
Self-assembling a natural small molecular inhibitor that shows aggregation-induced emission and potentiates antitumor efficacy†
Xiaona
Chen
,
Zhen
Hu
,
Liqian
Zhou
,
Fu
Zhang
,
Jianqin
Wan
and
Hangxiang
Wang
 *
*
The First Affiliated Hospital, Key Laboratory of Combined Multi-Organ Transplantation, Ministry of Public Health, School of Medicine, Zhejiang University, Hangzhou, 310003, P. R. China. E-mail: wanghx@zju.edu.cn
First published on 2nd November 2020
Abstract
Targeted therapy using small molecular inhibitors has been developed to rewire key signaling pathways in tumor cells, but these inhibitors have had mixed success in the clinic due to their poor pharmaceutical properties and suboptimal intratumoral concentrations. Here, we developed a “self-assembling natural molecular inhibitor” strategy to test the efficacy and feasibility of the water-insoluble agent dasatinib (DAS), a tyrosine kinase inhibitor, for cancer therapy. By exploiting a facile reprecipitation protocol, the DAS inhibitor self-assembled into soluble supramolecular nanoparticles (termed sDNPs) in aqueous solution, without an exogenous excipient. This strategy is applicable for generating systemically injectable and colloid-stable therapeutic nanoparticles of hydrophobic small-molecule inhibitors. Concurrently, during this process, we observed aggregation-induced emission (AIE) of fluorescence for this self-assembled DAS, which makes sDNPs suitable for bioimaging and tracing of cellular trafficking. Notably, in an orthotopic model of breast cancer, administration of sDNPs induced a durable inhibition of primary tumors and reduced the metastatic tumor burden, significantly surpassing the effects of the free DAS inhibitor after oral delivery. In addition, low toxicity was observed for this platform, with effective avoidance of immunotoxicity. To the best of our knowledge, our studies provide the first successful demonstration of self-assembling natural molecular inhibitors with AIE and highlight the feasibility of this approach for the preparation of therapeutic nanoparticles for highly lethal human cancers and many other diseases.
New conceptsMolecular targeting agents are widely used to rewire signaling pathways in tumor cells, but these inhibitors have had mixed success in the clinic. The efficacy of molecular therapies in patients has been greatly compromised by several limitations, such as poor aqueous solubility, low oral bioavailability, rapid metabolism in the body, and dose-limiting toxicity. By choosing a water-insoluble tyrosine kinase inhibitor, dasatinib (DAS), we here tested the validity of our “self-assembling natural molecular inhibitor” strategy for the generation of an injectable anticancer nanotherapy. Upon leveraging the self-assembly protocol, aggregation-induced emission (AIE) of fluorescence by the assembled DAS nanoparticles was unexpectedly observed. This represents the first example of a self-assembling natural molecular inhibitor that exhibits AIE characteristics, enabling its cellular imaging and trafficking tracing. The engineered nanoparticles composed of pure DAS inhibitor can inhibit local breast tumor growth and reduce the metastatic burden in mice more efficiently than the orally administered free DAS, while mitigating the immunotoxicity that is normally elicited by nanocarriers. Our self-assembling approach is radically novel in achieving a 100% drug loaded nanotherapy without requiring either tedious chemical modifications or exogenous excipients. This approach is also broadly applicable and can be applied to rescue other compounds that have limited efficacy due to pharmaceutical challenges. |
Introduction
Precision medicines that enable the selective regulation of specific pathways involved in tumor cells have led to the rapid emergence of next-generation therapies to treat cancer.1,2 Significant efforts have been invested in exploring molecular targeting agents as anticancer therapeutics via clinical testing of efficacy, and numerous molecular inhibitors have been clinically approved.3–5 However, these inhibitors have had only mixed success in improving survival outcomes in cancer patients.6,7 Most existing molecular inhibitors are water-insoluble and show poor pharmaceutical properties, which severely impair their bioavailability after oral administration.8 Therefore, the overall clinical efficacy of such inhibitors has been greatly compromised due to their insufficient intratumoral concentrations and dose-limiting toxicity. Harnessing the advantages of prodrug reconstitution and supramolecular nanoassembly, Sengupta et al. rationally formulated a PI3K inhibitor in liposomes, which enabled systemic injectability and improved drug bioavailability, as well as in vivo efficacy.9 Accumulating evidence suggests that new effective approaches are preferable to improve the therapeutic index for these pharmacologically potent inhibitors.10–13Supramolecular chemistry involves chemical systems that are constructed by the self-assembly of individual molecular subunits via noncovalent interactions, thus providing facile and cost-effective approaches for the creation of novel and inspiring nanostructures for biomedical applications.14,15 Couvreur et al. developed the “squalenoylation” technology that combines drug derivatization using squalene with amphiphilic self-assembly to construct injectable therapeutic nanoparticles.16,17 Taking advantage of a similar concept, we previously proposed a “PUFAylation” prodrug strategy for converting anticancer drugs (e.g., cabazitaxel, cisplatin, and gemcitabine) into self-assembling nanotherapeutics via conjugation with natural polyunsaturated fatty acids (PUFAs).18–23 By rendering the drug temporarily inactive through a cleavable linkage and assembling prodrug entities, toxicity is substantially alleviated, and drugs can be administered at higher doses.24,25 Despite these demonstrations of success, the strategies for pharmaceutical delivery of active payloads often entail complex synthetic schemes and/or exogenous matrices. Self-deliverable systems comprising pure drugs do not require exogenous materials and are thus able to completely avoid excipient-associated toxicity concerns.26,27 Such a delivery system should have great potential for translation to the clinic.
Here, we chose a water-insoluble small-molecule inhibitor, dasatinib (DAS), to test the validity of our “self-assembling natural molecular inhibitor” strategy. This strategy relies on neither tedious drug chemical modifications nor exogenous excipients for the fabrication of intravenously injectable nanotherapies. We found that the DAS agent was capable of self-organizing into colloid-stable nanosuspensions without any exogenous excipients, enabling intravenous injection. Moreover, for the first time, we unexpectedly observed aggregation-induced emission (AIE) by the self-assembled DAS nanoparticles, which enables imaging of cellular uptake. Mechanistic studies suggested that restriction of intramolecular motion (RIM) dominates the AIE characteristics of the self-assembled DAS molecules. In a preclinical 4T1 xenograft model of metastatic breast cancer, the administration of DAS nanoparticles produced a durable inhibition of primary tumors as well as a reduced metastatic tumor burden, which significantly surpassed the effects of orally delivered DAS. Thus, our results show that self-assembling molecular inhibitors could be a practical and expedient approach for the construction of effective and safe targeted therapeutic nanoparticles.
Results and discussion
Self-assembly of dasatinib and other molecular inhibitors
To assess whether DAS could self-assemble in aqueous media without additional matrices, we quickly injected a solution of DAS in dimethyl sulfoxide (DMSO) into deionized (DI) water under ultrasonication, and the solution was further dialyzed to remove DMSO. In this protocol, DMSO is simply used as a good organic solvent to solubilize the inhibitors. Upon addition to a poor solvent (i.e., water), the inhibitor amphiphiles underwent self-organization to form water-soluble nanostructures. This standard nanoprecipitation protocol allowed the formation of transparent solutions rather than precipitates. Fig. 1A shows the scanning electron microscopy (SEM) images of uniformly spherical nanostructures with an average diameter of ∼100 nm. We designated these self-assembled supramolecular DAS nanoparticles as sDNPs. Dynamic light scattering (DLS) measurements further confirmed that the average hydrodynamic diameter (DH) of sDNPs was ∼160 nm, with a narrow monomodal distribution (Fig. 1B) and positive zeta potentials (i.e., 29.7 ± 0.3 mV and 12.9 ± 0.2 mV in DI water and phosphate-buffered saline (PBS), respectively). When DAS was assembled into sDNPs, most of the DAS molecules (>96%) formed nanoaggregates, as evidenced by analyses using ultrafiltration and ultracentrifugation (Table S1, ESI†).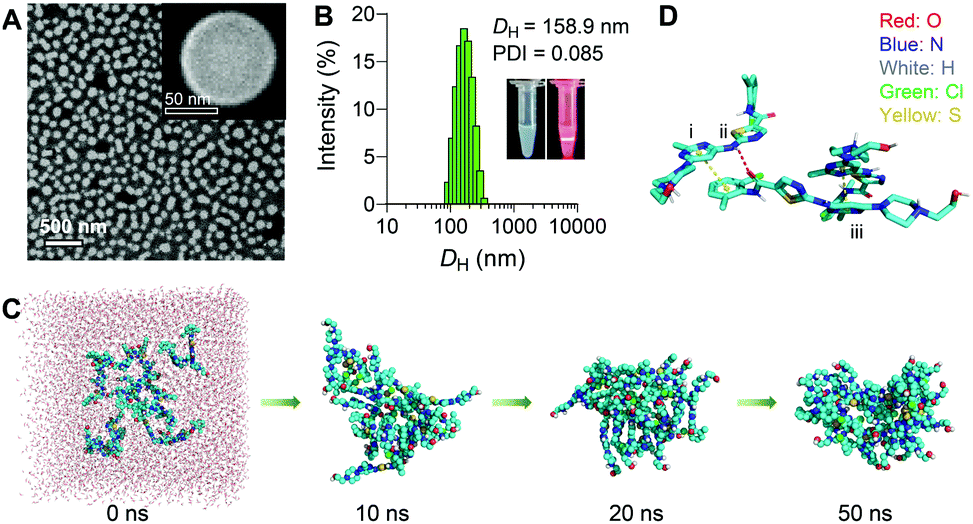 | ||
Fig. 1 Characterization of supramolecular dasatinib nanoparticles (sDNPs). (A) Scanning electron microscopy (SEM) image of sDNPs. Inset, enlarged nanoparticle. (B) Size distribution of the hydrodynamic diameter (DH) of sDNPs measured using dynamic light scattering (DLS) in water. Inset, photographs of the sDNP solution with the Tyndall effect. The polydispersity index, PDI. (C) Snapshots of the formation of DAS aggregates upon molecular dynamics simulation. (D) Noncovalent interactions observed: (i and iii) π–π interactions; (ii) intermolecular N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds. C hydrogen bonds. | ||
The interaction of nanoparticles with physiological systems is an important factor that affects the adsorption of blood proteins and the fate of intravenously administered nanoparticles. We thus examined whether sDNPs hold the ability to block nonspecific protein adsorption. In this experiment, three kinds of widely used formulations with different charges and surface chemistry were included for comparison (Table S2, ESI†).28 Upon exposure to bovine serum albumin (BSA) or bovine serum, the nanoparticles were precipitated by ultracentrifugation, and the adsorbed proteins were quantified (Fig. S1, ESI†). Interestingly, sDNPs adsorbed significantly less proteins than the positively charged EndoTAG-1 liposomes and exhibited comparable adsorption behaviors with those of PEGylated liposomes or polymeric micelles composed of poly(ethylene glycol)-block-poly(D,L-lactic acid) (e.g., PEG5k–PLA8k). While grafting particles with PEG is well known to suppress protein adsorption, a similar ‘stealth’ effect observed for the sDNPs could be attributed to the hydrophilic surface formed by the abundant external hydroxyl moieties.29 The stability test further suggested that sDNPs remained colloid-stable for one-week upon incubation in different media, including DI water, PBS (10 mM, pH7.4) and serum (5 and 10%, v/v) (Fig. S2, ESI†). Notably, using the molecular self-assembly protocol, the solubility of the DAS agent substantially increased from ∼8 × 10−3 to 7.9 g L−1 in DI water, which renders the sDNP platform injectable at therapeutically relevant doses. Due to the self-assembly of the pure drug, quantitative formulation efficiency and an exceptionally high drug loading of 100% were achieved.
Intrigued by this finding, we assessed whether this protocol could be extended to create soluble nanoassemblies of other inhibitors. We then chose a set of thirteen hydrophobic inhibitors with poor water solubility. Interestingly, this procedure identified seven compounds with the tendency to self-organize in water and form colloid-stable nanoparticles despite significant hydrophobicity (Fig. S3–S6 and Table S3, ESI†). These results suggest that our self-assembly approach is broadly applicable for a generation of self-deliverable injectable nanosystems for preclinical studies.
Molecular dynamics (MD) simulation was carried out to gain insight into the mechanism underlying the self-assembly of the DAS agent.30 Sixteen DAS molecules were included in a cuboid box with explicit water and subjected to 50 ns of MD simulations. In the initial state, the DAS molecules were randomly separated from each other (Fig. 1C). From the initial random configuration, a compact nanoaggregate was formed spontaneously after the simulations. We then explored the possible noncovalent interactions that dominate the self-assembly process. Typically, intermolecular N–H⋯OC hydrogen bonds were detected and were responsible for the alignment of internal stability (Fig. 1D, ii). In addition, π–π interactions and van der Waals interactions were observed between the aromatic rings (Fig. 1D, i and iii). Hydroxyl groups were exposed outside the cluster, stabilizing the entire nanosystem. Overall, these multiple interactions provided significant driving forces for the self-assembly of the DAS agent, contributing to the formation of stable nanoassemblies. These theoretical results were in agreement with the experimentally observed result that DAS was capable of exhibiting self-assembly behavior in water, as evidenced by SEM and DLS observations.
AIE and mechanistic studies
When we investigated the optical properties of sDNP solutions, we unexpectedly observed AIE behavior for this natural molecular inhibitor (Fig. 2A–D).31,32 When DAS was dissolved in DMSO, it was nonemissive, but it showed increasingly stronger fluorescence emission after the gradual addition of a poor solvent (water) (Fig. 2B). Moreover, the AIE feature of sDNPs in water/DMSO mixtures upon varying the water fraction was examined (Fig. 2C and D).33 DAS in DMSO solvent exhibited very weak emission, with the maximum intensity at 422 nm. Increasing the proportion of water intensified the emission (Fig. 2C), resulting from the formation of nanoaggregates.34,35 Finally, when the fraction of water reached 99%, the emission intensity was boosted further, with a 50-fold enhancement relative to that of DAS in the solution state (Fig. 2D). The UV absorption was also investigated, and a gradual decrease in the absorbance spectra was observed with increasing the water fraction (Fig. S7, ESI†). The fluorescence quantum yield (φf) was estimated using quinine sulfate as a standard.36 The calculated φf of DAS in the aggregated state increased to 0.15 from 0.03 in the solution state (Table S4, ESI†).37 Strikingly, compared with DAS powder, lyophilized sDNPs exhibited brighter emission under UV (365 nm) illumination due to the preservation of intensely compact structures (Fig. 2E). Furthermore, SEM observation revealed the spherical morphology of the self-assembled lyophilized sDNPs, whereas the DAS power exhibited amorphous structures (Fig. S8, ESI†).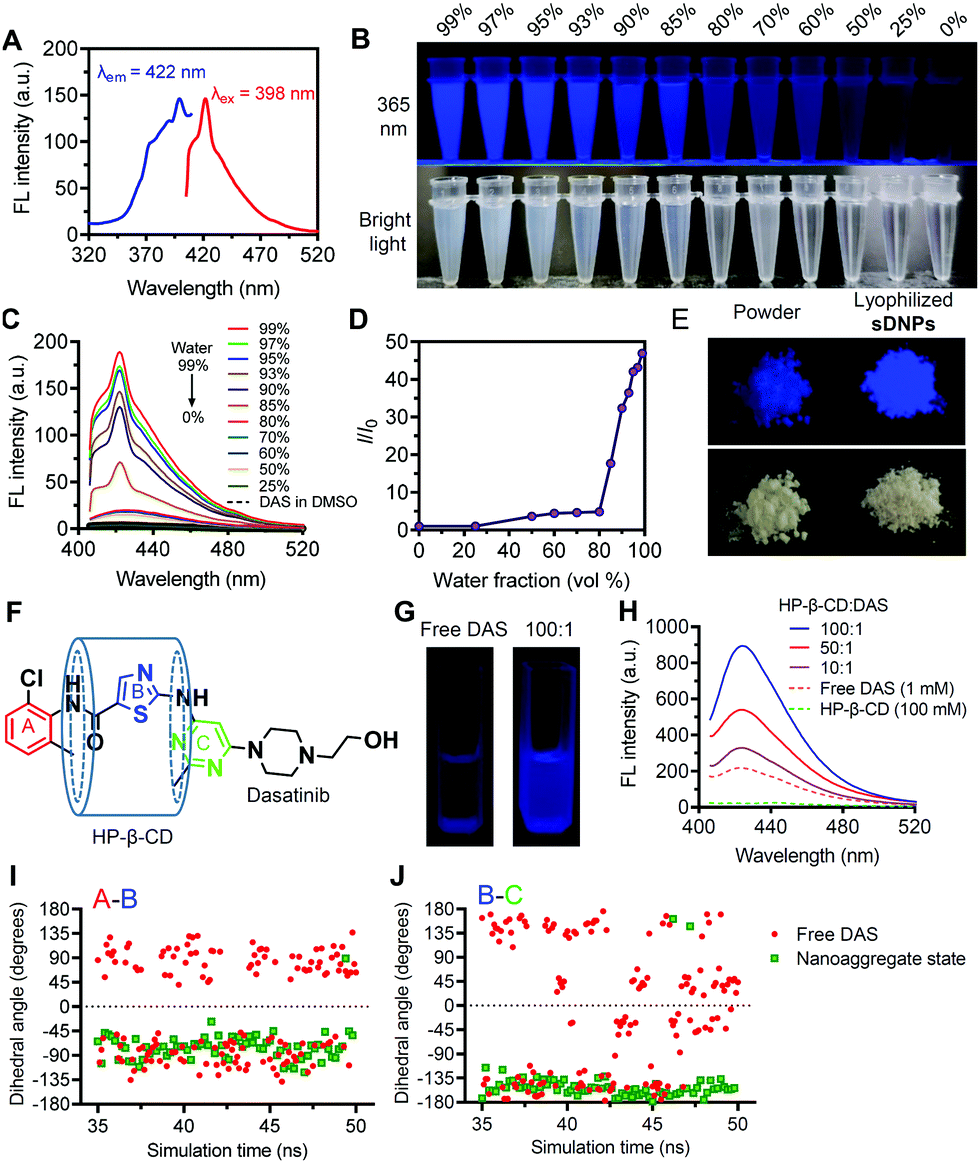 | ||
| Fig. 2 Characterization and mechanistic study of aggregation-induced emission (AIE) for self-assembled DAS agents. (A) Excitation and emission spectra of sDNPs. (B) Photographs of the DAS agent in water/DMSO mixtures with different water fractions (fw) taken under a UV lamp at 365 nm irradiation or ambient light. (C) Emission spectra of DAS solutions (showing fw). Excitation wavelength: 398 nm. (D) Plot of the relative emission intensity (I/I0) of DAS versus fw. Concentration: 0.5 mg mL−1. I0 = intensity at fw = 0%. (E) Digital images of the original solid powder of DAS and the lyophilized sDNPs taken under a UV lamp at 365 nm irradiation or bright light. (F) Schematic illustration of the complexation of HP-β-CD (host) and the DAS molecule (guest). (G) Photographs of DAS in DMSO with/without 100 mM HP-β-CD taken under UV irradiation. (H) Emission spectra of DAS in DMSO with different molar ratios of HP-β-CD. Observed dihedral angles between ring A (red) and ring B (blue) (I) and between ring B and ring C (green) (J) from one molecule in the free DAS state (red) or in the aggregated state (green), as acquired from the MD simulation. | ||
We attempted to elucidate the mechanism underlying the AIE effect of the DAS agents.38 The RIM mechanism is the primary contributor to the AIE effect for most AIE luminogens (AIEgens).39–43 Cyclodextrins (CDs) are a class of macrocyclic hosts with variable cavity sizes and the capacity to form host–guest complexes with many hydrophobic compounds. Owing to the potential interactions between β-CD and DAS drugs, we utilized β-CD (i.e., HP-β-CD) to restrict DAS molecular motion.44 Complexation of β-CD and DAS could confine all of the aromatic rings, thus making DAS emissive (Fig. 2F). As expected, upon the addition of HP-β-CD to the DMSO solution of DAS, we observed progressively increased fluorescence intensities (Fig. 2G and H). In addition, the temperature-dependent changes in the emission spectra of free DAS in DMSO were investigated, and the results showed that increasing the temperature from 20 to 80 °C gradually diminished the fluorescence intensity (Fig. S9, ESI†).
Furthermore, MD simulations enabled us to monitor the microscopic dynamics of DAS in each aggregated state at atomic resolution.45 Thus, key dihedral angles between the phenyl ring (ring A) and the thiophene ring (ring B) and between ring B and the pyrimidine ring (ring C) obtained from one representative molecule were recorded as a function of the MD simulation time (Fig. 2F). In the solution state, the dihedral angles of A/B and B/C in DAS are widely distributed between −180° and 180°, which results in energy consumption in the excited state because of the active molecular rotations (Fig. 2I and J). In contrast, the dihedral angles were restricted at approximately −90° (A/B) and −135° (B/C) in the aggregated state, suggesting a substantially constrained rotational motion of DAS molecules in sDNPs. Taken together, these data provide compelling evidence that DAS can indeed serve as an AIEgen induced by the RIM mechanism, and to the best of our knowledge, the DAS drug represents the first example of an AIE-active natural molecular inhibitor.
Mechanism of sDNP cellular uptake via endocytosis
We speculated that the cellular uptake and trafficking of sDNPs could be directly observed without additional labeling with fluorescent dyes because of their AIE characteristics. DU145 human prostate cells were exposed to sDNPs and visualized using confocal laser scanning microscopy (CLSM). A strong fluorescence signal ascribed to the self-assembled DAS molecules was observed, and the fluorescence intensity in the cells increased rapidly (Fig. 3A and B). Moreover, we found that sDNPs colocalized with LysoTracker Green in endo/lysosomes, as evidenced by significant merging in the CLSM images, suggesting the accumulation of sDNPs in endo/lysosomal compartments (Fig. 3A). Pearson's correlation coefficient (PCC) analysis was performed to evaluate the extent of colocalization (Fig. 3C). High PCC values (∼0.8) at 1 and 2 h were observed, which indicated positive colocalization and implied that the primary uptake pathway of sDNPs occurred via endocytosis. Furthermore, PCC values declined after 4 h, which should be attributable to nanoparticle escape from endo/lysosomes. Escape of therapeutic payloads from these compartments is essential for drug activity. During the self-assembly process, the hydrophilic piperazine moiety of DAS agents is exposed on the outside of the sDNP surface. This could endow the nanoparticles with proton-absorbing capability inside endosomes (pH 5–6) and lysosomes (pH 4–5), yielding a proton sponge effect to induce swelling and/or rupture of the organelle membrane.46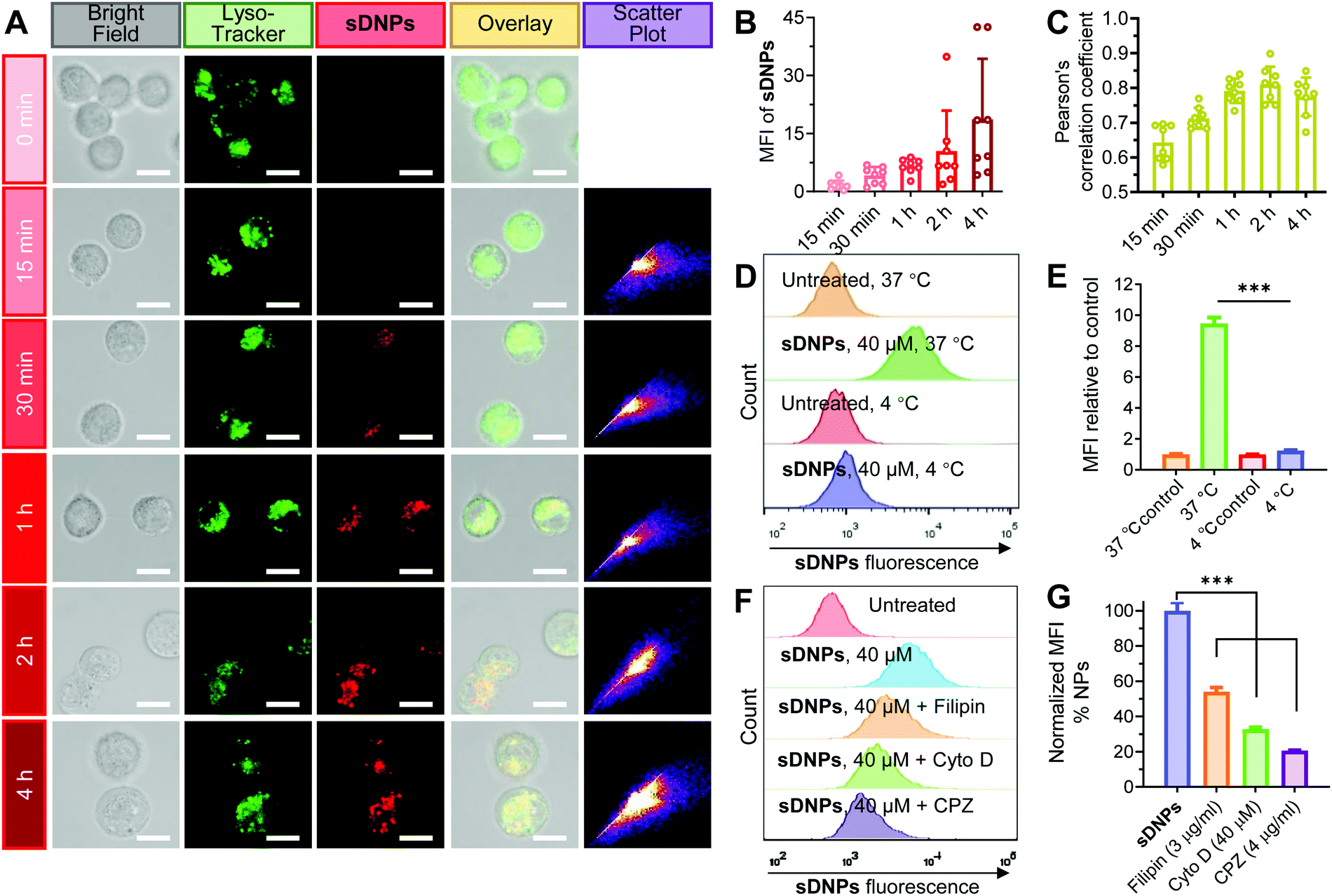 | ||
| Fig. 3 Intracellular uptake of sDNPs and their endocytic pathway in DU145 cells. (A) Representative images showing the colocalization of internalized sDNPs with an endo/lysosomal marker (LysoTracker Green DND-26; green) using confocal microscopy. Scale bar, 10 μm. For sDNPs, excitation wavelength: 405 nm; emission filter: 420–440 nm. (B) The mean fluorescence intensity of sDNPs and (C) Pearson's correlation coefficient (upon colocalization between sDNPs and LysoTracker) were analyzed using ImageJ. The data are presented as the mean ± S.D., quantified from eight independent fields of view. (D and E) The internalization efficiency of sDNPs in cells cultured at 37 °C or 4 °C using flow cytometry. (F and G) Fluorescence-activated cell sorting (FACS) analysis of DU145 cells incubated with sDNPs for 4 h in the absence or presence of endocytic inhibitors (CPZ, filipin and Cyto (D). The data are presented as the mean ± S.D. of three independent experiments. ***p < 0.001. | ||
The uptake process of the self-assembled nanoparticles was further investigated. Lowering the temperature of the cell cultures to 4 °C significantly impaired the uptake of sDNPs, supporting the energy-dependent endocytosis mechanism (Fig. 3D, E and Fig. S10, ESI†). To examine the detailed routes by which sDNPs are taken up, specific inhibitors that mediate the blockade of endocytic pathways were exploited.47 Filipin (an inhibitor of caveolae-dependent endocytosis), cytochalasin D (Cyto D; a macropinocytosis inhibitor), or chlorpromazine (CPZ; an inhibitor of clathrin-dependent endocytosis) was used to pretreat the cells for 30 min, and then the cells were incubated with sDNPs.48 Compared to that of the cells without inhibitor pretreatment, the uptake of sDNPs was dramatically reduced to 54.0%, 32.8% and 20.6% in the presence of filipin, Cyto D, and CPZ, respectively (Fig. 3F, G and Fig. S11A, ESI†). In addition, nanoparticle uptake was also attenuated by increased concentrations of CPZ (Fig. S11B and C, ESI†). Together, these results suggested that multiple endocytosis routes contributed to sDNP internalization but it was primarily dominated by the clathrin-mediated endocytic pathway. This is consistent with previous studies using nanomedicines to promote entry into cancer cells, in which multiple pathways were typically exploited for cellular uptake.49,50 Therefore, unlike free DAS drug, the sDNP platform is capable of relying on endocytic pathways to overcome barriers and facilitate cellular entry and subsequent effective endosomal escape to regain its activity.
Augmented in vitro activity endowed by the self-assembled DAS agent
Intrigued by whether the assembly of DAS could increase cytotoxic activity, we examined the viability of tumor cells using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) tetrazolium assay after exposure to the drugs. The half-maximal inhibitory concentration (IC50) was extrapolated from the dose–response curves (Fig. 4A, B and Table S5, ESI†). Compared to the free DAS agent, sDNPs exhibited slightly superior cytotoxicity towards DU145 human prostate cells and 4T1 murine breast cancer cells. We further reduced the drug incubation time to 24 h, and the cell apoptosis rate was examined using fluorescence-activated cell sorting (FACS) after dual staining with annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI). The results showed that sDNPs induced a 38.32% apoptosis rate in DU145 cells, compared to the rate of only 24.81% induced by free DAS at identical concentrations (Fig. 4C). This marked drug toxicity that triggered cell apoptosis was also confirmed in sDNP-treated 4T1 cells (Fig. 4D).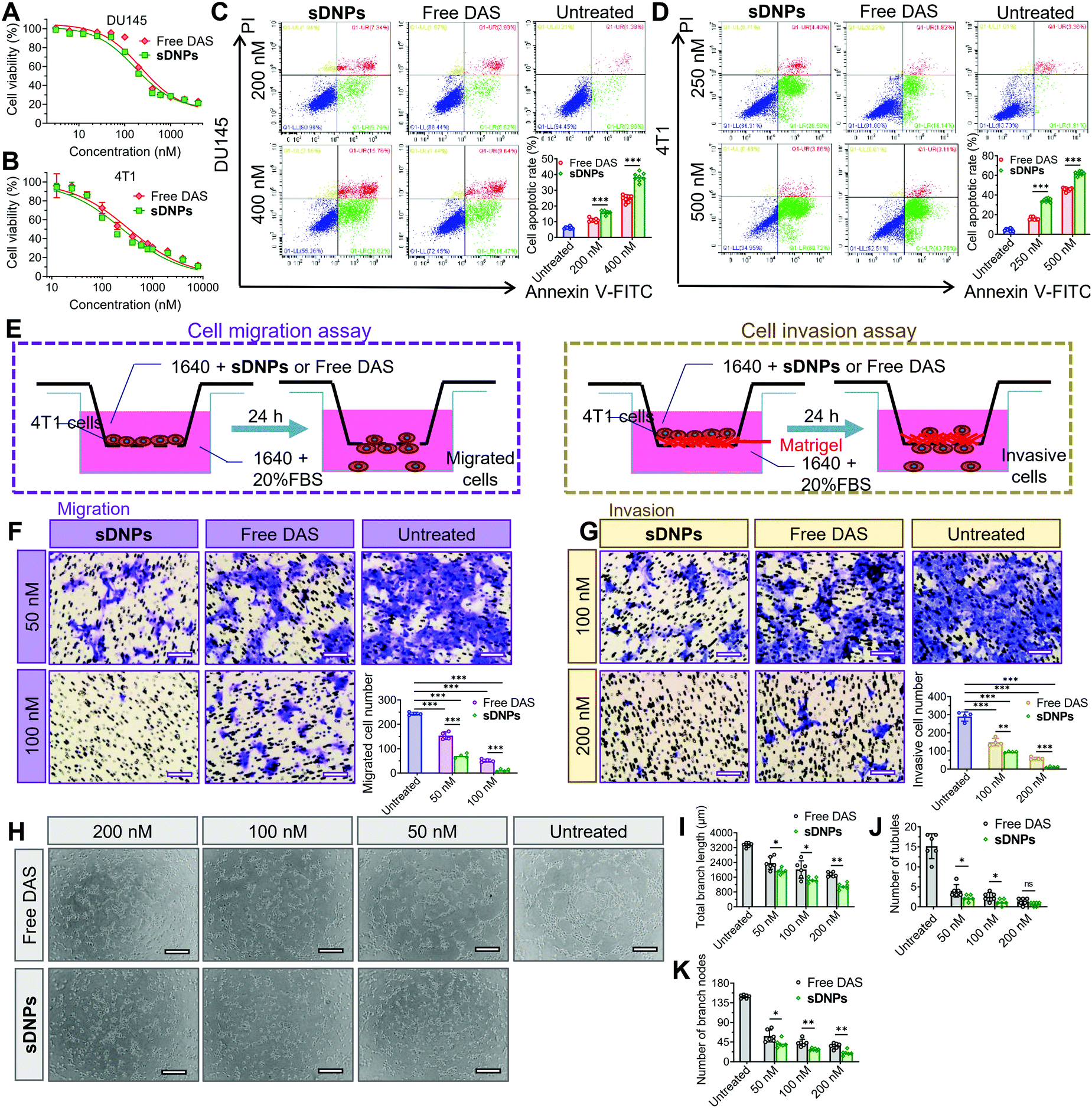 | ||
| Fig. 4 sDNPs outperformed free DAS, with enhanced antitumor activity and excellent inhibition of mobility and antiangiogenesis in vitro. The inhibitory effects of free DAS and sDNPs on cell viability in (A) DU145 cells and (B) 4T1 cells were evaluated using an MTT assay. Apoptosis assay of (C) DU145 cells and (D) 4T1 cells, as determined using FACS analysis (annexin V-FITC)/PI dual staining). (E) Schematic illustration of cell migration/invasion assays in 4T1 cells. Representative images of the (F) migration and (G) invasion assay results, with quantitative analysis corresponding to the images. Scale bars = 100 μm. 4T1 cells were incubated with PRMI 1640 medium containing sDNPs or free DAS for 24 h, and cells without drug treatment were used as the control group (n = 4). Tube formation of HUVECs treated with sDNPs or free DAS for 3 h. (H) Typical images of the tubular network were captured later. Quantitative results were evaluated by quantifying the (I) total branch length, (J) number of tubules and (K) number of branch nodes using ImageJ. The experiments were performed in quadruplicate, and the data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
We then focused on the activity of DAS as a multitargeted inhibitor of the Src family and BCR/ABL kinases, which are involved in multiple cellular processes, including mobility, metastasis and angiogenesis.51–53 To evaluate the inhibitory effect of sDNPs on cell motility, we performed a scratch wound-healing assay using human umbilical vein endothelial cells (HUVECs). In free DAS-treated HUVECs, the wound-healing rate was approximately 14.8%, showing less aggressive motility than that observed in untreated cells. Notably, treatment with sDNPs for 24 h was effective in substantially reducing healing, with a wound-healing rate of 5.50% (Fig. S12A and B, ESI†).
Subsequently, inhibition of cell migration and invasion was investigated using a Transwell assay to verify the effectiveness of the DAS agent delivered in nanoparticle form (Fig. 4E). The highly aggressive 4T1 breast cancer cell line was included in this experiment. As expected, the addition of the DAS agent reduced cell migration to the lower chamber relative to that of untreated cells. Cells treated with sDNPs showed significantly reduced migration compared to that of cells treated with DAS in its free form. The differences between the two treatment groups were statistically significant (Fig. 4F). The trend observed in the migration assay was similar to the results observed in the invasion assay; that is, sDNPs preserved the anti-invasive capacity and markedly impaired the ability of metastatic 4T1 cells to invade through the extracellular matrix (Fig. 4G).
DAS, a potent antiangiogenic inhibitor, suppresses VEGF-induced angiogenesis by blocking the Src signaling pathway.54 To further examine whether this activity was preserved in the sDNP platform, we conducted a tube formation assay in HUVECs. Upon the addition of free DAS or sDNPs, the capillary tubes were clearly destroyed compared to those in untreated cells; however, free DAS was less effective than sDNPs in the inhibition of tube-forming capacity (Fig. 4H). Consistently, sDNPs enhanced the antiangiogenic activity of DAS, as supported by the quantification of branch length and the number of tubules and nodes (Fig. 4I–K). Hence, nanoparticle delivery potentiates these in vitro activities more than delivery of DAS in its free drug form. This could presumably be due to the formation of positively charged nanoparticles, which facilitates cellular internalization.
Enhanced local tumor control and antimetastatic activity of injectable sDNPsin vivo
The clinical efficacy of molecular inhibitors has been greatly compromised due to several limitations, such as poor aqueous solubility, low oral bioavailability, and rapid metabolism in the body. Specifically, DAS is orally administered in a tablet form in the clinic due to its insolubility, and it generally shows poor absorption in the gastrointestinal tract and prominent first-pass metabolism.55 We hypothesized that injectable sDNPs could mitigate these obstacles.To test this hypothesis, the efficacy of sDNPs and orally delivered DAS was comparatively evaluated in a metastatic tumor xenograft mouse model. For this purpose, an orthotopic xenograft model was established by implanting murine 4T1 cells into the mammary fat pads of Balb/c mice. When the tumors reached ∼200 mm3 in volume, the mice were administered with free DAS via gavage or sDNPsvia intravenous injection five times every 2 days (q2dx5) at the same dosages. We compared antitumor activity by measuring the volume of the orthotopic tumors (Fig. 5A). Compared with saline treatment, sDNP treatment produced superior tumor growth inhibition of 54.8% and 72.7% at doses of 15 and 30 mg kg−1, respectively, in mouse tumors. Disappointingly, orally delivered DAS at these doses had very limited effects. We did not observe statistically significant differences between the saline and free DAS treatment groups. The weights of tumors excised at the end of the observation period further verified the improved efficacy of the nanodelivery of DAS (Fig. 5B and C).
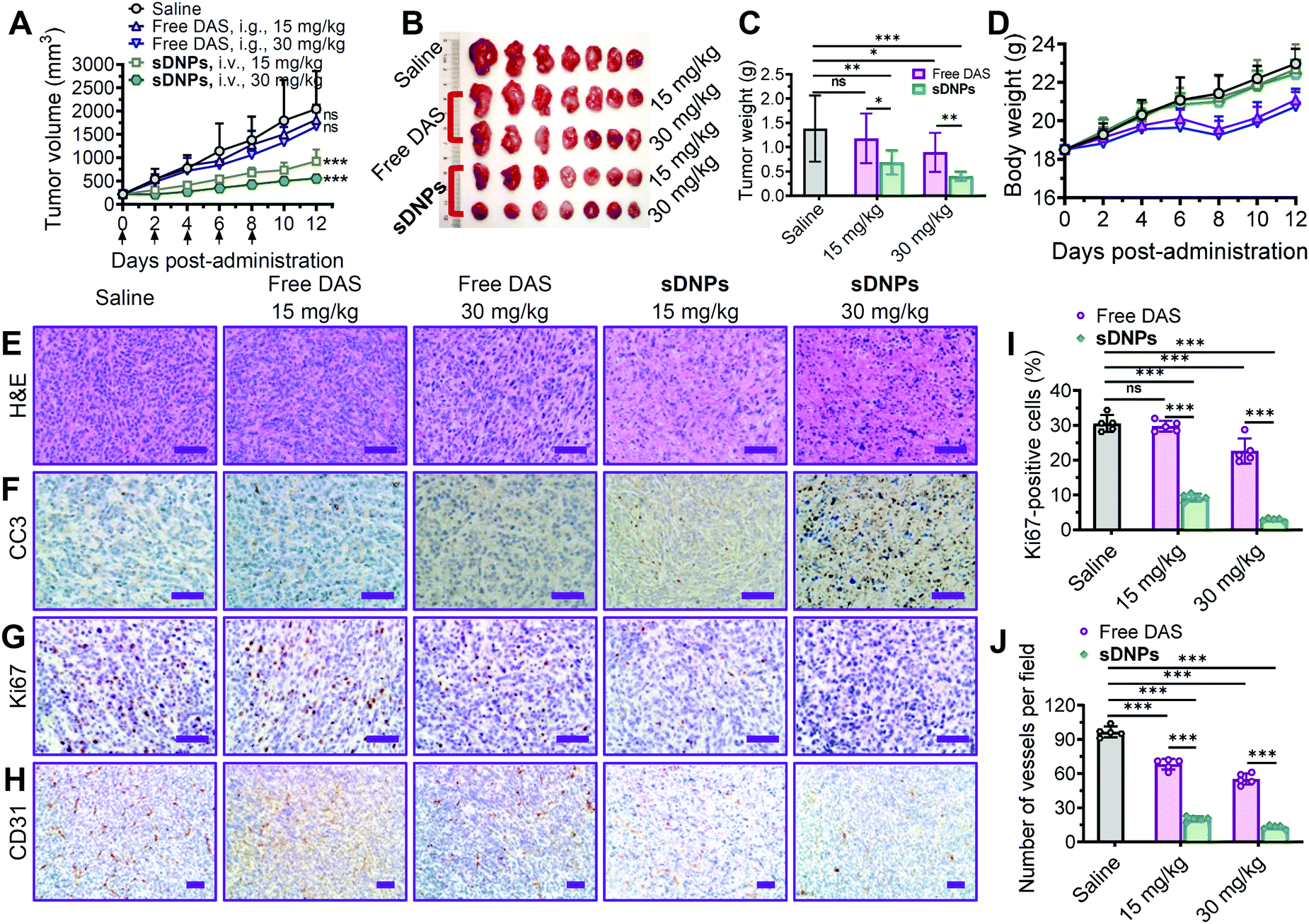 | ||
| Fig. 5 Parenteral treatment with sDNPs sufficiently inhibited the growth of primary tumors in 4T1 murine breast tumor-bearing Balb/c mice. (A) Tumor volume changes of the mice treated with different formulations (n = 7 per group). The tumors harvested from the mice at the end of the study were photographed (B) and weighed (C). (D) Body weight changes of the mice. (E–H) Representative images of H&E staining, cleaved caspase-3 (CC3) immunostaining, Ki67 immunohistochemistry, and CD31 labeling in 4T1 tumor sections. Scale bars: 50 μm. (I) The percentage of Ki67-positive cells was determined to evaluate cell proliferation. (J) Inhibition of angiogenesis in tumors was determined by immunostaining for the blood vessel marker CD31. Five random fields were evaluated from slices of primary tumor sections. | ||
Histological analyses were further conducted to assess antitumor efficacy. The superiority of sDNPs could be attributed to extensive intratumoral apoptosis, as evidenced by hematoxylin & eosin (H&E) staining (Fig. 5E) and immunohistochemical staining of cleaved caspase-3 (CC3) (Fig. 5F), along with strong blockage of proliferation, as indicated by Ki67 analysis (Fig. 5G and I). Furthermore, sDNP treatment remarkably reduced the density of vessels in tumors, as revealed by immunostaining for CD31, a biomarker of the microvasculature (Fig. 5H and J). Notably, the body weight of mice that received sDNP treatment remained stable, but weight loss was observed in the mice orally treated with DAS.
Of note, in an independent mouse model of human prostate DU145 cancer, intravenously injected sDNPs at 15 mg kg−1 outperformed orally administered free DAS (i.e., 15 or 30 mg kg−1) in terms of tumor growth suppression (Fig. S13, ESI†). The volume and weight of tumors excised at the end of the study as well as histological examination of the tumors supported the therapeutic efficacy of sDNPs (Fig. S14, ESI†). Furthermore, no loss of body weight was observed in mice treated with sDNPs, indicating that this treatment does not cause severe systemic toxicity.
Metastasis accounts for the majority of cancer-associated human mortality.56,57 Traditional chemotherapy is not capable of inhibiting metastasis but occasionally it promotes the dissemination of cancer cells to distant organs, leading to clinical failure. In vivo, orthotopic inoculation of murine 4T1 cells induces metastasis and rapid colonization of the lungs and liver. We thus tested the ability of sDNPs to reduce metastatic potential in this model. The mice were implanted with murine 4T1 cells, and after 1 week, they were administered with sDNPsvia the tail vein. To assess antimetastatic efficiency, the lungs and livers in each group were excised on day 28 after treatment. Many visible nodules were clearly observed in the lungs of the saline- or free DAS-treated mice (Fig. 6A). Strikingly, sDNPs greatly inhibited pulmonary and hepatic metastases at a low dose (15 mg kg−1). More impressively, the mice treated with a high dose of sDNPs presented the absence of signs of metastasis in both organs, supporting their potent inhibitory effect on cancer cell migration and invasion. Furthermore, histological analyses of lungs and livers using H&E staining revealed a consistent tendency (Fig. 6B, C and Fig. S15, ESI†). Of note, the metastatic area in the lungs was substantially reduced to 0.13% after treatment with sDNPs at a high dose (Fig. 6D). Moreover, the number of metastatic lesions in the livers of sDNP-treated mice was also remarkably inhibited compared to that of saline- or free drug-treated mice (Fig. 6E). Thus, these data support the desirable antimetastatic efficacy of the injectable sDNP platform.
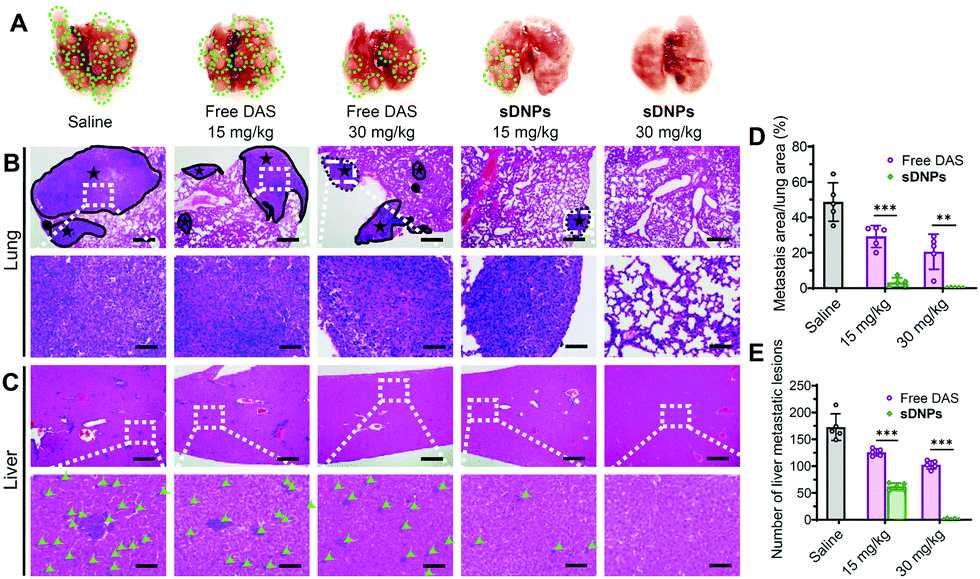 | ||
| Fig. 6 Inhibitory effects on lung and liver metastasis upon sDNP treatment of 4T1 breast tumor bearing mice. (A) Representative images of the lungs excised from each group. The green circles indicate visible metastatic nodules in the lung tissues. Histological analyses of metastatic lesions in (B) lungs (indicated by black solid circles) and (C) livers (indicated by green arrows) from each group. The lower images show an enlargement of the region in the white rectangle. Scale bars: 500 μm (upper row) and 100 μm (lower row). (D) Tumor area percentages in the lungs and (E) numbers of micrometastases in the livers quantified by H&E staining (n = 5 mice per group). The data are presented as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, nsp > 0.05. | ||
Alleviation of drug toxicity as a potential new generation of nanotherapeutics
The introduction of large quantities of carrier materials for the pharmaceutical delivery of potent therapeutics causes excipient-associated side effects, which impose an extra burden on patients. In addition, the undefined metabolism, clearance, and long-term toxicity of these materials greatly deter further clinical translation. To address these issues, minimizing the matrices used for drug delivery represents a promising solution. Hence, the self-assembling pure drug systems with 100% drug loading presented here could meet these requirements and increase translational capacity. Therefore, we investigated whether DAS delivered by nanoparticles can potentially reduce drug toxicity in vivo. When therapeutic nanoparticles are systemically injected, they encounter red blood cells (RBCs) and may potentially induce hemolysis. Thus, we evaluated the hemocompatibility of the sDNP scaffold using the hemolysis assay. RBCs were exposed to different concentrations of sDNPs, and the hemoglobin released from hemolyzed RBCs was measured. We detected only negligible hemolysis, with less than 1% hemolysis after treatment with sDNPs at concentrations as high as 0.6 mg mL−1 (i.e., equal to a 90 mg kg−1 treatment dose), demonstrating excellent hemocompatibility suitable for intravenous administration (Fig. S16, ESI†).The hydrophobicity of administered nanomaterials frequently causes immunotoxicity, which further deters the clinical translation of nanoparticle therapeutics.58–60 We therefore performed immunological studies by quantifying the levels of cytokine induced by sDNP administration. Synthetic aliphatic polyesters such as PEG5k–PLA8k, which are conventionally used in the delivery of hydrophobic drugs, were compared in this experimental setting. After administration of high doses of sDNPs (30 mg kg−1, q2dx5), serum samples from mice were subjected to a multiplex cytokine assay for measurement of a panel of inflammatory and proinflammatory cytokines/chemokines. As expected, blank PEG5k–PLA8k (not DAS fed) and DAS-loaded PEG5k–PLA8k nanoparticles triggered a substantial immune response by increasing the levels of cytokines/chemokines (e.g., the chemokines CCL11 and GM-CSF, the Th cytokines IFN-γ and IL-10 and the proinflammatory cytokines IL6 and TNF-α) on days 9 and 18 (Fig. S17, ESI†). This could be due to the hydrophobic PLA segments, which are expected to activate the immune response. Even on day 30 post-administration, we observed upregulated levels of IFN-γ, CCL-11, and GM-CSF, indicating long-term immunotoxicity. Very interestingly, when sDNPs were intravenously injected, the levels of these cytokines/chemokines remained within the normal range during the whole observation period. Overall, in contrast to traditional polymers, the noncovalent nanoassemblies prepared from therapeutic drugs degraded into small molecules only after performing the role of delivery reservoirs, which could be excreted from the body and benefit the alleviation of long-term immunotoxicity.
Conclusions
By exploiting the supramolecular self-assembly process, we have successfully transformed an insoluble molecular inhibitor into a more effective and less toxic therapeutic nanoparticle. The major advantages of the current sDNP platform include a uniform size distribution, colloid-stable nanoparticles, 100% drug loading, and simple nanoparticle preparation without either chemical modification or the use of carrier materials. In addition, upon forming nanoassemblies, the DAS inhibitor became fluorescently emissive. To the best of our knowledge, this represents the first example of a self-assembling natural molecular inhibitor that shows AIE characteristics, enabling cellular imaging and trafficking tracing of sDNPs without additional labeling with fluorescent dyes. The engineered sDNPs can inhibit local breast tumor growth and reduce the metastatic burden in mice more efficiently than the orally administered DAS inhibitor while mitigating the immunotoxicity that is normally elicited by nanocarriers. Although the administration of sDNPs was restricted to the parenteral route in this study, alternate methods for the delivery of these nanoassemblies, including oral gavage, deserve to be investigated. Finally, this approach is clinically feasible and can be applied to rescue other compounds that have limited efficacy due to pharmaceutical challenges.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Zhejiang Province Preeminence Youth Fund (LR19H160002) and the National Natural Science Foundation of China (82073296, 81773193, and 81571799).Notes and references
- S. Turajlic and C. Swanton, Nat. Rev. Drug Discovery, 2017, 16, 441–442 CrossRef CAS.
- G. K. Dy and A. A. Adjei, Ca-Cancer J. Clin., 2013, 63, 249–279 CrossRef.
- S. Klaeger, S. Heinzlmeir, M. Wilhelm, H. Polzer, B. Vick, P. A. Koenig, M. Reinecke, B. Ruprecht, S. Petzoldt, C. Meng, J. Zecha, K. Reiter, H. C. Qiao, D. Helm, H. Koch, M. Schoof, G. Canevari, E. Casale, S. R. Depaolini, A. Feuchtinger, Z. X. Wu, T. Schmidt, L. Rueckert, W. Becker, J. Huenges, A. K. Garz, B. O. Gohlke, D. P. Zolg, G. Kayser, T. Vooder, R. Preissner, H. Hahne, N. Tonisson, K. Kramer, K. Gotze, F. Bassermann, J. Schlegl, H. C. Ehrlich, S. Aiche, A. Walch, P. A. Greif, S. Schneider, E. R. Felder, J. Ruland, G. Medard, I. Jeremias, K. Spiekermann and B. Kuster, Science, 2017, 358, eaan4368 CrossRef.
- F. M. Ferguson and N. S. Gray, Nat. Rev. Drug Discovery, 2018, 17, 353–377 CrossRef CAS.
- N. Kong, W. Tao, X. Ling, J. Wang, Y. Xiao, S. Shi, X. Ji, A. Shajii, S. T. Gan, N. Y. Kim, D. G. Duda, T. Xie, O. C. Farokhzad and J. Shi, Sci. Transl. Med., 2019, 11, eaaw1565 CrossRef CAS.
- J. S. Logue and D. K. Morrison, Genes Dev., 2012, 26, 641–650 CrossRef CAS.
- R. Barouch-Bentov and K. Sauer, Expert Opin. Invest. Drugs, 2011, 20, 153–208 CrossRef CAS.
- M. Herbrink, B. Nuijen, J. H. Schellens and J. H. Beijnen, Cancer Treat. Rev., 2015, 41, 412–422 CrossRef CAS.
- A. A. Kulkarni, B. Roy, P. S. Rao, G. A. Wyant, A. Mahmoud, M. Ramachandran, P. Sengupta, A. Goldman, V. R. Kotamraju, S. Basu, R. A. Mashelkar, E. Ruoslahti, D. M. Dinulescu and S. Sengupta, Cancer Res., 2013, 73, 6987–6997 CrossRef CAS.
- A. S. Thakor and S. S. Gambhir, Ca-Cancer J. Clin., 2013, 63, 395–418 CrossRef.
- S. Quader and K. Kataoka, Mol. Ther., 2017, 25, 1501–1513 CrossRef CAS.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102–1110 CrossRef CAS.
- K. C. Nicolaou and S. Rigol, Angew. Chem., Int. Ed., 2019, 58, 11206–11241 CrossRef CAS.
- J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS.
- C. Q. Lan and S. L. Zhao, J. Mater. Chem. B, 2018, 6, 6685–6704 RSC.
- D. T. Bui, J. Nicolas, A. Maksimenko, D. Desmaele and P. Couvreur, Chem. Commun., 2014, 50, 5336–5338 RSC.
- L. Bildstein, C. Dubernet, V. Marsaud, H. Chacun, V. Nicolas, C. Gueutin, A. Sarasin, H. Benech, S. Lepetre-Mouelhi, D. Desmaele and P. Couvreur, J. Controlled Release, 2010, 147, 163–170 CrossRef CAS.
- H. Wang, Z. Lu, L. Wang, T. Guo, J. Wu, J. Wan, L. Zhou, H. Li, Z. Li, D. Jiang, P. Song, H. Xie, L. Zhou, X. Xu and S. Zheng, Cancer Res., 2017, 77, 6963–6974 CrossRef CAS.
- L. Wu, F. Zhang, X. Chen, J. Wan, Y. Wang, T. Li and H. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 3327–3340 CrossRef CAS.
- T. Fang, Z. Ye, J. Wu and H. Wang, Chem. Commun., 2018, 54, 9167–9170 RSC.
- H. X. Wang, H. Y. Xie, J. G. Wang, J. P. Wu, X. J. Ma, L. L. Li, X. Y. Wei, Q. Ling, P. H. Song, L. Zhou, X. Xu and S. S. Zheng, Adv. Funct. Mater., 2015, 25, 4956–4965 CrossRef CAS.
- B. B. Xie, J. Q. Wan, X. N. Chen, W. D. Han and H. X. Wang, Mol. Cancer Ther., 2020, 19, 822–834 CrossRef CAS.
- H. Xie, H. Zhu, K. Zhou, J. Wan, L. Zhang, Z. Yang, L. Zhou, X. Chen, X. Xu, S. Zheng and H. Wang, J. Controlled Release, 2020, 328, 237–250 CrossRef CAS.
- C. Y. Zhang, X. Y. Dong, J. Gao, W. J. Lin, Z. Liu and Z. J. Wang, Sci. Adv., 2019, 5, eaax7964 CrossRef CAS.
- L. L. Shi, Y. C. Wang, Q. C. Wang, Z. N. Jiang, L. L. Ren, Y. P. Yan, Z. X. Liu, J. Q. Wan, L. L. Huang, B. N. Cen, W. D. Han and H. X. Wang, J. Controlled Release, 2020, 324, 289–302 CrossRef CAS.
- S. Y. Qin, A. Q. Zhang, S. X. Cheng, L. Rong and X. Z. Zhang, Biomaterials, 2017, 112, 234–247 CrossRef CAS.
- Y. Li, Q. Zou, C. Yuan, S. Li, R. Xing and X. Yan, Angew. Chem., Int. Ed., 2018, 57, 17084–17088 CrossRef CAS.
- X. Q. Wei, L. Y. Hao, X. R. Shao, Q. Zhang, X. Q. Jia, Z. R. Zhang, Y. F. Lin and Q. Peng, ACS Appl. Mater. Interfaces, 2015, 7, 13367–13374 CrossRef CAS.
- Q. Peng and H. L. Mu, J. Controlled Release, 2016, 225, 121–132 CrossRef CAS.
- Q. Fan, L. Li, H. Xue, H. Zhou, L. Zhao, J. Liu, J. Mao, S. Wu, S. Zhang, C. Wu, X. Li, X. Zhou and J. Wang, Angew. Chem., Int. Ed., 2020, 132, 15253–15258 CrossRef.
- J. Li, J. X. Wang, H. X. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
- A. Nicol, W. Qin, R. T. K. Kwok, J. M. Burkhartsmeyer, Z. F. Zhu, H. F. Su, W. W. Luo, J. W. Y. Lam, J. Qian, K. S. Wong and B. Z. Tang, Chem. Sci., 2017, 8, 4634–4643 RSC.
- X. Liu, M. Li, T. Han, B. Cao, Z. Qiu, Y. Li, Q. Li, Y. Hu, Z. Liu, J. W. Y. Lam, X. Hu and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 11259–11268 CrossRef CAS.
- X. Cai and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 9868–9886 CrossRef CAS.
- J. Demasa and G. Crosby, J. Chem. Phys., 1968, 48, 4726 CrossRef.
- A. T. R. Williams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067–1071 RSC.
- J. Q. Shi, N. Chang, C. H. Li, J. Mei, C. M. Deng, X. L. Luo, Z. P. Liu, Z. S. Bo, Y. Q. Dong and B. Z. Tang, Chem. Commun., 2012, 48, 10675–10677 RSC.
- Z. Zhou, S. Xie, X. Chen, Y. Tu, J. Xiang, J. Wang, Z. He, Z. Zeng and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 9803–9807 CrossRef CAS.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
- H. Wang, E. G. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365–377 CrossRef CAS.
- T. Chen, S. Yang, J. Chai, Y. Song, J. Fan, B. Rao, H. Sheng, H. Yu and M. Zhu, Sci. Adv., 2017, 3, e1700956 CrossRef.
- L. Viglianti, N. L. C. Leung, N. Xie, X. G. Gu, H. H. Y. Sung, Q. Miao, I. D. Williams, E. Licandro and B. Z. Tang, Chem. Sci., 2017, 8, 2629–2639 RSC.
- Y. Gu, Z. Zhao, H. Su, P. Zhang, J. Liu, G. Niu, S. Li, Z. Wang, R. T. K. Kwok, X. L. Ni, J. Sun, A. Qin, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2018, 9, 6497–6502 RSC.
- D. Wang, M. M. S. Lee, W. Xu, G. Shan, X. Zheng, R. T. K. Kwok, J. W. Y. Lam, X. Hu and B. Z. Tang, Angew. Chem., Int. Ed., 2019, 58, 5628–5632 CrossRef CAS.
- M. Mohammadi, Z. Salmasi, M. Hashemi, F. Mosaffa, K. Abnous and M. Ramezani, Int. J. Pharm., 2015, 485, 50–60 CrossRef CAS.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS.
- Z. Li, C. Wang, H. Deng, J. Wu, H. Huang, R. Sun, H. Zhang, X. Xiong and M. Feng, Adv. Sci., 2019, 6, 1802057 CrossRef.
- N. P. Shah, C. Tran, F. Y. Lee, P. Chen, D. Norris and C. L. Sawyers, Science, 2004, 305, 399–401 CrossRef CAS.
- L. J. Lombardo, F. Y. Lee, P. Chen, D. Norris, J. C. Barrish, K. Behnia, S. Castaneda, L. A. Cornelius, J. Das, A. M. Doweyko, C. Fairchild, J. T. Hunt, I. Inigo, K. Johnston, A. Kamath, D. Kan, H. Klei, P. Marathe, S. Pang, R. Peterson, S. Pitt, G. L. Schieven, R. J. Schmidt, J. Tokarski, M. L. Wen, J. Wityak and R. M. Borzilleri, J. Med. Chem., 2004, 47, 6658–6661 CrossRef CAS.
- E. Weisberg, P. W. Manley, S. W. Cowan-Jacob, A. Hochhaus and J. D. Griffin, Nat. Rev. Cancer, 2007, 7, 345–356 CrossRef CAS.
- Y. R. Kim, Y. H. Kim, S. W. Kim, Y. J. Lee, D. E. Chae, K. A. Kim, Z. W. Lee, N. D. Kim, J. S. Choi, I. S. Choi and K. B. Lee, Chem. Commun., 2016, 52, 11764–11767 RSC.
- L. C. Kim, L. Song and E. B. Haura, Nat. Rev. Clin. Oncol., 2009, 6, 587–595 CrossRef CAS.
- A. V. Kamath, J. Wang, F. Y. Lee and P. H. Marathe, Cancer Chemother. Pharmacol., 2008, 61, 365–376 CrossRef CAS.
- R. D. Loberg, D. A. Bradley, S. A. Tomlins, A. M. Chinnaiyan and K. J. Pienta, Ca-Cancer J. Clin., 2007, 57, 225–241 CrossRef.
- C. Xie, X. Zhen, Q. Miao, Y. Lyu and K. Pu, Adv. Mater., 2018, 30, e1801331 CrossRef.
- M. A. Dobrovolskaia and S. E. McNeil, Nat. Nanotechnol., 2007, 2, 469–478 CrossRef CAS.
- G. Yu, X. Zhao, J. Zhou, Z. Mao, X. Huang, Z. Wang, B. Hua, Y. Liu, F. Zhang, Z. He, O. Jacobson, C. Gao, W. Wang, C. Yu, X. Zhu, F. Huang and X. Chen, J. Am. Chem. Soc., 2018, 140, 8005–8019 CrossRef CAS.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00469c |
| This journal is © The Royal Society of Chemistry 2021 |