
Selective photocatalytic production of CH4 using Zn-based polyoxometalate as a nonconventional CO2 reduction catalyst†
Nayeong
Kim
ab,
Jung Seung
Nam
cd,
Jinhyeong
Jo
a,
Junmo
Seong
c,
Hyunwoo
Kim
ab,
Youngkook
Kwon
 be,
Myoung Soo
Lah
c,
Jun Hee
Lee
*a,
Tae-Hyuk
Kwon
*cd and
Jungki
Ryu
*ab
be,
Myoung Soo
Lah
c,
Jun Hee
Lee
*a,
Tae-Hyuk
Kwon
*cd and
Jungki
Ryu
*ab
aDepartment of Energy Engineering, School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 44919, Republic of Korea. E-mail: jryu@unist.ac.kr; junhee@unist.ac.kr
bEmergent Hydrogen Technology R&D Center, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 44919, Republic of Korea
cDepartment of Chemistry, School of Nature Science, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 44919, Republic of Korea. E-mail: kwon90@unist.ac.kr
dCenter for Wave Energy Materials, School of Natural Science, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 44919, Republic of Korea
eSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 44919, Republic of Korea
First published on 19th February 2021
Abstract
Efficient and selective production of CH4 through the CO2 reduction reaction (CO2RR) is a challenging task due to the high amount of energy consumption and various reaction pathways. Here, we report the synthesis of Zn-based polyoxometalate (ZnPOM) and its application in the photocatalytic CO2RR. Unlike conventional Zn-based catalysts that produce CO, ZnPOM can selectively catalyze the production of CH4 in the presence of an Ir-based photosensitizer (TIr3) through the photocatalytic CO2RR. Photophysical and computation analyses suggest that selective photocatalytic production of CH4 using ZnPOM and TIr3 can be attributed to (1) the exceptionally fast transfer of photogenerated electrons from TIr3 to ZnPOM through the strong molecular interactions between them and (2) effective transfer of electrons from ZnPOM to *CO intermediates due to significant hybridization of their molecular orbitals. This study provides insights into the design of novel CO2RR catalysts for CH4 production beyond the limitations in conventional studies that focus on Cu-based materials.
New conceptsThere have been fundamental limitations to the selection of elements for the design and synthesis of CO2RR catalysts. For example, metallic Zn or Zn-based materials are known as CO producing catalysts, and CH4 producing catalysts have been limited to Cu-, Ru- and Ir-based materials. To the best of our knowledge, this is the first report that Zn-based materials including POMs can be used as a CH4 producing catalysts. Our study suggests that the overlap between the orbitals of catalytic elements and intermediate species (e.g., Zn in ZnPOM and *CO) plays a significant role in multistep electrochemical reactions such as the CO2RR. Conventionally, the underlying mechanism for the CO2RR has been explained mostly in terms of the binding energy between catalysts and intermediate species. This is also why there have been fundamental limitations on the selection of elements for the design and synthesis of CO2RR catalysts. In this regard, our study provides insights and broadens the material library for the design and synthesis of novel CO2RR catalysts, especially for the selective production of hydrocarbons including CH4. Considering that POMs can be considered as a 0-D nanomaterial with a well-defined structure, this study can offer a basis for understanding the structure-performance relationship of nanocatalysts. |
Introduction
The carbon dioxide (CO2) reduction reaction (CO2RR) into valuable chemicals using renewable energy resources is a promising technology for both ensuring sustainable growth and mitigating environmental problems.1–4 In principle, useful chemicals, such as carbon monoxide (CO), formic acid (HCOOH), and hydrocarbons, can be produced and consumed in a carbon-neutral manner by exploiting the CO2RR.5–7 Despite recent important progress, the CO2RR is a still challenging task that consumes a large amount of energy and results in low product yield/selectivity for a specific chemical. The difficulty stems from the high stability of CO2 molecules, the complexity of the CO2RR involving the transfer of multiple protons and electrons, and the presence of competing reactions.8–10 These problems necessitate the development of efficient and selective catalysts for the CO2RR.11–16 However, there have been fundamental limitations to the selection of elements in the design and synthesis of CO2RR catalysts (Scheme 1a). For example, most transition metals (coloured grey in Scheme 1a) favor hydrogen evolution reactions (HERs) rather than the CO2RR. Among diverse chemicals from the CO2RR, the production of hydrocarbons requires the formation of multiple stable intermediate states and the incorporation of more electrons and protons than other CO2RR products. For example, eight electrons and protons are required to produce CH4, whereas only two are required to produce CO or HCOOH. As a result, only a few elements, such as Cu,17 Ru,18 and Ir,19 are available for the design of the CO2RR catalysts for CH4 production (purple), whereas more diverse elements are available for CO (pink) and HCOOH (green). In this regard, the development of CO2RR catalysts for CH4 production has been mostly focused on Cu-based materials due to their low cost.20–22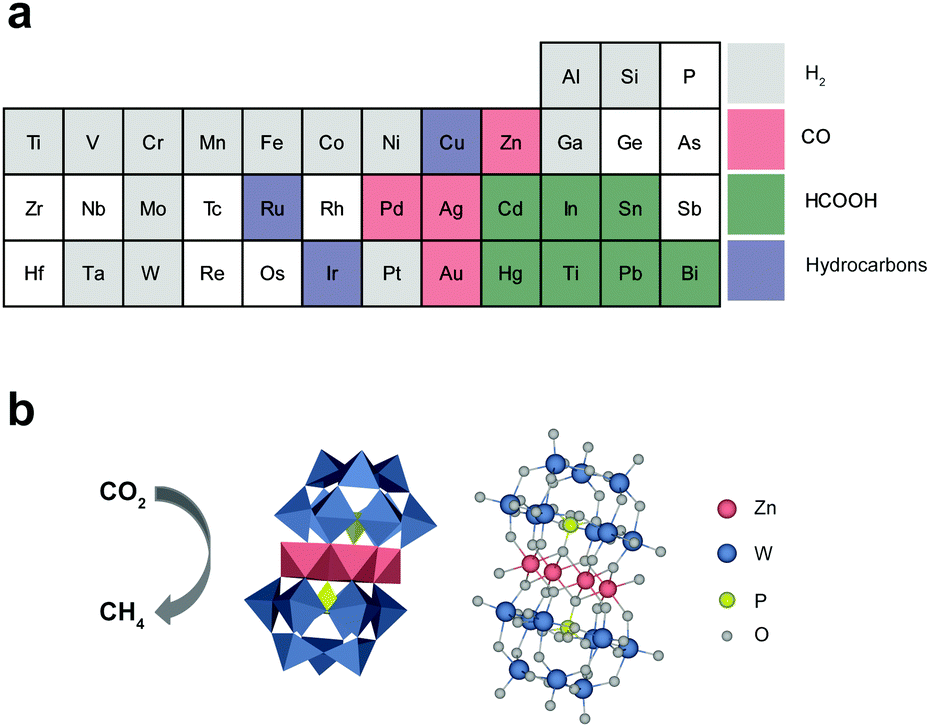 | ||
| Scheme 1 Production of various chemicals from the CO2RR. (a) Classification of the main chemical produced by each element in the periodic table through the CO2RR: hydrogen (grey), CO (pink), formic acid (green), and hydrocarbons (purple). (b) Structure of ZnPOM and its application as a CO2RR catalyst for the selective production of CH4. | ||
On the other hand, polyoxometalates (POMs) are considered promising electrocatalysts due to their molecular nature with uniform active sites, structural diversity, and rich electrochemistry.23 In particular, sandwich-type POMs have been demonstrated as efficient and robust multi-electron transfer catalysts because they can stably embed various transition metal ions between metal oxide clusters.24,25 For example, it is reported that POMs can serve as homogeneous water splitting catalysts: POMs with Co- and Ru-based active sites for oxygen evolution reactions26,27 and POMs with Mn-, Ni-, and Cu-based ones for the HER.28–30 In addition, POMs can rapidly scavenge photogenerated charge carriers from light harvesting materials through strong electrostatic interactions and allow effective photochemical water splitting.31–34 Despite such unique and promising features, POMs have rarely been studied as CO2RR catalysts; even POMs with a Cu-based active site were only tested for the HER.30 Among the reported POMs for the CO2RR, only a few POMs, which have highly complicated structures and are thus very difficult to synthesize, allow CH4 production while most POMs can catalyse CO production more efficiently than CH4.35–37
In this study, we first report the synthesis of a simple Zn-based POM (ZnPOM) and its application in a selective photochemical CO2RR to CH4. ZnPOM was readily prepared under mild conditions and its molecular structure was investigated using X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and extended X-ray absorption fine structure (EXAFS) analyses. ZnPOM showed catalytic activity for the CO2RR and allowed highly selective CH4 production under visible light irradiation in the presence of Ir-based photosensitizers (TIr3). The selectivity against CH4 was 97.8% when ZnPOM was used as a CO2RR catalyst (Scheme 1b). Static and time-resolved photoluminescence analyses revealed that photoexcited electrons of TIr3 photosensitizers rapidly transfer to ZnPOM. Density of states (DOS) calculations showed that ZnPOM can have strong electronic interactions with a bound CO intermediate (*CO) due to the significant overlap of the d-orbital of Zn in ZnPOM and the p-orbital of C in *CO, while metallic Zn cannot due to having no overlap. This explains how Zn—known as a CO-producing catalyst38,39—in ZnPOM can lead to selective production of CH4. This study provides insights for more flexible design and synthesis of CO2RR catalysts, especially for CH4 production.
Results and discussion
We synthesized ZnPOM with a molecular formula of [Zn4(PW9O34)2]10− for the CO2RR. There are a family of sandwich-type phosphotungstate POMs ([M(II)4(PW9O34)2]10−) embedded with various transition metal ions [M(II)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Co2+, Ni2+, Mn2+, Cu2+] (MPOM). However, to the best of our knowledge, there is no report on the synthesis of sandwich-type POMs with Zn ions. Conventionally, Zn- and Cu-based materials are known to produce CO and hydrocarbons, respectively, upon the CO2RR. Thus, we prepared ZnPOM and CuPOM and compared their catalytic activity for the CO2RR (Scheme 1b). It is expected that ZnPOM can be prepared by the same method for the synthesis of CuPOM, which was reported by Hill et al.,30 due to the similarity between Zn and Cu ions in terms of valence charge and ionic radius (74 and 73 pm, respectively).
Co2+, Ni2+, Mn2+, Cu2+] (MPOM). However, to the best of our knowledge, there is no report on the synthesis of sandwich-type POMs with Zn ions. Conventionally, Zn- and Cu-based materials are known to produce CO and hydrocarbons, respectively, upon the CO2RR. Thus, we prepared ZnPOM and CuPOM and compared their catalytic activity for the CO2RR (Scheme 1b). It is expected that ZnPOM can be prepared by the same method for the synthesis of CuPOM, which was reported by Hill et al.,30 due to the similarity between Zn and Cu ions in terms of valence charge and ionic radius (74 and 73 pm, respectively).
First, we investigated the properties and molecular structure of ZnPOM by using various characterization methods. The as-prepared ZnPOM was completely dissolved in an aqueous solution (Fig. S1, ESI†). According to dynamic light scattering and zeta potential (Fig. 1a) analyses, the average diameter and zeta potential of ZnPOM were 0.83 nm and −26.13 mV, respectively. These results are consistent with those of other sandwich-type POMs29 and confirm the molecular nature of ZnPOM rather than the particulate counterpart. XPS analysis confirmed the presence of Zn, W, K, and O atoms of ZnPOM (Fig. S2, ESI†). However, P was not identified possibly due to its relatively low content. Fourier-transform infrared (FT-IR) spectroscopy also showed peaks corresponding to P–O and W–O bands of sandwich-type POMs (Fig. S3, ESI†).30 To investigate the molecular structure of ZnPOM, we obtained crystallographic data using single crystal XRD after the crystallization of supersaturated ZnPOM from hot water (Table S1, ESI†). As expected, the crystal structure and atomic arrangement of ZnPOM (K10[Zn4(H2O)2(PW9O34)2]·24H2O) were almost identical to those of CuPOM, except for the types of transition metal ions embedded in metal-oxo clusters (i.e., Zn2+vs. Cu2+). The simulated XRD pattern from the single crystal XRD analysis well matched the measured powder XRD pattern (Fig. 1b). ZnPOM was further characterized by EXAFS analysis to check the coordination environment of Zn ions in ZnPOM (Fig. 1c and d, ESI†). The distances between Zn2+ and the nearest neighbouring elements in ZnPOM were 1.5 (Zn–O), 2.88 (Zn–W1), and 3.37 Å (Zn–W2), which were similar to those in CuPOM, respectively (Fig. S4, ESI†). All these results confirmed the successful synthesis of molecular CuPOM by the same method as that of ZnPOM. High purity of the synthesized ZnPOM (>99.9%) was confirmed by 31P NMR (Fig. S5, ESI†).
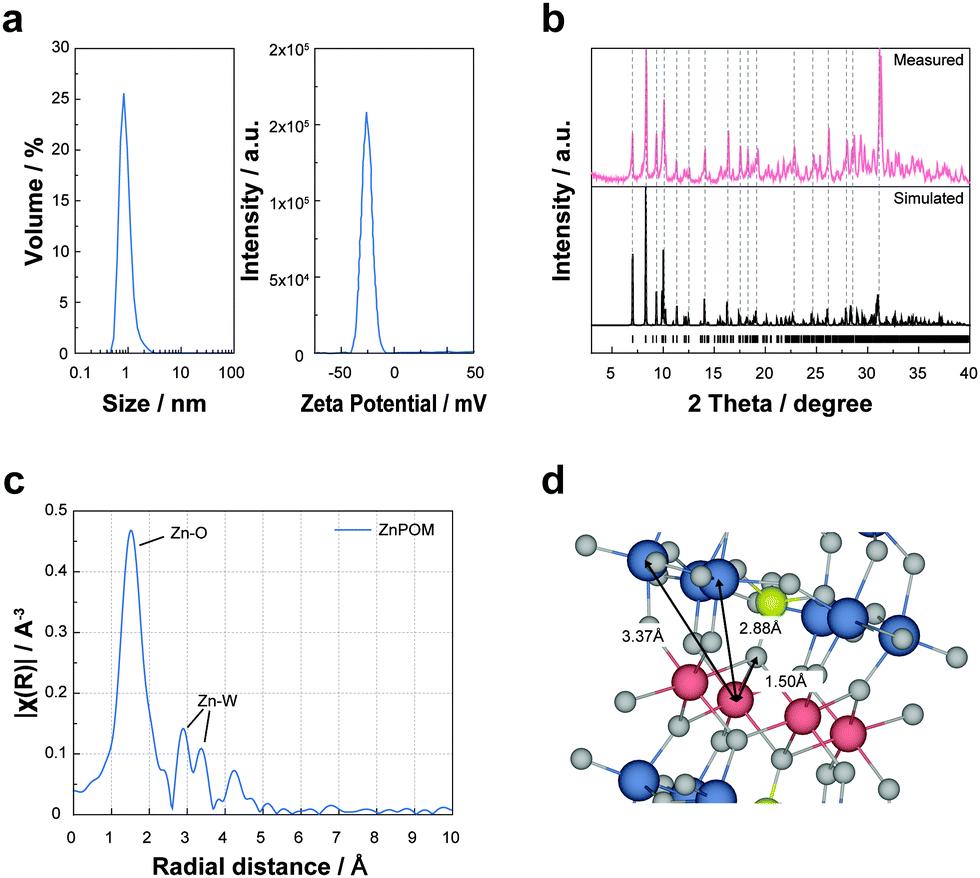 | ||
| Fig. 1 Characterization of ZnPOM. (a) Dynamic light scattering (left) and zeta potential (right) analyses of ZnPOM. (b) Simulated and measured XRD patterns of ZnPOM. (c) EXAFS analysis showing the distance between Zn and its nearest neighbouring elements in ZnPOM. (d) A graphical illustration showing the molecular structure of ZnPOM and coordination environment of Zn2+ ions in ZnPOM. | ||
Next, physicochemical properties of ZnPOM were investigated for the CO2RR. First, cyclic voltammetry (CV) of ZnPOM was conducted in 0.1 M KHCO3 saturated with Ar or CO2 gas (Fig. 2a). For both cases, there was rapid increase of current density without saturation at highly negative potentials possibly due to the HER. However, there was a pair of redox peaks at around −0.5 V vs. a reversible hydrogen electrode (RHE; −0.9 V vs. standard hydrogen electrode [SHE]) only when CO2 was purged, suggesting a possibility of ZnPOM as a CO2RR catalyst. Encouraged by these results, we carried out photochemical CO2RR using ZnPOM in the presence of Ir-based photosensitizers (TIr3) because ZnPOM has no or a negligible absorbance in the visible light region (Fig. 2b and Fig. S6, ESI†). TIr3 was chosen as a photosensitizer because it has sufficiently high reducing power for the CO2RR with the lowest unoccupied and highest occupied molecular orbital (LUMO and HOMO) levels of −3.08 and −5.62 eV vs. vacuum (−1.36 and 1.18 V vs. SHE), respectively.40 TIr3 had strong absorption in the UV and visible light region up to 500 nm. Interestingly, the absorbance of TIr3 with ZnPOM was significantly higher than their respective absorbance and arithmetic sum (Fig. 2b), suggesting strong molecular interactions between positively charged TIr3 and negatively charged ZnPOM. The wavelength-dependent apparent quantum yield of our photocatalytic CH4 production system showed the maximum efficiency at 400 nm and was in accordance with the absorbance spectra (Fig. S7, ESI†). CV analysis showed that TIr3 has no activity towards the CO2RR and can strongly interact with ZnPOM, as can be expected from the increase of current density of the redox pair at around −0.5 V vs. RHE in the presence of both (Fig. S8, ESI†). On the other hand, CuPOM had not only a negligible absorbance in the visible light region but also weak interactions with TIr3 in terms of absorbance (Fig. S9, ESI†).
 | ||
| Fig. 2 Photochemical CO2RR using ZnPOM as a catalyst and TIr3 as a photosensitizer. (a) CV analysis of ZnPOM in 0.1 M KHCO3 saturated with Ar or CO2 gas. (b) UV/visible absorbance spectra of each component and both in 0.1 M KHCO3. (c) Production and (d) selectivity of various chemicals upon photochemical CO2RR using 30 μM ZnPOM, 0.1 mM TIr3, and 0.05 M Na2SO3 under visible light irradiation. The selectivity of each product was calculated after 6 h of photochemical CO2RR by considering the number of electrons required to produce each. | ||
Photochemical reactions were conducted using ZnPOM (or CuPOM) to check its activity as a CO2RR catalyst without any external bias. TIr3 and Na2SO3 were used as a photosensitizer and a sacrificial electron donor, respectively. Under the optimum conditions with TIr3 at 0.1 mM, ZnPOM at 30 μM, and Na2SO3 at 50 mM, only CH4 was selectively produced with negligible or erroneous amounts of H2 and CO (Fig. S10, ESI†). Under continuous visible light irradiation, the amount of CH4 linearly increased and reached 4.16 μmol per unit mass of ZnPOM after 6 h (Fig. 2c). There were no other liquid products. Although the CH4 production yield of this study is not record-high, it was comparable to those in previous reports on the photocatalytic CO2RR (Table S2, ESI†). The product selectivity of ZnPOM towards a certain chemical (A) was calculated using the following equation that considers the number of electrons (n) required to make each product (Fig. 2d):
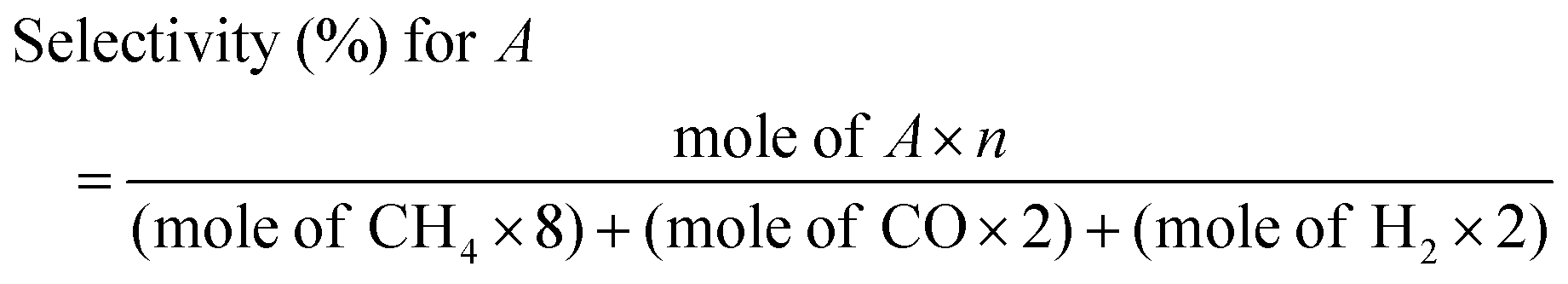
Static and dynamic photoluminescence quenching analyses were performed to confirm the electron transfer pathway in our photocatalytic system. Strong photoluminescence of TIr3 in the range of 500–700 nm was dramatically quenched as the concentration of ZnPOM increased (Fig. 3a). Photoluminescence of TIr3 disappeared immediately after the addition of ZnPOM, which can be observed even by the naked eye. Because the LUMO level of TIr3 (−0.8 V vs. RHE) is more negative than that of ZnPOM (−0.6 V vs. RHE), one can expect that photoexcited electrons from TIr3 can be transferred to ZnPOM, resulting in photoluminescence quenching (Fig. S8b and c, ESI†). To quantify the charge transfer dynamics, the photoluminescence lifetime of TIr3 was recorded in the absence and presence of an electron donor (Na2SO3) or ZnPOM (Fig. 3b). The photoluminescence lifetime of TIr3 dramatically decreased from 820 to 64 ns after the addition of ZnPOM, which well agrees with the static photoluminescence quenching results. In contrast, there was little change in the lifetime after the addition of Na2SO3, which rules out the reductive quenching pathway of TIr3. Thus, we can conclude that the main charge transfer pathway in this catalytic network follows the oxidative quenching pathway (Fig. 3c). Based on the ratio between the photoluminescence of TIr3 without (I0) and with ZnPOM (I), the bimolecular rate constant (KSV) was calculated from the Stern–Volmer equation (Fig. S15, ESI†). KSV for TIr3 and ZnPOM was 1.76 × 1011 M−1 s−1, which is remarkably fast compared to those in previously reported catalytic systems (Table S3, ESI†).
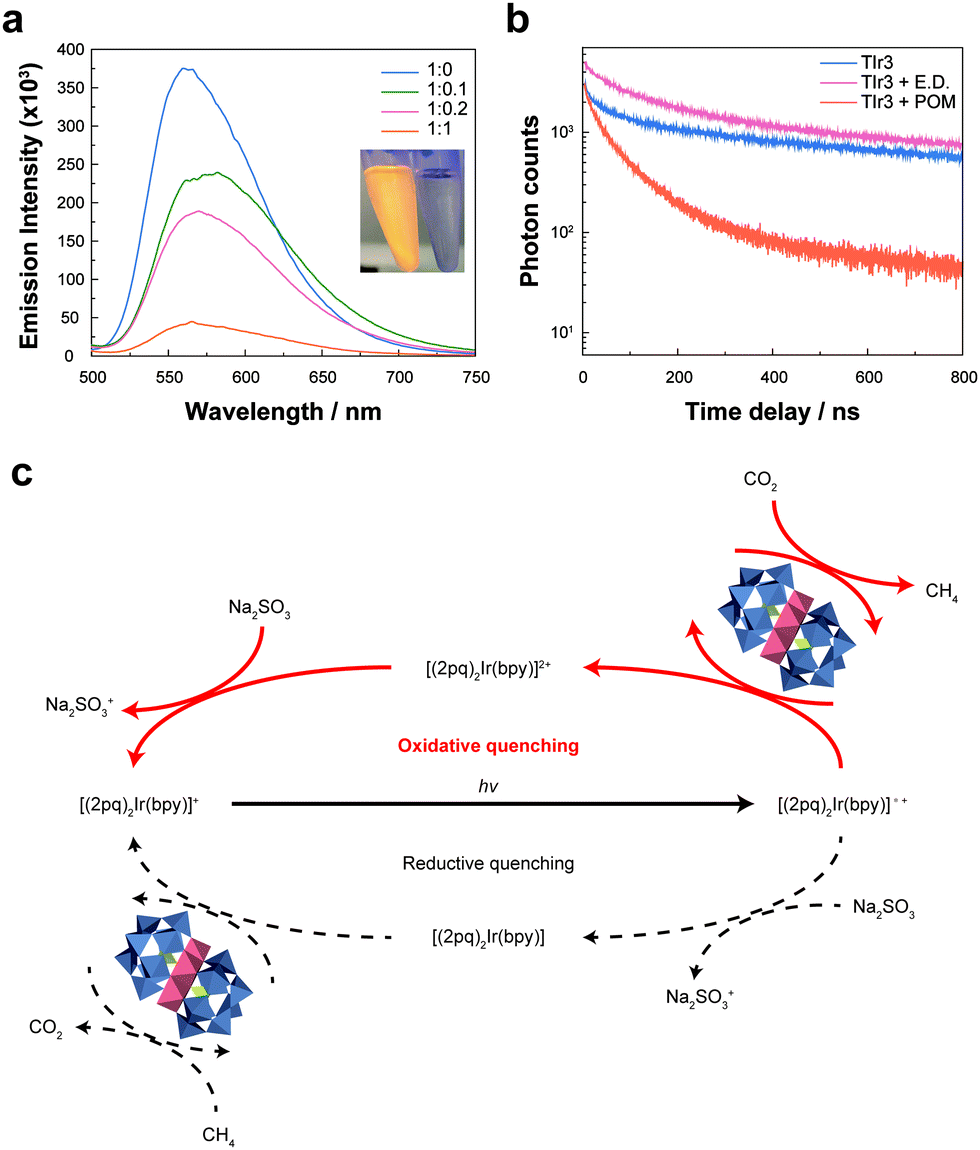 | ||
| Fig. 3 Photophysical analysis of photocatalytic CO2RR systems. (a) Static photoluminescence spectra of TIr3 (50 μM) without (blue line) and with ZnPOM at different concentration ratios of TIr3 and ZnPOM. The inset shows a photograph of TIr3 solutions under UV light without (left) and with (right) ZnPOM (50 μM). (b) Time-correlated single-photon counting for lifetime measurements of TIr3 without and with POM or sacrificial electron donor (E.D.) Na2SO3. (c) A suggested mechanistic pathway for the photocatalytic CO2RR to CH4 using ZnPOM and TIr3. | ||
DOS calculations from density functional theory were carried out to unveil the molecular mechanism for the selective production of CH4 by ZnPOM. Because metallic Zn and Zn-based materials are known to produce CO,41,42 we compared the DOS of Zn ions in ZnPOM with that of metallic Zn. In addition, one of the most important factors determining the products of the CO2RR is electronic interactions between catalysts and intermediate states.21,43 In particular, *CO is an important intermediate for the production of CH4. Our calculations showed that the DOS of the Zn atom (d-orbital) in ZnPOM significantly overlaps with that of the C atom (p-orbital) in *CO (Fig. 4a). In contrast, the DOS of the metallic Zn does not overlap with that of the C atom (p-orbital) in *CO (Fig. 4b). Thus, one can expect that the strong electronic hybridization between ZnPOM and the *CO intermediate allows the facile transfer of electrons for further reactions of *CO to CH4. To investigate the reaction mechanism more in detail, the binding energy between the key intermediate *CO and POMs was calculated (Fig. 4c). The binding energies of CuPOM, ZnPOM, and CoPOM were −0.48, −0.24, and +0.57 eV, respectively. Our results are consistent with the previous report on the relationship between the *CO binding energy and product selectivity in the CO2RR.1 While CuPOM strongly binding to *CO led to competition between the HER and CO2RR, CoPOM, which cannot easily bind with *CO, has poor activity for the CO2RR (Fig. S14, ESI†). In contrast, ZnPOM binding neither too strongly nor too weakly with *CO is advantageous for the selective production of CH4.
 | ||
| Fig. 4 DFT calculations of density of states and *CO binding energy of the respective materials. (a) The calculated DOS of the p-orbital of C in *CO and d-orbital of Zn in ZnPOM. (b) The calculated DOS of the p-orbital of C in *CO and d-orbital of Zn in metallic Zn. The insets show preferred products from the CO2RR by each catalyst (i.e., ZnPOM and Zn). (c) The calculated *CO binding energies for CuPOM, ZnPOM, and CoPOM and their implication in product selectivity for the CO2RR, according to the previous study.43 | ||
In this study, we first report the synthesis of ZnPOM as a CO2RR catalyst for the selective production of CH4 upon photocatalytic CO2RR. Conventionally, metallic Zn or Zn-based materials are known as CO producing catalysts, and CH4 producing catalysts have been limited to Cu-based materials. Although the efficiency of our catalyst should be further improved for practical application, this is the first report that Zn-based POMs can be used as a CH4 production catalyst. In this regard, this study provides insights and broadens the material library for the synthesis of novel CO2RR catalysts, especially for the selective production of hydrocarbons including CH4. Our analyses based on the absorbance and photoluminescence measurements suggest that strong molecular interactions exist between ZnPOM and TIr3. Different CO2RR activity of Zn—known to produce CO—and ZnPOM was also investigated with DOS calculations, which show significant overlap between the d-orbital of Zn2+ in ZnPOM and the p-orbital of C in *CO. Considering that *CO is an important intermediate for the production of CH4 and sandwich-type POMs can facilitate multiple electron transfer,27 these results explain why ZnPOM can selectively produce CH4 upon photochemical CO2RR.
Conclusions
To summarize, we first report the synthesis of ZnPOM and introduce a photochemical CO2RR system using ZnPOM as a CO2RR catalyst and TIr3 as a photosensitizer. According to the absorbance and quenching analyses, strong molecular interactions occurred between ZnPOM and TIr3, resulting in an exceptionally high bimolecular rate constant and rapid transfer of photoexcited electrons from TIr3 to ZnPOM. According to density functional theory calculations, ZnPOM can have electronic interactions with *CO due to the significant overlap of the DOS between Zn ions in ZnPOM and C in *CO. As a result, ZnPOM can selectively produce CH4 upon the photochemical CO2RR. Our results provide insights for the synthesis of cheap and efficient CO2RR catalysts using ZnPOM, especially for CH4 production.Experimental
Materials
ZnCl2 and KCl were purchased from Alfa Aesar (USA). Na2WO4·2H2O, acetic acid, and Na2SO3 were obtained from Sigma-Aldrich (USA). Na2HPO4 was purchased from VWR Life Science.Synthesis of [PW9O34]9−
Na2WO4·2H2O (120 g) was dissolved in 150 mL of deionized water. Then, 8.17 g of Na2HPO4 and 22 mL of acetic acid were added into the Na2WO4 solution. After 1 min, a white precipitate of [PW9O34]9− was formed, collected by vacuum filtration, and heated at 140 °C for 6 h in air.Synthesis of K10[Zn4(H2O)2(PW9O34)2]·24H2O
ZnCl2·2H2O (0.99 g) was dissolved in 12 mL of deionized water. Then, 5.1 g of [PW9O34]9− was added into the ZnCl2 solution and stirred for 10 min. After adding 1 g of KCl, a white precipitate of K10[Zn4(H2O)2(PW9O34)2]·24H2O formed and was collected by vacuum filtration.Synthesis of TIr3
TIr3 was prepared according to a previously reported paper.40 Briefly, IrCl3·H2O (298 mg, 1.00 mmol) and 2-phenylquinoline (431 mg, 2.10 mmol) were dissolved in a mixture of 2-methoxyethanol and H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v%) and refluxed for 24 h. After cooling to room temperature, distilled water was poured to form reddish brown precipitates. The precipitates were filtered and washed with distilled water, n-hexane, and cooled ether, respectively. The crude product of di-μ-chlorotetrakis[2-(2-quinolinyl-N)phenyl-C] diiridium(III) was acquired and used without any further purification (401 mg, 63.0%). Di-μ-chlorotetrakis[2-(2-quinolinyl-N)phenyl-C]diiridium(III) (150 mg, 0.123 mmol) and 2,2′-bipyridine (39 mg, 0.25 mmol) were dissolved in a mixture of dichloromethane and methanol (1:1, v/v%), refluxed overnight, concentrated in vacuo, and then re-dissolved in ethanol. Undissolved solid was filtered out and filtrate was concentrated in vacuo again. The orange product of TIr3 was obtained after the precipitation process with methylene chloride and n-hexane (137 mg, 70.2%).
:1, v/v%) and refluxed for 24 h. After cooling to room temperature, distilled water was poured to form reddish brown precipitates. The precipitates were filtered and washed with distilled water, n-hexane, and cooled ether, respectively. The crude product of di-μ-chlorotetrakis[2-(2-quinolinyl-N)phenyl-C] diiridium(III) was acquired and used without any further purification (401 mg, 63.0%). Di-μ-chlorotetrakis[2-(2-quinolinyl-N)phenyl-C]diiridium(III) (150 mg, 0.123 mmol) and 2,2′-bipyridine (39 mg, 0.25 mmol) were dissolved in a mixture of dichloromethane and methanol (1:1, v/v%), refluxed overnight, concentrated in vacuo, and then re-dissolved in ethanol. Undissolved solid was filtered out and filtrate was concentrated in vacuo again. The orange product of TIr3 was obtained after the precipitation process with methylene chloride and n-hexane (137 mg, 70.2%).
Elemental and structural characterization
The elemental composition of ZnPOM was characterized by K-alpha X-ray photoelectron spectroscopy (XPS) (Thermo Fisher Scientific, USA). FT-IR spectra were measured on a Varian 670-IR (Agilent Technologies, USA) using KBr pellets. UV/visible absorbance of each material was confirmed with a V-730 UV-Visible spectrophotometer (JASCO, Japan). For X-ray diffraction (XRD) analysis, microcrystals of ZnPOM were obtained by crystallization in the supersaturated solution. Single-crystal and powder XRD data were collected by an R-AXIS RAPID II (Rigaku, Japan) and a powder XRD D2 PHASER (Bruker, USA), respectively. Extended X-ray absorption fine structure (EXAFS) analysis was performed at the beamline 6D of the Pohang Light Source in the Pohang Accelerator Laboratory in Korea. The ZnPOM particle size was analysed with a Zeta sizer (Malvern, England). 31P NMR spectra were collected by an AVANCE III HD (Bruker, USA).Electrochemical and photochemical characterization
Cyclic voltammetry (CV) was carried out with a WMOG1000 multichannel potentiostat (WonA Tech CO. Ltd, Korea) in a three-electrode configuration: a working electrode, glassy carbon; a reference electrode, Ag/AgCl; a counter electrode, fluorine-doped tin oxide coated with 100 nm thick Pt; electrolyte, 0.1 M KHCO3 (pH 7.0) saturated with Ar or CO2 gas; and scan rate, 10 mV s−1. For the optimal photochemical CO2RR, 4 mL of a 0.1 M KHCO3 solution with 30 μM ZnPOM, 0.1 mM Ir-complex, and 0.05 M Na2SO3 in 0.1 M KHCO3 was used in a 10 mL vial. Gas products were collected from a sealed reaction vessel using an air-tight syringe (1 mL) and analysed with a GC-2010 plus gas chromatograph (Shimadzu Co., Japan) equipped with an FID and MICROPACKED ST column (Shinwa Chemical Industries Ltd, Japan). Product yields were calculated by dividing the amount of products by the amount of catalyst. Liquid products were measured using a Water 2410 HPLC (Waters, USA). A 300 W Xe lamp equipped with a 400 nm cut-on filter (100 mW cm−2) was used as a light source.Photoluminescence quenching analysis
TIr3 (50 μM) was mixed with POM at molar ratios of 1:0, 1:0.1, 1:0.5, and 1:1 in distilled water. The photoluminescence intensity of the prepared solutions was recorded with a FLS920 (Edinburgh Instruments, UK) and a PC1 (ISS, USA). Bimolecular rate constant (KSV) from photoluminescence quenching was calculated through Stern–Volmer plot by maximum photoluminescence intensity for each mixture ratio.
Time-correlated single photon counting for lifetime measurements
The photoluminescence lifetime of TIr3 was recorded with and without potential quenchers (e.g., ZnPOM or Na2SO3) using a FluoTime300 time-correlated single photon counting setup (TCSPC) (PicoQuant, Berlin, Germany). A LDH-D-C-450 pulsed diode laser coupled with a PDL 820 laser diode driver (<70 ps pulse width and 196 kHz–40 MHz repetition rate) was used as a photoexcitation source. The time-resolved exciton decay was measured with a PicoHarp 300E TCSPC module and a PMA-C 182-N-M photomultiplier tube. Fitting was carried out using FluoFit software to calculate the corresponding photoluminescence lifetimes.Density functional theory calculations
The Vienna Ab initio Simulation Package (VASP)44 was used to calculate relaxed structures and density of states. The generalized gradient approximation (GGA) method was adopted to describe the exchange correlation functional. The pseudopotentials generated under the projector-augmented wave (PAW) method45 were used and the energy cutoff for the plane wave basis set was set to 500 eV. The force criterion for optimizing the atomic positions was 5 × 10−3 eV Å−1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Climate Change Response (2017M1A2A2087813, and 2019M1A2A2065616), the Nano-Material Technology Development (2017M3A7B4052798), the Carbon to X (2020M3H7A1098231), the Science Research Center (SRC, 2016R1A5A1009405), and the Creative Materials Discovery (2017M3D1A1040828) programs through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT of Korea. This work was also supported by the Basic Science Research Program (2018R1D1A1A02046918) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education of Korea. This work was supported by the New Renewable Energy Core Technology Development Project of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) granted financial resources from the Ministry of Trade, Industry & Energy (No. 20183010013900). J. S. N. is thankful for the support of the ASAN Foundation Biomedical Science scholarship.Notes and references
- J. Hussain, H. Jónsson and E. Skúlason, ACS Catal., 2018, 8, 5240–5249 CrossRef CAS.
- F. Li, D. R. MacFarlane and J. Zhang, Nanoscale, 2018, 10, 6235–6260 RSC.
- Q. Lu and F. Jiao, Nano Energy, 2016, 29, 439–456 CrossRef CAS.
- D. Raciti and C. Wang, ACS Energy Lett., 2018, 3, 1545–1556 CrossRef CAS.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2019, 10, 1754–1768 CrossRef.
- M. Q. Yang and Y. J. Xu, Nanoscale Horiz., 2016, 1, 185–200 RSC.
- Y. Chen, C. Liu, S. Guo, T. Mu, L. Wei and Y. Lu, Green Energy Environ., 2020 DOI:10.1016/j.gee.2020.11.008.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- C. Xu, A. Vasileff, D. Wang, B. Jin, Y. Zheng and S.-Z. Qiao, Nanoscale Horiz., 2019, 4, 1411–1415 RSC.
- S. F. Ng, M. Y. L. Lau and W. J. Ong, Sol. RRL, 2020 DOI:10.1002/solr.202000535.
- Z. Qin, J. Wu, B. Li, T. Su and H. Ji, Acta Biochim. Biophys. Sin., 2020, 37, 2005020 Search PubMed.
- X. Jiao, K. Zheng, L. Liang, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2020, 49, 6592–6604 RSC.
- W. J. Ong, L. K. Putri and A. R. Mohamed, Chem. – Eur. J., 2020, 26, 9710–9748 CrossRef CAS PubMed.
- N. Zhang, C. Gao and Y. Xiong, J. Energy Chem., 2019, 37, 43–57 CrossRef.
- Y. Bo, C. Gao and Y. Xiong, Nanoscale, 2020, 12, 12196–12209 RSC.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans., 1989, 85, 2309–2326 RSC.
- C. Kim, S. Hyeon, J. Lee, W. D. Kim, D. C. Lee, J. Kim and H. Lee, Nat. Commun., 2018, 9, 3027 CrossRef PubMed.
- S. Park, D. Bezier and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11404–11407 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- M. Bonchio, M. Carraro, A. Sartorel, G. Scorrano and U. Kortz, J. Mol. Catal. B: Enzym., 2006, 251, 93–99 CrossRef CAS.
- J. M. Sumliner, H. Lv, J. Fielden, Y. V. Geletii and C. L. Hill, Eur. J. Inorg. Chem., 2014, 635–644 CrossRef CAS.
- S. Berardi, M. Carraro, A. Sartorel, G. Modugno and M. Bonchio, Isr. J. Chem., 2011, 51, 259–274 CrossRef CAS.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS.
- A. Sartorel, M. Carraro, G. Scorrano, R. De Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006–5007 CrossRef CAS PubMed.
- H. Lv, J. Song, H. Zhu, Y. V. Geletii, J. Bacsa, C. Zhao, T. Lian, D. G. Musaev and C. L. Hill, J. Catal., 2013, 307, 48–54 CrossRef CAS.
- H. Lv, W. Guo, K. Wu, Z. Chen, J. Bacsa, D. G. Musaev, Y. V. Geletii, S. M. Lauinger, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 14015–14018 CrossRef CAS PubMed.
- H. Lv, Y. Gao, W. Guo, S. M. Lauinger, Y. Chi, J. Bacsa, K. P. Sullivan, M. Wieliczko, D. G. Musaev and C. L. Hill, Inorg. Chem., 2016, 55, 6750–6758 CrossRef CAS PubMed.
- D. Jeon, H. Kim, C. Lee, Y. Han, M. Gu, B. S. Kim and J. Ryu, ACS Appl. Mater. Interfaces, 2017, 9, 40151–40161 CrossRef CAS PubMed.
- D. Jeon, N. Kim, S. Bae, Y. Han and J. Ryu, ACS Appl. Mater. Interfaces, 2018, 10, 8036–8044 CrossRef CAS.
- H. Kim, S. Bae, D. Jeon and J. Ryu, Green Chem., 2018, 20, 3732–3742 RSC.
- M. Orlandi, R. Argazzi, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and F. Scandola, Chem. Commun., 2010, 46, 3152–3154 RSC.
- S. Das, T. Balaraju, S. Barman, S. S. Sreejith, R. Pochamoni and S. Roy, Front. Chem., 2018, 6, 514 CrossRef CAS PubMed.
- S.-M. Liu, Z. Zhang, X. Li, H. Jia, M. Ren and S. Liu, Adv. Mater. Interfaces, 2018, 5, 1801062 CrossRef.
- S. L. Xie, J. Liu, L. Z. Dong, S. L. Li, Y. Q. Lan and Z. M. Su, Chem. Sci., 2019, 10, 185–190 RSC.
- J. Rosen, G. S. Hutchings, Q. Lu, R. V. Forest, A. Moore and F. Jiao, ACS Catal., 2015, 5, 4586–4591 CrossRef CAS.
- F. Quan, D. Zhong, H. Song, F. Jia and L. Zhang, J. Mater. Chem. A, 2015, 3, 16409–16413 RSC.
- J. S. Nam, M. G. Kang, J. Kang, S. Y. Park, S. J. Lee, H. T. Kim, J. K. Seo, O. H. Kwon, M. H. Lim, H. W. Rhee and T. H. Kwon, J. Am. Chem. Soc., 2016, 138, 10968–10977 CrossRef CAS PubMed.
- W. Luo, J. Zhang, M. Li and A. Züttel, ACS Catal., 2019, 9, 3783–3791 CrossRef CAS.
- Y. Lu, B. Han, C. Tian, J. Wu, D. Geng and D. Wang, Electrochem. Commun., 2018, 97, 87–90 CrossRef CAS.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2033397. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0nh00657b |
| This journal is © The Royal Society of Chemistry 2021 |