
Polarizability is a key parameter for molecular electronics†
Angélique
Gillet
a,
Sébastien
Cher
a,
Marine
Tassé
b,
Thomas
Blon
a,
Sandra
Alves
c,
Guillaume
Izzet
c,
Bruno
Chaudret
 a,
Anna
Proust
c,
Phillipe
Demont
d,
Florence
Volatron
*c and
Simon
Tricard
*a
a,
Anna
Proust
c,
Phillipe
Demont
d,
Florence
Volatron
*c and
Simon
Tricard
*a
aLaboratoire de Physique et Chimie des Nano-Objets, INSA, CNRS, Université de Toulouse, Toulouse, France. E-mail: tricard@insa-toulouse.fr
bLaboratoire de Chimie de Coordination, CNRS, Université de Toulouse, Toulouse, France
cInstitut Parisien de Chimie Moléculaire, CNRS, Sorbonne Université, Paris, France. E-mail: florence.volatron@sorbonne-universite.fr
dInstitut Carnot – Centre Inter-universitaire de Recherche et d’Ingénierie des Matériaux, INP-ENSIACET, CNRS, Université de Toulouse, Toulouse, France
First published on 18th January 2021
Abstract
Identifying descriptors that govern charge transport in molecular electronics is of prime importance for the elaboration of devices. The effects of molecule characteristics, such as size, bulkiness or charge, have been widely reported. Herein, we show that the molecule polarizability can be a crucial parameter to consider. To this end, platinum nanoparticle self-assemblies (PtNP SAs) are synthesized in solution, including a series of polyoxometalates (POMs). The charge of the POM unit can be modified according to the nature of the central heteroatom while keeping its size constant. POM hybrids that display remote terminal thiol functions strongly anchor the PtNP surface to form robust SAs. IV curves, recorded by conductive AFM, show a decrease in Coulomb blockade as the dielectric constant of the POMs increases. In this system, charge transport across molecular junctions can be interpreted as variations in polarizability, which is directly related to the dielectric constant.
New conceptsEven though molecular electronics has been developed for more than twenty years, and already explored using polyoxometalates, the electronic behavior in such systems has commonly been interpreted according to the electronic levels of the molecular constituents. The new concept here is to think about electron transport across molecules in terms of polarizability, which is the propensity of the electronic density of a molecule to reorganize under an electrical field. To vary polarizability only in a series of molecules, new hybrid polyoxometalates have been synthesized, whose physical properties have been adjusted by modifying the nature of their central heteroatom. As polarizability is directly linked to the dielectric constant, commonly used by several communities of physicist – e.g. in condensed matter, micro and nanoelectronics – the present work opens a new bridge between two ways of thinking about charge transport. |
One of the main objectives of molecular electronics is to relate the performance of devices to the structure and electronic state of molecules.1–3 A still open question is what molecular parameters are important to describe the transport of an electron across a molecule.4,5 The discussion started more than twenty years ago,6 and first considered resistive organic molecules as tunnel barriers, the shape of which could be tuned by the molecular functional moieties,7,8 although some examples showed it was not systematical.9–12 In a first approximation, the current depends exponentially on the length of the molecule, i.e. on the width of the tunnel barrier, as stated by Simmons's equation,13,14 but other considerations state that charge transfer processes should also be taken into account to describe thermally activated transport phenomena.15 Depending on the system, charge transport across molecules occurs though a competition between tunneling and hopping mechanisms.16,17 In the last mechanism, the electron gets relaxed and remains temporarily trapped in the accessible orbitals of the molecules, and must be thermally activated to hop on and off. By designing asymmetric molecules where the two mechanisms coexist, it has been possible to fabricate sophisticated molecular electronic components, e.g. rectifiers.18,19 Here, the nature and accessibility of the orbitals of the molecular junction are a clearly identified parameter that governs the electron transport mechanism, in addition to the molecule size and bulkiness.20 Some studies also reported that the charge of the molecule can have a significant effect.21,22 However, very few studies consider the polarizability of the molecule as a key parameter.23–25 Here, we show that the charge effect on electric transport through molecules in nanoparticle self-assemblies can be interpreted as variation of their dielectric constant, which is directly related to their polarizability.
Among the diverse approaches for studying molecular junctions, nanoparticle (NP) self-assemblies (SAs) with functional molecules have been extensively studied, as NPs can be considered as nanoelectrodes that connect molecules.26–30 In such systems, a large number of molecules are involved and only average behavior of the composite material is measured. The equivalent circuits are usually referred to as resistors and capacitors in series, the parameters of which vary as a function of the physico-chemical properties of both the molecules and the NPs.31 Several molecular effects have been reported to influence charge transport in NP SAs, such as e.g. the molecule length,32,33 redox state,34 conjugation,35 or spin state.36 Recently, we demonstrated that using ultra-small NPs (<2 nm), it was possible to observe Coulomb blockade at room temperature in NP SAs.37 This finding was of particular interest since such a blockade was observed at any scale of measurement: nano, micro, or macro. Charge transport was then determined by three parameters: the size of the NPs, the distance between them, and the polarizability of the molecules. However, even if we were able to dissociate effects of such three parameters, it remained very challenging to vary only one at a time. In the present study, we focused on polarizability effects, and thus designed the systems in order to keep the NP size constant (by using the same starting ultra-small PtNP), and the inter-particle distance constant (by choosing a series of molecules of the same size). Polarizability was varied by changing the chemical composition of the molecules.
Polyoxometalates (POMs) are polyanionic metal-oxides made of early transition metals (e.g. W, Mo) and often contain one or several central heteroatoms (e.g. B, P, Al). They have been exploited for use in molecular electronics, including memories,38,39 polarizers,40 rectifiers,41 or magnetic transistors.42 Electrical studies have been performed with POMs in diverse forms, such as for example thin layers,43 monolayers,44 dots on surfaces,45 or SAs with quantum dots.46 There, measurements were performed on assemblies of molecules, but thanks to their robustness, POMs are also suitable for experiments on single molecules,47e.g. either soldered in a junction48 or addressed under an STM tip.49 In the present study, we performed electrical measurements on SAs including PtNPs and POMs. POMs can be functionalized through the covalent anchorage of one or several organic moieties that will interact with the NP surface in order to form stable and robust SAs.46,50–52 In addition, isostructural POMs displaying different charges can be synthesized by changing the nature of their central heteroatom(s).53 In the present study, we developed a new series of POM hybrids, based on the Keggin structure, displaying remote thiol functions, where the central atom was varied from Al, to Si and P. The modification of the heteroatom tunes some of their physico-chemical properties such as their charge, and so their polarizability as they all have the same size.
Three POM hybrids of increasing charges, (TBA)3[PW11O39{O(SiC3H6SH)2}], (TBA)4[SiW11O39{O(SiC3H6SH)2}] and (TBA)5[AlW11O39{O(SiC3H6SH)2}], named POM-P, POM-Si and POM-Al, were synthesized by condensation of the organosilane (OCH3)3SiC3H6SH on the corresponding monolacunar POMs [XW11O39]n− in acidic medium (Fig. 1a – TBA stands for tetrabutylammonium (n-Bu)4N+). The three resulting POM hybrids were prepared as TBA salts to facilitate their dissolution in acetonitrile, in which the PtNPs were also soluble. They were characterized by NMR spectroscopy (Fig. S1–S4, ESI†), IR spectroscopy (Fig. S5, ESI†), mass spectrometry (Fig. S6–S8, ESI†), elemental analysis and cyclic voltammetry (CV – Fig. 1b). Detailed interpretation of the POM characterization is given in the supporting information and confirmed their purity and their increasing charges: 3 – for POM-P, 4 – for POM-Si and 5 – for POM-Al. In particular, the reversible and monoelectronic waves observed in CV attested to the absence of protonation. Moreover, the first reduction potential varied with the POM charge: the less charged POM-P (Ered = −0.43 V vs. SCE) was easier to reduce than POM-Si (Ered = −0.87 V vs. SCE) and POM-Al (Ered = −1.26 V vs. SCE). To support the study, a fourth POM hybrid called POM-AlH, which consisted of a protonated form of POM-Al, was also isolated. Its formula (TBA)4.1H0.9[AlW11O39{O(SiC3H6SH)2}] was assessed by combination of 1H NMR (Fig. S9, ESI†) and elemental analysis. The CV of POM-AlH displayed multiple waves, confirming its protonated state (Fig. S10, ESI†).
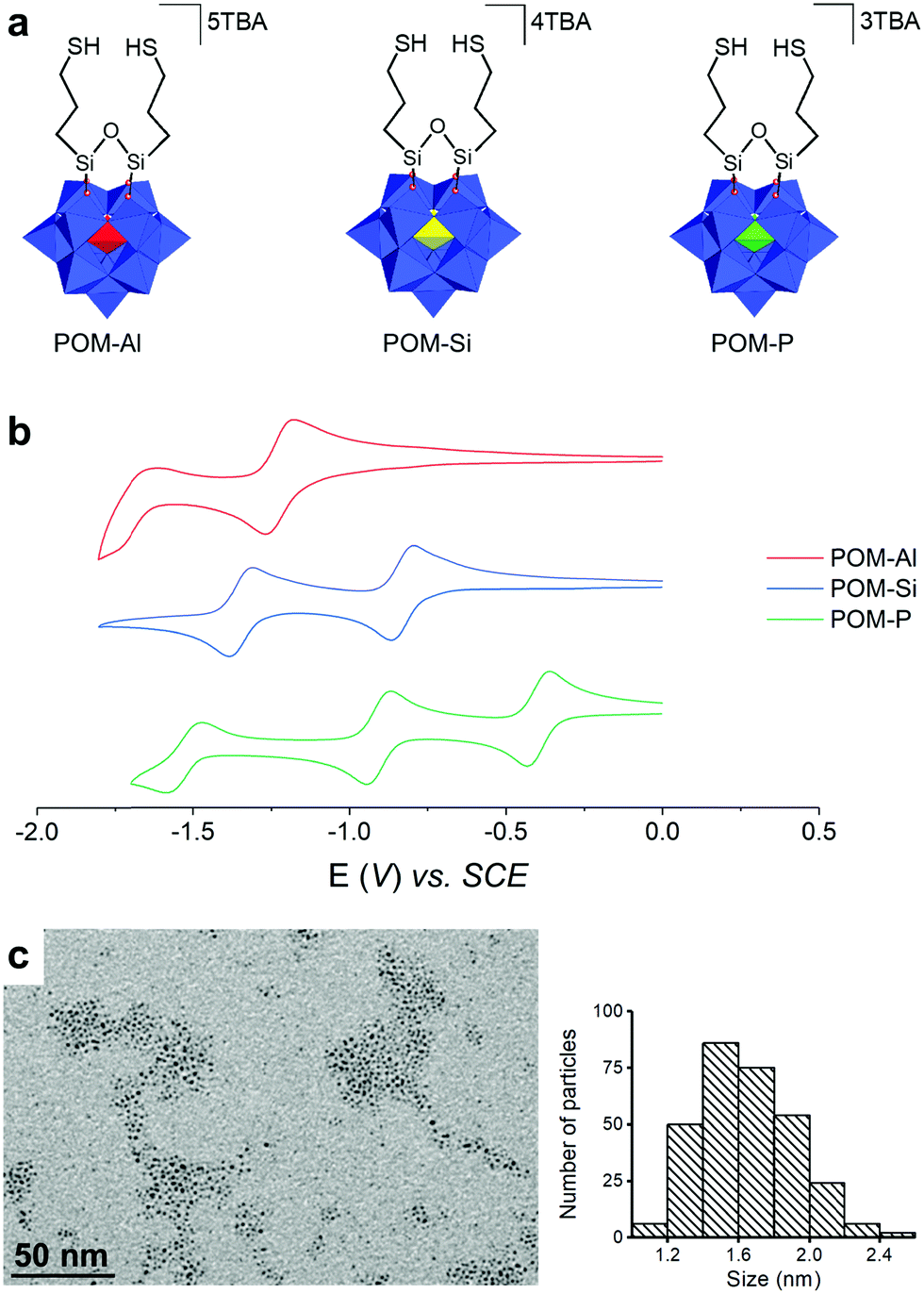 | ||
| Fig. 1 Building blocks. (a) Chemical structure of the POM-Al, POM-Si and POM-P polyanions. (b) Cyclic voltammograms at a glassy carbon electrode of POM-Al, POM-Si and POM-P (0.1 V s−1). (c) TEM picture and size distribution of the pristine ultra-small (1.7 ± 0.3 nm) Pt nanoparticles. | ||
The PtNPs were synthesized by decomposition of Pt2(dba)3 (dba = dibenzylideneacetone) under a carbon monoxide (CO) atmosphere in THF, followed by washing of the organic dba ligands with pentane, as previously reported.37,54 The obtained NPs were dissolved in acetonitrile, to have all the building blocks in the same solvent for SA. Transmission electron microscopy (TEM) pictures showed well-dispersed pristine NPs, with diameters of 1.7 ± 0.3 nm (Fig. 1c). Such NPs are particularly interesting since, in addition to their ultra-small sizes, they are “naked”, i.e. free of organic ligand, as they are only stabilized by CO from the synthesis and acetonitrile, a coordinating solvent. Solutions of PtNPs and of POM-Al, POM-Si, POM-P and POM-AlH were mixed together and agitated for two hours to lead to the SA-Al, SA-Si, SA-P, and SA-AlH SAs. The ratio between the quantity of POMs and the quantity of platinum atoms was chosen to be equal to 0.2, as it was previously demonstrated to be the equivalence ratio, when mostly all the ligands are coordinated at the NP surface.37 TEM images showed that microscopic rod-shaped SAs were formed, without any significant morphologic differences between the four systems (Fig. 2a–d). Each SA was constituted by an aggregation of the PtNPs associated with POMs (Fig. 2e and Fig. S11, ESI†).
 | ||
| Fig. 2 TEM pictures of self-assemblies of Pt nanoparticles and POMs: (a) SA-Al, (b) SA-Si, (c) SA-P, (d) SA-AlH, (e) zoomed-in view of SA-P, and (f) TEM picture of the mixture of Pt nanoparticles and unfunctionalized (TBA)3[PW12O40] polyanions. | ||
As already observed for SA between ultra-small PtNPs and molecular entities, a coordination interaction between the functional groups of the molecules and the NP surface was necessary to form coherent SAs.36,37,55 Here the presence of the thiol arms allows a strong coordination to the NP surface. Indeed, when we mixed the pristine NPs with unfunctionalized (TBA)3[PW12O40] polyanions, we did not observe any homogeneous SA, but aggregates of POMs decorated by adsorbed PtNPs (Fig. 2f). Fourier-transform infra-red (FT-IR) spectroscopy confirmed the interaction between the NPs and the POMs. The spectrum of the pristine NPs showed an intense peak at 2040 cm−1 corresponding to the vibration of terminal CO, and two smaller peaks at 1815 and 1883 cm−1 corresponding to vibrations of bridging CO (Fig. 3). A shift of the vibration of the terminal CO towards lower wavenumbers is a signature of the coordination of additional ligands at the NP surface, which turns out to be richer in electrons. Higher electronic density at the surface implies stronger back-donation from the NP to the antibonding orbitals of the CO molecule, and thus a weaker vibration.36,56 We can thus deduce that the shift from 2042 cm−1 in the pristine PtNPs to 2031 cm−1, 2034 cm−1, and 2036 cm−1 in SA-Al, SA-Si, and SA-P is a signature of electronic density donation from the POM hybrids to the NP surface; the higher the POM charge, the bigger the effect. Interestingly, we noticed that such a vibration was equal to 2035 cm−1 for SA-AlH, closer to the one of SA-Si than of SA-Al, thus suggesting that the trends in donation of electronic density from the POM hybrid to the NP surface depend on the global charge of the POM or POM/proton adduct more than the nature of the central atom. FT-IR measurements confirmed the absence of interaction for the unfunctionalized POM, as the CO terminal vibration was measured at 2041 cm−1, i.e. without any significant shift, in the mixture of the pristine NPs with (TBA)3[PW12O40] polyanions (Fig. S12, ESI†). The SA procedure was repeated with larger PtNPs of 2.0 ± 0.4 nm. We observed similar structuration in TEM (Fig. S13, ESI†), and the same trends in FT-IR (Fig. S14, ESI†). This new SA series with larger NPs is denoted as SA-Al-2, SA-Si-2, SA-P-2, and SA-AlH-2.
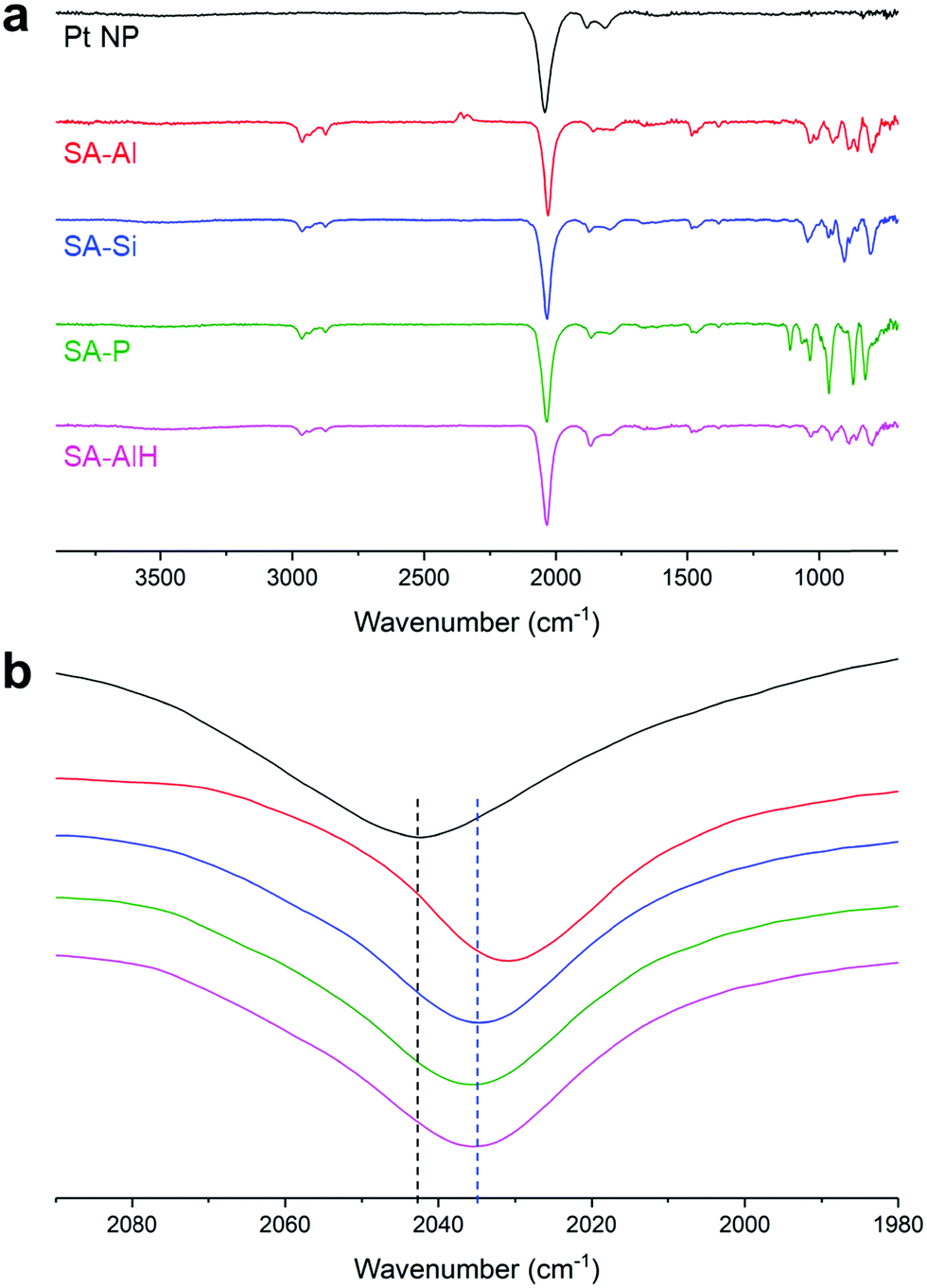 | ||
| Fig. 3 Infrared spectra of the pristine Pt nanoparticles and of the SA-Al, SA-Si, SA-P, and SA-AlH self-assemblies: (a) full spectra and (b) zoomed-in view of the terminal CO region (the baselines are shifted for clarity – the dashed lines are a guide for the eye); peak maxima: PtNP: 2042 cm−1, SA-Al: 2031 cm−1, SA-Si: 2034 cm−1, SA-P: 2036 cm−1, and SA-AlH: 2035 cm−1. | ||
Charge transport measurements were performed by conductive atomic force microscopy (CAFM), coupled to statistical analyses.37 The SAs were drop-cast on a gold surface and a large number of I–V curves (∼50) were measured by contacting the CAFM tip at different positions of individual objects. The I–V curves were normalized at 2 V and averaged to compare the current characteristics of one sample to another (Fig. 4a and b). The non-linear behavior in the I–V curves was the signature of the presence of Coulomb blockade at room temperature. Besides, for a given series, the non-linearity of the curve, i.e. the Coulomb blockade, decreased when the charge of the POM decreased. For example, the non-linearity of the I–V curve was more pronounced in SA-Al-2, than in SA-Si-2, than in SA-P-2. Characteristics of SA-AlH-2 were situated close to the one of SA-Si-2, confirming the FT-IR observation that the physico-chemical properties of SAs with POM-AlH were closer to the ones of POM-Si than to the ones of POM-Al.
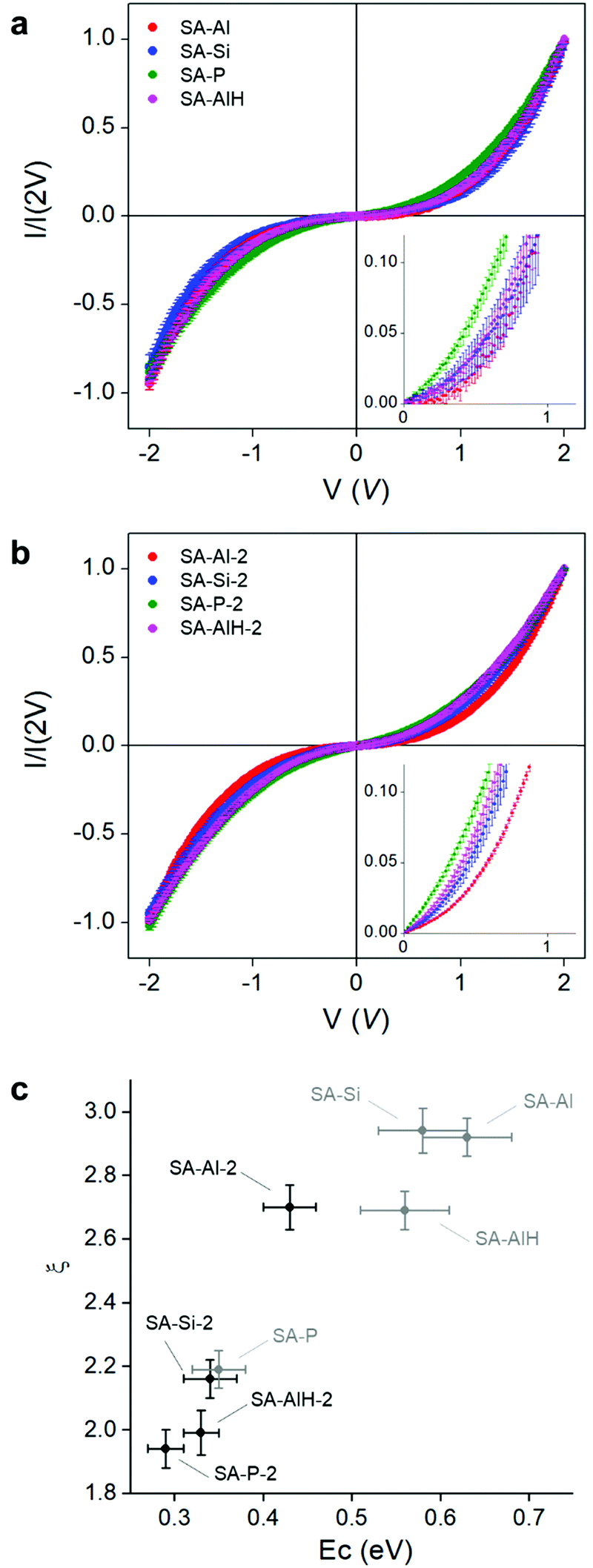 | ||
| Fig. 4 Charge transport measurement at the nanoscale, performed by conductive AFM on the (a) SA-Al, SA-Si, SA-P, SA-AlH (small nanoparticles) and (b) SA-Al-2, SA-Si-2, SA-P-2, SA-AlH-2 (large nanoparticles) self-assemblies, at room temperature; the curves are normalized at 2 V; insets: magnification on the 0–1 V regions. (c) Evolution of the power exponent ξ (fitted from the I–V characteristics with small nanoparticles – in grey – and with large nanoparticles – in black) as a function of the corresponding charging energies. | ||
The electron mobility in an NP SA in a Coulomb blockade regime is limited by the charging energy of an NP, defined as EC∼ e2/(2πεrε0d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(s/(s − d))), where e is the charge of the electron, ε0 the permittivity of vacuum, εr the dielectric constant of the medium surrounding the particles, d the particle diameter, and s the center-to-center distance between two particles.33,57 The s, d and εr parameters can be determined experimentally. The size d of the NPs was measured from the size distribution on the TEM pictures for each sample, and was equal on average to 1.6 and 2.0 nm for each series of SA (Fig. S11, S13 and Table S1, ESI†). Small angle X-ray scattering (SAXS) measurements were performed on SA-Si and SA-Si-2 SAs to determine the s values. Broad peaks centered at 0.26 and 0.22 Å−1 were observed for the SAs with small and large NPs, meaning that NPs were separated from each other by an average correlation distance s equal to 2.4 and 2.9 nm in each series (Fig. S15 and Table S1, ESI†).58 We can notice that for each series, the average interparticle distance s–d is about 0.8–0.9 nm, i.e. the same order of magnitude as the size of a Keggin-type POM. Finally, the dielectric constant of the POMs was determined by dielectric spectroscopy on powders of free molecules: εr = 3.10 for POM-Al, 3.36 for POM-Si, 3.97 for POM-P and 3.51 for POM-AlH (Table S1, ESI†). For the dielectric constant too, the properties of POM-AlH were close to the one of POM-Si, as observed in the IV curves and the IR spectra. As already demonstrated, even if no specific mechanistic model exists at room temperature for ultra-small NP SAs, current characteristics can phenomenologically be described by the equation I ∝ Vξ.37ξ is a scaling exponent related to current paths through the NP SA, and depends on the SA dimensionality.57,59 For example, it is generally found between 2 and 3.5 in three dimensional systems.60 We observed that the fitted ξ increased when the calculated EC increased (Fig. 4c and Table S1, ESI†). In the present series, the charging energy EC increases with the charge and decreases with the dielectric constant of the molecules. Indeed, charge and dielectric constant are linked to each other: the dielectric constant is the macroscopic parameter that reflects the microscopic polarizability of the molecules. As all the POMs have the same size, increasing the charge number corresponds to decreasing the polarizability.61
ln(s/(s − d))), where e is the charge of the electron, ε0 the permittivity of vacuum, εr the dielectric constant of the medium surrounding the particles, d the particle diameter, and s the center-to-center distance between two particles.33,57 The s, d and εr parameters can be determined experimentally. The size d of the NPs was measured from the size distribution on the TEM pictures for each sample, and was equal on average to 1.6 and 2.0 nm for each series of SA (Fig. S11, S13 and Table S1, ESI†). Small angle X-ray scattering (SAXS) measurements were performed on SA-Si and SA-Si-2 SAs to determine the s values. Broad peaks centered at 0.26 and 0.22 Å−1 were observed for the SAs with small and large NPs, meaning that NPs were separated from each other by an average correlation distance s equal to 2.4 and 2.9 nm in each series (Fig. S15 and Table S1, ESI†).58 We can notice that for each series, the average interparticle distance s–d is about 0.8–0.9 nm, i.e. the same order of magnitude as the size of a Keggin-type POM. Finally, the dielectric constant of the POMs was determined by dielectric spectroscopy on powders of free molecules: εr = 3.10 for POM-Al, 3.36 for POM-Si, 3.97 for POM-P and 3.51 for POM-AlH (Table S1, ESI†). For the dielectric constant too, the properties of POM-AlH were close to the one of POM-Si, as observed in the IV curves and the IR spectra. As already demonstrated, even if no specific mechanistic model exists at room temperature for ultra-small NP SAs, current characteristics can phenomenologically be described by the equation I ∝ Vξ.37ξ is a scaling exponent related to current paths through the NP SA, and depends on the SA dimensionality.57,59 For example, it is generally found between 2 and 3.5 in three dimensional systems.60 We observed that the fitted ξ increased when the calculated EC increased (Fig. 4c and Table S1, ESI†). In the present series, the charging energy EC increases with the charge and decreases with the dielectric constant of the molecules. Indeed, charge and dielectric constant are linked to each other: the dielectric constant is the macroscopic parameter that reflects the microscopic polarizability of the molecules. As all the POMs have the same size, increasing the charge number corresponds to decreasing the polarizability.61
If we now look at the current for a given voltage on the IV curves, we see that it increases from Al to Si to P, meaning that the charge transport rate increases. Indeed, if we consider a pure POM molecule, the first reduction potential measured by CV increased within this series (the POM is easier to reduce), reflecting a decrease of the LUMO energy, as calculated by DFT.53 As the Fermi energy of the PtNPs remains constant (since they are the same NPs) and below the POM LUMOs, the energies of theses orbitals are getting closer, which decreases the tunnel barrier of charge transfer across the POM. In addition, more redox peaks in the same CV window may indicate more possible conduction channels for charge transport, and thus higher conductivity. Although coherent with the experimental observations, such considerations on single POMs should be taken into account with caution: CV is measured on well-dispersed molecules in solution and DFT calculations are performed on molecules in the “gas phase”. But the composite material includes hundreds of PtNPs and POMs in series, and POMs are known to be very dependent on external conditions (nature of solvent, of the counter-cation, etc.). A new atomistic modelling framework would thus be needed to fully interpret the results presented here.
In summary, we synthesized a new series of Keggin-type POM hybrids of different charges: POM-Al (5-), POM-Si (4-), POM-P (3-) and POM-AlH (∼4-). Thanks to their thiol arms, it was possible to elaborate robust and homogeneous SAs with ultra-small PtNPs, on which we succeeded to perform CAFM charge transport measurements. The system was in a Coulomb blockade regime, at room temperature, and the key molecular parameter to describe charge transport was found to be the polarizability of the POM. Such a polarizability is directly linked to the dielectric constant of the molecular system, as described by the Clausius–Mossotti model, which might eventually be adapted to take into account the geometry of the systems at the nanometer scale.62 Besides, even if trends in charge transport can be considered in terms of LUMO energy levels,44 one should keep in mind that in an experimental setup, molecules are not standing by themselves and that molecular aspects cannot be fully decorrelated from the properties of the electrodes, the geometry of the leads and the associated Fermi electronic occupation distribution.63 The present study is about charge transport in ultra-small NP SA in the Coulomb blockade regime, but polarizability can definitely be considered in other approaches of molecular electronics and its variations can be an important parameter for rationalizing charge transport across molecular junctions.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge Josep M. Poblet for insightful discussion. Financial support from Agence Nationale de la Recherche (PhoCatSA grant ANR-10-LABX-0037-NEXT, and MOSC grant ANR-18-CE09-0007) is acknowledged. This study has been partially supported through the EUR grant NanoX no. ANR-17-EURE-0009 in the framework of the Programme des Investissements d’Avenir.References
- S. J. van der Molen, R. Naaman, E. Scheer, J. B. Neaton, A. Nitzan, D. Natelson, N. J. Tao, H. S. J. van der Zant, H. Mayor, M. Ruben, M. Reed and M. Calame, Nat. Nanotechnol., 2013, 8, 385–389 CrossRef PubMed.
- S. Elke and C. J. Carlos, Molecular Electronics: An Introduction To Theory And Experiment, 2nd edn, World Scientific, 2017 Search PubMed.
- P. T. Mathew and F. Fang, Engineering, 2018, 4, 760–771 CrossRef CAS.
- I. Kanelidis and T. Kraus, Beilstein J. Nanotechnol., 2017, 8, 2625–2639 CrossRef PubMed.
- M. Schwarze, C. Gaul, R. Scholz, F. Bussolotti, A. Hofacker, K. S. Schellhammer, B. Nell, B. D. Naab, Z. Bao, D. Spoltore, K. Vandewal, J. Widmer, S. Kera, N. Ueno, F. Ortmann and K. Leo, Nat. Mater., 2019, 18, 242–248 CrossRef CAS PubMed.
- A. Nitzan and M. A. Ratner, Science, 2003, 300, 1384–1389 CrossRef CAS PubMed.
- W. Y. Kim and K. S. Kim, Acc. Chem. Res., 2010, 43, 111–120 CrossRef CAS PubMed.
- N. Shivran, S. P. Koiry, C. Majumder, A. K. Chauhan, D. K. Aswal, S. Chattopadhyay and S. Mula, Phys. Chem. Chem. Phys., 2020, 22, 2098–2104 RSC.
- M. M. Thuo, W. F. Reus, F. C. Simeone, C. Kim, M. D. Schulz, H. J. Yoon and G. M. Whitesides, J. Am. Chem. Soc., 2012, 134, 10876–10884 CrossRef CAS.
- H. J. Yoon, N. D. Shapiro, K. M. Park, M. M. Thuo, S. Soh and G. M. Whitesides, Angew. Chem., Int. Ed., 2012, 51, 4658–4661 CrossRef CAS.
- H. J. Yoon, C. M. Bowers, M. Baghbanzadeh and G. M. Whitesides, J. Am. Chem. Soc., 2014, 136, 16–19 CrossRef CAS.
- G. D. Kong, M. Kim, H.-J. Jang, K.-C. Liao and H. J. Yoon, Phys. Chem. Chem. Phys., 2015, 17, 13804–13807 RSC.
- J. G. Simmons, J. Appl. Phys., 1963, 34, 238–239 CrossRef.
- M. M. Thuo, W. F. Reus, C. A. Nijhuis, J. R. Barber, C. Kim, M. D. Schulz and G. M. Whitesides, J. Am. Chem. Soc., 2011, 133, 2962–2975 CrossRef CAS PubMed.
- L. Yuan, L. Wang, A. R. Garrigues, L. Jiang, H. V. Annadata, M. Anguera Antonana, E. Barco and C. A. Nijhuis, Nat. Nanotechnol., 2018, 13, 322–329 CrossRef CAS PubMed.
- Q. Lu, K. Liu, H. Zhang, Z. Du, X. Wang and F. Wang, ACS Nano, 2009, 3, 3861–3868 CrossRef CAS PubMed.
- X. Song, B. Han, X. Yu and W. Hu, Chem. Phys., 2020, 528, 110514 CrossRef CAS.
- H. J. Yoon, K.-C. Liao, M. R. Lockett, S. W. Kwok, M. Baghbanzadeh and G. M. Whitesides, J. Am. Chem. Soc., 2014, 136, 17155–17162 CrossRef CAS PubMed.
- G. D. Kong, M. Kim, S. J. Cho and H. J. Yoon, Angew. Chem., Int. Ed., 2016, 55, 10307–10311 CrossRef CAS PubMed.
- L. O. Jones, M. A. Mosquera, M. A. Ratner and G. C. Schatz, J. Phys. Chem. C, 2020, 124, 3233–3241 CrossRef CAS.
- H. Nakanishi, K. J. M. Bishop, B. Kowalczyk, A. Nitzan, E. A. Weiss, K. V. Tretiakov, M. M. Apodaca, R. Klajn, J. F. Stoddart and B. A. Grzybowski, Nature, 2009, 460, 371–375 CrossRef CAS PubMed.
- H. Moreira, J. Grisolia, N. M. Sangeetha, N. Decorde, C. Farcau, B. Viallet, K. Chen, G. Viau and L. Ressier, Nanotechnology, 2013, 24, 095701 CrossRef PubMed.
- S. K. S. Mazinani, R. V. Meidanshahi, J. L. Palma, P. Tarakeshwar, T. Hansen, M. A. Ratner and V. Mujica, J. Phys. Chem. C, 2016, 120, 26054–26060 CrossRef CAS.
- M. Baghbanzadeh, L. Belding, L. Yuan, J. Park, M. H. Al-Sayah, C. M. Bowers and G. M. Whitesides, J. Am. Chem. Soc., 2019, 141, 8969–8980 CrossRef CAS PubMed.
- X. Chen, H. V. Annadata, B. Kretz, M. Zharnikov, X. Chi, X. Yu, D. A. Egger and C. A. Nijhuis, J. Phys. Chem. Lett., 2019, 10, 4142–4147 CrossRef CAS PubMed.
- A. Zabet-Khosousi and A.-A. Dhirani, Chem. Rev., 2008, 108, 4072–4124 CrossRef CAS PubMed.
- M. Pauly, J.-F. Dayen, D. Golubev, J.-B. Beaufrand, B. P. Pichon, B. Doudin and S. Bégin-Colin, Small, 2012, 8, 108–115 CrossRef CAS PubMed.
- Y. Zhou, S.-T. Han, Z.-X. Xu and V. A. L. Roy, Adv. Mater., 2012, 24, 1247–1251 CrossRef CAS PubMed.
- A. Nag, D. S. Chung, D. S. Dolzhnikov, N. M. Dimitrijevic, S. Chattopadhyay, T. Shibata and D. V. Talapin, J. Am. Chem. Soc., 2012, 134, 13604–13615 CrossRef CAS PubMed.
- J. Liao, S. Blok, S. J. van der Molen, S. Diefenbach, A. W. Holleitner, C. Schönenberger, A. Vladyka and M. Calame, Chem. Soc. Rev., 2015, 44, 999–1014 RSC.
- H. Nesser, J. Grisolia, A. Mlayah, T. Alnasser, D. Lagarde, B. Viallet and L. Ressier, Mater. Today Nano, 2018, 4, 38–45 CrossRef.
- D. Conklin, S. Nanayakkara, T.-H. Park, M. F. Lagadec, J. T. Stecher, M. J. Therien and D. A. Bonnell, Nano Lett., 2012, 12, 2414–2419 CrossRef CAS PubMed.
- J. Dugay, R. P. Tan, M. Ibrahim, C. Garcia, J. Carrey, L.-M. Lacroix, P.-F. Fazzini, G. Viau and M. Respaud, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 041406 CrossRef.
- J. Liao, J. S. Agustsson, S. Wu, C. Schönenberger, M. Calame, Y. Leroux, M. Mayor, O. Jeannin, Y.-F. Ran, S.-X. Liu and S. Decurtins, Nano Lett., 2010, 10, 759–764 CrossRef CAS PubMed.
- S. J. van der Molen, J. Liao, T. Kudernac, J. S. Agustsson, L. Bernard, M. Calame, B. J. van Wees, B. L. Feringa and C. Schönenberger, Nano Lett., 2009, 9, 76–80 CrossRef CAS PubMed.
- S. Usmani, M. Mikolasek, A. Gillet, J. Sanchez Costa, M. Rigoulet, B. Chaudret, A. Bousseksou, B. Lassalle-Kaiser, P. Demont, G. Molnár, L. Salmon, J. Carrey and S. Tricard, Nanoscale, 2020, 12, 8180–8187 RSC.
- S. Tricard, O. Said-Aizpuru, D. Bouzouita, S. Usmani, A. Gillet, M. Tassé, R. Poteau, G. Viau, P. Demont, J. Carrey and B. Chaudret, Mater. Horiz., 2017, 4, 487–492 RSC.
- C. Busche, L. Vilà-Nadal, J. Yan, H. N. Miras, D.-L. Long, V. P. Georgiev, A. Asenov, R. H. Pedersen, N. Gadegaard, M. M. Mirza, D. J. Paul, J. M. Poblet and L. Cronin, Nature, 2014, 515, 545–549 CrossRef CAS PubMed.
- X. Chen, P. Huang, X. Zhu, S. Zhuang, H. Zhu, J. Fu, A. S. Nissimagoudar, W. Li, X. Zhang, L. Zhou, Y. Wang, Z. Lv, Y. Zhou and S.-T. Han, Nanoscale Horiz., 2019, 4, 697–704 RSC.
- C. Kato, R. Machida, R. Maruyama, R. Tsunashima, X.-M. Ren, M. Kurmoo, K. Inoue and S. Nishihara, Angew. Chem., Int. Ed., 2018, 57, 13429–13432 CrossRef CAS PubMed.
- S. Sherif, G. Rubio-Bollinger, E. Pinilla-Cienfuegos, E. Coronado, J. C. Cuevas and N. Agraït, Nanotechnology, 2015, 26, 291001 CrossRef PubMed.
- J. de Bruijckere, P. Gehring, M. Palacios-Corella, M. Clemente-León, E. Coronado, J. Paaske, P. Hedegård and H. S. J. van der Zant, Phys. Rev. Lett., 2019, 122, 197701 CrossRef CAS PubMed.
- A. Balliou, M. Bouroushian, A. M. Douvas, G. Skoulatakis, S. Kennou and N. Glezos, Nanotechnology, 2018, 29, 275204 CrossRef PubMed.
- M. Laurans, K. Dalla Francesca, F. Volatron, G. Izzet, D. Guerin, D. Vuillaume, S. Lenfant and A. Proust, Nanoscale, 2018, 10, 17156–17165 RSC.
- K. Dalla Francesca, S. Lenfant, M. Laurans, F. Volatron, G. Izzet, V. Humblot, C. Methivier, D. Guerin, A. Proust and D. Vuillaume, Nanoscale, 2019, 11, 1863–1878 RSC.
- B. Martinez, C. Livache, E. Meriggio, X. Z. Xu, H. Cruguel, E. Lacaze, A. Proust, S. Ithurria, M. G. Silly, G. Cabailh, F. Volatron and E. Lhuillier, J. Phys. Chem. C, 2018, 122, 26680–26685 CrossRef CAS.
- K. Y. Monakhov, M. Moors and P. Kögerler, in Advances in Inorganic Chemistry, Elsevier, 2017, 69, pp. 251–286 Search PubMed.
- C. Wu, X. Qiao, C. M. Robertson, S. J. Higgins, C. Cai, R. J. Nichols and A. Vezzoli, Angew. Chem., Int. Ed., 2020, 59, 2–8 CrossRef.
- O. Linnenberg, M. Moors, A. Notario-Estévez, X. López, C. de Graaf, S. Peter, C. Baeumer, R. Waser and K. Y. Monakhov, J. Am. Chem. Soc., 2018, 140, 16635–16640 CrossRef CAS PubMed.
- G. Izzet, F. Volatron and A. Proust, Chem. Rec., 2017, 17, 250–266 CrossRef CAS PubMed.
- O. Makrygenni, E. Secret, A. Michel, D. Brouri, V. Dupuis, A. Proust, J.-M. Siaugue and R. Villanneau, J. Colloid Interface Sci., 2018, 514, 49–58 CrossRef CAS PubMed.
- C. Martin, K. Kastner, J. M. Cameron, E. Hampson, J. Alves Fernandes, E. K. Gibson, D. A. Walsh, V. Sans and G. N. Newton, Angew. Chem., Int. Ed., 2020, 59, 14331–14335 CrossRef CAS PubMed.
- I.-M. Mbomekallé, X. López, J. M. Poblet, F. Sécheresse, B. Keita and L. Nadjo, Inorg. Chem., 2010, 49, 7001–7006 CrossRef PubMed.
- S. Gomez, L. Erades, K. Philippot, B. Chaudret, V. Collière, O. Balmes and J.-O. Bovin, Chem. Commun., 2001, 1474–1475 RSC.
- G. Manai, H. Houimel, M. Rigoulet, A. Gillet, P.-F. Fazzini, A. Ibarra, S. Balor, P. Roblin, J. Esvan, Y. Coppel, B. Chaudret, C. Bonduelle and S. Tricard, Nat. Commun., 2020, 11, 2051 CrossRef CAS PubMed.
- C. Dablemont, P. Lang, C. Mangeney, J.-Y. Piquemal, V. Petkov, F. Herbst and G. Viau, Langmuir, 2008, 24, 5832–5841 CrossRef CAS PubMed.
- C. T. Black, Science, 2000, 290, 1131–1134 CrossRef CAS PubMed.
- N. Decorde, N. M. Sangeetha, B. Viallet, G. Viau, J. Grisolia, A. Coati, A. Vlad, Y. Garreau and L. Ressier, Nanoscale, 2014, 6, 15107–15116 RSC.
- A. A. Middleton and N. S. Wingreen, Phys. Rev. Lett., 1993, 71, 3198 CrossRef CAS PubMed.
- P. Yang, I. Arfaoui, T. Cren, N. Goubet and M.-P. Pileni, Nano Lett., 2012, 12, 2051–2055 CrossRef CAS PubMed.
- T. Buchecker, P. Schmid, S. Renaudineau, O. Diat, A. Proust, A. Pfitzner and P. Bauduin, Chem. Commun., 2018, 54, 1833–1836 RSC.
- A. Natan, N. Kuritz and L. Kronik, Adv. Funct. Mater., 2010, 20, 2077–2084 CrossRef CAS.
- A. R. Garrigues, L. Wang, E. del Barco and C. A. Nijhuis, Nat. Commun., 2016, 7, 11595 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary characterization data. See DOI: 10.1039/d0nh00583e |
| This journal is © The Royal Society of Chemistry 2021 |