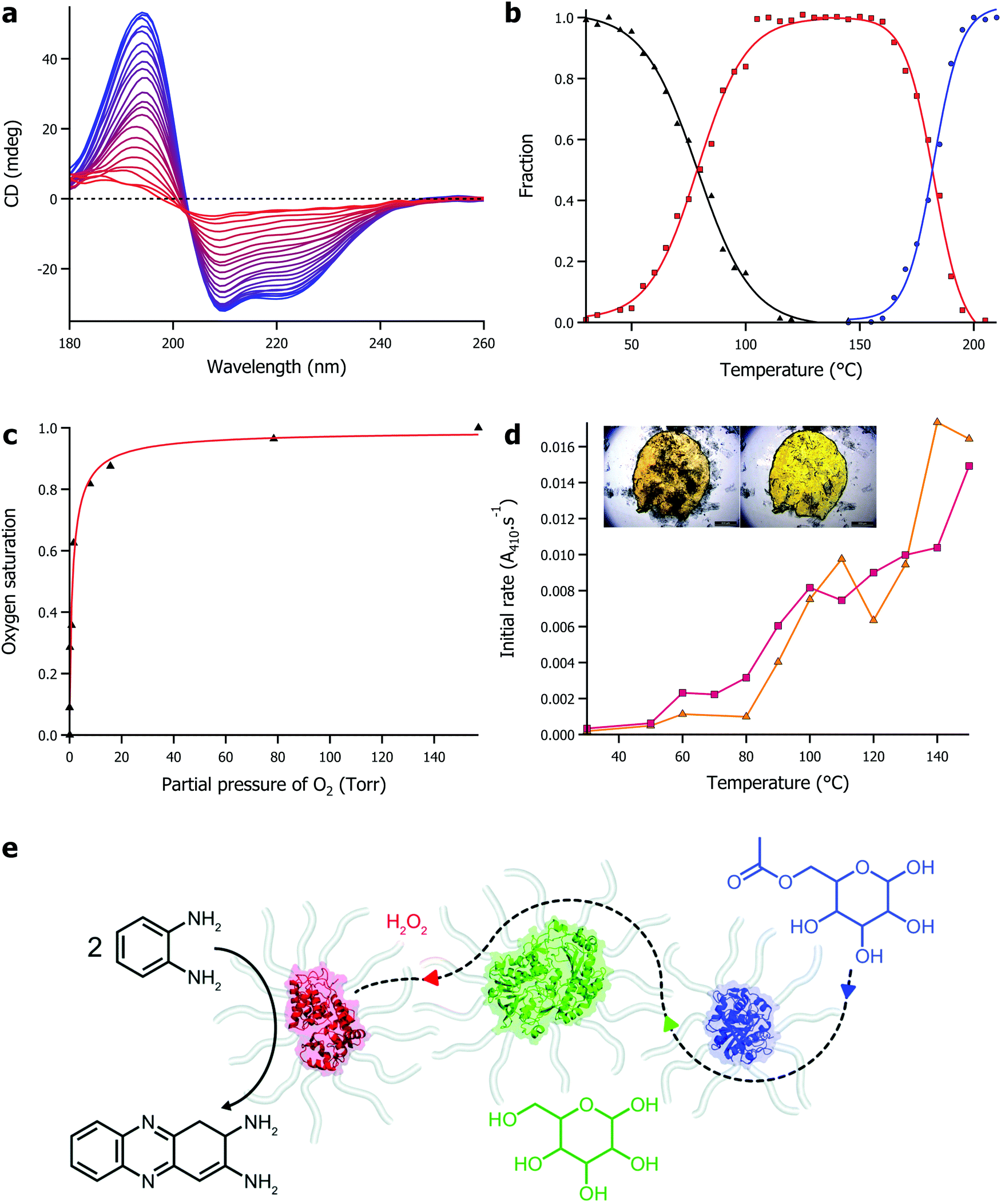 | ||
| Fig. 2 (a) Temperature dependent circular dichroism for solvent-free liquid myoglobin showing denaturation between 25 °C (blue) and 235 °C (red). Data originally presented in ref. 53. (b) Plot of fraction against temperature for native folded protein (black), reactive intermediate species (red), and denatured protein (blue) for solvent-free liquid lysozyme. Data originally presented in ref. 55. (c) Plot of oxygen saturation against partial pressure of oxygen for solvent-free liquid myoglobin, demonstrating biological function in the absence of water. Data originally presented in ref. 52. (d) Plot of initial activity against temperature for solvent-free lipases – Rhizomucor miehei (yellow), Thermomyces lanuginosus (pink) – showing hydrolysis of para-nitrophenyl palmitate in the absence of water. Inset shows microscopy images demonstrating solvent-free activity with release of para-nitrophenol (yellow colour) as a result of enzyme activity. Data originally presented in ref. 54. (e) Scheme showing solvent-free cascade involving horseradish peroxidase (red), glucose oxidase (green), and lipase (blue). Adapted under licence from D. L. Atkins, J. A. Berrocal, A. F. Mason and I. K. Voets, Nanoscale, 2019, 11, 19797 – Published by The Royal Society of Chemistry. | ||
The retention of biological function of proteins in the solvent-free liquid, in the absence of any water, was first observed in the reversible oxygen binding of myoglobin (Fig. 2c).52 This was an important proof of concept that demonstrated that not only did the modified protein have retained secondary structure in the solvent-free state, but it also had the global architecture and dynamics (later verified by EINS56) to sustain small molecule binding to a conserved active site within the protein. Development of solvent-free liquid proteins as a potential platform technology for biocatalysis required the demonstration of enzyme activity. Solvent-free liquid lipase was shown to maintain its ability to recognise its substrate, and perform its catalytic function in the absence of any water.54 Whilst the enzyme had lost 90% of its aqueous activity through the modification strategy, solvent-free lipase could still enzymatically cleave the ester bond in p-nitrophenyl butyrate and p-nitrophenyl palmitate in the absence of water, at ever increasing rates up to and including 150 °C, where a 90-fold increase in activity was observed compared to 30 °C (Fig. 2d). Pérez et al. later showed that it was possible to improve on the activity through careful consideration of the cationization process, doubling the activity of the solvent-free lipase as a result.66 Recently, solvent-free enzyme substrate scope has been expanded with Zhang et al. demonstrating phosphotriesterase activity67 and Mukhopadhayay et al. demonstrating protease activity.68 In addition, Atkins et al. showed that high thermal stability and enzyme activity could be conferred onto a mix of enzymes in a multicomponent solvent-free liquid.69 In this work the authors made a solvent-free liquid containing horseradish peroxidase, glucose oxidase, and lipase that was able to form 2,3-diaminophenazine from o-phenylenediamine mediated by a cascade reaction starting with acetylated glucose (Fig. 2e). Interestingly, the reaction did not proceed sufficiently until the temperature reached 80 °C, at which point diffusion of the various substrates was sufficient to sustain the sequential enzymatic reactions.
The development of solvent-free liquid proteins demonstrated that it was possible to create single enzyme constructs that were thermally stable, resistant to aggregation, and retained activity in the absence of water. Additionally, the surface modification significantly improves the solubility and stability of dry proteins in a broad range of non-aqueous solvents.60,70 As such, in the process of developing a new biomaterial, we had established a simple and effective platform for installing additional properties on to single protein molecules. Building on this, the modification strategy can be taken in two dominant directions. The first is to explore what new functionality can be installed onto protein surfaces. The Perriman Research Group have begun to show that this is an effective way of creating artificial membrane proteins that can re-engineer cell surfaces.71–73 The second, and the focus of the remainder of this article, centres on translating the enhanced stability of these protein nanoconjugates in non-aqueous environments to potential applications: demonstrating solvent-free liquid proteins as a platform biotechnology.
Ionic liquids and biocatalysis engineering
Given their enhanced stability against temperature and non-aqueous environments, solvent-free liquid proteins have potential as the ideal biomaterial for industrial biocatalysis. However, the one caveat to all of this: that with a typical viscosity at 30 °C of 1–5 kPa s (close to that of treacle/molasses), deploying protein liquids industrially would pose a challenge. However, inspired by the concept of biocatalysis engineering and the compatibility of protein liquids with a range of non-aqueous solvents, we turned to ionic liquids as a carrier solvent for the modified proteins. Ionic liquids–organic salts with melting temperatures typically below 100 °C and viscosities in the 10–100 mPa s range – are increasingly becoming popular as versatile solvents with chemical engineers as their near infinite combination of anions and cations allows for unprecedented reaction engineering possibilities.74 Particularly, ionic liquids have been shown to be advantageous in solubilizing otherwise recalcitrant substrates such as biopolymers (e.g. cellulose), providing avenues for broadening substrate scope. Additionally, their green and sustainable solvent credentials align well with moving towards a functional bioeconomy.75There has been a long standing body of research interfacing enzymes and ionic liquids. However, like many organic solvents, this has frequently been in conjunction with immobilization or dilution in large quantities of water.14 The alternative is use of ionic liquids that have been specifically developed to be enzyme friendly.76 However, the cost associated with developing and using bespoke ionic liquids will undoubtedly limit applications. The approach we took was to make protein-rich biofluids from common and ubiquitous ionic liquids and solvent-free liquid proteins. Specifically, we focussed on 1-ethyl-3-methylimidazolium based ionic liquids that are mass produced and available off-the-shelf from Sigma, UK. Using myoglobin, we showed that using solvent-free liquid proteins, it was possible to solvate dry proteins in both hydrophilic and hydrophobic ionic liquids up to 50 wt% (Fig. 3a).70 Furthermore, the protein retained secondary and tertiary structure in ionic liquids, with denaturation temperatures up to 55 °C higher than in water. This work was further investigated using the enzyme glucosidase in a greater range of ionic liquids.77 In this study, the thermal stability of glucosidase was improved by up to 79 °C compared to aqueous conditions, with a half denaturation temperature of 137 °C reported in the hydrophobic ionic liquid [bmpyrr][NTf2] (Fig. 3b). Improved thermal stability of glucosidase allowed for the shifting of the optimal window of glucosidase activity from 50 °C in aqueous environments, to 110 °C in the ionic liquid, which was concomitant with a 30-fold enhancement of the enzyme activity (Fig. 3c). Additionally, the superior solvent capabilities of ionic liquids towards (bio)polymer dissolution was used to demonstrate that glucosidase could release sugars from cellulose without the requirement of endo- and exo-cellulases typically found with glucosidase in cellulase cocktails (Fig. 3c and d). Mukhopadhayay et al. also demonstrated that ionic liquids could be used to improve the activity of solvent-free liquid α-chymotrypsin, further evidence to the effectiveness of the strategy.78
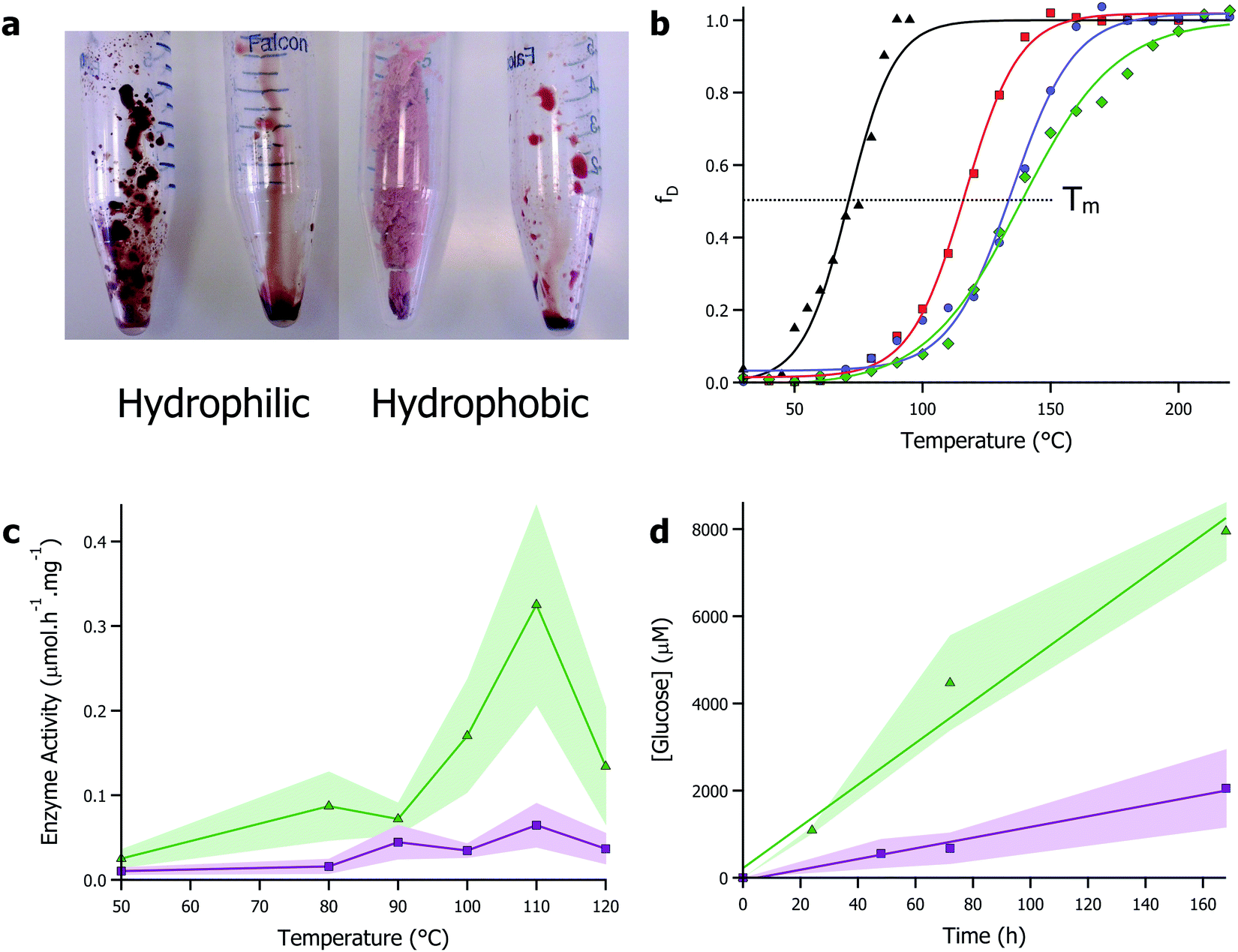 | ||
| Fig. 3 (a) Images showing solubility of solvent-free liquid myoglobin in hydrophilic ([bmpyrr][OTf]) and hydrophobic ([bmpyrr][NTf2]) ionic liquids. Images originally shown in ref. 70. (b) Plot of fraction denatured against temperature for modified glucosidase in aqueous buffer (black), [bmpyrr][OAc] (red), [bmpyrr][OTf] (blue), and [bmpyrr][NTf2]. Half denaturation temperature (Tm) increases significantly in the ionic liquids. (c) Glucosidase enzyme activity in ionic liquid against temperature demonstrating hydrolysis of cellobiose (green) and cellulose (purple). (d) Plot of glucose concentration against time for glucosidase in ionic liquid at 50 °C – linear plot demonstrates zero-order kinetics indicating enzyme is turning over in absence of water. Data for (b)–(d) originally presented in ref. 77. | ||
Soft-material design
The defining feature of solvent-free liquid proteins – particularly those of globular proteins and enzymes – is the sustained liquid phase of discrete dry proteins. The thermal stability and maintained activity of these materials has been the main focus of the research to date. However, the variety of 3-dimensional nanoscale architectures of proteins provide opportunities to design and control the properties of soft-materials. Solvent-free liquids of larger proteins such as glucose oxidase,59 protein assemblies such as ferritin,79 and viruses such as the M13 bacteriophage62 have all been shown – through crossed polarized optical microscopy (Fig. 4a) and scattering techniques – to have liquid crystalline phases in the temperature ranges between being a soft solid (<25 °C) and an isotropic liquid (>40 °C). For solvent-free glucose oxidase, due to the anisotropic nature of the protein and depending on the temperature conditions, liquid crystals with either spherulitic or dendritic morphologies could be formed.59 Combining solvent-free proteins with ionic liquids (or other non-aqueous solvents) provides additional design parameters for fine tuning soft materials. For example, the high stability and solubility of solvent-free M13 in a range of ionic liquids provides a genetically engineerable scaffold for development of new soft materials.62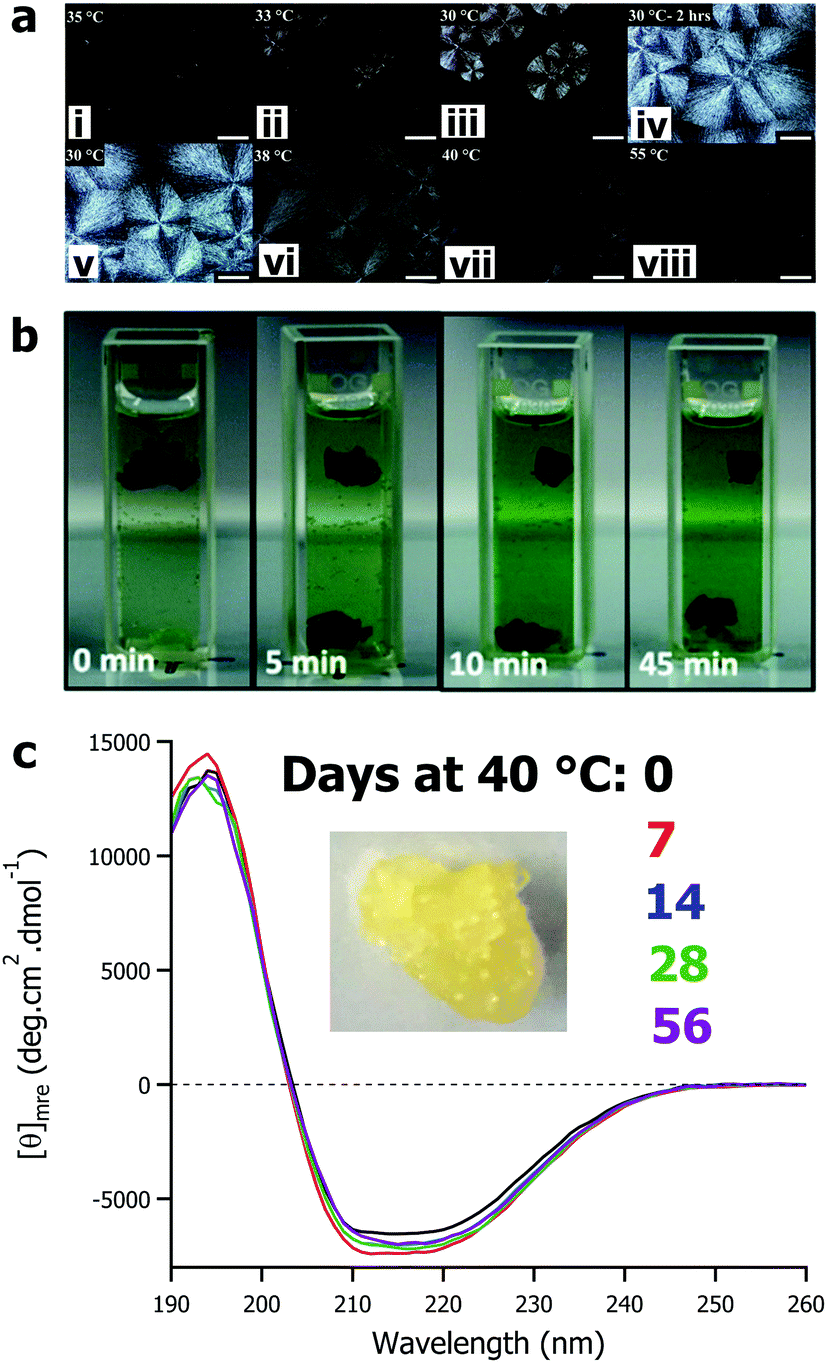 | ||
| Fig. 4 (a) Polarized optical microscope images of solvent-free glucose oxidase showing formation of spherulites upon cooling from 35 °C to 30 °C (i–iv) and formation of isotropic fluid phase on thermal annealing (v–vii). Adapted with permission from K. P. Sharma, Y. Zhang, M. R. Thomas, A. P. S. Brogan, A. W. Perriman and S. Mann, J. Phys. Chem. B, 2014, 118, 11573–11580. Copyright 2014 American Chemical Society. (b) Demonstration of enzyme cascade reaction of glucose oxidase and horseradish peroxidase films. Adapted with permission from T. Farrugia, A. W. Perriman, K. P. Sharma and S. Mann, Chem. Commun., 2017, 53, 2094–2097. Copyright 2017 Royal Society of Chemistry. (c) Circular dichroism spectra showing mean residue ellipticity of solvent-free liquid avidin after accelerated aging at 40 °C for up to 56 days. Data originally presented in ref. 82. | ||
Interestingly, through a slight change in surface modification – sub-stoichiometric surfactant addition – it is possible to engineer a different class of soft material: hierarchically assembled self-standing films.80,81 Here, film formation by surface modification is the kinetic product of protein–surfactant conjugation, whereas solvent-free liquid protein formation represents the thermodynamic product. Film formation proceeds via nanocluster growth and subsequent film formation driven by protein–protein interactions and terminated by passivation of the cluster surface by polymer surfactant coverage.80 Changing the rate of surfactant addition allowed control over the cluster size and even delineation between films and solvent-free liquids. The result is catalytically active enzyme films, where it is possible to fabricate reusable films of multiple enzymes that can sustain cascade reactions (Fig. 4b).81
Prospects and challenges
The current literature on solvent-free liquid proteins has predominately focussed on material development and characterization alongside potential application in non-aqueous biocatalysis. There is much left to be done in this arena, however there is also ample room to explore how we can utilise the high thermal stability and protection from aggregation beyond biocatalysis. Our recent work has begun exploring how this biomaterial derived surface modification strategy could be used to stabilize therapeutic proteins. Using the biotin binding protein avidin as a well characterised archetype, we demonstrated that solvent-free liquid avidin could allow for room temperature storage of this protein with no loss in structure for at least 160 days (Fig. 4c).82 Given the versatility of the modification strategy, it is not unreasonable to anticipate that this can be translated to therapeutic proteins (e.g. antibodies, growth factors, insulin, etc.) and deactivated viruses used in vaccination programs, amongst others. The significant improvement in the room temperature storage lifetime of these biomolecules would remove the requirement of the “cold chain” and could rapidly democratise access to life saving biotechnologies.Despite the advances made in developing solvent-free liquid proteins for non-aqueous biocatalysis and the initial forays into therapeutic protein stabilization, challenges remain. The chemical modification strategy appears to be ubiquitous with respect to protein size and function. However, the broad brush approach to cationization has on occasion lead to a reduction in activity, most notably in lipase54,66 and avidin.77 As such, optimisation of the process to ensure better retention of activity after chemical cationization is required. This could be achieved through a number of strategies both chemical (e.g. site-selective cationization) and biochemical (e.g. cationize the surface directly by changing amino acid sequence, or to engineer a more robust active site). Some recent work has begun to tackle this, with two approaches emerging. The first is based on controlling the chemistry of the modification: Deller et al. sought to control the kinetics of the cationization process to ensure active site preservation in thrombin,73 and Zhang et al. used a combination of anionic and cationic polymer surfactants to circumvent the requirement of covalent modification entirely.67 The second approach is realized through utilisation of synthetic biology to modify the protein itself to be more amenable to chemical modification. For example, Zhou et al. used site directed mutagenesis to ensure that chemical modification did not alter the active site of lipase, increasing subsequent activity as a result.83 These results are promising, however it is likely that further optimization will open up new possibilities, particularly in ensuring biomaterial formation remains versatile with respect to protein class. Looking forward, with respect to optimization of the biomaterial preparation, future research would do well to consider greater involvement of protein design and directed evolution, alongside increased biocompatibility of components with reversible modifications for downstream deployment in biomedical applications. To further demonstrate broad applicability of solvent-free liquid proteins, work should look to significantly expand the complexity of the enzyme reactions to be performed in the absence of water. Particularly, it would be instructive to establish whether (and how) chemical modification of enzymes to form solvent-free liquids, and the anhydrous environments used thereafter, affects the stereochemistry of products formed.
The combination of ionic liquids and solvent-free liquid proteins should also be explored further, particularly with reference to biocatalysis. The vast range of anions and cations that can be combined to yield ionic liquids has great potential in optimising enzyme activity, substrate selectivity, substrate scope, as well as taking advantage of the wide electrochemical window available in these solvents. Additionally, an area of investigation that has yet to be explored is how the negligible vapour pressure of ionic liquids (and solvent-free liquid proteins) can be exploited for product separation and for biocatalyst recycling. Furthermore, research involving technoeconomic analysis and life-cycle assessment of the use of enzymes stabilized in ionic liquids, alongside modelling of parameters for optimal activity conditions and solvent parameters (viscosity, polarity, etc.), would be of considerable use.
Conclusions
Solvent-free liquid proteins are a new class of biomaterial with a steadily growing literature base that demonstrates the expanding diversity of proteins, enzymes, and viruses that can form liquids. The surface modification platform that allows for the formation of the biomaterial also protects the protein structure against temperature and aggregation, with thermal stabilities observed as high as 180 °C. Proteins in the solvent-free liquid state also retain structural dynamics as if they were in water, which results in biological function that is maintained at temperatures far exceeding what is capable under aqueous conditions. These materials show high compatibility with a range of anhydrous solvents including both hydrophilic and hydrophobic ionic liquids. This enables a form of biocatalysis engineering whereby solvents can be tailored to target applications. In the case of glucosidase, stability in ionic liquids allowed for significantly enhanced activity and solvent-induced promiscuity of the enzyme towards cellulose, a feat not possible under aqueous conditions. Additionally, taking advantage of enhanced stability can allow for the development of thermally robust soft materials. Solvent-free liquid proteins therefore provide a platform to realise the full potential of biocatalysis in industrial settings, to develop new strategies to circumvent the cold chain for therapeutic distribution, and to fully design the next generation of functional biocompatible soft materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. P. S. B. would like to thank Dr Colleen Loynachan for discussion, critical feedback, and constant support.Notes and references
- A. Zaks and A. M. Klibanov, Science, 1984, 224, 1249–1251 CrossRef CAS PubMed.
- K. Griebenow and A. M. Klibanov, J. Am. Chem. Soc., 1996, 118, 11695–11700 CrossRef CAS.
- A. M. Klibanov, Nature, 2001, 409, 241–246 CrossRef CAS PubMed.
- D. B. Volkin, A. Staubli, R. Langer and A. M. Klibanov, Biotechnol. Bioeng., 1991, 37, 843–853 CrossRef CAS PubMed.
- P. P. Wangikar, P. C. Michels, D. S. Clark and J. S. Dordick, J. Am. Chem. Soc., 1997, 119, 70–76 CrossRef CAS.
- D. Bilanicová, A. Salis, B. W. Ninham and M. Monduzzi, J. Phys. Chem. B, 2008, 112, 12066–12072 CrossRef PubMed.
- D. S. Clark, Philos. Trans. R. Soc., B, 2004, 359, 1299–1307 CrossRef CAS PubMed.
- A. P. Borole and B. H. Davison, Appl. Biochem. Biotechnol., 2008, 146, 215–222 CrossRef CAS PubMed.
- C. N. Pace, S. Treviño, E. Prabhakaran and J. M. Scholtz, Philos. Trans. R. Soc., B, 2004, 359, 1225–1234 CrossRef CAS PubMed.
- C. Mattos and D. Ringe, Curr. Opin. Struct. Biol., 2001, 11, 761–764 CrossRef CAS PubMed.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- V. Stepankova, S. Bidmanova, T. Koudelakova, Z. Prokop, R. Chaloupkova and J. Damborsky, ACS Catal., 2013, 3, 2823–2836 CrossRef CAS.
- F. van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef CAS PubMed.
- T. Itoh, Chem. Rev., 2017, 117, 10567–10607 CrossRef CAS PubMed.
- A. S. Bommarius and M. F. Paye, Chem. Soc. Rev., 2013, 42, 6534–6565 RSC.
- L. Lancaster, W. Abdallah, S. Banta and I. Wheeldon, Chem. Soc. Rev., 2018, 47, 5177–5186 RSC.
- M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- K. Steiner and H. Schwab, Comput. Struct. Biotechnol. J., 2012, 2, e201209010 CrossRef PubMed.
- F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS PubMed.
- K. Chen and F. H. Arnold, Nat. Catal., 2020, 3, 203–213 CrossRef CAS.
- V. Erdmann, U. Mackfeld, D. Rother and A. Jakoblinnert, J. Biotechnol., 2014, 191, 106–112 CrossRef CAS PubMed.
- A. Jakoblinnert and D. Rother, Green Chem., 2014, 16, 3472–3482 RSC.
- J. Wachtmeister, A. Jakoblinnert, J. Kulig, H. Offermann and D. Rother, ChemCatChem, 2014, 6, 1051–1058 CrossRef CAS.
- J. Wachtmeister, P. Mennicken, A. Hunold and D. Rother, ChemCatChem, 2016, 8, 607–614 CrossRef CAS.
- C. Bernal, K. Rodríguez and R. Martínez, Biotechnol. Adv., 2018, 36, 1470–1480 CrossRef CAS PubMed.
- R. A. Sheldon and P. C. Pereira, Chem. Soc. Rev., 2017, 46, 2678–2691 RSC.
- X. Li, X. Cao, J. Xiong and J. Ge, Small, 2020, 16, 1902751 CrossRef CAS PubMed.
- G. Tavakoli, J. E. Armstrong, J. M. Naapuri, J. Deska and M. H. G. Prechtl, Chem. – Eur. J., 2019, 25, 6474–6481 CrossRef CAS PubMed.
- L. E. Heim, D. Thiel, C. Gedig, J. Deska and M. H. G. Prechtl, Angew. Chem., Int. Ed., 2015, 54, 10308–10312 CrossRef CAS PubMed.
- J. E. Dander, M. Giroud, S. Racine, E. R. Darzi, O. Alvizo, D. Entwistle and N. K. Garg, Commun. Chem., 2019, 2, 82 CrossRef PubMed.
- M. A. Emmanuel, N. R. Greenberg, D. G. Oblinsky and T. K. Hyster, Nature, 2016, 540, 414–417 CrossRef CAS PubMed.
- Y. Chen, P. Li, J. Zhou, C. T. Buru, L. Đorđević, P. Li, X. Zhang, M. M. Cetin, J. F. Stoddart, S. I. Stupp, M. R. Wasielewski and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 1768–1773 CrossRef CAS PubMed.
- Z. C. Litman, Y. Wang, H. Zhao and J. F. Hartwig, Nature, 2018, 560, 355–359 CrossRef CAS PubMed.
- C. Ji, J. Hou, K. Wang, Y. H. Ng and V. Chen, Angew. Chem., Int. Ed., 2017, 56, 9762–9766 CrossRef CAS PubMed.
- M. Yuan, M. J. Kummer, R. D. Milton, T. Quah and S. D. Minteer, ACS Catal., 2019, 9, 5486–5495 CrossRef CAS.
- K. Nakashima, T. Maruyama, N. Kamiya and M. Goto, Chem. Commun., 2005, 4297–4299 RSC.
- P. Thordarson, B. Le Droumaguet and K. Velonia, Appl. Microbiol. Biotechnol., 2006, 73, 243–254 CrossRef CAS PubMed.
- J. Ge, M. Yan, D. Lu, M. Zhang and Z. Liu, Biochem. Eng. J., 2007, 36, 93–99 CrossRef CAS.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45–53 RSC.
- L. A. Canalle, D. W. P. M. Löwik and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 329–353 RSC.
- J. H. Ko and H. D. Maynard, Chem. Soc. Rev., 2018, 47, 8998–9014 RSC.
- W. Gao, W. Liu, J. A. Mackay, M. R. Zalutsky, E. J. Toone and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15231–15236 CrossRef CAS PubMed.
- R. Falatach, C. McGlone, M. S. Al-Abdul-Wahid, S. Averick, R. C. Page, J. A. Berberich and D. Konkolewicz, Chem. Commun., 2015, 51, 5343–5346 RSC.
- M. Lucius, R. Falatach, C. McGlone, K. Makaroff, A. Danielson, C. Williams, J. C. Nix, D. Konkolewicz, R. C. Page and J. A. Berberich, Biomacromolecules, 2016, 17, 1123–1134 CrossRef CAS PubMed.
- B. Li, Z. Yuan, P. Zhang, A. Sinclair, P. Jain, K. Wu, C. Tsao, J. Xie, H. C. Hung, X. Lin, T. Bai and S. Jiang, Adv. Mater., 2018, 30, 1705728 CrossRef PubMed.
- B. Panganiban, B. Qiao, T. Jiang, C. Delre, M. M. Obadia, T. D. Nguyen, A. A. A. Smith, A. Hall, I. Sit, M. G. Crosby, P. B. Dennis, E. Drockenmuller, M. Olvera, D. Cruz and T. Xu, Science, 2018, 1243, 1239–1243 CrossRef PubMed.
- A. B. Bourlinos, R. Herrera, N. Chalkias, D. D. Jiang, Q. Zhang, L. A. Archer and E. P. Giannelis, Adv. Mater., 2005, 17, 234–236 CrossRef CAS.
- S. C. Warren, M. J. Banholzer and L. S. Slaughter, J. Am. Chem. Soc., 2006, 128, 12074–12075 CrossRef CAS.
- R. Rodriguez, R. Herrera, L. A. Archer and E. P. Giannelis, Adv. Mater., 2008, 20, 4353–4358 CrossRef CAS.
- A. W. Perriman and S. Mann, ACS Nano, 2011, 5, 6085–6091 CrossRef CAS PubMed.
- A. W. Perriman, A. P. S. Brogan, H. Cölfen, N. Tsoureas, G. R. Owen and S. Mann, Nat. Chem., 2010, 2, 622–626 CrossRef CAS PubMed.
- A. P. S. Brogan, G. Siligardi, R. Hussain, A. W. Perriman and S. Mann, Chem. Sci., 2012, 3, 1839–1846 RSC.
- A. P. S. Brogan, K. P. Sharma, A. W. Perriman and S. Mann, Nat. Commun., 2014, 5, 5058 CrossRef CAS PubMed.
- A. P. S. Brogan, K. P. Sharma, A. W. Perriman and S. Mann, J. Phys. Chem. B, 2013, 117, 8400–8407 CrossRef CAS PubMed.
- F.-X. Gallat, A. P. S. Brogan, Y. Fichou, N. McGrath, M. Moulin, M. Härtlein, J. Combet, J. Wuttke, S. Mann, G. Zaccai, C. J. Jackson, A. W. Perriman and M. Weik, J. Am. Chem. Soc., 2012, 132, 13168–13171 CrossRef PubMed.
- A. P. S. Brogan, R. B. Sessions, A. W. Perriman and S. Mann, J. Am. Chem. Soc., 2014, 136, 16824–16831 CrossRef CAS PubMed.
- K. P. Sharma, K. Bradley, A. P. S. Brogan, S. Mann, A. W. Perriman and D. J. Fermin, J. Am. Chem. Soc., 2013, 135, 18311–18314 CrossRef CAS PubMed.
- K. P. Sharma, Y. Zhang, M. R. Thomas, A. P. S. Brogan, A. W. Perriman and S. Mann, J. Phys. Chem. B, 2014, 118, 11573–11580 CrossRef CAS PubMed.
- Y. Zhang, A. J. Patil, A. W. Perriman and S. Mann, Chem. Commun., 2013, 49, 9561–9563 RSC.
- A. J. Patil, N. McGrath, J. E. Barclay, D. J. Evans, H. Cölfen, I. Manners, A. W. Perriman and S. Mann, Adv. Mater., 2012, 24, 4557–4563 CrossRef CAS PubMed.
- A. P. S. Brogan, N. Heldman, J. P. Hallett and A. M. Belcher, Chem. Commun., 2019, 55, 10752–10755 RSC.
- P.-R. Kommoju, Z.-W. Chen, R. C. Bruckner, F. S. Mathews and M. S. Jorns, Biochemistry, 2011, 50, 5521–5534 CrossRef CAS PubMed.
- Y. Ha, D. Shi, G. W. Small, E. C. Theil and N. M. Allewell, J. Biol. Inorg. Chem., 1999, 4, 243–256 CrossRef CAS PubMed.
- T. Lin, Z. Chen, R. Usha, C. V. Stauffacher, J.-B. Dai, T. Schmidt and J. E. Johnson, Virology, 1999, 265, 20–34 CrossRef CAS PubMed.
- B. Pérez, A. Coletta, J. N. Pedersen, S. V. Petersen, X. Periole, J. S. Pedersen, R. B. Sessions, Z. Guo, A. Perriman and B. Schiøtt, Sci. Rep., 2018, 8, 1–13 Search PubMed.
- W. H. Zhang, B. M. Carter, G. J. Day, N. Govan, C. Jackson and A. W. Perriman, Bioconjugate Chem., 2019, 30, 2771–2776 CrossRef CAS PubMed.
- A. Mukhopadhyay, T. Das, A. Datta and K. P. Sharma, Biomacromolecules, 2018, 19, 943–950 CrossRef CAS PubMed.
- D. L. Atkins, J. A. Berrocal, A. F. Mason and I. K. Voets, Nanoscale, 2019, 11, 19797–19805 RSC.
- A. P. S. Brogan and J. P. Hallett, J. Am. Chem. Soc., 2016, 138, 4494–4501 CrossRef CAS PubMed.
- W. Xiao, T. I. P. Green, X. Liang, R. C. Delint, G. Perry, M. S. Roberts, K. Le Vay, C. R. Back, R. Ascione, H. Wang, P. R. Race and A. W. Perriman, Chem. Sci., 2019, 10, 7610–7618 RSC.
- J. P. K. Armstrong, R. Shakur, J. P. Horne, S. C. Dickinson, C. T. Armstrong, K. Lau, J. Kadiwala, R. Lowe, A. Seddon, S. Mann, J. L. R. Anderson, A. W. Perriman and A. P. Hollander, Nat. Commun., 2015, 6, 7405 CrossRef CAS PubMed.
- R. C. Deller, T. Richardson, R. Richardson, L. Bevan, I. Zampetakis, F. Scarpa and A. W. Perriman, Nat. Commun., 2019, 10, 1887 CrossRef PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- J. Gorke, F. Srienc and R. Kazlauskas, Biotechnol. Bioprocess Eng., 2010, 15, 40–53 CrossRef CAS.
- A. P. S. Brogan, L. Bui-Le and J. P. Hallett, Nat. Chem., 2018, 10, 859–865 CrossRef CAS PubMed.
- A. Mukhopadhayay, D. Singh and K. P. Sharma, Biomacromolecules, 2020, 21, 867–877 CrossRef CAS PubMed.
- A. W. Perriman, H. Coelfen, R. W. Hughes, C. L. Barrie and S. Mann, Angew. Chem., Int. Ed., 2009, 48, 6242–6246 CrossRef CAS PubMed.
- K. P. Sharma, A. M. Collins, A. W. Perriman and S. Mann, Adv. Mater., 2013, 25, 2005–2010 CrossRef CAS PubMed.
- T. Farrugia, A. W. Perriman, K. P. Sharma and S. Mann, Chem. Commun., 2017, 53, 2094–2097 RSC.
- L. Bui-Le, A. P. S. Brogan and J. P. Hallett, Biotechnol. Bioeng., 2021, 118, 592–600 CrossRef CAS PubMed.
- Y. Zhou, N. C. Jones, J. Nedergaard Pedersen, B. Pérez, S. Vrønning Hoffmann, S. Vang Petersen, J. Skov Pedersen, A. Perriman, P. Kristensen, R. Gao and Z. Guo, ChemBioChem, 2019, 20, 1266–1272 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |