
Engineering a photosensitizer nanoplatform for amplified photodynamic immunotherapy via tumor microenvironment modulation†
Yaxin
Zhou
a,
Xiaomeng
Ren
a,
Zhaosheng
Hou
b,
Ningning
Wang
a,
Yue
Jiang
a and
Yuxia
Luan
 *a
*a
aDepartment of Pharmaceutics, Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, China. E-mail: yuxialuan@sdu.edu.cn
bCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan, 250014, China
First published on 31st October 2020
Abstract
Photosensitizer-based photodynamic therapy (PDT) can not only kill tumor cells by the generated cytotoxic reactive oxygen species (ROS), but also trigger immunogenic cell death (ICD) and activate an immune response for immunotherapy. However, such photodynamic immunotherapy suffers from major obstacles in the tumor microenvironment. The hypoxic microenvironment greatly weakens PDT, while the immunosuppressive tumor microenvironment caused by aberrant tumor blood vessels and indoleamine 2,3-dioxygenase (IDO) leads to a significant reduction in immunotherapy. To overcome these obstacles, herein, an engineered photosensitizer nanoplatform is designed for amplified photodynamic immunotherapy by integrating chlorin e6 (Ce6, a photosensitizer), axitinib (AXT, a tyrosine kinase inhibitor) and dextro-1-methyl tryptophan (1MT, an IDO inhibitor). In our nanoplatform, AXT improves the tumor microenvironment by normalizing tumor blood vessels, which not only promotes PDT by reducing the level of hypoxia of the tumor microenvironment, but also promotes immunotherapy through facilitating infiltration of immune effector cells into the tumor and reversing the immunosuppressive effect of vascular endothelial growth factor (VEGF). Moreover, 1MT effectively inhibits the activity of IDO, further reducing the immunosuppressive nature of the tumor microenvironment. Therefore, this nanoplatform demonstrates an amplified photodynamic immunotherapy via tumor microenvironment modulation, exhibiting outstanding therapeutic efficacy against tumor growth and metastasis with negligible side toxicity. The current concept of engineering photosensitizer nanoplatforms for overcoming photodynamic immunotherapy obstacles provides a promising strategy against tumors.
New conceptsTumor cells continuously secrete excessive vascular endothelial growth factor (VEGF), leading to the generation of aberrant vasculature, and ultimately promoting hypoxia in the tumor microenvironment. Therefore, aberrant vasculature is one major obstacle to photodynamic efficacy due to the resultant hypoxia. Moreover, tumor vasculature with dysfunctional blood flow prevents immune cells from deeply penetrating into the tumor. Furthermore, VEGF can not only increase immunosuppressive cell proliferation, but also suppress dendritic cell maturation and T cell function, thus significantly weakening immunotherapy. Even for activated immune cells already present inside the tumor, other immunosuppressive mechanisms still need to be overcome such as the overexpressed indoleamine 2,3-dioxygenase (IDO)-regulated immune response. Therefore, the immunosuppressive tumor microenvironment caused by aberrant tumor blood vessels and IDO is another major obstacle for immunotherapeutic efficacy in photodynamic immunotherapy. A rationally designed photosensitizer nanoplatform was, for the first time, utilized to amplify photodynamic immunotherapy by integrating chlorin e6, axitinib and dextro-1-methyl tryptophan with human serum albumin, which simultaneously maximized the photodynamic and immunotherapeutic capabilities to boost photodynamic immunotherapy via reducing the level of tumor hypoxia, promoting infiltration of immune effector cells and reversing the immunosuppressive effect of VEGF and IDO in the tumor microenvironment. The current method of engineering a photosensitizer nanoplatform for overcoming obstacles to photodynamic immunotherapy offers a promising strategy against tumors. |
1. Introduction
Photodynamic therapy (PDT) with the advantages of minimal invasiveness and low system toxicity is a promising therapeutic modality.1–4 During PDT, there are two ways to kill tumor cells. On one hand, photosensitizers with appropriate laser irradiation kill tumor cells directly by the produced cytotoxic reactive oxygen species (ROS).5 On the other hand, PDT-induced immunogenic cell death (ICD) stimulates tumor cells to release tumor-associated antigens (TAAs) and damage associated molecular patterns (DAMPs), thereby triggering dendritic cell (DC) maturation and activating an immune response for immunotherapy.6–8 Therefore, photosensitizer-based PDT itself can be called photodynamic immunotherapy. However, such photodynamic immunotherapy is extremely restricted by the major obstacle of the tumor microenvironment.9Oxygen is indispensable for ROS production in PDT, but hypoxia in the microenvironment greatly weakens the efficiency of ROS production in PDT.10–12 To obtain sufficient nutrients and oxygen to enable rapid growth and proliferation, tumor cells continuously secrete excessive vascular endothelial growth factor (VEGF), which results in an imbalance in the levels of proangiogenic and antiangiogenic factors, and rapidly generates aberrant vasculature.13–16 The aberrant vasculature is unevenly distributed, dilated and tortuous.17 The adjacent endothelial cells are flabbily connected to each other and pericytes surrounding blood vessels are separated from endothelial cells, leading to an inefficient blood supply.18,19 This ultimately promotes hypoxia in the tumor microenvironment. Thus, aberrant vasculature is a major barrier to preclinical PDT and clinical progress due to the resultant hypoxia.
The transport and infiltration of immune effector cells into tumor parenchyma is an essential step in the cancer immune cycle.20,21 The vascular network plays an important role in enabling immune cells to enter tumor tissue.17 However, aberrant vasculature with dysfunctional blood flow prevents immune cells from deeply penetrating into tumors.22,23 Moreover, studies have shown that VEGF can not only increase immunosuppressive cell proliferation such as tumor-associated macrophages (TAMs) and regulatory T cells (Tregs),24,25 but also suppress dendritic cell (DC) maturation and T cell function.26,27 Even if already inside the tumor, the activated immune cells still have to conquer other immunosuppressive mechanisms in the tumor microenvironment.17 For example, indoleamine 2,3-dioxygenase (IDO), which is overexpressed in most tumors, converts tryptophan (Trp) to kynurenine (Kyn) and other metabolites, thereby facilitating tolerance by suppressing the immune response.28–30 Therefore, the immunosuppressive tumor microenvironment caused by aberrant tumor blood vessels and IDO is the major obstacle for immunotherapy.
To conclude, the hypoxic microenvironment greatly weakens the efficiency of ROS production in PDT, while the immunosuppressive tumor microenvironment caused by aberrant tumor blood vessels and IDO severely limits the immunotherapeutic effectiveness. Therefore, the rational design of a photosensitizer-based nanoplatform to overcome the above major obstacles for achieving potent photodynamic immunotherapy is of great significance for tumor treatment. Until now, simultaneously amplifying both PDT and immunotherapy for realizing a potent photodynamic immunotherapy has rarely been reported.
Herein, we rationally designed a photosensitizer-based nanoplatform, which could achieve amplified photodynamic immunotherapy via promoting vascular normalization and reshaping the tumor microenvironment. Our nanoplatform (named CAM NPs) was constructed by the self-assembly of chlorin e6 (Ce6, a photosensitizer), axitinib (AXT, a tyrosine kinase inhibitor) and dextro-1-methyl tryptophan (1MT, an IDO inhibitor) with the help of human serum albumin (HSA) (Scheme 1). In our nanoplatform, hydrophobic pockets of biocompatible HSA facilitated the binding of hydrophobic molecules for effective delivery,31–34 while Ce6 stimulated an antitumor immune response under laser irradiation. Moreover, the HSA-based carriers can achieve enhanced accumulation at tumor sites via albumin-binding proteins overexpressed in various tumors.35,36 The process of proangiogenesis is dominated by the interaction between VEGF and its receptor (VEGFR, a receptor tyrosine kinase), while AXT could inhibit the activity of VEGFR,37–41 which promoted the normalization of blood vessels and improved tumor perfusion. This resulted in an increased delivery of therapeutic agents and oxygen into the tumors, which not only improved the efficiency of ROS production, but, more importantly, promoted the transport of activated immune cells into tumor parenchyma and enhanced the effectiveness of the immunotherapy. 1MT further improved the photodynamic therapy-induced immunotherapy by inhibiting the immunosuppressive IDO in the tumor microenvironment.42–44 Thus, the rational design of a photosensitizer-based nanoplatform that could simultaneously enhance PDT and PDT-induced immunotherapy by reducing the level of hypoxia and immunosuppressive nature of the tumor microenvironment was reported for the first time. Importantly, the as-prepared CAM NPs successfully suppressed the primary tumor, bilateral tumors and pulmonary metastasis, demonstrating a powerful photodynamic immunotherapy platform for fighting tumors.
 | ||
| Scheme 1 (a) Illustration of CAM NP preparation and (b) schematic diagram of tumor therapy based on CAM NPs by inducing an antitumor immune response, normalizing vasculature and regulating metabolism. | ||
2. Results and discussion
The CAM NPs were constructed by the self-assembly of Ce6, AXT and 1MT with the help of HSA. As can be seen from Fig. 1a (inset), a solution of CAM NPs with a turquoise color showed an obvious Tyndall effect under laser irradiation. Transmission electron microscopy (TEM) imaging demonstrated the spherical morphology of the CAM NPs with a size of 72.75 ± 27.64 nm (the inset in Fig. 1b). Dynamic light scattering (DLS) showed that the CAM NP size was about 170 nm (PDI = 0.279). The CAM NPs had a negative surface charge with the zeta potential of ∼10.5 mV (Fig. S1, ESI†). The CAM NPs were further characterized using FT-IR spectroscopy and UV-vis spectroscopy (Fig. S2, ESI,† and Fig. 1a). As can be seen from Fig. S2 (ESI†), the coexistence of typical FT-IR peaks of Ce6, AXT, 1MT and HSA confirmed the successful co-assembly of CAM NPs, and the slight shift of absorption peak due to the intermolecular hydrophobic interaction could also be observed in the FT-IR spectrum of the CAM NPs. As can be seen from Fig. 1a, CAM NPs exhibited typical UV-vis absorption with peaks at 666 nm (Ce6), 336 nm (AXT) and 285 nm (1MT), which further demonstrated the successful assembly of Ce6, AXT, 1MT with HSA. The drug loading contents of Ce6, AXT and 1MT in the CAM NPs were measured to be 12.6%, 9.5% and 8.9%, respectively. Besides, the stability of CAM NPs in different media was further characterized via monitoring their particle size. As shown in Fig. S3 (ESI†), the particle size of CAM NPs did not change significantly either in H2O, PBS or in 50% FBS/PBS (representing physiological serum conditions), which indicated the excellent stability of the prepared CAM NPs. The ROS generation ability of CAM NPs was determined using 1,3-diphenylisobenzofuran (DPBF) as the probe. As shown in Fig. 1c, the ability of CAM NPs to generate ROS was similar to that of free Ce6, indicating that the other components in the CAM NPs had little influence on the photodynamic efficacy of Ce6 in solution. | ||
| Fig. 1 (a) UV-vis spectra of 1MT, AXT, Ce6 and CAM NPs and photograph of the CAM NPs with irradiation (inset). (b) Size distribution (DLS) and TEM image of the CAM NPs (inset). Scale bar: 200 nm. (c) Normalized absorbance of DPBF at 424 nm in different groups. (d) FCM results of the cell uptake for free Ce6 and CAM NPs at different times. (e) Fluorescence microscopy images of intracellular ROS production of PBS (+), Ce6 (+) and CAM NPs (+). Scale bar: 50 μm. Cellular inhibition rate with different concentrations of (f) 1MT, AXT, Ce6 (+), and CAM NPs (+) and (g) Ce6 and CAM NPs on B16F10 cells. (h) The IDO inhibition efficiency in Kyn generation in B16F10 cells by free 1MT or CAM NPs. “(+)” represents the sample with laser irradiation. | ||
Moreover, the release behavior of 1MT and AXT at different pH (7.4, 6.5 and 5.0) was evaluated. As can be seen from Fig. S4 (ESI†), both 1MT and AXT exhibited pH-responsive release behaviors, with the maximum cumulative release at pH 5.0, a decrease at pH 6.5 and the minimum at pH 7.4. Since nitrogen-containing groups in 1MT and AXT could be easily protonated under acidic conditions, the hydrophobic interactions among CAM NPs would be attenuated, causing the disassembly of CAM NPs and the release of the drugs.
The cellular uptake property of CAM NPs by B16F10 melanoma cells was further examined by flow cytometry (FCM). The B16F10 cells, treated with CAM NPs, showed a significantly higher fluorescence intensity than that of free Ce6 at the corresponding time, verifying their superior internalization efficiency (Fig. 1d). The significantly high cellular uptake of CAM NPs could be attributed to the binding effect of their HSA component. It was reported that B16F10 cells overexpressed albumin-binding protein (secreted protein acidic and rich in cysteine, SPARC),45–47 which facilitated internalization of the albumin-based nanoplatform. For the CAM NPs (Fig. 1d), the highest internalization efficiency was found after 6 h of incubation, which was the optimal laser irradiation time for the in vitro laser-based studies. The fluorescence microscopy results for the cellular uptake of CAM NPs agreed well with the FCM results, further demonstrating their superior internalization efficiency (Fig. S5, ESI†).
The cellular uptake study of CAM NPs showed that the optimal laser irradiation for in vitro study should be performed at 6 h of incubation. The in vitro production level of ROS induced by PDT was further studied based on laser irradiation at 6 h of incubation. As can be seen from Fig. 1e and Table S1 (ESI†), the B16F10 cells, incubated with CAM NPs, displayed stronger fluorescence than the other groups, indicating their excellent cellular ROS production ability. Consistent with the results in Fig. 1c, the in vitro ROS production study further demonstrated that our CAM NPs were an efficient photodynamic nanoplatform. Moreover, the amount of generated ROS by CAM NPs without laser irradiation was negligible (Fig. S6 and S7, ESI†).
To understand the in vitro cytotoxicity of CAM NPs to B16F10 cells, MTT assays were performed with or without laser irradiation. For comparison, 1MT, AXT and Ce6 were also studied. As can be seen from Fig. 1f and g, the CAM NP group under laser irradiation demonstrated the highest cell inhibition rate compared to the other groups such as 1MT, AXT, Ce6 and Ce6 with laser irradiation, which was ascribed to their excellent internalization efficiency and ROS production ability. Without laser irradiation, CAM NPs also showed higher cytotoxicity than the Ce6 group, which resulted from the cytotoxicity effect of AXT.
IDO, overexpressed in most tumors, can convert Trp to Kyn and other metabolites, which results in suppression of the immune response. In order to understand whether our CAM NPs could suppress IDO in vitro, we further determined the concentration of Kyn in B16F10 cells after CAM NP incubation. As shown in Fig. 1h, the CAM NPs exhibited a similar IDO inhibitory activity to that of free 1MT, indicating that the CAM NPs were an efficient platform to inhibit the IDO pathway.
Exposure of calreticulin (CRT) on cell surface, the secretion of adenosine triphosphate (ATP) and the release of high mobility group box 1 (HMGB1) from the nucleus are the typical symbols of ICD. To study the PDT-induced ICD by our CAM NPs in vitro, CRT in B16F10 cells was firstly determined. As shown in Fig. 2a and Fig. S8 (ESI†), a larger amount of CRT exposed on the membrane was observed in CAM NP-treated cells after laser irradiation compared to other groups with or without the laser irradiation. The ATP secreted by tumor cells was further detected with an ATP analysis kit. As depicted in Fig. S9 (ESI†), the CAM NPs under irradiation caused higher ATP secretion in the cell culture medium; that is, 7.21-fold higher than that of the PBS group. Furthermore, immunofluorescence analysis of HMGB1 reported that HMGB1 was significantly released from cells treated with Ce6 and the CAM NPs under laser irradiation (Fig. 2b and Fig. S10, ESI†). These above results prove that CAM NPs with laser irradiation could cause excellent ICD for immunotherapy.
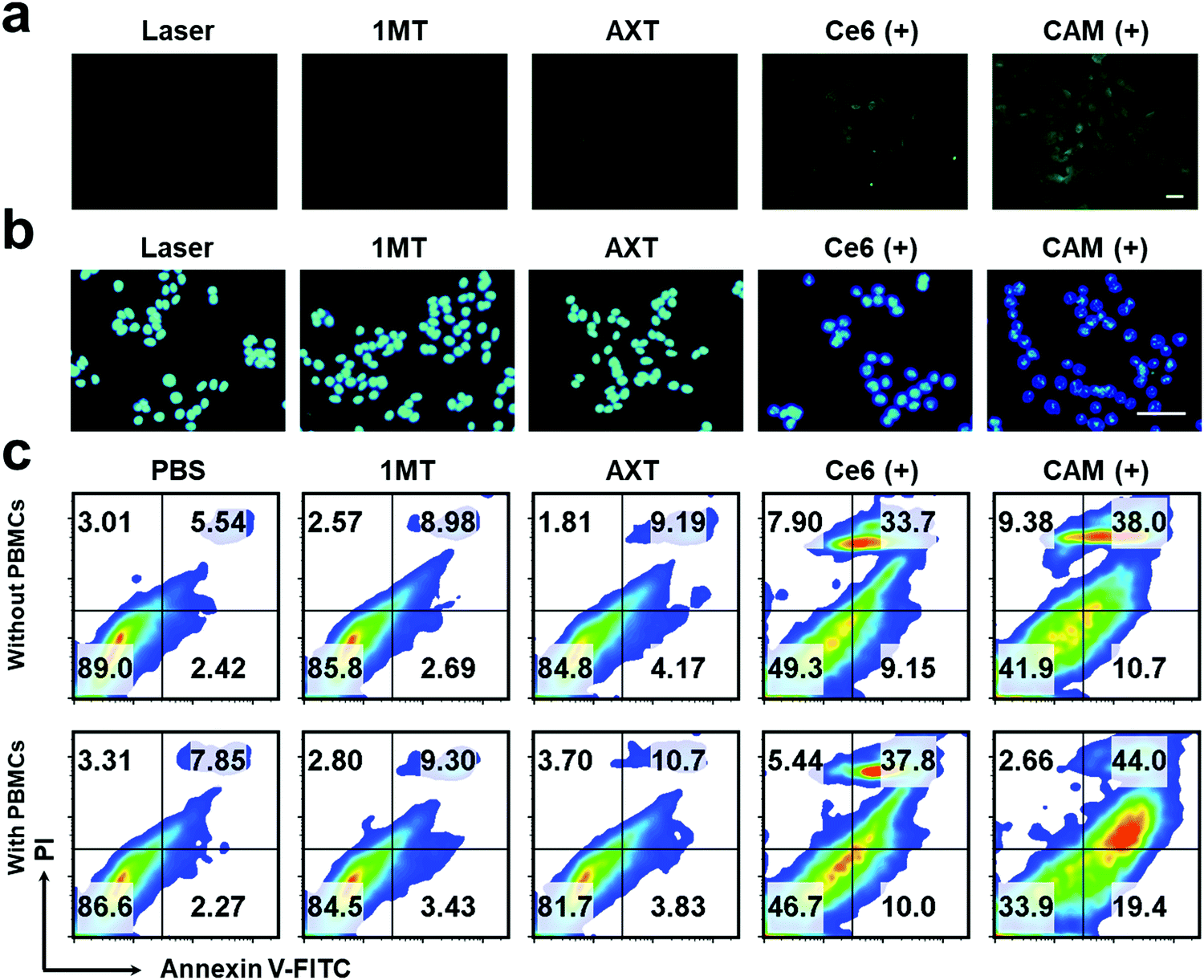 | ||
| Fig. 2 Fluorescence microscopy images of (a) CRT exposure and (b) HMGB1 release of B16F10 cells after various therapeutics. Scale bar: 20 μm. (c) FCM results of apoptosis of B16F10 cells with or without the PBMCs. “(+)” represents the sample with laser irradiation. | ||
The apoptosis of B16F10 cells induced by various treatments was further studied. The apoptotic rate of tumor cells treated with CAM NPs in the presence of laser irradiation was the highest (48.7%) among the studied groups, indicating that CAM NPs could effectively induce tumor cell apoptosis under laser irradiation (Fig. 2c). In contrast, the non-laser irradiated groups showed a much lower rate of apoptosis (Fig. S11, ESI†). We further studied the apoptosis rate of B16F10 cells that were first pretreated with various treatments and then incubated with peripheral blood mononuclear cells (PBMCs). For the group pretreated with CAM NPs under laser irradiation, it was found that the apoptosis rate of B16F10 cells with PBMCs remarkably increased to 63.4% in comparison with that without PBMCs (48.7%). It is known that PBMCs contain lymphocytes (natural killer cells, B cells and T cells), monocytes and DCs. After the B16F10 cells were pretreated with CAM NPs under laser irradiation, ICD occurred. The triggered ICD with the help of PBMCs would then result in the immune response to kill the tumor cells. The above results thus demonstrated that CAM NPs with laser irradiation could not only directly destroy tumor cells through producing ROS, but also activate immune effector cells for killing tumor cells. The above in vitro study thus indicated that CAM NPs could effectively kill tumor cells by PDT and successfully activate the immune response.
Inspired by the excellent results obtained in vitro, the antitumor effect of CAM NPs was further studied in vivo. First, the hemolysis ratio of CAM NPs was still less than 5% at a high concentration (1 mg mL−1), indicating that CAM NPs were biocompatible and suitable for intravenous administration (Fig. S12, ESI†). Then, we studied the biodistribution and intratumoral accumulation of CAM NPs in a B16F10 melanoma model. As presented in Fig. 3a, the total fluorescence intensity (TFI) at the tumor site was significantly stronger for the CAM NP group compared with the free Ce6 group. More importantly, the fluorescence signals lasted longer in the CAM NP group than in the free Ce6 group. The results thus demonstrated the superior accumulation of CAM NPs at the tumor site in vivo. The strong fluorescence intensity of CAM NPs in the liver and kidneys might be due to their metabolism by the liver and excretion by the kidneys (Table S2, ESI†). The higher accumulation of CAM NPs at the tumor site could be attributed to the enhanced permeability and retention (EPR) effect together with the pathway mediated by the albumin-binding proteins overexpressed in the tumors.48–50
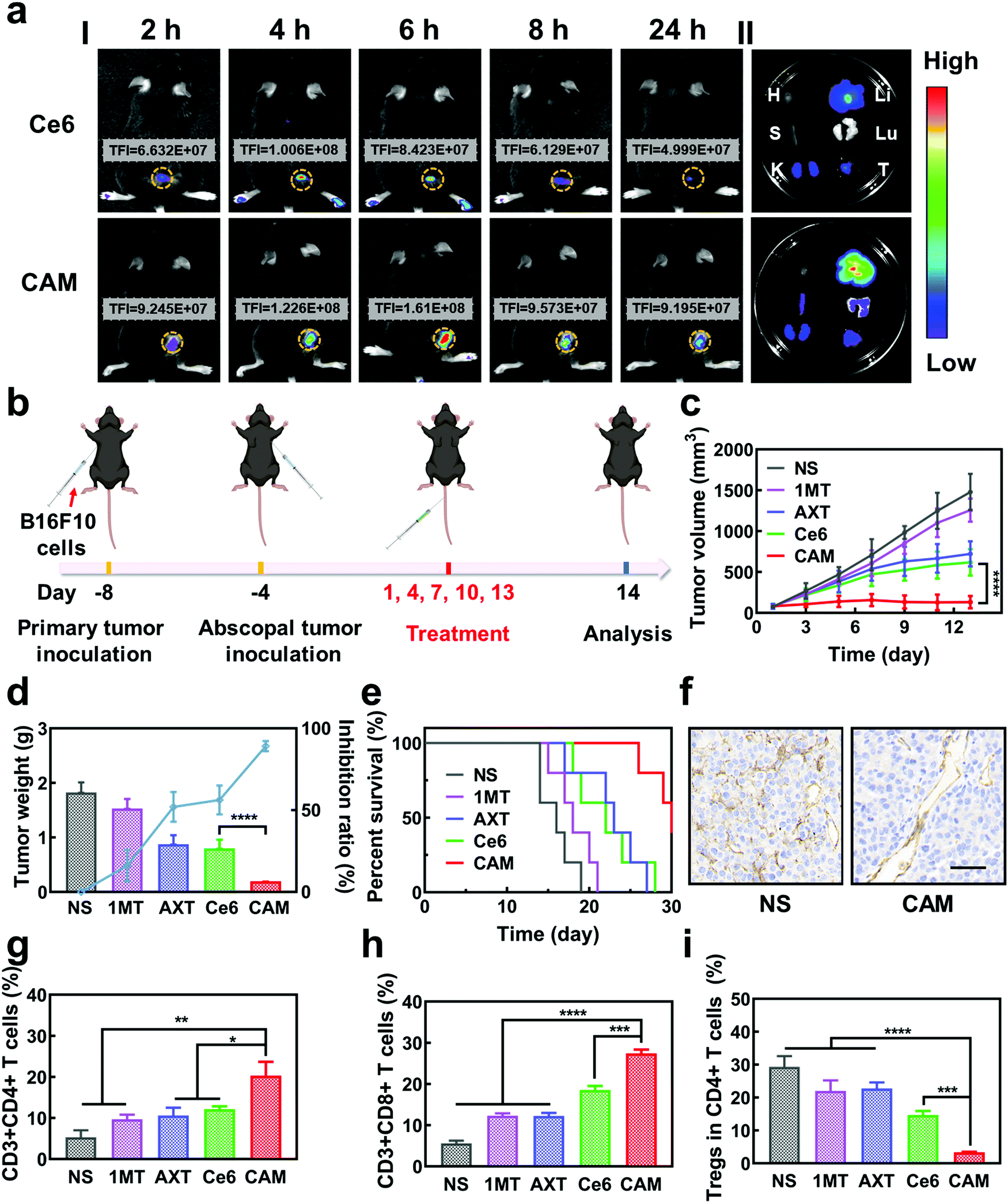 | ||
| Fig. 3 (a) Fluorescence photographs of (I) tumor-bearing mice after injection of Ce6 and CAM NPs and (II) main organs and tumors (H: heart, S: spleen, K: kidney, Li: liver, Lu: lung, and T: tumor) recovered after 24 h injection. (b) Schematic illustration of the bilateral experiment. Change in (c) primary tumor volume, (d) tumor inhibition rate, tumor weight and (e) survival rate of various treated groups (n = 5, mean ± SD, ****p < 0.0001, t-test). (f) IHC staining of CD31 in the primary tumor after different treatments. Scale bar: 50 μm. Relative populations of (g) Ths, (h) CTLs and (i) Tregs in primary tumors after different treatments (n = 3, mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001, t-test). | ||
The in vivo antitumor effect of CAM NPs in bilateral tumor models was then investigated (Fig. 3b). Both the right (primary tumor) and left (abscopal tumor) flank regions of C57BL/6 mice were injected with B16F10 cells subcutaneously to develop a bilateral mouse tumor model. The mice were injected with NS, 1MT, AXT, Ce6 or CAM NPs via the vena caudalis. After 6 h, laser irradiation was performed on the primary tumors, whereas the abscopal tumors were protected from the laser irradiation. The volume and weight of the primary tumors are shown in Fig. 3c and d. Tumor volume increased sharply with time in the NS group. The 1MT group showed weaker inhibition of tumor growth (inhibition rate of 16.1%) because of the lack of effective immune activation. Additionally, the AXT and Ce6 (laser irradiation) groups indicated moderate inhibition of tumor growth (inhibition rates of 52.1% and 56.2%, respectively). The CAM NP (laser irradiation) treatment demonstrated the highest inhibition of tumor growth, almost completely suppressing tumor growth. The survival curves demonstrated that CAM NPs significantly prolonged the survival of the mice compared to the other groups (Fig. 3e). Therefore, it was clear CAM NPs exhibited excellent therapeutic effects against tumors. Additionally, the mouse body weight in the CAM NP group showed no significant change during the treatment (Fig. S13, ESI†), and the results of hematoxylin–eosin (H&E) staining showed no significant pathological changes in the main organs (i.e. heart, spleen, liver, lungs, and kidneys) (Fig. S14, ESI†), which implied the good biocompatibility of CAM NPs.
To understand the antitumor mechanism of the CAM NPs, blood vessel density in the primary tumor tissue was examined. Vascular normalization is accompanied by CD31 (a sensitive marker of blood vessels, expressed in all blood vessels) downregulation.51–53 For the tumors treated with free Ce6-based PDT, the blood vessel density based on anti-CD31 immunohistochemistry (IHC) increased significantly compared to that of the NS group (Fig. 3f and Fig. S15, ESI†), which was attributed to the increased oxygen demand for PDT. However, all the AXT-containing groups (including AXT and CAM NPs) presented a more unobstructed vasculature, indicating the strong vascular normalization effect of AXT by inhibiting the activity of VEGFR. Therefore, these results indicated that the AXT delivered by CAM NPs significantly neutralizes the aberrant vasculature. Due to the vascular normalization, the hypoxic microenvironment could be improved by the enhanced blood flow, resulting in a boosted PDT effect. Moreover, vascular normalization can enable immune cells to enter tumor tissue to kill tumor cells. Particularly, the function of VEGF was greatly controlled. As a result, its negative effects on immunotherapy such as increasing immunosuppressive cell proliferation, suppressing DC maturation and inhibiting T cell function were blocked. Furthermore, 1MT in the CAM NPs could improve immunotherapeutic efficacy by inhibiting the immunosuppressive IDO in the tumor microenvironment. As a result, the photodynamic immunotherapy of the as-prepared CAM NPs was significantly amplified by simultaneously boosting their PDT and immunotherapeutic efficiency.
Previous studies have shown that cytotoxic T cells (CTLs, CD3+ CD8+) can directly destroy tumor cells, while helper T cells (Ths, CD3+ CD4+) regulate the immunity functions.54 Therefore, CTLs and Ths play a vital role in the antitumor immune response. However, Tregs (CD4+ FOXP3+) and TAMs (CD11b+ F4/80+) can suppress the tumor immune response.55,56 To understand the antitumor immune mechanism, the CTLs, Ths, Tregs and TAMs were characterized using FCM after various treatments such as NS, 1MT, AXT, Ce6 (laser irradiation) and CAM (laser irradiation). As can be seen from Fig. 3g and h, the proportion of tumor-infiltrating T lymphocytes (CTLs and Ths) in the CAM NP group was remarkably higher than those of the other groups in the primary tumor. Additionally, the populations of immunosuppressive cells (Tregs and TAMs) were greatly suppressed for the group treated by CAM NPs (Fig. 3i and Fig. S16, ESI†). Therefore, an excellent immune response was triggered for the CAM NP group, which resulted from the improvement of the tumor microenvironment via normalization of tumor vessels and IDO inhibition. Consequently, the primary tumor was almost completely destroyed by the present photodynamic immunotherapy nanoplatform.
DCs are typical sentinels of the immune system that can initiate and direct immune responses.57 To understand the level of maturity of DCs, DCs in inguinal-draining lymph nodes (LNs) were then characterized. The mature DCs were analyzed by staining with CD80 and CD86 markers. The results showed that CAM NPs boosted DC maturation more efficiently in LNs than other groups (Fig. 4a), demonstrating that CAM NPs could induce a much higher level of immune response. In order to further study the systemic immune response induced by CAM NPs, T lymphocytes in the spleen (the largest immune organ in the body) were collected. The results revealed that levels of CTLs and Ths were notably increased, while the levels of Tregs were significantly suppressed in the CAM NP groups (Fig. 4b–d and Fig. S17, ESI†). Moreover, the results of plasma concentrations of the crucial immunomodulatory cytokines verified that CAM NP treatment resulted in a higher level of tumor necrosis factor-alpha (TNF-α), interferon gamma (IFN-γ) and interleukins (IL-2 and IL-6) in comparison with the other groups (Fig. S18, ESI†). As a result, our CAM NPs had an excellent capability of promoting DC maturation, activating immune effector cells, reducing the number of suppressive immune cells and enhancing the levels of crucial immune cytokines, which accordingly resulted in an outstanding systemic immune response.
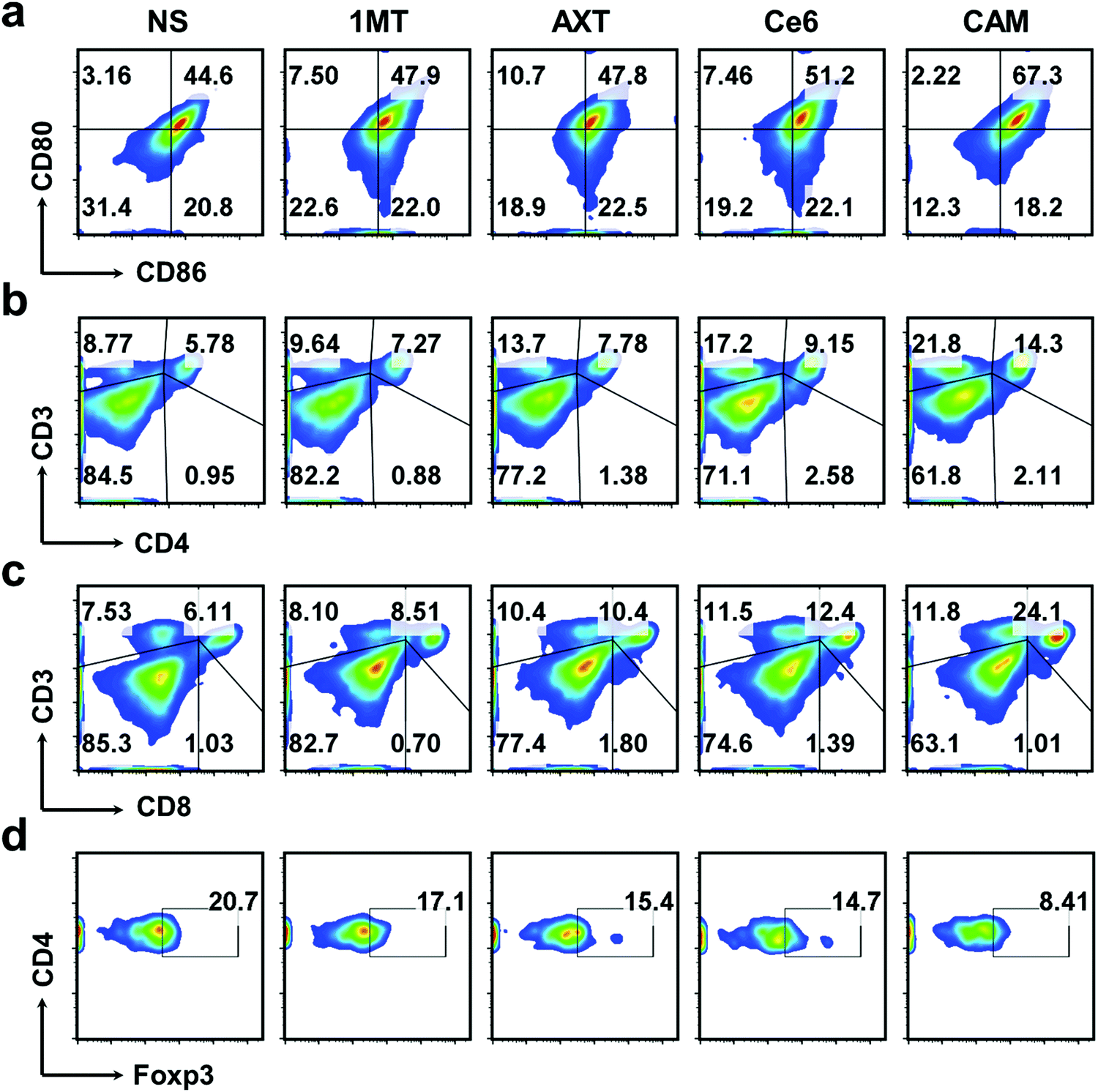 | ||
| Fig. 4 FCM results of (a) DC maturation and (b) Ths, (c) CTLs, and (d) Tregs in splenic lymphocytes of various treated mice. | ||
Metastasis of malignant tumors is often the main cause of cancer treatment failure. To further understand the antitumor immune effectiveness of CAM NPs for pre-existing metastatic tumors, abscopal tumors in bilateral models were studied. As depicted in Fig. S19 and S20 (ESI†), the growth of abscopal tumors in the CAM NP-treated mice was significantly inhibited compared to other groups, demonstrating the superiority of the CAM NP-based photodynamic immunotherapy. Additionally, up-regulation of the immunosupportive CTLs and Ths as well as down-regulation of the immunosuppressive Tregs and TAMs was observed in the abscopal tumors treated with CAM NPs (Fig. 5a–c and Fig. S21, ESI†), further indicating the CAM NPs could successfully amplify immunotherapy to fight abscopal tumor. Hence, our CAM NPs provide a robust nanoplatform to fight abscopal tumor growth through the activated systemic antitumor immune response, demonstrating its promising application in preventing metastasis of tumors.
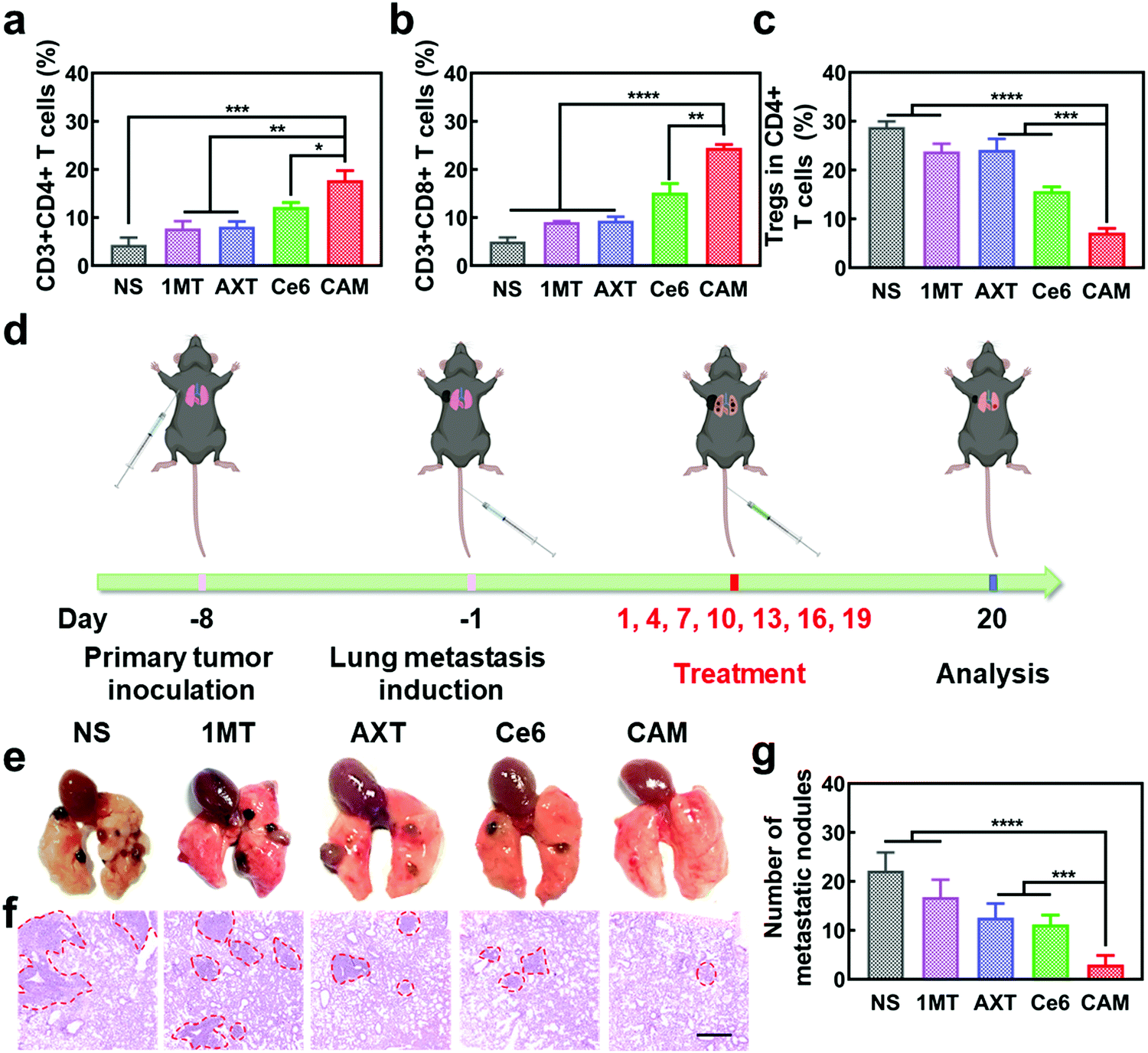 | ||
| Fig. 5 Relative populations of (a) Ths, (b) CTLs and (c) Tregs in abscopal tumors after different treatments (n = 3, mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001, t-test). (d) Schematic illustration of pulmonary metastatic experiment. (e) Camera images and (f) H&E staining photographs of the lung metastatic nodules of B16F10 tumors. Scale bar: 500 μm. (g) Numbers of lung metastatic nodules for each group (n = 3, mean ± SD, ***p < 0.001 and ****p < 0.0001, t-test). | ||
To further demonstrate the anti-metastatic effect of CAM NPs, a lung metastasis tumor-bearing mice model was established as a more aggressive experimental model (Fig. 5d). After various treatments, the lungs of mice were analyzed to study the metastatic lesions. The typical H&E staining assay of lung slices and lung images are presented in Fig. 5e and f. Severe lung metastasis was observed in the NS group. For the groups of 1MT, AXT and Ce6, there was also obvious metastasis although the metastasis was not as severe as that of the NS group. However, the CAM NP group displayed almost no metastasis since there were few obvious metastatic lesions in the lung. The numbers of lung metastatic nodules from each group are also presented in Fig. 5g, which agree well with the results of Fig. 5e. Therefore, the CAM NP-based photodynamic immunotherapy nanoplatform could effectively suppress the metastasis of tumors.
3. Conclusion
In summary, an innovative photosensitizer-based nanoplatform was rationally designed to amplify photodynamic immunotherapy by simultaneously boosting the efficacy of PDT and immunotherapy. The as-designed CAM NP nanoplatform had the advantages of enhancing accumulation in the tumor, promoting tumor vascular normalization and remodeling the tumor microenvironment. The HSA in the CAM NPs enhanced accumulation at tumor sites via albumin-binding proteins overexpressed in tumors. The AXT in the CAM NPs normalized tumor vasculature and improved tumor perfusion by inhibiting the activity of VEGFR, resulting in increased delivery of therapeutic agents and oxygen into tumors, which not only improved the efficiency of ROS production, but, more importantly, promoted the effectiveness of immunotherapy. The 1MT in the CAM NPs further improved the immunotherapy by inhibiting the immunosuppressive IDO in the tumor microenvironment. Our CAM NPs thus demonstrated an amplified photodynamic immunotherapy via tumor microenvironment modulation, exhibiting excellent therapeutic efficacy against tumor growth and metastasis, thereby providing a powerful therapeutic nanoplatform. The present engineering of a photosensitizer nanoplatform via rational design for overcoming photodynamic immunotherapy obstacles holds great promise for fighting tumors.4. Experimental section
Materials
Ce6 and Kyn were obtained from Beijing J & K Technology Co., Ltd. Trifluoroacetate (TFA), 2′,7′-dichlorofluorescein diacetate (DCFH-DA), p-dimethylaminobenzaldehyde, 1,3-diaphenylisobenzofuran (DPBF), HSA and AXT were bought from Aladdin Industrial Corporation. Trp and 1MT were provided by Sigma-Aldrich. Alexa Fluor 488-labeled goat anti-rabbit IgG, anti-HMGB1 and the ATP analysis kit were obtained from Beyotime Biotechnology Co., Ltd. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT), CRT rabbit monoclonal antibody (McAb), phosphate buffered saline (PBS) and the Annexin V-FITC/PI Apoptosis Detection Kit were purchased from Dalian Meilun Biological Technology Co., Ltd. Anti-CD3-APC, anti-CD4-FITC, anti-CD8a-PE, anti-FOXP3-Alexa Fluor 647, anti-CD80-APC, anti-CD86-PE, anti-CD45-FITC, anti-CD11b-PE, anti-F4/80-APC and LEGENDplex™ mouse Th1 Panel were all obtained from BioLegend.Preparation of CAM NPs
CAM NPs were prepared by self-assembly with the help of HSA. Briefly, 15 mg of 1MT, AXT and Ce6 were firstly dispersed separately in 1 mL of DMSO, respectively. An aqueous solution of 4 mg mL−1 HSA was impregnated with 300 μL of DMSO (containing 1.5 mg of 1MT, AXT and Ce6). The suspension was kept under stirring and sonication in the dark for 30 min. After that, the CAM NPs were obtained after dialysis against H2O (MWCO: 1000) to remove unbound molecules and organic solvents.Characterization of CAM NPs
Ce6 (1MT, AXT) levels were determined by UV-Vis spectrometry and the drug loading efficiency was calculated by the following equations. The size distributions and zeta potentials of the CAM NPs in aqueous solution were characterized by dynamic light scattering (DLS, Zetasizer Nano ZS90, Malvern). The morphologies were visualized by transmission electron microscopy (TEM, JEM-1400, Nippon electronics Co., Ltd). The composition of CAM NPs was further characterized using Fourier transform infrared (FT-IR) spectrometry (6700 FT-TR NXR FT-RAMAN, Nicolet). In addition, the physical stability of the CAM NPs in different media (H2O, PBS or 50% FBS/PBS) was estimated by DLS.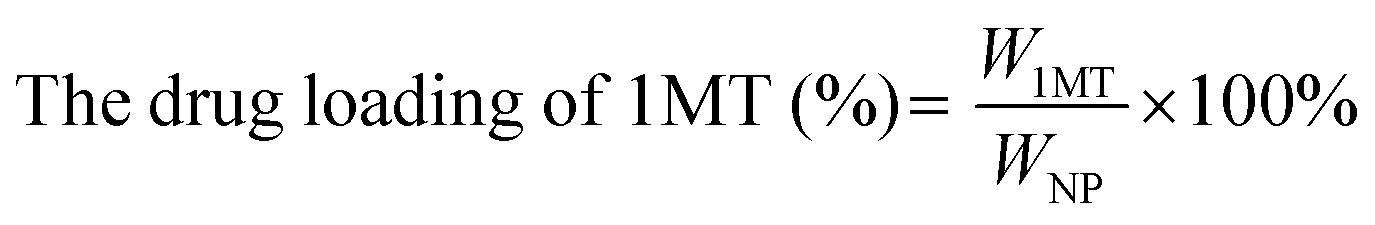 | (1) |
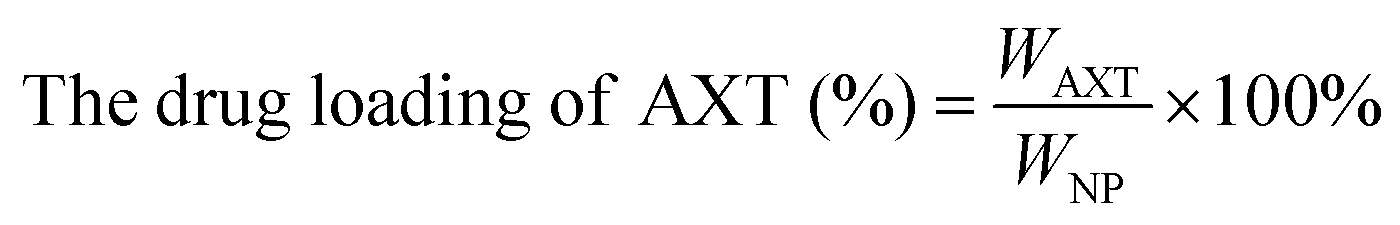 | (2) |
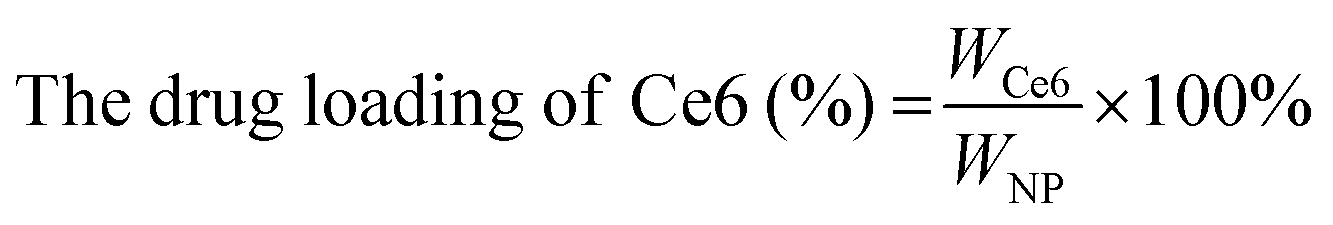 | (3) |
Singlet oxygen generation in vitro
The DPBF reagent was employed for the detection of generated cytotoxic ROS because the absorbance would decrease at 424 nm when DPBF reacted with ROS. Fresh DPBF solution (in DMF, 20 μL, 1.5 mg mL−1) was mixed with the samples (2 mL, 2 μg mL−1 for Ce6) in water and irradiated (660 nm, 100 mW cm−2). Characteristic absorbance at about 424 nm was measured by UV-vis spectrometry. The production efficiency of ROS was determined by the remaining DPBF (%), which was calculated by the following equation: remaining DPBF (%) = At/A0 × 100%. At represents the remaining DPBF absorbance after irradiating for t min, while A0 represents the absorbance without irradiation.In vitro release behavior of CAM NPs
The CAM NPs were put into a dialysis bag (MWCO: 1000 Da), then immersed in buffers (20 mL) of different pH (pH 5.0, 6.5 or 7.4), and oscillated gently (100 rpm) in a 37 °C constant temperature water bath. At a designated time, the dialysis medium was removed and an equal volume of fresh medium was added. Finally, the amount of drug released was measured by fluorospectrophotometry (F-7000, Hitachi High Technology).Cell culture
B16F10 melanoma cells were cultured in RMPI 1640 medium with fetal bovine serum (FBS, 10%), streptomycin (100 μg mL−1) and penicillin (100 U mL−1). Cells were incubated at 37 °C with 5.0% CO2.Cellular internalization analysis
B16F10 cells were cultured in 6-well plates (2 × 105 cells per well) overnight, and then treated with equal doses of free Ce6 or CAM NPs (equivalent to Ce6 at 15 μg mL−1) for 1, 2, 4, 6 and 8 h, respectively. Afterward, cells were washed and resuspended with PBS. The time-dependence of ingestion was quantitatively measured by FCM (BD FACS Aria III) and qualitatively measured by fluorescence microscopy (ECLIPSE-Ti, Nikon).Cytotoxicity assay
B16F10 cells were used to study the cytotoxicity of 1MT, AXT, Ce6 and CAM NPs at different concentrations by the classic MTT assay. B16F10 cells were seeded in 96-well plates (8 × 103 cells per well) overnight, and then treated with RMPI 1640 medium containing different formulations (1MT, AXT, Ce6 or CAM NPs). The concentrations of 1MT, AXT and Ce6 were consistent in all groups if included, varying from 1 to 8 μg mL−1 (1, 2, 4, 6 and 8 μg mL−1) for Ce6, 0.71 to 5.68 μg mL−1 (0.71, 1.42, 2.84, 4.26 and 5.68 μg mL−1) for 1MT and 0.76 to 6.08 μg mL−1 (0.76, 1.52, 3.04, 4.56 and 6.08 μg mL−1) for AXT for each group, respectively. After treatment for 6 h, cells were rinsed and cultured in fresh medium and then irradiated with laser (660 nm, 100 mW cm−2, and 5 min) or not. Finally, the cell viabilities were determined using MTT assay.Cellular ROS generation
B16F10 cells were cultured in 6-well plates (2 × 105 cells per well) overnight and then treated with Ce6 or CAM NP containing medium (equivalent to Ce6 at 2 μg mL−1). After 6 h treatment, cells were washed and then cultured in medium with DCFH-DA (20 μM) for 20 min, followed by laser irradiation (660 nm, 100 mW cm−2, and 5 min) or not. After washing with PBS thrice, the generation of ROS was quantitatively analyzed by FCM and qualitatively analyzed by fluorescence microscopy, respectively.IDO enzyme inhibition assay
B16F10 cells were seeded in 48-well plates (2 × 104 cells per well) overnight. Fresh medium containing free 1MT or CAM NPs at the designated concentration was added, respectively, into the wells. In addition, the medium also contained IFN-γ (50 ng mL−1) and L-Trp (100 μM) to stimulate the expression of IDO and provide sufficient reaction substrate, respectively. After 48 h incubation, the supernatant was mixed with TFA and incubated at 50 °C for 30 min to precipitate the protein. Then, after centrifugation (3000g, 10 min), the supernatant was mixed with acetic acid containing p-dimethylaminobenzaldehyde and measured at 480 nm wavelength using a microplate reader.ICD induced by CAM NPs
To investigate ICD induced by PDT, the expression of CRT, the distribution of HMGB1 and the secretion of ATP of B16F10 cells were examined in vitro. In order to study the exposed CRT on the cell surface, B16F10 cells were seeded in 6-well plates (2 × 105 cells per well) overnight, and then treated with 1MT, AXT, Ce6 or CAM NPs (equivalent to 0.71 μg mL−1 for 1MT, 0.76 μg mL−1 for AXT, and 1 μg mL−1 for Ce6) for 6 h. Next, the cells were washed with PBS thrice, fresh medium was added and then the cells were exposed to laser (100 mW cm−2, 660 nm, and 5 min) or not. After further incubation for 24 h, the cells were cultured with CRT rabbit McAb for 1 h, and then labeled with Alexa Fluor 488-goat anti-rabbit IgG for 1 h. Finally, the B16F10 cells were visualized by fluorescence microscopy.The distribution of HMGB1 in B16F10 cells was detected by immunofluorescence staining and visualized using fluorescence microscopy. Briefly, B16F10 cells were seeded in 6-well plates (2 × 105 cell per well), incubated overnight, and then treated with different samples. After 6 h, the medium was replaced with fresh drug-free medium, followed by laser irradiation (5 min, 660 nm, and 100 mW cm−2) or not. Then, the cells were washed with PBS, stained with Hoechst 33342 (15 min) and fixed by paraformaldehyde (4%, 15 min) at room temperature. After permeation with Triton X-100 (0.1%, 10 min), the cells were stained with anti-HMGB1 and Alexa Fluor 488-labeled goat anti-rabbit IgG in turn. Finally, the B16F10 cells were visualized and imaged using fluorescence microscopy.
The secretion of extracellular ATP was studied using an ATP analysis kit. Briefly, B16F10 cells seeded in 6-well plates (2 × 105 per well) were treated with different samples. After 6 h-treatment, the cells were subjected to laser irradiation (5 min, 660 nm, and 100 mW cm−2) or not. After centrifuging the cell lysate (12000 rpm, 5 min), the supernatant was tested with the ATP analysis kit.
Apoptosis analysis in vitro
B16F10 cells were cultured in 6-well plates (2 × 105 cells per well) overnight, and then incubated with 1MT, AXT, Ce6 or CAM NPs (the concentrations of 1MT, AXT and Ce6 were 1.42, 1.52 and 2 μg mL−1, respectively) for 6h. Afterwards, the cells were washed and cultured with complete medium (1 mL), followed by laser irradiation (5 min, 660 nm, and 100 mW cm−2) or not. Fresh medium (1 mL) with or without PBMCs (2 × 105 cells per well) was subsequently added into each well. After treatment for 48 h, the cells were obtained and marked with Annexin V-FITC and PI. Finally, apoptosis was detected by FCM. PBMCs were obtained by the Ficoll–Urografin density gradient method. In detail, an appropriate amount of Ficoll–Urografin (lymphocytes separation solution) was put into the bottom of a glass tube. Then, an equal volume of mouse peripheral blood was gently added to the upper layer of the separation solution. After centrifugation (2000 rpm, 20 min), the white film layer in the middle (PBMCs) was harvested, rinsed and ultimately incubated with complete medium.Hemolysis test
The hemolysis ratio of CAM NPs was evaluated using the erythrocytes extracted from a New Zealand white rabbit. Briefly, the erythrocytes were dispersed in normal saline (NS) to obtain a 2% erythrocyte suspension. Then, different concentrations of CAM NPs (0.15 mL) were mixed with the erythrocyte suspension (1.25 mL) and NS (1.10 mL) as the experimental group. At the same time, the erythrocyte suspension was mixed with distilled water and NS as positive and negative controls, respectively. After incubating at 37 °C for 3 h, the above samples were centrifuged (1500 rpm, 15 min) and the supernatant was measured using UV-vis spectrophotometry at 540 nm. The hemolysis ratio of CAM NPs was calculated by the following equation:where Asample, Anegative and Apositive represent the absorbance of the CAM NP group, negative group and positive group, respectively.
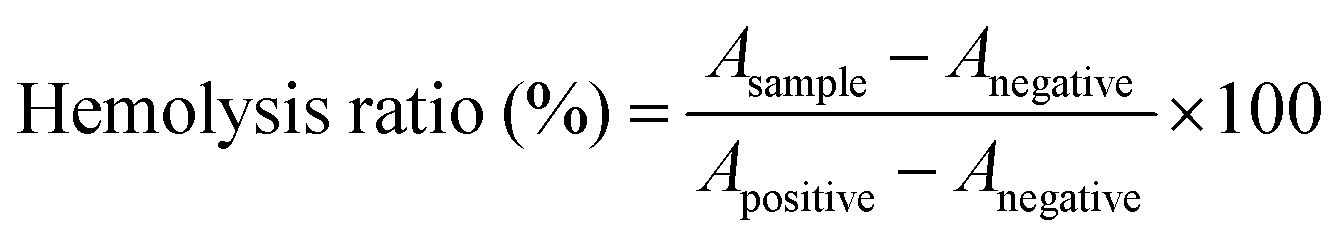
Animals
C57BL/6 mice (female, 6–8 weeks) were bought from Jinan Pengyue Experimental Animal Breeding Co., Ltd. All of the animal experiments were approved by Shandong University Animal Experiment Ethics Review and the Health Guide for the Care and Use of Laboratory Animals of National Institutes. All mice received care in accordance with international ethical guidelines.In vivo biodistribution analysis
1.0 × 106 B16F10 melanoma cells were injected subcutaneously into the right backs of the C57BL/6 mice. The B16F10 tumor-bearing mice were intravenously injected with free Ce6 or CAM NPs via the vena caudalis (equivalent to Ce6 at 3 mg kg−1), respectively, when the tumor reached about 100 mm3. An IVIS imaging system was used for fluorescence imaging at predetermined times (2, 4, 6, 8 and 24 h) after treatment. Finally, mice were sacrificed 24 h after the injection, and their major organs (kidneys, heart, spleen, liver, and lungs) and tumors were collected and imaged.Bilateral tumor model and treatment plan
To create the bilateral tumor model, 8.0 × 105 B16F10 cells were subcutaneously injected into the right anterior axillae (primary tumor) of the C57BL/6 female mice. Four days later, 8.0 × 105 tumor cells were injected subcutaneously into the mice's left forelimb (abscopal tumor). When the volume of primary tumors reached ∼80 mm3, the mice were randomly double-blindly divided into 5 groups, and injected with NS as a control, and 1MT, AXT, Ce6 and CAM NPs as experimental groups (equivalent to Ce6 at 3 mg kg−1, AXT at 2.3 mg kg−1 and 1MT at 2.1 mg kg−1). 6 h after injection, a laser (660 nm, 100 mW cm2, and 5 min) was applied to primary tumors, while the abscopal tumors were protected from irradiation. All the mice were treated every three days, and the body weights and tumor volumes were monitored every two days. The tumor volume was calculated as length × width2/2. After various treatments, the mice were sacrificed to investigate the anticancer effect and mechanism in vivo.In vivo antitumor immune response
After treatment, tumors, inguinal-draining lymph nodes, main organs and blood were harvested to verify the antitumor effect and mechanism. The collected tumors were weighed to determine the tumor inhibition rate. The main organs were stained with H&E to verify the biosafety. CD31 IHC was performed on the primary tumor slices to analyze the effect of blood vessel normalization. DCs (stained with anti-CD11c-FITC, anti-CD80-APC and anti-CD86-PE) in inguinal-draining lymph nodes, Ths (stained with anti-CD3-APC and anti-CD4-FITC), CTLs (stained with anti-CD3-APC and anti-CD8a-PE), Tregs (stained with anti-CD4-FITC and anti-FOXP3-Alexa Fluor 647) in spleens, and primary and abscopal tumors, and TAMs (stained with anti-CD45-FITC, anti-CD11b-PE and anti-F4/80-APC) in primary and abscopal tumors were all analyzed with FCM. The LEGENDplex™ mouse Th1 panel kit was used to quantify crucial cytokines like tumor necrosis factor alpha (TNF-α), interleukins (IL-2 and IL6) and interferon gamma (IFN-γ).Anti-metastasis analysis of pulmonary metastasis model
To create the lung metastatic tumor model, 8.0 × 105 B16F10 cells were injected subcutaneously into the right flank regions of 6–8 week-old C57BL/6 female mice. The mice were intravenously injected with 1.0 × 105 cells when the tumor reached about 80 mm3. After two days, the mice were divided randomly into 5 groups, and then treated in the same way as the bilateral tumor model. After 20 days of treatment, the mice lungs were harvested for H&E staining and metastatic lesion analysis.Statistical analysis
For comparison of two groups, unpaired Student's t-test was processed with the GraphPad Prism 8.0 software. Statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This project was financially supported by the National Natural Science Foundation of China (NSFC, No. 21872083 and 81903558) and Shandong Provincial Major Science and Technology Innovation Project (2018CXGC1411). The authors acknowledge the Pharmaceutical Biology Sharing Platform of Shandong University.References
- S. Z. F. Phua, G. Yang, W. Q. Lim, A. Verma, H. Chen, T. Thanabalu and Y. Zhao, ACS Nano, 2019, 13, 4742–4751 CrossRef CAS.
- D. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS.
- J. X. Fan, M. D. Liu, C. X. Li, S. Hong, D. W. Zheng, X. H. Liu, S. Chen, H. Cheng and X. Z. Zhang, Nanoscale Horiz., 2017, 2, 349–355 RSC.
- X. Li, S. Lee and J. Yoon, Chem. Soc. Rev., 2018, 47, 1174–1188 RSC.
- A. P. Castano, T. N. Demidova and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2005, 2, 91–106 CrossRef CAS.
- X. Duan, C. Chan and W. Lin, Angew. Chem., Int. Ed., 2019, 58, 670–680 CrossRef CAS.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS.
- Q. Li, D. Zhang, J. Zhang, Y. Jiang, A. Song, Z. Li and Y. Luan, Nano Lett., 2019, 19, 6647–6657 CrossRef CAS.
- J. H. Li, Y. Liu, X. N. Li, G. F. Liang, C. S. Ruan and K. Y. Cai, Nanoscale Horiz., 2020, 5, 350–358 RSC.
- Q. Chen, L. Feng, J. Liu, W. Zhu, Z. Dong, Y. Wu and Z. Liu, Adv. Mater., 2016, 28, 7129–7136 CrossRef CAS.
- G. L. Semenza, Nat. Rev. Cancer, 2003, 3, 721–732 CrossRef CAS.
- D. Y. Zhao, W. H. Tao, S. H. Li, L. X. Li, Y. X. Sun, G. T. Li, G. Wang, Y. Wang, B. Lin, C. Luo, Y. J. Wang, M. S. Cheng, Z. G. He and J. Sun, Nanoscale Horiz., 2020, 5, 886–894 RSC.
- G. Bergers and L. E. Benjamin, Nat. Rev. Cancer, 2003, 3, 401–410 CrossRef CAS.
- F. Fan, A. Schimming, D. Jaeger and K. Podar, J. Oncol., 2012, 281261 Search PubMed.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS.
- R. K. Jain, Science, 2005, 307, 58–62 CrossRef CAS.
- Y. Huang, B. Y. S. Kim, C. K. Chan, S. M. Hahn, I. L. Weissman and W. Jiang, Nat. Rev. Immunol., 2018, 18, 195–203 CrossRef CAS.
- Y. Huang, S. Goel, D. G. Duda, D. Fukumura and R. K. Jain, Cancer Res., 2013, 73, 2943–2948 CrossRef CAS.
- R. K. Jain, Cancer Cell, 2014, 26, 605–622 CrossRef CAS.
- D. S. Chen and I. Mellman, Immunity, 2013, 39, 1–10 CrossRef CAS.
- P. S. Hegde, J. J. Wallin and C. Mancao, Semin. Cancer Biol., 2018, 52, 117–124 CrossRef CAS.
- R. Ganss, B. Arnold and G. J. Hammerling, Eur. J. Immunol., 2004, 34, 2635–2641 CrossRef CAS.
- G. T. Motz and G. Coukos, Immunity, 2013, 39, 61–73 CrossRef CAS.
- J. Wada, H. Suzuki, R. Fuchino, A. Yamasaki, S. Nagai, K. Yanai, K. Koga, M. Nakamura, M. Tanaka, T. Morisaki and M. Katano, Anticancer Res., 2009, 29, 881–888 CAS.
- H. Laubli, P. Mueller, L. D'Amico, M. Buchi, A. S. Kashyap and A. Zippelius, Cancer Immunol. Immunother., 2018, 67, 815–824 CrossRef CAS.
- D. I. Gabrilovich, H. L. Chen, H. T. Cunningham, G. M. Meny, S. Nadaf, D. Kavanaugh and D. P. Carbone, Nat. Med., 1996, 2, 1096–1103 CrossRef CAS.
- N. G. Gavalas, M. Tsiatas, O. Tsitsilonis, E. Politi, K. Ioannou, A. C. Ziogas, A. Rodolakis, G. Vlahos, N. Thomakos, D. Haidopoulos, E. Terpos, A. Antsaklis, M. A. Dimopoulos and A. Bamias, Br. J. Cancer, 2012, 107, 1869–1875 CrossRef CAS.
- S. Loeb, A. Koenigsrainer, H. G. Rammensee, G. Opelz and P. Terness, Nat. Rev. Cancer, 2009, 9, 445–452 CrossRef CAS.
- G. C. Prendergast, C. Smith, S. Thomas, L. Mandik-Nayak, L. Laury-Kleintop, R. Metz and A. J. Muller, Cancer Immunol. Immunother., 2014, 63, 721–735 CrossRef CAS.
- D. H. Munn and A. L. Mellor, Trends Immunol., 2016, 37, 193–207 CrossRef CAS.
- S. S. Y. Lee, J. Li, J. N. Tai, T. L. Ratliff, K. Park and J. X. Cheng, ACS Nano, 2015, 9, 2420–2432 CrossRef CAS.
- Y. Gou, Y. Zhang, J. Qi, L. Kong, Z. Zhou, S. Liang, F. Yang and H. Liang, Chem. Biol. Drug Des., 2015, 86, 362–369 CrossRef CAS.
- A. C. van Leeuwen, T. Buckle, G. Bendle, L. Vermeeren, R. V. Olmos, H. G. van de Poel and F. W. B. van Leeuwen, J. Biomed. Opt., 2011, 16, 016004 CrossRef.
- M. Deng, L. Zhang, Y. Jiang and M. Liu, Angew. Chem., Int. Ed., 2016, 55, 15062–15066 CrossRef CAS.
- T. Lin, P. Zhao, Y. Jiang, Y. Tang, H. Jin, Z. Pan, H. He, V. C. Yang and Y. Huang, ACS Nano, 2016, 10, 9999–10012 CrossRef CAS.
- I. Altintas, R. Heukers, R. van der Meel, M. Lacombe, M. Amidi, P. M. P. V. B. E. Henegouwen, W. E. Hennink, R. M. Schiffelers and R. J. Kok, J. Controlled Release, 2013, 165, 110–118 CrossRef CAS.
- B. Escudier and M. Gore, Drugs R&D, 2011, 11, 113–126 CrossRef.
- S. Du Four, S. K. Maenhout, D. Benteyn, B. De Keersmaecker, J. Duerinck, K. Thielemans, B. Neyns and J. L. Aerts, Cancer Immunol. Immunother., 2016, 65, 727–740 CrossRef CAS.
- C. L. Peng, H. C. Lin, W. L. Chiang, Y. H. Shih, P. F. Chiang, T. Y. Luo, C. C. Cheng and M. J. Shieh, Photodiagn. Photodyn. Ther., 2018, 23, 111–118 CrossRef CAS.
- H. Min, J. Wang, Y. Qi, Y. Zhang, X. Han, Y. Xu, J. Xu, Y. Li, L. Chen, K. Cheng, G. Liu, N. Yang, Y. Li and G. Nie, Adv. Mater., 2019, 31, 1808200 CrossRef.
- S. Du Four, S. K. Maenhout, K. De Pierre, D. Renmans, S. P. Niclou, K. Thielemans, B. Neyns and J. L. Aerts, OncoImmunology, 2015, 4, e998107 CrossRef.
- D. H. Munn, M. Zhou, J. T. Attwood, I. Bondarev, S. J. Conway, B. Marshall, C. Brown and A. L. Mellor, Science, 1998, 281, 1191–1193 CrossRef CAS.
- D. H. Munn, E. Shafizadeh, J. T. Attwood, I. Bondarev, A. Pashine and A. L. Mellor, J. Exp. Med., 1999, 189, 1363–1372 CrossRef CAS.
- D. H. Munn and A. L. Mellor, J. Clin. Invest., 2007, 117, 1147–1154 CrossRef CAS.
- J. Park, B. Sun and Y. Yeo, J. Controlled Release, 2017, 263, 90–101 CrossRef CAS.
- B. Hoang, M. J. Ernsting, A. Roy, M. Murakami, E. Undzys and S. D. Li, Biomaterials, 2015, 59, 66–76 CrossRef CAS.
- C. Neuzillet, A. Tijeras-Raballand, J. Cros, S. Faivre, P. Hammel and E. Raymond, Cancer Metastasis Rev., 2013, 32, 585–602 CrossRef CAS.
- C. Zhou, X. Song, C. Guo, Y. Tan, J. Zhao, Q. Yang, D. Chen, T. Tan, X. Sun, T. Gong and Z. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 42534–42548 CrossRef CAS.
- M. Zhang, T. W. Herion, C. Timke, N. Han, K. Hauser, K. J. Weber, P. Peschke, U. Wirkner, M. Lahn and P. E. Huber, Neoplasia, 2011, 13, 537–549 CrossRef CAS.
- Q. Xu, J. Gu, Y. Lv, J. Yuan, N. Yang, J. Chen, C. Wang, X. Hou, X. Jia, L. Feng and G. Yin, Oncol. Lett., 2018, 15, 3437–3446 Search PubMed.
- W. J. Fu, J. Zhuo and L. K. Hu, Oncol. Lett., 2017, 13, 196–200 CrossRef CAS.
- Z. Liu and X. Chen, Chem. Soc. Rev., 2016, 45, 1432–1456 RSC.
- L. N. Guttlein, L. G. Benedetti, C. Fresno, R. G. Spallanzani, S. F. Mansilla, C. Rotondaro, X. L. Raffo Iraolagoitia, E. Salvatierra, A. I. Bravo, E. A. Fernandez, V. Gottifredi, N. W. Zwirner, A. S. Llera and O. L. Podhajcer, Mol. Cancer Res., 2017, 15, 304–316 CrossRef CAS.
- Q. Chen, L. Xu, C. Liang, C. Wang, R. Peng and Z. Liu, Nat. Commun., 2016, 7, 13193 CrossRef CAS.
- K. Shitara and H. Nishikawa, Ann. N. Y. Acad. Sci., 2018, 1417, 104–115 CrossRef CAS.
- J. Kim and J. S. Bae, Mediators Inflammation, 2016, 2016, 6058147 Search PubMed.
- J. Banchereau, F. Briere, C. Caux, J. Davoust, S. Lebecque, Y. T. Liu, B. Pulendran and K. Palucka, Annu. Rev. Immunol., 2000, 18, 767–811 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00480d |
| This journal is © The Royal Society of Chemistry 2021 |