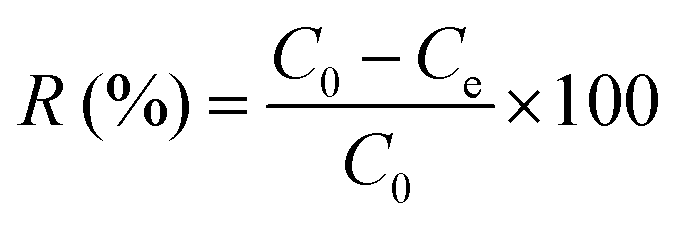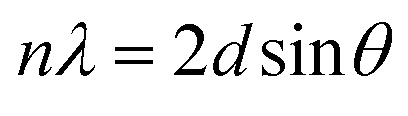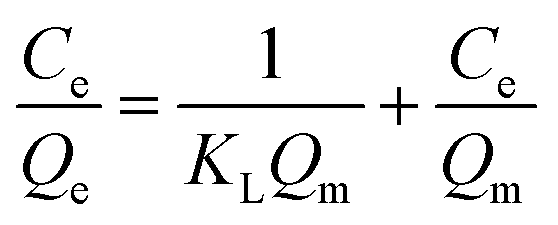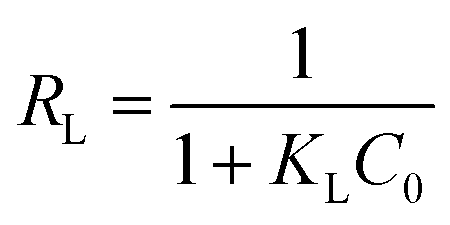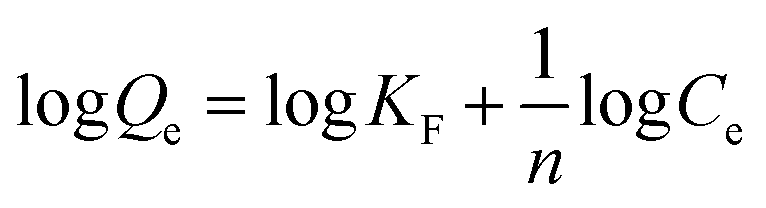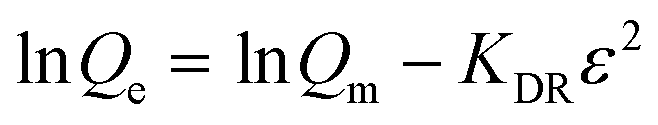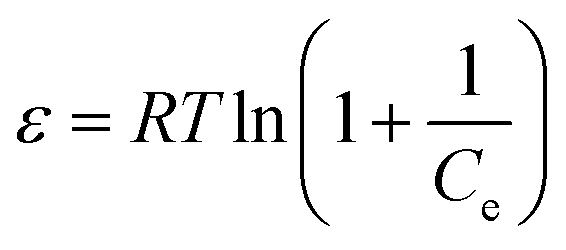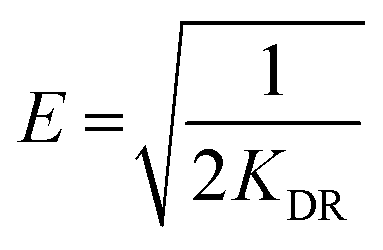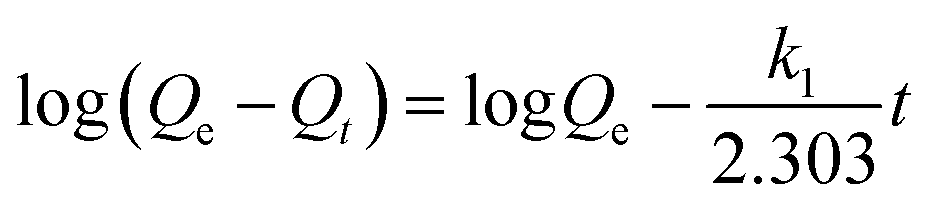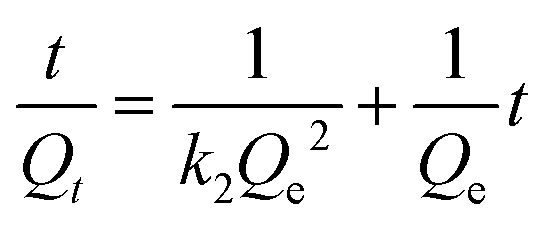DOI:
10.1039/D0NJ04277C
(Paper)
New J. Chem., 2021,
45, 268-281
Highly efficient and antibacterial ion exchanger based on graphene oxide for removal of chromate and nitrate from water: synthesis, characterization and application
Received
25th August 2020
, Accepted 18th November 2020
First published on 24th November 2020
Abstract
In this study, a new recyclable antibacterial anion exchanger based on graphene oxide (GO) and quaternary ammonium chloride (TMSQA) as a cross-linker/anion exchanger was synthesized and used for the separation of chromate and nitrate from water, which to the best of our knowledge has not been investigated before. The prepared anion exchanger was characterized by techniques such as FE-SEM, energy dispersive X-ray (EDX) analysis, FT-IR, TGA, XRD, and BET, and also its recyclability and antibacterial activity were investigated. The highest removal of NO3− and CrO42− in aqueous solutions containing 30 and 50 mg L−1 initial concentration of these ions, respectively, was 98% (29.3 mg g−1) and 100% (50 mg g−1) at pH 7 after 45 and 15 min contact time. The obtained experimental data were well fitted to the Langmuir isotherm and the pseudo-second-order rate kinetics. The exchanger (GO/TMSQA: 1/2) showed good antibacterial activity against Gram-positive B. subtilis and Gram-negative E. coli and showed no significant loss of exchangeability after four adsorption–desorption cycles. The removal of single valent nitrate was considerably inhibited in the presence of chromate, showing the superior binding of the bivalent chromate. The prepared anion exchanger has also shown suitable selectivity against sulfate and chloride ions as competing ions.
1. Introduction
Nowadays, groundwater quality is deteriorating as a result of increasing population, industrialization, agricultural activities, and environmental changes. Scientists have identified and introduced thousands of organic, inorganic, and biological compounds as contaminants of water resources. In the inorganic category, nitrates and chromates are dangerous pollutants for humans, the environment, and other aquatic organisms.1,2 The chromate and nitrate ions are completely soluble in water, they are transmitted easily in nature and because of their usage in industrial and manufacturing activities such as tanning and leather industries, paints, crafts, glass, electroplating, and agricultural activities, as well as their presence in chemical and animal fertilizers, and industrial and domestic wastewater, have been contaminating water sources. They can accumulate in the living organisms that cause severe poisoning, and are transmitted through the food cycle to other organisms. The chromium ion is present in two common oxidation modes: Cr(III), which is useful in moderate amounts, and Cr(VI), which is defined as a toxic and carcinogenic agent. Cr(VI) is found in the form of chromate (CrO42−) and dichromate (Cr2O72−) which depends on the pH of the solution.2–5
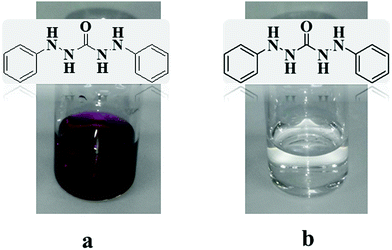
Nitrate and Cr(VI) ions, when present in more than a certain amount, can cause genetic mutations, stomach cancer, cutaneous diseases, respiratory, liver, and kidney problems, etc. For these reasons, the World Health Organization (WHO) has specified the maximum acceptable concentration of 50 mg L−1 for nitrate ions and 0.1 mg L−1 for chromium ions.6,7 Therefore, the anions before return to the environment should be removed from water sources. There are several elimination methods such as reverse osmosis, adsorption, chemical precipitation, biological degradation, electro-dialysis, and ion exchange.8,9 In the present work, the ion exchange method was chosen for its simplicity, effectiveness, cheapness, and reusability.1 In the ion exchange process one needs to select an appropriate adsorbent with the capability of ion exchange. Thus, according to the literature, quaternary ammonium compounds with chlorine anions are good candidates for the ion exchange process.10,11 These compounds with a positive charge on their surface and commonly known as antibacterial substances are active against both Gram-positive and Gram-negative bacteria.12–14 Nowadays, graphene oxide (GO) is modified by many organic compounds due to its specific surface area, low-cost, and numerous oxygen-containing functional groups such as hydroxyl and carboxyl groups. GO-Based sorbents can be prepared from graphite oxidation by potassium permanganate.15,16
The synthesis and application of graphene oxide-based materials have attracted considerable attention in environmental treatment strategies. In the previous work,8 we prepared an anion exchanger from gum tragacanth (GT) to remove nitrate ions from water. In the present study, we used graphene oxide (GO) as an abundant and inexpensive biodegradable and non-toxic material to synthesis a new antibacterial anion exchanger for the removal of CrO42− and NO3− ions from water. To the best of our knowledge, this work has not been reported before. In this study, the effect of interfering ions such as Cl− and SO42− on the removal of nitrate and chromate from water has also been investigated. In particular, the choice of water-dispersible GO is due to the presence of polar functional chemical groups such as epoxide and hydroxyl on the two sides of a single layer and carboxylic terminal groups at the edges which enables the formation of hydrogen bonds with hydrophilic molecules and gives rise to their chemical reactivity and formation of covalent linkages with TMSQA molecules. TMSQA was synthesized from the reaction between N-(trimethylsilyl)imidazole (NTMSI) and (3-chloropropyl)trimethoxysilane (CPTS), characterized, and used as a cross-linker in GO and also as an anion exchanger. The prepared GO-based anion exchanger was characterized by using FE-SEM, energy dispersive X-ray (EDX) analysis, FT-IR, TGA, X-ray diffraction (XRD), and BET, and tested for recyclability and antibacterial activity against Gram-negative and Gram-positive bacteria. We have also investigated the removal of chromium(VI) (CrO42−) and nitrate (NO3−) ions from aqueous solution at different pH values, initial anion concentration, exchanger dosage, and contact time, and in the presence of competing ions such as sulfate and chloride. Furthermore, the removal mechanism was evaluated by using several isotherms and kinetic models and the obtained results were compared with previous work.
2. Experimental section
2.1. Materials
Graphite (purity 99.5%), N-(trimethylsilyl)imidazole (NTMSI), (3-chloropropyl)trimethoxysilane (CPTS), potassium and sodium nitrate, toluene, ethanol, acetone, hydrochloric acid (37%), hydrogen peroxide (25%), potassium permanganate, sulfuric acid (98%), potassium sulfate, potassium chloride, and phosphoric acid (85%) were obtained from Merck Co. (Germany). Potassium chromate and 1,5-diphenylcarbazide were purchased from Fluka Co. All the purchased materials were utilized without any further purification.
2.2. Synthesis
2.2.1. Synthesis of trimethoxysilane quaternary ammonium salt (TMSQA).
NTMSI (3 mL, 20 mmol) was added into a 100 mL two-necked flask containing CPTS (11 mL, 60 mmol) and anhydrous toluene (10 mL, 94.4 mmol). The mixture was stirred at 110 °C for 24 h under an inert nitrogen atmosphere, as reported before.8 The reaction mixture was decanted into a mixture of acetonitrile and n-hexane three times. The n-hexane was discharged, and the acetonitrile phase contained product was evaporated. The sediment remaining was washed with diethylether (3 × 10 mL) several times and the obtained white precipitate was dried in a vacuum oven at 50 °C.
2.2.2. Preparation of graphene oxide.
Graphene oxide (GO) was synthesized by using the Hummers–Offerman method from 2.5 g of graphite powder and 1.25 g of NaNO3 in a flask containing acidic solution (H2SO4, 98%/H3PO4, 85%) (54/6 mL, 9/1 v/v) in an ice bath for 10 min. Then 7.5 g of KMnO4 was gently added to the mixture and stirred at 5 °C for 2 h. The temperature of the reaction mixture was raised gradually to the ambient temperature, and then to 40 °C for one hour and to 98 °C for one hour. After that, deionized water was added to the reaction mixture to give a volume of 200 mL and then the mixture was treated with 7.5 mL of hydrogen peroxide. The mixture was centrifuged and rinsed with deionized water and chlorine solution (5%) several times to achieve a neutral pH. The obtained solid was washed with acetone, centrifuged, and dried in a vacuum oven at 60 °C.17
2.2.3. Synthesis of the GO-based anion exchanger.
The synthesized GO (0.1 g) was first ultrasonicated in 30 mL of dry toluene for 45 min to achieve a dispersed suspension. Then, a certain amount of the synthetic ammonium salt (TMSQA) (0.05, 0.1, 0.2 g) was added to the prepared suspension of GO. The mixture was stirred at 110 °C for 48 h to induce the reaction between the hydroxyl groups of GO and trimethoxysilane of TMSQA.18 After completion of the reaction, the precipitate was separated, washed in distilled water first and then in ethanol several times, and dried in an oven at 50 °C.
2.3. Instruments
The Fourier transform infrared (FTIR) spectra of GO and TMSQA and GO-based anion exchanger were monitored in the range of 900 and 4000 cm−1 using a Bruker Tensor 27 FTIR spectrometer (Bruker, Karlsruhe, Germany). The morphology, elemental analysis, and percentage distribution of elements of the GO-based anion exchanger were evaluated using Field Emission Scanning Electron Micrographs (FESEM) (ZEISS Company, Germany) and energy dispersive X-Ray (EDX) mapping analysis. The X-ray diffraction (XRD) spectra were recorded on an XRD D8-Advance instrument (Bruker) using monochromatic CuKα (λ = 1.54060 Å) in 2θ = 5–80°. The thermal stability of the GO-based anion exchanger was evaluated by thermogravimetric analysis (TGA) using a TGA/DTA:STA 504 instrument at a heating rate of 10 °C min−1 and under a nitrogen atmosphere. The pH value adjustments were made by using a digital pH meter 766 calimatic (Knick). Ultraviolet-Visible (UV-Vis) spectra were collected to determine NO3− and CrO42− ion concentration in aqueous solution using a Unico 4802 UV-Vis double beam spectrophotometer. The Brunauer–Emmett–Teller (BET) analysis (model: belsorp mini II, Japan) was used to determine the specific surface area, pore volume, and pore size of the prepared anion exchanger. After degassing the samples at 100 °C, the N2 adsorption–desorption process was performed at 77 K.
2.4. Adsorption studies
A series of batch experiments were performed to investigate the separation of nitrate and chromate ions from water using the prepared GO-based anion exchanger. For this purpose, stock solutions of nitrate (1000 mg L−1) and chromate (500 mg L−1) were prepared from potassium nitrate and potassium chromate in deionized water, respectively. Various concentrations of chromate and nitrate ions were prepared by diluting the stock solutions with deionized water. The exchange of nitrate and chromate ions by the anion exchanger was investigated using 20 mL aqueous solution containing various concentrations of nitrate (20–100 mg L−1) and chromate (0.5–100 mg L−1) ions. The ion exchange experiments were carried out in 20 mL aqueous solution and the parameters affecting the exchange process such as the contact time (10–90 min), exchanger dosage (5–40 mg), initial ion concentration (20–100 mg L−1 for NO3− and 0.5–100 mg L−1 for CrO42−), and pH (3–11) were investigated. The pH of the solution was adjusted to the required value using 0.1 N NaOH or 0.1 N HCl solutions. When a particular parameter was under investigation, the other parameters were kept constant. The initial tests at the adjusted pH value indicated that the required time to exchange nitrate and chromate ions was 45 and 15 min, respectively. After the removal of the anion exchanger from the solution by centrifugation, the concentration of the residual ions in the solution was determined by using a UV-visible spectrophotometer. The conformity of the obtained data was checked with the standard curve of analyte solutions at the standard concentrations. The nitrate ion concentration was determined in acidic conditions by adding 0.4 mL of HCl (1 N) and measuring the absorption at 220 and 275 nm, using eqn (1):19| | | A(NO3−) = A(220nm) – 2A(275nm) | (1) |
The chromium ion concentration was measured at λ = 540 nm using the reported procedure.20,21 0.25 g of 1,5-diphenylcarbazide was dissolved in 50 mL of acetone, and each test cell contained 2 mL of chromate solution, 1 mL of 1,5-diphenylcarbazide solution, and 1 mL of sulfuric acid solution (0.2 N). The chromate ion concentration was also determined by using a UV-visible spectrophotometer. The presence of chromate ions was shown by a strong purple color (Fig. 1a) due to the formation of a complex between chromium(VI) and 1,5-diphenylcarbazide. The purple color completely disappeared and a clear solution was observed after using the anion exchanger (Fig. 1b). Furthermore, the existence of these two ions could be determined simultaneously through the UV-Vis spectrophotometer.2,22,23 The removal percentage (R%) of NO3− and CrO42− ions and the equilibrium capacity (Qe, mg g−1) of the ion exchange were calculated according to eqn (2) and (3), respectively:| | 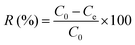 | (2) |
| | 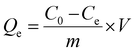 | (3) |
where C0 and Ce are the general symbols for the initial and equilibrium concentration of NO3− and CrO42− ions in solution, respectively, m is the weight of the adsorbent (g), and V is the volume of the solution (L). To verify the accuracy of the test data, each ion-exchange experiment was repeated three times to minimize the experimental error.
 |
| | Fig. 1 (a) 50 mg L−1 chromate solution in the presence of 1,5-diphenylcarbazide and (b) after addition of the anion exchanger and removal of chromium ions. | |
2.5. Regeneration of the GO-based anion exchanger
The regeneration and reuse of the exchanger are very important from the green chemistry perspective. Therefore, the effective reagents can be used such as alkaline and acidic treatment, to strip nitrate and chromate ions from the exhausted adsorbent.24–27 In this work, sodium hydroxide was used to study the desorption efficiency of the prepared anion exchanger. For this purpose, 0.03 and 0.02 g of exchanger in 20 mL of nitrate (30 mg L−1) and chromate (50 mg L−1) solution were stirred for 45 and 15 minutes, respectively. The exchangers were filtered and added to a 20 mL hydroxide solution (0.15 N) and stirred for 30 min. Finally, the exchangers were washed with deionized water twice.21,25 The treated exchangers were dried before their reuse in the exchange experiments.
2.6. Competing ions’ effect
Sulfate and chloride ions are the main ions competing with chromate and nitrate ions, which exist in high concentrations in industrial and agricultural wastewater.24,28 The presence of these ions in wastewater competes with the adsorption of chromate and nitrate anions during the exchange process. To investigate the effect of competing ions on the efficiency of the prepared ion exchanger, the experimental tests were carried out in separate aqueous solutions of nitrate and chromate anions and in solutions with the co-existence of these competing anions. The experiments were carried out in the 500–2000 mg L−1 range of sulfate and chloride concentration using 20 mg of exchanger in a 20 mL solution at pH 7, and the results are shown in Fig. 8b and c.
2.7. Antibacterial activity
The antibacterial performance of the prepared GO-based anion exchanger was studied in the presence of Gram-negative (E. coli) and Gram-positive (S. aureus, B. subtilis) bacteria using the Kirby–Bauer technique.29 Gentamicin and chloramphenicol were used as standard antibacterial drugs. Antimicrobial activity was assessed by measuring the diameter of the inhibition growth zone (mm) on the surface of the agar plates, and the results of this study were reported as mean ± standard deviation after three repetitions of the experiment.
3. Results and discussion
3.1. Synthesis and characterization of the GO-based anion exchanger
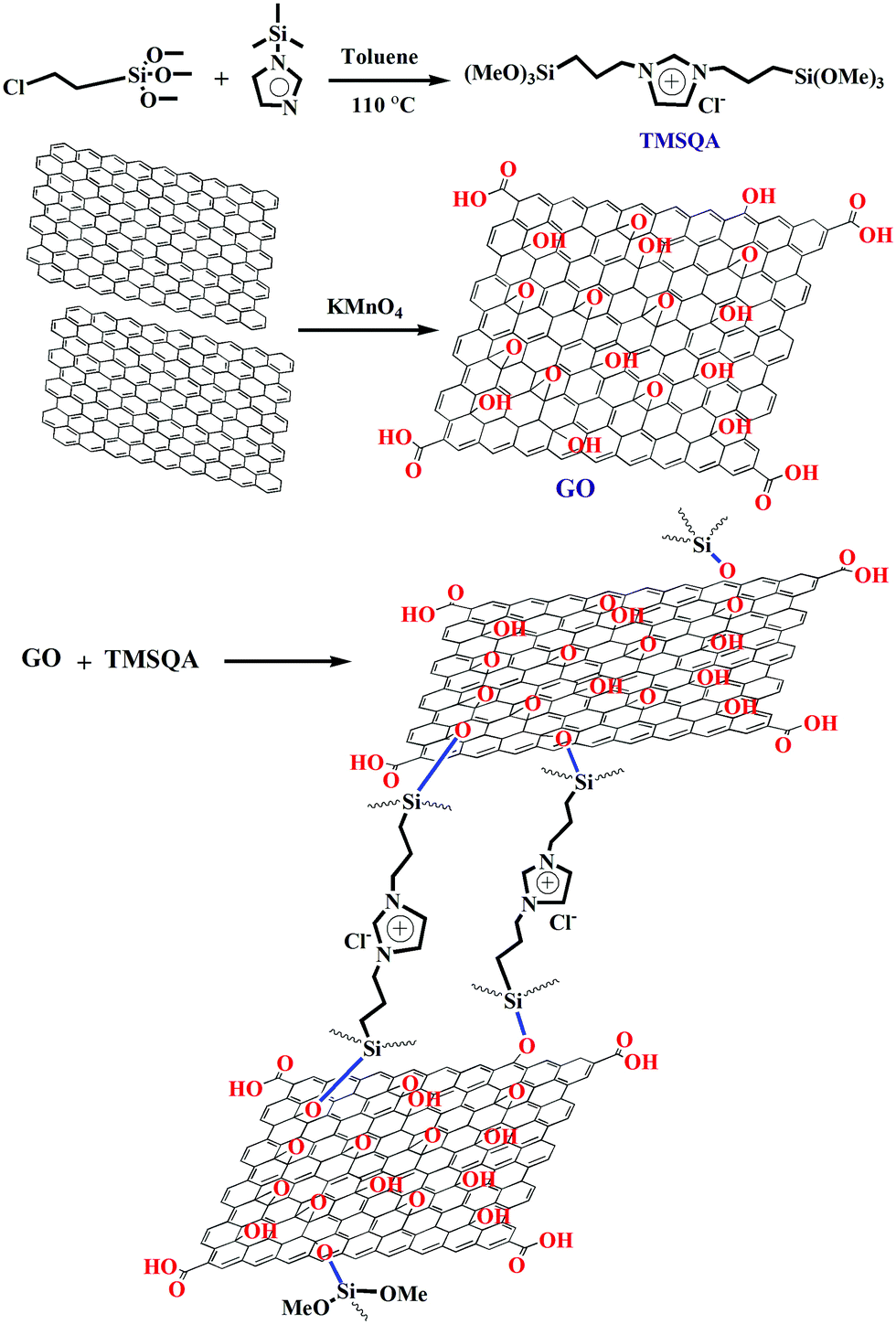
TMSQA was synthesized in toluene by the substitution reaction of CPTS with NTMSI. The nitrogen atom of imidazole is quaternized with chlorine to improve the ionic character of TMSQA. In this work, as shown schematically in Scheme 1, TMSQA molecules containing trimethoxysilane functional groups are able to react with the hydroxyl groups on the GO surface and form crosslinks between the GO plates, and the ion-exchangeable groups are also preserved in the quaternary ammonium chloride. To prepare the GO-based ion exchanger, a certain amount of TMSQA was added to the GO suspension and the mixture was heated up to 110 °C and stirred vigorously (for 48 h) to enhance the short-range interconnections among functional groups and the mobility of the formed macromeres. The crosslinking reactions lead to the formation of local networks and the viscosity of the reaction mixture progressively increases. The mobility of macromeres decreases as crosslinking proceeds, this allows closer functional groups to react more efficiently. The resulting physico-chemical condition promotes the ‘welding’ between the GO plates’ surfaces, giving rise to the three-dimensional macrostructure. The GO-based ion exchangers stabilize in the aqueous medium due to the interactions with the functional groups on the surface of the GO surface or the edges. The use of quaternary ammonium compounds as an anion exchanger for nitrate and chromate ions has also been reported in the work of E. Alcalde, K. Li, and Z. Kong.11,30,31 The adsorption of both tested anions (NO3− and CrO42−) by the pure TMSQA was approximately 100%. This suggests that the quaternary ammonium salts can remove anions from water through the ion exchange process and electrostatic attraction. The pure GO (unmodified) showed no measurable removal of the tested anions from the aqueous solution which can be due to the neutrality of GO or the presence of carboxylate anions on the surface which cause repulsion of the tested anions.
 |
| | Scheme 1 Synthesis of the GO-based anion exchanger and putative matrix chemical structure of crosslinking. | |
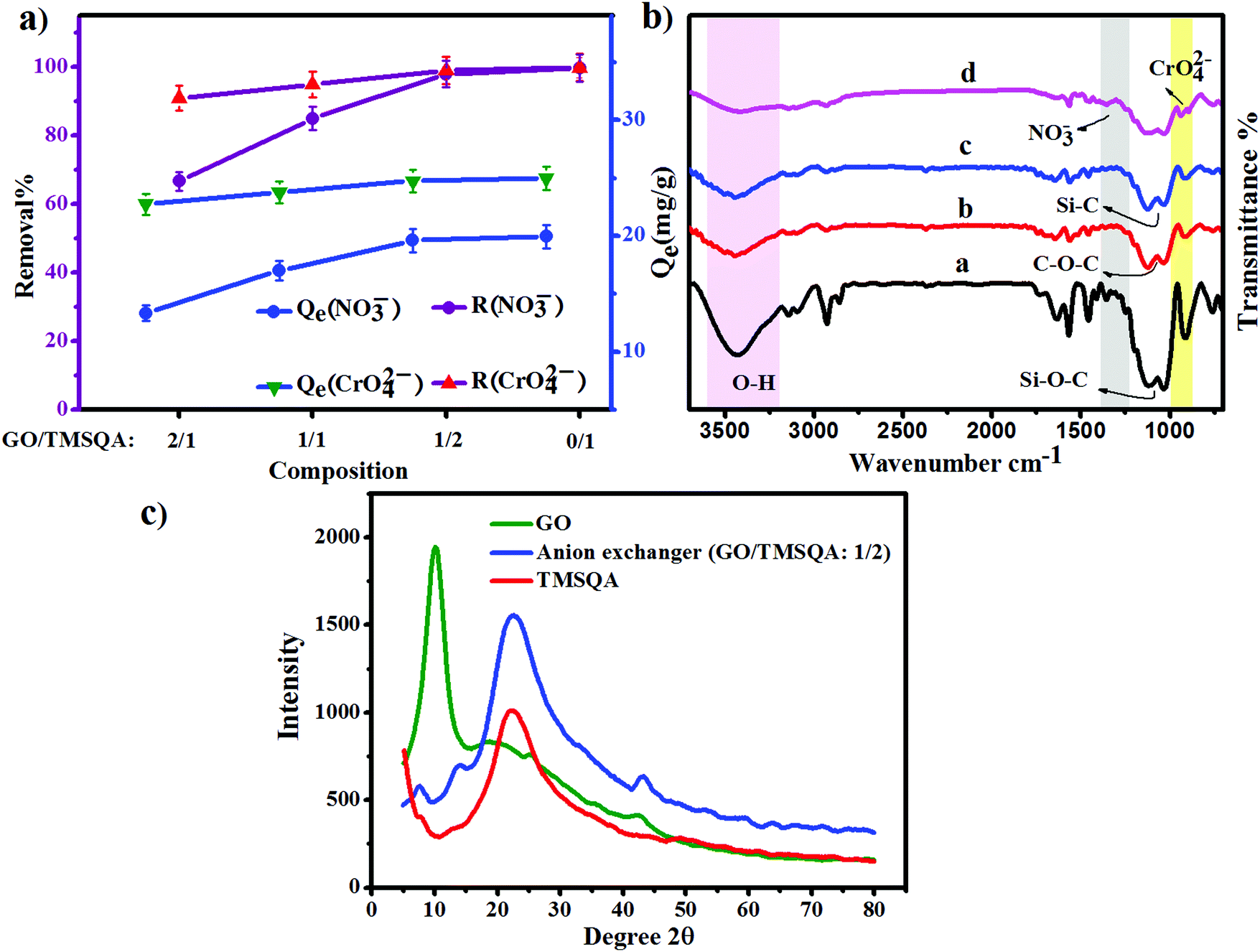
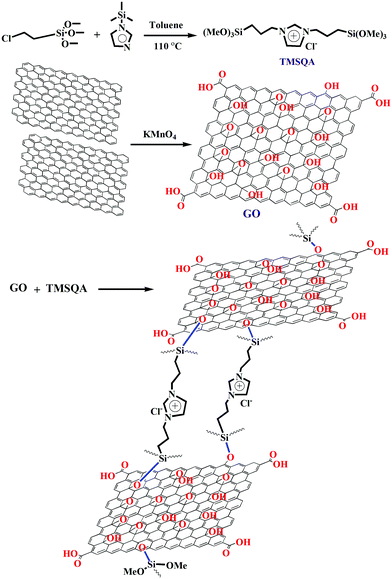
To obtain a material with reasonable anion exchangeability, different weight ratios of GO/TMSQA (2/1, 1/1, and 1/2) were investigated for the removal efficiency of NO3− and CrO42− ions using 20 mL aqueous solution containing 20 mg of exchanger, 20 mg L−1 NO3−, and 25 mg L−1 CrO42− for 30 and 20 min contact time, respectively, at pH 7. The results of this study are shown in Fig. 2a. As can be seen, the removal efficiency increased with the increasing amount of TMSQA, and the composition GO/TMSQA: 1/2 showed the highest exchange efficiency which can be due to the existence of a greater number of exchangeable ions (Cl−). This suggests that TMSQA used as a cross-linker for GO acted also as an efficient exchanger for the removal of the anions from the polluted water. In comparison with the composition GO/TMSQA: 2/1, the anion exchanger with the composition GO/TMSQA: 1/2 increased the removal percentage of the NO3− ions significantly (from 66.6% to 97%), while the removal percentage of chromate ions increased slightly (from 91% to 99%). The removal efficiency of chromate ions was not affected much by the composition of the exchanger. This can be due to the larger ionic radius of CrO42− ions.2 Therefore, the anion exchanger GO/TMSQA: 1/2 was selected for further experiments. These results also confirm that the main adsorption mechanism of the tested anions by the pure TMSQA and the GO modified with TMSQA is the electrostatic attraction which can be due to the positively charged species on the surface and in the matrix.11,30,31
 |
| | Fig. 2 (a) Effect of composition of the anion exchanger on removal percent; (b) FT-IR spectra: (a) TMSQA, (b) GO, (c and d) anion exchanger (GO/TMSQA: 1/2) before and after adsorption of nitrate and chromate; (c) XRD patterns of GO, anion exchanger (GO/TMSQA: 1/2) and TMSQA. | |
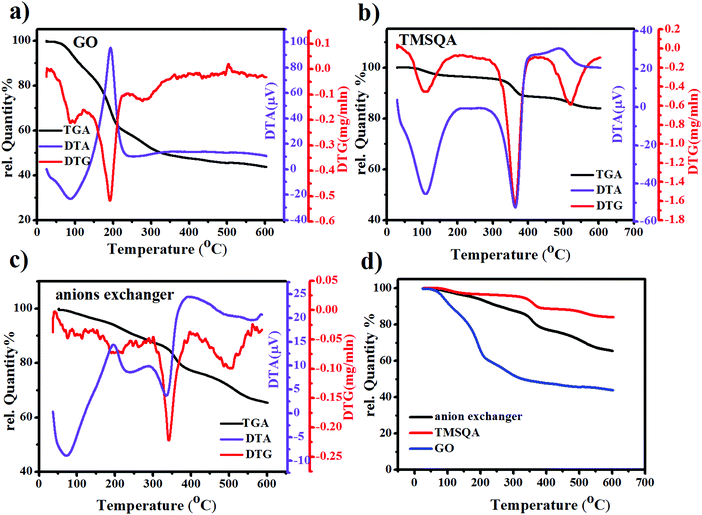
The synthesis of the anion exchanger was confirmed by using various identification techniques. The FT-IR spectra of GO, TMSQA, and the anion exchanger were monitored at wavelengths between 4000 and 700 cm−1 and are shown in Fig. 2b (a). The spectrum of TMSQA shows the characteristic absorption bands at 1030, 1100, 1377, and 1569 cm−1 which are related to the stretching vibration of Si–O–C, C–Si, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, and CC of the imidazole ring. The absorption band at 2900 cm−1 is related to the C–H stretching vibration and that at 3500 cm−1 to the O–H stretching vibration of the moisture absorbed on the surface. The characteristic absorption bands of GO in Fig. 2b (b) are related to the stretching vibration of –OH (3425 cm−1), CO (1720 cm−1), aromatic CC (1629 cm−1), and C–O of epoxy groups (1222 and 860 cm−1). As observed in Fig. 2b (c), the spectrum of the anion exchanger (GO/TMSQA: 1/2) shows overlapped absorption bands in the region of 1000–1250 cm−1 for C–Si and Si–O–C vibrations. The FT-IR spectrum in Fig. 2b (d) shows the structure of the same exchanger after simultaneous adsorption of NO3− and CrO42− ions. The absorption bands at 1384 and 938 cm−1 are related to the stretching vibration of single and double NO and CrO bonds of nitrate and chromate, respectively.32,33 The XRD spectrum of GO, TMSQA, and GO/TMSQA: 1/2 ion exchanger is shown in Fig. 2c. The d-spacing (diffraction peak) can be described as the interlayer spacing or as the distance between the plates of the atoms and is obtained by using the Bragg equation (eqn (4)).34,35 As shown in Fig. 2c, the XRD spectrum of GO indicates a peak at the scattering angle 2θ = 10° which can be correlated to a d-spacing of 0.87 nm and the crystallographic plane (001), which is related to the presence of oxygenated functional groups (epoxy and hydroxyl functional groups) in GO. TMSQA exhibited a peak at 2θ = 22° (Fig. 2c), indicating an amorphous structure. Furthermore the X-ray diffraction pattern of GO/TMSQA: 1/2 showed peaks at the scattering angle 2θ = 6.32, 16.84, and 22.95° corresponding to the d-spacing of 1.37, 0.53, and 0.39 nm, respectively. After the grafting reaction of GO with TMSQA, the diffraction peak of GO shifted from 2θ = 10.2 to 22.95° in the anion exchanger, and also the interlayer distance increased to 1.37 nm at scattering angle 2θ = 6.32°. This indicates that grafting of TMSQA can occur onto the GO sheets with cross-linking reactions.36 Also, the percentage of crystallinity (%Xcry) of the compounds was estimated using the XRD patterns by measuring the surface area under the sharp peaks (crystalline phase) and the surface area under the broad peaks (amorphous phase) using the following formula (eqn (5)):37
N, and CC of the imidazole ring. The absorption band at 2900 cm−1 is related to the C–H stretching vibration and that at 3500 cm−1 to the O–H stretching vibration of the moisture absorbed on the surface. The characteristic absorption bands of GO in Fig. 2b (b) are related to the stretching vibration of –OH (3425 cm−1), CO (1720 cm−1), aromatic CC (1629 cm−1), and C–O of epoxy groups (1222 and 860 cm−1). As observed in Fig. 2b (c), the spectrum of the anion exchanger (GO/TMSQA: 1/2) shows overlapped absorption bands in the region of 1000–1250 cm−1 for C–Si and Si–O–C vibrations. The FT-IR spectrum in Fig. 2b (d) shows the structure of the same exchanger after simultaneous adsorption of NO3− and CrO42− ions. The absorption bands at 1384 and 938 cm−1 are related to the stretching vibration of single and double NO and CrO bonds of nitrate and chromate, respectively.32,33 The XRD spectrum of GO, TMSQA, and GO/TMSQA: 1/2 ion exchanger is shown in Fig. 2c. The d-spacing (diffraction peak) can be described as the interlayer spacing or as the distance between the plates of the atoms and is obtained by using the Bragg equation (eqn (4)).34,35 As shown in Fig. 2c, the XRD spectrum of GO indicates a peak at the scattering angle 2θ = 10° which can be correlated to a d-spacing of 0.87 nm and the crystallographic plane (001), which is related to the presence of oxygenated functional groups (epoxy and hydroxyl functional groups) in GO. TMSQA exhibited a peak at 2θ = 22° (Fig. 2c), indicating an amorphous structure. Furthermore the X-ray diffraction pattern of GO/TMSQA: 1/2 showed peaks at the scattering angle 2θ = 6.32, 16.84, and 22.95° corresponding to the d-spacing of 1.37, 0.53, and 0.39 nm, respectively. After the grafting reaction of GO with TMSQA, the diffraction peak of GO shifted from 2θ = 10.2 to 22.95° in the anion exchanger, and also the interlayer distance increased to 1.37 nm at scattering angle 2θ = 6.32°. This indicates that grafting of TMSQA can occur onto the GO sheets with cross-linking reactions.36 Also, the percentage of crystallinity (%Xcry) of the compounds was estimated using the XRD patterns by measuring the surface area under the sharp peaks (crystalline phase) and the surface area under the broad peaks (amorphous phase) using the following formula (eqn (5)):37
| | 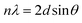 | (4) |
| | 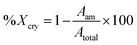 | (5) |
where
n,
λ,
d, and
θ are reflection, the wavelength of the incident radiation (nm), the inter-planar spacing (nm), and the incident angle, respectively. Moreover,
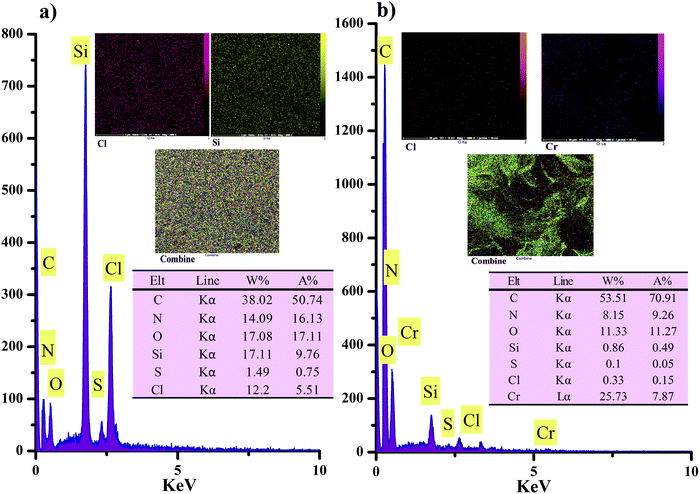
Atotal and
Aam are the total area of diffraction and amorphous peaks, respectively. The crystalline percentage of GO, TMSQA, and GO-based ion exchanger has been determined around 57.4, 14.5, and 46%, respectively. According to the XRD pattern, it is evident that a more amorphous structure can be suggested for the anion exchanger.
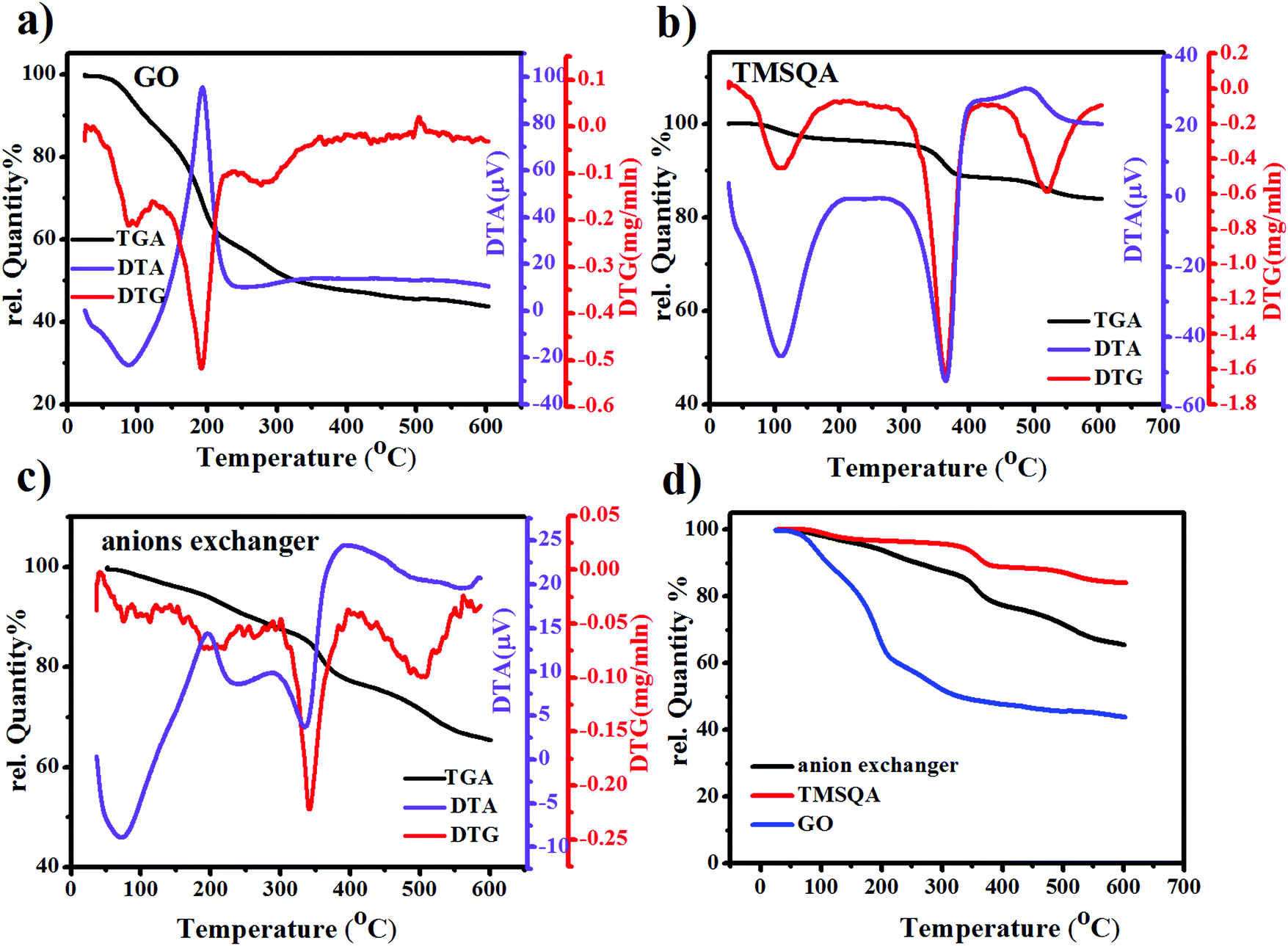
TGA, differential thermal analysis (DTA), and differential thermogravimetry (DTG) curves of TMSQA, GO, and GO-based ion exchanger (GO/TMSQA: 1/2) are shown in Fig. 3. Initially, a weight loss of ∼5 wt% is seen around 90 °C due to the evaporation of moisture and volatile compounds. In the TGA/DTA/DTG curves of GO, a weight loss of ∼25% was observed in the range of 150–400 °C due to the decomposition of oxygen functional groups such as carboxyl, hydroxyl, and carboxylate groups, which also appears as an exothermic peak in the DTA curve. The weight loss at temperatures above 300 °C can be attributed to the pyrolysis of the carbon skeleton. In the TGA/DTA/DTG curves of TMSQA, there is three-stage mass reduction at 110, 360, and 520 °C associated with the loss of adsorbed water and due to the decomposition of methyl groups and organic moieties, respectively. The residual weight at 600 °C is 82 wt%.8,16 TMSQA exhibited relatively high-temperature resistance which can be related to the symmetry in the structure. The TGA curve of the anion exchanger showed a steady weight loss of about 20 wt% from 180 °C up to 600 °C, and DTA and DTG plots showed that decomposition of the anion exchanger occurred in several successive steps at 210, 350, and above 500 °C which can be associated with the decomposition of oxygenated functional groups present in the network and condensation reaction of silane and carbon skeleton, respectively. The residual weight at 600 °C was 65 wt%.38–40 A comparison of the TGA curves shows that the combination of GO with ammonium salt resulted in higher thermal resistance, which could be due to the cross-linking reaction between the hydroxyl groups of GO and the trimethoxysilane groups in TMSQA. Therefore, in comparison with GO, the synthesized anion exchanger showed greater thermal stability with a slower decomposition rate up to 600 °C.
 |
| | Fig. 3 TGA/DTA/DTG curves of GO (a), TMSAQ (b), anion exchanger (GO/TMSQA: 1/2) (c), and the TGA curve of samples (d). | |
To evaluate the morphology of the as-synthesized anion exchanger (GO/TMSQA: 1/2), FESEM was employed. The morphological structure of GO and the prepared anion exchanger is shown under different magnifications in Fig. 4. According to the depicted image, the synthesized anion exchanger is 3D heterogeneous particles with a rough surface. Nanometer size particles of less than 50 nm can also be identified from the represented FESEM image.
 |
| | Fig. 4 FE-FESEM image of GO (a) and anion exchanger (GO/TMSQA: 1/2) (b) under different magnifications. | |
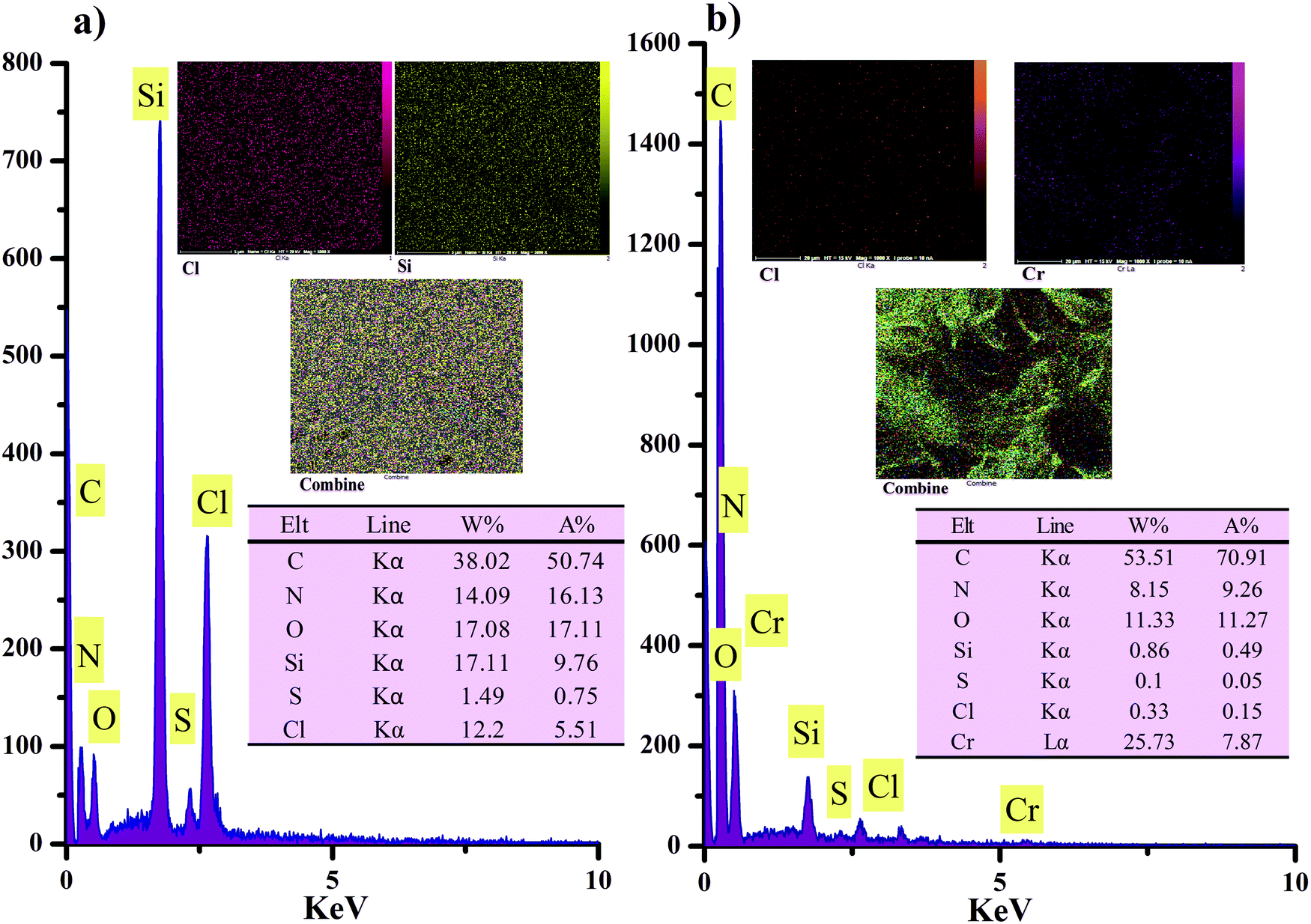
The elemental mapping images of the anion exchanger (Cl, Si and combine) and the EDS spectrum of the distribution of elements present of C (50.7%), O (17.11%), N (16.3%), Cl (5.51%), Si (9.76%), and S (0.75%) within the anion exchanger are presented in Fig. 5a. These observations suggest that TMSQA as a cross-linker has been grafted successfully onto the GO surface with the chlorine ions embedded within an insoluble gel network. Notably, the presence of the S element is related to the GO structure.17 The element mapping images and EDS spectrum of the exchanger after the adsorption of nitrate and chromate show the presence of all elements including the element chromium (7.87%). The intensity of chlorine ions has significantly reduced from 5.51 to 0.15% and the EDS spectrum of the distribution of all elements within the anion exchanger is presented in Fig. 5b. These results indicate that the exchange process of chlorine with the target anions may occur in the matrix of the exchanger.
 |
| | Fig. 5 EDS elemental mapping and EDS pattern of the anion exchanger (GO/TMSQA: 1/2): (a) before and (b) after adsorption of nitrate and chromate. | |
The results obtained from BET analysis of GO, GO/TMSQA: 1/1 and GO/TMSQA: 1/2 are summarized in Table 1, which are inferred from the comparison of the obtained data of the specific surface area. The results show that with increasing TMSQA amount, the specific surface area and the pore volume of the exchanger decrease, while the pore size of the samples increases. The BET results clearly show that the modification reaction of GO is well performed by the quaternary ammonium salt as a cross-linker, with the pore volume of the exchanger decreasing from 0.027 to 0.018 cm3 g−1 with the increase in the amount of the cross-linker. In other words, more cavities are filled and the amount of nitrogen gas uptake is reduced, and the specific surface area of the samples decreases from 11.21 to 3.88 m2 g−1. According to the BET data, it can be concluded that the adsorption process by the prepared ion exchanger is due to the presence of electrostatic interacting groups (quaternary ammonium chloride salt).
Table 1 The BET surface area analysis data
| Samples specific |
Surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Pore size (nm) |
| GO |
11.215 |
0.027204 |
9.703 |
| GO/TMSQA: 1/1 |
10.807 |
0.023857 |
10.681 |
| GO/TMSQA: 1/2 |
3.8843 |
0.017648 |
18.174 |
3.2. Adsorption study
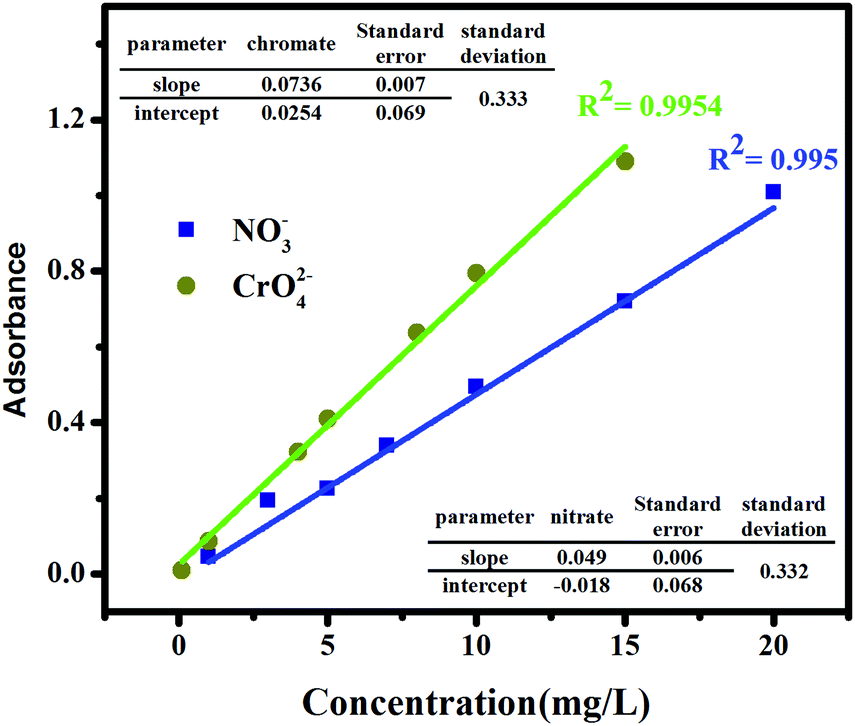
In this study, the ion exchange process of the prepared GO-based ion exchanger for the removal of nitrate and chromate anions was investigated. The anion exchange process is one of the major processes to purify aqueous solutions containing anions such as chromate and nitrate. Various factors affect the exchange capacity of the anion exchanger, such as exchanger composition, exchanger dosage, contact time, initial nitrate and chromium ion concentration, and pH of the solution. Batch studies were performed to evaluate the effect of these parameters using a certain amount of exchanger in contact with a 20 mL solution of nitrate and chromate ions. After equilibrium time, the solid phase was taken out from the solution and the concentration of ions remaining in the separate solutions was determined by using the calibration curves. The calibration curves, shown in Fig. 6, were obtained by plotting various concentrations of each of the anions versus their optical intensity, as explained in Section 2.4, using a UV-vis spectrometer.
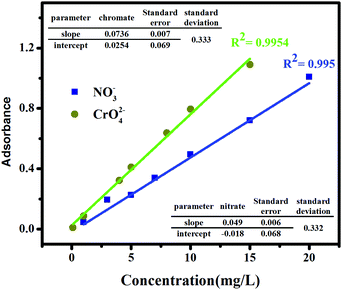 |
| | Fig. 6 Calibration curve of nitrate and chromate ions in water. | |
3.2.1. Effect of anion exchanger composition.
The effect of different weight ratios of GO/TMSQA (2/1, 1/1, and 1/2) in the anion exchanger was investigated to evaluate the removal efficiency of nitrate and chromate in 20 mL aqueous solution (pH 7) containing 20 mg of exchanger, 20 mg L−1 NO3− ions or 25 mg L−1 CrO42− ions, during 30 min or 20 min contact time, respectively, and at ambient temperature. The results are presented in Fig. 2a and explained in Section 3.1. According to the obtained results, the composition GO/TMSQA: 1/2 was selected as the most efficient composition for adsorption studies of chromate and nitrate ions.
3.2.2. Effect of initial nitrate and chromate concentration.
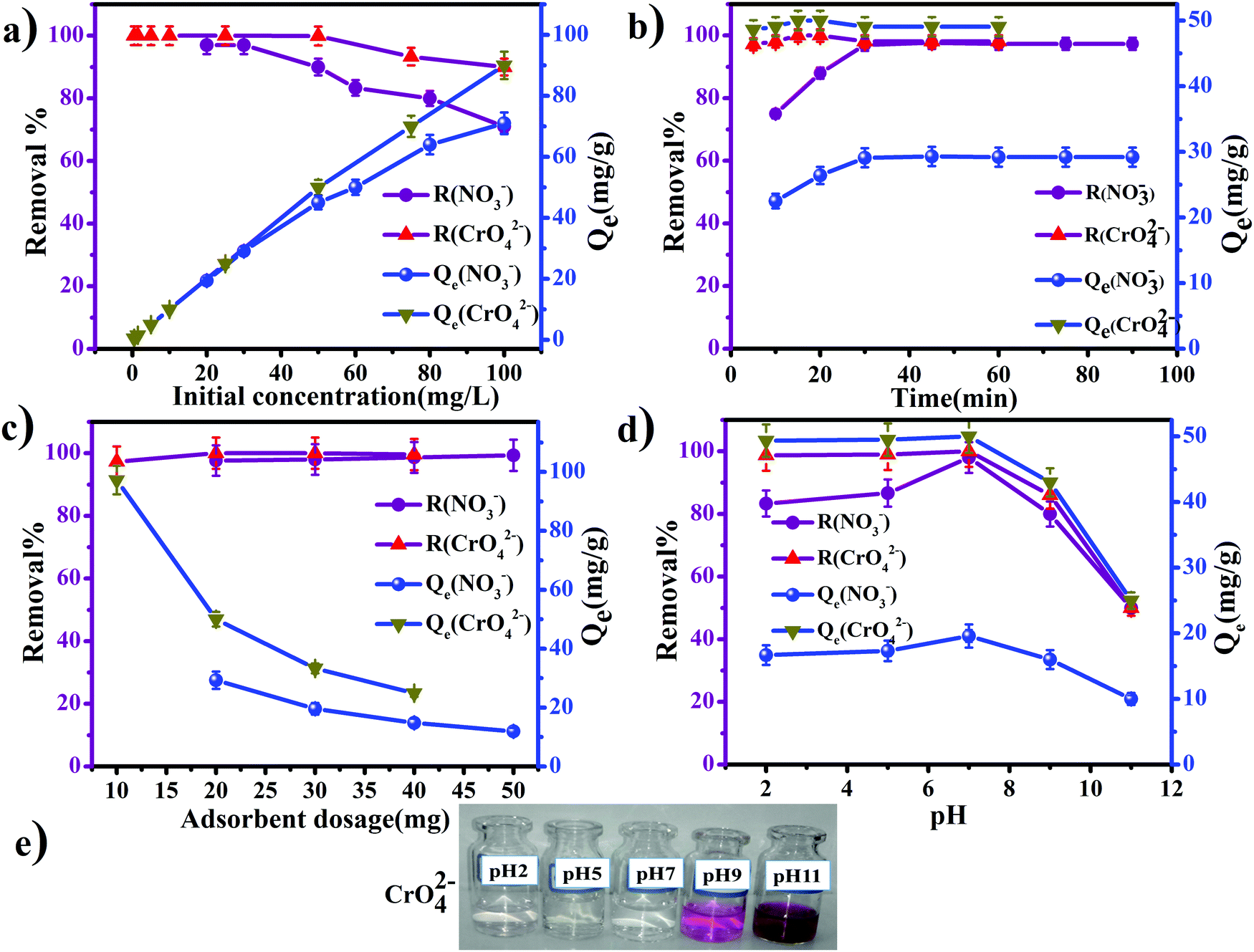
The exchange process of NO3− and CrO42− ions in 20 mL aqueous solution with chlorine of 20 mg of exchanger (GO/TMSQA: 1/2) was investigated at different initial concentrations of NO3− (20–100 mg L−1) and CrO42− (0.5–100 mg L−1) during 30 and 20 min contact time, respectively. The exchange percentage of nitrate and chromate ions is presented in Fig. 7a. As can be seen, with the increasing concentration of nitrate and chromate ions in the aqueous solution, the exchange capacity for nitrate ions increased from 19.4 to 71 mg g−1 when the concentration of nitrate increased to 100 mg L−1 and for chromate ions the exchange capacity increased from 0.5 to 90 mg g−1 when the concentration of chromate increased to 100 mg L−1.22 Therefore, the highest removal percentage of nitrate (97%) and chromate (99.9%) ions was achieved at the initial concentrations of 30 and 50 mg L−1, respectively. The number of CrO42− and NO3− ions which are transported from the aqueous solution to the bulk of the exchanger rises with increasing initial concentration and the interaction of the ions with the active sites of the exchanger is facilitated. As is evident from Fig. 7a, the percentage of nitrate ion removal decreased and that of chromate ions remained unchanged as the initial concentration increased. This is due to the decrease of active sites or the decrease of exchangeable ions as the initial concentration increases.
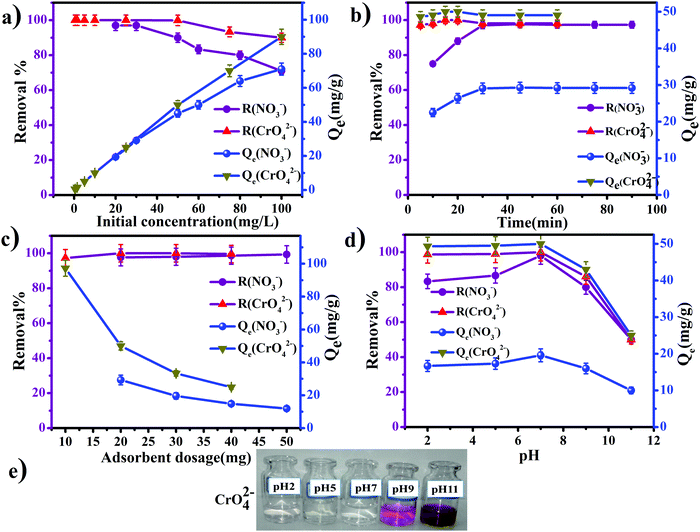 |
| | Fig. 7 Effect of (a) initial anion concentration, (b) contact time, (c) anion exchanger (GO/TMSQA: 1/2) dosage, and (d) pH on the removal% and equilibrium capacity Qe (mg g−1) of nitrate and chromate. (e) Pictures taken from samples showing the effect of pH = 2–11 on the removal efficiency of chromate. | |
3.2.3. Adsorption isotherms.
Adsorption isotherms explain how interactions between CrO42− and NO3− anions and active sites of the exchanger are and also evaluate a fundamental function for optimizing the ion exchange conditions. When the ion exchange reaches its equilibrium, the amount of CrO42− and NO3− ions exchanged at the exchange matrix is relevant to the amount of released chlorine. In this study, the isotherm models of Langmuir and Freundlich were investigated. To investigate the adsorption isotherms, an aqueous solution of nitrate and chromate containing an initial concentration ranging from 20 to 100 mg L−1 for nitrate and from 0.5 to 100 mg L−1 for chromate was used with 20 mg of exchanger (GO/TMSQA: 1/2) in 20 mL aqueous solution at ambient temperature and pH 7 for 30 and 20 min, respectively. The Langmuir and Freundlich adsorption isotherms are expressed mathematically by eqn (6) and (8), respectively:| | 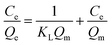 | (6) |
| | 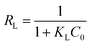 | (7) |
The variables Qe and Ce are the capacity and the concentration of anions exchanged at equilibrium, Qm is the highest exchange capacity of the anion exchanger (mg g−1), and KL is the Langmuir equilibrium constant which is calculated by plotting Ce/Qeversus Ce. The dimensionless separation factor (RL) defining a favorable process is obtained by eqn (7). The favorable and unfavorable adsorption process are represented by 0 < RL < 1 and RL > 1, respectively.41| | 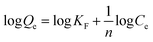 | (8) |
| | 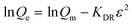 | (9) |
| | 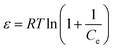 | (10) |
| | 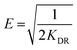 | (11) |
where KF and n are constants representing the adsorption capacity (mg g−1) and intensity, respectively, and are determined by plotting log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Qevs. logCe. The 1/n value between 0 and 1 evinces the desirable sorption.42 In the Dubinin–Radushkevich isotherm model, KDR and ε are the isotherm constants; KDR is obtained by plotting lnQevs. ε2 and sorption energy E (kJ mol−1) is determined from eqn (9)–(11). The E value can represent the type of chemical (E > 8) or physical absorption (E < 8).43 The results of the isotherm studies are summarized in Table 2. The best fit isotherm model is determined by the value of the regression coefficient (R2). As can be observed in Table 2, the results show that the obtained isotherm data for the removal of nitrate and chromate ions are fitted well with the Langmuir (R2 = 0.98) isotherms. The Langmuir model provides better fitting with the sorption data of the both anions in comparison with the Freundlich isotherm. The results of this study are similar to those reported by S. Samatya, X. Xu, P. Guzel, Y. Ye and A. Z. Moghaddam for the removal of these anions.22,44–47 The maximum monolayer adsorption capacity values determined from the Langmuir model (Qm,cal = 86.21 mg g−1 and 75.19 mg g−1) were close to the experimental results (Qm,exp = 90 and 71 mg g−1 for chromate and nitrate, respectively). When the average free energy is less than 8 kJ mol−1 (E < 8) in the Dubinin–Radushkevich isotherm, indicates abundant physical interactions between Cl− and the target anions ions (nitrate and chromate) on the prepared absorbent. The results of the isotherm studies are summarized in Table 2.
Qevs. logCe. The 1/n value between 0 and 1 evinces the desirable sorption.42 In the Dubinin–Radushkevich isotherm model, KDR and ε are the isotherm constants; KDR is obtained by plotting lnQevs. ε2 and sorption energy E (kJ mol−1) is determined from eqn (9)–(11). The E value can represent the type of chemical (E > 8) or physical absorption (E < 8).43 The results of the isotherm studies are summarized in Table 2. The best fit isotherm model is determined by the value of the regression coefficient (R2). As can be observed in Table 2, the results show that the obtained isotherm data for the removal of nitrate and chromate ions are fitted well with the Langmuir (R2 = 0.98) isotherms. The Langmuir model provides better fitting with the sorption data of the both anions in comparison with the Freundlich isotherm. The results of this study are similar to those reported by S. Samatya, X. Xu, P. Guzel, Y. Ye and A. Z. Moghaddam for the removal of these anions.22,44–47 The maximum monolayer adsorption capacity values determined from the Langmuir model (Qm,cal = 86.21 mg g−1 and 75.19 mg g−1) were close to the experimental results (Qm,exp = 90 and 71 mg g−1 for chromate and nitrate, respectively). When the average free energy is less than 8 kJ mol−1 (E < 8) in the Dubinin–Radushkevich isotherm, indicates abundant physical interactions between Cl− and the target anions ions (nitrate and chromate) on the prepared absorbent. The results of the isotherm studies are summarized in Table 2.
Table 2 Parameter values in the isotherm models for exchanging CrO42− and NO3− ions
| Model |
Parameters |
Value of parameters |
| Chromate |
Nitrate |
| Langmuir |
Q
mcal. (mg g−1) |
86.21 |
75.19 |
|
q
mexp. (mg g−1) |
90 |
71 |
|
K
L (L g−1) |
8.92 |
0.37 |
|
R
L
2
|
0.98 |
0.98 |
|
|
| Freundlich |
K
F (L g−1) |
60.71 |
26.097 |
|
n
|
2.58 |
3.25 |
| 1/n |
0.39 |
0.308 |
|
R
F
2
|
0.76 |
0.96 |
|
|
| Dubinin–Radushkevich |
K
DR (mol2 kJ−2) |
0.01 |
0.2 |
|
q
mcal. (mg g−1) |
115.7 |
57.22 |
|
E (kJ mol−1) |
0.07 |
1.58 |
|
R
DR
2
|
0.91 |
0.90 |
3.2.4. Effect of contact time.
The effect of time on the removal efficiency of CrO42− and NO3− ions was studied from 5 to 60 min and from 10 to 90 min, respectively, by keeping other parameters constant (20 mg of anion exchanger in 20 mL of aqueous solution, pH = 7, initial concentration of NO3− ions 30 mg L−1 and for CrO42− ions 50 mg L−1). From the results shown in Fig. 7b, the nitrate removal percentage increased from 75% to 97.6%, but the chromate removal percentage remained constant during the 90 min experiment. The optimum time for the maximum removal capacity of nitrate ions (29.3 mg g−1, 97.3% efficiency) was 45 min and for the chromate ions (50 mg g−1, 100% efficiency) it was 15 min. The exchange rate for the removal of nitrate and chromate ions by the anion exchanger was investigated by two equations: pseudo-first-order model (eqn (12), for the initial rapid phase) and pseudo-second-order model (eqn (13), chemisorption is the rate-limiting step):| | 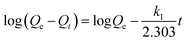 | (12) |
| | 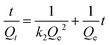 | (13) |
where Qt and Qe are the exchange capacity at time t and at equilibrium, respectively. k1 and k2 are obtained from the slope and intercept of the plot of log (Qe − Qt) vs. t (eqn (12)) and t/(Qt) versus t (eqn (13)), respectively.41 The data obtained from this study are tabulated in Table 3. The R value is a criterion for confirming the type of the absorption kinetics model. The maximum value of the correlation coefficient (R2) is 0.999 and 1 for CrO42− and NO3− ions, so the pseudo-second-order kinetics model is considered as the desirable model for the exchange of the anions by the prepared anion exchanger. The obtained kinetic results reveal the closeness of the values of the calculated and experimental exchange capacity (Qe,exp = 50 and 29.3 mg g−1; Qe,cal = 49.02 and 29.24 mg g−1 for chromate and nitrate, respectively). Therefore, the chemical exchange is suggested to be the main mechanism and controls the exchange rate of CrO42− and NO3− ions with Cl− in the anion exchanger.48 It should be noted that, in all calculations, absorption efficiencies above 99.99% were considered to be approximately 100%.
Table 3 Parameters and coefficients of the kinetics models for chromate and nitrate removal
| Kinetics model |
Parameter |
Chromate |
Nitrate |
| Pseudo-first-order model |
k
1 (min−1) |
0.004 |
0.025 |
|
Q
e,cal (mg g−1) |
1.12 |
1.256 |
|
R
2
|
0.31 |
0.034 |
|
|
| Pseudo-second-order model |
k
2 (g mg−1 min−1) |
0.16 |
2.92 |
|
Q
e,cal (mg g−1) |
49.02 |
29.24 |
|
Q
e,exp (mg g−1) |
50 |
29.3 |
|
R
2
|
0.999 |
1 |
3.2.5. Effect of ion exchanger dosage.
The amount of exchanger is another important factor influencing exchange efficiency. Therefore, to evaluate the effect of the exchanger dosage on the removal efficiency, various amounts of the anion exchanger (GT/TMSQA: 1/2) were used (10–50 mg) under optimum conditions: 20 mL of aqueous solution containing 50 mg L−1 chromate and 30 mg L−1 nitrate tested for 15 and 45 min, respectively, at room temperature and pH 7. As the results in Fig. 7c show, the removal percentage of both anions did not change when the amount of the exchanger increased from 10 to 50 mg. However, the exchange capacity for both anions decreased with the increasing amount of the anion exchanger. The CrO42− and NO3− ions that remained in the solution were determined by increasing the amount of the anion exchanger. The decrease in the exchange capacity showed that less and less amount of the exchange anions (CrO42− and NO3− ions) remains in the solution as the active sites for exchange (Cl−) increase.49 Therefore, 30 and 20 mg anion exchanger in 20 mL solution was used as the optimum dosage for CrO42− and NO3− ion removal, respectively.
3.2.6. Effect of solution pH.
The chromate ions can exist as various species depending on the pH values and the total concentration of chromium (Cr(0)) in solution is less than 1 M, that included CrO4−, HCrO4−, NaCrO4− and Cr2O7− species. The predominant species in an acidic medium are HCrO4− and CrO42− with a minor contribution of Cr2O72− ions, while CrO42− and HCrO4− ions are predominant in neutral conditions and CrO42− and minor ions from NaCrO4− are predominant in alkaline conditions. According to the reported articles, the predominant form in 4 < pH < 10 is CrO42− species in solution.50,51 In the following, the effect of solution pH on the absorption process of chromate and nitrate ions in the aqueous solution is examined. One of the effective parameters in the anion exchange process is the solution pH which affects the surface characteristics and ionization/dissociation of the ion exchanger. The influence of the solution pH in the range of 2–11 was investigated in 20 mL of aqueous solution containing 50 and 30 mg L−1 chromate and nitrate ions, and 20 and 30 mg of anion exchanger (GO/TMSQA: 1/2) for 15 and 45 min contact time, respectively. The results of pH influence on the exchange of CrO42− and NO3− ions with Cl− ions are shown in Fig. 7d. The highest removal efficiency and adsorption capacity of CrO42− and NO3− ions were attained at pH 7. As illustrated, the removal percentage of nitrate ions and the capacity increased from 80% to almost 100% and for chromate ions the removal percentage remained unchanged (100%) with increasing the solution pH from 2 to 7. Then the removal percentage and the capacity started to decrease sharply for both anions with a further increase in pH up to 11. As can be seen in Fig. 7e, the color of the solution in the presence of 1,5-diphenylcarbazide changes in acidic and basic environments. The process of color change represents the effect of pH on the removal of chromate, which had the highest efficiency in acidic and neutral conditions. The exchange efficiency of ions is reduced sharply in the basic environment because of the presence of hydroxyl ions in the system which causes electrostatic repulsion and competition between hydroxyl ions and target ions. Therefore, the ion exchange process for both CrO42− and NO3− ions decreased drastically in the alkaline solution, especially at pH 11. The reduced removal efficiency of chromate ions is also clearly seen by the dark purple color in the pictures shown in Fig. 7e. In acidic medium, the ion exchange capacity and the removal efficiency of nitrate are reduced more than chromate ions owing to the presence of chloride ions in the acid chloride solution. These Cl− ions in solution compete with the nitrate and chromate ions for the exchange process. Moreover, nitrate and chromate ions also become protonated by hydrogen ions in the acidic solution which reduces the ion exchange process on the anion exchanger and decreases the exchange capacity (Qe) of these ions.52
3.2.7. Simultaneous removal of nitrate and chromate.
Several research papers have reported the removal of chromate or nitrate using different techniques such as adsorption, chemical precipitation, biological degradation, electro-dialysis, and ion exchange. However, there are no reports of removing chromate and nitrate simultaneously from an aqueous solution using a GO-based anion exchanger. In this study, the effect of the co-existence of nitrate and chromium anions in the aqueous solution on their simultaneous removal by the anion exchanger (GO/TMSQA: 1/2) was investigated. The experimental conditions for the simultaneous removal were 30 mg L−1 nitrate and 50 mg L−1 chromate with 20 mg (and 30 mg) of anion exchanger in a 20 mL aqueous solution of pH 7 and ambient temperature for 45 min. The simultaneous removal characteristics of both anions were analyzed by using a UV-vis spectrophotometer.2,22,23 The results in Table 4 show the exchange capacity and the removal efficiency in separate solutions of NO3− and CrO42− anions and in a solution with the co-existence of these anions.
Table 4 Effect of exchanger dosage on the simultaneous removal of nitrate (30 mg L−1) and chromate (50 mg L−1) in 20 mL solution
| Ions |
Adsorbent dosage (mg) |
In a separate exchange |
In a simultaneous exchange |
|
R% |
Q
e (mg g−1) |
R% |
Q
e (mg g−1) |
| NO3− |
20 |
97.6 |
29.3 |
78.32 |
23.5 |
| 30 |
98 |
19.6 |
88.53 |
17.71 |
|
|
| CrO42− |
20 |
100 |
50 |
99.62 |
49.81 |
| 30 |
100 |
33.33 |
99.76 |
33.25 |
As can be seen in this table, the removal efficiency of CrO42− anions is not affected by the presence of NO3− anions. Also, the removal efficiency of chromate in the presence of nitrate did not change using a 20 or 30 mg exchanger. However, the removal efficiency of the nitrate decreased considerably in the presence and the absence of chromate on changing the amount of the anion exchanger from 20 to 30 mg. It has been reported that compounds with a higher valence state reduce the removal efficiency of compounds with a lower valence state.2 In the present study, the results show that chromate removal was not inhibited significantly by the presence of nitrate, while nitrate removal was reduced considerably by the presence of chromate. Therefore, in a solution of NO3− and CrO42−, chromate anions bound to the exchanger preferentially, and the binding of nitrate began to increase after chromate binding reached its saturation. The Bivalent chromate can bound to the anion exchanger superior to the monovalent nitrate ions.
3.2.8. Evaluation of recycling and reusing the anion exchanger.
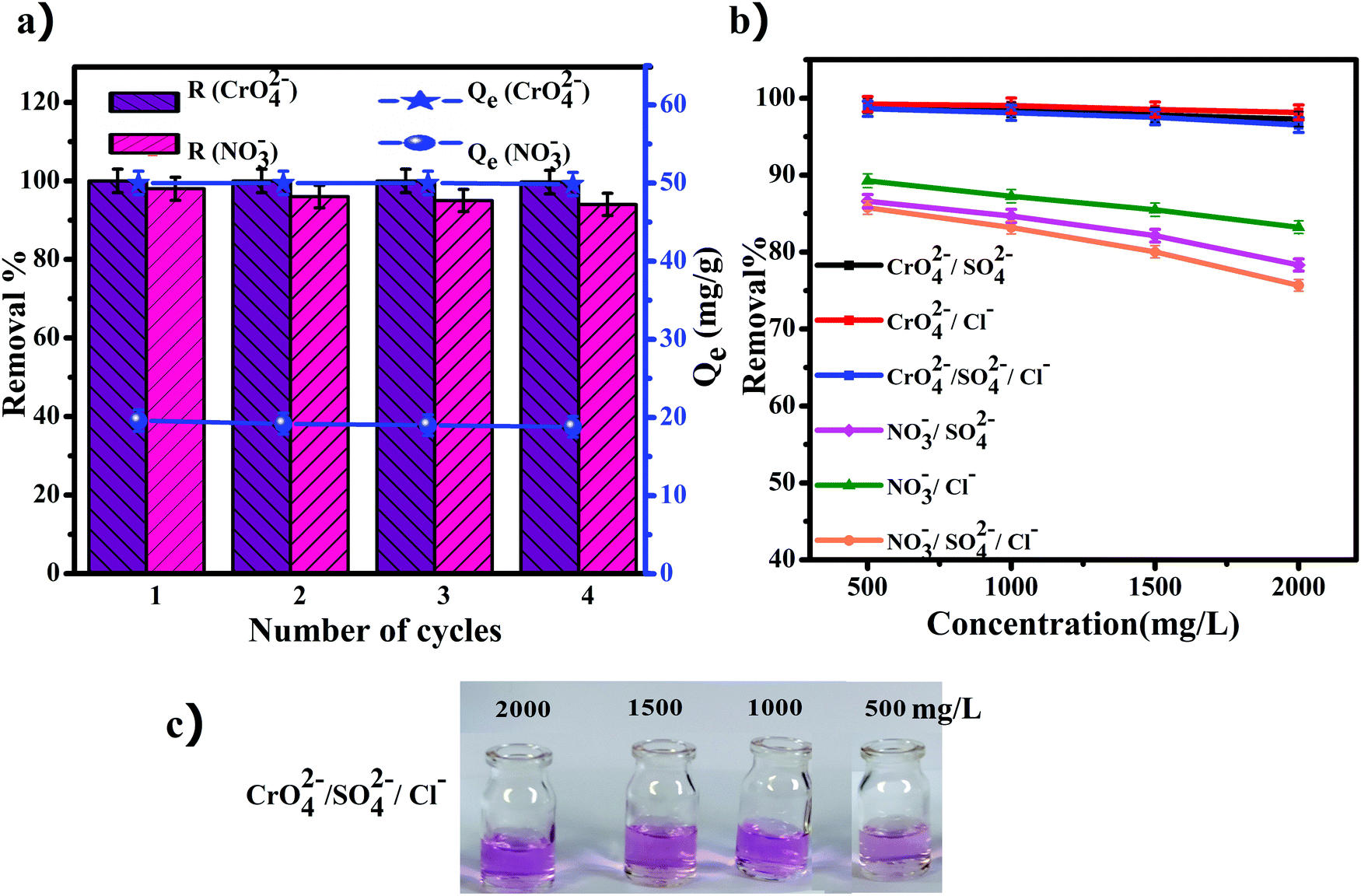
In recent years, recyclable and reusable adsorbents are an increasingly important issue due to environmental regulations and reduction of the cost of production. The prepared anion exchanger was examined for recyclability and reusability via desorption of the adsorbed anions in sodium hydroxide. The obtained results after four cycles of regeneration and reuse are shown in Fig. 8a. During the desorption process, the adsorbed nitrate and chromate anions are replaced with hydroxyl ions and the released anions are converted to sodium nitrate and sodium chromate soluble salts. Therefore, the anion exchanger is free from the initially loaded nitrate and chromate anions and was immersed in the solution of sodium chloride for conversion to its chloride form. These investigations were performed in four recovery cycles for each of the nitrate and chromate ions separately. As the results in Fig. 8a show, after the fourth adsorption/desorption process, the removal efficiency of chromate ions changed from 100 to 99.7% (the capacity decreased from 50 to 49.85 mg g−1), while the removal efficiency for the nitrate ions decreased slowly to 88%.
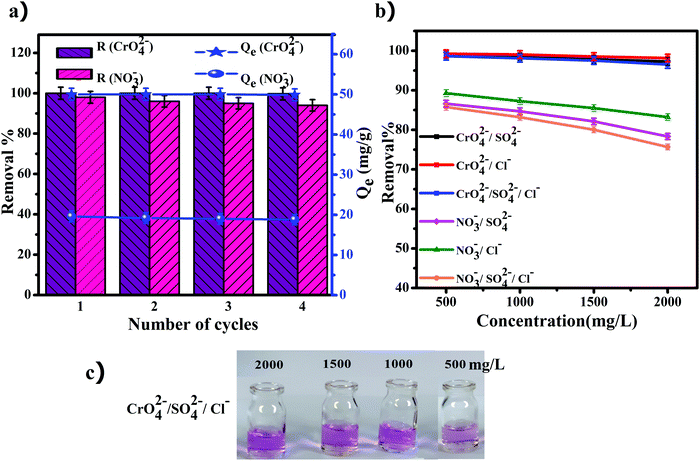 |
| | Fig. 8 (a) Regeneration of the anion exchanger (GO/TMSQA: 1/2), (b) effect of competing ion concentration on the removal percent of nitrate and chromate, and (c) solutions of different concentrations of competing ions (500–2000 mg L−1) after adsorption of chromate. | |
3.2.9. Selectivity tests.
The removal of nitrate and chromate by the prepared ion exchanger was investigated in the presence of different concentrations (500–2000 mg L−1) of chloride and sulfate ions. The maximum allowable concentrations for sulfate and chloride ions in drinking water by the World Health Organization are 400 and 250 mg L−1, respectively.53 The removal percentage of nitrate and chromate in the presence of various concentrations of the competing ions is shown in Fig. 8b and the pictures taken from the experiment are shown in Fig. 8c. As can be seen in Fig. 8b, the results show that the removal percentage of chromate did not change in the presence of the tested competing ions. On the other hand, the removal percentage of nitrate showed a ∼10% decrease in the presence of the competing ions. The reduction in the presence of sulfate ions is more than the reduction in the presence of chloride ions, which can be due to the more negative charged anions of sulfates.
3.2.10. Comparison with other anion exchange materials.
The exchange capacity of nitrate and chromium anions by the prepared anion exchanger (GO/TMSQA: 1/2), at the optimal dosage and contact time, was compared with that of the other adsorbents (Table 5). As can be seen, the exchange capacity of the as-prepared GO-based anion exchanger for removal of these two anions is more than or comparable with that of the other adsorbents.
Table 5 Comparison of removal of nitrate and chromate with other exchangers
| Adsorbent |
Ions |
Q
e (mg g−1) |
Contact time (min) |
Weight (g L−1) |
Ref. |
| Anion exchanger based on GO (GO/TMSQA) |
Chromium |
50 |
15 |
1 |
This work |
| Nitrate |
29.3 |
45 |
1.5 |
| SBPIL |
Chromium |
40.8 |
120 |
2.25 |
32
|
| Treated waste newspaper (TWNP) |
Chromium |
5.28 |
60 |
3 |
4
|
| Pumice SBA-15 |
Nitrate |
15.6 |
250 |
2.85 |
54
|
| 12.3 |
100 |
|
| ALR-AE resin |
Nitrate |
40.5 |
10 |
2 |
45
|
| Amberlite IRA |
Nitrate |
14.8 |
60 |
1.25 |
55
|
| Bioadsorbent GT/TMSQA |
Nitrate |
19.95 |
20 |
2 |
8
|
3.3. Antibacterial activity evaluation
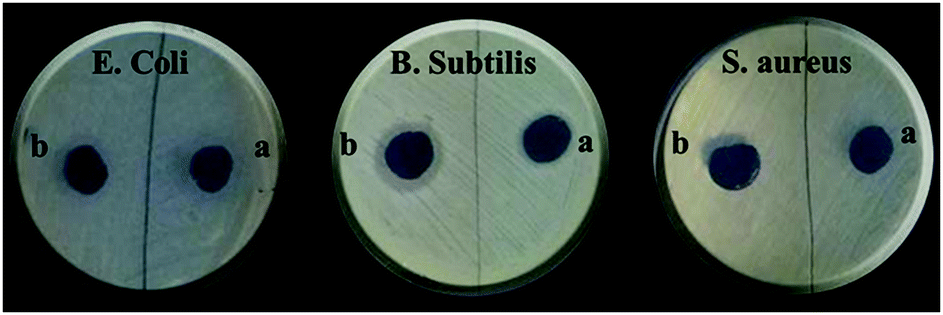
The positive charge on the surface of quaternary ammonium salts was responsible their antibacterial ability. Their mechanism of action is as follows: the quaternary ammonium compounds penetrate the bacterial cell wall, causing cell membrane damage and leakage of the intracellular fluid, eventually causing cell death.56 The antibacterial properties of the anion exchanger were studied by using the Kirby–Bauer technique and two standard drugs gentamicin and chloramphenicol were used for comparison. As can be observed in Fig. 9 and from the obtained data in Table 6, the sample of GO/TMSQA: 1/2 composition showed higher antimicrobial activity against E. coli and B. subtilis bacteria in comparison with the samples of GO/TMSQA: 2/1 and GO/TMSQA: 1/1 composition. This can be due to the higher amount of TMSQA in the GO/TMSQA: 2/1 exchanger than in the other two samples. However, also the sample with GO/TMSQA: 1/2 composition showed a relatively acceptable antibacterial activity against E. coli and B. subtilis under the test conditions compared with the standard drugs, but did not show activity against S. aureus bacteria. The sample GO/TMSQA: 1/1 showed some antibacterial activity against Gram-positive B. subtilis but showed no activity against E. coli and S. aureus. Therefore, the anion exchanger with a high amount of quaternary ammonium salt in the network structure, GO/TMSQA: 1/2, showed acceptable antibacterial activity and can be recommended for water purification.
 |
| | Fig. 9 The inhibition zone test for the prepared anion exchanger: (a) GO/TMSQA: 1/1, (b) GO/TMSQA: 1/2 against Gram-negative (E. coli) and Gram-positive bacteria (B. subtilis and S. aureus). | |
Table 6 Antibacterial activity of the anion exchangers using the Kirby– Bauer technique (zone of growth inhibition, mm)
| Test strain |
Zone of growth inhibition (mm)a |
| GO/TMSQA (weight ratio) (2 mg per disc) |
Gentamicin (10 μg per disc) |
Chloramphenicol (30 μg per disc) |
| 2/1 |
1/1 |
1/2 |
|
The diameter of the film: 10 mm, Mueller–Hinton agar plate.
No effect.
|
|
E. coli
|
NEb |
NE |
17.0 ± 0.7 |
19.6 ± 1.1 |
20.7 ± 1.5 |
|
S. aureus
|
NE |
NE |
NE |
20.3 ± 1.5 |
21.7 ± 0.6 |
|
B. subtilis
|
NE |
13.0 ± 0.7 |
17.0 ± 0.7 |
26.0 ± 1.7 |
22.3 ± 1.2 |
4. Conclusion
In this paper, for the first time, the preparation of a GO-based anion exchanger using quaternary ammonium chloride (TMSQA) as a cross-linker and an anion exchanger is reported to separate nitrate and chromate from polluted water in separate solutions, in the co-existence solution, and in the presence of interfering ions. The primary investigation showed that the anion exchanger with the weight ratio of GO/TMSQA: 1/2 has the highest exchange capacity and was selected for further experiments. The results showed a removal efficiency of 97.3% (29.3 mg g−1) for NO3− and 100% (50 mg g−1) for CrO42− ions under optimized conditions: 30 and 50 mg L−1 initial concentration of NO3− and CrO42− ions using 1.5 and 1 g L−1 anion exchanger during 45 and 15 min contact time, respectively, in a 20 mL aqueous solution of pH 7 and at room temperature. The exchange process for both anions followed the pseudo-second-order kinetics model and the Langmuir isotherm model. Therefore, according to the accepted kinetics and isotherm models and due to the FT-IR and EDX analyses before and after the adsorption, it can be concluded that the adsorption mechanism of the tested anions is ion-exchange and electrostatic attraction. The results from the solution with the co-existence of anions showed that the bivalent CrO42− ions reduced the removal efficiency of single valent NO3− ions from 29.3 to 22.9 mg g−1. This can be due to the superior binding of the chromate in comparison to the binding of single valent nitrate. Also, the removal efficiency of chromate did not change in the presence of sulfate and chloride ions as interfering ions, while the removal efficiency of nitrate reduced ∼22%. These results indicate the appropriate selectivity by the anion exchanger even in the presence of a high concentration of interfering ions (competing ions). The prepared anion exchanger showed suitable antibacterial activity against E. coli and B. subtilis. The regeneration tests showed that the as-prepared anion exchanger is reusable, which is an important economic issue, and is also suitable for column application, related to the use of solid-like materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The antibacterial tests were performed in the Microbiology Laboratory at the University of Mazandaran (Iran). The authors would like to thank Dr Mojtaba Mohseni who operated the tests and for his helpful discussions. The authors would also like to thank Dr Seyed Mojtaba Amininasab from the Faculty of science, University of Kurdistan (Iran) for his useful guidance in conducting this research.
References
- A. Bhatnagar and M. Sillanpää, Chem. Eng. J., 2011, 168, 493–504 CrossRef CAS.
- K. Baek and J.-W. Yang, J. Hazard. Mater., 2004, 108, 119–123 CrossRef CAS.
- K. Chauhan, J. Kaur, P. Singh, P. Sharma, P. Sharma and G. S. Chauhan, Ind. Eng. Chem. Res., 2016, 55, 2507–2519 CrossRef CAS.
- M. H. Dehghani, D. Sanaei, I. Ali and A. Bhatnagar, J. Mol. Liq., 2016, 215, 671–679 CrossRef CAS.
- Ü. Daniş and B. Keskinler, Desalination, 2009, 249, 1356–1364 CrossRef.
- M. H. Ward, R. R. Jones, J. D. Brender, T. M. De Kok, P. J. Weyer, B. T. Nolan, C. M. Villanueva, S. G. Van Breda, R. R. J. Id, J. D. Brender, T. M. De Kok, P. J. Weyer, B. T. Nolan, C. M. Villanueva and S. G. Van Breda, Int. J. Environ. Res. Public Health, 2018, 15, 1557 CrossRef.
- H. Sun, J. Brocato, M. Costa and O. C. Exposure, Curr. Environ. Health Rep., 2015, 2, 295–303 CrossRef CAS.
- M. Shojaipour, M. Ghaemy, S. Mojtaba and S. M. Amininasab, Carbohydr. Polym., 2020, 227, 115367 CrossRef CAS.
- K. Cheballah, A. Sahmoune, K. Messaoudi, N. Drouiche and H. Lounici, Chem. Eng. Process., 2015, 96, 94–99 CrossRef CAS.
- J. W. Palko, D. I. Oyarzun, B. Ha, M. Stadermann and J. G. Santiago, Chem. Eng. J., 2018, 334, 1289–1296 CrossRef CAS.
- E. Alcalde, I. Dinarès, A. Ibáñez and N. Mesquida, Molecules, 2012, 17, 4007–4027 CrossRef CAS.
- S. Buffet-bataillon, P. Tattevin, M. Bonnaure-mallet and A. Jolivet-gougeon, Int. J. Antimicrob. Agents, 2012, 39, 381–389 CrossRef CAS.
- J. Wang, G. Wang, T. Wu, D. Wang, Y. Yuan, J. Wang, T. Liu, L. Wang and J. Qiu, ACS Sustainable Chem. Eng., 2018, 6, 17204–17210 CrossRef CAS.
- E. Obłak, A. Piecuch, K. Guz-Regner, E. Dworniczek, E. Obłąk, A. Piecuch, K. Guz-Regner and E. Dworniczek, FEMS Microbiol. Lett., 2014, 350, 190–198 CrossRef.
- M. Lv, L. Yan, C. Liu, C. Su, Q. Zhou, X. Zhang, Y. Lan, Y. Zheng, L. Lai and X. Liu, Chem. Eng. J., 2018, 349, 791–799 CrossRef CAS.
- M. Mohammadian, R. Sahraei and M. Ghaemy, Chemosphere, 2019, 225, 259–269 CrossRef CAS.
- J. Song, X. Wang and C.-T. Chang, J. Nanomater., 2014, 2014, 276143 Search PubMed.
- J. Safari, S. Gandomi-Ravandi and S. Ashiri, New J. Chem., 2016, 40, 512–520 RSC.
- H. R. Nodeh, H. Sereshti, E. Z. Afsharian and N. Nouri, J. Environ. Manage., 2017, 197, 265–274 CrossRef.
- D. Homa, E. Haile and A. P. Washe, Int. J. Anal. Chem., 2016, 2016, 7214932 Search PubMed.
- P. K. Saw, A. K. Prajapati and M. K. Mondal, J. Mol. Liq., 2018, 269, 101–109 CrossRef CAS.
- Y. Ye, Y. Ren, J. Zhu, J. Wang and B. Li, Desalin. Water Treat., 2016, 57, 26427–26439 CrossRef CAS.
- A. Rezaee, H. Hossini, H. Masoumbeigi and R. D. C. Soltani, Int. J. Environ. Sci. Dev., 2011, 2, 294–298 CrossRef.
- G. Kabir and S. E. Ogbeide, Int. J. Environ. Res., 2008, 2, 377–384 Search PubMed.
- I. Aswin Kumar and N. Viswanathan, J. Chem. Eng. Data, 2018, 63, 147–158 CrossRef CAS.
- B.-U. U. Bae, Y.-H. H. Jung, W.-W. W. Han and H.-S. S. Shin, Water Res., 2002, 36, 3330–3340 CrossRef CAS.
- A. S. K. Kumar, S. Kalidhasan, V. Rajesh and N. Rajesh, Ind. Eng. Chem. Res., 2012, 51, 58–69 CrossRef CAS.
- P. Shahbazi, F. Vaezi, A. H. Mahvi, K. Naddaffi and A. R. Rahmani, J. Res. Health Sci., 2010, 10, 91–97 CAS.
- R. Baharfar, S. Asghari, S. Rassi and M. Mohseni, Res. Chem. Intermed., 2015, 41, 6975–6984 CrossRef CAS.
- K. Li, P. Li, J. Cai, S. Xiao, H. Yang and A. Li, Chemosphere, 2016, 154, 310–318 CrossRef CAS.
- Z. Y. Kong, J. F. Wei, Y. H. Li, N. N. Liu, H. Zhang, Y. Zhang and L. Cui, Chem. Eng. J., 2014, 254, 365–373 CrossRef CAS.
- A. G. Khiratkar, S. S. Kumar and P. R. Bhagat, RSC Adv., 2016, 6, 37757–37764 RSC.
- A. Alighardashi, Z. Kashitarash Esfahani, F. Najafi, A. Afkhami and N. Hassani, Environ. Processes, 2018, 5, 41–64 CrossRef CAS.
- T. N. Blanton and D. Majumdar, Powder Diffr., 2012, 27, 104–107 CrossRef CAS.
- Y. Sheng, X. Tang, E. Peng and J. Xue, J. Mater. Chem. B, 2013, 1, 512–521 RSC.
- Z. Sekhavat Pour, M. Ghaemy, Z. S. Pour and M. Ghaemy, Compos. Sci. Technol., 2016, 136, 145–157 CrossRef CAS.
- P. Ahvenainen, I. Kontro and K. Svedström, Cellulose, 2016, 23, 1073–1086 CrossRef CAS.
- Z. Yu, L. Lv, Y. Ma, H. Di, Y. He and G. M. Sheldrick, RSC Adv., 2016, 6, 18217–18226 RSC.
- S. L. Qiu, C. S. Wang, Y. T. Wang, C. G. Liu, X. Y. Chen, H. F. Xie, Y. A. Huang and R. S. Cheng, Express Polym. Lett., 2011, 5, 809–818 CrossRef CAS.
- I. M. Khan, K. Alam, M. J. Alam and M. Ahmad, New J. Chem., 2019, 43, 9039–9051 RSC.
- P. Karthikeyan, H. A. T. Banu, S. Meenakshi, H. Ali, T. Banu and S. Meenakshi, Int, J. Biol. Macromol., 2019, 124, 492–504 CrossRef CAS.
- A. Khaled, A. El Nemr, A. El-Sikaily, O. Abdelwahab, A. El Nemr, A. El-Sikaily and O. Abdelwahab, J. Hazard. Mater., 2009, 165, 100–110 CrossRef CAS.
- Q. Hu and Z. Zhang, J. Mol. Liq., 2019, 277, 646–648 CrossRef CAS.
- S. Samatya, N. Kabay, Ü. Yüksel, M. Arda and M. Yüksel, React. Funct. Polym., 2006, 66, 1206–1214 CrossRef CAS.
- X. Xu, B. Gao, Y. Zhao, S. Chen, X. Tan, Q. Yue, J. Lin and Y. Wang, J. Hazard. Mater., 2012, 203, 86–92 CrossRef.
- P. Guzel, Y. A. Aydın, N. D. Aksoy and N. Deveci Aksoy, Int. J. Environ. Sci. Technol., 2016, 13, 1277–1288 CrossRef CAS.
- A. Z. Moghaddam, M. E. Jazi, A. Allahrasani, M. Khazaei, M. R. Ganjali, M. R. Saeb and V. Vatanpour, ChemistrySelect, 2020, 5, 5404–5413 CrossRef CAS.
- C. Kan, A. H. Ibe, K. K. P. Rivera, R. O. Arazo, M. D. G. de Luna, K. Katrina, P. Rivera, R. O. Arazo, M. Daniel and G. De Luna, Sustainable Environ. Res., 2017, 27, 163–171 CrossRef CAS.
- K. S. Padmavathy, G. Madhu and P. V. Haseena, Procedia Technol., 2016, 24, 585–594 CrossRef.
- M. Ghashghaee, V. Farzaneh and O. Article, Iran. J. Toxicol., 2016, 10, 15–21 CrossRef CAS.
- A. H. Meena and Y. Arai, Geochem. Trans., 2016, 17, 1 CrossRef.
- I. A. Kumar and N. Viswanathan, Ind. Eng. Chem. Res., 2018, 57, 14470–14481 CrossRef CAS.
-
D. N. R. Iowa, Iowa Dep. Nat. Resour. Consult. Packag., 2009, p. 79.
- J. Y. Kim, M. S. Balathanigaimani and H. Moon, Water, Air, Soil Pollut., 2015, 226, 431–442 CrossRef.
- M. Chabani, A. Amrane and A. Bensmaili, Chem. Eng. J., 2006, 125, 111–117 CrossRef CAS.
- K.-S. S. Huang, C.-H. H. Yang, S.-L. L. Huang, C.-Y. Y. Chen, Y.-Y. Y. Lu and Y.-S. S. Lin, Int. J. Mol. Sci., 2016, 17, 1578 CrossRef.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Mousa
Ghaemy
and
Mousa
Ghaemy