
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel application of electrochemical bipolar exfoliated graphene for highly sensitive disposable label-free cancer biomarker aptasensors†
Shahrzad
Forouzanfar
a,
Iman
Khakpour
 b,
Fahmida
Alam
a,
Nezih
Pala
a and
Chunlei
Wang
*bc
b,
Fahmida
Alam
a,
Nezih
Pala
a and
Chunlei
Wang
*bc
aDepartment of Electrical and Computer Engineering, Florida International University, USA
bDepartment of Mechanical and Materials Engineering, Florida International University, USA
cCenter for Study of Matter at Extreme Conditions, Florida International University, USA
First published on 12th August 2021
Abstract
Label-free aptasensors can be a promising point-of-care biosensor for detecting various cancer diseases due to their selectivity, sensitivity, and lower cost of production and operation. In this study, a highly sensitive aptasensor based on gold-covered polyethylene terephthalate electrodes (PET/Au) decorated with bipolar exfoliated graphene is proposed as a possible contender for disposable label-free aptasensor applications. Bipolar electrochemical exfoliation enables simultaneous exfoliation, reduction, and deposition of graphene nanosheets on prospective electrodes. Our comparative study confirms that the bipolar exfoliated graphene deposited on the negative feeding electrode (i.e., reduced graphene oxide) possesses better electrochemical properties for aptasensing. The optimized aptasensor based on bipolar exfoliated graphene deposited on PET/Au electrodes exhibits a highly sensitive response of 4.07 μA log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c−1 (unit of c, pM) which is linear in the range of 0.0007–20 nM, and has a low limit of detection of 0.65 pM (S/N = 3). The aptasensor establishes highly selective performance with a stability of 91.2% after 6 days. This study demonstrates that bipolar electrochemistry is a simple yet efficient technique that could provide high-quality graphene for biosensing applications. Considering its simplicity and efficiency, the BPE technique promises the development of feasible and affordable lab-on-chip and point-of-care cancer diagnosis technologies.
c−1 (unit of c, pM) which is linear in the range of 0.0007–20 nM, and has a low limit of detection of 0.65 pM (S/N = 3). The aptasensor establishes highly selective performance with a stability of 91.2% after 6 days. This study demonstrates that bipolar electrochemistry is a simple yet efficient technique that could provide high-quality graphene for biosensing applications. Considering its simplicity and efficiency, the BPE technique promises the development of feasible and affordable lab-on-chip and point-of-care cancer diagnosis technologies.
Introduction
Cancer is one of the significant causes of death, killing millions of people every year.1 Most cancers have a high fatality rate because cancer is diagnosed when it has significantly progressed and invaded many vital organs.2 The main reason for such late diagnosis is that patients with cancer diseases have no physical symptoms at the early stages of the disease.3 Many cancer biomarkers associated with various cancers have been discovered.2 Among the discovered cancer biomarkers, platelet-derived growth factor-BB (PDGF-BB) is known to have a potent role in the growth and metastasis of several solid malignant tumors, and its levels in the blood increase under the influence of cancer.4 Numerous studies have reported the involvement of PDGF-BB in the development and lymphatic metastasis of various cancers, including breast,5 pancreatic,6 prostate,7 ovarian,8 and liver.9 For example, a concentration above 1.72 nM (the equivalent of 42 ng mL−1) is confirmed for patients with lung cancer,10 although there is no Food and Drug Administration (FDA) official report for the healthy and cancerous levels of PDGF-BB in human blood. The significant role of PDGF-BB in the development and metastasis of different cancer diseases highlights the importance of developing point-of-care (POC) biosensors to detect the PDGF-BB biomarker. PDGF-BB POC biosensors can be of immense help for the early diagnosis of many cancers associated with solid tumors such as breast, brain, prostate, ovarian, and lung cancers.The price of sensing units is one of the major concerns in developing feasible POC biosensors. The envisioned sensing unit should be accurate and yet inexpensive enough to rationalize its installment for on-site applications. Combining label-free detection with economically efficient manufacturing techniques can be a viable option for developing versatile sensing systems to detect and monitor different bio-targets, including PDGF-BB. Thus far, noticeable attention has been devoted to developing cheap and disposable electrochemical biosensors. Various substrates such as paper, polymers (e.g., polyethylene terephthalate), and screen-printed carbon electrodes have been used for developing disposable biosensors.11–15 For instance, Wang et al. demonstrated disposable paper-based label-free cancer biomarker aptasensors in which paper-based electrodes were functionalized with amino-treated graphene, gold nanoparticles, and Prussian blue. The proposed disposable aptasensors showed good sensing performances with linear ranges of 0.01–500 ng mL−1 for carcinoembryonic antigens and a limit of detection (LoD) of 2 pg mL−1.11 Yaman et al. proposed label-free cancer cell aptasensors based on pencil graphite electrodes. The developed aptasensors were functionalized with diphenyl alaninamide-based peptide nanoparticles and DLD-1 cancer cell affinity aptamers. The disposable pencil graphite aptasensor exhibited a good linear range ranging from 2 × 102 to 2.0 × 105 cells per mL with a LoD of 100 cells per mL.16 The proposed disposable label-free cancer aptasensors illustrate the high potential of this class of aptasensors as cancer biomarker disposable POC aptasensors.
Furthermore, the emergence of the COVID-19 pandemic in early 2020 highlighted the importance of fast, reliable, and affordable POC biosensors suitable for the detection of various biotargets (e.g., SARS-CoV-2). The integration of artificial intelligence (AI) and internet of things (IoT) with POC pathogen biosensors has shown that POC biosensors can be effectively used for performing personalized diagnostics under desired conditions and at desired locations, helping the management of the epidemic in an economic and timely manner.17–19 Hence, the authors believe that integration of AI and IoT with cancer biomarker POC biosensors could be an effective strategy to make a significant impact on the diagnosis and management of cancer.
Since the discovery of graphene in the early 2000s, it has attracted massive attention in various domains because of its unique features.20 The large surface area, high thermal conductivity and charge mobility, the wide electrochemical window, and unconventional superconductivity of graphene made this material a potent candidate for the development of biosensors.21,22 The conventional application of graphene for biosensors involves synthesis of graphene with wet chemical processes (e.g., Hummers) or electrochemical approaches, reduction of exfoliated graphene oxide (GO) to reduced graphene oxide (rGO), and its deposition on the desired biosensing electrode.23 Wet chemical synthesis requires harsh oxidizing agents such as KMnO4, NaNO3, and KClO3 in strong acidic agents such as hydrohalic acid and hydrazine.24–27 The application of harsh chemicals can be limited by using electrochemical approaches; however, there could be non-desired functional groups and basal plane damage during anodic exfoliation.28–30 Furthermore, a comparative study by Kauling et al. demonstrated that commercially available graphene (from 60 different vendors worldwide) has a low graphene content (less than 10% in most products and not more than 50% in the highest quality products available ), which illustrates the reason why commercial graphene-based devices are not commonly available yet.31 Nonetheless, conventional methods for synthesis and deposition of GO and rGO cannot simultaneously exfoliate, reduce, and deposit GO and rGO in an environmentally friendly fashion. Such a combination can be achieved via bipolar electrochemistry.32 In addition, Khakpour et al. demonstrated that the bipolar exfoliated rGO has a vertically aligned morphology, which could eliminate the necessity of using spacers such as carbon nanotubes to prevent the aggregation of graphene nano-flakes.33–35
Herein, the novel application of bipolar exfoliation of graphene for a disposable label-free PDGF-BB aptasensor is presented. There are no reports on applying bipolar exfoliated (BPE) graphene for biosensor applications to the best of our knowledge. The bipolar exfoliation method presented in this study is a direct single-step of exfoliation, deposition, and reduction of graphene utilizing a graphite rod in deionized water (DIW). The process of bipolar exfoliation includes applying a constant electric field between two conductive feeder electrodes, which aggregates in opposite polarizations of the graphite rods. Consequently, if the apparent electrical field is strong enough, simultaneous faradaic reactions of reduction at the cathodic pole and oxidation at the anodic pole would occur.32 Thus far, BPE has been demonstrated for various applications due to the simplicity of the setup and lower cost of the process.33,36–38
In this study, BPE-graphene was deposited on stainless steel (SS) electrodes and gold-covered polyethylene terephthalate electrodes (PET/Au) for a comparative study and as the proposed disposable biosensor, respectively. The analysis confirmed that the BPE-rGO deposited on the cathodic (i.e., negative) feeding electrode is highly suitable for aptasensing applications. Thus, PET/Au was used as the negative feeding electrode. The amino-terminated PDGF-BB affinity aptamers were covalently immobilized on BPE-graphene deposited on SS and PET/Au electrodes. The BPE-graphene crystal structure and its morphology were studied via transmission electron microscopy (TEM) and scanning electron microscopy (SEM), respectively. The characteristics of the fabricated BPE-based aptasensors were studied using Fourier-transform infrared spectroscopy (FTIR). Electrochemical and sensing performance analyses were conducted using differential pulse voltammetry (DPV) and cyclic voltammetry (CV). The results revealed that the bipolar exfoliation provides high-quality rGO with promising characteristics suitable for aptasensors. The low cost and high efficiency of the process developed in this study are highly appealing features for POC biosensors. Several reasons—including the use of easily accessible and low-priced materials (i.e., graphite rods and DIW), the merging of several steps from other commonly practiced graphene biosensing application methods (i.e., exfoliation, reduction, and deposition) into one single step, and the elimination of the need for spacers such as CNTs due to the morphology of synthesized graphene (i.e., vertically aligned nanosheets)—make this process novel, economical, and highly efficient. Furthermore, the BPE technique was proven to be both versatile and amenable to mass production, thus making it a promising technique for various biotechnology applications, including but not limited to disposable cancer aptasensors and POC microdevices.
Results and discussion
In this study, a bipolar exfoliation cell illustrated in Fig. 1a has been used for exfoliation, reduction, and deposition of graphene nanosheets on SS and PET/Au electrodes. The bipolar mechanism details have been discussed in previous studies.32,33,39 Briefly, a 45 V direct current (DC) voltage was applied through the 2 × 2 cm feeding electrodes with a 9 cm distance to induce an electric field of 5 V cm−1. The resistivity of DIW used for bipolar exfoliation was 17 MΩ. During bipolar exfoliation, two half-reactions of water electrolysis happen at the feeding electrodes: (1) reduction of two water molecules to one gaseous hydrogen molecule (2H+ + 2e− → H2) and hydroxyl ions (OH−) at the cathode and (2) oxidation of hydroxyl ions (OH−) to oxygen gas and water molecules (4OH− → 2H2O + O2 + 4e−) at the anode. Furthermore, assuming that the potential drop is negligible at the electrodes and linear in solution, the electric field induces a sufficient potential difference (>1.5 V) between the two ends of the graphite rod to stimulate faradaic reactions at the side surfaces of the graphite rod. This potential difference helps in the oxidation of graphite into GO at the anodic pole of the graphite rod (indicated as δ+ in Fig. 1a). | ||
| Fig. 1 Schematic illustration of a (a) bipolar exfoliation cell, (b) development of PET/Au/rGOApt PDGF-BB aptasensors, (c) target incubation, and a (d) three-electrode electrochemical cell with a Ag/AgCl reference electrode, Pt wire counter electrode, and PET/Au/rGOApt in 5 mL aqueous electrolytes of 0.1 M PBS/5 mM K3Fe(CN)6/0.05–1 M KCl. | ||
Prior to immobilizing aptamers, each electrode was examined with SEM to confirm the uniform deposition of rGO or GO on the electrodes. Next, amino-modified PDGF-BB affinity aptamers were immobilized on SS and PET/Au electrodes modified with BPE-graphene (Fig. 1b). The aptasensors were incubated with the desired PDGF-BB concentration at various temperatures ranging from 20 °C to 50 °C for between 10 and 150 minutes (Fig. 1c). The optimum incubation temperature was found to be 30 °C, and the optimum reaction time for target molecules to bind with aptamers was found to be 40 minutes. A three-electrode setup illustrated in Fig. 1d was used for CV and DPV evaluations of the electrodes. 5 mL aqueous electrolytes of 0.1 M PBS, 5 mM – K3Fe(CN)6, and different KCl concentrations in the range of 50 mM to 1 M were utilized for electrochemical analysis.
Electrochemical and FTIR characterization studies were conducted on four different BPE-treated SS electrodes referred to as SS/GO, SS/rGO, SS/GOApt, and SS/rGOApt, which represent the samples of GO deposited on the SS electrode (i.e., positive feeding electrode), rGO on deposited the SS electrode (i.e., negative feeding electrode), an aptamer immobilized on the SS/GO electrode, and an aptamer immobilized on SS/rGO electrode, respectively. An aptamer concentration of 15 μM was used to fabricate SS/GOApt, SS/rGOApt, and PET/Au/rGOApt electrodes studied in the following subsections except for PET/Au/rGOApt electrodes prepared for defining the optimum concentration of the aptamer. An optimum aptamer concentration of 10 μM was utilized to fabricate PET/Au/rGOApt electrodes studied in subsections 2.4. All BPE-treated electrodes had an effective area of 0.5 cm2 confined via bee's wax to minimize side reactions.
Materials characterization of BPE treated electrodes
The SEM image of BPE-rGO deposited on SS electrodes after 24 hours of deposition is presented in Fig. 2a. The SEM image of rGO deposited on negative feeding electrodes clearly shows a porous morphology with vertically aligned structures with a pore size of approximately 100 nm. This porous morphology is highly suitable for various applications, including electrochemical biosensors and energy storage microdevices. On the other hand, the SEM image of GO deposited on positive feeding electrodes (Fig. S1†) shows bulky plane structures with cracks implying restacking of GO during the deposition stage. | ||
Fig. 2 (a) SEM images of BPE-rGO deposited on the negative feeding electrode. (b) TEM image (SAED patterns in the inset, yellow circles are associated with 〈1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 00〉 planes and green circles are associated with 〈210〉 planes). (c) HRTEM image of BPE-rGO. 00〉 planes and green circles are associated with 〈210〉 planes). (c) HRTEM image of BPE-rGO. | ||
The TEM and selected area electron diffraction (SAED) images of BPE-rGO deposited on the TEM mesh are given in Fig. 2b. The TEM image shows that thin sheets of rGO (with overlaps or folds) with a span of approximately 400 nm are formed. Single crystalline SAED patterns confirm that less defective graphene sheets are formed during the bipolar exfoliation. Only a few diffraction spots beside the spots related to the crystal are formed, and no superlattice arrays are identified in the deposited graphene. Therefore, it can be concluded that the BPE-rGO is reduced and has high purity.40,41
Moreover, it has been reported that when the intensity of spots diffracted from the 〈210〉 planes is higher than the intensity of spots diffracted from the 〈100〉 planes, there is more than one layer stacked in the graphene material. It can be seen in the SAED pattern of the TEM image given in Fig. 2b that spots diffracted from the 〈210〉 planes (marked with green circles) have lower intensity than spots diffracted from the 〈100〉 planes (marked with yellow circles). The difference in the intensity of spots diffracted from the 〈210〉 planes and 〈100〉 planes suggests that the graphene deposited on the negative feeding electrode has a few layers.40,41 An interplanar spacing (also known as the d-spacing) of 0.2 nm is measured from the high-resolution TEM (HRTEM) image presented in Fig. 2c, in which the measured interplanar spacing is smaller than the typical value of GO.40,41
FTIR characterization was used to analyse the spectral data of the BPE-rGO and BPE-GO samples deposited on SS electrodes, including the formation of amide binding after aptamer immobilization. The FTIR spectra of the background, SS/GO, SS/rGO, SS/GOApt, and SS/rGOApt are represented in Fig. 3. The three samples of SS/GO, SS/GOApt, and SS/rGOApt showed a broad peak between 2800 and 3200 cm−1, which is ascribed to O–H stretching.42 The noteworthy peaks for all samples were identified at 1430 and 1600 cm−1, ascribed to C–O bending and aromatic C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching, respectively.43 Furthermore, all the samples showed peaks at 1100–1300 cm−1, which are attributed to sp3 C–H bending.42
C stretching, respectively.43 Furthermore, all the samples showed peaks at 1100–1300 cm−1, which are attributed to sp3 C–H bending.42
 | ||
| Fig. 3 FTIR spectra of SS/GO, SS/rGO, SS/GOApt, and SS/rGOApt electrodes. | ||
The FTIR spectrum of SS/GOApt and SS/rGOApt confirms a peak at 1571 cm−1, associated with the amide II band, representing covalent bonding of PDGF-BB aptamers with BPE-graphene.42 The FTIR results confirm that the locally formed carboxyl groups on the graphene surfaces can be used directly to immobilize the amino-terminated biorecognition agents. The immobilization efficiency can also be improved using oxygen-plasma etching to increase the percentages of carboxyl groups on the surface of the BPE-graphene.1,44
Electrochemical analysis of BPE-graphene samples
Herein, the electrochemical analysis of BPE-graphene based aptasensors at different stages of development is presented. The analysis intended to compare the different electrochemical properties of BPE-graphene deposited on negative and positive SS electrodes. Two hypotheses for comparative analysis of the BPE-based aptasensors were considered; first, the electrodes based on BPE-rGO exhibit better electrochemical performance than BPE-GO; second, trapped target molecules (i.e., the formation of the target–aptamer complex) elevates the charge-transfer resistance by blocking the passing of redox species to the aptasensor electrode, which can be traced as decreasing current measured via the DPV method. A sample of 100 pM PDGF-BB (designated as T) was used as a target sample for electrochemical analysis purposes.In Fig. 4a, the CV curves measured for bare SS electrodes, four samples BPE treated electrodes, and subsequent aptasensors are presented. The CV curve of BPE-graphene deposited on the negative feeding electrode (i.e., SS/rGO) shows the highest peak currents and the largest CV curve area. The areal capacitances were calculated from CV curves using eqn (1):45
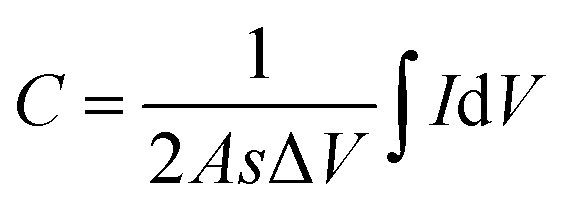 | (1) |
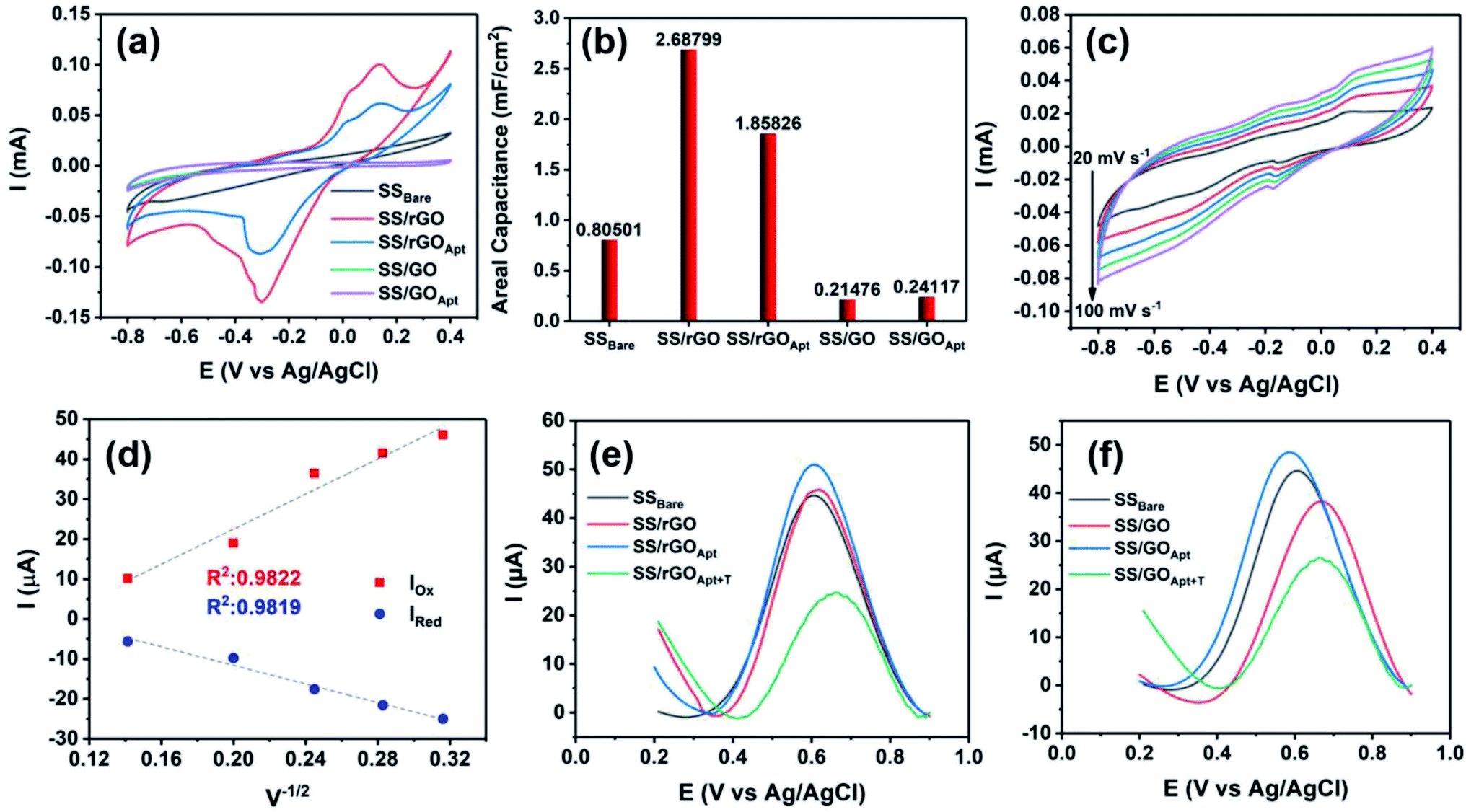 | ||
| Fig. 4 (a) CV (40 mV s−1) plots of bare SS and BPE-graphene deposited on SS electrodes before and after aptamer immobilization. (b) The areal capacitance calculated from CV plots. (c) The CV plots of the SS/rGO electrode at different scan rates in the range of 10–100 mV s−1. (d) Calibration curves of reduction and oxidation peak currents versus the square root of scan rates. (e) The DPV plots of BPE-based aptasensors based on graphene deposited on the negative feeding electrode at various development stages and the sensing responses of the aptasensors to 100 pM PDGF-BB. (f) The DPV plots of BPE-based aptasensors based on graphene deposited on positive feeding electrodes at various development stages and the responses of the aptasensors to 100 pM PDGF-BB. | ||
In order to estimate the electrochemically active areas of BPE-rGO and BPE-GO using the Randles–Sevcik equation, the correlation of CV peak currents and the square root of the scan rate were studied to confirm the occurrence of a diffused-controlled process. The peak currents of CV measurements conducted on SS/rGO and SS/GO proportionally increased with the increase of scan rates at scan rates of 10–100 mV s−1 (CV curves are given in Fig. 4c and S2,† respectively). The CV peak currents of SS/rGO linearly increased with respect to the increase of the square root of the scan rates (Fig. 4d) with correlation coefficients (R2) of 0.9819 and 0.9822 for reduction peak currents and oxidation peak currents, respectively. Similarly, the linear dependence of the reduction and oxidation peak currents of SS/GO had R2 values of 0.9723 and 0.9891 for reduction and oxidation peak currents, respectively (the inset calibration plots in Fig. S2†). With the confirmation of the occurrence of a diffused-controlled process, the active surface areas of the BPE-rGO and BPE-GO samples were calculated using the Randles–Sevcik equation at room temperature (T = 295 K) (eqn (2)).46
| IP = (2.69 × 105)n3/2ACD1/2ν1/2 | (2) |
The DPV responses of BPE-rGO and BPE-GO aptasensors at different development stages are given in Fig. 4e and f, respectively. The DPV response of SS/rGO shows a similar peak current to SSBare (i.e., SS electrode without any modifications) with values of 45.8 μA and 44.6 μA for SS/rGO and SSBare, respectively. These responses suggest a good electrochemical conductivity of deposited rGO on the SS electrode. However, the DPV response of SS/GO shows a decrease in peak current with a value of 37.9 μA, because of the structure of GO and the functional groups covering the surface of SS/GO. The DPV responses of SS/rGOApt and SS/GOApt show increased peak currents with values of 51 μA and 48.4 μA, respectively. The increase in peak currents after aptamer immobilization can be explained by the engagement of negatively charged carboxylate groups in covalent immobilization of aptamers and extinguished remaining functional groups by a Tween-20 quencher.47 The SS/rGOApt and SS/GOApt electrodes incubated with target molecules (i.e., SS/rGOApt+T and SS/GOApt+T) have considerably lower peak currents with values of 24 μA for SS/rGOApt+T and 26.5 μA for SS/GOApt+T, respectively. The decrease in peak currents is due to the isolative properties of the PDGF-BB oncoproteins, which decreases the passing of the redox species to the SS/rGOApt and SS/GOApt electrodes.48 Therefore, the DPV characterization of BPE-graphene-covered electrodes confirmed the successful entrapment of PDGF-BB oncoproteins on the surfaces of the envisioned aptasensors and that seizing of the target proteins decreases the peak current.
Parameter optimization and comparison of aptasensors based on rGO and GO
Several factors are known to affect the performance of electrochemical aptasensors.2 In this study, the effects of five parameters on the responses of BPE-graphene based aptasensors, including the reaction-time, the temperature of incubation, the pH of the electrolyte, the concentration of the supporting salt (i.e., KCl), and the binding aptamer concentration, have been analysed. Among the analysed parameters, the effect of aptamer concentration was studied explicitly for PET/Au-based aptasensors since the other four parameters are predominantly dependent on the aptamer DNA structures, incubation condition, and measurement medium.The optimum reaction time in aptasensors is referred to as the minimum time required for forming the aptamer–target complex that is both efficient and sufficiently quick. To find the optimum reaction time, the SS/rGOApt samples were incubated with 100 pM PDGF-BB for different periods ranging from 10–120 minutes and analysed by measuring the peak current of the DPV plot. As shown in Fig. 5a, the peak current decreases by increasing the incubation time up to 40 minutes and saturates afterward. Therefore, the optimum reaction time was found to be 40 minutes.
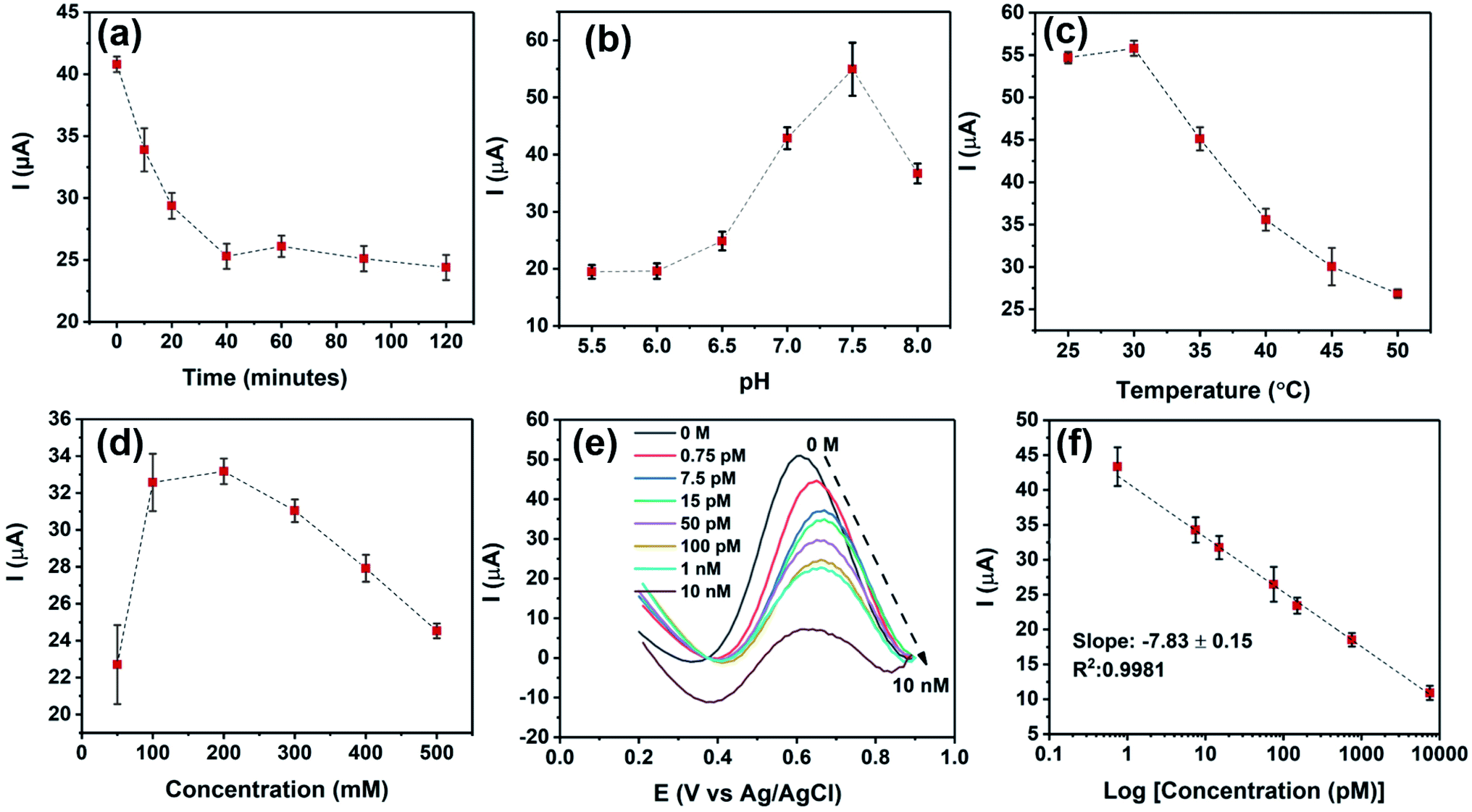 | ||
| Fig. 5 (a) Peak currents obtained from DPV curves measured in response to 100 pM PDGF-BB for defining the optimum reaction times. (b) Peak currents obtained from DPV curves in response to 100 pM PDGF-BB for analyzing the effect of pH on the peak current. (c) Peak currents obtained from DPV curves measured in response to 100 pM PDGF-BB for studying the effect of incubation temperature. (d) Peak currents obtained from DPV curves measured in response to 100 pM PDGF-BB for studying the effect of KCl concentration on the peak current. (e) DPV response of the SS/rGOApt electrode to PDGF-BB ranging from 0–10 nM. (f) Obtained calibration curve of peak currents measured from DPV responses. | ||
The effect of pH on DPV measurements was investigated by conducting DPV measurements on SS/rGOApt incubated with 100 pM PDGF-BB in the electrolyte of 0.2 M KCl/5 mM K3Fe(CN)6/0.1 M PBS with different pH values in the 5.5–8 range. As given in Fig. 5b, the peak response value was recorded at pH 7.5; thus, the sensing measurements were conducted in the electrolyte with pH 7.5. The incubation temperature effect on the peak current was studied by incubating an SS/rGOApt electrode with 100 pM PDGF-BB at different temperatures ranging from 25 °C to 50 °C. The attained results presented in Fig. 5c show that the highest peak current was achieved with incubation at 30 °C; hence, 30 °C was used as the optimum incubation temperature. The result of optimum supporting salt concentration analysis is presented in Fig. 5d. The highest peak current was recorded in the electrolyte with 200 mM KCl; therefore, 200 mM was chosen as the optimum KCl concentration.
The responses of SS/rGOApt to 0–10 nM PDGF-BB measured under optimum conditions are given in Fig. 5e. The peak current of DPV curves at E ≈ 0.6 V (vs. Ag/AgCl) decreases proportionally to the PDGF-BB concentration increase. The observed trend in peak current results from the increased charged transfer resistance due to the isolative properties of PDGF-BB oncoproteins.48 The calibration curve of peak currents measured from DPV curves is presented in Fig. 5f. The linear dependence of peak currents to the logarithm of PDGF-BB concentrations has a slop of −7.84 μA logc−1 and a R2 of 0.9981.
The correlation of the logarithm of the PDGF-BB concentration and SS/rGOApt peak current can be evaluated as follows (eqn (3)):
| i = 41.05 − 7.84logc−1, r = 0.9977 | (3) |
c−1. The LoD for SS/rGOApt aptasensors based on DPV measurements was estimated to be 0.53 pM based on the linear regression (eqn (4)),49 | (4) |
The responses of SS/GOApt to 0–10 nM PDGF-BB measured under optimum conditions reached their saturation point at a PDGF-BB concentration of 150 pM (see Fig. S3†). The most probable reason for the narrow range of SS/GOApt is the low amount of immobilized aptamers due to the limited active area. The sensing analysis of SS/rGOApt and SS/GOApt demonstrated that the BPE-graphene deposited on negative feeding electrodes, because of its highly porous morphology and higher quality, provides a more suitable material for biosensing application purposes. Hence, the PET/Au based aptasensors were developed using BPE-rGO.
Analysis of the PDGF-BB aptasensor based on BPE treated PET/Au electrodes
The comparative study on BPE-rGO and BPE-GO confirmed that the BPE-rGO provides a superior electrode material for envisioned label-free aptasensors. Therefore, the PET/Au electrodes were used as a negative feeding electrode in bipolar exfoliation cells. SEM analysis, confirmed the successful deposition of BPE-graphene on PET/Au electrodes (Fig. 6a). Herein, performance analysis of the aptasensors based on BPE-treaded PET/Au electrodes is given.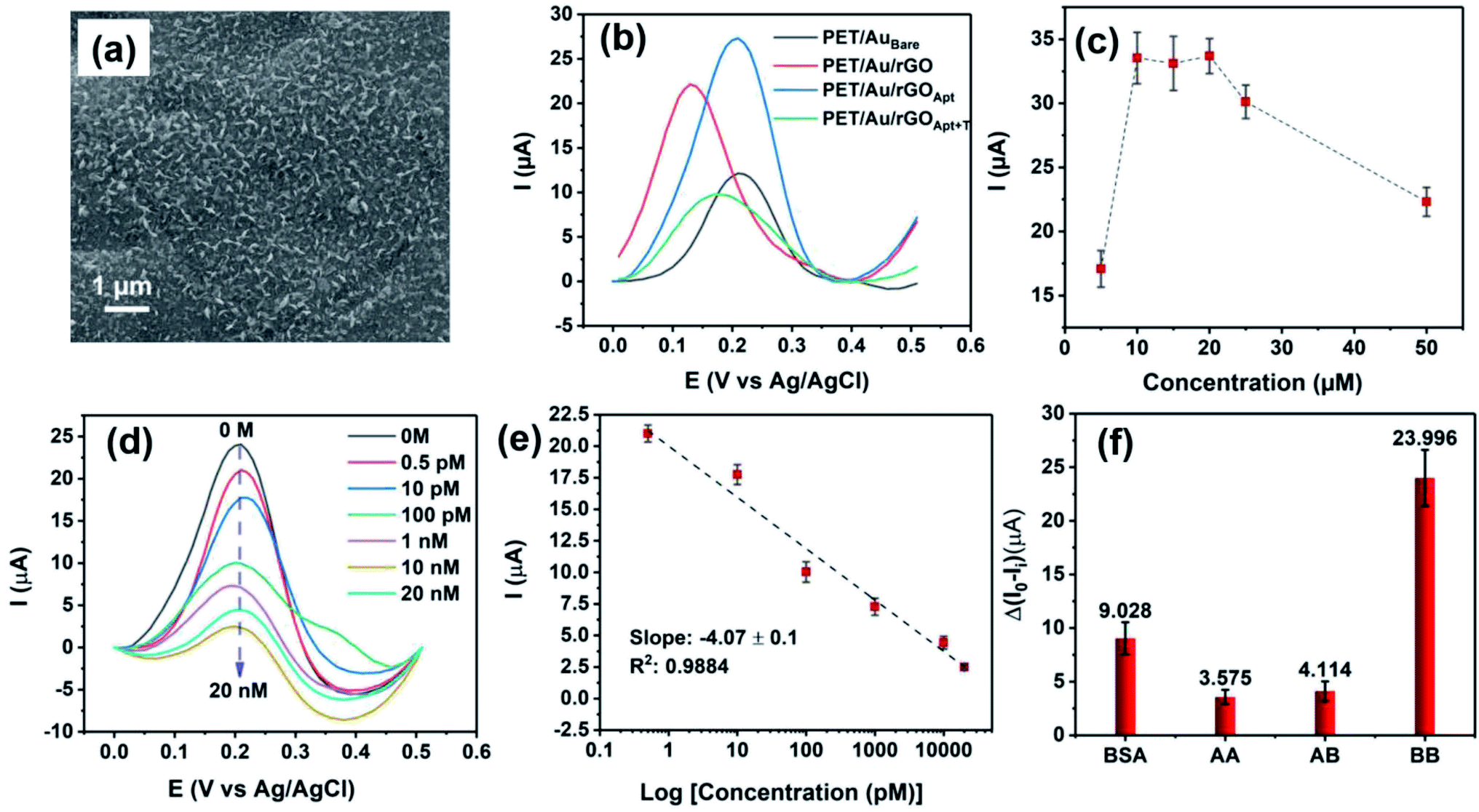 | ||
| Fig. 6 (a) SEM image of BPE-graphene deposited on the PET/Au negative feeding electrode. (b) The DPV plots of PET/Au-based aptasensors based on graphene deposited on the negative feeding electrode at various development stages and the response of the aptasensor to 100 pM PDGF-BB. (c) Peak currents obtained from DPV curves measured in response to 100 pM PDGF-BB for studying the effect of aptamer concentration. (d) DPV curves of the PET/Au/rGOApt electrode response to PDGF-BB ranging from 0–20 nM. (e) Calibration curve for peak currents measured from DPV curves. (f) DPV peak current measured in response of PET/Au/rGOApt electrodes to 10 nM PDGF-AA, 1 μg mL−1 bovine serum albumin (BSA), 10 nM PDGF-AB, and 100 pM PDGF-BB. | ||
In Fig. 6b, the DPV plots of aptasensors based on BPE-treaded PET/Au electrodes at different development stages are presented. The DPV results show a similar performance pattern to the DPV analysis of SS based electrodes, in which the deposition of BPE-rGO increased the DPV peak current of BPE-treaded PET/Au electrodes (designated as PET/Au/rGO) with a value of 22.2 μA compared to bare PET/Au electrodes (designated as PET/AuBare) with a peak current value of 12.04 μA. The DPV plot of PET/Au/rGO electrodes after aptamer immobilization (designated as PET/Au/rGOApt) shows an increase in peak current with a value of 27.45 μA, as discussed in Section 2.2.
The optimum aptamer concentration for PET/Au/rGO-based aptasensors was analyzed by measuring the DVP response of PET/Au/rGO based aptasensors prepared with a specific aptamer concentration to 100 pM PDGF-BB. A separate PET/Au/rGOApt electrode was prepared using an aptamer concentration ranging from 0.5–50 μM. As shown in Fig. 6c, the PET/Au/rGOApt aptasensor showed its peak DPV response in a sample prepared with an aptamer concentration of 10 μM, and the peak current saturated afterward. Hence, the 10 μM aptamer concentration was utilized for sensing performance analysis.
The responses of PET/Au/rGOApt to 0–20 nM PDGF-BB measured under optimum conditions are given in Fig. 6d. Similar to the sensing performances of aptasensors based on SS electrodes, the peak current of DPV curves decreases proportionally to the increase of PDGF-BB concentration. The calibration curve of peak currents measured from DPV curves is presented in Fig. 6e. The linear dependence of peak currents on the logarithm of PDGF-BB concentrations has a slop of −4.07 μA logc−1 and a R2 of 0.9884. The correlation of the PET/Au/rGOApt peak current with the logarithm of the PDGF-BB concentration can be assessed as follows (eqn (5)):
| i = 20.06 − 4.07logc−1, r = 0.9854 | (5) |
c−1. The LoD for PET/Au/rGOApt aptasensors based on DPV measurements was calculated as 0.65 pM based on the linear regression (eqn (4)) in which S was calculated as 0.174 μA (n = 10), and b was calculated as 0.81 μA c−1.
The selectivity of the PET/Au/rGOApt electrodes was investigated via recording the response of the aptasensors to 1 μg mL−1 bovine serum albumin, and 10 nM of PDGF-BB oncoprotein isomers (i.e., PDGF-AB and PDGF-AA) along with 100 pM PDGF-BB. The concentrations of interference agents were chosen to be approximately 100 times higher than the concentrations of the target molecules. The DPV peak currents measured for selectivity are presented in Fig. 6f, in which the peak current was measured from the DPV curves and differentiated from the aptasensor's blank response. The DPV peak current of the PET/Au/rGOApt aptasensor to PDGF-BB was 6.71 and 5.83 times higher than that of PDGF-AA and PDGF-AB aptasensors, respectively. The higher response of the PET/Au/rGOApt aptasensor to BSA than PDGF-AA and PDGF-AB is attributed to the bigger size of this protein and higher physical adsorption of BSA on the surface of the PET/Au/rGOApt aptasensor.
The storage stability of the PET/Au/rGOApt aptasensor was analyzed by conducting continuous electrochemical measurements on PET/Au/rGOApt in response to 100 pM PDGF-BB every day. The DPV peak current of 25.4 μA of PET/Au/rGOApt to 100 pM PDGF-BB after 6 days was 91.02% of the initial capacitive response (23.12 μA), which implies that the envisioned aptasensor has adequate storage stability.
Table 1 summarizes the recently developed electrochemical PDGF-BB aptasensors. The current aptasensor exhibited a wide linear range. In comparison to similar labeled and label-free gold based electrodes, noticeable improvement in the linear range has been achieved. The linear response range of envisioned aptasensor based BPE-treaded PET/Au covers the healthy levels of PDGF-BB as well as elevated levels in patients with cancer diseases.52,53 Furthermore, the proposed aptasensors are sufficiently stable. It is worth noting that the attained LoD of detection of 0.65 pM (the equivalent of 1.29 pg mL−1) is adequately lower than the minimum cut-off point of 0.1 ng mL−1 for the healthy levels of PDGF-BB in human serums.54 In general, the proposed aptasensor provides a wide linear range with efficient accuracy (i.e., limit of detection) suitable for determining PDGF-BB levels in healthy patients and patients with cancer diseases.
| Electrode | Modification | Technique | Detection strategy | LoD | Linear range | Stability | Ref. |
|---|---|---|---|---|---|---|---|
| a GCE: glassy carbon electrode; Cu-MOFs: Cu-based metal–organic frameworks; TpBD-COFs: 1,3,5-triformylphloroglucinol (Tp), benzidine (BD), covalent organic frameworks (COFs); HAP-NPs: hydroxyapatite nanoparticles; SWV: square wave voltammetry; C-MEMS: carbon-microelectromechanical systems; EIS: electrochemical impedance spectroscopy; AuNPs: gold nanoparticles. | |||||||
| GCE | Cu-MOFs/TpBD-COFs | DPV | Label-free | 0.034 pg mL−1 | 0.0001–60 ng mL−1 | N/A | 50 |
| HAP-NPs | SWV | Labeled: HAP-NPs | 50 fg mL−1 | 0.1 pg mL−1 to 10 ng mL−1 | N/A | 51 | |
| C-MEMS thin film | Oxygen plasma etching | CV | Label-free | 7 pM | 0.01–50 nM | 90.34% after 10 days | 47 |
| EIS | 1.9 pM | 0.005–50 nM | |||||
| Au | AuNPs | SWV | Labeled: α-cyclodextrin | 0.52 nM | 0.52–1.52 nM | 96.4% after 7 days | 52 |
| Graphene doped with silver nanoclusters | EIS | Label-free | 26.5 fM | 32.3 fM to 1.61 pM | 96.7% after 10 days | 53 | |
| SS | BPE rGO | DPV | Label-free | 0.53 pM | 0.005–10 nM | 91.2% after 6 days | This work |
| PET/Au | 0.65 pM | 0.0007–20 nM | |||||
The envisioned disposable biosensor was a suggested concept to study the BPE-graphene compatibility for biotechnology applications. The simplicity of electrochemical bipolar exfoliation of graphene and the environmental friendliness of the process enabled high potential for other various applications, including but not limited to biofuel cells, batteries, and supercapacitors. It is worth noting that the envisioned technology requires further studies in order to be applicable for clinical settings. Several topics can be considered such as investigating the performance of the sensor in response to real samples collected from human blood (e.g., the effect of biofoulents) and possible integration of anti-biofouling materials on to the envisioned sensor, the robustness of the sensor under harsh and uncontrolled conditions (e.g., exposure to high or very low temperatures), and the challenges related to mass-production. Furthermore, integration of sensors with AI and IoT is another interesting topic for future studies.
Conclusion
The comparative study on BPE-graphene deposited on negative and positive electrodes confirmed that the BPE-graphene deposited on the negative feeding electrode (i.e., rGO) is a more suitable candidate for biosensing applications. The SEM and TEM analysis revealed that BPE-graphene deposited on the negative feeding electrode was porous graphene with a pore size of 100 nM and consisted of single-layer graphene nanosheets. The electrochemical and biosensing performance analysis confirmed that the BPE-rGO is a more suitable active material for label-free aptasensing than BPE-GO. The fabricated label-free electrochemical PDGF-BB aptasensor based on BPE-rGO showed a noticeably better sensing performance than the similar BPE-GO based aptasensor. The PET/Au/rGOApt aptasensor showed a wide linear range of 0.0007–20 nM, high sensitivity of 4.07 μA logc−1 (unit of c, pM), and low limit of detection of 0.65 pM (S/N = 3). This aptasensor demonstrated that electrochemical bipolar exfoliation of graphene is a highly promising technique for developing highly sensitive, reliable and inexpensive biosensors for disposable sensor platforms.
Experimental
Materials and reagents
The PDGF-BB binding aptamer (ssDNA, amino linker-5′-C6-CAG GCT ACG GCA CGT AGA GCA TCA CCA TGA TCC TG-3′2) was purchased from HPLC purification from ThermoFisher Scientific, USA. Tris-ethylenediaminetetraacetic acid (TE) buffer, ethanol, acetone, phosphate-buffered saline (1 M and pH 8) (PBS), a 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride linker (EDC), a N-hydroxysuccinimide linker (NHS), hydrochloric acid (HCl), polyoxyethylene (20) sorbitan monolaurate (Tween-20), KCl, and K3Fe(CN)6 were purchased from ThermoFisher Scientific, USA. Trehalose, bovine serum albumin (BSA), platelet-derived growth factor-AA, AB, and BB were purchased from Sigma Aldrich, USA. A NANO™ SU-8 25 negative photoresist was purchased from Microchem, USA. Graphite rods (3 cm in length and 6.15 mm in diameter, Ultra “F” purity 99.9995%) were purchased from Fisher Scientific, USA. The 316 stainless steel (SS) electrodes were purchased from Maudlin Inc., USA. All chemicals were of analytical grade. Milli-Q (Sigma Aldrich, USA) deionized water was used in this study.Apparatus
A Bio-Logic versatile multichannel potentiostat (VMP3) was used for the electrochemical analysis. A JEOL SEM 6330 and Philips CM-200 FEG TEM were used to obtain the SEM and TEM images of the deposited materials, respectably. An Agilent Technologies N6705A dc Power Analyzer was used for applying a DC voltage of 45 V across the feeding electrodes. An Ag/AgCl (KCl saturated) and a Pt wire were used as the reference electrode and working electrode in all electrochemical measurements, respectively. A 713 Metrohm pH meter was used for measuring the pH of electrolytes. A JASCO FTIR 4100 was used to conduct FTIR analysis.Preparing PDGF-BB aptasensors based on BPE treated SS and PET/Au electrodes
Prior to aptamer immobilization, the BPE-treated SS and PET/Au electrodes were washed with PBS and DIW. The stock solution of binding aptamers was diluted to the desired concentration in aqueous solution of 0.1 M TE buffer to the final volume of 20 μL. Next, 20 μL of 20 mg mL−1 NHS and 10 μL of 20 mg mL−1 EDC were added to the aptamer solution and incubated at 25 °C for 30 minutes. This step is recommended in the oligonucleotide datasheet released by ThermoFisher Scientific for activating the amino tags of the aptamers. Next, the BPE-treated electrodes were dipped in a small sealed container and incubated overnight at 25 °C. Following the incubation, the electrodes were washed completely in DIW to remove any unattached binding aptamers. Next, the electrodes with the immobilized aptamer were dipped in an aqueous solution of 1% (v/v) Tween-20 + 0.1 M PBS to block the blank areas of the aptasensors.Electrochemical characterization of PDGF-BB aptasensors
The CV measurements were conducted at a voltage window of −0.8 V to 0.4 V versus the reference electrode with scan rates of 10–100 mV s−1 and cycle repeating of n = 20. DPV measurements were conducted in a voltage range of 0.2 to 0.9 V (vs. Ag/AgCl) for SS based electrodes and −0.05 to 0.5 V (vs. Ag/AgCl) for PET/Au based electrodes with a pulse height of 200 mV and pulse width of 100 ms with n = 10 numbers of repetitions. The electrolyte pH was attuned using HCl to attain electrolytes with a pH of 8–5.5.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (NSF) projects with the award no. 1611088 and 1648451. The authors would like to thank the staff members of the Advanced Materials Engineering Research Institute (AMERI) at Florida International University.References
- S. Forouzanfar, F. Alam, I. Khakpour, A. R. Baboukani, N. Pala and C. Wang, IEEE Sensor. J., 2020, 20, 8965–8972 CAS.
- S. Forouzanfar, F. Alam, N. Pala and C. Wang, J. Electrochem. Soc., 2020, 167, 067511 CrossRef CAS.
- S. K. Chatterjee and B. R. Zetter, Future Oncol., 2005, 1(1), 37–50 CrossRef CAS PubMed.
- V. Penmatsa, A. R. Ruslinda, M. Beidaghi, H. Kawarada and C. Wang, Biosens. Bioelectron., 2013, 39, 118–123 CrossRef CAS PubMed.
- B. Yi, P. J. Williams, M. Niewolna, Y. Wang and T. Yoneda, Cancer Res., 2002, 62, 917–923 CAS.
- J. Kawaguchi, S. Adachi, I. Yasuda, T. Yamauchi, T. Yoshioka, M. Itani, O. Kozawa and H. Moriwaki, Oncol. Rep., 2012, 27, 935–939 CrossRef CAS PubMed.
- J. Cheng, H. Ye, Z. Liu, C. Xu, Z. Zhang, Y. Liu and Y. Sun, J. Cell. Biochem., 2013, 114, 1510–1518 CrossRef CAS PubMed.
- A. M. Cimpean, I. M. Cobec, R. A. Ceaușu, R. Popescu, A. Tudor and M. Raica, Cancer Genomics Proteomics, 2016, 13, 511–517 CrossRef CAS PubMed.
- C. K. Lau, Z. F. Yang, D. W. Ho, M. N. Ng, G. C. Yeoh, R. T. Poon and S. T. Fan, Clin. Cancer Res., 2009, 15, 3462–3471 CrossRef CAS PubMed.
- R. Ma, Q. Yang, S. Cao, S. Liu, H. Cao, H. Xu, J. Wu and J. Feng, OncoTargets Ther., 2020, 13, 1883 CrossRef CAS PubMed.
- Y. Wang, J. Luo, J. Liu, S. Sun, Y. Xiong, Y. Ma, S. Yan, Y. Yang, H. Yin and X. Cai, Biosens. Bioelectron., 2019, 136, 84–90 CrossRef CAS PubMed.
- F. Alam, A. H. Jalal, S. Forouzanfar, M. Karabiyik, A. R. Baboukani and N. Pala, IEEE Sensor. J., 2020, 20, 5102–5109 CAS.
- S. Carvajal, S. N. Fera, A. L. Jones, T. A. Baldo, I. M. Mosa, J. F. Rusling and C. E. Krause, Biosens. Bioelectron., 2018, 104, 158–162 CrossRef CAS PubMed.
- F. Alam, A. H. Jalal, S. Forouzanfar, M. M. Hasan and N. Pala, Smart Biomedical and Physiological Sensor Technology XVIII, 2021, vol. 11757, p. 1175708 Search PubMed.
- F. Alam, M. M. Hasan, M. R. Siddiquee, S. Forouzanfar, A. H. Jalal and N. Pala, Smart Biomedical and Physiological Sensor Technology XVIII, 2021, vol. 11757, p. 117570H Search PubMed.
- Y. T. Yaman, Ö. Akbal, G. Bolat, B. Bozdogan, E. B. Denkbas and S. Abaci, Biosens. Bioelectron., 2018, 104, 50–57 CrossRef CAS PubMed.
- A. K. Kaushik, J. S. Dhau, H. Gohel, Y. K. Mishra, B. Kateb, N.-Y. Kim and D. Y. Goswami, ACS Appl. Bio Mater., 2020, 3, 7306–7325 CrossRef CAS.
- M. A. Mujawar, H. Gohel, S. K. Bhardwaj, S. Srinivasan, N. Hickman and A. Kaushik, Mater. Today Chem., 2020, 17, 100306 CrossRef CAS PubMed.
- S. Jain, M. Nehra, R. Kumar, N. Dilbaghi, T. Y. Hu, S. Kumar, A. Kaushik and C.-z. Li, Biosens. Bioelectron., 2021, 179, 113074 CrossRef CAS PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. Booth, V. Khotkevich, S. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- S. Azzouzi, L. Rotariu, A. M. Benito, W. K. Maser, M. B. Ali and C. Bala, Biosens. Bioelectron., 2015, 69, 280–286 CrossRef CAS PubMed.
- Q. Chen, T. Sun, X. Song, Q. Ran, C. Yu, J. Yang, H. Feng, L. Yu and D. Wei, Nanotechnology, 2017, 28, 315501 CrossRef PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- P. Mahanandia, F. Simon, G. Heinrich and K. K. Nanda, Chem. Commun., 2014, 50, 4613–4615 RSC.
- J. Chen, B. Yao, C. Li and G. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- S. Pei, J. Zhao, J. Du, W. Ren and H.-M. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- K. S. Rao, J. Senthilnathan, Y.-F. Liu and M. Yoshimura, Sci. Rep., 2014, 4, 1–6 Search PubMed.
- P. Yu, S. E. Lowe, G. P. Simon and Y. L. Zhong, Curr. Opin. Colloid Interface Sci., 2015, 20, 329–338 CrossRef CAS.
- A. Abdelkader, A. Cooper, R. Dryfe and I. Kinloch, Nanoscale, 2015, 7, 6944–6956 RSC.
- A. P. Kauling, A. T. Seefeldt, D. P. Pisoni, R. C. Pradeep, R. Bentini, R. V. Oliveira, K. S. Novoselov and A. H. Castro Neto, Adv. Mater., 2018, 30, 1803784 CrossRef PubMed.
- A. Allagui, M. A. Abdelkareem, H. Alawadhi and A. S. Elwakil, Sci. Rep., 2016, 6, 21282 CrossRef PubMed.
- I. Khakpour, A. Rabiei Baboukani, A. Allagui and C. Wang, ACS Appl. Energy Mater., 2019, 2, 4813–4820 CrossRef CAS.
- J. L. Suter, R. C. Sinclair and P. V. Coveney, Adv. Mater., 2020, 32, 2003213 CrossRef CAS PubMed.
- I. Khakpour, A. R. Baboukani, A. Allagui, A. A. Hachicha and C. Wang, Nanotechnology, 2021, 32, 345603 CrossRef PubMed.
- S. E. Fosdick, K. N. Knust, K. Scida and R. M. Crooks, Angew. Chem., Int. Ed., 2013, 52, 10438–10456 CrossRef CAS PubMed.
- L. Koefoed, S. U. Pedersen and K. Daasbjerg, Curr. Opin. Electrochem., 2017, 2, 13–17 CrossRef CAS.
- A. R. Baboukani, I. Khakpour, V. Drozd, A. Allagui and C. Wang, J. Mater. Chem. A, 2019, 7, 25548–25556 RSC.
- A. Allagui, J. M. Ashraf, M. Khalil, M. A. Abdelkareem, A. S. Elwakil and H. Alawadhi, ChemElectroChem, 2017, 4, 2084–2090 CrossRef CAS.
- N. R. Wilson, P. A. Pandey, R. Beanland, R. J. Young, I. A. Kinloch, L. Gong, Z. Liu, K. Suenaga, J. P. Rourke and S. J. York, ACS Nano, 2009, 3, 2547–2556 CrossRef CAS PubMed.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S.-J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS.
- E.-Y. Choi, T. H. Han, J. Hong, J. E. Kim, S. H. Lee, H. W. Kim and S. O. Kim, J. Mater. Chem., 2010, 20, 1907–1912 RSC.
- J. Coates, Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation, 2006 Search PubMed.
- V. Penmatsa, H. Kawarada, Y. Song and C. Wang, Mater. Sci. Res. India, 2014, 11, 01–08 Search PubMed.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- J. B. Allen and R. F. Larry, Electrochemical Methods Fundamentals and Applications, John Wiley & Sons, 2001 Search PubMed.
- S. Forouzanfar, F. Alam, N. Pala and C. Wang, Biosens. Bioelectron., 2020, 170, 112598 CrossRef CAS PubMed.
- X. Liu, H.-L. Shuai and K.-J. Huang, Anal. Methods, 2015, 7, 8277–8284 RSC.
- A. Shrivastava and V. Gupta, Chron. Young Sci., 2011, 2, 21–25 CrossRef.
- Y. Li, Z. Liu, W. Lu, M. Zhao, H. Xiao, T. Hu, J. Ma, Z. Zheng, J. Jia and H. Wu, Analyst, 2021, 146, 979–988 RSC.
- W. Jiang, D. Tian, L. Zhang, Q. Guo, Y. Cui and M. Yang, Microchim. Acta, 2017, 184, 4375–4381 CrossRef CAS.
- M. Hasanzadeh, N. Razmi, A. Mokhtarzadeh, N. Shadjou and S. Mahboob, Int. J. Biol. Macromol., 2018, 108, 69–80 CrossRef CAS PubMed.
- Z. Zhang, C. Guo, S. Zhang, L. He, M. Wang, D. Peng, J. Tian and S. Fang, Biosens. Bioelectron., 2017, 89, 735–742 CrossRef CAS PubMed.
- K. Leitzel, W. Bryce, J. Tomita, G. Manderino, I. Tribby, A. Thomason, M. Billingsley, E. Podczaski, H. Harvey and M. Bartholomew, Cancer Res., 1991, 51, 4149–4154 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00470k |
| This journal is © The Royal Society of Chemistry 2021 |