
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular level insights into the direct health impacts of some organic aerosol components†
Fatemeh
Keshavarz
 ab
ab
aInstitute for Atmospheric and Earth System Research, Faculty of Science, University of Helsinki, FI-00014 Helsinki, Finland. E-mail: fatemeh.keshavarz@helsinki.fi
bDepartment of Physics, School of Engineering Science, LUT University, FI-53851 Lappeenranta, Finland. E-mail: fatemeh.keshavarz@lut.fi
First published on 10th March 2021
Abstract
Organic aerosols (OAs) comprise 20–90 w% of atmospheric aerosols. While they have some positive impacts (e.g. acting as precipitation nuclei), they can pose some negative impacts on human health. The health impacts range from pulmonary inflammation to suppression of defense mechanisms, cancer and death. To understand how OA components can affect human body, this study employed a combination of in silico techniques: molecular docking, quantum chemical analysis of interactions, thermodynamics and kinetics of the reaction of OA components with DNA and oxidative stress mechanism, in addition to theoretical evaluation of their absorption, distribution, metabolism and excretion (ADME) properties. As OAs contain many components, this study just focused on benzo[a]pyrene (BaP), benzo[a]pyrene diol epoxide (BPDE), the cyclopentanoperhydrophenanthrene (CPPP) sterane, 2,6-dimethyl-1,4-benzoquinone (DMBQ), hopane (HOP), 9,10-phenanthrenequinone (PQ), and an OA model (fulvic acid; FA), which are all known for their negative health impacts. According to the ADME predictions, the low bioavailability of FA would limit its negative impacts. However, the ADME estimations and docking simulations suggested that BPDE, DMBQ and PQ have significant impact in affecting human health negatively: they can be feasibly distributed throughout human body by human serum albumin, they can likely permeate cell membranes, the blood–brain barrier and the gastrointestinal tract, and they show higher DNA binding efficiencies compared to the other OA components. Based on the quantum chemical calculations, BPDE, DMBQ and PQ can interact with the adenosine nucleobases of DNA, but they cannot directly form DNA adducts because of the nonspontaneous and kinetically infeasible nature of the corresponding reaction routes. Similarly, they cannot directly cause oxidative stress. However, in reference to the experimental evidence offered by earlier studies, their metabolites can result in both oxidative stress and carcinogenicity.
Introduction
Organic aerosols (OAs) comprise a large proportion of atmospheric aerosols1 ranging from 20 to 60 w% in continental mid-latitudes to about 90 w% in tropical forested areas.2 They not only control the energy budget, biogeochemical cycles (including precipitation patterns) and climate of Earth, but also pose adverse health effects.3–6 Particularly, as a significant portion of particulate matter (PM), OAs can be responsible for a portion of the 3 million deaths per year caused by PM.7An important point is that the health impacts of aerosols depend on many parameters such as their particle size, organic compounds, transition metals and specific surface.8,9 Therefore, their health effects are highly sensitive to their physiochemical properties. However, generally speaking, exposure to aerosol particles can induce pulmonary inflammation, lung cancer, oxidative stress caused by organic compounds or transition metals, inflammation and immune impacts initiated by biological aerosol components, covalent modification of enzymes and other key intracellular proteins, regulation of airway reactivity and heart rate variability, stimulation of autonomic nervous system activity, enhanced response to environmental allergens, and suppression of defense mechanisms.9–11 Some other health implications of PM are related to reproductive systems and include low birth weight, preterm labor, growth retardation and female infertility.12 All these health risks increase on prolonged exposure to PM.13
While most studies have considered the health impacts of PM rather than focusing on OAs, the hazardous nature of some OA components has been the subject of ongoing research. The most outstanding examples are quinones (such as 9,10-phenanthrenequinone (PQ) and 2,6-dimethyl-1,4-benzoquinone (DMBQ)), polycyclic aromatic hydrocarbons (PAHs; e.g. benzo[a]pyrene (BaP)), triterpenes (e.g. hopanes (HOP)) and phenanthrenes (like the cyclopentanoperhydrophenanthrene (CPPP) sterane).
PQ represents the quinones found in diesel exhaust particles, and it is believed to induce cytotoxic effects through the generation of reactive oxygen species (ROSs; i.e. oxidative stress).14 Similarly, DMBQ in ambient particles leads to oxidative stress. However, the contribution of DMBQ to the toxicity of PM should be lower than that of PQ.15 BaP has been assessed as the most genotoxic, mutagenic and carcinogenic component of PM.16–18 These properties are owed to its ability to form adducts with DNA.19 BaP is mainly found in its oxygenated forms (e.g. (+)-benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide (BPDE)), and its oxygenated forms might be responsible for its genotoxicity.20 Finally, hopanes and steranes, which are fossil fuel markers,21 are linked to toxicity but no plausible mechanism has been proposed for them.22
To the best of our knowledge, no study has evaluated the molecule-level impacts of OAs. However, some details about the molecule-level effect of several quinones on oxidative stress (e.g. the reaction of tetrachloro-o-benzoquinone with H2O223 and the redox mechanism of several quinones24) and DNA (see ref. 25–27) have been studied. To this end, we consider an OA model, PQ, DMBQ, BaP, BPDE, the simplest HOP and CPPP, and analyze their corresponding absorption, distribution, metabolism and excretion (ADME) properties. While the metabolic response to OAs is completely complicated, we assess their direct impacts and potential toxicity by analyzing their interaction with human serum albumin (HSA) and DNA through molecular docking, in addition to concerning their mechanistic role in oxidative stress and DNA alkylation using quantum chemical approaches. The docking and quantum chemical investigations are devised to focus on an isolated process. However, the ADME properties are used to study the health impacts of the OA components at a broader scope. The ADME results can offer pharmacokinetic/physiological details beyond extensive quantum chemical or molecular docking simulations because they can follow metabolic networks rather than a single isolated reaction.
The main working hypothesis of the study is that the applied molecular modeling tools can qualitatively describe what will happen to the OA components at their first encounter with important biomolecules. However, because of the complexity and vast range of bioprocesses, all plausible bioprocesses are not studied. Particularly, the metabolic products of the OA components and their reaction/interaction with the evaluated biomolecular targets are neglected, and only the direct impacts of the OA components have been concerned. Furthermore, although all the adopted techniques are strong and advantageous to some extent, each has some shortcomings which are addressed along with the results. Therefore, we suppose that the results are qualitatively reliable, but the quantitative results might suffer from some level of inaccuracy. In addition, we encourage the use of the findings of this study as a starting point for a more comprehensive experimental/computational study of the health impacts.
HSA is eventually selected because it is the transportation vehicle in human body, and it controls the distribution and metabolism of different organic compounds (like drugs).28,29 DNA is chosen as the principal target of genotoxicity. For comparison purposes, the same analyses are done on cis-platin (CPT) and ThioTEPA (N,N′,N′′-triethylenephosphoramide) (globally known anticancer drugs both forming adducts with DNA)30,31 and ibuprofenate (IBPPA; isobutyl phenyl propionic acid or (RS)-2-(4-(2-methylpropyl)phenyl)propanoic acid) (a famous anti-inflammatory drug with no impact on DNA)32 as reference compounds. Fig. 1 shows the chemical structure of the evaluated compounds.
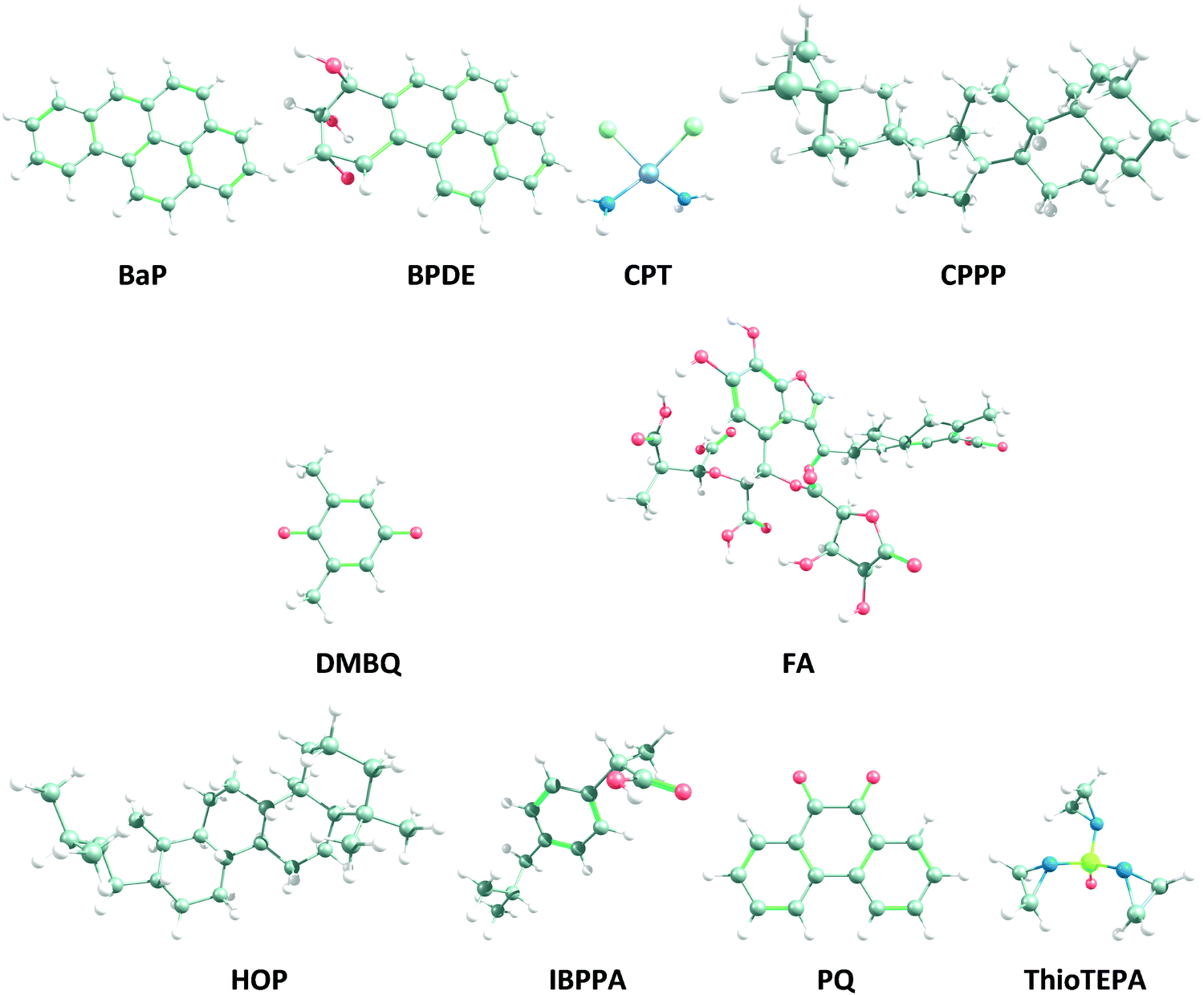 | ||
| Fig. 1 Geometries of the model OA (FA), PQ, DMBQ, BaP, BPDE, HOP, CPPP, CPT, ThioTEPA and IBPPA optimized at the ωB97X-D/def2-TZVP level of theory, in water (see Fig. S1, ESI,† for the 2D structures). The carbon, hydrogen, oxygen, nitrogen, chloride, sulfur and platinum atoms are represented as grey, white, red, blue, light green, yellowish and light purple balls, respectively. | ||
Results and discussion
ADME properties
Table 1 reports the ADME properties of the aerosol components and the reference drugs. It should be noted that these results are obtained through machine learning based protocols, and they are not completely reliable without further experimental verification. According to the results, just FA is slightly hydrophilic (negative log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Po/w), while the other compounds are lipophilic (positive logPo/w). As cell membranes are made of lipid-type structures (i.e. phospholipid bilayers), all the evaluated compounds, except FA, should be able to pass through cell membranes passively (rather than diffusing through membrane proteins). Also, just FA and ThioTEPA have substantial water solubility (WS). Regardless of solubility and hydrophilicity, the ADME results predict BPDE, DMBQ, IBPPA, PQ and ThioTEPA as the compounds that can be absorbed through the gastrointestinal system. ThioTEPA has been approved as a drug that can significantly affect (toxify) the gastrointestinal tract.33 Also, the long-term use of IBPPA has been associated with gastrointestinal ulceration.34 Therefore, BPDE, DMBQ and PQ are likely to damage the gastrointestinal tract in a similar way. In addition, the blood–brain barrier (BBB) permeation results outline that BPDE, DMBQ and PQ can permeate the BBB like the IBPPA anti-inflammatory/painkiller agent. As these compounds are lipophilic and have the potential of crossing the BBB, they might affect the central nervous system.35 However, both DMBQ and PQ are quinones, and some quinones, e.g. pyrroloquinoline quinone,36 have neuroprotective effects on the nervous system, meaning that they can prevent/reduce damages to the nervous system. Consequently, the predicted BBB permeation should not be considered as a negative health impact by organic aerosols without experimental evidence. None of the compounds can pass through skin (negative logKp) as expected. However, there is experimental evidence for the entrance of OAs and their components through lungs.37 Also, as BPDE, DMBQ, IBPPA, PQ and ThioTEPA are predicted to pass through the gastrointestinal tract, processing of OA contaminated drinks and foods by the gastric system can be considered as a potential route for their entrance to cellular areas.
Po/w), while the other compounds are lipophilic (positive logPo/w). As cell membranes are made of lipid-type structures (i.e. phospholipid bilayers), all the evaluated compounds, except FA, should be able to pass through cell membranes passively (rather than diffusing through membrane proteins). Also, just FA and ThioTEPA have substantial water solubility (WS). Regardless of solubility and hydrophilicity, the ADME results predict BPDE, DMBQ, IBPPA, PQ and ThioTEPA as the compounds that can be absorbed through the gastrointestinal system. ThioTEPA has been approved as a drug that can significantly affect (toxify) the gastrointestinal tract.33 Also, the long-term use of IBPPA has been associated with gastrointestinal ulceration.34 Therefore, BPDE, DMBQ and PQ are likely to damage the gastrointestinal tract in a similar way. In addition, the blood–brain barrier (BBB) permeation results outline that BPDE, DMBQ and PQ can permeate the BBB like the IBPPA anti-inflammatory/painkiller agent. As these compounds are lipophilic and have the potential of crossing the BBB, they might affect the central nervous system.35 However, both DMBQ and PQ are quinones, and some quinones, e.g. pyrroloquinoline quinone,36 have neuroprotective effects on the nervous system, meaning that they can prevent/reduce damages to the nervous system. Consequently, the predicted BBB permeation should not be considered as a negative health impact by organic aerosols without experimental evidence. None of the compounds can pass through skin (negative logKp) as expected. However, there is experimental evidence for the entrance of OAs and their components through lungs.37 Also, as BPDE, DMBQ, IBPPA, PQ and ThioTEPA are predicted to pass through the gastrointestinal tract, processing of OA contaminated drinks and foods by the gastric system can be considered as a potential route for their entrance to cellular areas.
| Molecule | BaP | BPDE | CPT* | DMBQ | FA | HOP | IBPPA* | PQ | CPPP | ThioTEPA* |
|---|---|---|---|---|---|---|---|---|---|---|
|
a MW: molecular weight in g mol−1; NRB: number of rotatable bonds; MR: molar refractivity as a measure of polarizability; TPSA: total polar surface area in Å2; logPo/w: octanol–water partition as a lipophilicity measure; WS: water solubility in g L−1; GIA: gastrointestinal absorption; BBB: blood–brain barrier permeant; Pgp: P-glycoprotein substrate; CYP1A2: cytochrome P450 1A2 inhibitor; CYP2C19: cytochrome P450 2C19 inhibitor; CYP2C9: cytochrome P450 2C9 inhibitor; CYP2D6: cytochrome P450 2D6 inhibitor; CYP3A4: cytochrome P450 3A4 inhibitor; logKp: skin permeation in cm s−1; and BS: bioavailability score. The reference drugs are distinguished by an asterisk.
|
||||||||||
| MW | 252.31 | 303.33 | 298.03 | 136.15 | 774.63 | 370.65 | 206.28 | 208.21 | 372.67 | 201.21 |
| NRB | 1 | 0 | 0 | 0 | 14 | 1 | 4 | 0 | 5 | 3 |
| MR | 87.65 | 88.34 | 19.23 | 37.91 | 176.76 | 120.81 | 62.18 | 60.54 | 122.67 | 64.96 |
| TPSA | 0.00 | 52.99 | 52.04 | 34.14 | 339.23 | 0.00 | 37.30 | 34.14 | 0.00 | 35.91 |
| logPo/w |
5.34 | 1.86 | — | 1.21 | −0.26 | 7.56 | 3.01 | 2.46 | 8.21 | 0.51 |
| WS | 1.38 × 10−6 | 1.08 × 10−2 | — | 3.66 | 3.08 × 102 | 6.34 × 10−4 | 7.49 × 10−2 | 1.17 × 10−3 | 2.43 × 10−5 | 5.59 × 102 |
| GIA | Low | High | — | High | Low | Low | High | High | Low | High |
| BBB | No | Yes | — | Yes | No | No | Yes | Yes | No | No |
| Pgp | No | Yes | — | No | Yes | No | No | No | No | Yes |
| CYP1A2 | Yes | Yes | — | No | No | Yes | No | Yes | No | No |
| CYP2C19 | No | No | — | No | No | No | No | Yes | No | No |
| CYP2C9 | No | No | — | No | No | Yes | No | No | No | No |
| CYP2D6 | No | Yes | — | No | No | No | No | No | No | No |
| CYP3A4 | No | No | — | No | No | No | No | No | No | No |
| logKp |
−3.6 | −6.74 | — | −6.26 | −10.95 | −1.26 | −5.07 | −5.78 | −0.61 | −7.12 |
| BS | 0.55 | 0.55 | — | 0.55 | 0.11 | 0.55 | 0.56 | 0.55 | 0.55 | 0.55 |
Among the tested compounds, BPDE, FA and ThioTEPA are proposed as P-glycoprotein (a drug transporter) substrates by theoretical ADME estimations, which means that their pharmacokinetic properties would likely change in human body. However, the bioavailability of FA is estimated to be low, meaning that just a small fraction of it can potentially circulate among cells and organs systematically. Therefore, the metabolism of FA and its interaction with other metabolites should be limited. In the case of ThioTEPA, being a P-glycoprotein substrate can be related to tumor chemoresistance.38 Therefore, this result can imply that P-glycoprotein can likely alter the health impacts of BPDE including the inhibition of the cytochrome P450 1A2 and 2D6 enzymes. The negative or positive effect of P-glycoprotein on the health impacts of BPDE requires experimental assessment. Also, BaP, HOP and PQ are predicted to inhibit cytochrome P450 1A2. Similarly, HOP and PQ are predicted to respectively inhibit cytochromes P450 2C9 and P450 2C19. All these enzymes are involved in the metabolism of drugs and physiologically important compounds.39–41 Therefore, BaP, BPDE, HOP and PQ seem to have the potential to negatively affect the metabolism of some pharmaceutics and metabolic agents.
DNA and HSA docking
The docking results are summarized in Table 2 and Tables S1, S2 and depicted in Fig. S2–S4 (ESI†). According to Table S1 (ESI†), the strongest interaction (i.e. the lowest interaction energy) with HSA refers to FA (the largest compound) and the weakest interaction refers to CPT (the smallest compound). None of the tested compounds bind to HSA through electrostatic interactions, while hydrogen bonding plays a significant role in the binding of BPDE, CPT, DMBQ, FA and IBPPA and steric (van der Waals) interactions are the main drivers of the compounds binding to HSA. The dominance of steric interactions can be attributed to the hydrophobicity (lipophilicity) of most ligands (see the positive values of logPo/w in Table 1) and the hydrophobicity of most HSA domains.
| Ligand | Interaction energy | Electrostatic interaction | Hydrogen bonding | LE | Binding site |
|---|---|---|---|---|---|
| a The energy and ligand efficiency (LE = interaction energy/number of non-hydrogen atoms) values are unitless. | |||||
| BaP | −156.9 | 0.0 | 0.0 | −6.96 | CAA/GTT |
| BPDE | −160.0 | 0.0 | 0.0 | −7.84 | ATT/TAA |
| CPPP | −102.9 | 0.0 | 0.0 | −3.81 | AAT/TTA |
| CPT | −52.2 | 0.0 | 0.0 | −10.45 | CA/GT |
| DMBQ | −89.9 | 0.0 | 0.0 | −8.99 | TTT/AAA |
| FA | −231.5 | 0.0 | 0.0 | −4.21 | AATA/TTAT |
| HOP | −110.4 | 0.0 | 0.0 | −4.09 | TAA/ATT |
| IBPPA | −114.8 | 0.0 | 0.0 | −7.65 | AAA/TTT |
| PQ | −132.8 | 0.0 | 0.0 | −8.30 | TTT/AAA |
| ThioTEPA | −74.0 | 0.0 | 0.0 | −6.73 | TA/AT |
In physiological conditions, ligand binding is not just governed by the sum of molecular interactions. In competition with other ligands, the ligand with the highest diffusion rate and the highest concentration would bind to the target more efficiently. Larger molecules diffuse slower toward the target, and the ligand with the highest concentration in the microenvironment has the highest potential of binding. To account for diffusion contribution and eliminate the effect of molecular size on the binding of the ligands, ligand efficiency (LE) was calculated as the interaction energy divided by the number of non-hydrogen atoms of the compound.42 The LE values showed that CPPP, HOP and FA have the lowest HSA binding efficiencies, whereas BaP, BPDE, CPT, DMBQ, IBPPA and PQ have noticeable LE values (LE < −5.00) and a high potential of transportation to all cellular targets by HSA.
Similar to HSA binding, Table 2 and Table S2 (ESI†) indicate the lowest (strongest interaction) and highest DNA binding energies, respectively, for FA and CPT, and the involvement of no electrostatic interaction. Here, no hydrogen bonding interaction can be observed. Therefore, the docking results indicate that the DNA–ligand binding of the studied compounds should be mainly driven by steric interactions. However, one should note that the employed docking simulations are based on semi-flexible treatment of ligand binding to the targets, and the calculated interaction energies/types are based on classical force field parameters. Therefore, the docking results might be biased with inaccurate estimation of atomic parameters and intermolecular interactions, leading to underestimation of electrostatic and hydrogen-bonding interactions.
For both DNA strands, the best LE values (LE < −6.00) are obtained for BaP, BPDE, CPT, DMBQ, IBPPA, PQ and ThioTEPA, which are the compounds giving HSA LE values lower than −5.00. Particularly, the CPT anticancer drug gives the best ligand efficiency, and the ThioTEPA alkylating agent shows a fairly noticeable LE value. These two reference compounds were neglected in the quantum mechanical calculations because their DNA cleavage mechanisms are reported elsewhere,31,43 and just BaP, BPDE, DMBQ, IBPPA and PQ were concerned in further evaluation of the OA–DNA interaction and oxidative stress.
Mutagenicity potential/DNA interaction
To increase the accuracy of the DNA/ligand interaction analysis and to identify any missing interaction type/pairs, NBO analysis was performed. NBO analysis goes beyond classical force fields and atom pairs, and it utilizes wave functions to calculate orbital properties and follow the trends of charge transfer between them. In this way, it can specifically distinguish hydrogen bonds. The results of Natural Bond-Orbital (NBO) analysis on the interactions of BaP, BPDE, DMBQ, IBPPA and PQ with 3US0 are outlined in Table S3 (ESI†), where higher E(2) stabilization energy values are indicative of stronger interactions. 3US0 is selected as the DNA target because it interacts with the case compounds more strongly, relative to 4AWL (see Table 2 and Table S2, ESI†). Although the docking results of none of the compounds predict hydrogen bonding interactions, the quantum chemistry based NBO results identify very weak to strong hydrogen bonding interactions in the form of charge transfer from the lone-pair (LP) orbital of proton donors (such as O and N atoms) to the antibonding (σ*) orbital of proton acceptors (e.g. OH and CH). The BaP interaction with the T nucleobase of 3US0 involves a very weak hydrogen bond. Also, a weak hydrogen bond is observed between PQ and an A nucleobase. DMBQ and IBPPA do not form any hydrogen bonds with 3US0. However, a strong hydrogen bonding network helps BPDE stabilize along the A and T nucleotides. On the other hand, both docking-based interaction energies and E(2) results show that BPDE and BaP can interact with 3US0 the strongest, whereas DMBQ can interact with DNA weakly, relative to the other tested compounds. This increases the DNA adduct formation potential of BPDE (and its BaP non-oxidized form), but it cannot be directly interpreted as the experimentally observed BaP carcinogenicity19 without further theoretical and experimental evaluations. In addition, the results suggest that BaP, BPDE, DMBQ and PQ mainly interact with the A nucleobases of DNA, while IBPPA just interacts significantly with the deoxyribose moiety of the A, C and T nucleotides. This is in line with the experimental findings that report that IBPPA has no significant negative impact on DNA,44 or the studies that show it prevents DNA damage.45 Also, it should be noted that A nucleobases are more susceptible to mutagenic lesion formation when interacting with some carbonyl containing compounds (such as formaldehyde46), and BaP, BPDE, DMBQ and PQ interact efficiently with the A nucleobases. This opens up a question about the possible contribution of such interaction mode to their mutagenicity, which should be answered through more accurate calculations (such as QM/MM molecular dynamics simulations or quantum chemical modeling) or experimental evidences. To answer this question, we used quantum chemical calculations to re-evaluate the binding of BaP, BPDE, DMBQ and PQ to their corresponding DNA binding site (according to the docking and NBO results) by full geometry optimization of the binding pairs and re-calculation of their binding energies under cell-like conditions. Also, we conducted quantum chemistry level mechanism exploration to assess any DNA alkylating/non-alkylating reaction between these OA components and DNA.Based on the NBO results, more accurate interaction energies were calculated only for BaP, BPDE, DMBQ and PQ by cropping the effective DNA binding sites (i.e. the main interacting nucleotides). The ZPE-corrected and Gibbs free interaction energies, along with the optimized structures, are shown in Fig. 2. According to the obtained values, the interactions of BPDE, DMBQ and PQ with the nucleotides are slightly nonspontaneous (positive Gibbs free energies), but the BaP interaction is slightly spontaneous. Therefore, the binding of these compounds to DNA cannot be completely neglected, while it cannot be thermodynamically highly plausible. Nevertheless, the potential mutagenicity and carcinogenicity of the OA components were mechanistically assessed by considering their roles in DNA alkylation (adduct formation), DNA cleavage or any other structural changes in DNA. However, exhaustive reaction path scanning and TS predictions failed to propose any mechanism.
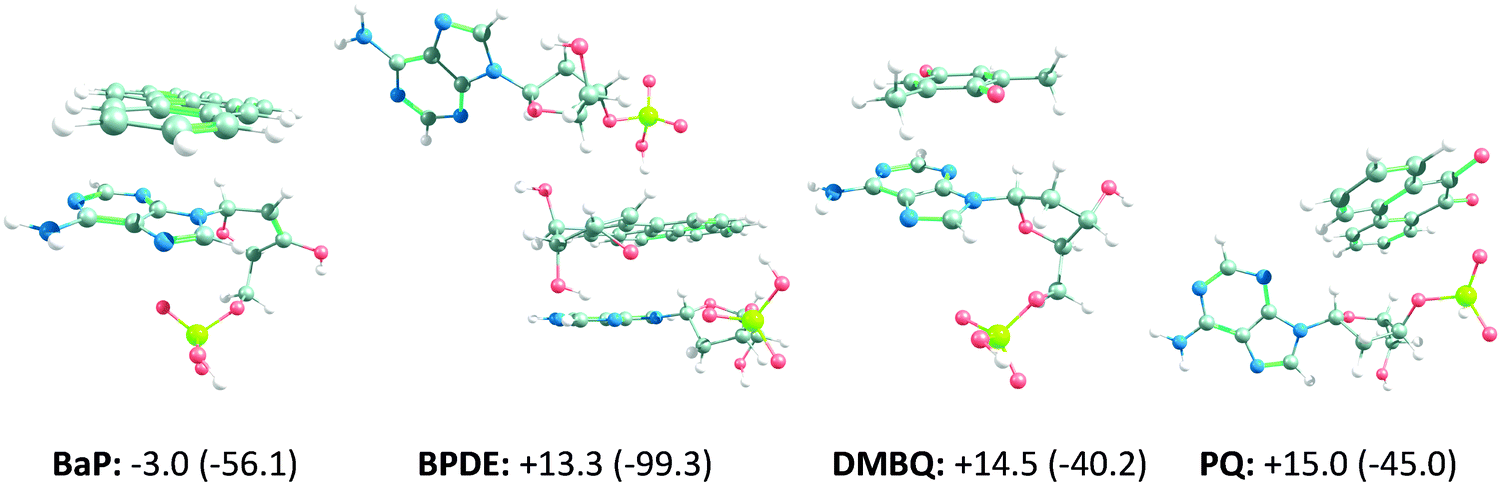 | ||
| Fig. 2 Geometry and binding Gibbs free energy (and ZPE-corrected interaction energy) of the ligand–nucleotide complexes retrieved from NBO analysis. The energies are in kJ mol−1. The complexes, nucleotides and ligands are optimized and energy analyzed under cellular conditions while conserving their conformational state in their corresponding ligand–nucleotide complexes. | ||
Because many experimental studies have associated BPDE with genotoxicity, mutagenicity and carcinogenicity,16–18,20 two common mechanisms were further studied for adduct formation between DNA and BPDE, DMBQ and PQ; type-2 nucleophilic substitution (SN2; formation of one bond between two reactants simultaneously with the cleavage of a bond in one of the reactants) similar to the alkylation mode of the ThioTEPA anti-cancer agent;31 and Michael addition (MA; nucleophilic addition of a nucleophile to a carbonyl compound) between the electron deficient carbon of quinones and the exocyclic NH2 moiety of nucleobases.47 Here, just an A nucleobase was targeted to exclude steric restrictions and simplify the calculations. The SN2 scenario (bond formation between the OA components and the A nucleobase simultaneously with opening of the epoxy functional group in BPDE, or conversion of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond to a C–O− single bond) was rejected by the scanning and TS calculations, and every time the predicted TSs converted into the individual reactants.
O double bond to a C–O− single bond) was rejected by the scanning and TS calculations, and every time the predicted TSs converted into the individual reactants.
The MA scenario was more successful, and a reaction path was approved for DMBQ (R1) and PQ (R2) which is shown in Fig. 3. Also, a TS was found for adduct formation between BPDE and the A nucleobase at the ωB97X-D/3-21g level and verified using IRC calculations ((R3); see Fig. S5, ESI,† for the TS, and the connecting pre-reaction complex and product). But the TS could not be validated at the ωB97X-D/def2-TZVP level. Notably, in reactions (R1)–(R3), “A” stands for the adenine (A) nucleobase. To assure that the reaction path observed for BPDE at the ωB97X-D/3-21g level is an artifact of low-level calculations, the TS was re-optimized at the BVP86/def2-TZVP, M06-2X/def2-TZVP and B3LYP/def2-TZVP levels. All alternative level calculations failed, asserting the absence of any DNA adduct formation route for BPDE. However, there might be still some reaction paths involved in mutagenicity induction but missing from our calculations. Some experiments have stated that it is the metabolites of BaP and BPDE (like phenols and quinines) that can induce mutagenic, teratogenic and cytotoxic effects48–51 through ROS generation.52
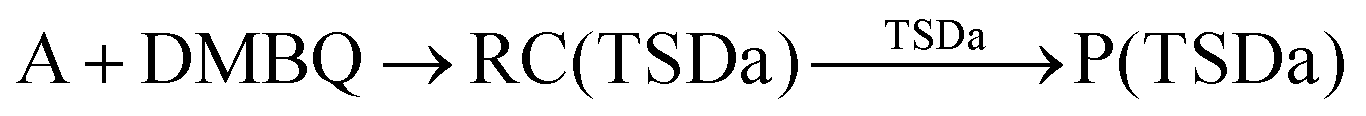 | (R1) |
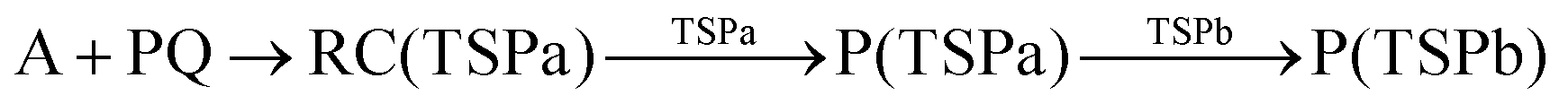 | (R2) |
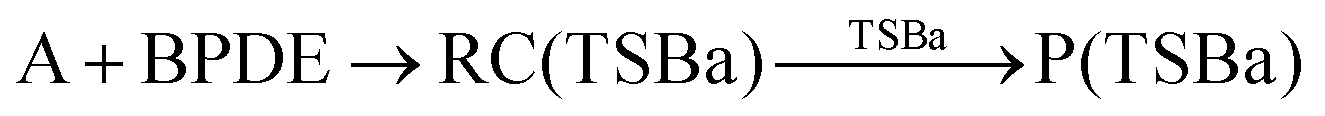 | (R3:unapproved reaction) |
 | ||
| Fig. 3 Mechanism of DNA adduct formation by DMBQ and PQ. The Gibbs free energies (and ZPE-corrected energy values) are calculated relative to the corresponding reactants in kJ mol−1. The carbon, hydrogen, oxygen, and nitrogen atoms are represented as grey, white, red, and blue balls, respectively. | ||
As all our quantum chemical evaluations suggest, the tested OA components are incapable of causing mutagenicity directly through the evaluated mechanisms under cellular conditions. However, some mechanistic schemes might be missing from our calculations. Particularly, as the ADME results predicted, binding to P-glycoprotein might convert BPDE to secondary metabolites, which can end up in carcinogenic effects. Also, experimental studies on some quinones47,53,54 have attributed DNA adduct formation to quinone metabolites. Therefore, while our results decline a direct DNA effect by the OA components, the possibility of un-examined reaction mechanisms and the indirect effect of OA metabolites on DNA should not be ignored without sufficient computational and/or experimental evidence.
Oxidative stress
According to Squadrito et al.,55 the first step in oxidative stress induced by different organic compounds is hydride transfer from NADPH to the organic compound in a nucleophilic substitution reaction (simplified as (R4) with OC being the organic compound and the transferred hydride being represented by h−). Among the studied OA components, the quinones (PQ and DMBQ), BPDE and FA can offer an electrophilic site for acceptance of hydrides, but FA has low bioavailability based on the HSA docking and ADME estimations. Therefore, the oxidative stress mechanism was just followed for PQ, DMBQ and BPDE. Fig. 4, 5 and Fig. S5 (ESI†) depict the minimum energy reaction paths of the observed mechanism, which takes place in five individual steps. In the first step (Fig. 4; (R5)–(R7)), NADPH and DMBQ/BPDE diffuse toward each other to form RC complexes, respectively, named RC(TSD1) and RC(TSB1). For PQ, no stable RC forms. Then, a hydride transfers from NADPH to the carbon in the carbonyl groups of PQ and DMBQ, or the oxirane carbon of BPDE. Simultaneously, the attacked bond cleaves, forming a single bond between the attacked carbon and a (now) negatively charged oxygen atom along with NADP+ in the form of a post-reaction complex (PC). The products then diffuse apart. Under cell-like conditions, the Gibbs free energy barriers of this step for BPDE (TSB1), DMBQ (TSD1) and PQ (TSP1) are, respectively, 118.7, 109.7 and 98.3 kJ mol−1 high, meaning that this reaction is the most feasible for PQ. However, in general, these reaction steps are not kinetically feasible, unless side metabolic reactions provide sufficient energy for surpassing the corresponding barriers.| NADPH + OC → NADP+ + (OC)h− | (R4) |
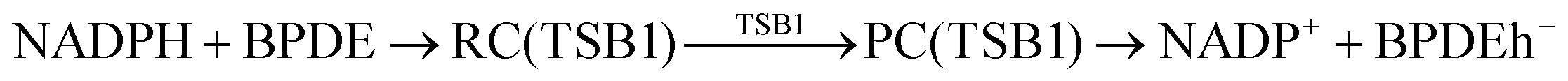 | (R5) |
 | (R6) |
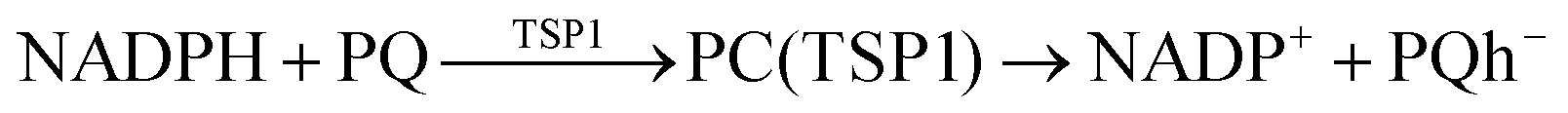 | (R7) |
| H+ + (OC)h− → (OC)p | (R8) |
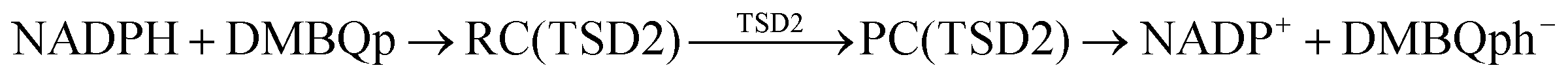 | (R9) |
 | (R10) |
| H+ + (OC)ph− → (OC)pp | (R11) |
| O2− + BPDEp → RC(BPDEp + O2) → PC(BPDEp + O2) → BPDEr− + H2O2 | (R12) |
| O2− + DMBQpp → RC(DMBQpp + O2) → PC(DMBQpp + O2) → DMBQr− + H2O2 | (R13) |
| O2− + PQpp → RC(PQpp + O2) → PC(PQpp + O2) → PQr− + H2O2 | (R14) |
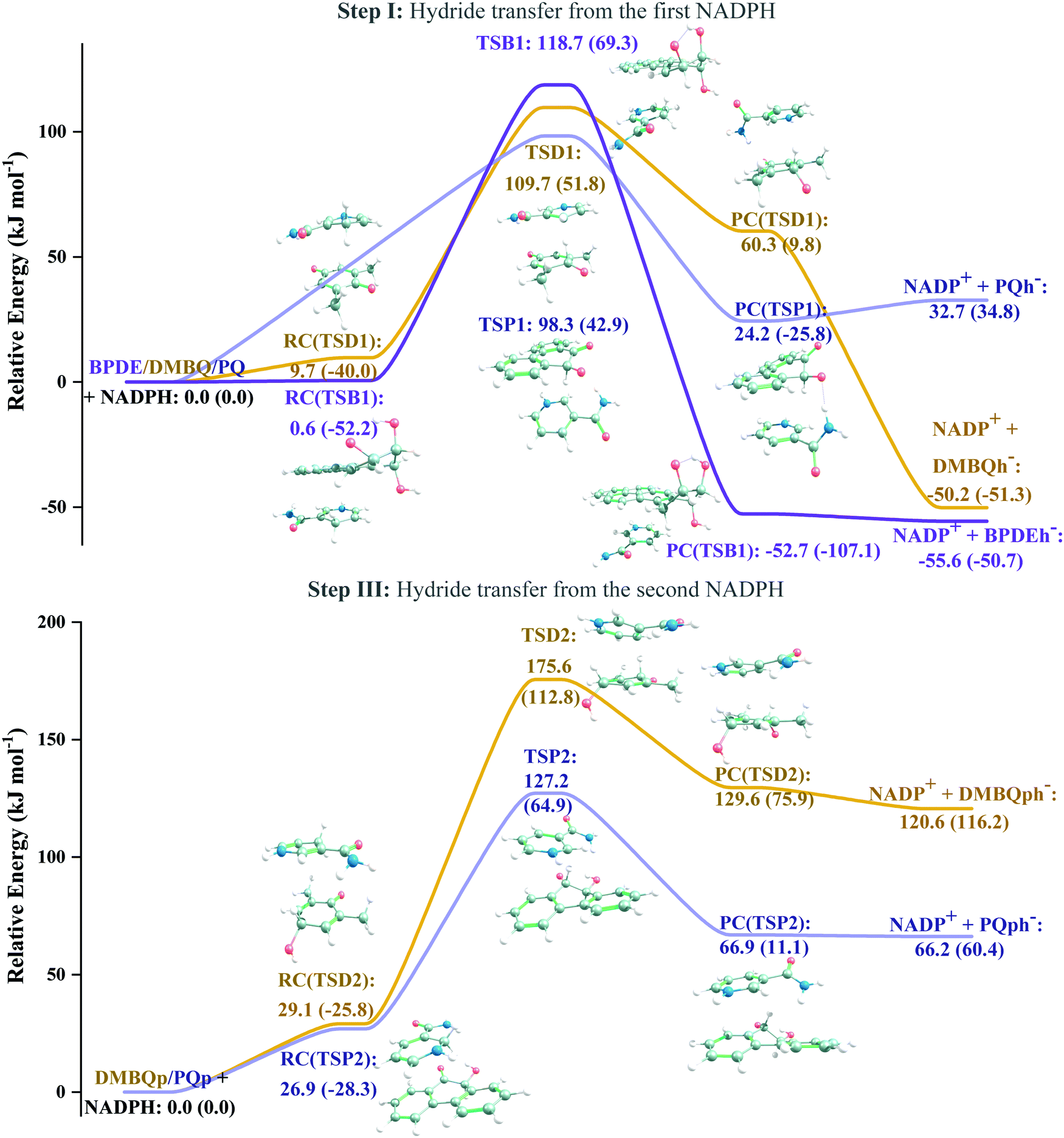 | ||
| Fig. 4 Steps I and III of the oxidative stress mechanism initiated by the organic aerosol components. The Gibbs free energies (and ZPE-corrected energy values) of the BPDE, DMBQ and PQ reaction sets are calculated relative to the corresponding reactants in kJ mol−1. The carbon, hydrogen, oxygen, and nitrogen atoms are represented as grey, white, red, and blue balls, respectively. | ||
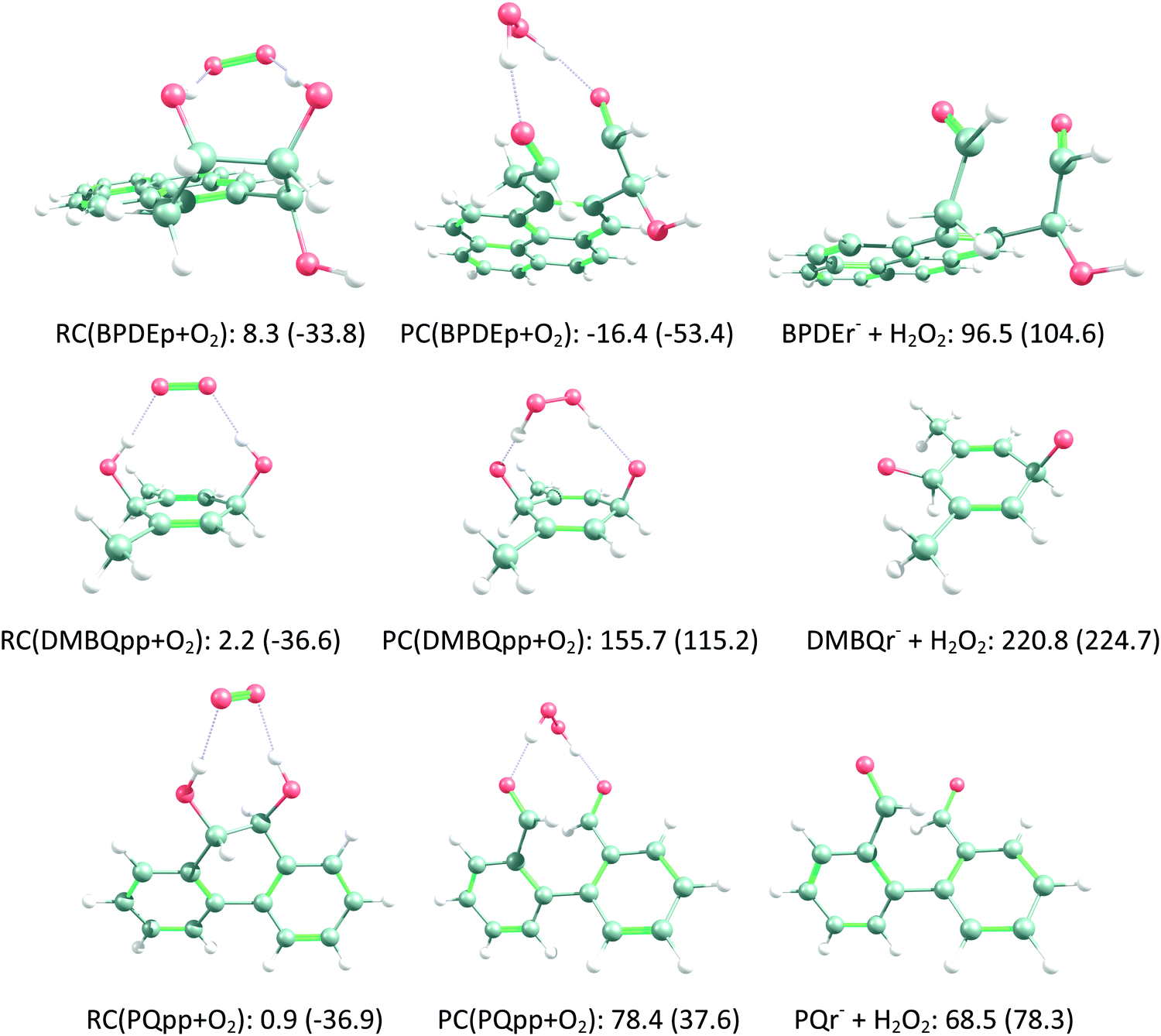 | ||
| Fig. 5 Step V of the oxidative stress mechanism (H2O2 generation) initiated by the organic aerosol components. The Gibbs free energies (and ZPE-corrected energy values) of the BPDE, DMBQ and PQ reaction sets are calculated relative to the corresponding reactants in kJ mol−1. The carbon, hydrogen, oxygen, and nitrogen atoms are represented as grey, white, red, and blue balls, respectively. | ||
Conclusion
The theoretically examined reaction schemes indicated that the studied OA components are unable to have any direct negative impacts on human health through oxidative stress or DNA alkylation. However, there might be some potential direct/non-direct reaction mechanisms that have not been considered in this study. Particularly, as suggested by earlier experimental studies, the OA metabolites can pose some inevitable health impacts. Based on the ADME predictions, the OA components cannot enter human body through skin. However, as BPDE, DMBQ and PQ enter the system of human body, they can likely permeate cellular membranes, the gastrointestinal tract and the blood–brain barrier, affecting both the gastric and nervous systems. The consequence of their permeation is not necessarily negative, and they should be studied in detail using experimental techniques. Also, as BPDE and PQ (along with BaP and HOP) were predicted to have the potential of inhibiting some cytochrome enzymes, and they can be easily transported by HSA to all metabolism centers, their plausible negative effects on the metabolism of pharmaceutics and metabolic agents should be addressed by further experimental or computational research. Lastly, this study serves as a first step toward understanding the health impacts of organic aerosols at the molecular level, and further in vitro and in vivo experiments are recommended to complement the results and enhance our understanding of the impacts.Computational details
OA model
Fulvic acid (FA) was selected to represent OAs because (1) it mimics both the aliphatic and aromatic structures in the polycarboxylic acid fraction of OAs,61 (2) it displays the functionality and behavior of organic PM fragments,62 (3) 6–11% of the total organic mass extracted from dust aerosols is composed of humic and fulvic acids,63 and (4) it has been accepted as a suitable surrogate for highly oxidized OAs64 and a proxy for atmospheric PM.65,66 Among various FA chemical structures, the model structure selected and geometry optimized by Sadhu et al.67 were adopted. This OA model (Fig. 1) is composed of aromatic and non-aromatic moieties, a quinone group, phenolic and heterocyclic hydroxyl functionalities, and multiple carboxylate groups.ADME properties
The ADME properties of the selected OA components (PQ, DMBQ, BaP, BPDE, HOP and CPPP), the reference drugs (CPT, ThioTEPA and IBPPA) and the OA model were evaluated using SwissADME.68 SwissADME is a web tool that applies machine learning principles, and offers fast and robust models for prediction of the pharmacokinetic, physiochemical and medicinal properties of various molecules. In other words, it describes any input molecule (read as a 2D structure or SMILES notation) by a set of molecular descriptors, and then it cross-matches the descriptors with those of molecules with known properties to predict their ADME performance.Molecular docking
For analysis of the interaction of PQ, DMBQ, BaP, BPDE, HOP, CPPP, CPT, ThioTEPA, IBPPA and FA with DNA and HSA, molecular docking simulations were performed. The target DNA structures included a DNA structure complexed with the NF-Y transcription factor (pdb (protein data bank)69 code: 4AWL;70 25 nucleobase pairs, sequences: TTCTGAGCCAATCACCGAGCTCGAT and ATCGAGCTCGGTGATTGGCTCAGAA) and a free DNA domain with the pdb code 3US071 (22 nucleobase pairs with the AAACATGTTTATAAACATGTTT and AAACATGTTTATAAACATGTTT sequences). For HSA, chain A of the 2BXD72 protein structure was taken. The structures of the DNA strands and HSA (with the warfarin ligand removed from the 2BXD crystal) equilibrated in cell-like conditions (using molecular dynamics simulations with identical procedures and similar parameters) were, respectively, adopted from the studies of Keshavarz and Mohammad-Aghaei73 and Alavianmehr et al.74The docking simulations were performed using Molegro Virtual Docker (MVD) 5.0 software,75,76 adding the optimized structures (aqueous-phase; see Quantum chemical calculations) of FA, the OA components and the reference drugs as ligands, and considering the ligand-free DNA and HSA structures. Each ligand/target docking simulation was performed by ignoring the possibility of the presence of any other ligand in the binding site. During the simulations, the ligand structures, the DNA strands, and the side chain of HSA were structurally flexible. First, the top ten sterically available regions (probable binding sites; called cavities) of the targets were identified. As MVD basically detects proteins as the docking target and identifies DNA as a cofactor, the cavity searching was manually directed toward the DNA strands by adjusting the cavity search space on them. After that, a spherical search grid with 0.30 Å resolution was adjusted on all identified cavities, and the docking search was restrained to the search grid. The search grid covered almost all parts of the DNA and HSA targets. For each ligand, the docking run was repeated 10 times. At each run, 1500 pose iterations were applied to a maximum of 80 poses (max size) and stopped at the energy threshold of 100 (unitless). One top molecular pose was returned from each run for further analysis. At the end of pose generation, the ligand structure was energy-minimized in the binding site to enhance the energy results. This resulted in 10 of the most energetically feasible ligand/target binding configurations for each ligand and target. After completion of the docking runs, the 10 poses of each ligand corresponding to the 10 runs (ligand/target binding configurations) were inspected visually, the binding site hosting the highest number of strongly binding poses (i.e. the poses giving the lowest MolDock and Rerank scores) was distinguished as the main binding site, and the interaction of the pose giving the lowest binding energy (the overall interaction energy, in addition to the MolDock and Rerank energies) was analyzed with respect to the strength of interaction and the factors affecting the interaction. A description of the docking algorithm (the hybrid guided differential evolution algorithm) and the scoring functions (which are based on piecewise linear potentials) can be found in ref. 75.
Quantum chemical calculations
All quantum chemical calculations were carried out using Gaussian 16 revision A.03.77 For all computations, the ωB97X-D density functional was used in combination with the def2-TZVP78,79 basis set to provide long-range exchange–correlation correction, high accuracy of thermodynamics and chemical kinetics analyses,80–82 and cost-effectiveness.83 The application of the ωB97X-D/def2-TZVP level of theory was justified by taking the experimentally resolved structures of PQ,84 DMBQ,85 HOP,86 CPT,87 and IBPPA,88 optimizing them and comparing the optimized structures to the experimental geometries. As seen in Fig. S7 (ESI†), the selected computational level gives bond lengths as accurate as 0.04 Å for the organic compounds. However, its accuracy error for reproducing the Pt–N bond length of CPT is 0.23 Å, and it predicts the N–Pt–N and Cl–Pt–Cl bond angles with, respectively, 17.1° and 6.8° error. This can be a result of the inefficiency of the ωB97X-D/def2-TZVP level in precise treatment of the Pt transition metal. Therefore, the performance of the selected computational level is not completely acceptable for quantum chemical simulations of CPT, but it can be reliably used for modeling of the studied organic compounds. Notably, experimental structures were not available for all studied compounds. In the case of ThioTEPA, the starting structure was taken from former theoretical studies.31,74 For BaP and BPDE, the overall structure has no specific conformational flexibility, and no reference starting structure was needed. However, CPPP could pose various conformers with close energy values. Because of the nature of its structure (having multiple alkyl and carbocycle groups), we supposed the barrier toward conformational changes to be low in energy and focused on just one conformer (shown in Fig. 1) for simplicity of the calculations.To simulate cellular conditions, cellular ionic strength should be considered. However, as cellular ionic strength varies with extracellular environment,89 all geometry optimizations, (harmonic) frequency and energy calculations (at 310 K and 1 atm), and mechanism explorations were performed in the pure water solvent. We suppose that this approximation is adequate for description of the microenvironment of the evaluated targets, and it does not introduce a large source of error in the energy profiles considering the non-ionic nature of the studied OA components. The aqueous phase was modeled using the SMD polarizable continuum model,90 which has been commonly used as the best solvent model in many recent studies, e.g. see ref. 91–95. This implicit model can outperform explicit water treatments96 and accurately predict reaction free energy barriers.97,98 Accordingly, all energy values were directly extracted from the aqueous-phase properties, with the Gibbs free energies and zero-pint energy (ZPE) corrections being calculated using harmonic oscillator and rigid-rotor partition functions.
For analysis of the OA interaction with DNA, a subset of the studied compounds was selected based on their ligand efficiencies (interactions with DNA and HSA during the docking simulations) and their mutagenicity potential (i.e. BaP and BPDE16–18). Then, the ligand/DNA interactions were analyzed through natural bond-orbital (NBO) analysis on the ligand/DNA complexes obtained from molecular docking (just the binding site and a few additional nucleotides). After the analysis of the NBO results, the main interacting nucleobases were identified, and the ligand/nucleobase complexes were used for mechanism exploration.
To study oxidative stress, we focused on the mechanism suggested by Squadrito et al.,55 which leads to the generation of the O2− radical (and OH; through H2O2 formation) by the intervention of nicotinamide adenine dinucleotide phosphate (NADPH; H− transfer to the organic compounds). The most stable conformers of NADPH and NADP+ were taken from the study of Cao et al.99 and re-optimized under cell-like conditions at the ωB97X-D/def2-TZVP level (see Fig. S8, ESI,† for their optimized structures). To reduce computational costs, we replaced the full NADPH and NADP+ structures with simple models. Fig. S9 (ESI†) compares the geometries of the models with the full experimental and theoretical NADPH and NADP+ structures and confirms that the models resemble the structural features of NADPH and NADP+. Also, the cellular activity of the models was found fairly acceptable by calculating the Gibbs free energy (G) of H− loss from NADPH (NADPH → NADP+ + H−) in cell-like conditions (i.e. aqueous phase, 310 K and 1 atm; 167.9 kJ mol−1 for the model vs. 152.2 kJ mol−1 for NADPH).
After NADPH/NADP+ model approval, configurational sampling100 was used to obtain the lowest energy NADPH/organic compound/water pre-reaction complexes. For this purpose, the aqueous-phase NADPH model, water and organic compounds were imported to the ABCluster 1.4 program101,102 and 200 local minima complexes were created using the artificial bee colony (ABC) algorithm, 700 initial guesses, 200 generation rounds and 4 scout bees. To sort the generated complexes and determine the local minima (LM) structures, the energy of the complexes was computed as the sum of coulombic interactions (using atomic polar tensor (APT) atomic charges; ωB97X-D/def2-TZVP) and Lennard–Jones interactions (CHARMM36 force field103,104). Next, all LM structures were re-optimized using the XTB 6.0.1105 program at the GFN-xTB106 semi-empirical level. Then, a maximum of 35 non-identical structures meeting the 100 kJ mol−1 energy threshold relative to the most stable complex were filtered and re-optimized at the ωB97X-D/def2-TZVP level. The complex giving the lowest G value was chosen as the global minima (GM) structure and processed to the reaction mechanism explorations, along with three to five low-lying LM structures.
For mechanism investigation, various reaction coordinates of the ligand/nucleobase complexes or organic compound/NADPH/water systems were scanned. When no transition state (TS) or reaction intermediate was found, several TSs were guessed. In extreme cases, when a TS was suspected but no TS could be found by the two other approaches, the Synchronous Transit-Guided Quasi-Newton (STQN) method107 (i.e. the QST2 feature of the Gaussian package) was applied to search for the “missing” TSs. Regardless of the approach, all structures were optimized and their harmonic frequencies were computed to label them as intermediates/products/reactions (no imaginary frequencies) or TSs (with imaginary frequency). After that, intrinsic reaction coordinate (IRC)108,109 analysis was carried out on the TSs to follow their corresponding reaction paths and connect them to their associated reactant and product sets.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The computational resources of the CSC-IT Center for Science in Espoo (Finland) and the useful comments of Hanna Vehkamäki and Theo Kurtén are highly appreciated.References
- J. Wang, G. Wang, C. Wu, J. Li, C. Cao, J. Li, Y. Xie, S. Ge, J. Chen and L. Zeng, et al., Enhanced aqueous-phase formation of secondary organic aerosols due to the regional biomass burning over North China Plain, Environ. Pollut., 2020, 256, 113401 CrossRef CAS PubMed.
- A. G. Carlton, C. Wiedinmyer and J. H. Kroll, A review of secondary organic aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 2009, 9(14), 4987–5005 CrossRef CAS.
- M. O. Andreae and P. J. Crutzen, Atmospheric aerosols: biogeochemical sources and role in atmospheric chemistry, Science, 1997, 276(5315), 1052–1058 CrossRef CAS.
- S. Geddes, J. Zahardis and G. A. Petrucci, Chemical transformations of peptide containing fine particles: oxidative processing, accretion reactions and implications to the atmospheric fate of cell-derived materials in organic aerosol, J. Atmos. Chem., 2009, 63(3), 187–202 CrossRef CAS.
- U. Baltensperger, M. Kalberer, J. Dommen, D. Paulsen, M. Rami Alfarra, H. Coe, R. Fisseha, A. Gascho, M. Gysel and S. Nyeki, et al., Secondary organic aerosols from anthropogenic and biogenic precursors, Faraday Discuss., 2005, 130, 265–278 RSC.
- D. J. Jacob, D. L. Mauzerall, J. Martínez Fernández and W. T. Pennell, Global change and air quality, Technical Challenges of Multipollutant Air Quality Management, Springer, Dordrecht, 2011 Search PubMed.
- W.-H. Lien, P. Opiyo Owili, M. Adoyo Muga and T. H. Lin, Ambient particulate matter exposure and under-five and maternal deaths in Asia, Int. J. Environ. Res. Public Health, 2019, 16(20), 3855 CrossRef CAS PubMed.
- K. Donaldson, V. Stone, P. J. A. Borm, L. A. Jimenez, P. S. Gilmour, R. P. F. Schins, A. M. Knaapen, I. Rahman, S. P. Faux and D. M. Brown, et al., Oxidative stress and calcium signaling in the adverse effects of environmental particles (PM10), Free Radical Biol. Med., 2003, 34(11), 1369–1382 CrossRef CAS PubMed.
- U. Pöschl, Atmospheric aerosols: composition, transformation, climate and health effects, Angew. Chem., Int. Ed., 2005, 44(46), 7520–7540 CrossRef PubMed.
- J. A. Bernstein, N. Alexis, C. Barnes, I. L. Bernstein, A. Nel, D. Peden, D. Diaz-Sanchez, S. M. Tarlo and P. B. Williams, Health effects of air pollution, J. Allergy Clin. Immunol., 2004, 114(5), 1116–1123 CrossRef PubMed.
- T. V. Vu and R. M. Harrison, Chemical and physical properties of indoor aerosols, Indoor Air Pollut., 2019, 48, 66 CAS.
- O. Udagawa, A. Furuyama, K. Imai, Y. Fujitani and S. Hirano, Effects of diesel exhaust-derived secondary organic aerosol (SOA) on oocytes: Potential risks to meiotic maturation, Reprod. Toxicol., 2018, 75, 56–64 CrossRef CAS PubMed.
- G. Kiesewetter, W. Schoepp, C. Heyes and M. Amann, Modelling PM2. 5 impact indicators in Europe: health effects and legal compliance, Environ. Model. Software, 2015, 74, 201–211 CrossRef.
- M. Yang, H. Ahmed, W. Wu, B. Jiang and Z. Jia, Cytotoxicity of air pollutant 9,10-phenanthrenequinone: role of reactive oxygen species and redox signaling, BioMed Res. Int., 2018, 2018, 9523968 Search PubMed.
- T. Ito, K. Bekki, Y. Fujitani and S. Hirano, The toxicological analysis of secondary organic aerosol in human lung epithelial cells and macrophages, Environ. Sci. Pollut. Res., 2019, 1–9 Search PubMed.
- N. Zhang, C. Lin, X. Huang, A. Kolbanovskiy, B. E. Hingerty, S. Amin, S. Broyde, N. E. Geacintov and D. J. Patel, Methylation of cytosine at C5 in a CpG sequence context causes a conformational switch of a benzo [a] pyrene diol epoxide-N2-guanine adduct in DNA from a minor groove alignment to intercalation with base displacement, J. Mol. Biol., 2005, 346(4), 951–965 CrossRef CAS PubMed.
- K. Dimitriou and P. Kassomenos, The influence of specific atmospheric circulation types on PM10-bound benzo (a) pyrene inhalation related lung cancer risk in Barcelona, Spain, Environ. Int., 2018, 112, 107–114 CrossRef CAS PubMed.
- Y.-C. Lin, F.-C. Chou, Y.-C. Li, S.-R. Jhang and S. Shangdiar, Effect of air pollutants and toxic emissions from various mileage of motorcycles and aerosol related carcinogenicity and mutagenicity assessment, J. Hazard. Mater., 2019, 365, 771–777 CrossRef CAS PubMed.
- S. Zhou, L. W. Y. Yeung, M. W. Forbes, S. Mabury and J. P. D. Abbatt, Epoxide formation from heterogeneous oxidation of benzo [a] pyrene with gas-phase ozone and indoor air, Environ. Sci.: Processes Impacts, 2017, 19(10), 1292–1299 RSC.
- M. Shiraiwa, K. Ueda, A. Pozzer, G. Lammel, C. J. Kampf, A. Fushimi, S. Enami, A. M. Arangio, J. Fröhlich-Nowoisky and Y. Fujitani, et al., Aerosol health effects from molecular to global scales, Environ. Sci. Technol., 2017, 51(23), 13545–13567 CrossRef CAS PubMed.
- Z. Y. Khan, J. Kettler, H. A. Khwaja, I. I. Naqvi, A. Malik and E. A. Stone, Organic aerosol characterization and source identification in Karachi, Pakistan, Aerosol Air Qual. Res., 2018, 18(10), 2550–2564 CrossRef CAS.
- J. L. Mauderly and J. C. Chow, Health effects of organic aerosols, Inhalation Toxicol., 2008, 20(3), 257–288 CrossRef CAS PubMed.
- P. Li, C. Guo, W. Feng, Q. Sun and W. Wang, A DFT study on the reaction mechanism between tetrachloro-o-benzoquinone and H2O2 and an alternative reaction approach to produce the hydroxyl radical, RSC Adv., 2017, 7(37), 22919–22926 RSC.
- R. S. Borges, A. S. Carneiro, T. G. Barros, C. A. L. Barros, A. M. J. Chaves Neto and A. B. F. da Silva, Understanding the cytotoxicity or cytoprotective effects of biological and synthetic quinone derivatives by redox mechanism, J. Mol. Model., 2014, 20(12), 2541–2547 CrossRef PubMed.
- S. P. Mayalarp, R. H. J. Hargreaves, J. Butler, C. C. O'Hare and J. A. Hartley, Cross-linking and sequence specific alkylation of DNA by aziridinylquinones. 1. Quinone methides, J. Med. Chem., 1996, 39(2), 531–537 CrossRef CAS PubMed.
- K. Kolšek, J. Mavri and M. Sollner, Dolenc, Reactivity of bisphenol A-3, 4-quinone with DNA. A quantum chemical study, Toxicol. In Vitro, 2012, 26(1), 102–106 CrossRef PubMed.
- B. Jawad, L. Poudel, R. Podgornik, N. F. Steinmetz and W. Y. Ching, Molecular mechanism and binding free energy of doxorubicin intercalation in DNA, Phys. Chem. Chem. Phys., 2019, 21(7), 3877–3893 RSC.
- X. M. He and D. C. Carter, Atomic structure and chemistry of human serum albumin, Nature, 1992, 358(6383), 209–215 CrossRef CAS PubMed.
- P. N. Naik, S. A. Chimatadar and S. T. Nandibewoor, Interaction between a potent corticosteroid drug–dexamethasone with bovine serum albumin and human serum albumin: a fluorescence quenching and Fourier transformation infrared spectroscopy study, J. Photochem. Photobiol., B, 2010, 100(3), 147–159 CrossRef CAS PubMed.
- E. L. Mamenta, E. E. Poma, W. K. Kaufmann, D. A. Delmastro, H. L. Grady and S. G. Chaney, Enhanced replicative bypass of platinum-DNA adducts in cisplatin-resistant human ovarian carcinoma cell lines, Cancer Res., 1994, 54(13), 3500–3505 CAS.
- H. Torabifard and A. Fattahi, Mechanisms and kinetics of thiotepa and tepa hydrolysis: DFT study, J. Mol. Model., 2012, 18(8), 3563–3576 CrossRef CAS PubMed.
- I. Vasincu, M. Apotrosoaei, C. Tuchilus, A. T. Pânzariu, O. Dragostin, D. Lupaşcu and L. Profire, New derivatives of aryl-propionic acid. Synthesis and biological evaluation, Rev. Med.-Chir. Soc. Med. Nat. Iasi, 2013, 117, 532–537 Search PubMed.
- H. M. Lazarus, M. D. Reed, T. R. Spitzer, M. Salah Rabaa and J. L. Blumer, High-dose iv thiotepa and cryopreserved autologous bone marrow transplantation for therapy of refractory cancer, Cancer Treat. Rep., 1987, 71(7), 689–695 CAS.
- M. Amir and S. Kumar, Synthesis and evaluation of anti-inflammatory, analgesic, ulcerogenic and lipid peroxidation properties of ibuprofen derivatives, Acta Pharm., 2007, 57(1), 31–45 CAS.
- D. Kaushik, S. Ahmad Khan, G. Chawla and S. Kumar, N′-[(5-chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)methylene] 2/4-substituted hydrazides: Synthesis and anticonvulsant activity, Eur. J. Med. Chem., 2010, 45(9), 3943–3949 CrossRef CAS PubMed.
- N. Shanan, A. Ghasemi Gharagoz, R. Abdel-Kader and H.-G. Breitinger, The effect of pyrroloquinoline quinone and resveratrol on the survival and regeneration of cerebellar granular neurons, Neurosci. Lett., 2019, 694, 192–197 CrossRef CAS PubMed.
- P. H. Chowdhury, Q. He, T. Lasitza Male, W. H. Brune, Y. Rudich and M. Pardo, Exposure of lung epithelial cells to photochemically aged secondary organic aerosol shows increased toxic effects, Environ. Sci. Technol. Lett., 2018, 5(7), 424–430 CrossRef CAS.
- S. T. Pan, Z. L. Li, Z. X. He, J. X. Qiu and S. F. Zhou, Molecular mechanisms for tumour resistance to chemotherapy, Clin. Exp. Pharmacol. Physiol., 2016, 43(8), 723–737 CrossRef CAS PubMed.
- M. S. Faber and U. Fuhr, Time response of cytochrome P450 1A2 activity on cessation of heavy smoking, Clin. Pharmacol. Ther., 2004, 76(2), 178–184 CrossRef CAS PubMed.
- P. Rowland, F. E. Blaney, M. G. Smyth, J. J. Jones, V. R. Leydon, A. K. Oxbrow, C. J. Lewis, M. G. Tennant, S. Modi and D. S. Eggleston, et al., Crystal structure of human cytochrome P450 2D6, J. Biol. Chem., 2006, 281(11), 7614–7622 CrossRef CAS PubMed.
- P. A. Williams, J. Cosme, A. Ward, H. C. Angove, D. M. Vinković and H. Jhoti, Crystal structure of human cytochrome P450 2C9 with bound warfarin, Nature, 2003, 424(6947), 464–468 CrossRef CAS PubMed.
- C. Abad-Zapatero, Ligand efficiency indices for effective drug discovery, Expert Opin. Drug Discovery, 2007, 2(4), 469–488 CrossRef CAS PubMed.
- P. Carloni, M. Sprik and W. Andreoni, Key steps of the cis-platin-DNA interaction: density functional theory-based molecular dynamics simulations, J. Phys. Chem. B, 2000, 104(4), 823–835 CrossRef CAS.
- F. Roodbari, N. Abedi and A. R. Talebi, Early and late effects of Ibuprofen on mouse sperm parameters, chromatin condensation, and DNA integrity in mice, Iran. J. Reprod. Med., 2015, 13(11), 703–710 Search PubMed.
- M. G. Barbosa, B. C. Jorge, J. Stein, D. A. Santos Ferreira, A. C. da Silva Barreto, A. C. Casali Reis, S. Da Silva Moreira, L. C. De Lima Inocencio, L. F. Benitez Macorini and A. C. Arena, Pre-pubertal exposure to ibuprofen impairs sperm parameters in male adult rats and compromises the next generation, J. Toxicol. Environ. Health, Part A, 2020, 1–14 Search PubMed.
- K. A. Wilson, J. L. Garden, N. T. Wetmore, L. R. Felske and S. D. Wetmore, DFT and MD studies of formaldehyde-derived DNA adducts: molecular-level insights into the differential mispairing potentials of the adenine, cytosine, and guanine lesions, J. Phys. Chem. A, 2019, 123(29), 6229–6240 CrossRef CAS PubMed.
- Y. Lai, M. Lu, X. Gao, H. Wu and Z. Cai, New evidence for toxicity of polybrominated diphenyl ethers: DNA adduct formation from quinone metabolites, Environ. Sci. Technol., 2011, 45(24), 10720–10727 CrossRef CAS PubMed.
- M. K. Buening, P. G. Wislocki, W. Levin, H. Yagi, D. R. Thakker, H. Akagi, M. Koreeda, D. M. Jerina and A. H. Conney, Tumorigenicity of the optical enantiomers of the diastereomeric benzo [a] pyrene 7, 8-diol-9, 10-epoxides in newborn mice: exceptional activity of (+)-7beta, 8alpha-dihydroxy-9alpha, 10alpha-epoxy-7, 8, 9, 10-tetrahydrobenzo [a] pyrene, Proc. Natl. Acad. Sci. U. S. A., 1978, 75(11), 5358–5361 CrossRef CAS PubMed.
- H. V. Gelboin, Benzo [alpha] pyrene metabolism, activation and carcinogenesis: role and regulation of mixed-function oxidases and related enzymes, Physiol. Rev., 1980, 60(4), 1107–1166 CrossRef CAS PubMed.
- R. Barhoumi, Y. Mouneimne, I. Awooda, S. H. Safe, K. C. Donnelly and R. C. Burghardt, Characterization of calcium oscillations in normal and benzo [a] pyrene-treated clone 9 cells, Toxicol. Sci., 2002, 68(2), 444–450 CrossRef CAS PubMed.
- P. V. Kiruthiga, R. Beema Shafreen, S. Karutha Pandian, S. Arun, S. Govindu and K. Pandima, Devi, Protective effect of silymarin on erythrocyte haemolysate against benzo (a) pyrene and exogenous reactive oxygen species (H2O2) induced oxidative stress, Chemosphere, 2007, 68(8), 1511–1518 CrossRef CAS PubMed.
- T. M. Penning, S. Tsuyoshi Ohnishi, T. Ohnishi and R. G. Harvey, Generation of reactive oxygen species during the enzymatic oxidation of polycyclic aromatic hydrocarbon trans-dihydrodiols catalyzed by dihydrodiol dehydrogenase, Chem. Res. Toxicol., 1996, 9(1), 84–92 Search PubMed.
- H. Zhao, J. Wei, L. Xiang and Z. Cai, Mass spectrometry investigation of DNA adduct formation from bisphenol A quinone metabolite and MCF-7 cell DNA, Talanta, 2018, 182, 583–589 CrossRef CAS PubMed.
- I. Klopčič and M. Dolenc, Chemicals and drugs forming reactive quinone and quinone imine metabolites, Chem. Res. Toxicol., 2018, 32(1), 1–34 Search PubMed.
- G. Squadrito, R. Cueto, A. Deutsch, W. A. Pryor and B. Dellinger, Damage to human DNA by redox active radicals on PM-2.5, J. Free Radicals Biol. Med., 2001, 31(9), 1132–1138 Search PubMed.
- I. M. Kennedy, The health effects of combustion-generated aerosols, Proc. Combust. Inst., 2007, 31(2), 2757–2770 CrossRef.
- L. Wang, R. Atkinson and J. Arey, Formation of 9, 10-phenanthrenequinone by atmospheric gas-phase reactions of phenanthrene, Atmos. Environ., 2007, 41(10), 2025–2035 CrossRef CAS.
- M. Oginuma, H. Shimada and Y. Imamura, Involvement of carbonyl reductase in superoxide formation through redox cycling of adrenochrome and 9, 10-phenanthrenequinone in pig heart, Chem. – Biol. Interact., 2005, 155(3), 148–154 CrossRef CAS PubMed.
- J. L. Bolton, M. A. Trush, T. M. Penning, G. Dryhurst and T. J. Monks, Role of quinones in toxicology, Chem. Res. Toxicol., 2000, 13(3), 135–160 Search PubMed.
- Y. Demir, M. S. Özaslan, H. E. Duran, Ö. İrfan Küfrevioğlu and Ş. Beydemir, Inhibition effects of quinones on aldose reductase: antidiabetic properties, Environ. Toxicol. Pharmacol., 2019, 70, 103195 CrossRef CAS PubMed.
- S. Fuzzi, S. Decesari, M. C. Facchini, E. Matta, M. Mircea and E. Tagliavini, A simplified model of the water soluble organic component of atmospheric aerosols, Geophys. Res. Lett., 2001, 28(21), 4079–4082 CrossRef CAS.
- G. McFiggans, M. Rami Alfarra, J. Allan, K. Bower, H. Coe, M. Cubison, D. Topping, P. Williams, S. Decesari, C. Facchini and S. Fuzzi, Simplification of the representation of the organic component of atmospheric particulates, Faraday Discuss., 2005, 130, 341–362 RSC.
- C. D. Hatch, K. M. Gierlus, J. D. Schuttlefield and V. H. Grassian, Water adsorption and cloud condensation nuclei activity of calcite and calcite coated with model humic and fulvic acids, Atmos. Environ., 2008, 42(22), 5672–5684 CrossRef CAS.
- S. H. Kessler, T. Nah, K. E. Daumit, J. D. Smith, S. R. Leone, C. E. Kolb, D. R. Worsnop, K. R. Wilson and J. H. Kroll, OH-initiated heterogeneous aging of highly oxidized organic aerosol, J. Phys. Chem. A, 2012, 116(24), 6358–6365 CrossRef CAS PubMed.
- D. Kwon, M. J. Sovers, V. H. Grassian, P. D. Kleiber and M. A. Young, Optical properties of humic material standards: solution phase and aerosol measurements, ACS Earth Space Chem., 2018, 2(11), 1102–1111 CrossRef CAS.
- N. Borduas-Dedekind, R. Ossola, R. O. David, L. S. Boynton, V. Weichlinger, Z. A. Kanji and K. McNeill, Photomineralization mechanism changes the ability of dissolved organic matter to activate cloud droplets and to nucleate ice crystals, Atmos. Chem. Phys., 2019, 19(19), 12397–12412 CrossRef CAS.
- B. Sadhu, M. Sundararajan and T. Bandyopadhyay, Water-mediated differential binding of strontium and cesium cations in fulvic acid, J. Phys. Chem. B, 2015, 119(34), 10989–10997 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, The protein data bank, Nucleic Acids Res., 2000, 28(1), 235–242 CrossRef CAS PubMed.
- M. Nardini, N. Gnesutta, G. Donati, R. Gatta, C. Forni, A. Fossati, C. Vonrhein, D. Moras, C. Romier and M. Bolognesi, et al., Sequence-specific transcription factor NF-Y displays histone-like DNA binding and H2B-like ubiquitination, Cell, 2013, 152(1–2), 132–143 CrossRef CAS PubMed.
- C. Chen, N. Gorlatova and O. Herzberg, Pliable DNA conformation of response elements bound to transcription factor p63, J. Biol. Chem., 2012, 287(10), 7477–7486 CrossRef CAS PubMed.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, Structural basis of the drug-binding specificity of human serum albumin, J. Mol. Biol., 2005, 353(1), 38–52 CrossRef CAS PubMed.
- F. Keshavarz and D. Mohammad-Aghaie, Dual-target anticancer drug candidates: Rational design and simulation studies, Phys. Chem. Res., 2015, 3(2), 125–143 Search PubMed.
- M. M. Alavianmehr, R. Yousefi, F. Keshavarz and D. Mohammad-Aghaie, Probing the binding of thioTEPA to human serum albumin using spectroscopic and molecular simulation approaches, Can. J. Chem., 2014, 92(11), 1066–1073 CrossRef CAS.
- R. Thomsen and M. H. Christensen, MolDock: a new technique for high-accuracy molecular docking, J. Med. Chem., 2006, 49(11), 3315–3321 CrossRef CAS PubMed.
- A. P. S. Molegro, MVD 5.0 Molegro Virtual Docker, DK-8000 Aarhus C, Denmark, 2011 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian 6, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8(9), 1057–1065 RSC.
- W.-L. Chen, C.-M. Hsieh, L. Yang, C.-C. Hsu and S.-T. Lin, A critical evaluation on the performance of COSMO-SAC models for vapor–liquid and liquid–liquid equilibrium predictions based on different quantum chemical calculations, Ind. Eng. Chem. Res., 2016, 55(34), 9312–9322 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10(44), 6615–6620 RSC.
- D. K. Singh, B. Rathke, J. Kiefer and A. Materny, Molecular structure and interactions in the ionic liquid 1-ethyl-3-methylimidazolium trifluoromethanesulfonate, J. Phys. Chem. A, 2016, 120(31), 6274–6286 CrossRef CAS PubMed.
- J. M. Voss, B. M. Marsh, J. Zhou and E. Garand, Interaction between ionic liquid cation and water: infrared predissociation study of [bmim]+·(H2O)n clusters, Phys. Chem. Chem. Phys., 2016, 18(28), 18905–18913 RSC.
- S. Y. Matsuzaki, M. Gotoh and A. Kuboyama, Molecular and Crystal Structure of the α and β Forms of 9, 10-Phenanthrenequinone, Mol. Cryst. Liq. Cryst., 1987, 142, 127–139 CrossRef CAS.
- D. Rabinovich and G. M. J. Schmidt, 387. Topochemistry. Part V. The crystal Structure of 2, 5-Dimethyl-1, 4-Benzoquinone, J. Chem. Soc., 1964, 2030–2040 RSC.
- G. W. Smith, The Crystal and Molecular Structure of 29-nor-17αH-Hopane, C29H50, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 526–530 CrossRef.
- P. Carloni, M. Sprik and W. Andreoni, Key Steps of the Cis-Platin-DNA Interaction: Density Functional Theory-Based Molecular Dynamics Simulations, J. Phys. Chem. B, 2000, 104, 823–835 CrossRef CAS.
- A. A. Freer, J. M. Bunyan, N. Shankland and D. B. Sheen, Structure of (S)-(+)-Ibuprofen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 1378–1380 CrossRef.
- B. Liu, B. Poolman and A. J. Boersma, Ionic strength sensing in living cells, ACS Chem. Biol., 2017, 12(10), 2510–2514 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. J. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- K. Kříž and J. Řezáč, Reparametrization of the COSMO solvent model for semiempirical methods PM6 and PM7, J. Chem. Inf. Model., 2019, 59(1), 229–235 CrossRef PubMed.
- P. P. Fehér and A. Stirling, Assessment of reactivities with explicit and implicit solvent models: QM/MM and gas-phase evaluation of three different Ag-catalysed furan ring formation routes, New J. Chem., 2009, 43(39), 15706–15713 RSC.
- G. Mahmoudzadeh, R. Ghiasi and H. Pasdar, Solvent influence on structure and electronic properties of Si2Me4: a computational investigation using PCM-SCRF method, Russ, J. Phys. Chem. A, 2019, 93(11), 2244–2249 Search PubMed.
- R. Mera-Adasme, M. Caroli Rezende and M. Domínguez, On the physical-chemical nature of solvent polarizability and dipolarity, Spectrochim. Acta, Part A, 2020, 229, 118008 CrossRef CAS PubMed.
- I. G. Shenderovich and G. B. Denisov, Adduct under field—A qualitative approach to account for solvent effect on hydrogen bonding, Molecules, 2020, 25(3), 436 CrossRef CAS PubMed.
- J. Chen, Y. Shao and J. Ho, Are explicit solvent models more accurate than implicit solvent models? A case study on the Menschutkin reaction, J. Phys. Chem. A, 2019, 123(26), 5580–5589 CrossRef CAS PubMed.
- E. L. M. Miguel, C. I. L. Santos, C. M. Silva and J. R. Pliego Jr, How accurate is the SMD model for predicting free energy barriers for nucleophilic substitution reactions in polar protic and dipolar aprotic solvents?, J. Braz. Chem. Soc., 2016, 27(11), 2055–2061 CAS.
- H. Ostovari, E. Zahedi, I. Sarvi and A. Shiroudi, Kinetic and mechanistic insight into the formation of amphetamine using the Leuckart–Wallach reaction and interaction of the drug with GpC·CpG base-pair step of DNA: a DFT study, Monatsh. Chem., 2018, 149(6), 1045–1057 CrossRef CAS.
- X. Cao, L. Wu, J. Zhang and M. Dolg, Density functional studies of coenzyme NADPH and its oxidized form NADP+: structures, UV-Vis spectra, and the oxidation mechanism of NADPH, J. Comput. Chem., 2020, 41(4), 305–316 CrossRef CAS PubMed.
- J. Kubecka, V. Besel, T. Kurtén, N. Myllys and H. Vehkamäki, Configurational sampling of noncovalent (atmospheric) molecular clusters: sulfuric acid and guanidine, J. Phys. Chem. A, 2019, 123(28), 6022–6033 CrossRef CAS PubMed.
- J. Zhang and M. Dolg, ABCluster: the artificial bee colony algorithm for cluster global optimization, Phys. Chem. Chem. Phys., 2015, 17(37), 24173–24181 RSC.
- J. Zhang and M. Dolg, Global optimization of clusters of rigid molecules using the artificial bee colony algorithm, Phys. Chem. Chem. Phys., 2016, 18(4), 3003–3010 RSC.
- A. D. MacKerell Jr, D. Bashford, M. L. D. R. Bellott, R. L. Dunbrack Jr, J. D. Evanseck, M. J. Field and S. Fischer, et al., All-atom empirical potential for molecular modeling and dynamics studies of proteins, J. Phys. Chem. B, 1998, 102(18), 3586–3616 CrossRef PubMed.
- I. S. Gutiérrez, F.-Y. Lin, K. Vanommeslaeghe, J. A. Lemkul, K. A. Armacost, C. L. Brooks III and A. D. MacKerell Jr, Parametrization of halogen bonds in the CHARMM general force field: Improved treatment of ligand–protein interactions, Bioorg. Med. Chem., 2016, 24(20), 4812–4825 CrossRef PubMed.
- S. Grimme, C. Bannwarth and P. Shushkov, A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z = 1–86), J. Chem. Theory Comput., 2017, 13(5), 1989–2009 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions, J. Chem. Theory Comput., 2019, 15(3), 1652–1671 CrossRef CAS PubMed.
- C. Peng and H. B. Schlegel, Combining synchronous transit and quasi-Newton methods to find transition states, Isr. J. Chem., 1993, 33(4), 449–454 CrossRef CAS.
- K. Fukui, The path of chemical reactions-the IRC approach, Acc. Chem. Res., 1981, 14(12), 363–368 CrossRef CAS.
- H. P. Hratchian and H. B. Schlegel, Theory and Applications of Computational Chemistry: The First 40 Years, Elsevier, Amsterdam, 2005, pp. 195–249 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The output files for all reported structures, the results of method validation, molecular docking and NBO analysis, steps II and IV of the oxidative stress mechanism, and the disapproved BPDE–DNA adduct formation reaction mechanism. See DOI: 10.1039/d1nj00231g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |