
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNonlinear optical properties of diaromatic stilbene, butadiene and thiophene derivatives†
Esa
Kukkonen
 a,
Elmeri
Lahtinen
a,
Pasi
Myllyperkiö
b,
Matti
Haukka
a and
Jari
Konu
*a
a,
Elmeri
Lahtinen
a,
Pasi
Myllyperkiö
b,
Matti
Haukka
a and
Jari
Konu
*a
aDepartment of Chemistry, University of Jyväskylä, P. O. Box 35, FI-40014 Jyväskylä, Finland. E-mail: jari.a.konu@jyu.fi; Tel: +358-40-805-4406
bNanoscience Center, University of Jyväskylä, P. O. Box 35, FI-40014 Jyväskylä, Finland
First published on 19th March 2021
Abstract
Series of highly polar stilbene (1a–e), diphenylbutadiene (2a–c) and phenylethenylthiophene (3a–c) derivatives were prepared via Horner–Wadsworth–Emmons method with a view to produce new and efficient materials for second harmonic generation (SHG) in the solid-state. The single-crystal X-ray structures of compounds 1–3 reveal extensive polymorphism and a peculiar photodimerization of the 2-chloro-3,4-dimethoxy-4′-nitrostilbene derivative 1a to afford two polymorphs of tetra-aryl cyclobutane 4. The stilbene congeners 2-chloro-3,4-dimethoxy-4′-nitrostilbene (1a·non-centro), 5-bromo-2-hydroxy-3-nitro-4′-nitrostilbene (1b) and 4-dimethylamino-4′-nitrostilbene (1e), as well as 4′-fluoro-4′′-nitro-1,4-diphenyl-1,3-butadiene (2a) present the ideal, non-centrosymmetric arrangement of the chromophores for nonlinear optical (NLO) activity. Compounds 1b and 2a exhibit only relatively low intensity for second harmonic generation (0.04 and 0.18 times that of urea reference, respectively), while the stilbene polymorph 1a·non-centro shows NLO activity of over 32 times that of urea. In addition, the conjugated diaromatic compounds 1–3 display fluorescence behaviour in CH2Cl2 solutions with the exception of stilbene derivative 1b.
Introduction
The study of nonlinear optical (NLO) materials has gained increasing interest especially owing to their significance in laser technology and optoelectronics. Ultrafast electro-optical switches, optical signal processing and computing, data storage, and photonic technologies are just some of the various applications where these materials have become indispensable.1–8 One of the most sought-after nonlinear properties for these applications is the second harmonic generation (SHG), a phenomenon in which the material combines two photons into one, creating radiation with twice the frequency of the original. The fundamental requirement for a material to have SHG activity is to possess non-zero second-order nonlinear susceptibility χ(2), which necessitates the compound to have non-zero first-order hyperpolarizability β at the molecular level.9 It is highly beneficial if the NLO active material crystallizes in a non-centrosymmetric space group (lack of inversion symmetry) to avoid the effective cancelation of hyperpolarizability, even though centrosymmetric crystals have also been reported to display SHG through processes involving intermolecular interactions.10,11Although most of the commercially available NLO crystals are based on inorganic compounds such as β-barium borate (BBO) and potassium titanyl phosphate (KTP), primarily owing to their high chemical and physical stability,12–14 the academic research on new NLO active materials has largely focused on organic “push–pull” molecules for which high SHG activities have been observed frequently.15–18 A group of compounds consistently showing remarkable SHG efficiencies are diarylethenes, commonly known as stilbenes (compound 1, Scheme 1). They have been studied extensively by both theoretical and experimental methods,19,20 and, for example, 3-methyl-4-methoxy-4′-nitrostilbene (MMONS, Scheme 1), the most efficient second order NLO material reported to date with the SHG efficiency of up to 1250 times of urea,21 belongs to this group of conjugated systems. Additionally, a variety of heteroaromatic, diarylethene-based systems where one or both aryl moieties have been replaced with thiophene, thiazole, or pyrrole rings have also been investigated.22–25 The primary aim of these studies was to increase the hyperpolarizability (β) of the chromophores by altering the charge-transfer properties. While the electric field induced second harmonic generation (EFISHG) measurements of especially thiophene containing diarylethenes in solution have shown to improve the NLO properties at molecular level,22–25 the information on SHG activity of these systems in the solid-state, requiring the most advantageous non-centrosymmetric molecular packing in the crystal lattice, is much more scarce.
 | ||
| Scheme 1 Synthesis of the stilbene (1), diphenylbutadiene (2), and phenylethenylthiophene (3) derivatives. | ||
We recently reported a novel method for producing NLO active lenses by stereolithographic (SLA) 3D printing technique.26 This new approach avoids the often tedious and time-consuming process of growing large single crystals by utilizing microcrystalline powders of NLO active components similarly to the Kurtz–Perry powder method for determination of relative SHG intensities.27 At the same time, the protective layer of photopolymer resin used in the SLA 3D printing could enable the use of labile organic chromophores as NLO active units.26 In this context and with the above considerations in mind, we have now investigated the conjugated, –NO2 substituted stilbene-based systems with the goal of producing new NLO active materials in the solid-state by the means of (1) changing the functional groups at the electron-donating end of stilbene, (2) extending the chain length between the aromatic groups to afford diarylbutadiene derivatives, and (3) changing one of the phenyl groups to thiophene. Consequently, we report here the synthesis, spectroscopic (1H NMR, IR, UV-Visible and fluorescence) and structural characterization (single-crystal XRD), and the SHG properties (Kurtz–Perry powder method)27 of stilbene (1a–e), diphenylbutadiene (2a–c), and phenylethenylthiophene (3a–c) derivatives (Scheme 1).
Experimental methods
Reagents and general procedures
The reactions were carried out in air. All the starting materials and solvents were purchased from commercial sources. Solvents were dried over 3 Å molecular sieves and the remaining reagents were used without further purification: diethyl(4-nitrobenzyl)phosphonate (TCI Chemicals, >97.0%), 4-methoxy-3-methylbenzaldehyde (Sigma-Aldrich, 99%), 2-chloro-3,4-dimethoxybenzaldehyde (TCI Chemicals, >98.0%), 5-bromo-3-nitrosalicylaldehyde (TCI Chemicals, >97.0%), 4-bromothiophene-2-carboxaldehyde (TCI Chemicals, >98.0%), 5-methylthiophene-2-carboxaldehyde (TCI Chemicals, >97.0%), 4-fluorocinnamaldehyde (TCI Chemicals, >95.0%), sodium hydroxide (VWR Chemicals, 99%), 4-anisaldehyde (Merck, 99%), 3,4-dimethoxybenzaldehyde (Aldrich, 99%), 5-(methylthio)thiophenecarboxaldehyde (TCI Chemicals, >98.0%), 4-methoxycinnamaldehyde (Sigma-Aldrich, ≥98%), 4-dimethylaminocinnamaldehyde (TCI Chemicals, >98.0%), CH2Cl2 (VWR Chemicals, 99%), chloroform (Fisher Chemical, analytical reagent grade), acetone (Mallinckrodt, >99.5%), Ethanol (Altia, >99.5%). Elemental analyses were performed by analytical services at the Department of Chemistry, University of Jyväskylä.Spectroscopic methods
The 1H NMR spectra were obtained in CD2Cl2 at 30 °C on Bruker Avance III 300 spectrometer operating at 300.15 MHz. 1H NMR spectra are referenced to the solvent signal and the chemical shifts are reported relative to (CH3)4Si. The IR spectra were measured with Bruker Alpha FTIR spectrometer. The extinction coefficients (molar absorptivity) were determined with PerkinElmer Lambda 650 spectrophotometer by measuring the absorbances of 1 mM and 10 μM solutions in CH2Cl2 (800 to 250 nm). The fluorescence spectra of the compounds were recorded on a Varian Cary Eclipse spectrophotometer from 0.1–10 μM solutions in CH2Cl2.Nonlinear optical (NLO) measurements
The nonlinear optical properties of compounds 1a–e, 2a–c, and 3a–c were measured with Kurtz–Perry powder method27 by using femtosecond laser radiation at 1030 nm. In addition, the second harmonic generation (SHG) was double-checked by using spectrally resolving detection scheme using Kurtz–Perry sample geometry. For these experiments, both femtosecond and nanosecond laser pulses were used. Femtosecond experiments were performed with the setup consisting an amplified femtosecond laser source at 1030 nm (Pharos, Light Conversion Ltd) running at 600 kHz repetition rate. Generated SHG signal was detected by using a high-resolution spectrometer equipped with 300 mm spectrograph (Acton SpectraPro 300i) with spectroscopy CCD detector (Newton DU971N-BV, Andor). Nano-second laser pulses at 100 Hz repetition rate and at 1030 nm and 1060 nm were taken from the ns-OPO (Ekspla Ltd.). Spectrum of the SHG signal was recorded by using time-gated ICCD detector (ISTAR, Andor) with 150 mm spectrograph. (Acton SpectraPro 150i). The crystalline samples, apart from 1b, were sieved to a particle size of <125 μm and placed into capillary tubes for the determination of relative SHG intensity of the compounds. A capillary tube with urea sieved to the same particle size was used as a reference.Anal. calcd (%): C, 60.10; H, 4.41; N, 4.38 found: C, 59.54; H, 4.19; N, 4.40%. 1H NMR (CD2Cl2, 30 °C): δ 8.21 [m, 2H, –C6H4NO2], δ 7.68 [m, 2H, –C6H4NO2], δ 7.65 [d, 1H, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–, 3J (1H,1H) = 16 Hz], δ 7.49 [m, 1H, –C6H2], δ 7.05 [d, 1H, –CHCH–, 3J (1H,1H) = 17 Hz], δ 6.93 [m, 1H, –C6H2], δ 3.91 [s, 3H, –OCH3], δ 3.85 [s, 3H, –OCH3]. X-Ray quality crystals of the two polymorphs of compound 1a (1a·centro and 1a·non-centro) as well as two polymorphs of the subsequent dimerization products (4·Pcell and 4·Ccell, cf. main text) were obtained by slow evaporation of ethanol/chloroform and ethanol/dichloromethane solutions at 23 °C.
CH–, 3J (1H,1H) = 16 Hz], δ 7.49 [m, 1H, –C6H2], δ 7.05 [d, 1H, –CHCH–, 3J (1H,1H) = 17 Hz], δ 6.93 [m, 1H, –C6H2], δ 3.91 [s, 3H, –OCH3], δ 3.85 [s, 3H, –OCH3]. X-Ray quality crystals of the two polymorphs of compound 1a (1a·centro and 1a·non-centro) as well as two polymorphs of the subsequent dimerization products (4·Pcell and 4·Ccell, cf. main text) were obtained by slow evaporation of ethanol/chloroform and ethanol/dichloromethane solutions at 23 °C.
Anal. calcd (%): C, 46.05; H, 2.48; N, 7.67 found: C, 44.98; H, 2.64; N, 7.86%. 1H NMR (CD3CN, 30 °C): δ 11.10 [s, 1H, –OH], δ 8.24 [m, 3H, –C6H4NO2 (2H) and –C6H2 (1H)], δ 8.04 [m, 1H, –C6H2], δ 7.72 [m, 2H, –C6H4NO2], δ 7.58 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 7.32 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz]. X-Ray quality crystals of compound 1b were obtained by slow evaporation of ethanol/chloroform solution at 23 °C.
Anal. calcd (%): C, 70.57; H, 5.13; N, 5.49 found: C, 69.83; H, 4.82; N, 5.52%. 1H NMR (CD2Cl2, 30 °C): δ 8.19 [m, 2H, –C6H4NO2], δ 7.63 [m, 2H, –C6H4NO2], δ 7.52 [m, 2H, –C6H4OMe], δ 7.27 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 7.05 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 6.94 [m, 2H, –C6H4OMe], δ 3.84 [s, 3H, –OCH3]. X-Ray quality crystals of compound 1c were obtained by slow evaporation of methanol solution at 23 °C.
Anal. calcd (%): C, 67.36; H, 5.30; N, 4.91 found: C, 66.46; H, 5.08; N, 4.81%. 1H NMR (CD2Cl2, 30 °C): δ 8.20 [m, 2H, –C6H4NO2], δ 7.64 [m, 2H, –C6H4NO2], δ 7.26 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 7.12 [m, 2H, –C6H3], δ 7.05 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 6.90 [m, 2H, –C6H3O], δ 3.91 [s, 3H, –OCH3] δ 3.87 [s, 3H, –OCH3]. X-Ray quality crystals of compound 1d were obtained by slow evaporation of methanol solution at 23 °C.
Anal. calcd (%): C, 71.62; H, 6.01; N, 10.44 found: C, 70.25; H, 5.81; N, 10.32%. 1H NMR (CD2Cl2, 30 °C): δ 8.17 [m, 2H, –C6H4NO2], δ 7.59 [m, 2H, –C6H4NO2], δ 7.45 [m, 2H, –C6H4NMe2], δ 7.24 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 6.96 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 6.72 [m, 2H, –C6H4NMe2], δ 3.01 [s, 6H, –N(CH3)2]. X-Ray quality crystals of the compound 1e were obtained by slow evaporation of methanol solution at 23 °C.
Anal. calcd (%): C, 71.36; H, 4.49; N, 5.20 found: C, 70.05; H, 4.33; N, 5.18%. 1H NMR (CD2Cl2, 30 °C): δ 8.18 [m, 2H, –C6H4NO2], δ 7.58 [m, 2H, –C6H4NO2], δ 7.47 [m, 2H, –C6H4OMe], δ 7.18–6.71 [m, 6H, –C6H4OMe & –CHCH–]. X-Ray quality crystals of compound 2a were obtained by slow evaporation of acetone/water solution at 23 °C.
Anal. calcd (%): C, 72.58; H, 5.38; N, 4.98 found: C, 71.36; H, 5.27; N, 5.01%. 1H NMR (CD2Cl2, 30 °C): δ 8.17 [m, 2H, –C6H4NO2], δ 7.57 [m, 2H, –C6H4NO2], δ 7.43 [m, 2H, –C6H4OMe], δ 7.14 [q, 1H, –CHCH–], δ 6.90 [m, 3H, –C6H4OMe & –CHCH–], δ 6.78 [d, 1H, –CHCH–, 3J (1H,1H) = 15 Hz], δ 6.69 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 3.82 [s, 3H, –OCH3]. X-Ray quality crystals of compound 2b were obtained by slow evaporation of dichloromethane solution at 23 °C and the structure was confirmed to be analogous to that reported previously31 by unit cell measurement.
Anal. calcd (%): C, 73.44; H, 6.16; N, 9.52 found: C, 72.83; H, 6.07; N, 9.47%. 1H NMR (CD2Cl2, 30 °C): δ 8.15 [m, 2H, –C6H4NO2], δ 7.54 [m, 2H, –C6H4NO2], δ 7.36 [m, 2H, –C6H4NMe2], δ 7.14 [q, 1H, –CHCH–], δ 6.87–6.68 [m, 4H, –C6H4NMe2 & –CHCH–], δ 6.62 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 2.99 [s, 6H, –N(CH3)2]. X-Ray quality crystals of compound 2c were obtained by slow evaporation of dichloromethane solution at 23 °C and the structure was confirmed to be analogous to that reported previously32 by unit cell measurement.
Anal. calcd (%): C, 46.47; H, 2.60; N, 4.52 found: C, 45.91; H, 2.62; N, 4.53%. 1H NMR (CD2Cl2, 30 °C): δ 8.20 [m, 2H, –C6H4NO2], δ 7.62 [m, 2H, –C6H4NO2], δ 7.34 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 7.23 [m, 1H, –C4H2S], δ 7.12 [m, 1H, –C4H2S], δ 7.01 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz]. X-Ray quality crystals of compound 3a·mono were obtained by slow evaporation of ethanol/chloroform solution at 23 °C, while the crystals of 3a·ortho were obtained by slow evaporation of toluene solution at 23 °C.
Anal. calcd (%): C, 63.65; H, 4.52; N, 5.71 found: C, 62.70; H, 4.40; N, 5.59%. 1H NMR (CD2Cl2, 30 °C): δ 8.18 [m, 2H, –C6H4NO2], δ 7.58 [m, 2H, –C6H4NO2], δ 7.35 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 6.99 [m, 1H, –C4H2S], δ 6.84 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 6.72 [m, 1H, –C4H2S], δ 2.51 [s, 3H, –CH3]. X-Ray quality crystals of compound 3b were obtained by slow evaporation of ethanol/chloroform solution at 23 °C.
Anal. calcd (%): C, 56.29; H, 4.00; N, 5.05 found: C, 55.53; H, 3.97; N, 5.05%. 1H NMR (CD2Cl2, 30 °C): δ 8.19 [m, 2H, –C6H4NO2], δ 7.59 [m, 2H, –C6H4NO2], δ 7.33 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 7.04 [m, 1H, –C4H2S], δ 6.96 [m, 1H, –C4H2S], δ 6.87 [d, 1H, –CHCH–, 3J (1H,1H) = 16 Hz], δ 2.55 [s, 3H, –CH3]. X-Ray quality crystals of compound 3c were obtained by slow evaporation of dichloromethane solution at 23 °C.
Results and discussion
Synthesis and crystal structures of compounds 1–4
Horner–Wadsworth–Emmons synthetic method33 was used to synthesize the compounds 1–3 (Scheme 1). Equimolar amounts of aromatic aldehyde and phosphonate were reacted in basic conditions to yield spectroscopically and analytically pure products in good yields (cf. ESI† for the 1H NMR and IR spectra). The reactions resulted in the precipitation of pure products with the exception of compound 1b which was extracted with a mixture of CH2Cl2 and H2O followed by evaporation of the organic phase. All the compounds 1–3 possess –NO2 group at the electron withdrawing end of the push–pull molecules. Various substituents at the electron donating end were selected with a view to have an effect on the charge-transfer properties and therefore the molecular hyperpolarizability as well as to increase the possibility for the ideal, non-centrosymmetric packing of the chromophores in the crystal lattice (Scheme 1).Single-crystal X-ray structures of stilbene derivatives
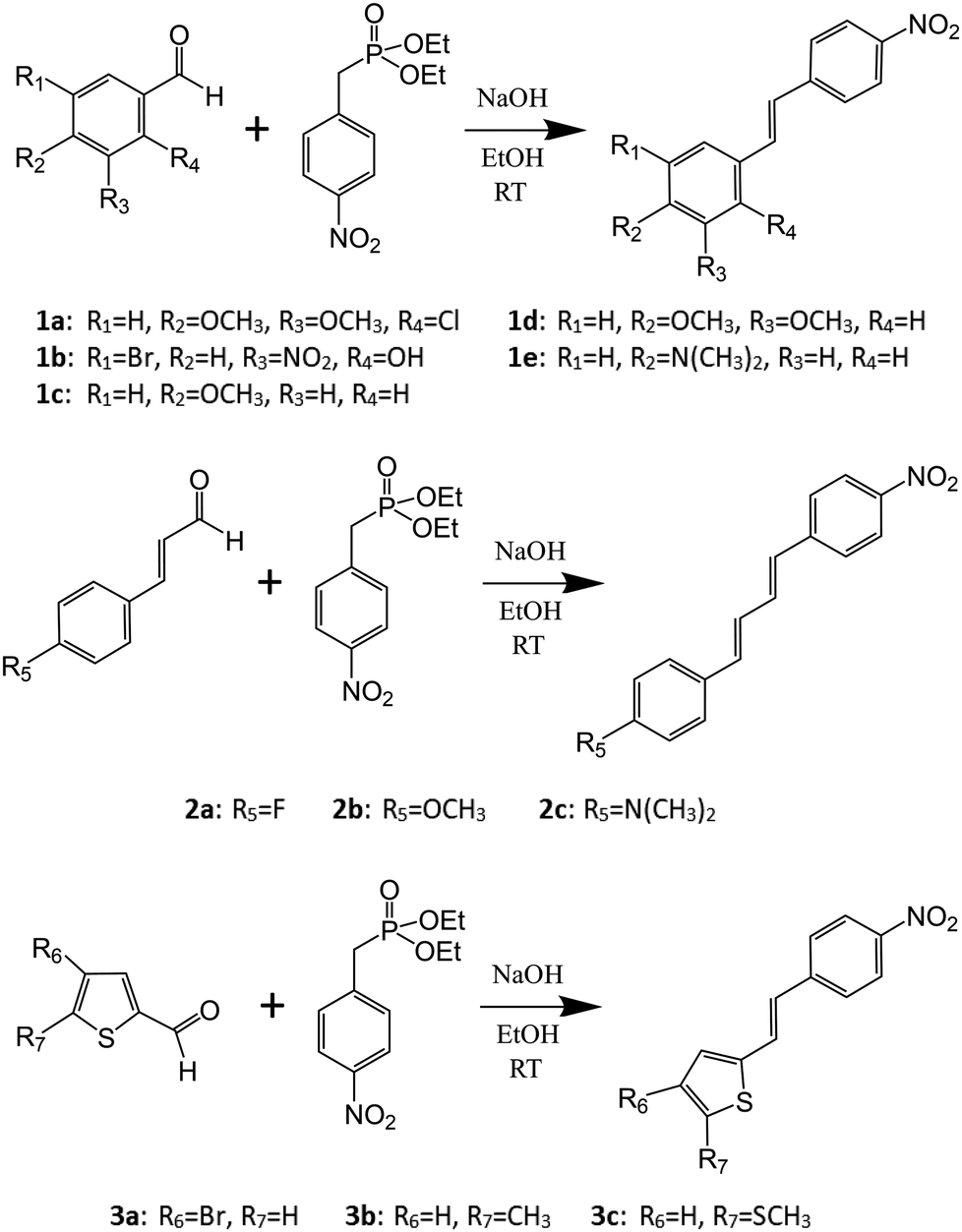
Similarly to many reported stilbene derivatives,20,21,34,35 the 2-chloro-3,4-dimethoxy-4′-nitrostilbene compound 1a shows polymorphism by crystallization in both centrosymmetric (P21/c, 1a·centro) and non-centrosymmetric (P21, 1a·non-centro) space groups (Fig. 1, cf. ESI† for molecular figures of all the structures with atomic numbering schemes and the tables of pertinent bond parameters). Analogously to 3-methyl-4-methoxy-4′-nitrostilbene (MMONS),34 the discrete molecules in the centrosymmetric form 1a·centro are nearly planar with ca. 3.8° angle between the aryl rings. By contrast, more pronounced twisting is notable in the non-centrosymmetric polymorph 1a·non-centro as evidenced by the corresponding angle of ca. 19.2° in the molecule. The primary driving force of the crystal packing in both polymorphs of 1a seems to be π–π interactions, and while in 1a·centro this leads to a herringbone-like linear arrangement of molecular chains (Fig. 1a), in 1a·non-centro half of the molecules are rotated by ca. 90° (Fig. 1b). Consequently, only 1a·non-centro displays intermolecular Cl⋯ONO interactions of 3.27 Å whereas the remaining close contacts (e.g. MeO⋯H, NO2⋯H) are virtually insignificant in both polymorphs.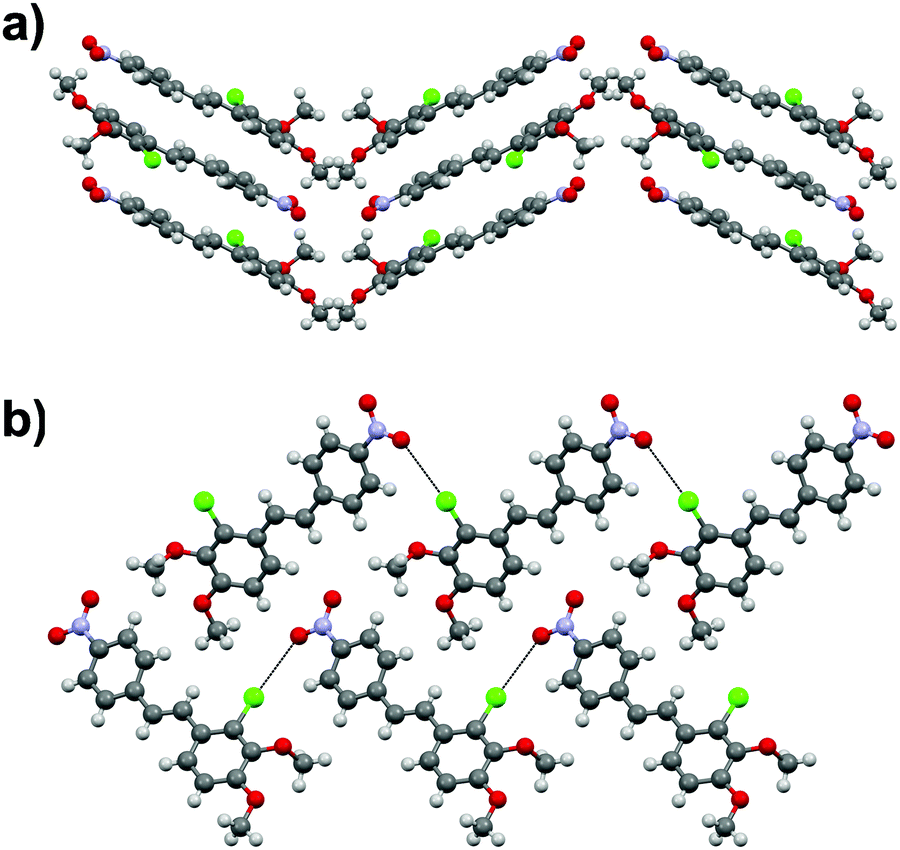 | ||
| Fig. 1 Molecular packing in (a) 1a·centro (along c-axis) and (b) 1a·non-centro (along a-axis). | ||
Stilbene 1a was also noted to go through, presumably, a photodimerization process of the ethenyl linkage to afford tetra-aryl cyclobutane compound 4 (Fig. 2). Analogous behaviour has been previously observed with other stilbenes and diaromatic olefins where a distance of ca. 3.5–4.2 Å between the central CC bonds of adjacent molecules facilitates the solid state photodimerization process.36–38 Suitable alignment of molecules and a distance of ca. 3.7 Å between the double bonds can be found in 1a·centro whereas no such arrangement is observable in 1a·non-centro therefore suggesting that the former polymorph is the primary source for the formation of tetra-aryl cyclobutane dimer 4.
 | ||
| Fig. 2 The difference in phenyl ring and substituent orientations between P-cell (red) and C-cell (blue) tetra-aryl cyclobutanes 4. | ||
The cyclobutane dimer 4 also shows polymorphism with crystallization in two monoclinic space groups, P21/n (4·Pcell) and C2/c (4·Ccell). The difference in crystal packing can also be seen in a slightly different orientation of the phenyl groups around the central cyclobutane unit (Fig. 2). The dimerization results in the expected lengthening of the double bond in the ethenyl part of the original monomers from 1.333(2)–1.333(4) Å in 1a·non-centro and 1a·centro to 1.546(2)–1.558(2) Å in 4·Pcell and 4·Ccell. In addition, the C–C bonds between the two stilbene units in cyclobutane rings in 4·Pcell and 4·Ccell are slightly longer than within each stilbene unit at 1.583(2)–1.600(2) Å vs. 1.546(2)–1.558(2) Å, respectively (cf. ESI†).
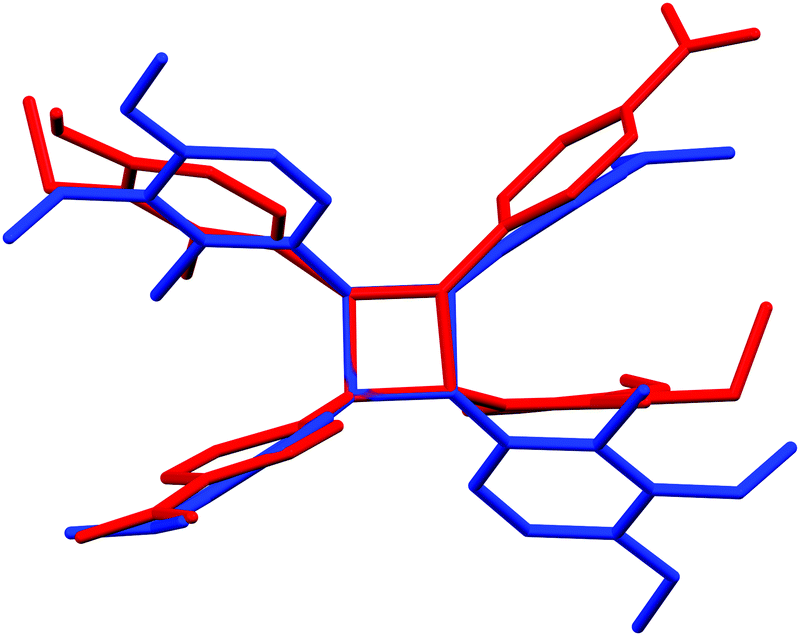
Stilbene derivative 1b with –OH, –Br and two –NO2 substituents crystallizes in a non-centrosymmetric space group Cc. The phenyl rings within each molecule are strongly twisted compared to both polymorphs of 1a with an angle of 29.7° between the aryl groups (Fig. 3a). Similarly to 1a, the crystal packing in 1b can be primarily contributed to π–π interactions even though weak Br⋯ONO halogen contacts of ca. 3.51 and 3.37 Å, analogous to the Cl⋯ONO contacts in 1a·non-centro, also exist. The two orientations of independent molecules in 1b (Fig. 3b) and those observed in 1a·non-centro (Fig. 1b) also bear a close resemblance.
 | ||
| Fig. 3 (a) Crystal packing in compound 1b showing the orientation of the molecules within each layer (along a-axis) and (b) side view of the layer revealing the molecular distortion. | ||
Stilbene 1c with –NO2 and –MeO substituents displays three independent molecules in the crystal lattice arranged in a rather random manner in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Fig. 4a). In contrast to the structures of 1a and 1b, there is an apparent lack of π–π interactions in 1c. Instead, one of the three independent molecules forms centrosymmetric dimers through weak H⋯OMe hydrogen bonds between the MeO group and one of the hydrogens of the phenyl rings (Fig. 4b).
(Fig. 4a). In contrast to the structures of 1a and 1b, there is an apparent lack of π–π interactions in 1c. Instead, one of the three independent molecules forms centrosymmetric dimers through weak H⋯OMe hydrogen bonds between the MeO group and one of the hydrogens of the phenyl rings (Fig. 4b).
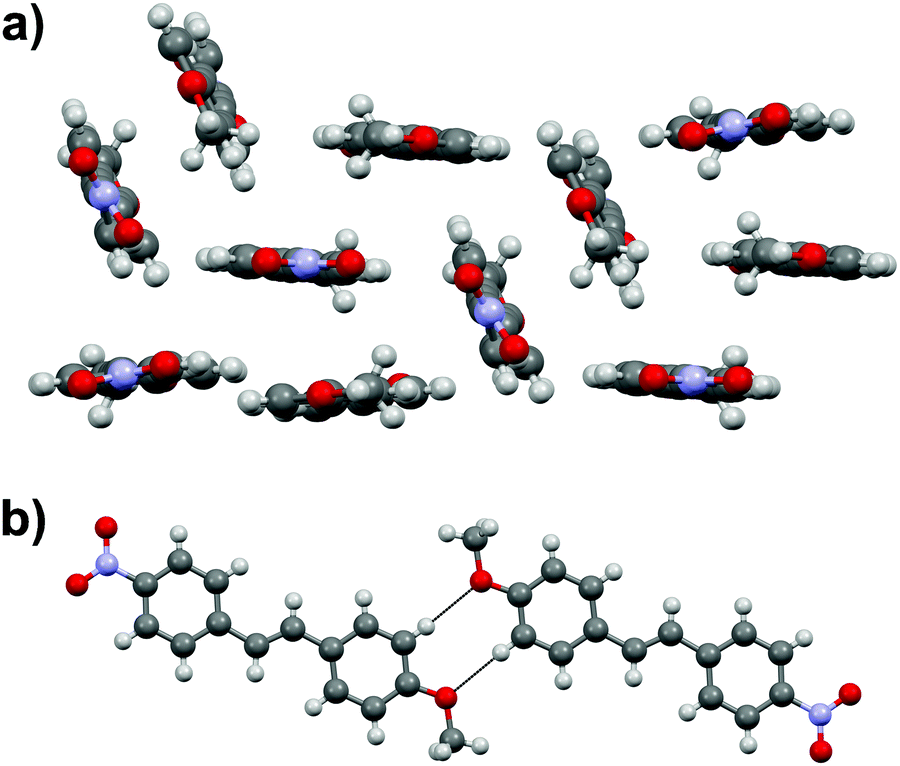 | ||
| Fig. 4 (a) Orientation of stilbene molecules in the crystal lattice, and (b) close contacts in 1c. | ||
Stilbene derivative 1d with one –NO2 and two –MeO groups crystallizes in a centrosymmetric space group P21/c. The aromatic rings within each molecule are heavily twisted with an angle of 43.7° between the rings (Fig. 5a). The molecules in 1d form dimeric units through MeO⋯H3CO interactions (Fig. 5b) while the overall arrangement of molecules in the crystal lattice is rather arbitrary.
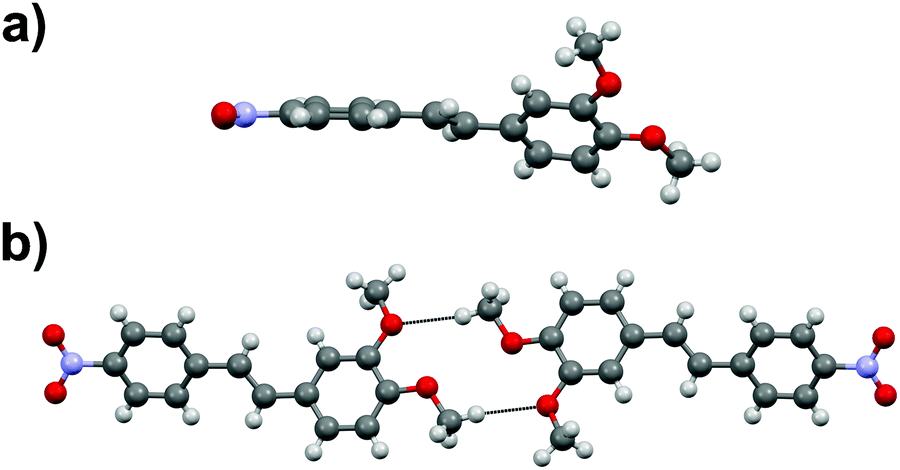 | ||
| Fig. 5 (a) Rotation of the phenyl rings, and (b) the dimeric unit in 1d. | ||
Stilbene congener 1e with –NO2 and –NMe2 substituents crystallizes in a non-centrosymmetric space group P21. The structure shows disorder in which the molecules are organized in two orientations in the crystal lattice in 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 ratio (cf. ESI† for a molecular figure of disorders in 1e). The slightly twisted stilbene molecules form planes along a-axis, and chains through weak NO2⋯(H3C)N hydrogen bonding along c-axis with alternating chain directions in the planes (Fig. 6).
:40 ratio (cf. ESI† for a molecular figure of disorders in 1e). The slightly twisted stilbene molecules form planes along a-axis, and chains through weak NO2⋯(H3C)N hydrogen bonding along c-axis with alternating chain directions in the planes (Fig. 6).
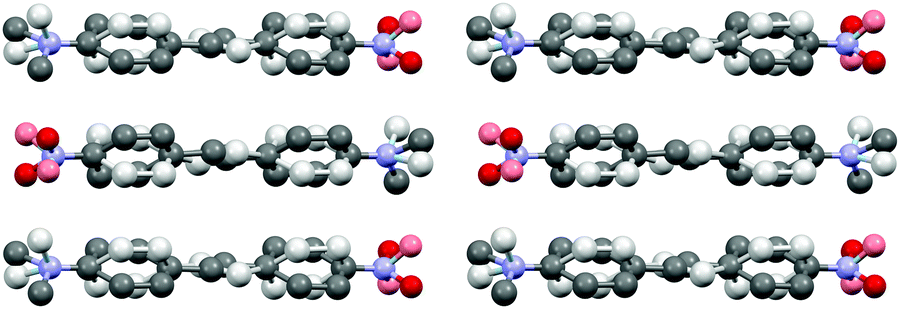 | ||
| Fig. 6 Molecular packing of stilbene derivative 1e viewed along a-axis (H atoms removed for clarity, disorder indicated with lighter colours). | ||
Single-crystal X-ray structures of diphenylbutadiene derivatives
The diphenylbutadiene 2a crystallizes in non-centrosymmetric space group Pc. Despite multiple crystallizations and several data collections, the crystal structure consistently shows orientational disorder where adjacent molecules are placed opposite directions in head-to-tail fashion (cf. ESI† for a molecular figure of disorder in 2a).39 The slightly twisted diphenylbutadienes in 2a form planes along a-axis with alternating orientation of the molecules within each plane, and a herringbone style layering between the planes (Fig. 7). The crystal structures of diphenylbutadienes 2b and 2c have been reported previously (CSD entries YIQJEQ and EDUSAA).31,32 The former crystallizes in centrosymmetric space group P with head-to-tail organization of the molecules. Despite the increase in chain length compared to stilbenes derivatives 1a–e, the conjugated molecules in 2b are notably planar (Fig. 8) forming a sheet-like structure with π–π interactions leading to a relatively short distance of 3.70 Å between centroids of the phenyl rings. The diphenylbutadiene 2c crystallizes in centrosymmetric space group P21/c and, analogously to 2b, with markedly planar geometry within the molecules. In contrast to 2b, however, there is some angling of the molecules between the layers (Fig. 8c), and therefore no π–π interactions can be observed. Despite the extensive crystallization efforts, no polymorphism and discovery of new, non-centrosymmetric crystal structures of 2b and 2c were observed.
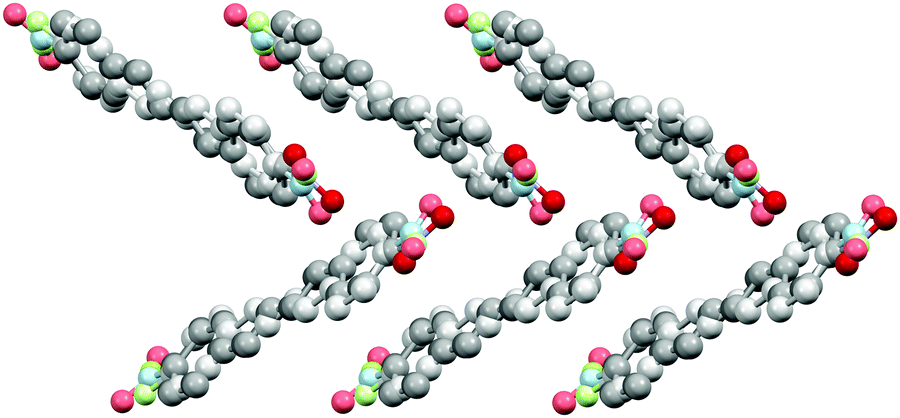 | ||
| Fig. 7 Crystal structure of 2a with herringbone-style layers viewed along a-axis (H atoms removed for clarity, disorder indicated with lighter colours). | ||
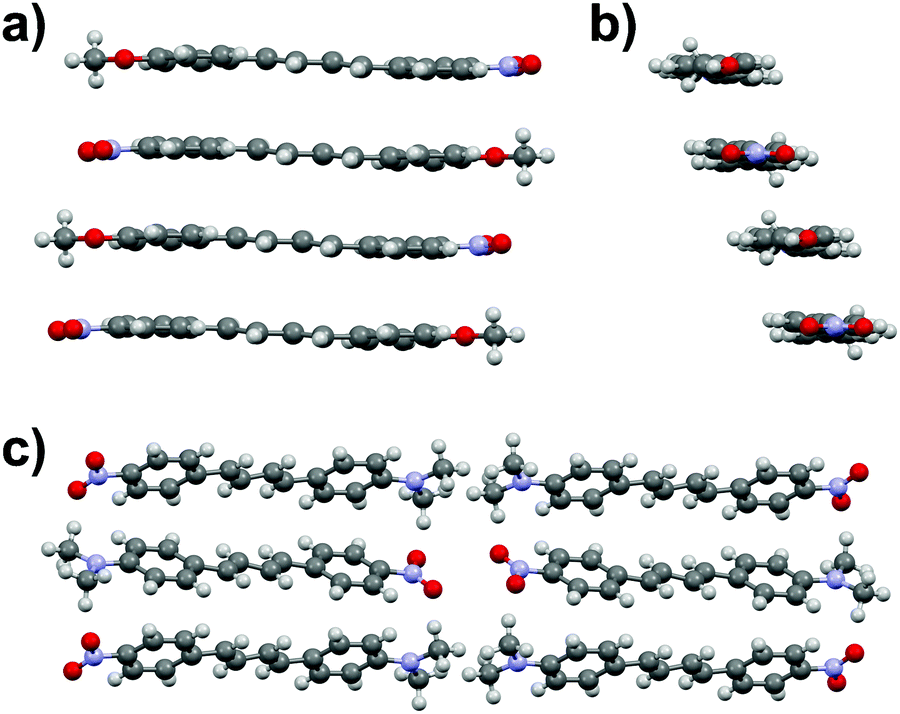 | ||
| Fig. 8 (a) Side view and (b) end view of molecular arrangement in 2b (CSD entry “YIQJEQ”),31 and (c) molecular arrangement in 2c (CSD entry “EDUSAA”).32 Disorder in 2c has been removed for clarity. | ||
Single-crystal X-ray structures of phenylethenylthiophene derivatives
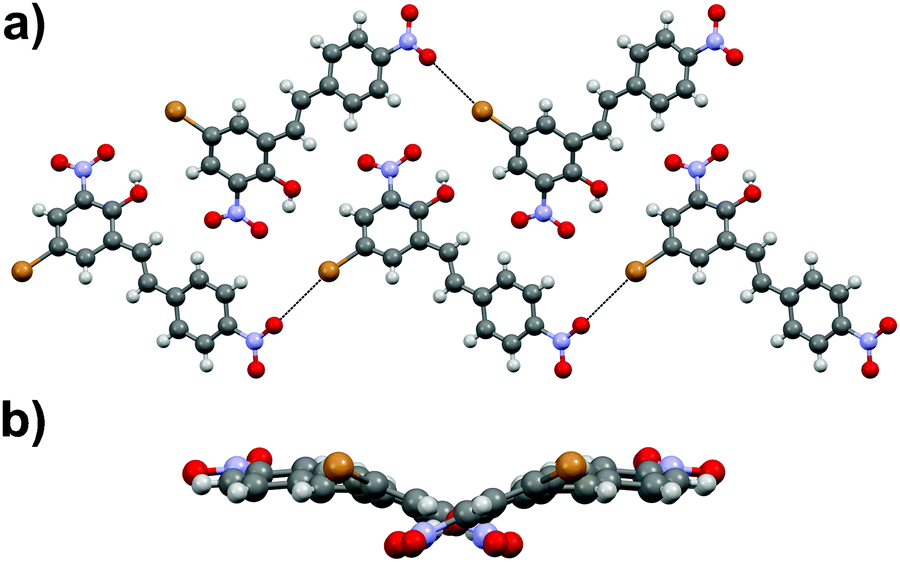
Similarly to stilbene 1a, the phenylethenylthiophene 3a displays polymorphism by crystallizing in two centrosymmetric space groups, monoclinic P21/c (3a·mono) and orthorhombic Pbca (3a·ortho). Most notably, the phenyl and thiophene rings in 3a·mono are significantly twisted (by 12.4° and 17.5° in two independent molecules, respectively) while the analogous twisting in 3a·ortho is less pronounced (4.7°). Both polymorphs exhibit significant halogen bonding through NO2⋯Br contacts of ca. 3.06 and 3.00 Å in 3a·mono and 3a·ortho, respectively. Overall, the molecules form layered structure in 3a·mono (Fig. 9a) whereas in 3a·ortho the distortion between layers is notable (Fig. 9b).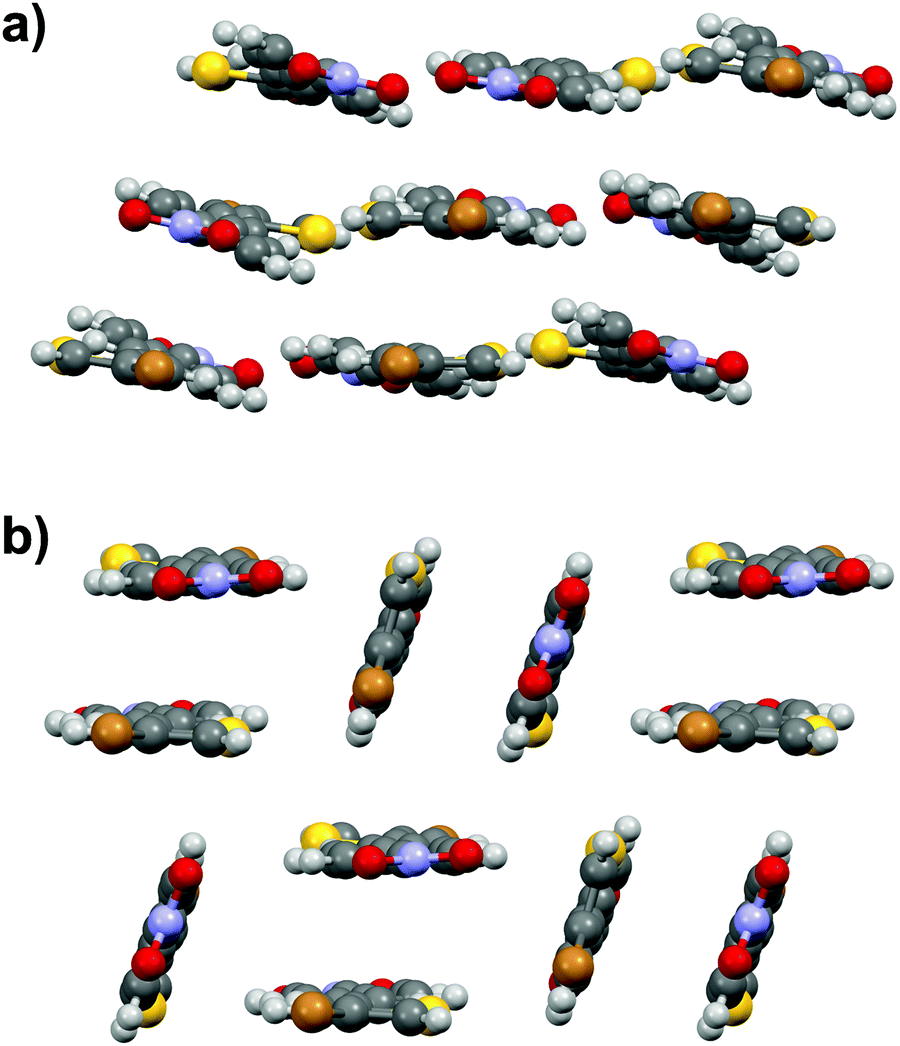 | ||
| Fig. 9 (a) Crystal structure of 3a·mono viewed along b-axis, and (b) side view of 3a·ortho. | ||
Phenylethenylthiophene derivative 3b crystallizes in a centrosymmetric space group P21/n (Fig. 10a). The crystal structure displays two independent molecules in the unit cell with only slight deviation from planarity (ca. 6.8° and 5.7° angles between the aromatic rings, respectively). One of the two molecules in asymmetric unit exhibits significant rotation of the –NO2 substituent with respect to the phenyl ring (3.6° vs. 27.8° in the two distinct molecules). This behaviour is similar to 3a·mono, in which the –NO2 groups show various degrees of rotation (3.2° and 13.7° degrees), whereas in 3a·ortho all the substituents are virtually planar with respect to the phenyl and thiophene rings. The weak NO2⋯H3C interactions result in chain formation with alternating directions and V-shaped geometry along the chains (Fig. 10b).
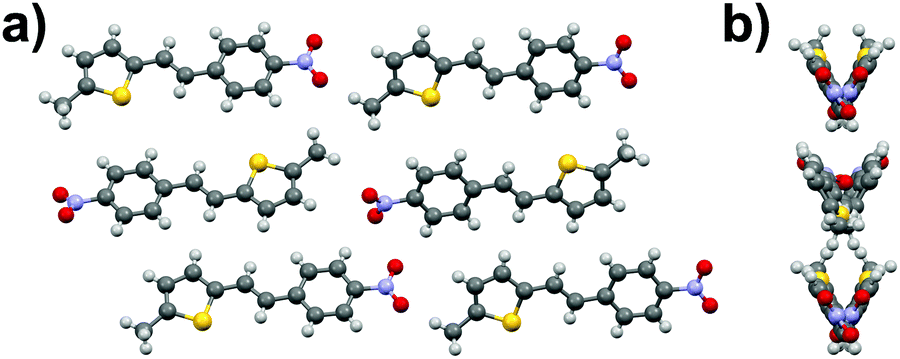 | ||
| Fig. 10 (a) Crystal structure of compound 3b viewed along b-axis, and (b) side view of the layers showing the V-shaped arrangement. | ||
Phenylethenylthiophene 3c crystallizes in centrosymmetric space group P21/n. The molecular chains form herringbone-style planes with altering direction of the chains within each layer (Fig. 11). The only intermolecular interactions are the weak NO2⋯H3CS contacts between the two substituents.
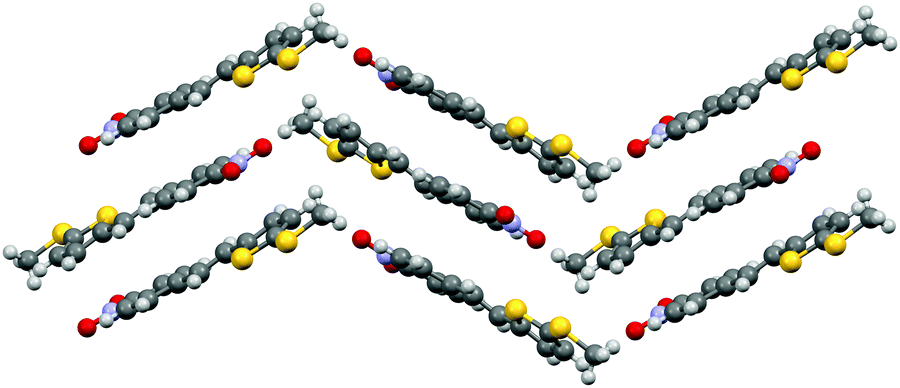 | ||
| Fig. 11 Herringbone style layers of 3c viewed along a-axis. | ||
Nonlinear optical properties of compounds 1a–e, 2a–c and 3a–c
The nonlinear optical properties of compounds 1a–e, 2a–c, and 3a–c were initially measured with Kurtz–Perry powder method27 by using femtosecond laser radiation at 1030 nm. In these experiments the SHG efficiency of MMONS (Scheme 1) displayed intensity of 61 times compared to that of urea (Table 1), far from the reported literatures values of ca. 750–1250 times urea.21 In view of the possible competing processes, such as two-photon fluorescence, the SHG intensities of MMONS, urea and the most promising new stilbene derivative, 1a·non-centro, were also measured by using nanosecond laser radiation at both 1030 and 1060 nm to closely reproduce the literature experiments (Table 1 and Fig. 12).21 While the switch from femtosecond to nanosecond laser source did increase the SHG intensity of MMONS from 61 to ca. 200 times to that of urea, the change of the wavelength from 1030 to 1060 nm did not show significant effect on the SHG intensity consistently with the solid-state absorption maxima of MMONS reported at lower wavelength (346 and 474 nm)21 than either of the SHG signals at 515 and 530 nm. However, the SHG intensity of MMONS still remained notably lower than the literature values (200 vs. 750–1250 times of urea) possibly owing to the fragmentation of fragile MMONS samples into too small microcrystalline material in the sieving process to reduce the SHG intensity,21 or because of partial contamination of the samples with inactive polymorphs of MMONS.34 The latter, however, was rendered unlikely by space group determinations of several individual crystals and by X-ray powder measurements.| Femtosecond laser radiation at 1030 nm (laser power: b1.3 mW, c6.2 mW) | |||||
|---|---|---|---|---|---|
| Samplea | Signal areab (a.u.) | Relative areab (times urea) | Sampleb | Signal areac (a.u.) | Relative areac (times urea) |
| a Crystalline samples sieved to particle size of <125 μm. | |||||
| Urea | 31000 |
1 | Urea | 397800 |
1 |
| MMONS | 1890000 |
61.0 | 1b | 15600 |
0.04 |
| 1a·non-centro | 169700 |
5.5 | 2a | 71300 |
0.18 |
| Nanosecond laser radiation at d1030 and e1060 nm | |||||
|---|---|---|---|---|---|
| Samplea | Signal aread (a.u.) | Relative area (times urea)d | Sampled | Signal areae (a.u.) | Relative areae (times urea) |
| Urea | 4760 | 1 | Urea | 7120 | 1 |
| MMONS | 977100 |
205 | MMONS | 1498300 |
210 |
| 1a·non-centro | 36640 |
7.7 | 1a·non-centro | 230650 |
32.4 |
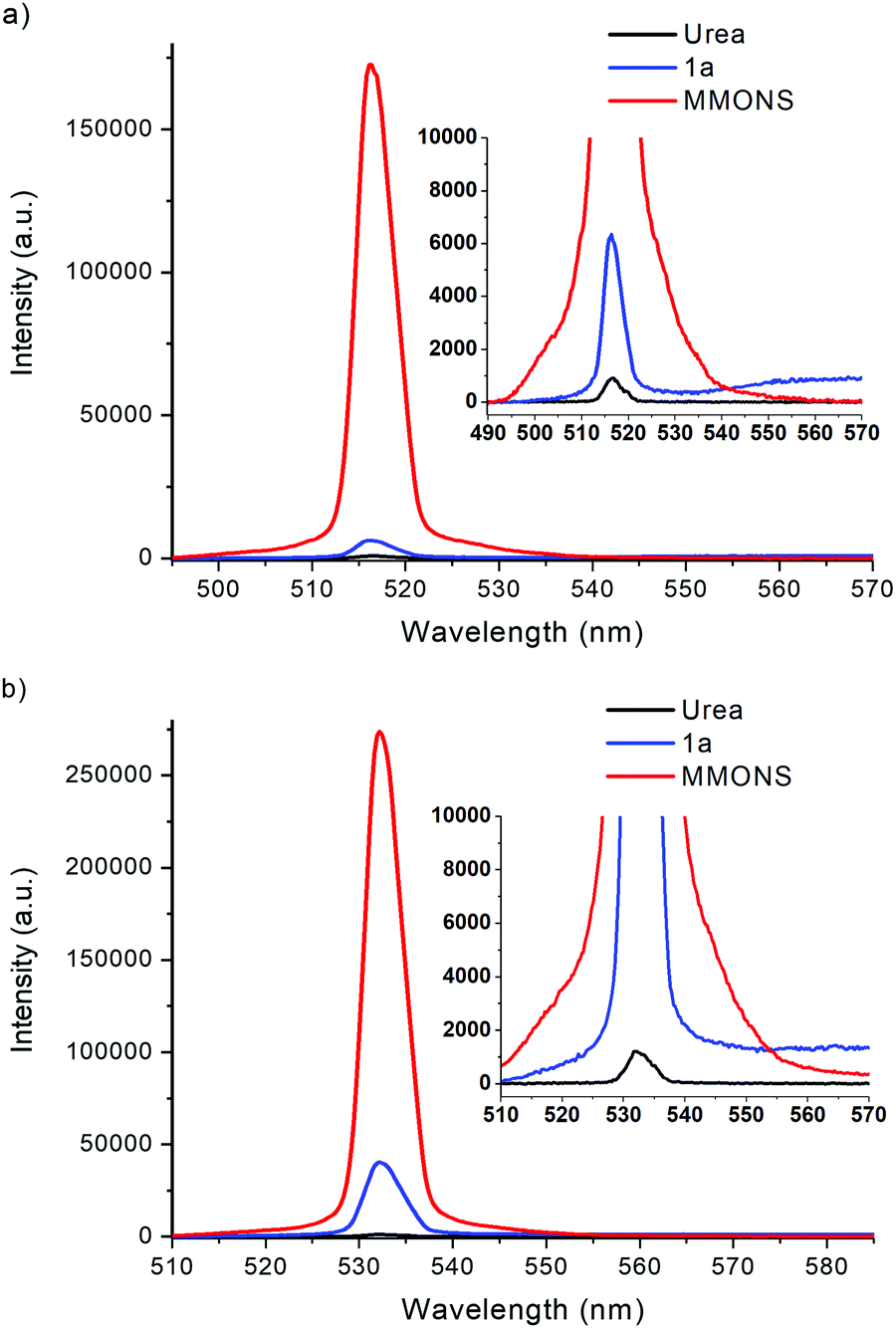 | ||
| Fig. 12 SHG signals of 1a·non-centro, MMONS and urea generated at (a) 1030 nm and (b) 1060 nm with nanosecond laser source. | ||
Hand-picked crystalline sample of the non-centrosymmetric polymorph 1a·non-centro displays SHG intensity of ca. 5.5 times to that of urea in the femtosecond laser experiments at 1030 nm. The SHG intensity of 1a·non-centro increases to 7.7 and, significantly, 32.4 times to that of urea when nanosecond laser source is used at 1030 and 1060 nm wavelength, respectively. In all measurements the microcrystalline sample of 1a·non-centro shows detectable broad fluorescence indicating that some SHG efficiency is lost due to electronic excitation of the material, namely two-photon absorption of the laser radiation or absorption of the generated SH signal. The absorbance at 515 nm is virtually non-existing in CH2Cl2 solution (see below), but in the microcrystalline sample signal faces multiple interactions increasing absorption significantly. This may explain the observed difference in the intensity of SHG signal between two fundamental wavelengths. In addition, fluorescence is not evident to be two-photon induced process as the intensity is not clearly dependent on the duration of laser pulse, suggesting that the generated SHG signal is to some extent absorbed in the sample.
The stilbene derivatives 1b and 1e also display non-centrosymmetric crystal packing in the solid state with the space groups Cc and P21, respectively. However, only negligible SHG activity is observed for compound 1b (0.04 times that of urea), and virtually no SHG activity is detected for 1e with femtosecond laser experiments at 1030 nm. The stilbene derivative 1c has also been reported to give rise to SHG signal, and polymorphism by crystallization in both centrosymmetric and non-centrosymmetric triclinic space groups.20,40,41 However, our investigation revealed only centrosymmetric space group (P) and no SHG activity for 1c.42
While the stilbene derivatives 1a–e expectedly display varying degree of SHG activity in the solid state depending on the efficiency of crystal packing, the alterations in the chain length between aryl groups to give diarylbutadienes 2 and changing one of the aryl groups to thiophene in compounds 3 resulted primarily in centrosymmetric arrangements of the chromophores despite the extensive crystallization efforts to obtain non-centrosymmetric polymorphs. Consequently, among all the derivatives of 2 and 3, only the diarylbutadiene compound 2a shows SHG intensity (0.18 times of urea) consistently with the non-centrosymmetric space group Pc.
It has been suggested that the highest recorded SHG activity of 3-methyl-4-methoxy-4′-nitrostilbene (MMONS) arises partly from the strong π–π interactions that enhance the intermolecular charge-transfer and, consequently, the second order polarizability.21 In accordance, MMONS displays distances as short as ca. 3.56 Å between the phenyl ring centroids despite the two distinct orientations of the molecules. The same distances in stilbene derivatives 1a·non-centro and 1b are ca. 4.22 and 3.82 Å, respectively, even though it is the former chromophore that displays higher SHG intensity. This is likely a result of the better arrangement of polar centres in the crystal lattice of 1a·non-centro and the presence of electron withdrawing –NO2 group also in the “push” part of the chromophore 1b. In contrast to the aforementioned compounds, the non-centrosymmetric stilbene derivative 1e does not show significant π–π interactions, which is possibly contributing to the virtual absence of SHG activity.
Optical spectroscopy of compounds 1–3
The absorption spectra of compounds 1–3 were measured in CH2Cl2 at room temperature (Table 2 and Fig. 13). All compounds display one high and one low energy absorption maximum in solution, albeit the former is observable only as a small shoulder with the solvent signal in the case of stilbene derivatives 1a and 1c. Additionally, stilbene congener 1b, the only compound with –NO2 group at both ends of the molecule, displays a distinct shoulder also at 410 nm. The high energy (low wavelength) absorption maxima show relatively narrow range of 278–303 nm for stilbene derivatives 1, 289–322 nm for diphenylbutadiene compounds 2, and 272–291 nm for phenylethenylthiophene species 3, while the corresponding low energy (high wavelength) bands exhibit significantly broader range of 336–439 nm for 1a–e, 381–452 nm for 2a–c and 358–397 nm for 3a–c. Expectedly, all the extinction coefficient values of ca. 9–37 × 103 M−1 cm−1 are in the range typically observed for π → π* transition. Given the wider wavelength range observed for the low energy absorption maxima, these bands are likely reflecting the modifications made by substituent alterations at the electron donating part of compounds 1–3 whereas the high energy bands in the absorption spectra are contributed to the electron withdrawing, –NO2 substituted end of the conjugated push–pull systems where no alterations were made within each series. Consistently, both diphenylbutadiene (2) and phenylethenylthiophene (3) derivatives exhibit a steady red shift in the low energy absorption maximum following the approximate order of increasing electron donating power of the substituents: F (2a, 381 nm) < OCH3 (2b, 401 nm) < NMe2 (2c, 452 nm), and Br (3a, 358 nm) < CH3 (3b, 390 nm) < SCH3 (3c, 397 nm), respectively. Similarly, stilbene derivative 1e with the strongest electron donating group (NMe2) gives the lowest energy absorption maximum of the series at 439 nm.| Absorption bandsa | |||||
|---|---|---|---|---|---|
| λ Abs (nm) | ε (M−1 cm−1) | λ Abs (nm) | ε (M−1 cm−1) | ||
| a 1 mM and 10 μM sol. in CH2Cl2 at 23 °C. b Absorption maximum. c Extinction coefficient. d 0.1–10 μM solutions in CH2Cl2 at 23 °C. e Excitation maximum. f Emission maximum. g Stokes shift in wavenumbers calculated by the equation (107/λAbs) − (107/λEm). | |||||
| 1a | 365 | 17200 |
2a | 381 | 23350 |
| 289 | 10700 |
||||
| 1b | 336 | 23800 |
2b | 401 | 36450 |
| 278 | 13700 |
301 | 16350 |
||
| 1c | 376 | 23100 |
2c | 452 | 35350 |
| 322 | 18450 |
||||
| 1d | 385 | 24350 |
3a | 358 | 10600 |
| 280 | 13000 |
272 | 13750 |
||
| 1e | 439 | 23850 |
3b | 390 | 16300 |
| 303 | 12750 |
278 | 10800 |
||
| 3c | 397 | 27350 |
|||
| 291 | 8850 | ||||
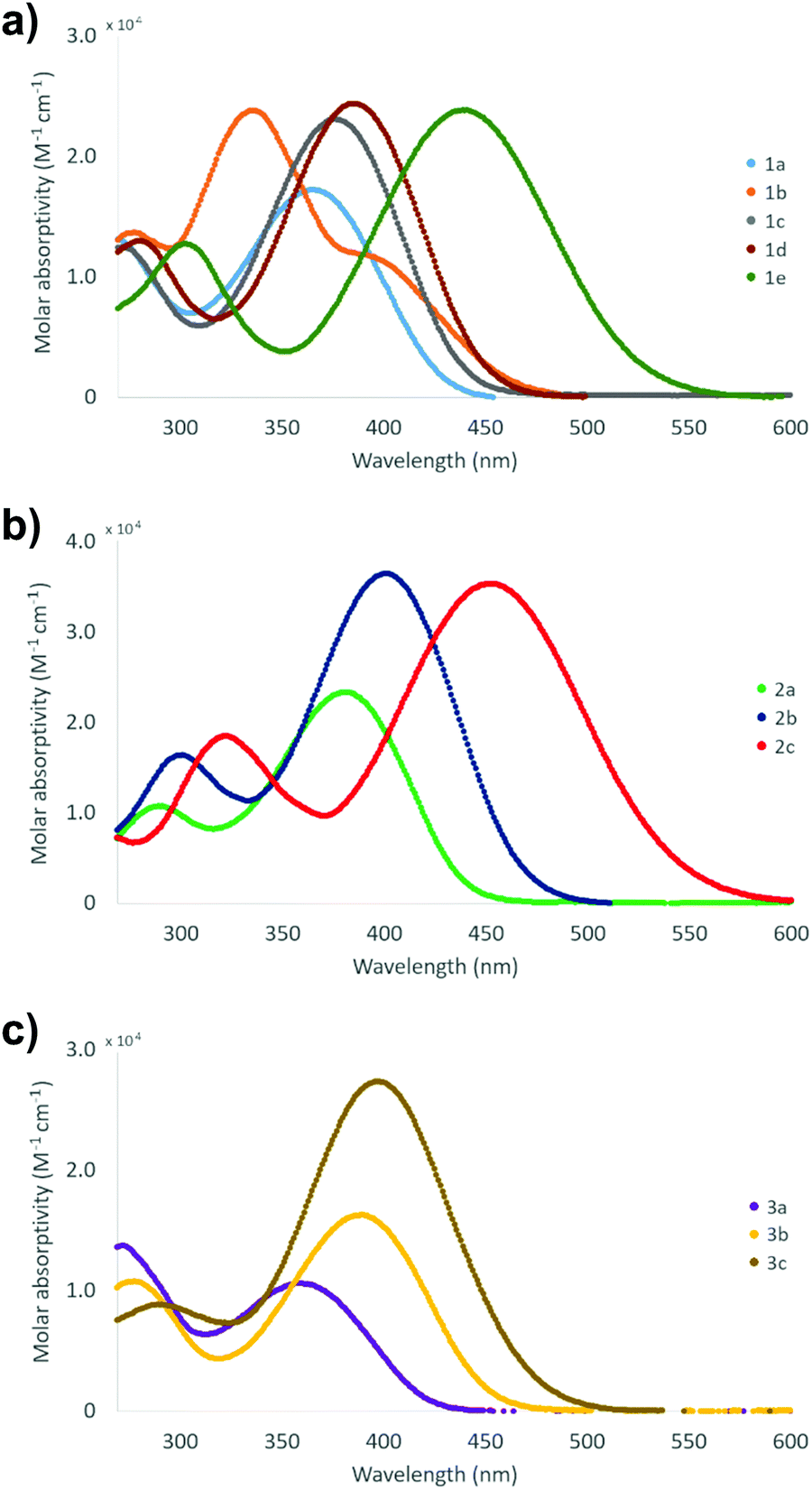 | ||
| Fig. 13 Visible spectra of compounds (a) 1a–e, (b) 2a–c and (c) 3a–c (1 mM and 10 μM sol. in CH2Cl2 at 23 °C). | ||
The fluorescent nature of stilbene and its derivatives is well established and various aspects of their optical, photophysical and photochemical properties have been exhaustively studied.43–47 Consistently with the high optical activity, the stilbene (1), diphenylbutadiene (2) and phenylethenylthiophene (3) derivatives in this investigation all show fluorescent properties with the exception of compound 1b (Table 2 and Fig. 14). In contrast to the two absorption bands, only single maximum with poorly resolved shoulders is observed in the excitation spectra of all the compounds 1–3 in CH2Cl2 at room temperature. However, the wavelength of the excitation maximum of each derivative displays close correlation with the corresponding low energy absorption band. Similarly to the absorption spectra, both the excitation and emission spectra of compounds 1–3 exhibit more pronounced red shift together with the increasing electron donating power of the substituent(s) within each series. Accordingly, the –NMe2 containing derivatives 1e and 2c give rise to the highest wavelength excitation and emission maxima. The large Stokes red shift values expectedly indicate high dipole moment in the excited state for all the compounds 1–3 in CH2Cl2 as has been previously demonstrated, for example, for compound 1e.43
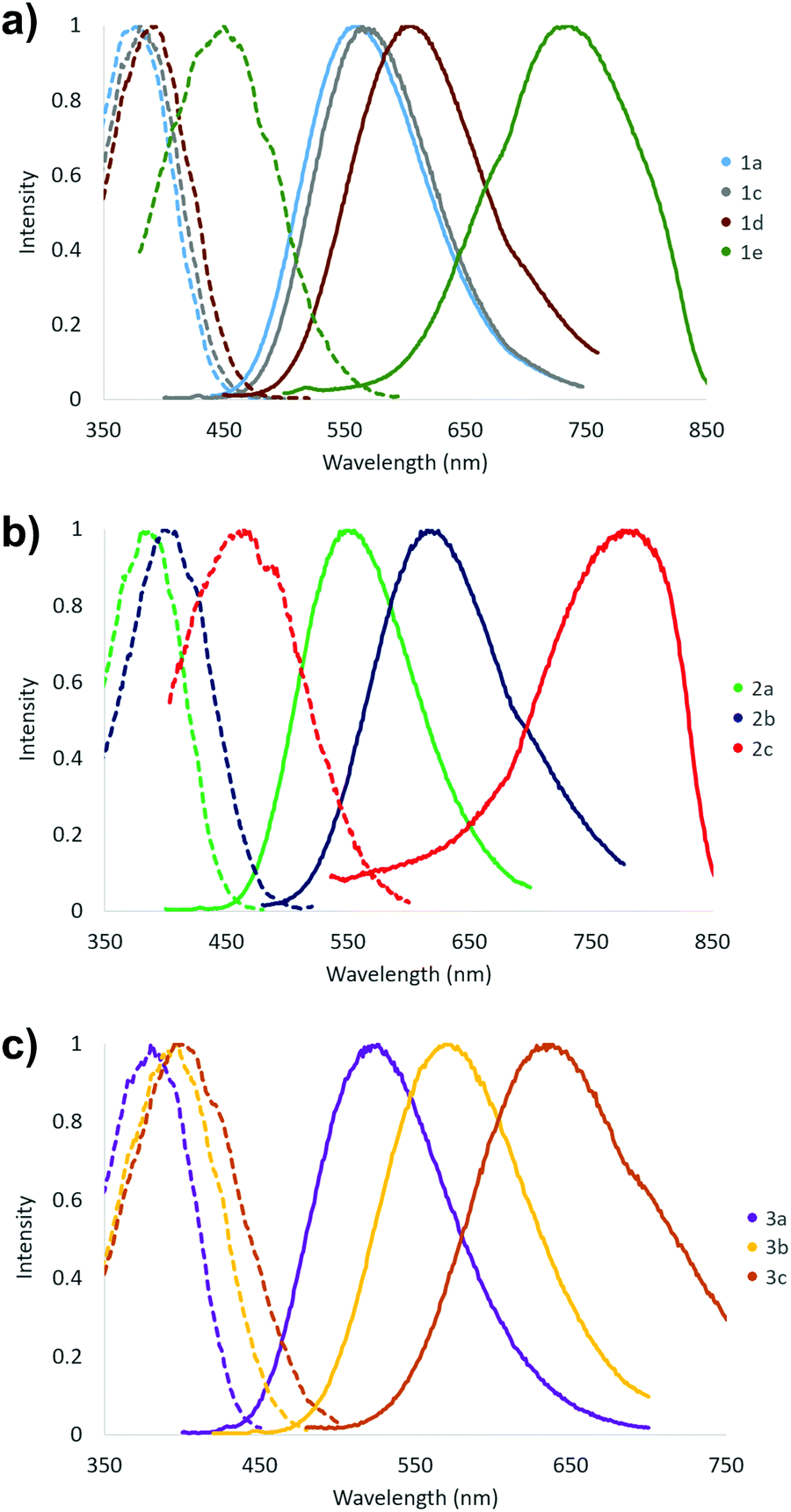 | ||
| Fig. 14 Fluorescence spectra of compounds (a) 1a, 1c–1e, (b) 2a–c and (c) 3a–c (emission spectra: solid line, excitation spectra: dotted line, 0.1–10 μM sol. in CH2Cl2 at 23 °C). | ||
Conclusions
We report here the preparation and characterization of conjugated stilbene, diphenylbutadiene and phenylethenylthiophene based diaromatic compounds and their SHG activities measured from the crystalline samples by the Kurtz–Perry powder method. The most significant activity was observed for the stilbene 1a, which produces SHG signal with intensity ca. 32 times stronger than that of the urea reference when nanosecond laser radiation at 1060 nm is applied. The stilbene derivative 1b and diphenylbutadiene 2a also display SHG activity, albeit with significantly lower intensity compared to 1a. The often high SHG intensities reported for stilbene derivatives have been contributed, in part, to the strong π–π interactions that enhance the intermolecular charge-transfer and, consequently, the second order polarizability.21 Consistently, the stilbene derivative 1a exhibits somewhat weaker π–π interactions compared to 3-methyl-4-methoxy-4′-nitrostilbene (MMONS, Scheme 1) with significantly higher SHG intensity.Our efforts to further investigate the effect of weak interactions to the NLO properties by changing the functional groups at the electron-donating end of stilbene, the extended chain length between the aromatic groups in diarylbutadienes and with thiophene derivatives were hampered by the extensive polymorphism that resulted in mainly centrosymmetric, NLO inactive chromophores despite the comprehensive crystallization efforts. Even the stilbene 1a with notable SHG activity also crystallizes in centrosymmetric, inactive space group and goes through, presumably, a photodimerization process to form two polymorphs of the tetra-aryl cyclobutane 4. Nevertheless, the high SHG intensities of especially 3-methyl-4-methoxy-4′-nitrostilbene (MMONS) and congeneric derivative 1a make them attractive chromophores for future studies to produce NLO active lenses by stereolithographic 3D printing technique.26 Furthermore, the current study illustrates the versatility of the structural, chemical and optical properties in these diaromatic, conjugated systems as also evidenced by their fluorescent nature in solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Finnish Cultural Foundation and the Magnus Ehrnrooth Foundation.References
- S. Tao, T. Miyagoe, A. Maeda, H. Matsuzaki, H. Ohtsu, M. Hasegawa, S. Takaishi, M. Yamashita and H. Okamoto, Adv. Mater., 2007, 19, 2707–2710 CrossRef CAS.
- J. M. Hales, S. Zheng, S. Barlow, S. R. Marder and J. W. Perry, J. Am. Chem. Soc., 2006, 128, 11362–11363 CrossRef CAS PubMed.
- B. Champagne, A. Plaquet, J.-L. Pozzo, V. Rodriguez and F. Castet, J. Am. Chem. Soc., 2012, 134, 8101–8103 CrossRef CAS PubMed.
- J. N. Zhang, X. Q. Wang, Q. Ren, H. L. Yang, J. W. Chen, Q. Sun, G. C. Li and T. B. Li, Opt. Laser Technol., 2011, 43, 679–682 CrossRef CAS.
- S. Di Bella, C. Dragonetti, M. Pizzotti, D. Roberto, F. Tessore and R. Ugo, in Molecular Organometallic Materials for Optics, Topics in Organometallic Chemistry, ed. H. Le Bozec and V. Guerchais, Coordination and Organometallic Complexes as Second-Order Nonlinear Optical Molecular Materials, Springer, Berlin, Heidelberg, ch. 1, vol. 28, 2010, pp. 1–55 Search PubMed.
- K. B. Manjunatha, R. Dileep, G. Umesh and B. R. Bhat, Opt. Laser Technol., 2013, 52, 103–108 CrossRef CAS.
- V. Nalla, R. Medishetty, Y. Wang, Z. Bai, H. Sun, J. Wei and J. J. Vittal, IUCrJ, 2015, 2, 317–321 CrossRef CAS PubMed.
- C.-F. Sun, C.-L. Hu, X. Xu, J.-B. Ling, T. Hu, F. Kong, X.-F. Long and J.-G. Mao, J. Am. Chem. Soc., 2009, 131, 9486–9487 CrossRef CAS PubMed.
- R. W. Boyd, Nonlinear Optics, Academic Press, 3rd edn, 2008 Search PubMed.
- G. J. Ashwell, G. Jefferies, D. G. Hamilton, D. E. Lynch, M. P. S. Roberts, G. S. Bahra and C. R. Brown, Nature, 1995, 375, 385–388 CrossRef CAS.
- G. J. Ashwell, Adv. Mater., 1996, 8, 248–250 CrossRef CAS.
- C. Chuantian, W. Bochang, J. Aidong and Y. Guiming, Sci. Sin., Ser. B, 1985, 28, 235–243 Search PubMed.
- F. C. Zumsteg, J. D. Bierlein and T. E. Gier, J. Appl. Phys., 1976, 47, 4980–4985 CrossRef CAS.
- J. D. Bierlein and H. Vanherzeele, J. Opt. Soc. Am. B, 1989, 6, 622 CrossRef CAS.
- I. Ledoux, C. Lepers, A. Périgaud, J. Badan and J. Zyss, Opt. Commun., 1990, 80, 149–154 CrossRef CAS.
- S. R. Marder, J. W. Perry and C. P. Yakymyshyn, Chem. Mater., 1994, 6, 1137–1147 CrossRef CAS.
- P. S. Patil, S. M. Dharmaprakash, H.-K. Fun and M. S. Karthikeyan, J. Cryst. Growth, 2006, 297, 111–116 CrossRef CAS.
- B. F. Levine, C. G. Bethea, C. D. Thurmond, R. T. Lynch and J. L. Bernstein, J. Appl. Phys., 1979, 50, 2523–2527 CrossRef CAS.
- L. T. Cheng, W. Tam, S. H. Stevenson, G. R. Meredith, G. Rikken and S. R. Marder, J. Phys. Chem., 1991, 95, 10631–10643 CrossRef CAS.
- Y. Wang, W. Tam, S. H. Stevenson, R. A. Clement and J. Calabrese, Chem. Phys. Lett., 1988, 148, 136–141 CrossRef CAS.
- W. Tam, B. Guerin, J. C. Calabrese and S. H. Stevenson, Chem. Phys. Lett., 1989, 154, 93–96 CrossRef CAS.
- P. R. Varanasi, A. K.-Y. Jen, J. Chandrasekhar, I. N. N. Namboothiri and A. Rathna, J. Am. Chem. Soc., 1996, 118, 12443–12448 CrossRef.
- A. K.-Y. Jen, Y. Cai, P. V. Bedworth and S. R. Marder, Adv. Mater., 1997, 9, 132–135 CrossRef CAS.
- K. Mandal, T. Kar, P. K. Nandi and S. P. Bhattacharyya, Chem. Phys. Lett., 2003, 376, 116–124 CrossRef CAS.
- V. P. Rao, A. K.-Y. Jen, K. Y. Wong and K. J. Drost, Tetrahedron Lett., 1993, 34, 1747–1750 CrossRef CAS.
- E. Kukkonen, E. Lahtinen, P. Myllyperkiö, J. Konu and M. Haukka, ACS Omega, 2018, 3, 11558–11561 CrossRef CAS PubMed.
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798–3813 CrossRef CAS.
- CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxfordshire, England.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- Crystal structure of compound 2b has been deposited to CCDC database: S. P. Kelley, K. Yang, N. Corretjer, S. Gadban, R. Glaser, M. M. Kozak and B. A. Hathaway, CSD Private Commun, 2018, CCDC 1879519.
- M. Rawal, K. E. Garrett, L. E. Johnson, W. Kaminsky, E. Jucov, D. P. Shelton, T. Timofeeva, B. E. Eichinger, A. F. Tillack, B. H. Robinson, D. L. Elder and L. R. Dalton, J. Opt. Soc. Am. B, 2016, 33, E160 CrossRef CAS.
- W. S. Wadsworth and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733–1738 CrossRef CAS.
- P. Munshi, B. W. Skelton, J. J. McKinnon and M. A. Spackman, CrystEngComm, 2008, 10, 197–206 RSC.
- S. N. Oliver, P. Pantelis and P. L. Dunn, Appl. Phys. Lett., 1990, 56, 307–309 CrossRef CAS.
- G. Marras, P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and A. Vij, New J. Chem., 2006, 30, 1397 RSC.
- M. A. Sinnwell and L. R. MacGillivray, Angew. Chem., Int. Ed., 2016, 55, 3477–3480 CrossRef CAS PubMed.
- G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 3641–3649 CrossRef CAS.
- Analogous disorder with detailed examination of correct space group has been reported for (trans)-4-chloro-4′-nitrostilbene: N.-R. Behrnd, G. Labat, P. Venugopalan, J. Hulliger and H.-B. Bürgi, CrystEngComm, 2010, 12, 4101–4108 RSC.
- Preliminary single-crystal and powder X-ray studies with non-centrosymmetric space group P1 and some SHG activity has been reported for compound 1c: P. M. Dinakaran, G. Bhagavannarayana and S. Kalainathan, Spectrochim. Acta, Part A, 2012, 97, 995–1001 CrossRef CAS PubMed ; these results were subsequently challenged, cf. ref. 41.
- B. R. Srinivasan, Z. Tylczynski and V. S. Nadkarni, Opt. Mater., 2013, 35, 1616–1618 CrossRef.
- An analogous crystal structure of compound 1c was deposited to CCDC database during the time of writing this contribution: T. H. Chunhua, A. C. Soegiarto and M. D. Ward, CSD Private Commun., 2020, CCDC 1976072.
- H. Gruen and H. Goerner, J. Phys. Chem., 1989, 93, 7144–7152 CrossRef CAS.
- D. Pines, E. Pines and W. Rettig, J. Phys. Chem. A, 2003, 107, 236–242 CrossRef CAS.
- T. Nakabayashi, M. Wahadoszamen and N. Ohta, J. Am. Chem. Soc., 2005, 127, 7041–7052 CrossRef CAS PubMed.
- R. Lapouyade, K. Czeschka, W. Majenz, W. Rettig, E. Gilabert and C. Rulliere, J. Phys. Chem., 1992, 96, 9643–9650 CrossRef CAS.
- D. Schulte-Frohlinde, H. Blume and H. Güsten, J. Phys. Chem., 1962, 66, 2486–2491 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A pdf file containing tables of crystallographic data (Tables S1–S3) and pertinent bond parameters (Tables S4–S16, together with molecular figures for atomic numbering scheme) as well as 1H NMR (Fig. S1–S11) and IR spectra (Fig. S12–S22) for all compounds. This material is available free of charge. CCDC 2058507–2058519 for compounds 1a·centro, 1a·non-centro, 1b, 1c, 1d, 1e, 4·Pcell, 4·Ccell, 2a, 3a·mono, 3a·ortho, 3b, and 3c. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1nj00456e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |