
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlavin-adenine-dinucleotide gold complex nanoparticles: chemical modeling design, physico-chemical assessment and perspectives in nanomedicine†
Celia
Arib
a,
Nadia
Bouchemal
a,
Maria
Barile
b,
Didier
Paleni
 c,
Nadia
Djaker
a,
Nathalie
Dupont
a and
Jolanda
Spadavecchia
*a
c,
Nadia
Djaker
a,
Nathalie
Dupont
a and
Jolanda
Spadavecchia
*a
aCNRS, UMR 7244, CSPBAT, Laboratoire de Chimie, Structures et Propriétés de Biomatériaux et d’Agents Thérapeutiques, Université Paris 13, Sorbonne Paris Nord, 1 Rue Chablis 93000, Bobigny, France. E-mail: jolanda.spadavecchia@univ-paris13.fr
bDept. of Biosciences, Biotechnology and Biopharmaceutics, University of Bari “Aldo Moro”, Via Orabona 470126, Bari, Italy
cBioEVEN start-up, 75 Rue de Lourmel 75015, Paris
First published on 19th August 2021
Abstract
Flavoproteins play an important role in the regulatory process of cell life, and they are involved in several redox reactions that regulate the metabolism of carbohydrates, amino acids, and lipids. The development of effective drug delivery systems is one of the major challenges in the fight against cancer. This study involves a nanomedicine pathway to encapsulate the cofactor flavin adenine dinucleotide (FAD) using polymeric gold nanoparticles (PEG-AuNPs) through two chemical methods of functionalization (chelation (IN); carbodiimide chemistry (ON)). These hybrid gold nanoparticles and their precursors were characterized by analytical techniques (Raman, UV-Vis, and H1-NMR spectroscopy and transmission electron microscopy (TEM)) which confirmed the grafting of the cofactor agent. The results of the computational studies (Density Functional Theory (DFT)) were in agreement with the experimental observations. We also monitored the interaction of our hybrid nanoparticle systems with small aptamers (APT) in order to validate the hypotheses on the biomolecular mechanisms and also investigate their biological efficiency on pancreatic cancer cells (MIAPaCa-2 cells).
Introduction
Flavin adenine dinucleotide (FAD) (Scheme S1 in the ESI†), synthesized from riboflavin (also known as vitamin B2), is a redox cofactor that ensures the functionality of flavoenzymes that are involved in the oxidative metabolism of carbohydrates, amino acids, and fatty acids, and in mitochondrial electron transport.1 The flavin ring system, called isoalloxazine, is the main place for the catalytic function of FAD and is used as a pharmacophore to design new drugs.2 Among flavins, only FAD has conformational flexibility and exists mainly in two forms, ‘stacked’ (closed) and ‘unstacked’ (open or extended) along with another intermediate conformer, partially stacked.3 FAD exhibits conformational versatility even inside naturally occurring flavoproteins. In some of them FAD exists in the ‘unstacked’ conformer4 and in others the ‘stacked’ conformer is preferred.5,6,7 However, reasons behind such orientations inside the protein nanocavity are scarcely available8 though researchers have successfully monitored the behavior of FAD in cells.3b,9,10 Up to now, many theoretical studies have been dedicated to the elucidation of this conformational versatility directly in relation with the FAD bioefficiency.1b,11 During oral administration, the bioavailability of FAD is highly limited because the molecule is fragile in alkaline medium and breaks down very quickly in the stomach before it can reach the bloodstream. In order to improve the bioavailability and stability of flavoenzymes, a combination with AuNPs has been considered to circumvent the therapeutic side effects. Therefore, the environmentally friendly approach of synthesizing AuNPs using natural macromolecules has gained increasing interest in recent decades.10,12a,b In recent years, J. Spadavecchia et al. developed an original synthetic route to obtain hybrid gold nanoparticles associated with drugs and/or biomolecules, and then decorated with a biopolymer according to the “IN” methodology.13–16 On the basis of these findings, J. Spadavecchia and D. Paleni et al. recently reported the role of the FAD cofactor as a hybrid theranostic complex in cancer metabolism as an adjuvant and anticancer drug.17,18 In the present study, the first aim is to examine the most recent development of novel nanomedicines for biological applications by using FAD complexed to gold ions to obtain nanoparticles (FAD IN PEG-AuNPs), compared to carbodiimide chemistry in which the cofactor (FAD) is grafted onto the surface of pegylated gold nanoparticles (FAD ON PEG-AuNPs). The experimental and theoretical results are presented in parallel. Starting from the unstacked and stacked conformations available in the bibliography, FAD alone, complexed with {Au(III)Cl2}+ (ref. 1a), in the presence of 4 to 8 gold atom small clusters to simulate the “Method IN” and associated with 1 and 2 PEG functions to simulate “Method ON” have been simulated using DFT and TD-DFT theoretical computations in implicit water solvation conditions. Molecular descriptors and electrostatic potential maps are used for fundamental reactivity hypothesis. Comparisons between the theoretical results and experimental spectroscopic UV visible and Raman data are also considered. For the second aim, we studied the interaction of our hybrid nanoparticle systems with small aptamers in order to validate the hypotheses on the bimolecular mechanisms and investigate their biomolecular efficiency after internalization in pancreatic cancer MIA PaCa-2 cells to confirm their excellent properties as a theranostic agent (third aim). This study highlights the potential of a chemical approach combined with the experimental and theoretical results to investigate FAD–polymer–gold interactions in relation to drug delivery and drug targeting activity.Results and discussion
Formation mechanism of the FAD–PEG diacid complex (FAD–PEG)
The first part of this study is taken from a recent patent and paper,18 in which we demonstrated, for the first time, the power of the FAD PEG-diacid Complex (FAD–PEG) as an adjuvant in chemotherapy. For this aim, we designed a lot of experiments in which FAD–PEG was applied as a complex in the nucleation of pegylated gold nanoparticles confirmed by HPLC chromatography (Fig. S1-A in the ESI†). The active isoalloxazine ring of flavin cofactors (FAD) induces one- or two-electron redox transitions in a wide range of biochemical reactions pertinent to cell bioenergetics, protein folding, and cellular redox homeostasis.10,11The aim of this study was to apply a FAD–PEG diacid complex as a promoter of hybrid gold nanoparticles under specific conditions of the reaction. For this purpose, in the first step, we mixed FAD molecules and PEG diacid in water solution at room temperature under specific pH conditions (pH 7.2) (Scheme 1).The absorption spectra of flavins is characterized by two intense π–π* transition bands originating from the isoalloxazine chromophore corresponding to the ground state (S0) to the S1 state (λabs: 450 nm) and S2 (λabs: 375 nm) transitions, respectively28 (blue line in Fig. 1A). When PEG diacid (PEG) interacts with the FAD molecule in aqueous solution, a chemical-physical change occurred, as shown in the UV-Vis spectra (black line in Fig. 1A). The principal optical modification in the spectra is due to the increase of both the bands (372 nm; 449 nm) towards probe encapsulation in PEG chains due to the electronic transition associated with the amino ions of FAD and diacid groups of polymers upon complexation. The DLS and potential zeta measurements confirmed the successful synthetic reaction of formulation (FAD–PEG) (Table 1). The trend of increasing intensity is due to a higher population of ‘unstacked’ conformers of FAD in PEG diacid aqueous solution.28 The spectral shift observed indicates that FAD gets encapsulated in PEG diacid. However, the positively charged FAD may intercalate at the interface region in which it can elude the repulsion from the negatively charged carboxylic group. The consequent FAD conformational change reduces the tendency of the electron transfer from the adenine moiety to the isoalloxazine ring.18,29
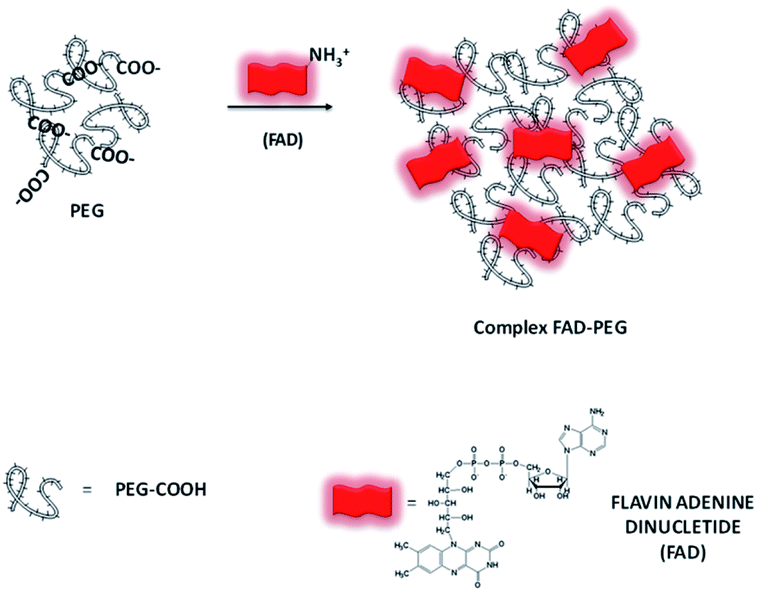 | ||
| Scheme 1 Schematic representation of the hybrid complex formation (FAD–PEG) (please note that drawings are not to scale and are not intended to be representative of the full sample composition).18 | ||
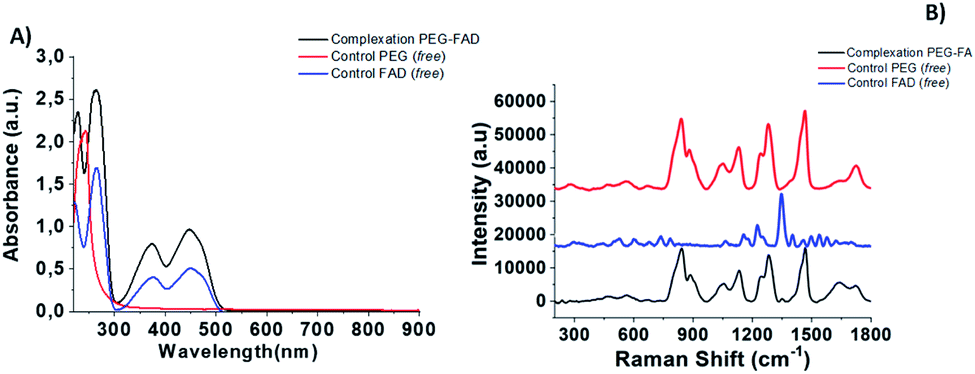 | ||
| Fig. 1 (A) Normalized UV-Vis absorption and (B) Raman spectra of PEG-FAD formulation (black line), PEG (red line) and FAD (blue line) as controls. Experimental conditions: λexc = 785 nm; laser power 20 mW; accumulation time 180 s. | ||
| Synthetic product | Zeta potential (mV) | Hydrodynamic diameter (nm) | PDI |
|---|---|---|---|
| FAD ON PEG-AuNPs | −30 ± 2 | 46 ± 1 | 0.256 |
| FAD IN PEG-AuNPs | −36 ± 1 | 91 ± 1.2 | 0.156 |
| FAD–PEG | 1 ± 2 | 152 ± 2.3 | 0.233 |
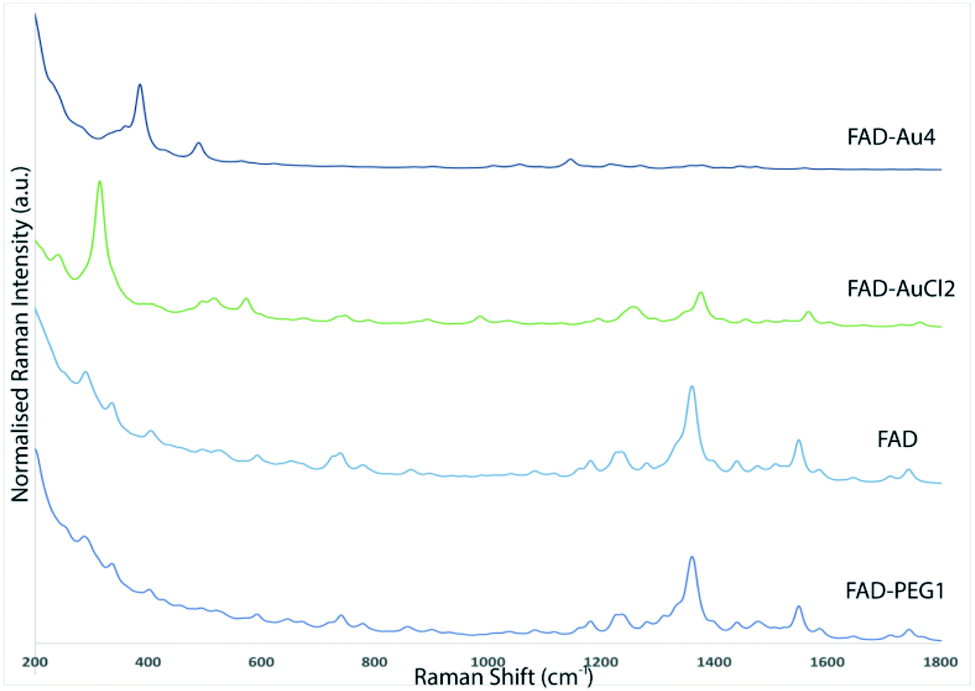
The Raman spectra of free PEG molecules showed characteristic peaks at 1137 cm−1, 1270 cm−1, and 1455 cm−1 corresponding to the vibrations of C–O–H, C–O–C and C–O chemical groups, respectively (Fig. 1B, redline). After FAD complexation, distinct bands with diverse relative intensities appear: one peak at 1120 cm−1 and 1250 cm−1 characteristic to the C–O vibration, a peak at 1330 cm−1 which corresponds to the N–O group and one at 1420 cm−1 related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group, confirming the successful complexation. The Raman spectra also confirm the spectroscopic behavior of FAD molecules as the control (Fig. 1B, blue line).
C group, confirming the successful complexation. The Raman spectra also confirm the spectroscopic behavior of FAD molecules as the control (Fig. 1B, blue line).
Formation mechanism of FAD ON PEG-AuNPs and FAD IN PEG-AuNPs
There is strong evidence in the literature that biological systems commonly reduce free metal ions.30 It is exhaustively demonstrated that the FAD-dependent enzyme glutathione reductase (GR) catalyzes the NADPH-dependent reduction of AuCl4−, forming gold nanoparticles at the active site that are tightly bound through the catalytic cysteines.30c In the last few years, Spadavecchia et al. developed two methods of synthesis, in which the first method consists of conjugating biomolecules (protein, antibody, peptides, and aptamers) onto pegylated gold nanoparticles (PEG-AuNPs) by carbodiimide chemistry (method ON).31In the second methodology (Method IN), the biomolecules are primarily complexed with gold salt in order to improve their stability, biocompatibility and efficiency.13 Actually, we studied the feasibility of FAD grafting onto the surface of PEG-AuNPs by carbodiimide chemistry (method ON) and a chelation bond to form PEGylated Au(III)-FAD (method IN) in order to improve the biochemical and optical characteristics of biomolecules. In the first method (ON), we proceed to the synthesis of PEG-AuNPs, and then FAD was grafted onto the surface of pegylated nanoparticles via the formation of amide links between the COOH groups present on the surface of the PEG-AuNPs and the NH2 groups at the 4th position of FAD prior to activation with EDC/NHS32 (Scheme 2). In the second method (IN), FAD participates in the nucleation of AuNPs through complexation and electrostatic interaction between its phosphate and amino groups with chloride auric ions (FAD–AuCl2−) and dicarboxylic pegylated chains (Scheme 3). Additional PEG-diacid increases the kinetics of reduction by complexation of Au ions, monitoring the final growth process of nanoparticles (FAD IN PEG-AuNPs) after reduction with NaBH4 (Scheme 3). Chemically, the pKa of FAD/FADH2 is equal to 6.7. At pH 7, half of the molecules are phosphated They form relatively stable radicals that participate in the activation of oxygen and reacts with thiols. During the formation of the gold complex (FAD–Au3+), the FAD molecule is in the dehydrogenate form at pH 7. After pegylation and reduction with NaBH4, FAD was embedded in a stable form in PEG diacid chains to form FAD IN PEG-AuNPs, which are more stable at acid pH. Colloid formation and growth are controlled by the amphiphilic character of the PEG and FAD molecules. The intermediate and final products of our synthetic procedure were extensively characterized by UV-Vis absorption spectroscopy, TEM, Raman Spectroscopy and 1H-NMR. Computational modeling was also carried out in order to fully understand the reaction mechanism and the interactions occurring during gold–FAD complexation and PEG addition.
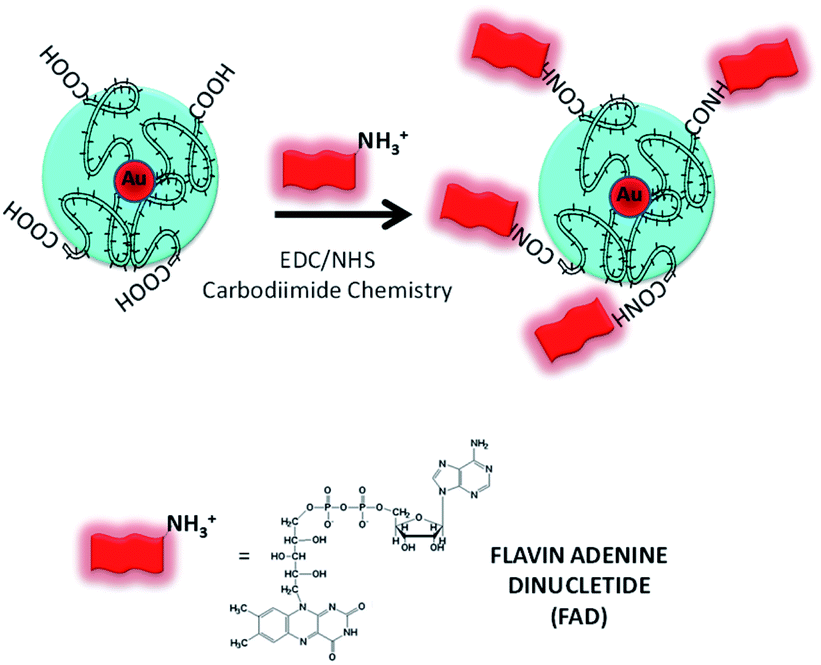 | ||
| Scheme 2 Schematic representation of the synthesis of FAD ON PEG-AuNPs via carbodiimide chemistry (please note that drawings are not to scale and are not intended to be representative of the full sample composition). | ||
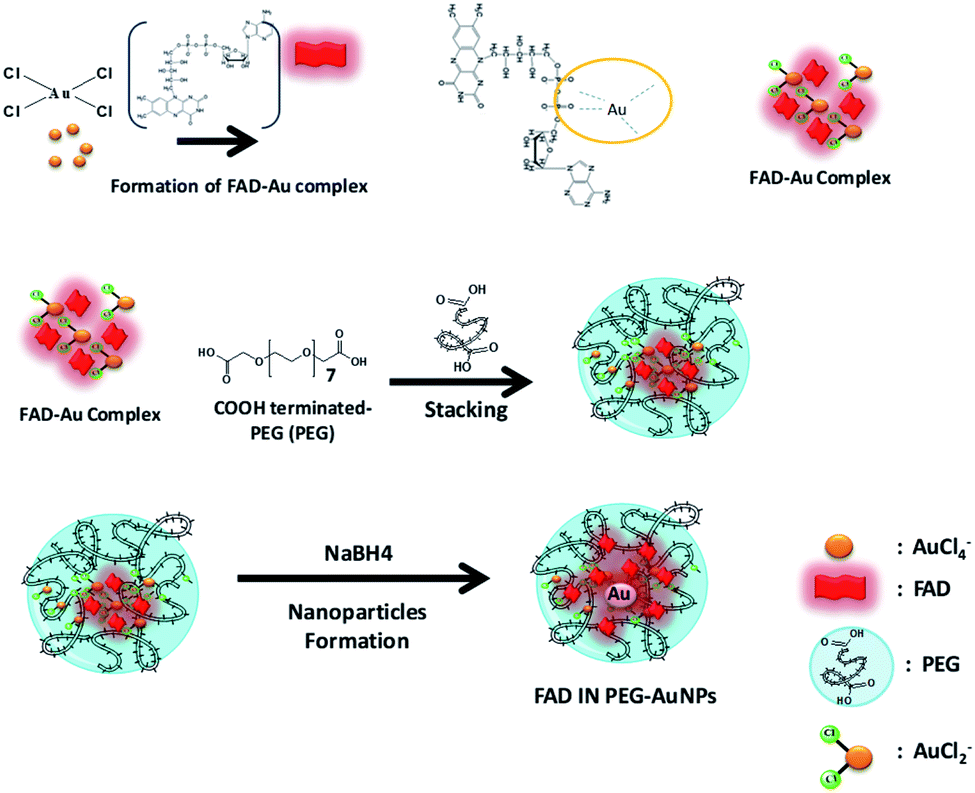 | ||
| Scheme 3 Schematic representation of the synthesis of FAD IN PEG-AuNPs via a three-step chelation procedure (method IN). (Please note that drawings are not in scale and are not intended to be representative of the full sample composition). | ||
Theoretical study on FAD–PEG diacid (FAD–PEG), FAD–AuCl3 and FAD–Au(x) complexes
Quantum chemical computations can lead to different types of results. First, we will consider the geometrical parameters and electronic features deduced from DFT optimization and HF population analysis such as the frontier orbitals HOMO and LUMO energies, energy gap, dipolar moment, global hardness, Mulliken atomic charges and molecular polarizability alpha.33 Secondly, simulations of TD-DFT UV/visible and DFT Raman spectra will be considered. All molecular files of isolated FAD, with two negative charges, in closed and open conformations based upon the Kuppuraj et al. method have been considered.11a The numbering scheme corresponds to Scheme 4.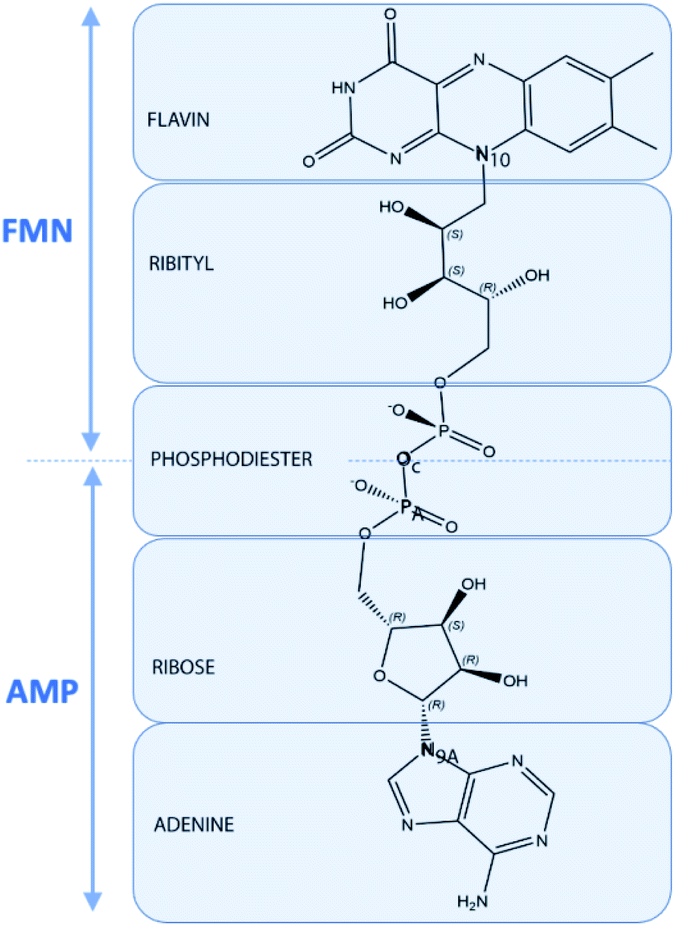 | ||
| Scheme 4 Representation of the structure of FAD in terms of FMN, AMP, flavin, isoalloxazine, ribityl, phosphodiester, ribos and adenine moieties including atom labeling used in the text. | ||
Closed FAD structures result in stacking interactions involving adenine and isoalloxazine moieties. Previous molecular dynamics studies of FAD in gas phase and in implicit or explicit water indicate that the unbound coenzyme is found to be flexible with a distance between adenine and the isoalloxazine rings ranging from 4 to 16 Å without any preference.34 As a first approach, geometrical considerations attributed to the open and closed conformations can be based on the “backbone” N9A–N10 distance and on the virtual dihedral angle N9A–PA–P–N10, corresponding respectively to the distance between adenine and the isoalloxazine rings and their orientation relative to the pyrophosphate moiety axis. Most of the closed FAD conformations exhibit N9A–N10 distances < 8 Å, whereas N9A–N10 distances > 11 Å in a range from 15 to 16 Å is preferred for open conformation. Most bound FAD structures exhibit near trans orientation, with a high dihedral virtual angle value near 180° corresponding to an opposite orientation of adenine and isoalloxazine rings relative to the PA–OC–P moiety axis. In water, the isolated FAD molecule dihedral virtual angle value fluctuates, indicating nonpreferred orientation.11a
Optimisation of different starting FAD conformations in gas phase and in implicit water gives rise to extended conformations with a N9A–N10 distance = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 588 Å and a virtual dihedral angle N9A–PA–P–N10 = 146.99° for the final optimized structure in iefpcm water used for further computations.
588 Å and a virtual dihedral angle N9A–PA–P–N10 = 146.99° for the final optimized structure in iefpcm water used for further computations.
In “Method IN ′′, the tetrachloroaurate ions AuCl4− first react with FAD forming complex reaction intermediates. The FAD–AuCl2 hypothetic structure consists in the association of FAD with AuCl2+via the phosphate moieties. The following steps of “Method IN” consist in the direct formation and growth of AuNPs by reduction in the presence of surrounded PEG molecules. To simulate the premices of “Method IN” and associated electronic effects, two different systems FAD–Au4 and FAD–Au8 are considered.
These structures combine small Au4 and Au8 clusters and a FAD molecule via two phosphate functions. Geometries of the clustered structures are directly inspired by publications.35
In “Method ON”, AuNPs are primarily prepared and stabilized in the presence of PEG molecules. Finally, FAD is added by pegylation. In this case, the electronic properties of FAD are more influenced by PEG molecules than by AuNPs. Two more systems involving one or two PEG motifs in association with the adenine part of the FAD molecule have been simulated and respectively named FAD–PEG1 and FAD–PEG2 in the following parts of the text. PEG motifs do not influence the geometrical and electronic structure compared to FAD alone. FAD–PEG1 and FAD–PEG2 give similar results and only FAD–PEG1 will be considered further in the text. As a first hypothesis, FAD electronic properties, and so the FAD activity, are supposed to be less influenced by the presence of AuNPs in “Method ON”. All the structures considered in this study are presented in Fig. 2.
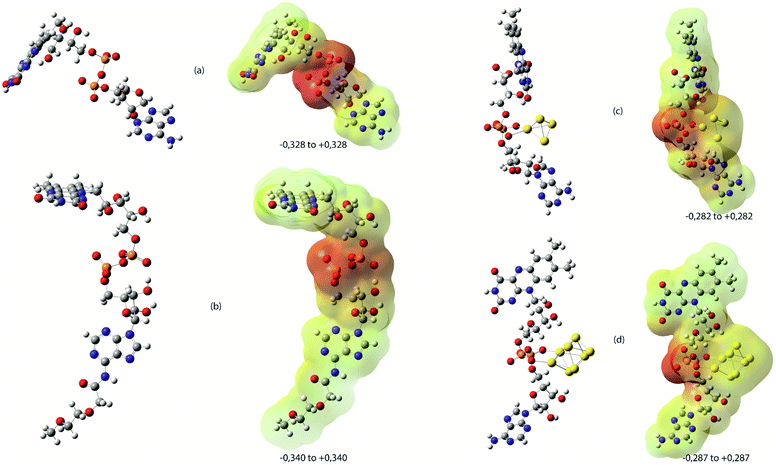 | ||
| Fig. 2 DFT optimised structures in water at the B3LYP level of theory considered in the theoretical study and the corresponding electrostatic potential mapped on total density (MESP) computed by the HF method as recommended. (a)FAD alone, (b) FAD–PEG1, (c) FAD–Au4 and (d) FAD–Au8 systems. On MESP, the electrostatic potential is represented with a colour scale from red colour corresponding to a negative value (extreme values are indicated for each MESP representation in blue colour corresponding to the most positive value and yellow color for neutral values). | ||
All chemical descriptors are available in Tables S-B1 and S-B2 given in the ESI.† As an intermediate species in “Method IN”, FAD–AuCl2 will be only considered for UV-Vis and Raman spectral comparisons.
Considering the geometrical parameters, method IN and method ON have no significant effect. All structures exhibit N9A–N10 distances > 11 Å corresponding to extended conformations. Considering the virtual dihedral angle N9A–PA–P–N10, FAD–PEG1 (173.35°) and FAD–Au8 (173.65°) exhibit a clear trans conformation contrary to FAD (146.99°) and FAD–Au4 (129.596°).
Considering the electronic features, relevant characteristics imply differences in terms of chemical reactivity between the “Method IN” and “Method ON” complexed form of FAD–AuNPs. The dipolar moment in Debye results from a non-uniform charge distribution on the various atoms in a given molecule. It is frequently considered to describe the intermolecular non-bonded interactions such as dipole–dipole interactions. The dipole moment of FAD–PEG1 (17.6172D) is approximately half of the FAD dipole moment (31.4391D). In the presence of small gold clusters, the dipole moments tend to increase up to 41.7459D in the case of FAD–Au4 and then decrease to a value of 25.7008D in FAD–Au8.
As polarizability measures the ability of electron density to be influenced by an external stimulus, the harder a molecule is, the lower is its average polarizability. Computed total α polarizabilities evolve from approx 700 Bohr3 for harder systems FAD and FAD–PEG to more than 1200 Bohr3 for FAD–Au4 and FAD–Au8. In the presence of small gold clusters, FAD systems tend to soften with the electron density more easily influenced by an external stimulus. The frontier molecular orbitals, the HOMO and LUMO, guide the interaction of a molecule with other species. A representation of the HOMO and LUMO is available in Fig. S-B1 to S-B4.†
In FAD, FAD–PEG and FAD–Au systems, the LUMO is located on the flavin moiety with an energy level near −3.1 eV. The smaller the value of the LUMO energy, the smaller the resistance to accept electrons.
In FAD, the HOMO is located on the adenine moiety with an energy level of −6.27 eV. In FAD–PEG1, the HOMO is located in the isoalloxazine moiety as the LUMO with an energy level of 6.56 eV. In FAD–Au4 (−4.24 eV) and FAD–Au8 (−4.75 eV), the HOMO is located on the gold metal cluster with higher values of HOMO energies related to the greater ease of donating electrons to the unoccupied orbital of a receptor. According to Koopman's theory, the ionization energy I and electron affinity A can be directly expressed through absolute values of HOMO and LUMO energies, respectively. The gap ΔE in energy between the HOMO and LUMO is an important stability index and thus an indicator of chemical reactivity. According to the hard–soft acid–base principle of Pearson,33d the hardness η is directly related to the gap energy. Thus, FAD–Au4 and FAD–Au8 are supposed to be more reactive and softener than FAD and FAD–PEGs. The electronegativity index and electrophilicity index also decrease in the presence of gold clusters in the direct proximity of FAD.
Considering Mulliken charges computed for some atoms, the PA atom tends to be more positive in the presence of gold clusters (1.636 in FAD, 1.695 in FAD–Au4, and 1.703 in FAD–Au8); the same phenomenon is observed for the P atom (1.642 in FAD, 1.684 in FAD–Au4, and 1.726 in FAD–Au8) whereas N9A tends to slightly become less negative (−0.586 in FAD, −0.569 in FAD–Au4, and −0.573 in FAD–Au8).
The molecular electrostatic potential surface MEPS is a plot of the electrostatic potential mapped on the total density surface (Fig. 2). Different values of the electrostatic potential on the surface are represented by a colour scale: red colour represents a negative electrostatic potential, or major electrophilic centers, whereas the most positive electrostatic potential regions are supposed to be represented in blue, and yellow regions represent a neutral electrostatic potential. Red regions are located in the pyrophosphate moiety with a regular circular repartition in the case of FAD and FAD–PEG1 while the red region is located on the opposite part of the gold cluster in FAD–Au4 and FAD–Au8. Raman spectra have been simulated and are represented with nonscaled frequencies and normalised relative intensities in Fig. 3.
 | ||
| Fig. 3 Graphical superimposition of the calculated Raman spectra at the B3LYP/6-311G(d,p)/LANL2DZ level of theory for FAD–PEG1, FAD, FAD–AuCl2, and FAD–Au4 from the bottom to the top of the figure. Frequencies are not scaled. | ||
It is noticeable that interactions between AuNPs and PEG are not taken into account in our simplified simulation protocol.
FAD and FAD–PEG1 Raman spectra are totally superimposable and not easily discernible. Two fingerprints are available in the FAD–AuCl2 spectrum: O–Au–Cl vibrations appear around 317 cm−1 and a doublet relative to the O–P–O–Au–Cl region appears around 520 and 580 cm−1. When following FAD-IN AuNP synthesis, these fingerprints are supposed to appear instead of the case of the “Method ON” protocol when no FAD–AuCl2 intermediate species are supposed to exist.
Two fingerprints are available in the FAD–Au4 spectrum: O–Au–P–O vibrations around 390 cm−1 and Au–O stretching vibrations around 500 cm−1. In “Method ON”, the first step consists in the formation of AuNps in the presence of PEG molecules with the appearance of such O–Au bands in the same regions around 400 cm−1 and 500 cm−1. Thus FAD pegylation does not imply any relevant modification of the Raman spectra at lower frequencies.
TD-DFT UV computed visible transition spectra are presented in Fig. 4 with arbitrary normalised intensities. Charge transfer is not considered in the simulation.
 | ||
| Fig. 4 Graphical superimposition of the calculated UV visible spectra at the TD-DFT/RB3LYP/6-311G(d,p)/LANL2DZ level of theory for FAD–PEG2, FAD–PEG1, FAD, FAD–AuCl2, FAD Au4 and FAD–Au8 from the bottom to the top of the figure. | ||
FAD, FAD–PEG1 and FAD–PEG2 spectra are totally superimposable with a more intensive electronic transition of about 420 nm and not easily discernible. Details of the electronic transitions are given in Tables S-B3 to S-B7† and OM involved in the most intensive transitions are presented in Fig. S-B1 to S-B4.† In the presence of gold atoms, a red shift of the more intensive transition is observed up to 600 nm and can be considered as the fingerprint of the FAD–IN AuNP system.
FAD grafted nanoparticles: physicochemical evaluation
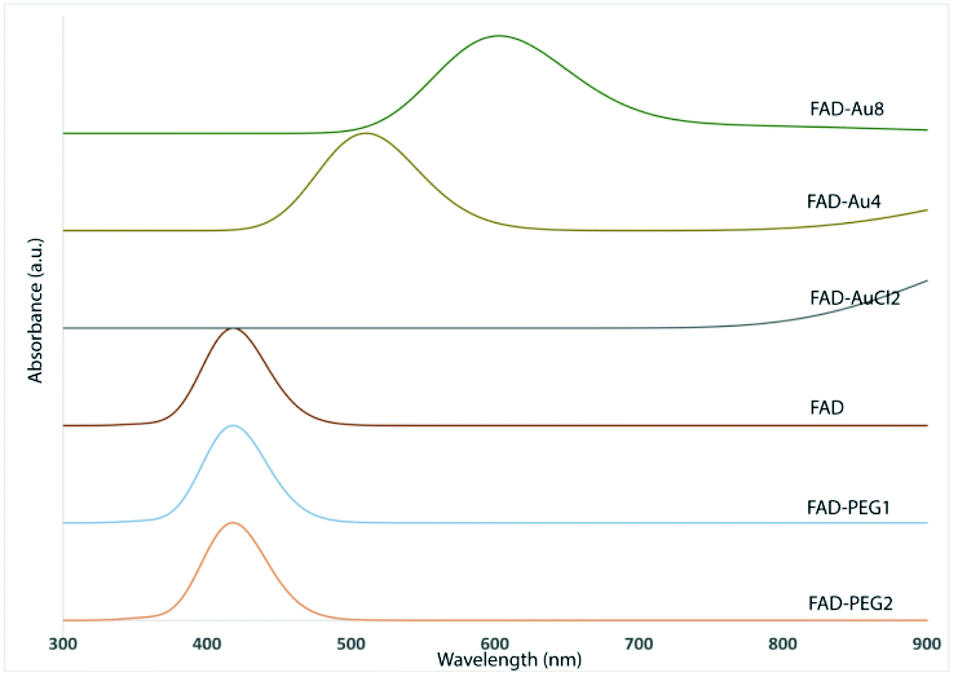
Previously, D. Schott et al.30c proved that flavoenzymes employ an electron-transfer mechanism to reduce Au3+ ions to form a gold nanoparticle tightly bound to the active site. Other authors have shown that enzymes can catalyze independent reactions whose products reduce metals in the solution.36 These cofactors, participate in the nucleation and growth of redox active metallic nanoparticles that are protected by sequestration in the enzyme active site.The UV-visible spectra of PEG-AuNPs showed a surface plasmon band at 533 nm (Fig. 5A, red line). After FAD grafting onto the PEG-AuNPs (FAD ON PEG-AuNPs) through carbodiimide chemistry, the plasmon band is red-shifted to 543 nm (Fig. 5A, blue line). The red-shift can be explained by the modification of the dielectric environment of the nanoparticles, thus corroborating the successful grafting of FAD onto the surface of AuNPs. Concerning the complexation (method IN), the red shift is higher (to 555 nm) (Fig. 5A, pink line).The red-shifted spectrum also suggests the electron transfer between AuCl4− and FADH2. The reoxidation of FADH2 by AuCl4− is rapid (over within 5 s), suggesting the direct electron transfer from the active site bound flavin to AuCl4−. These results confirm the change of the steric conformation of FAD with the chelation method.
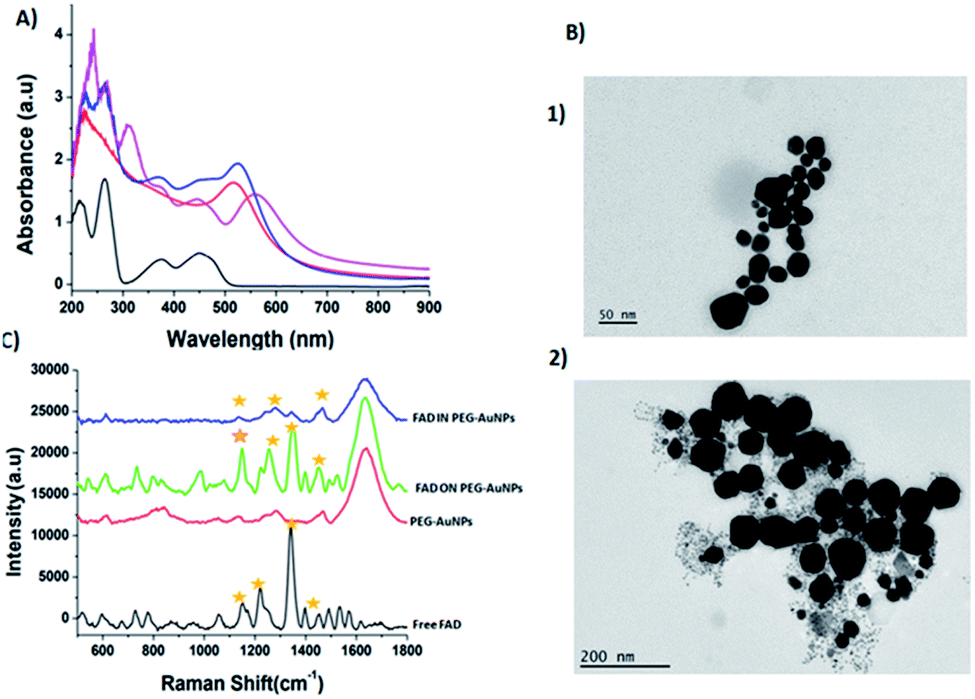 | ||
| Fig. 5 (A) Normalized UV-Vis absorption spectra of PEG-AuNPs (red line), FAD ON PEG-AuNPs (blue line) and FAD IN PEG-AuNPs (pink line). (B) TEM images (scale bars: 50 nm; 200 nm) of FAD ON PEG-AuNPs (panel 1) and FAD IN PEG-AuNPs (panel 2); (C) Raman spectra of FAD ON PEG-AuNPs (green line) and FAD IN PEG-AuNP (blue line) products compared to those of free PEG-AuNPs (red line) and FAD free (black line) as controls. The yellow label inset shows the characteristics peaks and the success of reaction. Experimental conditions: λexc = 660 nm; laser power 20 mW; accumulation time 180 s. | ||
This performance has a direct relationship with the steric conformation of the FAD molecule on the PEG-AuNP surface.16 With the carbodiimide (EDC/NHS) functionalization, FAD is situated directly on the gold surface decorated with a PEG layer, while for the chelation method, FAD will be in the core of gold, between the surface embedded by the PEG layer. Moreover the UV-Vis spectra of FAD IN PEG-AuNPs remain unaltered after storage at room temperature, confirming the formation of stable colloidal solution (Fig. S1-B in the ESI†). The TEM images of FAD ON PEG-AuNPs (Fig. 5B panel 2) and FADIN PEG-AuNPs (Fig. 5B panel 1) show a spherical shape, with a diameter around 46 ± 1 nm for FAD ON PEG-AuNPs and 91 ± 1.2 nm for FAD IN PEG-AuNPs, compared with PEG-AuNPs as the control31a with an average size of 20 ± 1 nm and the positively charged FAD–PEG complex of about 152 ± 2.3 nm (Table 1).Therefore, we assumed that FAD IN PEG-AuNPs are a better candidate in terms of stability. For this purpose we chose FAD IN PEG-AuNPs to proceed to the next experiments on active targeting and drug release.
The Raman spectroscopy results also showed the successful functionalization of FAD onto and/or into PEG-AuNPs (Fig. 5C). The Raman spectra of free gold nanoparticles (PEG-AuNPs) display few bands attributed to the PEG molecules (the broad band perceived around 1600 cm−1 on the Raman spectra is attributed to water). The detection of the fingerprint of PEG-COOH on the AuNP surface was displayed by the Raman bands at 1137 cm−1, 1270 cm−1, and 1455 cm−1, corresponding to the vibrations of C–O–H, C–O–C and C–O chemical groups, respectively (Fig. 5C red line). After FAD grafting, novel bands appeared for both grafting methods used (Fig. 5C green line (FAD ON PEG-AuNPs); Fig. 5C blue line (FAD IN PEG-AuNPs)). Distinct bands of the grafted AuNPs but with diverse relative intensities: two peaks at 1120 cm−1 and 1250 cm−1 specific to the vibration of the C–O group, one at 1330 cm−1 which corresponds to the N–O group and one at 1420 cm−1 specific to the CC group. These bands correspond to FAD. If we compare the Raman results of the bioconjugation of FAD with PEG-AuNPs (FAD ON PEG-AuNPs; FAD IN PEG-AuNPs), it affirms that the chemical and steric change of FAD depending of the grafting method affects the conformation of FAD. Specifically, the bands are more marked in FAD ON PEG-AuNPs compared to FAD IN PEG-AuNPs, affirming a different steric disposition of FAD.
1H-NMR characterization
Fig. S2 in the ESI† shows respectively, the 1H NMR spectra of FAD (a) and the complexed PEG/FAD–AuNPs (b).The total 1H and 13C assignment of FAD is presented in Table S1 in the ESI.†
As shown in Fig. S2 and S2 bis extension1,† the major change between the FAD spectra (a) and the PEG/FAD–Au spectra (b) is located in the aromatic peak area (7.55 to 8.3 ppm), suggesting that FAD was complexed to Au by forming bonds between the aromatic protons of FAD. This is confirmed by the measure of the relaxation time T1 (reported in Table S1†) where the values are larger for aromatic protons of PEG/FAD–AuNP compared to those of FAD alone.
One of the possibilities offered by NMR is its ability to assess all molecular motions over a very wide time range, at the atomic scale. This dynamic study can be related to the size of the studied structure. Longitudinal relaxation is related to energy exchanges between nuclei and their environment. It is characterized by the longitudinal relaxation time T1. The relaxation rate R1, inversely proportional to T1, depends on the speed of the rotational movements of the molecule. As the size of the molecule increases, relaxation increases. Thus, the measurement of the relaxation parameters makes it possible to obtain information on the movements of the molecules and therefore on their size.
Therefore, in our study, measurements of T1 relaxation times show a slowing of movement of FAD in the presence of PEG-Au, indicating that FAD and PEG could be very “close” to each other.
Unfortunately, the typical peaks corresponding to PEG were also observed at 3.64 (s, 14H) and 3.66 to 3.70 ppm (m, 4H), at the same chemical shift area of FAD, hiding the peaks of a part of the molecule.
No change was observed between the FAD–Au complex and PEG-FAD–Au spectra (not shown here), confirming that covalent bonds are not implicated in the reaction process.
FAD loading and release of FAD IN PEG-AuNPs
The loading ratio of FAD into PEG-AuNPs (FAD IN PEG-AuNPs) was confirmed by the absorption peaks at 450 nm and 375 nm, characteristic of FAD (ref NTO). The loading ability was estimated to be 68% with 8 μg present in 3.6 × 10−7 mol of NPs. FAD IN PEG-AuNP releases are pH- and time-dependent. As previously discussed for other biomolecules and drugs,13,37 the FAD release under acidic conditions is due to the protonation of carboxylate groups of PEG molecules at acidic pH. In our case, we assume that the ionic interaction of FAD with PEG chains enhances the water solubility of the FAD molecules and consequently their release.Previous studies13 have showed the difference in the chemical conformation and drug release on varying the pH from 7.0 to 4.0. Thus, the FAD IN PEG–AuNPs were characterized by UV-visible (S3 A-A1 in the ESI†) and Raman spectroscopy (S3 B-B1 in the ESI†) following incubation for 72 h in PBS at 37 °C in order to confirm the (1) drug release and (2) assess variations in the chemical orientation of FAD at pH 4.0 (when FAD is protonated, positively charged, and carrying the NH3+ group) (S3 A1-B1 in the ESI†) and pH 7.0 (S3 A-B in the ESI†) (when FAD is partially deprotonated at the amino group with pKa 8.2). The Raman bands at 1120 and 1420 cm−1 were supervised to assess FAD release from AuNPs. The intensity of the band at 1420 cm−1 decreases over time after 72 h (S3 B 1 in the ESI†). These results were confirmed by UV visible and also showed that FAD release was pH and time dependent. We conclude that, under acidic conditions, FAD release is faster.
Aptamer targeting (APT-FAD IN PEG–AuNPs): spectroscopic characterization and biomolecular hypothesis
Aptamers (APTs) are functional small single-stranded oligonucleotides (DNA) that exhibit high affinity to target molecules such as proteins or small analytes.38 Based on the specificity and the binding affinity, aptamers can be used in industrial and medical applications.39 To improve the therapeutic applications of our nanoparticles, it's necessary to study the interaction between the aptamer and FAD. Previously, we studied the interaction between a specific aptamer and protein manganese superoxide dismutase (MnSOD) as a liver cancer biomarker.40 Herein, we report the behavior of aptamer molecules on FAD pegylated gold nanoparticles (FAD IN PEG-AuNPs) after interaction. Under basic conditions, FAD IN PEG-AuNPs were negative charged and reacted with aptamers by electrostatic interactions between NH3+ and the carboxylic group of pegylated polymers.The Aptamer (APT) binding to PEG-AuNPs and FAD IN PEG-AuNPs was confirmed by UV-Vis and Raman spectroscopy (Fig. 6 A and B). As first, APT conjugation onto PEG-AuNPs as the control displays a red-shift of the absorption band from 533 nm (Fig. 6A black line) to 555 nm (Fig. 6A blue line), and a second band at 260 nm assigned to the APT was observed. The successful conjugation of the APT onto FAD IN PEG-AuNPs (Fig. 6A red line) was validated by a strong decrease of the UV-Vis absorption spectra of FAD IN PEG-AuNPs with the disappearance of the characteristic peaks of FAD molecules at 450 nm and 375 nm, respectively. Several spectral differences can be noticed in the Raman spectra before (black line) and after the APT grafting (red line) onto FAD IN PEG-AuNPs (Fig. 6B). We observe the bands at 820 cm−1 and 1058 cm−1, typical to the sugar and phosphate groups (PO43−) or a peak at 1371 cm−1 which corresponds to the thymine base specific to the aptamer31b (Fig. 6B red line).
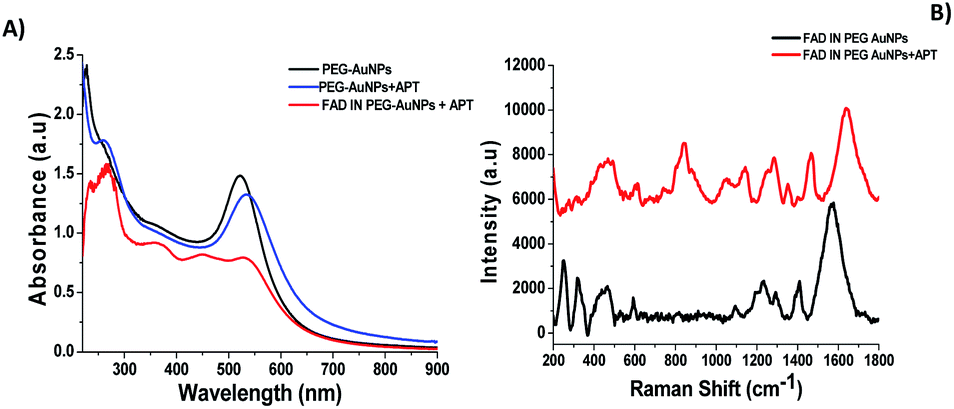 | ||
| Fig. 6 (A) Normalized UV-Vis absorption of FAD IN PEG-AuNPs + APT (red line), PEG-AuNPs + APT (blue line) and PEG-AuNPs (black line) as controls; (B) Raman spectra of FAD IN PEG-AuNPs (black line) and FAD IN APT PEG-AuNPs (red line). Experimental conditions: λexc = 785 nm; laser power 20 mW; accumulation time 180 s. | ||
Its high intensity is due to the good alignment of the aptamer with the Au surface of nanoparticles,41 contrary to the lower signal recorded for the case of the free thymine DNA in water.31b After grafting of the APT onto FAD IN PEG AuNPs, we also observed a disapparition of a double peak in the range of 200–300 cm−1, corresponding to the Au–O–C and Au–Cl, O–O stretching vibrations. This spectroscopic behavior confirms a different steric and chemical configuration of the APT on FAD IN PEG-AuNPs and its interaction with FAD molecules. Fig. 7A shows a dynamic behaviour concerning the interaction of the APT and FAD IN PEG-AuNPs within 24 h. We can observe a progressive decrease of the UV-Vis absorption spectra of FAD IN PEG-AuNPs with a progressive disappearance of peaks at 450 and 350 nm, characteristic of FAD after 24 h. This behavior will be depicted as the biomolecular intercalation and interaction process within FAD IN PEG AuNPs and APT molecules with the consequent release of the FAD–Au complex under experimental conditions (Fig. 7B).
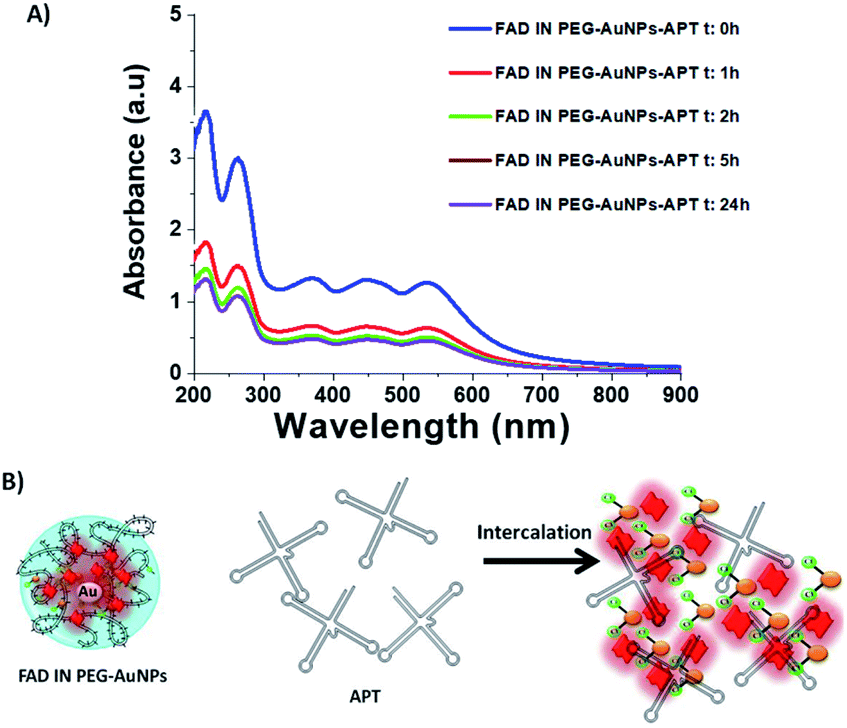 | ||
| Fig. 7 (A) UV Vis absorption spectra of FAD IN PEG-AuNPs and dynamic behaviour of the interaction of the APT and FAD IN PEG-AuNPs within 24 h; (B) schematic biomolecular intercalation and interaction process within FAD IN PEG AuNPs and APT molecules. | ||
We hypothesise that when our NPs (FAD IN PEG-AuNPs) interact with DNA strands the tautomeric alloxazine mutations are reduced and break the bond with adenine (Scheme 5).
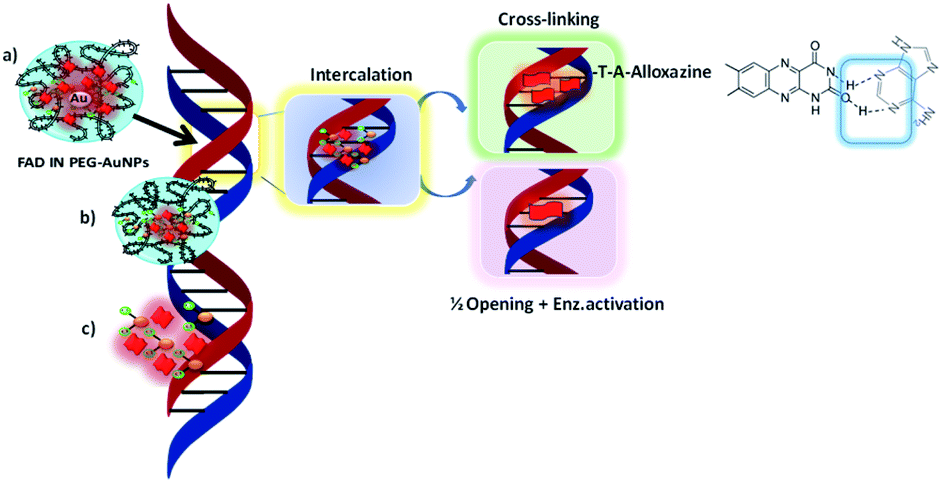 | ||
| Scheme 5 Schematic hypothesis of intercalation between FAD IN PEG-AuNPs and DNA strands. | ||
FAD chelated to pegylated gold nanoparticles (FAD IN PEG-AuNPs) assumes a different steric and chemical configuration compared to free FAD. As a consequence, FAD can modify the allosteric characteristics as a vector of enzymatic reactions, such as the activation of the ubiquitin-catalyst targeting the destruction of proteins, which is known to bind to histone H2A or to activate transcription by RNA polymerase.42 These inter-chain bridges can be established at the level of poly A DNA sequences43 through rapid positioning on DNA, and feeders would allow high gene amplification and the synthesis of ribosomal segmental DNA. Other types of chemical bonds of DNA can be envisaged, both at the level of different bases and in the configuration of the bridge: anti complementary bonds and single stranded bridges. Similarly, nicotinamide–dinucleotides can establish pH-dependent bridges by N+–O− ionic bonding with the phosphate groups of the opposite strand, or with the end groups of protein modulating chromosomal activity.
Cytotoxicity of FAD gold complex nanoparticles in pancreatic cells
FAD is a redox cofactor guaranteeing the activity of many flavoenzymes situated in the mitochondria but also pertinent to nuclear redox characteristics.1a Isoalloxazine is a part of mono and dinucleotides FMN and FAD notable in the transport of hydrogen.44 Previously it has been hypnotized that in eukaryotes FAD biosynthesis occurred only in the cytosol.45 However, using cell fractionation procedures and enzymatic activity measurements, M. Barile et al. established the presence of FAD activity in the mitochondria of a rat liver.1a More recently, the existence of several FAD isoforms with specific subcellular localization has been corroborated by biochemical approaches. Thanks to its capacity to interact with the genetic code, FAD is considered as a nucleo-cytoplasmatic mediator.46 In order to evaluate the potential cytotoxic effect of hybrid gold FAD nanoparticles, pancreatic cells, MIA PaCa-2 cells, were incubated in a medium with pegylated nanoparticles (PEG-AuNPs i.e. control), FAD–PEG, FAD–AuCl4 as complex-gold-complex precursors and hybrid gold nanoparticles, respectively (FAD ON PEG-AuNPs; FAD IN PEG-AuNPs).The therapeutic agent FAD was present and the final concentration was 40 μM. Cell viability was maintained for up to 48 hours.The cytotoxicity of FAD IN PEG-AuNPs and FAD ON PEG-AuNPs in MIA PaCa-2 cells was measured using the 3-(4,5-dimethyl-2- thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) test (Fig. 8). We found significant cell death of MIA PaCa-2 cells when treated with FAD IN PEG-AuNPs (88%), FAD ON PEG-AuNPs (85%) and FAD–PEG (86%). On the other side, there is no inhibition of proliferation for the exposed cells to FAD alone and PEG-AuNPs (at the same concentration to that of the nanoparticle). All these experiments were carried out at a fixed concentration of FAD which is 40 μM. We have fixed 40 μM as this is the concentration of FAD usually used in clinics. Fig. 8 shows that FAD–PEG, FAD IN-PEG-AuNPs and FAD ON-PEG-AuNPs at 40 μM concentration of FAD inhibit the proliferation of pancreatic cancer cells. We can observe that the cell death with FAD–PEG, FADON-PEG-AuNPs and FADIN-PEG-AuNPs is about 89%, whereas the cell death with FAD alone is only 43%.
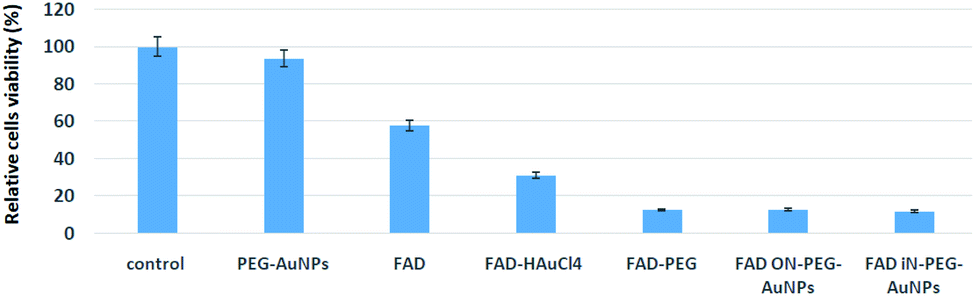 | ||
| Fig. 8 Cytotoxicity of FAD IN PEG AuNPs and FAD ON PEG AuNPs in MIA PaCa-2 cells. | ||
The efficiency of FAD ON-PEG-AuNPs and FAD IN-PEG-AuNPs can be due to the facile release of the drug (FAD) inside the cells as the drug is encapsulated in AuNPs and PEG.
To sum up, the cytotoxicity results of FAD ON-PEG-AuNPs, FAD IN-PEG-AuNPs and FAD–PEG were translated to cell viability. The results show that FAD is more efficient when it's complexed with PEG or encapsulated in AuNPs. The main interesting result from the cytotoxicity study is that cell death with FAD complexed with or encapsulated in AuNPs is 57/11 = 5 times more efficient than that with FAD alone, demonstrating the high potential provided by the PEG and AuNPs to vectorize the antitumoral molecules into the cells.
We assume that after nanoparticle cell internalization, FAD was released into the cytoplasm as the FAD–Au complex throughout migration in the PEG chains, blocking the binding of aminoacyl-tRNA to the mRNA-ribosome complex with noticeable alteration in the driving force and electrolytic conditions. Consequently, the synergic effect of FAD as a complex improves the inhibition of glucose 6 phosphate dehydrogenase47 and simultaneous repair and consequently the improvement of anticancer activity (Scheme 6).
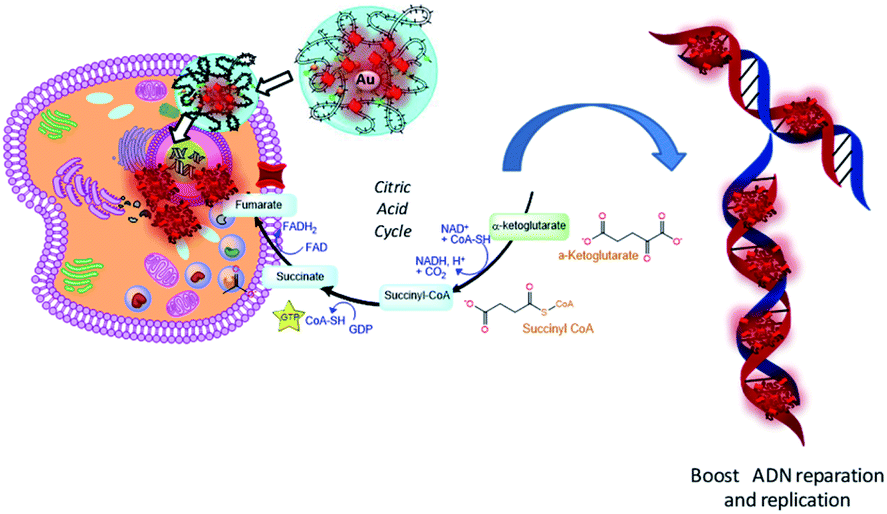 | ||
| Scheme 6 Schematic hypothesis of FAD IN PEG-AuNPs and FAD–PEG diacid complex effects on the metabolic and glycolysis pathway in cancer cells. | ||
Experimental
Materials and methods
Tetrachloroauric acid (HAuCl4·6H2O), sodium borohydride (NaBH4), dicarboxylic poly ethylene glycol (PEG)-600 (PEG-diacid), N-hydroxysuccinimide (NHS), 1-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), phosphate-buffered saline (PBS), and flavin adenine dinucleotide (FAD) are provided by FISHER SCIENTIFIC at maximum purity grade. All solvents were used without any further purification. Experiments were carried out at room temperature if not specified otherwise.Aptamer (APT)
The sequence of the aptamer purchased from Eurogentec and dispersed in PBS buffer (pH = 9) is as follows: 5′HS-(C6)-TTTTTTTTTTTTTTTT-CTTCTCTAGCTGAATAACCGGAAGTAACTCATCGTTTCGATGAGTTACTTCCGGTTATTCAGCTAGAGAAG 3′.The spacer included here is only to increase the distance of the active part of the aptamer from the gold surface and provide higher flexibility to the active part of the aptamer.
Formulation of the FAD–PEG diacid complex (FAD–PEG)
The synthesis of the FAD–PEG complex was recently described18Synthesis of pegylated gold nanoparticles (PEG-AuNPs)
The synthesis of PEG-AuNPs was described previously.19FAD grafting onto nanoparticles (FAD ON PEG-AuNPs and FAD IN PEG-AuNPs)
In this study, we used two strategies of bioconjugation in order to evaluate the different steric arrangements of FAD molecules onto pegylated gold nanoparticles (PEG-AuNPs).FAD grafting onto PEG-AuNPs by carbodiimide cross-linker chemistry (FAD ON PEG-AuNPs)
The first grafting strategy consists of the immobilization of the FAD molecules on the PEG-AuNP surface by carbodiimide bonds19 according to the previously described procedure (Scheme 1). Briefly, 20 μL of EDC/NHS (40/10 w/w ratio) aqueous solution was added into 4.5 mL PEG-AuNP dispersion (42 nM). After 40 min, 0.5 mL of FAD solution (0.4 mM in water MiliQ) were added in 5 mL of the reaction mixture and stirred for 2 h at room temperature. The FAD ON PEG-AuNPs thus obtained were centrifuged two times at 9.000 rpm for 30 min to remove excess non-conjugated FAD.FAD grafting into PEG-AuNPs by chelation chemistry (FAD IN PEG-AuNPs)
The synthesis of FAD IN PEG-AuNP colloids included three main steps. 20 mL of HAuCl4 aqueous solution (2.5 × 10−4 M) was added to FAD solution (2.5 mL, 0, 4 mM in water) and aged for 10 min. After 10 min, 250 μL of dicarboxylic PEG (PEG-COOH) was added and mixed by magnetic stirring for 10 min at room temperature. Finally, 600 μL of aqueous 0.01 M NaBH4 was added at once. The formation of the FAD IN PEG-AuNPs was observed as an instantaneous colour change of the solution from pale yellow to bright pink purple after addition of the reducing agent. Products of each synthetic step were stored at 27–29 °C and characterized by UV-Vis spectroscopy, transmission electron microscopy (TEM), and Raman spectroscopy. The “as-prepared” FAD IN PEG-AuNP solution was centrifuged at 9.000 rpm for 15 min three times; then, the supernatant was discarded. The pellet was redispersed two times in an equivalent amount of water to remove excess non-conjugated dicarboxylic PEG and FAD, respectively.Stability of FAD IN PEG-AuNPs in DMEM
The stability of FAD IN PEG-AuNPs and FAD ON PEG-AuNPs was monitored by UV Vis spectroscopy. All nanoparticles were dissolved in Dulbecco’s modified Eagle's medium (DMEM) and stored for 72 h (Fig. S1B in the ESI†).Physico-chemical characterization
All the measurements were performed in triplicate in order to validate the reproducibility of the synthetic and analytical procedures.UV/Vis absorption
The absorption spectra were recorded using a PerkinElmer Lambda UV/Vis 900 spectrophotometer in plastic cuvettes with an optical path of 10 mm. The wavelength range was 200–900 nm.Transmission electron microscopy (TEM)
TEM images were acquired with a JEOL JEM 1011 microscope (JEOL, USA) at an accelerating voltage of 100 kV. 2 μL of the particle suspension was placed on a carbon coated copper grid (Smethurst High-Light Ltd) and dried at room temperature.Raman Spectroscopy
The Raman spectra were recorded on an Xplora spectrometer (Horiba Scientifics-France) using an excitation wavelength of 785 nm (diode laser) at room temperature. For measurements in solution, a macro-objective with a focal length of 40 mm (NA = 0.18) was used in the backscattering configuration. The achieved spectral resolution is close to 2 cm−1.1H-NMR
NMR experiments were conducted on a Bruker AVANCE III spectrometer operating at 500 MHz with a 5 mm gradient indirect detection probe, at a probe temperature of 300 K. For FAD, FAD–AuCl4, FAD–PEG and FAD IN PEG-AuNPs, 500 μL aliquots were directly mixed with 100 μL deuterium oxide (D2O) 99.96% (Eurisotop) and 50 μL DSS like internal reference. The samples were placed in 5 mm diameter tubes for 1H-NMR analysis. Chemical shifts (ν, in ppm) were determined with 1D and 2D experiments expressed relative to the NMR signal DSS (internal reference). To measure the relaxation parameter T1 (R1 = 1/T1), the pulse sequence used here is an inversion recovery classical sequence.20Dynamic light scattering (DLS)
The size measurements were performed using a Zetasizer NANOZS (Malvern Instruments, Malvern, UK) equipped with a He–Ne laser (633 nm and a fixed scattering angle of 173°) at room temperature (see the ESI†).Zeta potential measurements
The zeta potential of all hybrid gold nanoparticles dispersed in water was measured using the electrophoretic mode of a Zetasizer NANOZS (Malvern Instruments Ltd, UK) (see the ESI†).FAD loading efficiency
The amount of FAD incorporated into FAD–PEG polymeric chains and FAD IN-PEG-AuNPs was measured by UV-Vis absorption spectroscopy. The absorption at 450 nm and 376 nm was used to extrapolate FAD concentrations based on a calibration curve. The FAD loading efficiency was calculated as follows (eqn (1)):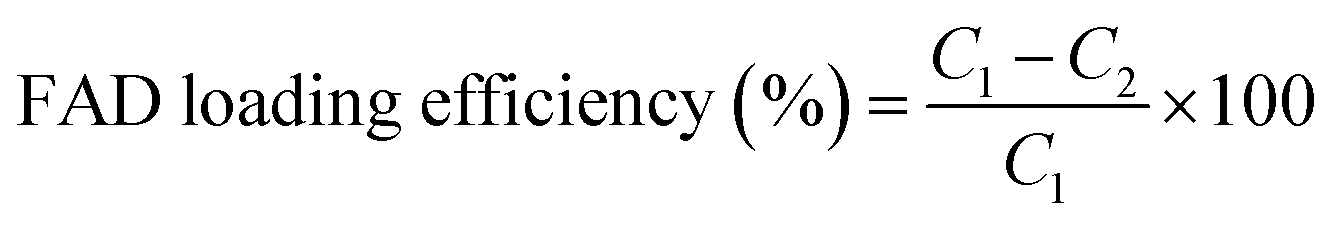 | (1) |
000 NMWL, (Millipore, USA).
HPLC chromatography
All HPLC measurements were performed at Pr. Maria Barile's laboratory (University of Bari).FAD release from the FAD–PEG diacid-complex and FAD IN PEG-AuNPs
FAD release was evaluated at a physiological temperature (37 °C). FAD IN PEG-AuNPs were dispersed at a concentration of 1.5 × 1010 particles per mL in 2.5 mL PBS. Dialysis was performed with membrane tubing with dialysis tubing Spectra/Por 3 (molecular weight cut-off 3500 Da, Serva Electrophoresis, Germany) with continuous stirring (150 rpm). FAD release was evaluated in a 24-fold donor/acceptor volume ratio to mimic the significant dilution of the suspension in the organism under physiological conditions. pH was adjusted to 7.0 or 4.0 to evaluate the pH-dependence on the drug release. To measure the amount of FAD released, 1 mL was periodically removed from the dialysis bath and replaced with an equivalent volume of PBS. The amount of released FAD was determined by UV-Vis absorption spectroscopy at 450 nm and 376 nm. The concentration of FAD released from FAD IN PEG-AuNPs was expressed as a percentage of the total FAD present in the sample (eqn (2)) and plotted as a function of time. | (2) |
DFT calculations
All computations have been realized according to previously described procedures.13,15All molecular calculations have been performed primarily in the gas phase and achieved in implicit water using the IEFPCM continuum model,21 as implemented in the Gaussian 16 suite of program.22 All geometries were fully optimized without any symmetry constraints using density functional theory at the hybrid functional B3LYP level.23 The B3LYP method with the 6-311g (d,p) basis set is commonly used in predicting the vibrational spectra of polyaromatic molecules.24
The standard split valence basis set 6-311G (d,p) was used for all atoms, except for Au, which was treated by the relativistic effective core potential (ECP) and associated valence double-ξ basis set of Hay and Wadt (abbreviated as LANL2DZ).25 After structural optimization, vibrational frequencies and Raman scattering were computed at the same level of density functional theory. It has been checked that no negative frequencies have been observed, indicating a realistic final optimized structure.
GaussSum 3.0 (ref. 26) has been used to generate Raman intensities with a 785 nm excitation wavelength and full width at a half-maximum parameter of 20 cm−1. UV-Vis transitions were finally evaluated in the gas phase and in water with a PCM model using the time-dependent (TD) approach27 at the same level of DFT. In addition, electrostatic potential (ESP) and the corresponding contour maps (MESP) have been considered for the structures using the HF method. In order to emphasize the differences between the different atomic sites, a red to blue colored scale has been considered from the negative to positive electrostatic potential values.
Please note, only relevant results are mentioned in the main text, whereas most of the computed data are reported in the part S-Bas of the ESI.†
In vitro biological activity of FAD IN PEG-AuNPs
Cells were either left under basal conditions or exposed to PEG-AuNPs, FAD ON PEG-AuNPs (40 μM) and FAD IN PEG-AuNPs (40 μM) for 48 hours at 37 °C to assess cytotoxicity using 3-(4,5-dimethyl-2-410 thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay.
Conclusions
In our study, we conceived a new hybrid nanovector based on the immune cofactor flavin adenine dinucleotide (FAD), comparing two chemical methods of functionalization (method IN and method ON). The successful chemical methodology was assessed by analytical techniques (Raman, UV-Vis, and 1H-NMR spectroscopy and transmission electron microscopy (TEM)). By considering simplified molecular systems in order to discern electronic and geometrical effects of method IN and method ON, theoretical studies have given a hypothesis concerning the difference in terms of chemical reactivity between FAD IN PEG-AuNPs and FAD ON PEG-AuNPs. In FAD IN PEG-AuNPs-, neighbouring gold atoms tend to increase the polarizabilty of the systems and thus their ability to be influenced by an external electronic stimulus. There is also a decrease in the energy gap and thus a higher reactivity. Computations of Raman and UV/visible electronic transition spectra confirm the presence of specific fingerprints, observed in the experimental data, for the “Method IN” protocol, involving a lower frequency Raman band around 317 cm−1 and a red shift for the main transition. We also investigate their biological efficiency in pancreatic cancer cells (MIAPaCa-2 cells). The next step, in a future paper, consists in molecular dynamics simulations of different systems, involving aptamers, to have a better understanding of the evolution and interactions in time and space. Finally, the design and assessment of FAD hybrid gold nanoparticles as nanovectors open new possibilities in drug-delivery and cancer therapy.Author contributions
Celia Arib: experimental synthesis and physical–chemical and biological charcterization; Nadia Bouchemal: 1H-NMR characterization; Maria Barile: HPLC measurements and interpretation; Didier Paleni: concept; Nadia Djaker: Raman spectroscopy interpretation; Nathalie Dupont: modeling study and redaction; Jolanda Spadavecchia: chemical design, concept and redaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All computations were done on the Sorbonne Paris Nord University MAGI supercomputer. The authors thank Nicolas Greneche for his help for computing job launching and management. We also thank the NMR-PF facility (University Sorbonne Paris Nord, France) and BIOEVEN s.r.l. for financing Celia Arib (CA)'s thesis.Notes and references
- (a) T. A. Giancaspero, G. Busco, C. Panebianco, C. Carmone, A. Miccolis, G. M. Liuzzi, M. Colella and M. Barile, J. Biol. Chem., 2013, 288, 29069–29080 CrossRef CAS PubMed; (b) J. C. Molano-Arevalo, D. R. Hernandez, W. G. Gonzalez, J. Miksovska, M. E. Ridgeway, M. A. Park and F. Fernandez-Lima, Anal. Chem., 2014, 86, 10223–10230 CrossRef CAS PubMed.
- (a) D. Schaller, D. Šribar, T. Noonan, L. Deng, T. N. Nguyen, S. Pach, D. Machalz, M. Bermudez and G. Wolber, WIREs Computational Molecular Science, 2020, 10, e1468 CAS; (b) I. Lans, E. Anoz-Carbonell, K. Palacio-Rodríguez, J. A. Aínsa, M. Medina and P. Cossio, PLoS Comput. Biol., 2020, 16(8), e1007898 CrossRef CAS PubMed.
- (a) G. Weber, Biochem. J., 1950, 47, 114–121 CrossRef CAS PubMed; (b) S. D. M. Islam, T. Susdorf, A. Penzkofer and P. Hegemann, Chem. Phys., 2003, 295, 137–149 CrossRef CAS.
- (a) J. J. Kim, M. Wang and R. Paschke, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 7523–7527 CrossRef CAS PubMed; (b) J. S. Grinstead, S.-T. D. Hsu, W. Laan, A. M. J. J. Bonvin, K. J. Hellingwerf, R. Boelens and R. Kaptein, ChemBioChem, 2006, 7, 187–193 CrossRef CAS PubMed; (c) S. D. Pegan, M. Sturdy, G. Ferry, P. Delagrange, J. A. Boutin and A. D. Mesecar, Protein Sci., 2011, 20, 1182–1195 CrossRef CAS PubMed.
- R. Pokorny, T. Klar, U. Hennecke, T. Carell, A. Batschauer and L.-O. Essen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 21023–21027 CrossRef CAS.
- J. Brazard, A. Usman, F. Lacombat, C. Ley, M. M. Martin, P. Plaza, L. Mony, M. Heijde, G. Zabulon and C. Bowler, J. Am. Chem. Soc., 2010, 132, 4935–4945 CrossRef CAS PubMed.
- Y. Huang, R. Baxter, B. S. Smith, C. L. Partch, C. L. Colbert and J. Deisenhofer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17701–17706 CrossRef CAS PubMed.
- A. Sengupta, W. D. Sasikala, A. Mukherjee and P. Hazra, ChemPhysChem, 2012, 13, 2142–2153 CrossRef CAS PubMed.
- M. S. Islam, M. Honma, T. Nakabayashi, M. Kinjo and N. Ohta, Int. J. Mol. Sci., 2013, 14, 1952–1963 CrossRef CAS PubMed.
- Y. Wu, L. Li, L. Frank, J. Wagner, P. Andreozzi, B. Hammer, M. D'Alicarnasso, M. Pelliccia, W. Liu, S. Chakrabortty, S. Krol, J. Simon, K. Landfester, S. L. Kuan, F. Stellacci, K. Müllen, F. Kreppel and T. Weil, ACS Nano, 2019, 13, 8749–8759 CrossRef CAS PubMed.
- (a) G. Kuppuraj, D. Kruise and K. Yura, J. Phys. Chem. B, 2014, 118, 13486–13497 CrossRef CAS PubMed; (b) P. A. W. F. Van den Berg, Y.-Q. Wang, A. E. Mark, H. J. C. Berendsen and A. J. W. G. Visser, J. Phys. Chem. B, 2002, 106, 8858–8869 CrossRef CAS; (c) M.-Y. Li, Y.-Q. Wang, Y.-L. Ying and Y.-T. Long, Chem. Sci., 2019, 10, 10400–10404 RSC.
- (a) Z. A. Nurakhmetova, A. N. Azhkeyeva, I. A. Klassen and G. S. Tatykhanova, Polymers, 2020, 12, 2625 CrossRef CAS; (b) Q. Liu, F. Aouidat, P. Sacco, E. Marsich, N. Djaker and J. Spadavecchia, Colloids Surf., B, 2020, 185, 15 Search PubMed; (c) V. Torresan, D. Forrer, A. Guadagnini, D. Badocco, P. Pastore, M. Casarin, A. Selloni, D. Coral, M. Ceolin, M. B. Fernández van Raap, A. Busato, P. Marzola, A. E. Spinelli and V. Amendola, ACS Nano, 2020, 14(10), 12840–12853 CrossRef CAS PubMed; (d) L. Li, S. Fu, C. Chen, X. Wang, C. Fu, S. Wang, W. Guo, X. Yu, X. Zhang, Z. Liu, J. Qiu and H. Liu, ACS Nano, 2016, 10(7), 7094–7105 CrossRef CAS PubMed.
- H. Moustaoui, D. Movia, N. Dupont, N. Bouchemal, S. Casale, N. Djaker, P. Savarin, A. Prina-Mello, M. L. de la Chapelle and J. Spadavecchia, ACS Appl. Mater. Interfaces, 2016, 8, 19946–19957 CrossRef CAS PubMed.
- M. Monteil, H. Moustaoui, G. Picardi, F. Aouidat, N. Djaker, M. L. de La Chapelle, M. Lecouvey and J. Spadavecchia, J. Colloid Interface Sci., 2018, 513, 205–213 CrossRef CAS PubMed.
- C. Barbey, N. Bouchemal, P. Retailleau, N. Dupont and J. Spadavecchia, ACS Omega, 2021, 6, 1235–1245 CrossRef CAS PubMed.
- C. Arib, J. Spadavecchia and M. L. de la Chapelle, Sci. Rep., 2021, 11, 3208 CrossRef CAS PubMed.
- (a) J. S. Didier Paleni, PCT national yer, PCT/EP2019/079693 2019; (b) J. S. Didier Paleni, WO International, WO/2020/089310, 2020.
- C. Arib, H. Liu, Q. Liu, A.-M. Cieutat, D. Paleni, X. Li and J. Spadavecchia, Nanotheranostics, 2021, 5, 405–416 CrossRef PubMed.
- J. Spadavecchia, D. Movia, C. Moore, C. M. Maguire, H. Moustaoui, S. Casale, Y. Volkov and A. Prina-Mello, Int. J. Nanomed., 2016, 11, 791–822 CrossRef CAS PubMed.
- M. A. Kennedy and P. D. Ellis, Concepts Magn. Reson., 1989, 1, 35–47 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Molecular Modeling and Synthesis of Ethyl Benzyl Carbamates as Possible Ixodicide Activity, Wallingford, C. T., 2016 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (c) A. D. Becke, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS.
- G. Shakila, S. Periandy and S. Ramalingam, J. At., Mol., Opt. Phys., 2011, 2011, 512841 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- A. Sengupta, K. Gavvala, R. K. Koninti, H. Chaudhuri and P. Hazra, Chem. Phys. Lett., 2013, 584, 67–73 CrossRef CAS.
- D. F. A. R. Dourado, M. Swart and A. T. P. Carvalho, Chem. –Eur. J., 2018, 24, 5246–5252 CrossRef CAS PubMed.
- (a) T. Barkay, S. M. Miller and A. O. Summers, FEMS Microbiol. Rev., 2003, 27, 355–384 CrossRef CAS PubMed; (b) J. R. Lloyd, D. R. Lovley and L. E. Macaskie, Adv. Appl. Microbiol., 2003, 53, 85–128 CAS; (c) D. Scott, M. Toney and M. Muzikár, J. Am. Chem. Soc., 2008, 130, 865–874 CrossRef CAS PubMed.
- (a) J. Spadavecchia, D. Movia, C. Moore, C. M. Maguire, H. Moustaoui, S. Casale, Y. Volkov and A. Prina-Mello, Int. J. Nanomed., 2016, 11, 791–822 CrossRef CAS; (b) R. Dekhili, K. Cherni, H. Liu, X. Li, N. Djaker and J. Spadavecchia, ACS Omega, 2020, 5, 13851–13859 CrossRef CAS PubMed.
- J. Bolley, E. Guenin, N. Lievre, M. Lecouvey, M. Soussan, Y. Lalatonne and L. Motte, Langmuir, 2013, 29, 14639–14647 CrossRef CAS PubMed.
- (a) P. Sarmah and R. C. Deka, J. Mol. Model., 2010, 16, 411–418 CrossRef CAS PubMed; (b) P. Sarmah and R. C. Deka, J. Comput.-Aided Mol. Des., 2009, 23, 343–354 CrossRef CAS PubMed; (c) W. Boufas, N. Dupont, M. Berredjem, K. Berrezag, I. Becheker, H. Berredjem and N.-E. Aouf, J. Mol. Struct., 2014, 1074, 180 CrossRef CAS; (d) R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- J. Hahn, M.-E. Michel-Beyerle and N. Rösch, J. Mol. Model., 1998, 4, 73–82 CrossRef CAS.
- (a) M. Khatun, R. S. Majumdar and A. Anoop, Frontiers in Chemistry, 2019, 7, 644 CrossRef CAS PubMed; (b) S. Goel, K. A. Velizhanin, A. Piryatinski, S. Tretiak and S. A. Ivanov, J. Phys. Chem. Lett., 2010, 1, 927–931 CrossRef CAS; (c) L. Achou, Y. Al-Sayad and A. Doghmane, Nano-Struct. Nano-Objects, 2019, 19, 100376 CrossRef CAS.
- I. Willner, R. Baron and B. Willner, Adv. Mater., 2006, 18, 1109–1120 CrossRef CAS.
- H. Liu, P. Jiang, Z. Li, X. Li, N. Djaker and J. Spadavecchia, Part. Part. Syst. Charact., 2018, 35, 1800082 CrossRef.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS PubMed.
- P. Röthlisberger, C. Gasse and M. Hollenstein, Int. J. Mol. Sci., 2017, 18, 2430 CrossRef PubMed.
- C. Arib, Q. Liu, N. Djaker, W. Fu, M. Lamy de la Chapelle and J. Spadavecchia, Plasmonics, 2019, 14, 1029–1038 CrossRef CAS.
- H. He, J. Dai, Z. Duan, B. Zheng, Y. Meng, Y. Guo and X. Dan, Sci. Rep., 2016, 6, 30878 CrossRef CAS PubMed.
- M. S. Santisteban, M. Hang and M. M. Smith, Mol. Cell. Biol., 2011, 31, 1848–1860 CrossRef CAS.
- A. Srivastava, R. Timsina, S. Heo, S. W. Dewage, S. Kirmizialtin and X. Qiu, Nucleic Acids Res., 2020, 48, 7018–7026 CAS.
- J. T. Pinto and A. J. L. Cooper, Adv. Nutr., 2014, 5, 144–163 CrossRef CAS PubMed.
- E. M. Torchetti, C. Brizio, M. Colella, M. Galluccio, T. A. Giancaspero, C. Indiveri, M. Roberti and M. Barile, Mitochondrion, 2010, 10, 263–273 CrossRef CAS PubMed.
- M. Darguzyte, N. Drude, T. Lammers and F. Kiessling, Cancers, 2020, 12, 295 CrossRef CAS PubMed.
- R. C. Stanton, IUBMB Life, 2012, 64, 362–369 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00444a |
| This journal is © The Royal Society of Chemistry 2021 |